
Mannich-type allylic C–H functionalization of enol silyl ethers under photoredox–thiol hybrid catalysis†
Tsubasa
Nakashima
,
Kohsuke
Ohmatsu
 and
Takashi
Ooi
*
and
Takashi
Ooi
*
Institute of Transformative Bio-Molecules (WPI-ITbM) and Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Chikusa, Nagoya 464-8601, Japan. E-mail: tooi@chembio.nagoya-u.ac.jp
First published on 23rd September 2020
Abstract
The synergy of an Ir-based photosensitizer with mild oxidizing ability and a thiol catalyst enables efficient allylic C–H functionalization of enol silyl ethers with imines under visible light irradiation. Subsequent transformations of the aminoalkylated enol silyl ethers allow for the facile construction of unique molecular frameworks such as functionalized octahydroisoindol-4-one.
Introduction
Aminocarbonyls serve as valuable building blocks for the assembly of biologically active and medicinally relevant organic molecules, especially those containing this structural motif as the core component.1 Hence, there is strong demand for the development of reliable yet practical methods for rapid access to a diverse array of nitrogen-containing carbonyl compounds in the fields of synthetic chemistry and pharmaceutical science.2 One of the most versatile reaction manifolds in this regard is the Mannich-type bond connection, including vinylogous Mannich addition, wherein a wide variety of carbonyl nucleophiles and imine electrophiles can be employed for the construction of β- or δ-aminocarbonyl architectures.3To access γ-aminocarbonyl compounds via the nucleophilic addition of carbonyl compounds to imines, activation of the inherently electrophilic or inert carbonyl β-carbons as nucleophiles is necessary. Although this mode of bond formation is attractive given the potential diversity in the structures of accessible γ-aminocarbonyl compounds, research on the development of strategies for enabling the requisite nucleophilic activation of carbonyl β-carbons has met with limited success.4,5 A representative means for implementing the nucleophilic activation of carbonyl β-carbons relies on the polarity inversion of α,β-unsaturated aldehydes using N-heterocyclic carbenes (NHC) as catalysts.4 Activation of the β-sp3 carbons in saturated carbonyl substrates was also achieved, although the scope was limited to β-arylpropionic acid esters (Scheme 1a).5
 | ||
| Scheme 1 (a) NHC-catalyzed β-Mannich-type addition–cyclization reaction; (b) β-Mannich-type reaction by photoredox–enamine catalysis; (c) allylic Mannich-type addition of enol silyl ethers. | ||
Recently, the synergy of photoredox and other molecular catalysts has created a basis for devising methodologies that allow for the formal activation of carbonyl β-carbons. In the seminal contribution by MacMillan and coworkers, the single-electron oxidation of an enamine and subsequent deprotonation of the resulting radical cation generated an intermediate with a nucleophilic carbon radical at the allylic position.6 This strategy has been successfully applied to establish β-Mannich-type reactions of saturated cyclic ketones with imines (Scheme 1b).7 On the other hand, we have introduced a photoredox–Brønsted base co-catalyzed allylic C–H alkylation of enol silyl ethers with electron-deficient olefins.8 One of the salient features of this protocol is the broad substrate scope, as a series of cyclic and acyclic ketone-derived enol silyl ethers undergo allylic C–H alkylation to afford β-functionalized ketone derivatives. Herein, we extend the allylic C–H functionalization strategy to the reaction of enol silyl ethers with imines (Scheme 1c). The present transformation and subsequent derivatization of the resulting functionalized enol silyl ethers provide facile access to a range of γ-aminocarbonyl compounds.
