
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Aryl mercaptoacetamides as potential multi-target inhibitors of metallo-β-lactamases (MBLs) and the virulence factor LasB from Pseudomonas aeruginosa†
Samir
Yahiaoui‡
 a,
Katrin
Voos‡
b,
Jörg
Haupenthal
a,
Thomas A.
Wichelhaus
c,
Denia
Frank
c,
Lilia
Weizel
d,
Marco
Rotter
d,
Steffen
Brunst
d,
Jan S.
Kramer
d,
Ewgenij
Proschak
d,
Christian
Ducho
*b and
Anna K. H.
Hirsch
*ae
a,
Katrin
Voos‡
b,
Jörg
Haupenthal
a,
Thomas A.
Wichelhaus
c,
Denia
Frank
c,
Lilia
Weizel
d,
Marco
Rotter
d,
Steffen
Brunst
d,
Jan S.
Kramer
d,
Ewgenij
Proschak
d,
Christian
Ducho
*b and
Anna K. H.
Hirsch
*ae
aHelmholtz Institute for Pharmaceutical Research Saarland (HIPS) – Helmholtz Centre for Infection Research (HZI), Campus E8 1, 66123 Saarbrücken, Germany. E-mail: Anna.Hirsch@Helmholtz-hips.de
bDepartment of Pharmacy, Pharmaceutical and Medicinal Chemistry, Saarland University, Campus C2 3, 66123 Saarbrücken, Germany. E-mail: Christian.Ducho@Uni-Saarland.de
cInstitute of Medical Microbiology and Infection Control, University Hospital Frankfurt, Paul-Ehrlich-Straße 40, 60596 Frankfurt, Germany
dInstitute of Pharmaceutical Chemistry, Goethe University Frankfurt, Max-von-Laue-Straße 9, 60438, Frankfurt, Germany
eDepartment of Pharmacy, Saarland University, Campus Building E8 1, 66123 Saarbrücken, Germany
First published on 29th July 2021
Abstract
Increasing antimicrobial resistance is evolving to be one of the major threats to public health. To reduce the selection pressure and thus to avoid a fast development of resistance, novel approaches aim to target bacterial virulence instead of growth. Another strategy is to restore the activity of antibiotics already in clinical use. This can be achieved by the inhibition of resistance factors such as metallo-β-lactamases (MBLs). Since MBLs can cleave almost all β-lactam antibiotics, including the “last resort” carbapenems, their inhibition is of utmost importance. Here, we report on the synthesis and in vitro evaluation of N-aryl mercaptoacetamides as inhibitors of both clinically relevant MBLs and the virulence factor LasB from Pseudomonas aeruginosa. All tested N-aryl mercaptoacetamides showed low micromolar to submicromolar activities on the tested enzymes IMP-7, NDM-1 and VIM-1. The two most promising compounds were further examined in NDM-1 expressing Klebsiella pneumoniae isolates, where they restored the full activity of imipenem. Together with their LasB-inhibitory activity in the micromolar range, this class of compounds can now serve as a starting point for a multi-target inhibitor approach against both bacterial resistance and virulence, which is unprecedented in antibacterial drug discovery.
Introduction
Due to the emergence of drug-resistant bacteria and the lack of new antibiotics in the pipeline, novel effective and innovative treatment options are urgently needed, in particular to cure infections with Gram-negative pathogens. One of the highly problematic Gram-negative bacteria is Pseudomonas aeruginosa, which has been assigned critical priority by the World Health Organization (WHO).1 This opportunistic pathogen is responsible for fatal lung infections in cystic fibrosis patients and many hospital-acquired infections.2,3P. aeruginosa is a versatile human pathogen that easily acquires resistance against multiple classes of antibacterial agents, including aminoglycosides, quinolones, polymyxins, and β-lactams.4,5The β-lactams are still the most commonly used group of antibiotics.6 One of the main mechanisms of resistance in Gram-negative pathogens including P. aeruginosa is the production of β-lactamases.7–9 These enzymes can inactivate β-lactam antibiotics by the hydrolysis of their four-membered β-lactam ring.10,11 Based on their mechanism of action, two main groups of β-lactamases (BLs) were identified: the serine-β-lactamases (SBLs) and the metallo-β-lactamases (MBLs).
SBLs act via a nucleophilic serine residue that covalently binds to the carbonyl unit of the β-lactam ring to achieve its hydrolysis. They have a variable substrate spectrum ranging from small-, broad- to extended-spectrum, and even carbapenems can be degraded by some SBLs. Additionally, several effective SBL inhibitors (e.g., clavulanic acid) are already in clinical use in combination with β-lactam antibiotics to overcome resistance.12
MBLs can be divided into classes B1 to B3 which differ in their amino acid sequence homology and, as a result, their zinc coordination.13 Class B2 (e.g., CphA) has only one catalytically active zinc ion and a very narrow substrate profile,14 while classes B1 (e.g., NDM) and B3 (e.g., AIM) coordinate two zinc ions and show a very broad substrate spectrum.14–16 In particular the B1 class is of high clinical relevance.17–19 So far, two MBL inhibitors having a cyclic boronate as core structure (see Fig. 1) are in clinical trials. QPX7728 is in phase I (NCT04380207, alone and with QPX2014) and VNRX-5133 (taniborbactam) is in phase III in combination with cefepime (NCT03840148).20–24 However, no clinically approved MBL inhibitors have been reported to date.25,26
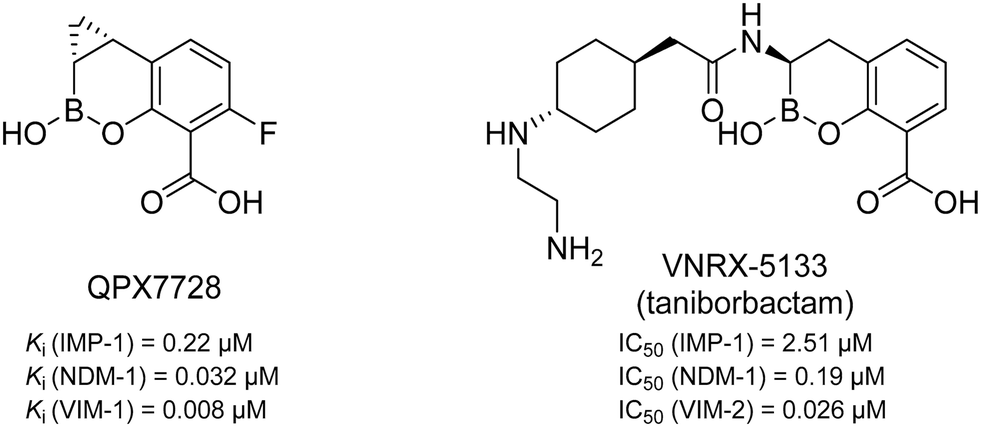 | ||
| Fig. 1 Chemical structures and reported Ki/IC50 values against different MBLs of QPX772820,21 and VNRX-5133 (taniborbactam),22–24 two cyclic boronates that are in clinical trials. | ||
It is known that bacterial pathogenesis and infectivity are mediated by microbial components called virulence factors, which play pivotal roles in adhesion, biofilm formation, invasion of host tissues, and immune evasion.27,28 Targeting these factors is a newly emerging approach in the field of anti-infective drug discovery. This so-called antivirulence strategy is intended to disarm the bacteria rather than killing them. Several bacterial virulence factors have been identified including toxins, extracellular enzymes needed for cell invasion and biofilm formation, cell-surface proteins that mediate bacterial attachment, secretion systems, and quorum-sensing systems that regulate bacterial cell-to-cell communication.27,29–31