Results and discussion
For the preliminary experiment, we selected cyclohexanone-derived enol silyl ether 1a and N-methanesulfonyl imine 2a as model substrates and subjected them to the reaction conditions previously optimized for the allylic C–H alkylation (Scheme 2).8 Thus, [Ir(dFCF3ppy)2(4,4′-dCF3bpy)]PF6 (4a in Fig. 1, 2 mol%) and 2,4,6-collidine (10 mol%) were employed as the photoredox and Brønsted base catalysts, respectively, to generate an allylic radical from 1a (Eox = 1.52 V vs. SCE) via a single-electron oxidation–deprotonation sequence in the presence of 2a as a radical acceptor in 1,2-dichloroethane under visible light irradiation. In this trial, however, the desired Mannich-type allylic C–H functionalization did not occur at all. We inferred that the addition of the 1a-derived allylic radical to 2a would be unfavorable because of the relative instability of the nitrogen-centered radical generated after carbon–carbon bond formation.9 Because aromatic substituents with electron-donating groups effectively stabilize nitrogen radicals,9aN-aryl imines, such as N-PMP imines, would be more suitable electrophilic partners for promoting the nucleophilic radical addition. Nevertheless, the corresponding products bearing an N-arylamino functionality would have lower oxidation potential than that of enol silyl ethers (the oxidation potential of anilines is typically below 1.0 V vs. SCE),10 thus being incompatible with the catalysis of 4a . These assumptions led us to reconsider the catalytic system for realizing the Mannich-type allylic C–H alkylation of enol silyl ethers.
. These assumptions led us to reconsider the catalytic system for realizing the Mannich-type allylic C–H alkylation of enol silyl ethers.
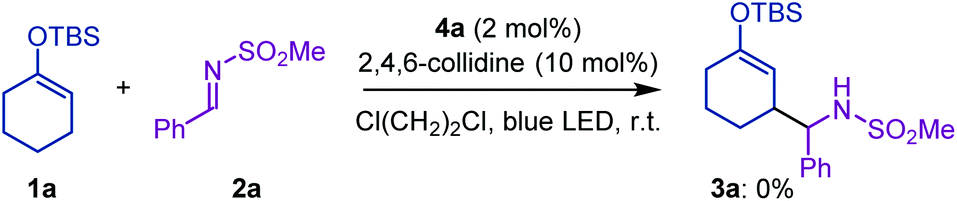 | ||
| Scheme 2 Preliminary investigation of Mannich-type C–H alkylation of enol silyl ether. | ||
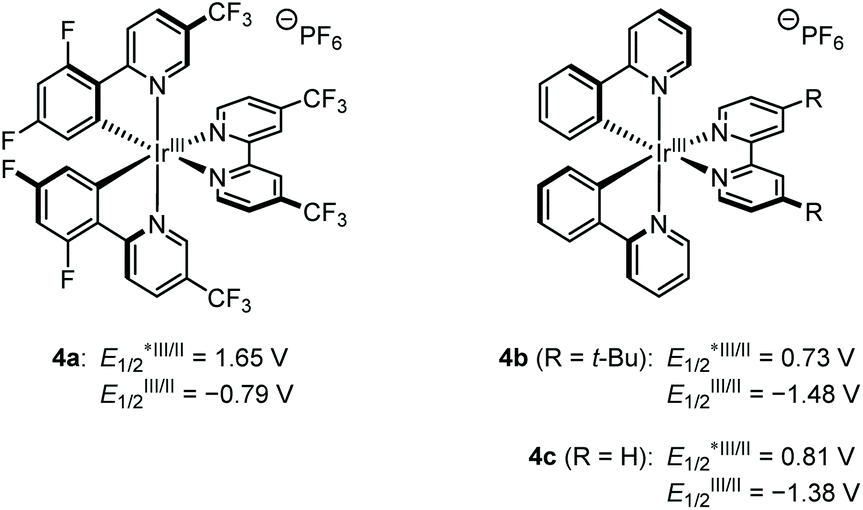 | ||
| Fig. 1 Photoredox catalysts used in this study. | ||
Considering the weak bond dissociation energy of allylic C–H bonds (about 80 kcal mol−1)11 and the leading examples of photoredox-catalyzed hydrogen-atom transfer (HAT) C–H functionalization with imines,12,13 we envisioned that a hybrid of a photosensitizer with mild oxidizing ability and a thiol catalyst would be effective for the generation of an allylic radical from enol silyl ethers via HAT, while accommodating N-aryl imines as acceptors. Toward this end, we attempted the reaction of 1a with N-PMP imine 2b under the influence of [Ir(ppy)2(dtbbpy)]PF6 (4b) (2 mol%), triisopropylsilylthiol (20 mol%), and 2,4,6-collidine (10 mol%) in acetonitrile under irradiation by a blue LED, which involved the catalytic generation of a thiyl radical as a HAT-active species via proton-coupled electron transfer (Table 1, entry 1).14 As expected, this attempt afforded the desired aminoalkylation product 3b in a moderate yield (46%). The parallel reaction of 1a with N-sulfonyl imine 2a under otherwise identical conditions did not furnish the product (entry 2), confirming the appropriate reactivity of N-aryl imines. Subsequent evaluation of the effect of the Brønsted base revealed that lithium acetate significantly improved the reaction efficiency (entries 3–6).12a On the other hand, switching the photocatalyst to 4c, which is a slightly