One of the major virulence factors of P. aeruginosa is the elastase LasB. This extracellular zinc-containing protease is responsible for the invasion of host tissues, biofilm formation, and immune evasion. It has been demonstrated that LasB can cleave important components of the connective tissues such as elastin and collagen.32,33 The protease was also shown to promote biofilm formation through the regulation of rhamnolipids.34 Moreover, it was found to alter the human immune response by degrading host defense components such as cytokines,35 antimicrobial peptides (AMP),36 and surfactant proteins. These multiple roles of LasB in the infection process of P. aeruginosa make this protease an attractive antivirulence target.37,38
Recently, we have demonstrated the potential of thiocarbamates as prodrugs of N-aryl mercaptoacetamides (Scheme 1) as promising inhibitors of several bacterial proteases. Interestingly, these compounds are potent inhibitors of P. aeruginosa LasB and collagenases from Clostridium and Bacillus species.39,40 Regarding LasB, in addition to the elucidation of the binding mode of our hit compound (1, Scheme 1), we demonstrated the in vivo efficacy of the same derivative in a Galleria mellonella larvae infection model. Moreover, compound 1 shows no cytotoxic effects on HepG2, HEK293 and A549 cells (see ESI,† Table S1) and its thiocarbamate prodrug 1a (see Scheme 1) remarkably displays high selectivity over several human matrix metalloproteases (MMPs).39
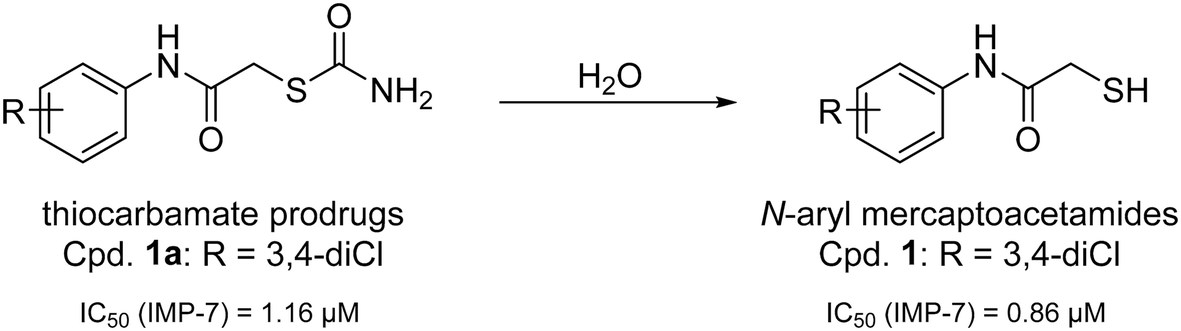 | ||
| Scheme 1 Hydrolysis of thiocarbamate prodrug 1a to its active thiol form 1 and their corresponding IC50 values reported for the inhibition of IMP-7.39 | ||
It has been demonstrated that several thiol-containing drugs such as captopril (an angiotensin converting enzyme (ACE) inhibitor which is used for the treatment of hypertension), thiorphan (the active metabolite of racecadotril used for the treatment of diarrhoea), and tiopronin (which is used in patients with cystinuria, Fig. 2), display promising in vitro inhibitory activities toward different MBLs.41 The shown three inhibitors all consist of the same core structure, that is, a 2-substituted (2-mercaptoacetyl)- or (2-mercatopropanoyl)glycine. Substantial synthetic work has already been done on this scaffold to obtain novel MBL inhibitors, and co-crystallization experiments have been reported.42–46
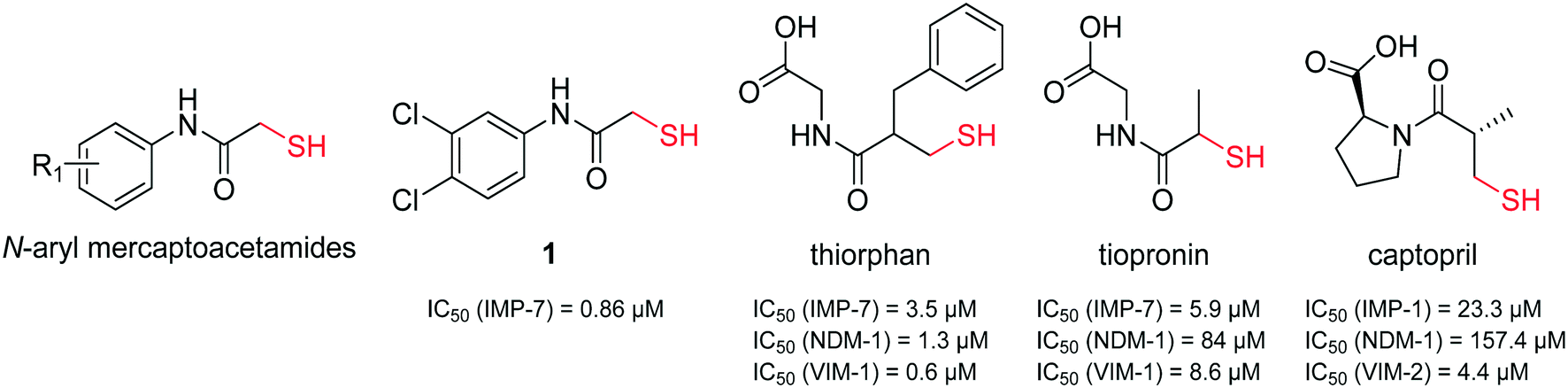 | ||
| Fig. 2 Chemical structures and reported IC50 values of N-aryl mercaptoacetamides, compound 1,39 thiorphan,41 tiopronin41 and captopril47 for the inhibition of different MBLs. | ||
Inspired by this finding, and with respect to our slightly similar mercaptoacetamide core structure, we explored the effect of our thiol-based LasB inhibitor (compound 1) toward a class B1 MBL called imipenemase 7 (IMP-7). Interestingly, our LasB inhibitor showed a strong inhibitory effect with a sub-micromolar IC50 value (IC50 = 0.86 ± 0.06 μM).39
Based on the encouraging inhibitory activity of compound 1 against IMP-7, this study aimed to further investigate the inhibitory profile of the N-aryl mercaptoacetamide class against various MBLs. Aside from that, another goal was to explore the potential of using this class as multi-target inhibitors tackling both virulence and bacterial resistance. In this work, we report on the design, synthesis, and in vitro evaluation of 13 N-aryl mercaptoacetamide derivatives for inhibition of three clinically important and transferrable MBLs of the class B1: IMP-7, Verona integrin encoded metallo-β-lactamase 1 (VIM-1) and New Delhi metallo-β-lactamase 1 (NDM-1). Furthermore, we tested the two most active compounds in combination with the carbapenem antibiotic imipenem against a clinical NDM-1-expressing Klebsiella pneumoniae isolate and confirmed their activity on the bacterial level. The selectivity over nine potential human off-targets was additionally confirmed.
Results and discussion
Synthesis of N-aryl mercaptoacetamides
Here, we report on the biological evaluation of 13 N-aryl mercaptoacetamides bearing various polar and nonpolar substituents on the aryl moiety. We have previously demonstrated that by variation of the substitution pattern on the aryl ring (and thus the electron density and H-bonding interactions), we either obtain LasB or ColH inhibitors. Additionally, the phenyl ring seems to be crucial for the selectivity of these inhibitors over human MMPs as potential off-targets.39,40 Therefore, the impact of the same structural modifications on the inhibition of MBLs should also be investigated. Eight out of the 13 compounds were newly synthesised, while five had already been published as LasB or ColH inhibitors.39,40 The majority of all our previously reported N-aryl mercaptoacetamides were tested against LasB and ColH in their thiocarbamate prodrug form.39 However, the co-crystal structure revealed the free thiol as the active form. This finding was confirmed by stability tests and in vitro assays.39,40 In this work, we therefore synthesised the free-thiol forms of four compounds, which had previously been tested against LasB and ColH as thiocarbamates, as well as four novel mercaptoacetamide derivatives. The synthetic access to the mercaptoacetamide derivatives is outlined in Scheme 2.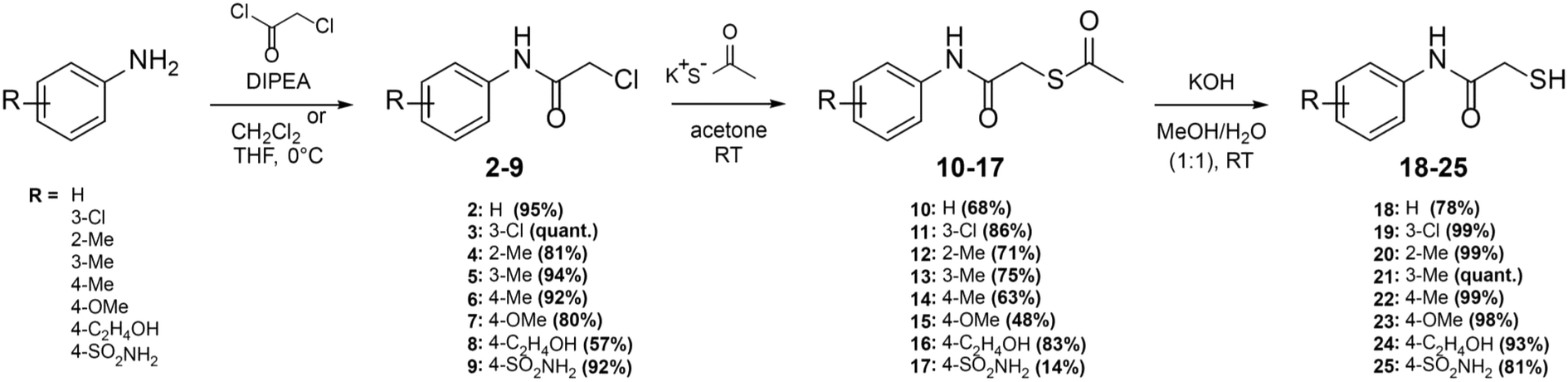 | ||
| Scheme 2 Synthesis of the thiols 18–25 as potential multi-target inhibitors of LasB and metallo-β-lactamases. | ||
A three-step synthetic route afforded target compounds 18–25, starting from the corresponding aniline derivatives. In a first step, an amide formation was carried out by dropwise addition of chloroacetyl chloride to a cooled solution of the corresponding aniline and DIPEA in CH2Cl2 or THF. Second, an SN2 reaction of the alkyl chlorides 2–9 with potassium thioacetate in acetone led to the thioacetates 10–17. Compounds 10–17 were hydrolysed with aqueous KOH in methanol to afford the thiol target molecules 18–25. These as well as the previously synthesised compounds 1 and 26–29 (Fig. 3) were then evaluated with respect to their biological activities.
 | ||
| Fig. 3 Previously reported thiols 26–29 as potential multi-target inhibitors of bacterial virulence and resistance.39,40 | ||
N-Aryl mercaptoacetamides as broad-spectrum MBL inhibitors
To explore the potential of our thiol-based class to inhibit MBLs, we first tested the synthesised compounds 18–25 as well as the previously reported compounds 1 and 26–29 (Fig. 3) in vitro for their inhibitory potency against NDM-1, VIM-1 and IMP-7.39,40 Based on their in vitro activity, we then selected several derivatives for further evaluation in bacteria in combination with imipenem.MBL activity assays
We used an activity assay based on kinetic fluorescence using fluorocillin as a substrate41 to evaluate the inhibitory activity of the mercaptoacetamide derivatives against NDM-1, VIM-1 and IMP-7. The results can be found in Table 1.|
|
||||||
|---|---|---|---|---|---|---|
| Cp. | R | Thiol IC50a [μM] IMP-7 | Thiol IC50a [μM] NDM-1 | Thiol IC50a [μM] VIM-1 | Thiol IC50a [μM] LasB | Thiocarbamate IC50a [μM] LasB |
| a Means of at least two independent measurements ± standard deviation. | ||||||
| 1 | 3,4-DiCl | 0.86 ± 0.06 | 0.65 ± 0.04 | 2.2 ± 0.3 | 6.6 ± 0.3 (ref. 39) | 6.2 ± 0.3 |
| 18 | H | 4.7 ± 0.7 | 3.6 ± 0.9 | 5.1 ± 1.3 | 39 ± 3 | — |
| 26 | 2-Cl | 1.9 ± 0.5 | 1.8 ± 0.1 | 2.3 ± 0.2 | 11 ± 1 | 14 ± 1 |
| 19 | 3-Cl | 4.1 ± 1.6 | 2.7 ± 0.4 | 3.6 ± 1.2 | 14 ± 2 | 19 ± 1 |
| 27 | 4-Cl | 2.4 ± 0.3 | 2.1 ± 0.1 | 4.7 ± 1.8 | 21 ± 1 (ref. 39) | 16 ± 1 |
| 20 | 2-Me | 6.4 ± 2.0 | 6.0 ± 0.5 | 4.9 ± 1.3 | 14 ± 1 | — |
| 21 | 3-Me | 4.4 ± 1.5 | 3.4 ± 0.2 | 4.1 ± 0.8 | 38 ± 3 | 48 ± 2 |
| 22 | 4-Me | 5.4 ± 1.5 | 3.4 ± 0.3 | 5.0 ± 1.6 | 19 ± 1 | 36 ± 1 |
| 23 | 4-OMe | 8.6 ± 1.8 | 5.0 ± 2.1 | 5.9 ± 1.3 | 24 ± 1 | 48 ± 1 |
| 24 | 4-C2H4OH | 11 ± 4 | 6.7 ± 3.0 | 5.9 ± 2.0 | 52 ± 3 | — |
| 25 | 4-SO2NH2 | 7.7 ± 1.2 | 6.3 ± 1.5 | 4.1 ± 0.9 | 54 ± 3 | — |
| 28 | 4-Ac | 5.4 ± 2.5 | 1.5 ± 0.2 | 3.5 ± 0.8 | 72 ± 5 | 73 ± 3 |
| 29 | 3-Cl-2,6-diMe | 2.7 ± 0.7 | 8.2 ± 2.6 | 4.0 ± 0.7 | 12 ± 1 (ref. 39) | — |
Remarkably, all derivatives were active against all three MBL enzymes and consequently displayed broad-spectrum effects in vitro (Table 1). Compound 1 showed a sub-micromolar activity against both IMP-7 (IC50 = 0.86 ± 0.06 μM)39 and NDM-1 (IC50 = 0.65 ± 0.04 μM) and an IC50 of 2.2 ± 0.3 μM against VIM-1. All the other derivatives showed low micromolar activities against the three tested MBLs. Regarding many previously reported MBL inhibitors (a selection can be found in Fig. 2 and 4),41–43,48–50 our compounds can be considered equipotent. Only the class of cyclic boronates (e.g.34 (Fig. 4), QPX7728, and VNRX-5133 (Fig. 1)) was significantly more active against MBLs (with IC50 values in the low nanomolar range) and has already two candidates in clinical trials.20,23,50,51
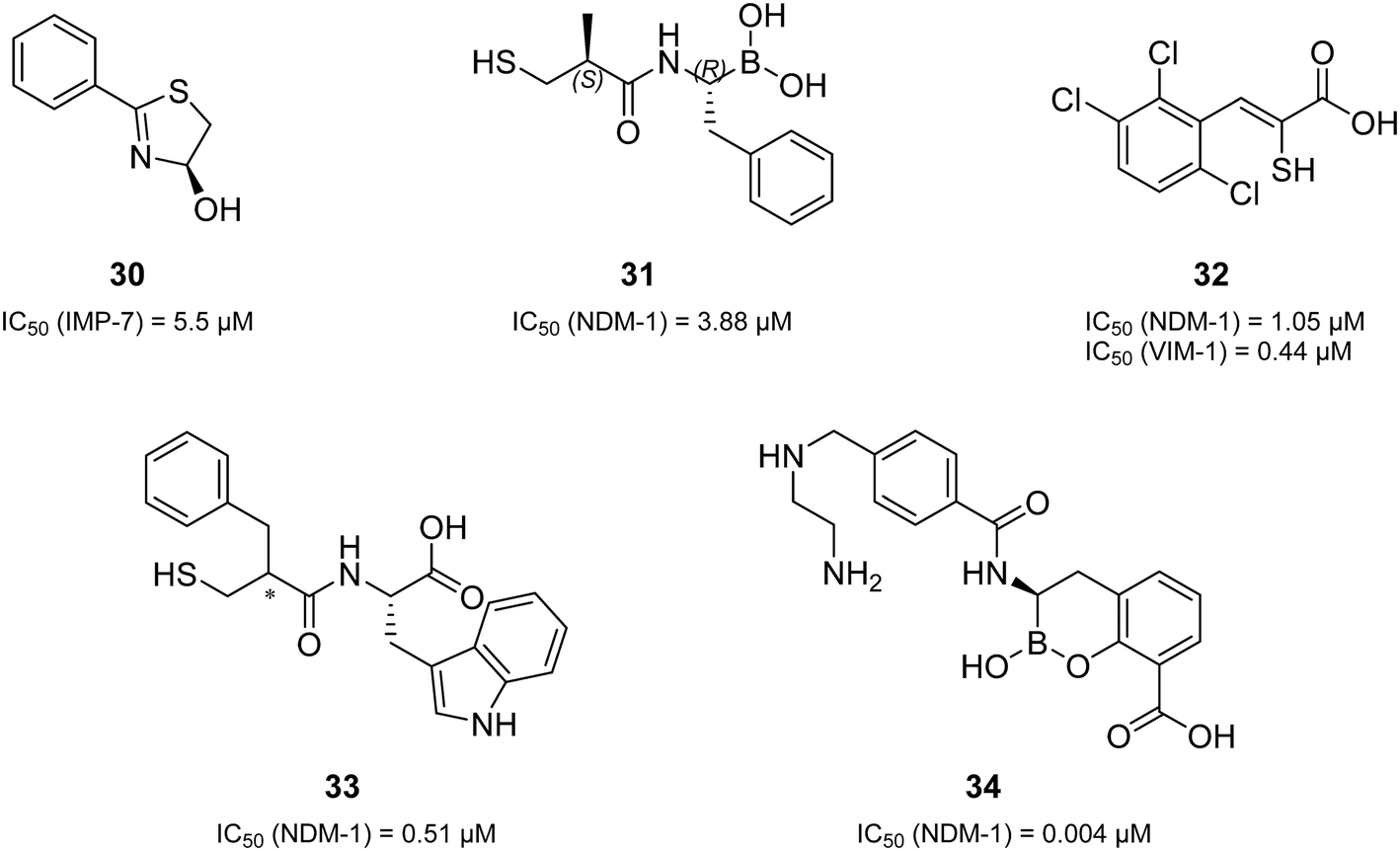 | ||
| Fig. 4 A selection of the structural variations of reported MBL inhibitors and their corresponding inhibitory activities in vitro (30,4831,4332,4933,42 and 3450). | ||
In comparison to compound 1, all structural modifications of the aryl moiety resulted in a moderate loss of activity against all MBLs. Regarding IMP-7, the IC50 values range from 1.9 μM (compound 26) to 11 μM (compound 24) with better tolerance for the nonpolar substituents. The most potent derivative against NDM-1 was compound 28 (1.5 μM), and as observed for IMP-7, the nonpolar groups were slightly preferred. Concerning VIM-1, all compounds showed comparable activities with IC50 values ranging between 2.3 μM (26) and 5.9 μM (24). Additionally, for mono-substituted compounds featuring a chlorine or methyl group, the position of the substituent did not have a significant effect on the biological response.