stronger oxidant in its excited state and a weaker reductant in its reduced form, led to a notable decrease in the reaction conversion (entry 7). A decrease in the yield of 3b was also observed when the reaction was conducted with methyl thioglycolate as a thiol catalyst (entry 8). Screening of solvents revealed that acetonitrile was the optimal choice (entries 9–11). Eventually, we found that slightly increasing the amount of 1a (1.5 equiv.) had a beneficial impact on the reactivity profile, furnishing 3b in a satisfactory yield (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | 4 | Base | Solvent | Yieldb (%) |
a Unless otherwise indicated, the reactions were performed with 0.12 mmol of 1a and 0.10 mmol of 2b with photocatalyst 4 (2 mol%), i-Pr3SiSH (20 mol%), and base (10 mol%) in solvent (1.0 mL) at ambient temperature under an argon atmosphere with light irradiation (blue LED, 750 W m−2).
b
NMR yield with styrene as an internal standard. The value in parentheses is the isolated yield. Diastereomeric ratio (d.r.) of 3b ranged from 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1.2:1.
c With 2a instead of 2b.
d With MeO2CCH2 SH instead of i-Pr3SiSH.
e With 0.15 mmol of 1a. :1 to 1.2:1.
c With 2a instead of 2b.
d With MeO2CCH2 SH instead of i-Pr3SiSH.
e With 0.15 mmol of 1a.
|
||||
| 1 | 4b | 2,4,6-Collidine | MeCN | 46 |
| 2c | 4b | 2,4,6-Collidine | MeCN | 0 |
| 3 | 4b | KOAc | MeCN | 21 |
| 4 | 4b | NaOAc | MeCN | 43 |
| 5 | 4b | LiOAc | MeCN | 66 |
| 6 | 4b | Li2CO3 | MeCN | 16 |
| 7 | 4c | LiOAc | MeCN | 41 |
| 8d | 4b | LiOAc | MeCN | 20 |
| 9 | 4b | LiOAc | Acetone | 49 |
| 10 | 4b | LiOAc | PhCF3 | 11 |
| 11 | 4b | LiOAc | EtCN | 53 |
| 12e | 4b | LiOAc | MeCN | 87 (79) |
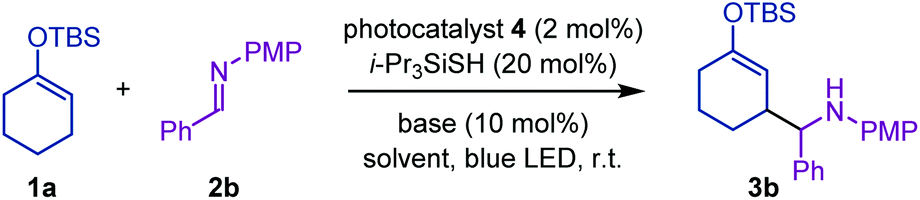
With the optimized reaction conditions in hand, we next explored the substrate scope of this Mannich-type C–H alkylation of enol silyl ethers. As shown in Fig. 2, N-PMP imines bearing ortho-, meta-, or para-methylphenyl substituents underwent radical addition to give the corresponding products 3c–3e in moderate to high yields. Sterically demanding mesityl-substituted imine and fused-aromatic imines, typically 2-naphthylimine, were also amenable to the present hybrid catalytic system, affording 3f and 3g, respectively. Imines with additional functional groups such as ether, thioether, and pyridine proved to be good substrates for furnishing the Mannich-type adducts 3h–3k in moderate to good yields.15 Moreover, the reaction with the imine bearing the N-ortho-methoxyphenyl group as a radical acceptor proceeded smoothly to yield 3l. With respect to enol silyl ethers, cyclohexanone-derived triethylsilyl and triisopropylsilyl ethers were tolerated, leading to the formation of 3m and 3n. Other cyclic enol silyl ethers with five- and seven-membered rings, as well as those with oxygen-containing six-membered rings, were also suitable candidates for the nucleophilic components and the corresponding adducts 3o–3q were obtained in good yields. Notably, various acyclic ketone-derived enol silyl ethers and butyryl pyrazole-derived ketene silyl hemiaminal could be converted into aminoalkylated products 3r–3w with moderate to high efficiencies. In all cases, diastereoselectivity in the construction of two contiguous stereocenters was of a negligible degree.