Growth-inhibition assays
Encouraged by the promising in vitro activities of our N-aryl mercaptoacetamides, we investigated the effects of compounds 1 and 26 to restore the antibacterial effect of imipenem (ESI,† Fig. S1) in a growth inhibition assay. As we wanted to investigate MBL activity without the influence of LasB inhibition, and as our compounds were the most active on NDM-1, we selected a clinical imipenem-resistant K. pneumoniae isolate (strain T2301) that is able to express NDM-1 but not LasB. K. pneumoniae is one of the known ESKAPE pathogens and is rated critical by the WHO on their global priority list of antibiotic-resistant bacteria.52,53 Nosocomial infections with K. pneumoniae have caused several outbreaks in multiple locations with very high mortality rates, i.e., up to 72%.54–56 NDM-1 (and other antibiotic resistance genes) spread between several pathogens via horizontal transfer,57–59 and NDM-1-expressing K. pneumoniae have caused several outbreaks in hospitals in the last decade.60–62 Growth curves of the bacterial isolate with imipenem were determined in the presence and absence of the named compounds (two separate experiments, see Fig. 5A and B). Compounds 1 and 26 were selected based on their promising activities against MBLs.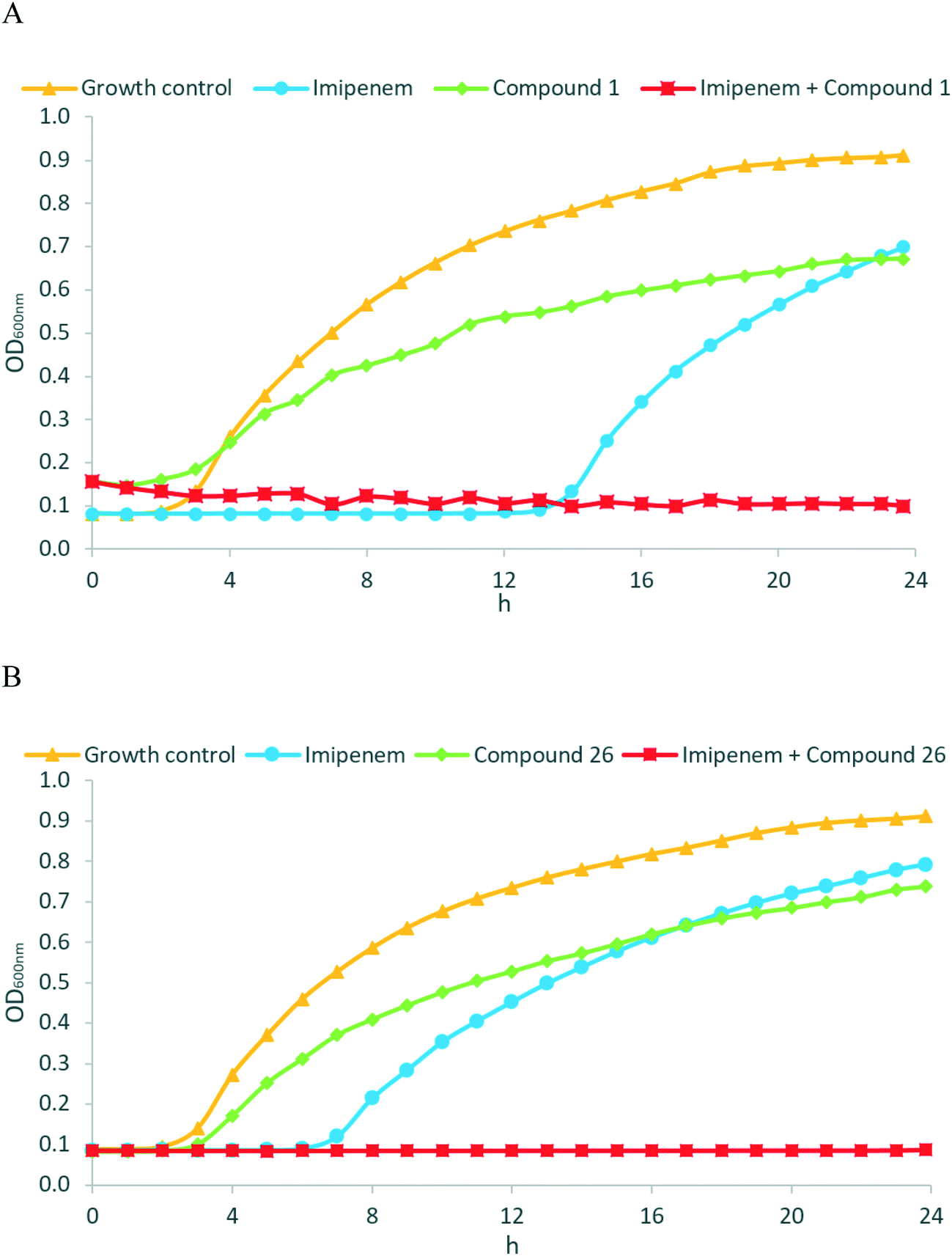 | ||
| Fig. 5 Growth-inhibition assay. Growth curves of NDM-1-expressing K. pneumoniae (T2301) over time (h) in the absence and presence of imipenem at 8 μg mL−1 (i.e., 0.5× MIC) ± MBL inhibitors at 50 μg mL−1 ( = 21 μM (1) or 25 μM (26)); compound 1 (A) and compound 26 (B). | ||
As shown in the growth curves in Fig. 5, the presence of imipenem (at 8 μg mL−1, i.e., 0.5× MIC) delayed the initiation of bacterial growth by a few hours, but after 24 hours, no relevant differences between the growth of imipenem-treated bacteria and the imipenem-free control were observed. Remarkably, when MBL inhibitors 1 (Fig. 5A, at 50 μg mL−1 ( = 21 μM)) or 26 (Fig. 5B, at 50 μg mL−1 ( = 25 μM)) were combined with imipenem (at 8 μg mL−1), the inhibitory potential of imipenem was fully restored, and no growth of bacteria occurred over 24 hours. MBL inhibitors alone did not exhibit any intrinsic antibacterial activity at 50 μg mL−1. As an additional control experiment, we employed EDTA as an MBL inhibitor, with MBL inhibition being caused by the metal-chelating properties of EDTA (ESI,† Fig. S2). The resultant growth curves were rather similar to those obtained with MBL inhibitors 1 and 26, respectively. Overall, this highlights the potency of the reported MBL inhibitors not just on the enzymatic, but also on the cellular level, making them attractive candidates for further development.
N-Aryl mercaptoacetamides as multi-target inhibitors of LasB and MBLs
The promising antibacterial effects of N-aryl mercaptoacetamides in combination with imipenem (Fig. 5) prompted us to further investigate the potential of this class as multi-target inhibitors targeting both virulence and resistance. Therefore, all synthesised compounds were evaluated for their potential inhibition of LasB as a bacterial virulence factor. Inhibitory activity towards ColH was not determined, as the best two compounds on MBLs (1 and 26) had already been tested in their thiocarbamate form against ColH and did just show micromolar activities, which is more than three orders of magnitude less potent than the ColH-inhibiting hit compound 28.40The evaluation of compounds 1, 19, 21, 26, and 27 in a functional LasB inhibition assay expectedly39,40 showed similar activities compared to their thiocarbamate prodrug derivatives (Table 1). However, compounds 22 and 23 were two-fold more active than their thiocarbamate congeners. Regarding the novel mercaptoacetamides, the unsubstituted derivative 18 as well as the 2-methyl compound 20 showed decreased IC50 values relative to compound 1. Moreover, as expected based on our previous studies,39 compounds 24 and 25 were the least active derivatives in the series due to the presence of polar substituents that are unfavourable for LasB inhibition.
Selectivity over other proteases
As most of our compounds all show activity on LasB and the three tested MBLs, and as it is known that 28 additionally is a nanomolar collagenase H (ColH) inhibitor,40 it is crucial to exclude that our compounds are just non-selective zinc-chelators. Thiocarbamates 1a and 28a had already been evaluated for their selectivity over a range of six MMPs which had been chosen with respect to their structural variations in their S1′ binding pockets.39,40 We decided to test our hit compound 1 accordingly and, additionally, expand the selectivity panel with two histone deacetylases (HDACs) and the tumour necrosis factor α converting enzyme (TACE). Those three enzymes are all zinc-dependent proteases.63–65 HDACs play a crucial role in epigenetic regulation by modification of gene expression and thus in cell proliferation.66,67 TACE, also known as “a disintegrin and metalloprotease 17” (ADAM17), is important for a functional immune system, for the nervous system and tissue generation.68–71 Therefore, these enzymes represent important antitargets. As controls, batimastat (ESI,† Fig. S3) as unselective MMP inhibitor, trichostatin A (ESI,† Fig. S4), a known inhibitor of HDACs, and ilomastat as TACE inhibitor (ESI,† Fig. S5) were chosen. Compound 1 was tested at a concentration of 100 μM for its inhibitory activity on those nine enzymes. The results are reported in Table 2.| 1 inhibition at 100 μMa [%] | Batimastat IC50a [nM] | Trichostatin A inhibition at 0.5 μMa [%] | |
|---|---|---|---|
| a Means and SD of at least two independent measurements are displayed; n.d. = not determined; n.i. = no inhibition (if ≤10%). | |||
| MMP-1 | n.i. | 2.2 ± 0.1 | n.d. |
| MMP-2 | n.i. | 1.8 ± 0.1 | n.d. |
| MMP-3 | n.i. | 5.6 ± 0.9 | n.d. |
| MMP-7 | n.i. | 7.0 ± 0.2 | n.d. |
| MMP-8 | n.i. | 0.7 ± 0.2 | n.d. |
| MMP-14 | n.i. | 2.8 ± 0.2 | n.d. |
| TACE | 40 ± 4 | n.d. | n.d. |
| HDAC-3 | 9 ± 7 | n.d. | 94 ± 3 |
| HDAC-8 | 11 ± 5 | n.d. | 95 ± 2 |
Compound 1 showed no inhibition of the six tested MMPs and the two HDACs. A slight inhibition of TACE (40 ± 4%) at a rather high concentration of 100 μM was found, but this still corresponds to a selectivity factor of more than five, which should be considered sufficient in particular in this early phase of development.
Cytotoxicity assays
It had already been demonstrated in our previous studies that compound 1 did not show any cytotoxicity against the three tested cell lines HepG2, HEK293 and A549 at a concentration of 100 μM. The results can be found in the ESI,† Table S1.39,73Additionally, compound 1 already had been subjected to a more complex in vivo toxicity model in zebrafish embryos (ESI,† Table S2).73 Here the maximum tolerated concentration (MTC) was 10 μM. At higher concentrations (e.g. 30 μM), the compound precipitated in this assay and the zebrafish embryos died.73 Further studies to determine whether the toxicity derived from the compound's in vivo effect itself or from the formation of crystals will be performed in the course of future hit optimisation.
Conclusions
In summary, based on the encouraging inhibitory activity of a previously reported LasB inhibitor (compound 1) against IMP-7, we designed, synthesised and evaluated a series of N-aryl mercaptoacetamide derivatives against the three metallo-β-lactamases IMP-7, VIM-1 and NDM-1. Interestingly, all tested compounds showed promising inhibition of all three MBLs. Compound 1 displayed the strongest inhibitory effects with submicromolar IC50 values toward both IMP-7 and NDM-1, and low micromolar activity against VIM-1.To confirm the potential of our mercaptoacetamide derivatives as MBL inhibitors, compounds 1 and 26 were tested in combination with imipenem against a clinical NDM-1-expressing K. pneumoniae isolate. Remarkably, at 50 μg mL−1 (∼20 μM), both compounds were able to restore the activity of imipenem. Additionally, compound 1 showed high selectivity over a total of nine potential off-targets (six MMPs, TACE and two HDACs) at a concentration of 100 μM, at which it already previously had demonstrated to also have no cytotoxic effects on human cell lines.39,73 These promising effects of our N-aryl mercaptoacetamides against MBLs, their previously confirmed anti-LasB properties and their demonstrated high selectivity over human off-targets inspires the use of this class as multi-target inhibitors tackling both virulence and resistance of pathogens. This is a new approach that is unprecedented in antibacterial drug discovery.
Although further investigations will be needed to improve the inhibitory potencies of our inhibitors, our results demonstrate an attractive new multi-targeting approach, which can enhance the efficacy of currently used antibiotics against resistant MBL-expressing pathogens. In addition to restoring the sensitivity to antimicrobial agents, the effect of the reported compounds on virulence factors can make bacteria less pathogenic and more vulnerable towards immune response and/or low doses of antibiotics. Even though no improvement of the in vitro inhibitory activities of compound 1 has been achieved so far, the reported results indicate that some structural variations in the aryl moiety appear to be feasible. This opens the way to a future optimisation of this compound class, also with respect to ADME-T properties.
Experimental section
Chemistry
Compounds 1,3926,4027,39,402840 and 2939 were previously reported. All other compounds were synthesised according to Scheme 1 as described in detail in the ESI.†IMP-7, NDM-1 and VIM-1 in vitro inhibition assays
The assays were performed according to a previously described procedure.41 They were carried out at room temperature in black polystyrol 96-well plates (Corning) using fluorocillin (prepared as described by Rukavishnikov et al.74), as substrate. The fluorescence emitted by the fluorescent product difluorofluorescein was kinetically monitored using a Tecan fluorescent plate reader Infinite F200. IC50 values were calculated using data obtained from measurements with at least six different inhibitor concentrations, applying a sigmoidal dose–response (variable slope with four parameters) equation using GraphPad Prism 5 (GraphPad software, La Jolla, CA, USA) software.Growth inhibition assay
A computerised incubator (Tecan Infinite M200 pro, Crailsheim, Germany) was used to obtain the growth curves over a time course. Optical density (OD600) was measured for evaluating bacterial (i.e., K. pneumoniae NDM-1) density in suspension. Prior to each experiment, bacteria were cultured to the exponential phase (OD600 = 0.6–0.8) in cation-adjusted Mueller Hinton broth (Becton Dickinson, Heidelberg, Germany). 5 μL of diluted bacterial suspension and 100 μL of cation-adjusted Mueller Hinton broth either with or without imipenem (Arcos Organics, Schwerte, Germany) and/or the respective MBL inhibitor were added into wells of a 96 well plate to give a final bacterial inoculum of approx. 5 × 105 CFU mL−1. The temperature was adjusted to 37 °C. A permanent shaking speed of 450 rpm was used and only interrupted before OD600 measurement. The optical densities of samples were recorded on-line every 10 minutes for 24 hours. Experiments were determined in triplicate. Representative growth curves are shown. For quality control, bacterial strain E. coli ATCC 25922 was used.LasB in vitro inhibition assay
The purification of LasB as well as the performance, measurement and evaluation of the FRET-based in vitro inhibition assay were carried out as previously described by Kany et al.39MMP in vitro inhibition assay
The assays were performed as previously described.39,40TACE in vitro inhibition assay
ADAM-17 (TACE) inhibitor screening assay kit was purchased from Sigma-Aldrich. The assay was performed according to the guidelines of the manufacturer. Fluorescence signals were measured in a CLARIOstar plate reader (BMG Labtech).HDAC in vitro inhibition assay
HDAC3 and HDAC8 inhibitor screening kits were purchased from Sigma-Aldrich. The assay was performed according to the guidelines of the manufacturer.Author contributions
Samir Yahiaoui, investigation, writing – original draft; Katrin Voos, investigation, validation, visualization, writing – original draft; Jörg Haupenthal, conceptualization, project administration, validation, writing – review & editing; Thomas A. Wichelhaus, project administration, resources, supervision, validation, visualization, writing – review & editing; Denia Frank, investigation; Lilia Weizel, investigation; Marco Rotter, investigation; Steffen Brunst, investigation; Jan S. Kramer, investigation; Ewgenij Proschak, project administration, supervision, validation, writing – review & editing; Christian Ducho, conceptualization, project administration, supervision, validation, writing – review & editing; Anna K. H. Hirsch, conceptualization, funding acquisition, project administration, supervision, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Jeannine Jung and Jelena Konstantinović for excellent technical support. A. K. H. Hirsch gratefully acknowledges funding from the Helmholtz-Association's Initiative and Networking Fund. E. Proschak thanks German Research Foundation (DFG, Heisenberg-Professur PR1405/7-1) for financial support.Notes and references
- World Health Organization, Antibacterial Agents in Clinical Development, in An Analysis of the Antibacterial Clinical Development Pipeline, Including Tuberculosis, The World Health Organization, Geneva, 2017 Search PubMed.