 | ||
| Fig. 2 Scope of substrates. Isolated yields are indicated. Diastereomeric ratios (d.r.) of the products were 1.0:1–1.3:1. See the ESI† for details. | ||
Finally, the synthetic utility of this Mannich-type C–H functionalization of enol silyl ethers was demonstrated through the derivatization of aminoalkylated products (Scheme 3). For instance, the Mukaiyama–Mannich cyclization of 3b with formaldehyde proceeded smoothly under the action of a catalytic amount of diarylborinate16 to form fused bicyclic product 5. The octahydroisoindol-4-one structure of 5 is often encountered in natural products and biologically active compounds.17 In addition, the treatment of 3w with trifluoroacetic acid facilitated the protonation of the enol silyl ether and sequential cyclization to furnish β,γ-disubstituted γ-lactam 6 in a high yield.
 | ||
| Scheme 3 Derivatization of aminoalkylated enol silyl ethers. | ||
Conclusions
We have developed a Mannich-type allylic C–H functionalization of enol silyl ethers with N-aryl imines by exploiting photoredox–thiol hybrid catalysis. This catalytic system is applicable to a series of cyclic and acyclic ketone-derived enol silyl ethers, thus providing facile access to diverse γ-aminocarbonyl derivatives. We believe that this study expands the utility of enol silyl ethers for the efficient and diverse synthesis of functionalized carbonyl compounds on demand.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI Grant Number JP17H06446 in Hybrid Catalysis for Enabling Molecular Synthesis on Demand and JP18H01972.Notes and references
-
A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Willey-VCH, 2008 Search PubMed
.
- R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS
- For selected recent reviews, see:
(a) Y. Shi, Q. Wang and S. Gao, Org. Chem. Front., 2018, 5, 1049–1066 RSC
- For examples of β-functionalization of α,β-unsaturated aldehydes with imines using N-heterocyclic carbene (NHC) as a catalyst, see:
(a) M. He and J. W. Bode, Org. Lett., 2005, 7, 3131–3134 CrossRef CAS
- Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, Nat. Chem., 2013, 5, 835–839 CrossRef CAS
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS
- J. L. Jeffrey, F. R. Petronijević and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 8404–8407 CrossRef CAS
- K. Ohmatsu, T. Nakashima, M. Sato and T. Ooi, Nat. Commun., 2019, 10, 2706 CrossRef
-
(a) J. Hioe, D. Šakić, V. Vrček and H. Zipse, Org. Biomol. Chem., 2015, 13, 157–169 RSC
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS
-
(a) S. L. Khursan, D. A. Mikhailov, V. M. Yanborisov and D. I. Borisov, React. Kinet. Catal. Lett., 1997, 61, 91–95 CrossRef CAS
-
(a) D. Hager and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986–16989 CrossRef CAS
- J. A. Leitch, T. Rossolini, T. Rogova, J. A. P. Maitland and D. J. Dixon, ACS Catal., 2020, 10, 2009–2025 CrossRef CAS
-
(a) R. Knowles and H. Yayla, Synlett, 2014, 25, 2819–2826 CrossRef
- The present Mannich-type reaction was not tolerant to aliphatic imines. For instance, the reactions of 1a with pivalaldehyde- or cyclohexanecarboxaldehyde-derived imines resulted in no reaction or the formation of a complex mixture, including the parent 4-methoxyaniline and aldehyde generated through imine hydrolysis.
- Y. Tanaka, T. Hasui and M. Suginome, Synlett, 2008, 1239–1242 CAS
-
(a) Y.-J. Li, H.-M. Huang, H.-Q. Dong, J.-H. Jia, L. Han, Q. Ye and J.-R. Gao, J. Org. Chem., 2013, 78, 9424–9430 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01862g |
| This journal is © The Royal Society of Chemistry 2021 |