- R. E. W. Hancock and D. P. Speert, Antibiotic Resistance in Pseudomonas Aeruginosa: Mechanisms and Impact on Treatment, Drug Resist. Updates, 2000, 3(4), 247–255, DOI:10.1054/drup.2000.0152.
- D. M. Ramsey and D. J. Wozniak, Understanding the Control of Pseudomonas Aeruginosa Alginate Synthesis and the Prospects for Management of Chronic Infections in Cystic Fibrosis: P. Aeruginosa Pathogenesis, Mol. Microbiol., 2005, 56(2), 309–322, DOI:10.1111/j.1365-2958.2005.04552.x.
- Z. Pang, R. Raudonis, B. R. Glick, T.-J. Lin and Z. Cheng, Antibiotic Resistance in Pseudomonas Aeruginosa: Mechanisms and Alternative Therapeutic Strategies, Biotechnol. Adv., 2019, 37(1), 177–192, DOI:10.1016/j.biotechadv.2018.11.013.
- M. Bassetti, A. Vena, A. Croxatto, E. Righi and B. Guery, How to Manage Pseudomonas Aeruginosa Infections, Drugs Context, 2018, 7, 1–18, DOI:10.7573/dic.212527.
- K. Bush and P. A. Bradford, β-Lactams and β-Lactamase Inhibitors: An Overview, Cold Spring Harb. Perspect. Med., 2016, 6(8), a025247, DOI:10.1101/cshperspect.a025247.
- D. J. Wolter and P. D. Lister, Mechanisms of β-Lactam Resistance Among Pseudomonas Aeruginosa, Curr. Pharm. Des., 2013, 19, 209–222, DOI:10.2174/138161213804070311.
- D. Rawat and D. Nair, Extended-Spectrum β-Lactamases in Gram Negative Bacteria, J. Global Infect. Dis., 2010, 2(3), 263–274, DOI:10.4103/0974-777X.68531.
- A. A. Medeiros, Evolution and Dissemination of β-Lactamases Accelerated by Generations of β-Lactam Antibiotics, Clin. Infect. Dis., 1997, 24(Supplement 1), S19–S45, DOI:10.1093/clinids/24.supplement_1.s19.
- A. A. Medeiros, β-Lactamases: Quality and Resistance, Clin. Microbiol. Infect., 1997, 3, 4S2–4S9, DOI:10.1016/S1198-743X(14)65030-8.
- M. I. Page and A. P. Laws, The Mechanism of Catalysis and the Inhibition of β-Lactamases, Chem. Commun., 1998, 1609–1617, 10.1039/A803578D.
- K. A. Toussaint and J. C. Gallagher, β-Lactam/β-Lactamase Inhibitor Combinations: From Then to Now, Ann. Pharmacother., 2015, 49(1), 86–98, DOI:10.1177/1060028014556652.
- T. Palzkill, Metallo-β-Lactamase Structure and Function, Ann. N. Y. Acad. Sci., 2013, 1277(1), 91–104, DOI:10.1111/j.1749-6632.2012.06796.x.
- C. Bebrone, Metallo-β-Lactamases (Classification, Activity, Genetic Organization, Structure, Zinc Coordination) and Their Superfamily, Biochem. Pharmacol., 2007, 74(12), 1686–1701, DOI:10.1016/j.bcp.2007.05.021.
- M. I. Abboud, C. Damblon, J. Brem, N. Smargiasso, P. Mercuri, B. Gilbert, A. M. Rydzik, T. D. W. Claridge, C. J. Schofield and J.-M. Frère, Interaction of Avibactam with Class B Metallo-β-Lactamases, Antimicrob. Agents Chemother., 2016, 60(10), 5655–5662, DOI:10.1128/AAC.00897-16.
- M.-N. Lisa, A. R. Palacios, M. Aitha, M. M. González, D. M. Moreno, M. W. Crowder, R. A. Bonomo, J. Spencer, D. L. Tierney, L. I. Llarrull and A. J. A. Vila, General Reaction Mechanism for Carbapenem Hydrolysis by Mononuclear and Binuclear Metallo-β-Lactamases, Nat. Commun., 2017, 8(1), 538, DOI:10.1038/s41467-017-00601-9.
- P. Nordmann, L. Poirel, T. R. Walsh and D. M. Livermore, The Emerging NDM Carbapenemases, Trends Microbiol., 2011, 19(12), 588–595, DOI:10.1016/j.tim.2011.09.005.
- M. F. Mojica, R. A. Bonomo and W. Fast, B1-Metallo-β-Lactamases: Where Do We Stand?, Curr. Drug Targets, 2016, 17(9), 1029–1050, DOI:10.2174/1389450116666151001105622.
- A. U. Khan, L. Maryam and R. Zarrilli, Structure, Genetics and Worldwide Spread of New Delhi Metallo-β-Lactamase (NDM): A Threat to Public Health, BMC Microbiol., 2017, 17(1), 101, DOI:10.1186/s12866-017-1012-8.
- S. J. Hecker, K. R. Reddy, O. Lomovskaya, D. C. Griffith, D. Rubio-Aparicio, K. Nelson, R. Tsivkovski, D. Sun, M. Sabet, Z. Tarazi, J. Parkinson, M. Totrov, S. H. Boyer, T. W. Glinka, O. A. Pemberton, Y. Chen and M. N. Dudley, Discovery of Cyclic Boronic Acid QPX7728, an Ultrabroad-Spectrum Inhibitor of Serine and Metallo-β-Lactamases, J. Med. Chem., 2020, 63(14), 7491–7507, DOI:10.1021/acs.jmedchem.9b01976.
- O. Lomovskaya, D. Rubio-Aparicio, K. Nelson, D. Sun, R. Tsivkovski, M. Castanheira, J. Lindley, J. Loutit and M. Dudley, In Vitro Activity of the Ultrabroad-Spectrum Beta-Lactamase Inhibitor QPX7728 in Combination with Multiple Beta-Lactam Antibiotics against Pseudomonas Aeruginosa, Antimicrob. Agents Chemother., 2021, 65(6), e00210-21, DOI:10.1128/AAC.00210-21.
- J. C. Hamrick, J.-D. Docquier, T. Uehara, C. L. Myers, D. A. Six, C. L. Chatwin, K. J. John, S. F. Vernacchio, S. M. Cusick, R. E. L. Trout, C. Pozzi, F. De Luca, M. Benvenuti, S. Mangani, B. Liu, R. W. Jackson, G. Moeck, L. Xerri, C. J. Burns, D. C. Pevear and D. M. Daigle, VNRX-5133 (Taniborbactam), a Broad-Spectrum Inhibitor of Serine- and Metallo-β-Lactamases, Restores Activity of Cefepime in Enterobacterales and Pseudomonas Aeruginosa, Antimicrob. Agents Chemother., 2020, 64(3), e01963-19, DOI:10.1128/AAC.01963-19.
- B. Liu, R. E. L. Trout, G.-H. Chu, D. McGarry, R. W. Jackson, J. C. Hamrick, D. M. Daigle, S. M. Cusick, C. Pozzi, F. De Luca, M. Benvenuti, S. Mangani, J.-D. Docquier, W. J. Weiss, D. C. Pevear, L. Xerri and C. J. Burns, Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-β-Lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections, J. Med. Chem., 2020, 63(6), 2789–2801, DOI:10.1021/acs.jmedchem.9b01518.
- S. Mushtaq, A. Vickers, M. Doumith, M. J. Ellington, N. Woodford and D. M. Livermore, Activity of β-Lactam/Taniborbactam (VNRX-5133) Combinations against Carbapenem-Resistant Gram-Negative Bacteria, J. Antimicrob. Chemother., 2021, 76(1), 160–170, DOI:10.1093/jac/dkaa391.
- Faridoon and U. I. Nazar, An Update on the Status of Potent Inhibitors of Metallo-β-Lactamases, Sci. Pharm., 2013, 81(2), 309–328, DOI:10.3797/scipharm.1302-08.
- Z. Cheng, C. A. Thomas, A. R. Joyner, R. L. Kimble, A. M. Sturgill, N.-Y. Tran, M. R. Vulcan, S. A. Klinsky, D. J. Orea, C. R. Platt, F. Cao, B. Li, Q. Yang, C. J. Yurkiewicz, W. Fast and M. W. Crowder, MBLinhibitors.Com, a Website Resource Offering Information and Expertise for the Continued Development of Metallo-β-Lactamase Inhibitors, Biomolecules, 2020, 10(3), 459, DOI:10.3390/biom10030459.
- T. J. Foster, J. A. Geoghegan, V. K. Ganesh and M. Höök, Adhesion, Invasion and Evasion: The Many Functions of the Surface Proteins of Staphylococcus Aureus, Nat. Rev. Microbiol., 2014, 12(1), 49–62, DOI:10.1038/nrmicro3161.
- J. W. Newman, R. V. Floyd and J. L. Fothergill, The Contribution of Pseudomonas Aeruginosa Virulence Factors and Host Factors in the Establishment of Urinary Tract Infections, FEMS Microbiol. Lett., 2017, 364(15), 1–11, DOI:10.1093/femsle/fnx124.
- K. Tam and V. J. Torres, Staphylococcus aureus Secreted Toxins and Extracellular Enzymes, in Gram-Positive Pathogens, ed. V. A. Fischetti, R. P. Novick, J. J. Ferretti, D. A. Portnoy, M. Braunstein and J. I. Rood, ASM Press, Washington, DC, USA, 2019, pp. 640–668, DOI:10.1128/9781683670131.ch40.
- S. T. Rutherford and B. L. Bassler, Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control, Cold Spring Harb. Perspect. Med., 2012, 2(11), a012427–a012427, DOI:10.1101/cshperspect.a012427.
- M. B. Calvert, V. R. Jumde and A. Titz, Pathoblockers or Antivirulence Drugs as a New Option for the Treatment of Bacterial Infections, Beilstein J. Org. Chem., 2018, 14, 2607–2617, DOI:10.3762/bjoc.14.239.
- B. Wretlind and O. R. Pavlovskis, Pseudomonas Aeruginosa Elastase and Its Role in Pseudomonas Infections, Clin. Infect. Dis., 1983, 5(Supplement_5), S998–S1004, DOI:10.1093/clinids/5.Supplement_5.S998.
- L. W. Heck, K. Morihara, W. B. McRae and E. J. Miller, Specific Cleavage of Human Type III and IV Collagens by Pseudomonas Aeruginosa Elastase, Infect. Immun., 1986, 51(1), 115–118, DOI:10.1128/IAI.51.1.115-118.1986.
- H. Yu, X. He, W. Xie, J. Xiong, H. Sheng, S. Guo, C. Huang, D. Zhang and K. Zhang, Elastase LasB of Pseudomonas Aeruginosa Promotes Biofilm Formation Partly through Rhamnolipid-Mediated Regulation, Can. J. Microbiol., 2014, 60(4), 227–235, DOI:10.1139/cjm-2013-0667.
- M. Parmely, A. Gale, M. Clabaugh, R. Horvat and W. W. Zhou, Proteolytic Inactivation of Cytokines by Pseudomonas Aeruginosa, Infect. Immun., 1990, 58(9), 3009–3014, DOI:10.1128/IAI.58.9.3009-3014.1990.
- A. Schmidtchen, I.-M. Frick, E. Andersson, H. Tapper and L. Björck, Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37, Mol. Microbiol., 2002, 46(1), 157–168, DOI:10.1046/j.1365-2958.2002.03146.x.
- Z. Kuang, Y. Hao, B. E. Walling, J. L. Jeffries, D. E. Ohman and G. W. Lau, Pseudomonas Aeruginosa Elastase Provides an Escape from Phagocytosis by Degrading the Pulmonary Surfactant Protein-A, PLoS One, 2011, 6(11), e27091, DOI:10.1371/journal.pone.0027091.
- J. F. Alcorn and J. R. Wright, Degradation of Pulmonary Surfactant Protein D by Pseudomonas Aeruginosa Elastase Abrogates Innate Immune Function, J. Biol. Chem., 2004, 279(29), 30871–30879, DOI:10.1074/jbc.M400796200.
- A. M. Kany, A. Sikandar, J. Haupenthal, S. Yahiaoui, C. K. Maurer, E. Proschak, J. Köhnke and R. W. Hartmann, Binding Mode Characterization and Early in Vivo Evaluation of Fragment-Like Thiols as Inhibitors of the Virulence Factor LasB from Pseudomonas Aeruginosa, ACS Infect. Dis., 2018, 4(6), 988–997, DOI:10.1021/acsinfecdis.8b00010.
- E. Schönauer, A. M. Kany, J. Haupenthal, K. Hüsecken, I. J. Hoppe, K. Voos, S. Yahiaoui, B. Elsässer, C. Ducho, H. Brandstetter and R. W. Hartmann, Discovery of a Potent Inhibitor Class with High Selectivity toward Clostridial Collagenases, J. Am. Chem. Soc., 2017, 139(36), 12696–12703, DOI:10.1021/jacs.7b06935.
- F.-M. Klingler, T. A. Wichelhaus, D. Frank, J. Cuesta-Bernal, J. El-Delik, H. F. Müller, H. Sjuts, S. Göttig, A. Koenigs, K. M. Pos, D. Pogoryelov and E. Proschak, Approved Drugs Containing Thiols as Inhibitors of Metallo-β-Lactamases: Strategy To Combat Multidrug-Resistant Bacteria, J. Med. Chem., 2015, 58(8), 3626–3630, DOI:10.1021/jm501844d.
- J. Ma, Q. Cao, S. M. McLeod, K. Ferguson, N. Gao, A. L. Breeze and J. Hu, Target-Based Whole-Cell Screening by 1 H NMR Spectroscopy, Angew. Chem., Int. Ed., 2015, 54(16), 4764–4767, DOI:10.1002/anie.201410701.
- Y.-L. Wang, S. Liu, Z.-J. Yu, Y. Lei, M.-Y. Huang, Y.-H. Yan, Q. Ma, Y. Zheng, H. Deng, Y. Sun, C. Wu, Y. Yu, Q. Chen, Z. Wang, Y. Wu and G.-B. Li, Structure-Based Development of (1-(3′-Mercaptopropanamido)Methyl)Boronic Acid Derived Broad-Spectrum, Dual-Action Inhibitors of Metallo- and Serine-β-Lactamases, J. Med. Chem., 2019, 62(15), 7160–7184, DOI:10.1021/acs.jmedchem.9b00735.
- Y. Yan, G. Li and G. Li, Principles and Current Strategies Targeting Metallo-β-lactamase Mediated Antibacterial Resistance, Med. Res. Rev., 2020, 40(5), 1558–1592, DOI:10.1002/med.21665.
- S. Liu, L. Jing, Z.-J. Yu, C. Wu, Y. Zheng, E. Zhang, Q. Chen, Y. Yu, L. Guo, Y. Wu and G.-B. Li, (( S )-3-Mercapto-2-Methylpropanamido)Acetic Acid Derivatives as Metallo-β-Lactamase Inhibitors: Synthesis, Kinetic and Crystallographic Studies, Eur. J. Med. Chem., 2018, 145, 649–660, DOI:10.1016/j.ejmech.2018.01.032.
- G.-B. Li, J. Brem, R. Lesniak, M. I. Abboud, C. T. Lohans, I. J. Clifton, S.-Y. Yang, J.-C. Jiménez-Castellanos, M. B. Avison, J. Spencer, M. A. McDonough and C. J. Schofield, Crystallographic Analyses of Isoquinoline Complexes Reveal a New Mode of Metallo-β-Lactamase Inhibition, Chem. Commun., 2017, 53(43), 5806–5809, 10.1039/C7CC02394D.
- J. Brem, S. S. van Berkel, D. Zollman, S. Y. Lee, O. Gileadi, P. J. McHugh, T. R. Walsh, M. A. McDonough and C. J. Schofield, Structural Basis of Metallo-β-Lactamase Inhibition by Captopril Stereoisomers, Antimicrob. Agents Chemother., 2016, 60(1), 142–150, DOI:10.1128/AAC.01335-15.
- P. Chen, L. B. Horton, R. L. Mikulski, L. Deng, S. Sundriyal, T. Palzkill and Y. Song, 2-Substituted 4,5-Dihydrothiazole-4-Carboxylic Acids Are Novel Inhibitors of Metallo-β-Lactamases, Bioorg. Med. Chem. Lett., 2012, 22(19), 6229–6232, DOI:10.1016/j.bmcl.2012.08.012.
- J. Brem, S. S. van Berkel, W. Aik, A. M. Rydzik, M. B. Avison, I. Pettinati, K.-D. Umland, A. Kawamura, J. Spencer, T. D. W. Claridge, M. A. McDonough and C. J. Schofield, Rhodanine Hydrolysis Leads to Potent Thioenolate Mediated Metallo-β-Lactamase Inhibition, Nat. Chem., 2014, 6(12), 1084–1090, DOI:10.1038/nchem.2110.
- J. Brem, R. Cain, S. Cahill, M. A. McDonough, I. J. Clifton, J.-C. Jiménez-Castellanos, M. B. Avison, J. Spencer, C. W. G. Fishwick and C. J. Schofield, Structural Basis of Metallo-β-Lactamase, Serine-β-Lactamase and Penicillin-Binding Protein Inhibition by Cyclic Boronates, Nat. Commun., 2016, 7(1), 12406, DOI:10.1038/ncomms12406.
- S. T. Cahill, R. Cain, D. Y. Wang, C. T. Lohans, D. W. Wareham, H. P. Oswin, J. Mohammed, J. Spencer, C. W. G. Fishwick, M. A. McDonough, C. J. Schofield and J. Brem, Cyclic Boronates Inhibit All Classes of β-Lactamases, Antimicrob. Agents Chemother., 2017, 61(4), e02260-16, DOI:10.1128/AAC.02260-16.
- E. Tacconelli, N. Magrini, Y. Carmeli, S. Harbarth, G. Kahlmeter, J. Kluytmans, M. Mendelson, C. Pulcini, N. Singh and U. Theuretzbacher, Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics, World Health Organization, 2017, vol. 27, pp. 318–327 Search PubMed.
- L. B. Rice, Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE, J. Infect. Dis., 2008, 197(8), 1079–1081, DOI:10.1086/533452.
- A. Borer, L. Saidel-Odes, K. Riesenberg, S. Eskira, N. Peled, R. Nativ, F. Schlaeffer and M. Sherf, Attributable Mortality Rate for Carbapenem-Resistant Klebsiella Pneumoniae Bacteremia, Infect. Control Hosp. Epidemiol., 2009, 30(10), 972–976 CrossRef PubMed.
- E. B. Hirsch and V. H. Tam, Detection and Treatment Options for Klebsiella Pneumoniae Carbapenemases (KPCs): An Emerging Cause of Multidrug-Resistant Infection, J. Antimicrob. Chemother., 2010, 65(6), 1119–1125, DOI:10.1093/jac/dkq108.
- P. Nordmann, G. Cuzon and T. Naas, The Real Threat of Klebsiella Pneumoniae Carbapenemase-Producing Bacteria, Lancet Infect. Dis., 2009, 9(4), 228–236, DOI:10.1016/S1473-3099(09)70054-4.
- S. Chatterjee, A. Mondal, S. Mitra and S. Basu, Acinetobacter Baumannii Transfers the BlaNDM-1 Gene via Outer Membrane Vesicles, J. Antimicrob. Chemother., 2017, 72(8), 2201–2207, DOI:10.1093/jac/dkx131.
- T. Krahn, D. Wibberg, I. Maus, A. Winkler, S. Bontron, A. Sczyrba, P. Nordmann, A. Pühler, L. Poirel and A. Schlüter, Intraspecies Transfer of the Chromosomal Acinetobacter Baumannii Bla NDM-1 Carbapenemase Gene, Antimicrob. Agents Chemother., 2016, 60(5), 3032–3040, DOI:10.1128/AAC.00124-16.
- H. Lee, J. Shin, Y.-J. Chung, M. Park, K. J. Kang, J. Y. Baek, D. Shin, D. R. Chung, K. R. Peck, J.-H. Song and K. S. Ko, Co-Introduction of Plasmids Harbouring the Carbapenemase Genes, BlaNDM-1 and BlaOXA-232, Increases Fitness and Virulence of Bacterial Host, J. Biomed. Sci., 2020, 27(1), 8, DOI:10.1186/s12929-019-0603-0.
- L. Poirel, Z. Al Maskari, F. Al Rashdi, S. Bernabeu and P. Nordmann, NDM-1-Producing Klebsiella Pneumoniae Isolated in the Sultanate of Oman, J. Antimicrob. Chemother., 2011, 66(2), 304–306, DOI:10.1093/jac/dkq428.
- E. Voulgari, C. Gartzonika, G. Vrioni, L. Politi, E. Priavali, S. Levidiotou-Stefanou and A. Tsakris, The Balkan Region: NDM-1-Producing Klebsiella Pneumoniae ST11 Clonal Strain Causing Outbreaks in Greece, J. Antimicrob. Chemother., 2014, 69(8), 2091–2097, DOI:10.1093/jac/dku105.
- J. A. Escobar Pérez, N. M. Olarte Escobar, B. Castro-Cardozo, I. A. Valderrama Márquez, M. I. Garzón Aguilar, L. Martinez de la Barrera, E. R. Barrero Barreto, R. A. Marquez-Ortiz, M. V. Moncada Guayazán and N. Vanegas Gómez, Outbreak of NDM-1-Producing Klebsiella Pneumoniae in a Neonatal Unit in Colombia, Antimicrob. Agents Chemother., 2013, 57(4), 1957–1960, DOI:10.1128/AAC.01447-12.
- A. Vannini, C. Volpari, G. Filocamo, E. C. Casavola, M. Brunetti, D. Renzoni, P. Chakravarty, C. Paolini, R. De Francesco, P. Gallinari, C. Steinkuhler and S. Di Marco, Crystal Structure of a Eukaryotic Zinc-Dependent Histone Deacetylase, Human HDAC8, Complexed with a Hydroxamic Acid Inhibitor, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(42), 15064–15069, DOI:10.1073/pnas.0404603101.
- K. Maskos, C. Fernandez-Catalan, R. Huber, G. P. Bourenkov, H. Bartunik, G. A. Ellestad, P. Reddy, M. F. Wolfson, C. T. Rauch, B. J. Castner, R. Davis, H. R. G. Clarke, M. Petersen, J. N. Fitzner, D. P. Cerretti, C. J. March, R. J. Paxton, R. A. Black and W. Bode, Crystal Structure of the Catalytic Domain of Human Tumor Necrosis Factor-α-Converting Enzyme, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(7), 3408–3412, DOI:10.1073/pnas.95.7.3408.
- P. J. Watson, L. Fairall, G. M. Santos and J. W. R. Schwabe, Structure of HDAC3 Bound to Co-Repressor and Inositol Tetraphosphate, Nature, 2012, 481(7381), 335–340, DOI:10.1038/nature10728.
- K. Struhl, Histone Acetylation and Transcriptional Regulatory Mechanisms, Genes Dev., 1998, 12, 599–606 CrossRef CAS PubMed.
- B. G. Pogo, V. G. Allfrey and A. E. Mirsky, RNA Synthesis and Histone Acetylation during the Course of Gene Activation in Lymphocytes, Proc. Natl. Acad. Sci. U. S. A., 1966, 55(4), 805–812, DOI:10.1073/pnas.55.4.805.
- F. Zunke and S. Rose-John, The Shedding Protease ADAM17: Physiology and Pathophysiology, Biochim. Biophys. Acta, Mol. Cell Res., 2017, 1864(11), 2059–2070, DOI:10.1016/j.bbamcr.2017.07.001.
- M. Gooz, ADAM-17: The Enzyme That Does It All, Crit. Rev. Biochem. Mol. Biol., 2010, 45(2), 146–169, DOI:10.3109/10409231003628015.
- J. Scheller, A. Chalaris, C. Garbers and S. Rose-John, ADAM17: A Molecular Switch to Control Inflammation and Tissue Regeneration, Trends Immunol., 2011, 32(8), 380–387, DOI:10.1016/j.it.2011.05.005.
- R. Menghini, L. Fiorentino, V. Casagrande, R. Lauro and M. Federici, The Role of ADAM17 in Metabolic Inflammation, Atherosclerosis, 2013, 228(1), 12–17, DOI:10.1016/j.atherosclerosis.2013.01.024.
- A. M. Kany, A. Sikandar, S. Yahiaoui, J. Haupenthal, I. Walter, M. Empting, J. Köhnke and R. W. Hartmann, Tackling Pseudomonas Aeruginosa Virulence by a Hydroxamic Acid-Based LasB Inhibitor, ACS Chem. Biol., 2018, 13(9), 2449–2455, DOI:10.1021/acschembio.8b00257.
- J. Konstantinović, S. Yahiaoui, A. Alhayek, J. Haupenthal, E. Schönauer, A. Andreas, A. M. Kany, R. Mueller, J. Köhnke, F. Berger, M. Bischoff, R. W. Hartmann, H. Brandstetter and A. K. H. Hirsch, N-Aryl-3-Mercaptosuccinimides as Antivirulence Agents Targeting Pseudomonas Aeruginosa Elastase and Clostridium Collagenases, J. Med. Chem., 2020, 63(15), 8359–8368, DOI:10.1021/acs.jmedchem.0c00584.
- A. Rukavishnikov, K. R. Gee, I. Johnson and S. Corry, Fluorogenic Cephalosporin Substrates for β-Lactamase TEM-1, Anal. Biochem., 2011, 419(1), 9–16, DOI:10.1016/j.ab.2011.07.020.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00187f |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |