 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the background and local contribution of the oxidants in London and Bangkok†
M. Anwar H.
Khan
 *a,
Rayne
Holland
a,
Amy
Foulds
a,
James C.
Matthews
a,
Sanjee
Panditharatne
a,
Michael E.
Jenkin
b,
Douglas
Lowe
c,
Panida
Navasumrit
d,
Carl J.
Percival
e and
Dudley E.
Shallcross
af
*a,
Rayne
Holland
a,
Amy
Foulds
a,
James C.
Matthews
a,
Sanjee
Panditharatne
a,
Michael E.
Jenkin
b,
Douglas
Lowe
c,
Panida
Navasumrit
d,
Carl J.
Percival
e and
Dudley E.
Shallcross
af
aSchool of Chemistry, University of Bristol, Cantock’s Close, Bristol, BS8 1TS, UK. E-mail: anwar.khan@bristol.ac.uk
bAtmospheric Chemistry Services, Okehampton, Devon EX20 4QB, UK
cSchool of Earth, Atmospheric and Environmental Science, The University of Manchester, Manchester M13 9PL, UK
dLaboratories of Environmental Toxicology/Chemical Carcinogenesis, Chulabhorn Research Institute, Laksi, Bangkok 10210, Thailand
eNASA Jet Propulsion Laboratory, California Institute of Technology, 4800 Oak Grove Dr, Pasadena, CA 91109, USA
fDepartment of Chemistry, University of the Western Cape, Robert Sobukwe Road, Bellville, 7375, South Africa
First published on 15th October 2020
Abstract
The contribution of NOx emissions and background O3 to the sources and partitioning of the oxidants [OX (= O3 + NO2)] at the Marylebone Road site in London during the 2000s and 2010s has been investigated to see the impact of the control measures or technology changes inline with the London Mayor’s Air Quality Strategy. The abatement of the pollution emissions has an impact on the trends of local and background oxidants, [OX]L and [OX]B, decreasing by 1.4% per year and 0.4% per year, respectively from 2000 to 2019. We also extend our study to three roadside sites (Din Daeng, Thonburi and Chokchai) in another megacity, Bangkok, to compare [OX]L and [OX]B and their behavioural changes with respect to the Marylebone Road site. [OX]L and [OX]B at the Marylebone Road site (0.21[NOx] and 32 ppbv) are comparable with the roadside sites of Thailand (0.12[NOx] to 0.26[NOx] and 29 to 32 ppbv). The seasonal variation of [OX]B levels displays a spring maximum for London, which is due to the higher northern hemispheric ozone baseline, but a maximum during the dry season is found for Bangkok which is likely due to regional-scale long-range transport from the Asian continent. The diurnal variations of [OX]L for both London and Bangkok roadside sites confirm the dominance of the oxidants from road transport emissions, which are found to be higher throughout the daytime. WRF-Chem-CRI model simulations of the distribution of [OX] showed that the model performed well for London background sites when predicting [OX] levels compared with the measured [OX] levels suggesting that the model is treating the chemistry of the oxidants correctly. However, there are large discrepancies for the model–measurement [OX] levels at the traffic site because of the difficulties in the modelling of [OX] at large road networks in megacities for the complex sub grid-scale dynamics that are taking place, both in terms of atmospheric processes and time-varying sources, such as traffic volumes. For roadside sites in Bangkok, the trend in changes of [OX] is predicted by the model correctly but overestimated in absolute magnitude. We suggest that this large deviation is likely to be due to discrepancies in the EDGAR emission inventory (emission overestimates) beyond the resolution of the model.
Introduction
Nitrogen dioxide (NO2) and ozone (O3) are key urban air pollutants with well-documented public health impacts.1 It is well established that the chemistry of O3, NO2 and nitric oxide (NO) is strongly coupled by the following null cycle, typically on the timescale of a few minutes under most ambient conditions.2,3| NO + O3 → NO2 + O2 | (1) |
| NO2 + hν (+ O2) → NO + O3 | (2) |
Because of this rapid interconversion, the term “oxidant” (hereafter denoted OX) has sometimes been used as a collective term for NO2 and O3,4,5 similarly to NOx being the well-established collective term for NO and NO2. Reactions (1) and (2) therefore partition NOx between its component forms of NO and NO2, and OX between its component forms of O3 and NO2, but conserves both NOx and OX.
Reported analyses of ambient data have shown that the concentration of OX at a given location, [OX], can be described in terms of the combination of a background (NOx-independent) source and a local (NOx-dependent) source, denoted here as [OX]B and [OX]L, respectively,5,6 as also apparent from the example data shown in Fig. 1.
| [OX] = [OX]B + [OX]L | (i) |
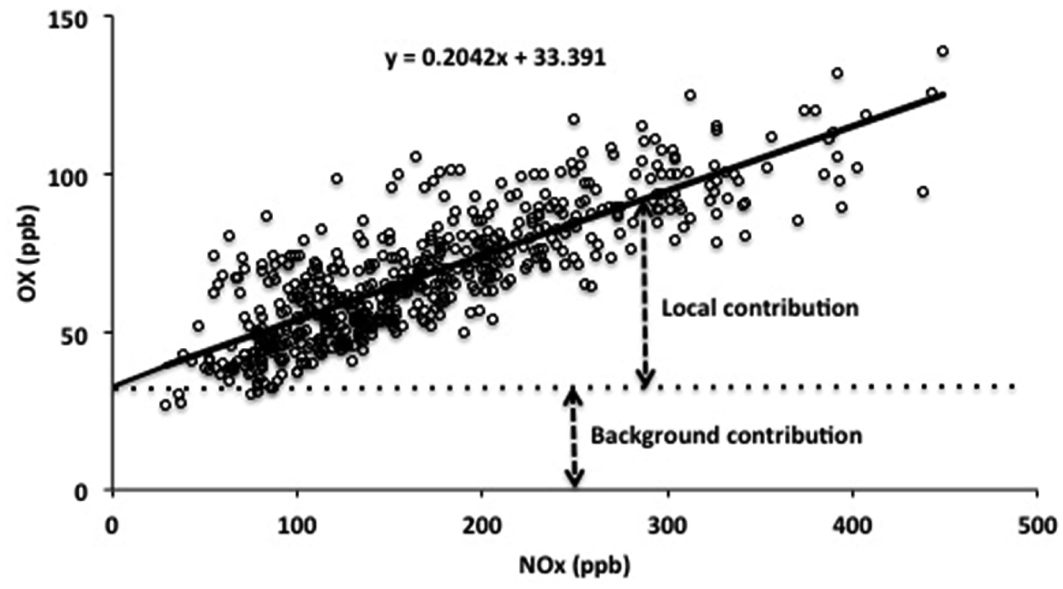 | ||
| Fig. 1 Variation of hourly OX with NOx at the Marylebone Road site for the month of August 2012. | ||
[OX]L is believed to be mainly derived from primary emissions of NO2, particularly at roadside and kerbside locations, such that the slope of the [OX] vs. [NOx] relationship provides an estimate of the volumetric fraction of NOx emitted as NO2.6,7 [OX]B provides a quantification of the background [O3] which would exist at the given location in the notional absence of NOx. It can therefore be regarded as the global (hemispheric) baseline O3 level, modified by regional-scale processes (i.e. deposition and chemistry) that can either remove or produce OX. Analyses of this type have been carried out in a number of studies,5,6,8,9 with values of [OX]B and [OX]L reported for a variety of locations and time periods.
There is a substantial spatial variability in the levels of NO2 and O3 in urban areas, resulting from localised sources of NOx and its relatively short life-time.10–12 The dominant NOx sources are road traffic and static combustion in domestic, commercial, and industrial processes.13–15 The combined health effect of NO2 and O3 has been reported to be elevated compared with their individual effects, which has led to interest in the processes controlling OX levels in the urban areas.16,17 A recent study showed that the OX can enhance the chronic health risks of fine particulate matter (PM2.5),18 thus the reduction of OX levels could be beneficial to reduce the public health impacts of PM2.5.
Currently there are 33 megacities (cities with population of more than 10 million) declared by United Nations in 2018 and the number is likely to increase to 43 by 2030.19 London is one of the megacities in the world where air quality is an issue of increasing public concern. For example, levels of NO2 at roadside and kerbside locations in London have exceeded the EU annual mean limit value of 40 μg m−3 on many occasions.20 The U.K. has progressively adopted EU emissions control measures for both NOx and volatile organic compounds (VOCs) since the early 1990s, with potential impacts on the magnitude of [OX]L and [OX]B.21–23 We therefore investigate the time dependence of these oxidant components at the London Marylebone Road kerbside site, over the time period 2000–2019, to examine the effects of the control measures.
Thailand is one of the rapid economically developing and urbanised countries, which has led to the emergence of a megacity (e.g. Bangkok) and concomitant problems of poor air quality. The transportation, electricity generation and manufacturing industries are considered to be the major sources of air pollutants in Bangkok.24 A recent study showed that NOx levels in Bangkok are mostly below the National Ambient Air Quality Standards (NAAQS), but, O3 levels in Bangkok frequently exceed the NAAQS.25 Uttamang et al.25 analysed the Bangkok metropolitan region pollutant data for 2010–2014 and found that both local and background OX contributions are responsible for increasing the concentration of O3. We update the work of Uttamang et al.25 using more calendar year-coverage data (2005–2018), highlighting roadside and kerbside locations in Bangkok and compare the results with those of the Marylebone Road site in London to investigate any systematic differences in the magnitudes and time dependences of the oxidant components for the two different scenarios (midaltitude vs. tropics).
Megacities are immense sources of air pollutants, with large impacts on regional and global atmospheric chemistry.26 In addition to local sources, ozone precursors (NOx, volatile organic compounds (VOCs)) can also be transported over long distances from the source and cause ozone formation in the downwind locations.27 The balance between local and long-range transport effects are assumed to depend strongly on regional meteorological and geographical differences. The representation of chemistry and accurate ozone precursor emission inventories in the model is very important to reproduce O3 globally and regionally. Recently, we used a mesoscale non-hydrostatic 3-D meteorological model, WRF-Chem-CRI, to simulate the distribution of important trace gases (e.g. NOx, O3) over North-West Europe and found reasonable agreement with measurement data of urban and rural areas of the U.K.28 In this study, we use the similar model set-up to conduct month-long simulations of O3 and NOx during summer and winter months of 2012 over the U.K. and week-long simulations of O3 and NOx during January, May and September months of 2017 over Thailand which is then evaluated with Automatic Urban and Rural Network (AURN), Department for Environment, Food and Rural Affairs (DEFRA), measurement data for London and Pollution Control Department (PCD) measurement data for Bangkok. We compare the modelled and measured OX data of London sites with different environments (e.g. suburban background, urban background, urban traffic, and urban industrial) to investigate how the WRF-Chem model predicts background OX in these sites.
Methods
Site description
Marylebone Road is a kerbside site in central London, which is located next to a busy six-lane road in a street canyon with traffic flows of over 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 vehicles per day with frequent congestion.29 For model comparison, we selected five sites with different environmental conditions: two urban background (Westminster, 17 m away from the nearest road; Bloomsbury, 25 m away from the nearest road), one suburban background (Eltham, 25 m away from the nearest road), one urban industrial (Harlington, 10 m away from the nearest road) and one urban traffic (Marylebone Road, 1 m away from the road).
000 vehicles per day with frequent congestion.29 For model comparison, we selected five sites with different environmental conditions: two urban background (Westminster, 17 m away from the nearest road; Bloomsbury, 25 m away from the nearest road), one suburban background (Eltham, 25 m away from the nearest road), one urban industrial (Harlington, 10 m away from the nearest road) and one urban traffic (Marylebone Road, 1 m away from the road).
Three roadside monitoring sites: Kheha Chumchon Din Daeng, Chokchai Police Station and Thonburi Electricity Authority substation site in Bangkok operated by PCD were chosen based on traffic density and traffic flow. The Din Daeng site is located in central Bangkok, next to a busy six-lane road. South of the measurement site are tall apartment buildings, but to the north is a school and grassy spaces. Thonburi Electricity Authority substation site is next to a six-lane road, which is based in an industrial area with a canal to the west, a nearby gas station to the east and a residential area to the north. Chokchai is next to an eight-lane road, which can accumulate air pollutants due to frequent congestion. The site is not located in the business centre district, but the inlet of the sampling site is very close to a bus stop.
Measurement technique
NOx and O3 have been monitored simultaneously at the Marylebone Road since July 1997 as part of the AURN set up by DEFRA. Throughout the AURN, measurements were made with commercial instrumentation using recognised techniques (i.e. photometry is used for O3 measurements and chemiluminescence is used for NOx measurements) as described by DEFRA.30 However, it is important to note that these analysers are subject to potential interferences from several other oxidised nitrogen species, which can be erroneously reported as NO2.31,32 Despite this, these species are only likely to have a significant impact at rural or remote locations where NOx concentrations are reduced and so-called NOz components abound, due to their formation from NOx processing (e.g. PANs). Thus, [NOz]/[NOx] increases with chemical processing time, so the interferences are not important to consider at roadside or kerbside locations. The uncertainty (expressed at a 95% confidence level) of the measured data for NOx and O3 was around 15%.33 Measurements for these species are reported at hourly resolution and are openly available from the U.K. Air Information Resource (http://uk-air.defra.gov.uk/data/data_selector). This study considers the hourly mean OX and NOx data for the years 2000–2019 inclusive where data has been divided by hour of the day for each month of the year. The data coverage at this site is reasonably consistent except 2011 with the majority of months showing well over the 80% coverage required for inclusion in this study.For the Bangkok roadside sites, NO and NO2 measurements were performed using chemiluminescence detection (Thermo Scientific 42i). The working range of the instrument is 0–500 ppb to 0–20 ppm with a minimum detection limit <0.5 ppb and the precision of the instrument is 0.5 ppb (<1%). O3 was analysed by using UV absorption photometry detection spectroscopy (Thermo Scientific 49i). The working range of the instrument is 0–500 ppb to 0–10 ppm and the instrument has a detection limit of <0.6 ppb and with a precision of <1%. The single point calibration for detectors was performed every 15 days and the multi-point calibration with 3 span levels (20%, 40% and 80%) was performed every 90 days. The data was accepted with the span drift better than ±10% of full scale for the NO2 detector and ±10% of full scale for the O3 detector and zero drift better ±5 ppb for both NO2 and O3 detectors. The hourly data of NOx and O3 measurements over the period from January 2005 to December 2014 for Din Daeng and Chokchai and January 2005 to December 2018 for Thonburi was retrieved from offline source of PCD (http://www.pcd.go.th). The monthly data of NOx and O3 have been computed for [OX] calculation if more than 70% of the data was captured over the individual month (see ESI Fig. S1† for valid data coverage of each site).
Background and local OX calculations
Using the approach of Jenkin,6,7 the ‘background’ and ‘local’ contributions to the total oxidant, [OX]B and [OX]L, are estimated from a linear fit of [OX] against [NOx]. The annual NOx mixing ratios vary over a wide range of values with diurnal and seasonal variations. Thus, we used the hourly data of NOx and OX for each measurement site in London and Bangkok to generate the [NOx] vs. [OX] plots on a month-by-month basis. From here, statistics for the linear relationship were calculated using the ‘least squares’ method to give exact values from gradient (local NOx-dependent contribution), intercept (background NOx-independent contribution) and standard errors for both values. This data was compiled and averaged accordingly to give insights into the monthly and hourly dependencies of [OX]B and [OX]L and how their contributions and trends vary over time.Modelling
We used a mesoscale non-hydrostatic 3-D meteorological model, WRF-Chem-CRI, which is an online fully coupled chemistry transport model.34 The chemistry and aerosol components, along with the prognostic meteorological variables, are integrated over the same timestep as the transport processes, using the same advection and physical parameterisations.34 The meteorological, physical and chemical parameterizations of the model are described in detail elsewhere.28,35 The meteorology was driven by the European Centre for Medium Range Weather Forecasts (ECMWF) ERA-Interim reanalysis data.36 The chemical boundary conditions were provided from MOZART-4 global model.37 Biogenic emissions were calculated online by the Model of Emissions of Gases and Aerosols from Nature (MEGAN) (https://www2.acom.ucar.edu/modeling/model-emissions-gases-and-aerosols-nature-megan).38 The MEGAN model calculates the biogenic emissions of the species from terrestrial ecosystems with a resolution of 1 km × 1 km and is driven by land cover by vegetation, environmental factors (e.g. temperature, humidity, solar intensity, soil moisture) and atmospheric chemical composition. The anthropogenic emissions for the London study have been sourced from a combination of the UK National Atmospheric Emissions Inventory (NAEI) (https://naei.beis.gov.uk) and the European The Netherlands Organisation for Applied Scientific Research, Monitoring Atmospheric Composition and Climate’s emission inventory (TNO-MACC-IIIT).39 The NAEI data for the year 2012 with a resolution of 1 km × 1 km was used in this study. The NAEI includes seven direct greenhouse gases addressed at the Kyoto summit, as well as other trace gases believed to be of atmospheric significance: NOx, CO, non-methane volatile organic compounds (NMVOCs) and SO2. Where data from the NAEI was not available, the TNO was employed to fill the gaps. The TNO data with a resolution of 0.125° × 0.0625° for the year 2011 was used in the study. Scaling factors based on those built for the EMEP model are applied to the anthropogenic emission inventories in order to account for daily, weekly and monthly differences.40 The anthropogenic emissions for the Bangkok study were extracted from the global Emission Database for Global Atmospheric Research with Task Force on Hemispheric Transport of Air Pollution (EDGAR v4.3.2) emission inventories for 2010 at 0.1° × 0.1° resolution.41,42 These datasets are provided as monthly means giving the seasonal cycles. Daily activity cycles, and speciation of NMVOC emissions (see ESI, Table S1†), have been imposed (see details in Chen et al.43 and Lowe).44 The chemistry scheme used is CRI (Common Representatives Intermediate Mechanism), a chemical mechanism describing gas-phase transformations of methane and 25 NMVOCs, via 232 chemical species participating in 638 simulated reactions.28The model domain for the London study covers North-West Europe with a 15 km horizontal resolution with a size of 134 (E–W) by 146 (N–S) grid cells and 41 vertical levels with enhanced resolution within the planetary boundary layer. Two scenarios were run using this domain from 00:00 UTC on 30 July 2012 to 00:00 UTC 24 August 2012 and from 00:00 UTC on 8 January 2012 to 00:00 UTC on 12 February 2012 covering summer and winter months, respectively. Because of the long time period in the scenarios, the re-initialisation of the model meteorology was performed every 3 days using ECMWF meteorology to minimize the errors in the modelled meteorology (bias from model to actual). The Bangkok study region included Thailand, Vietnam, Laos and Cambodia (8°N–22°N and 94°E–108°E, see ESI Fig. S2†) centred at 15°N and 101°E with 15 km horizontal resolution and 41 vertical levels. Three scenarios were run using this domain for the periods of 1–7 January 2017 (winter season), 1–7 May 2017 (summer season) and 1–7 September 2017 (rainy season) with 2 additional days for spin-up.
Results and discussion
Local and background oxidants and their contribution and trend
The comparison of the average ‘local’ and ‘background’ contribution to the OX levels, [OX]L and [OX]B, in the Marylebone Road (2000–2019), Din Daeng (2005–2014), Chokchai (2005–2014) and Thonburi (2005–2018) with the other studies is shown in Table 1. The variability of [OX]L for London and Bangkok for different studies could be due to the control strategy response (e.g. changing the fleet technology by introducing Euro-classes and alternative-fuelled vehicles and/or adopting a range of regulations) for reducing vehicle emissions over time. The [OX]L and [OX]B levels in the Marylebone Road found in the study are comparable with the study of Jenkin6 who analysed the [OX]L and [OX]B in London rural, urban and kerbside sites over 1992–2001. The large difference of [OX]L levels in the study from the Jenkin6 study can be explained by the larger fraction of NOx emitted as NO2 in the roadside site than that in the rural, urban and kerbside sites. For Bangkok roadside sites, we found average [OX]B levels of 32 ppb, 31 ppb, and 29 ppb for Din Daeng, Chokchai and Thonburi, respectively, but Uttamang et al.25 reported significantly higher average [OX]B of 48 ppb and 95 ppb for non-episodes ([O3] < 100 ppb) and episodes ([O3] > 100 ppb), respectively. This is to be expected for Bangkok where our study analysed 24 hour data of NOx and O3 during 2005–2018 for Thonburi and 2005–2014 for Din Daeng and Chokchai, but the Uttamang et al.25 study considered only daytime data during 2010–2014 for Din Daeng and Thonburi. The higher [OX]L levels at Bangkok roadside sites in this study compared with the Uttamang et al.25 study are caused by the fact that the Bangkok data set is a much longer data set, and the initial years of the study (2005–2009) resulted in much higher local pollution levels as emissions control technologies or vehicle fleet composition were not implemented during this time period.| Site | Site information | Time period | [OX ]L (ppb) | [OX]B (ppb) | Ref. |
|---|---|---|---|---|---|
| Din Daeng, Bangkok | Roadside | 2005–2014 | 0.12 ± 0.03[NOx] | 32.0 ± 4.4 | This study |
| Thonburi, Bangkok | Roadside | 2005–2018 | 0.26 ± 0.11[NOx] | 29.1 ± 5.4 | This study |
| Chokchai, Bangkok | Roadside | 2005–2014 | 0.18 ± 0.08[NOx] | 31.4 ± 5.5 | This study |
| Marylebone Road, London | Roadside | 2000–2009 | 0.22 ± 0.02[NOx] | 32.4 ± 3.2 | This study |
| 2010–2019 | 0.20 ± 0.03[NOx] | 32.1 ± 3.1 | |||
| Bangkok | Roadside suburb | 2010–2014 | 0.13[NOx] | 53.9 | 25 |
| 2010–2014 | 0.31[NOx] | 47.0 | |||
| Greater London | Rural, urban & kerbside | 1992–2001 | 0.10[NOx] | 34.7 | 6 |
| Greater London | Rural, urban & kerbside | June 98 and June 99 | 0.10[NOx] | 38.2 | 5 |
| Buenos Aires, Argentina | Urban background | Aug–Sep 2011 | 0.10[NOx] | 22.0 | 9 |
| Delhi, India | Urban background | Sep 2010–Aug 2012 | 0.54[NOx] | 28.9 | 45 |
We averaged the hourly gradient from the plot of [NOx] vs. [OX] for each month and then multiplied by the monthly average NOx levels to calculate the monthly [OX]L mixing ratios. The summation of [OX]L and [OX]B for each month is found to be very similar with the summation of O3 and NO2 for both London and Bangkok roadside sites (correlation coefficient ∼ 1, r2 = 0.99 for each site, ESI Fig. S3†). At the Marylebone Road, the average [OX]L over the last 20 years contributes a significant amount of 25.0 ± 3.2 ppb (44% to the total OX levels, Fig. 2b). A reduction of contribution of average [OX]L from the 2000s (28.0 ± 3.1 ppb; 46% to the total OX) to the 2010s (22.0 ± 3.2 ppb; 41% to the total OX) is found. However, our results still indicate that there is a significant local pollution source (e.g. the fraction of NOx emitted as NO2) in the Marylebone Road site (see ESI Fig. S4a†) that contributes to a large fraction of [OX]L levels during the 2010s. At the Bangkok roadside sites, the contribution of average [OX]L to the total oxidants is lower (e.g. 14.5 ± 5.0 ppb (31% to the total OX, Fig. 3b) for Din Daeng; 12.5 ± 6.0 ppb (29% to the total OX, Fig. 3d) for Chokchai and 14.1 ± 6.0 ppb (32% to the total OX, Fig. 3f) for Thonburi) than the Marylebone Road. The aspect ratio (ratio of building height to street width, H/W) comparison (Marylebone Road ∼1.0,46 Bangkok roadside sites ∼0.4 (ref. 47)) suggests that street canyons are much deeper in the Marylebone Road than that in the roadside sites of Bangkok. Thus the street canyons in Marylebone Road are not as well ventilated as the comparatively open locations in the Bangkok roadside sites, which is a possible reason for increased [OX]L in the Marylebone Road.48,49 However, the pollution levels defined by the fraction of NOx emitted as NO2 in Bangkok roadside sites are highly variable with lower in Din Daeng compared with Thonburi and Chokchai roadside sites (ESI Fig. S4b†). This might be explained by the mobile sources (e.g. vehicles) in Bangkok metropolitan region,25 which is highly dependent on traffic density and traffic flow. The NOx emissions from vehicles are dependent on fuel type and speed of the vehicles,50 which can be responsible for the variation of the [OX]L for different roadside sites in Bangkok.
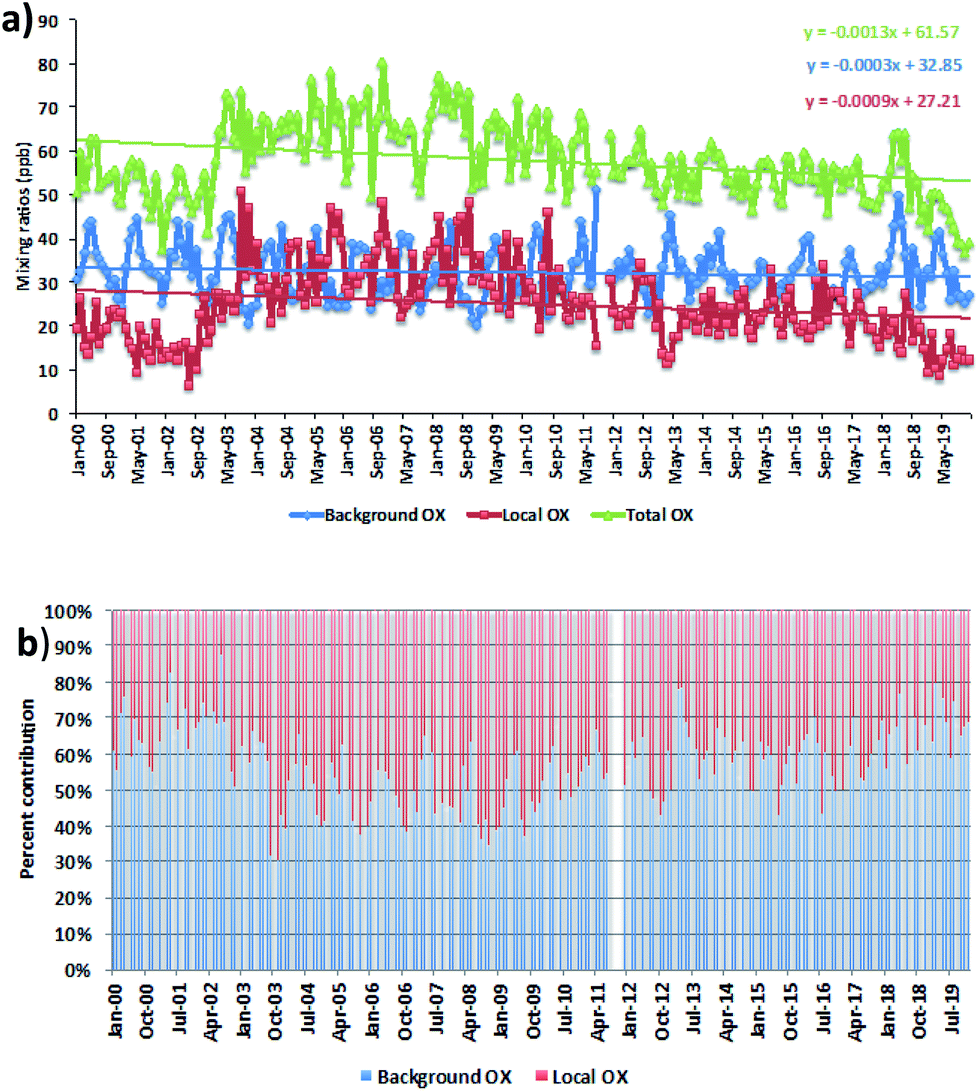 | ||
| Fig. 2 (a) The trends and (b) the percent contribution of oxidant levels in the Marylebone Road for the period of 2000–2019. Statistical significance is based on a p < 0.001 and the trends are reported with 95% confidence intervals. | ||
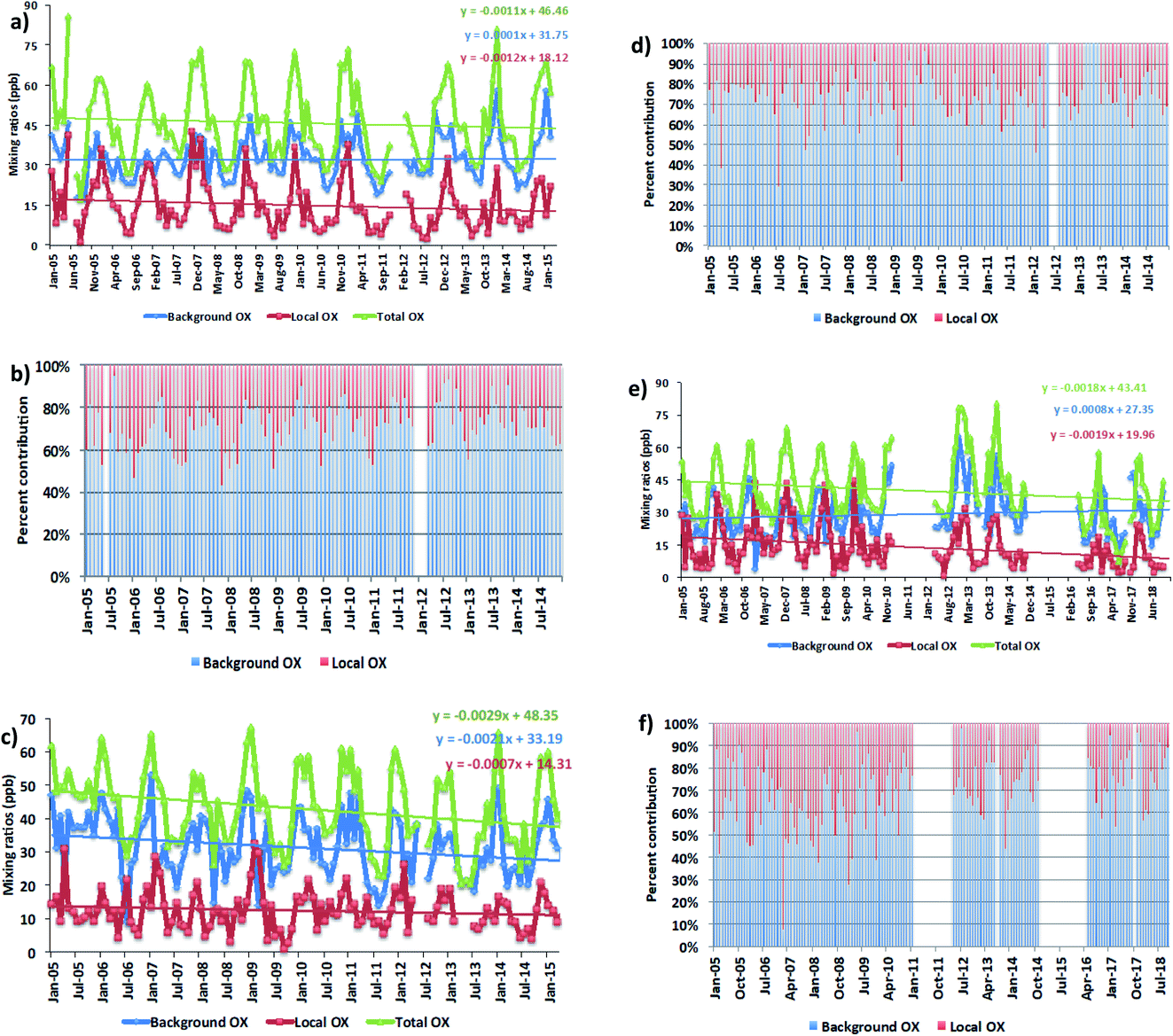 | ||
| Fig. 3 The trends (a, c and e) and the percent contribution (b, d and f) of oxidant levels in Din Daeng (2005–2014), Chokchai (2005–2014) and Thonburi (2005–2018), respectively. Statistical significance is based on a p < 0.001 and the trends are reported with 95% confidence intervals. | ||
For the Marylebone Road, decreasing trends of both [OX]L and [OX]B levels, by 0.33 ppb per year (1.4% per year, p < 0.001) and 0.11 ppb per year (0.4% per year, p < 0.001), respectively, have been found for the period of 2000 to 2019 (Fig. 2a). The local Δ[OX] has been found to have an upward trend (2.12 ppb per year, 8.9% per year, p < 0.001) for the 2000s, but a downward trend (1.17 ppb per year, 5.4% per year, p < 0.001) for the 2010s (see ESI Fig. S5†). An increasing trend of [OX]L from 2002–2005 is seen, which can be explained by the absolute increase in the percentage of NOx emitted from vehicles as NO2 (Fig. S4a†). In response to the Euro III control in the U.K., we found an increase of 5.2% per year NOx emitted as NO2 in the Marylebone Road from 2001 to 2005. The increased introduction of Euro-III diesel vehicles fitted with oxidation catalysts or the fitting of catalytically regenerative particle traps to buses can produce this extra amount of NO2,51 which could be responsible for increasing [OX]L levels from 2001 to 2005 by 9.8% per year. In response to the Euro IV–VI and Euro 4–6 legislations, a large number of policy initiatives were introduced in line with the London Mayor’s Air Quality Strategy (e.g. the roll out of new hybrid and electric vehicles or zero-emission vehicles), thus changing the composition of the vehicle fleet, which is a direct consequence of the Low and Ultra Low Emission Zone legislature and inclusion of the Congestion Charge Zone across London to improve air quality by reducing NOx and particulate matter (PM) levels.21,52 On-road emissions from Euro IV heavy vehicles and buses showed a reduction in the primary NO2 fraction resulting in NO2 emissions from 2010 to 2014 in London.53,54 Furthermore, it has also been shown that the emissions from aged Euro 4 and 5 diesel cars reduced the ratio of NO2/NOx.53 These control strategies could be the main reasons to see a decrease of 1.5% per year NOx emitted as NO2 from 2010 to 2019. This is consistent with the study of Font et al.55 and this NO2 decrease is likely to contribute to the decrement of [OX]L in the 2010s. It can be concluded that the adopted emission control in recent years has been successful in the abatement of pollutant emissions, which reduced [OX]L levels at a rate of 5.4% per year from 27.2 ppb (2000s) to 15.6 ppb (2010s).
The [OX]B in London is strongly dependent on the mixing ratios of O3 on the northern hemispheric ozone baseline.7,56 We found very similar decreasing trends of [OX]B in the 2010s as shown for northern hemispheric mid-latitude baseline O3 by Derwent et al.57 The relatively constant northern hemispheric ozone baseline during the 2000s is reported in the Derwent et al.57 study, but we found a higher downward trend of the Δ[OX]B with 0.55 ppb per year (1.6%, p < 0.001) in the 2000s compared with 0.08 ppb per year (0.3%, p = 0.05) in the 2010s. The O3 can be formed from regional-scale photochemical processing of VOCs and NOx over north-west Europe during the anti-cyclonic heat-wave period58 and which can then be transported to the south and east of the U.K. making an additional contribution of [OX]B at the Marylebone Road. The significant reductions of the European anthropogenic emissions of NOx, VOCs in response to precursor emissions controls (e.g. 1999 Gothenburg Protocol) from 2000 to 2009 can be responsible for reducing regional photochemical ozone production.59 This is likely to be the main reason for the decreasing [OX]B trend during the 2000s.
For Bangkok roadside sites, similar downward trends for [OX]L levels are found in Din Daeng with 0.44 ppb per year (2.8% per year, p = 0.001), in Thonburi with 0.69 ppb per year (4.6%, p < 0.001) and in Chokchai with 0.26 ppb per year (2.0%, p = 0.004) (Fig. 3a, c and e). From 2007–2015, the registered vehicle numbers in Bangkok metropolitan area increased from 6.2 million to approximately 10 million, but the annual local pollutants (e.g. NOx, CO, NMVOCs) emissions from vehicles have significantly decreased due to the policy implemented on road transport activities.60 Thailand has subscribed to the Euro systems of light duty vehicle emissions standard since 1997, but have adopted the more intensive enforcement of fuel and engine standard levels for light duty vehicles from the Euro 2 standard level in 1999 to the Euro 4 standard level in 2013 (with a stricter limit on NOx and VOCs emissions from gasoline engines and on CO, NOx, VOCs + NOx and PM emissions from diesel engines adopted).61 These policies have had some impact on decreasing NOx emitted as NO2 by 1.8% per year (Din Daeng; 2005–2014), 0.6% per year (Chokchai; 2005–2014), and 1.0% per year (Thonburi; 2005–2018) (see Fig. S4b†).
For [OX]B levels, the upward trends are found in Din Daeng (0.04 ppb per year, 0.1% per year, p = 0.18) and in Thonburi (0.97 ppb per year, 2.9%, p = 0.19), but the changes are not significant in the 95% confidence interval (Fig. 3a and e). However, a downward trend of [OX]B is seen in Chokchai (0.77 ppb per year, 2.6% per year, p < 0.001) (Fig. 3c). Although the NOx emissions have decreased since 2011, the O3 concentration increases in the Asian continental air are still significant due to the continued increase in VOC emissions.62 Thus, the decreasing local NOx emissions are not the only effective way to reduce the oxidant levels due to the regional air pollution transport, which can increase the [OX]B levels in Bangkok. The different [OX]B trends for London and Bangkok roadside sites can be explained by meteorological variability and hemispheric transport of pollution.63
Seasonal variation of local and background OX
For the Marylebone Road, the [OX]B has an annual cycle with a maximum in May (41 ppb), influenced by the spring maximum northern hemispheric ozone baseline and a minimum in November (27 ppb) (Fig. 4a). More generally, high NOx is associated with low boundary layer heights due to weak convective activities and turbulent mixing in the winter, which can increase the loss of O3 and NO2 by deposition to the surface, thereby reducing [OX]B in winter months. The previous study at the Marylebone Road showed the seasonal variation of [OX]B with a maximum during spring (43 ppb in April) and a minimum during autumn (30 ppb in October).7 From the 2000s to the 2010s, the spring [OX]B is reduced by ∼1 ppb but the summer [OX]B is reduced by ∼3 ppb (see Fig. 4a) which could be explained by the reduced photochemical O3 production as a result of reduced European regional O3 precursor emissions. The number of extreme heat waves in Europe is fewer in the 2010s than in the 2000s.64 In summer, heat waves are common and [OX]B levels are expected to increase due to an increase in regional-scale photochemical events.56 However, the significant control of O3 precursor emissions in recent years decreased the [OX]B formation from European polluted air mass arriving at Marylebone Road. This confirms the success of the ozone precursor emission control strategies in the abatement of oxidant emissions/production. | ||
| Fig. 4 The seasonal cycles of (a) background OX and (b) local OX contributions in the Marylebone Road (2000–2019). | ||
For [OX]L, the seasonal variability will depend on the level of the primary pollutants, NO2 and the prevalent local photochemistry, thus a peak of [OX]L is found during summer months (Fig. 4b). The contribution of [OX]L is found to be higher during the 2000s compared with the 2010s which can be explained by the extra emissions of NO2 due to the introduction of EURO III light duty diesel vehicles (fitted with oxidation catalysts) into the vehicle fleet and the fitting of catalytically regenerative particle traps to bus fleets.30 Significantly high [OX]L is seen in the 2000s during winter (when a shallow inversion layer is formed) which is most likely due to the combination of extra emissions of NO2 and stagnant air.
For Bangkok, the seasonal variations of [OX]B and [OX]L (Fig. 5) show that high mixing ratios were found during dry months (November to April) with low mixing ratios during wet months (June to September). The differences in the meteorological conditions (solar radiation, temperature, cloud cover) between wet and dry seasons affect the photochemical production of OX. The dry season is linked to a more stable atmosphere and less intensive wet removal of oxidants65 resulting in higher [OX]L and [OX]B levels. The long-range transport from the Asian continent can contribute to [OX]B concentrations in Thailand,66 which plays an important role in seasonal fluctuations of OX in Bangkok.67 There is a slight increase of [OX]B during April–May (Fig. 5a), which could be due to the transport of ozone precursors from the burning region.68
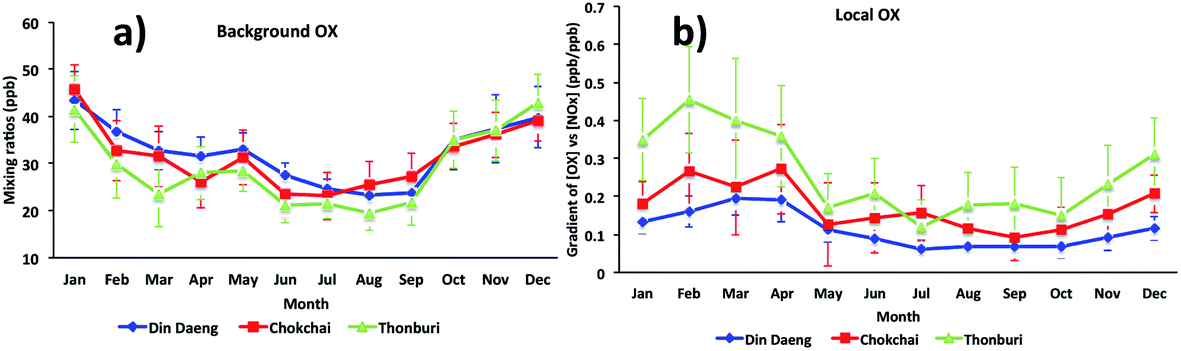 | ||
| Fig. 5 The seasonal cycles of (a) background OX and (b) local OX contributions in Bangkok roadside sites (Din Daeng, 2005–2014; Chokchai, 2005–2014; Thonburi, 2005–2018). | ||
Diurnal variation of local and background OX
The diurnal cycles of [OX]B in London and Bangkok roadside sites are found to reach a maximum during the afternoon (Fig. 6 and 7) which is attributed to a deepening boundary layer causing entrainment of ozone-rich air from aloft. The diurnal cycle is very consistent with that in similar studies for London7 and Bangkok25 roadside sites. | ||
| Fig. 6 The diurnal cycles of (a) background and (b) local oxidant levels in the Marylebone Road (2000–2019). | ||
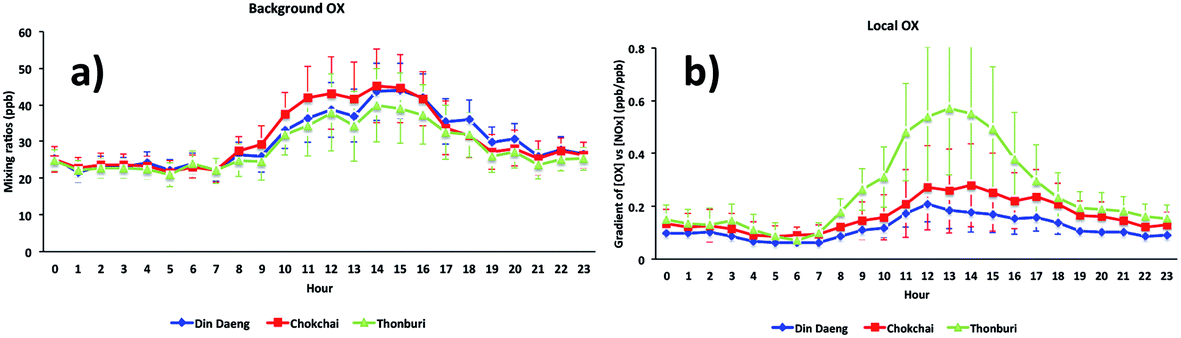 | ||
| Fig. 7 The diurnal cycles of (a) background and (b) local oxidant levels in Bangkok roadside sites (Din Daeng, 2005–2014; Chokchai, 2005–2014; Thonburi, 2005–2018). | ||
The diurnal variation of [OX]L is related to transport emissions of NO2. Thus, the levels of [OX]L at the Marylebone Road and Bangkok roadside sites are high throughout the daytime (Fig. 6 and 7). The diesel NOx emissions peak during the middle of the day.69 The NO2/NOx emission ratio is high for diesel engines which can increase even further with lower vehicle speeds.30 The high [OX]L in the Marylebone Road during the 2000s can be explained by the introduction of EURO III light duty diesel vehicles (fitted with oxidation catalysts) into the vehicle fleet. Thonburi is an industrial area, so the frequent movement of diesel engine heavy vehicles in this area can emit a significant amount of NOx as NO2 resulting in higher [OX]L levels compared with Din Daeng and Chokchai. The traffic at both Din Daeng and Chokchai throughout the day is busy, but, due to the close proximity of a bus stop at the Chokchai monitoring site, buses regularly stop and idle which could be responsible for enhanced [OX]L levels compared with that in Din Daeng. After sunset, the formation of [OX]L at both London and Bangkok roadside sites is inhibited and the planetary boundary layer becomes more stable resulting in [OX]L reduction through chemical reaction (e.g. O3 + NO2 → NO3 + O2) and physical processes (e.g. dry deposition).70 Since NO3 is building up in the night-time and rapidly photolysed during the day, this process regenerates local oxidants in early daylight hours.
Modelling of OX
We simulated model [OX = (NO2 + O3)] mixing ratios and compared with the measurements at the five ground sites in the centre of London. The results showed a general trend in peak [OX] which is well represented during summer months (Fig. 8). The correlation coefficients for the Westminster, Harlington, Eltham and Bloomsbury sites are high (r ∼ 0.8) suggesting that the model captures the [OX] trend very well. The model–measurement agreement is also reasonably good as the biases and root mean square errors (RMSE) for all these sites are within ∼10 ppb (Table 2). The Westminster and Bloomsbury sites are strongly influenced by local traffic, which cause increased biases and RMSEs compared with the Eltham and Harlington sites. The small correlation coefficient (r = 0.4) and high bias (−36 ppb) and RMSE (40 ppb) for the Marylebone Road suggests that the model agreement with measurement for this site is relatively poor compared with other London sites. This is likely to be a consequence of the monitoring station being in close proximity to a busy six-lane road in a street canyon, which regularly experiences high traffic volumes and prolonged periods of congestion. The site is subject to a well-characterized canyon vortex leading to concentrations dominated by wind direction. Although running at a relatively fine resolution of 15 km by 15 km, WRF-Chem-CRI is unable to pick up the local sources and sub grid-scale chemical dynamics, which drive the air quality at street level sites. Dispersion models such as ADMS take time-varying local sources (such as traffic at street level) into account, as well as incorporating features such as building, complex terrain, which alter the dynamics and thus composition of the surrounding atmosphere. Overall, the model is generally able to reproduce the [OX] levels in London to a reasonable extent and provide an accurate representation of air quality in complex urban environments.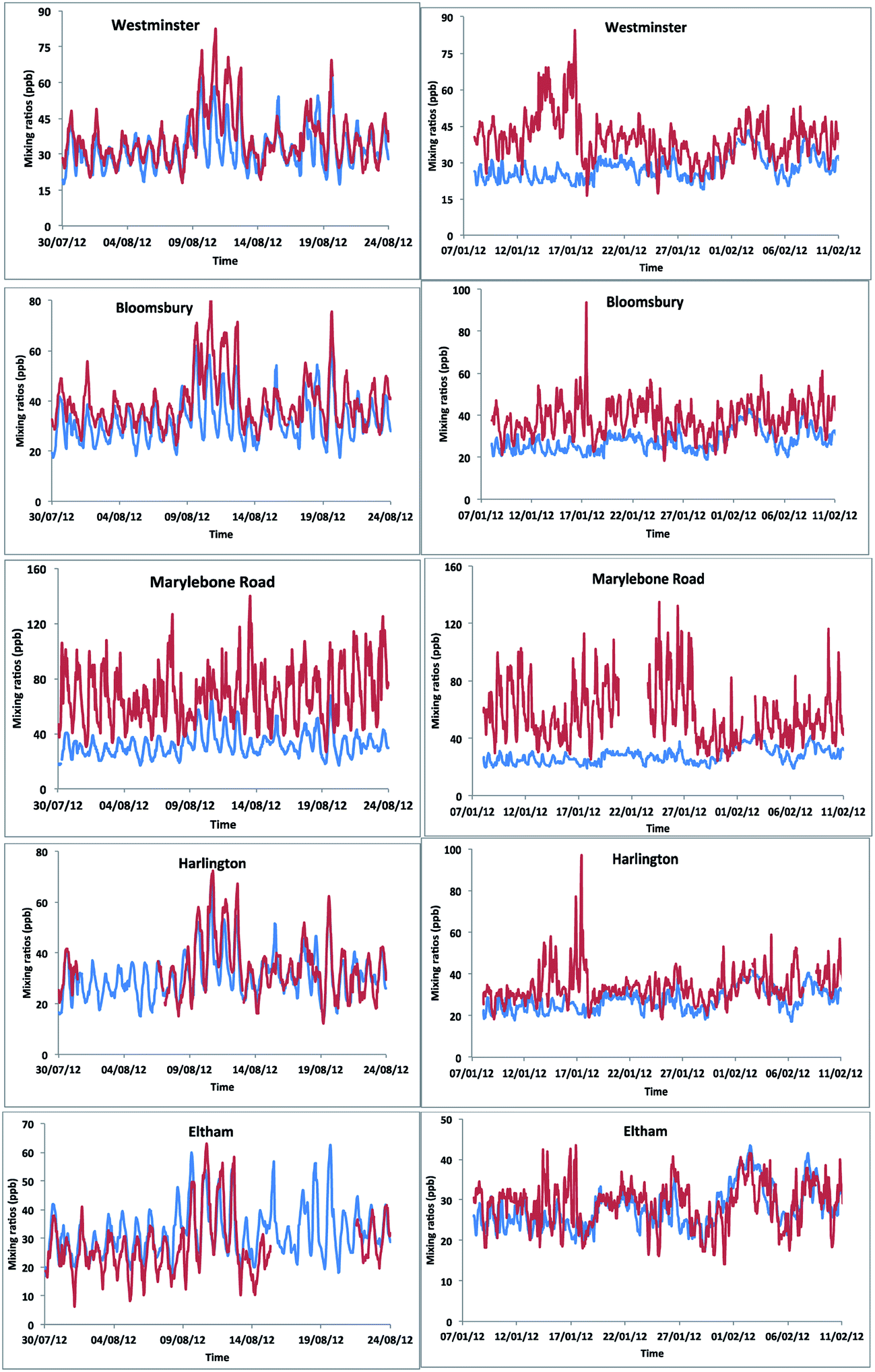 | ||
| Fig. 8 The model–measurement comparison of [OX] levels in London sites for the period of 30 July to 24 August 2012 (left side figures) and 8 January to 11 February 2012 (right side figures). The red and blue lines represent measurement and modelled data, respectively. | ||
| Site | Summer | Winter | ||||
|---|---|---|---|---|---|---|
| Bias (ppb) | RMSE (ppb) | Pearson correlation | Bias (ppb) | RMSE (ppb) | Pearson correlation | |
| Marylebone Road | −35.8 | 40.3 | 0.41 | −28.6 | 34.9 | 0.05 |
| Westminster | −4.2 | 7.9 | 0.78 | −12.0 | 15.7 | 0.07 |
| Harlington | −1.9 | 6.7 | 0.81 | −7.0 | 10.9 | 0.27 |
| Eltham | 4.2 | 7.5 | 0.78 | −1.2 | 5.3 | 0.52 |
| Bloomsbury | −7.1 | 9.7 | 0.77 | −10.9 | 13.3 | 0.37 |
There are many discrepancies in the [OX] peaks during winter months. The NOx emissions from vehicles are higher in winter months compared with summer months, thus the increased emissions fraction of NOx in the form of NO2 from vehicles can have a direct local-scale impact on the levels of [OX]. Except the Eltham site (a suburban site which experiences very low traffic volumes compared with other sites), the model–measurement agreements for all other sites have poor correlation coefficients, biases and RMSEs during winter months compared with summer months. This is likely to be a result of the resolution of the model, which is not enough to resolve the local effects (e.g. fraction of NOx emitted as NO2 from vehicle). In the model, the local sources are spread out across the grid cell, resulting in diffusion of the emissions and leading to lower simulated mixing ratios of [OX]. There have also been studies which have found significant underestimations (30–40%) of traffic-related NO2 emissions in the inventory data, NAEI, used by WRF-Chem-CRI.71 As traffic (diesel vehicles in particular) is a significant source of NO2 in urban environments such as London, the observed underestimation in modelled [OX] mixing ratios seen at London sites is therefore likely to be a consequence of the underestimation of traffic-related emissions in the inventory data used in the model.
WRF-Chem-CRI is able to reproduce the diurnal variations of background [OX] at London sites, with good agreement between mixing ratios in the modelled and measured data (biases/RMSE < 10 ppb and correlation coefficient > 0.5) (Fig. 9 and Table 3). In general, the [OX]B is underestimated by the model, which is likely due to the under-prediction of the diesel related VOCs used in the model.72 Furthermore, the VOCs used in the model were not constrained using the AURN dataset which can partly contribute the imbalances between model–measurement. However, there are times when peak [OX]B is overestimated by the model (e.g. summer time in Westminster, Bloomsbury and Eltham), which could be due to the regional photochemical events occurring below the resolution of the model. Westminster and Bloomsbury are urban background sites, but they are very close (within 2 km) to the traffic sites (e.g. Marylebone Road) and Eltham is a suburban site in London, which is also not far from the Marylebone Road (∼14 km). These sites lay on a large concentration gradient, thus the WRF-Chem model with 15 km × 15 km resolution was unable to distinguish between points beyond the areas. This can also explain the large model–measurement [OX]B deviation for the Marylebone Road site compared with the other London sites.
 | ||
| Fig. 9 The model–measurement comparison of background [OX] levels in London sites for the period of (a) 30 July to 24 August 2012 and (b) 8 January to 11 February 2012 (right side figure). The red, blue, black, yellow, green lines represent Marylebone Road, Bloomsbury, Eltham, Westminster and Harlington, respectively. The solid lines are measurement data and broken lines are modelled data. | ||
| Site | Summer | Winter | ||||
|---|---|---|---|---|---|---|
| Bias (ppb) | RMSE (ppb) | Pearson correlation | Bias (ppb) | RMSE (ppb) | Pearson correlation | |
| Marylebone Road | −9.9 | 12.3 | 0.83 | −7.2 | 7.8 | 0.66 |
| Westminster | 1.7 | 3.8 | 0.72 | −3.9 | 4.2 | 0.68 |
| Harlington | −4.2 | 6.6 | 0.68 | −2.2 | 2.5 | 0.69 |
| Eltham | 6.4 | 7.5 | 0.83 | −1.1 | 1.7 | 0.81 |
| Bloomsbury | 0.8 | 2.5 | 0.88 | −2.9 | 3.1 | 0.90 |
The model–measurement comparison plots (Fig. 10) for different months (January, May and September) show that the model matched the changes in [OX] levels with measurement very well for the Bangkok roadside site Thonburi with observed peaks and troughs at nearly similar times (correlation coefficients, r > 0.5). However, there is a large overprediction of model [OX] values with biases and RMSEs of ∼10 and ∼15 ppb for January, ∼17 and ∼27 ppb for May, and ∼21 and ∼26 ppb for September (Fig. 10 and Table 4) which is most likely arising from inaccurate NOx emissions (traffic, industrial) in the inventory data. Models are used to predict and forecast the effects of changes in emissions. They therefore require accurate emission inventory data and complete representations of physical and chemical atmospheric processes in order to do this as well as possible. There have been noted issues with emissions inventories (RETRO, INTEX-B, MACCity, SEAC4RS) most recently which cause an over-prediction of O3 mixing ratios in Thailand for March and December.73 The spatial distribution of the regional emissions, with hot spots seen in Bangkok, implemented in EDGAR is based on population, which does not reflect the real emission scenarios of Thailand. Although Bangkok has a large population, the stationary sources of emissions are likely to be overestimated in the EDGAR emission inventory. To improve the model–measurement deviation, the development of an alternative regional emission inventory based on emission control technology for stationary and mobile sources is necessary.
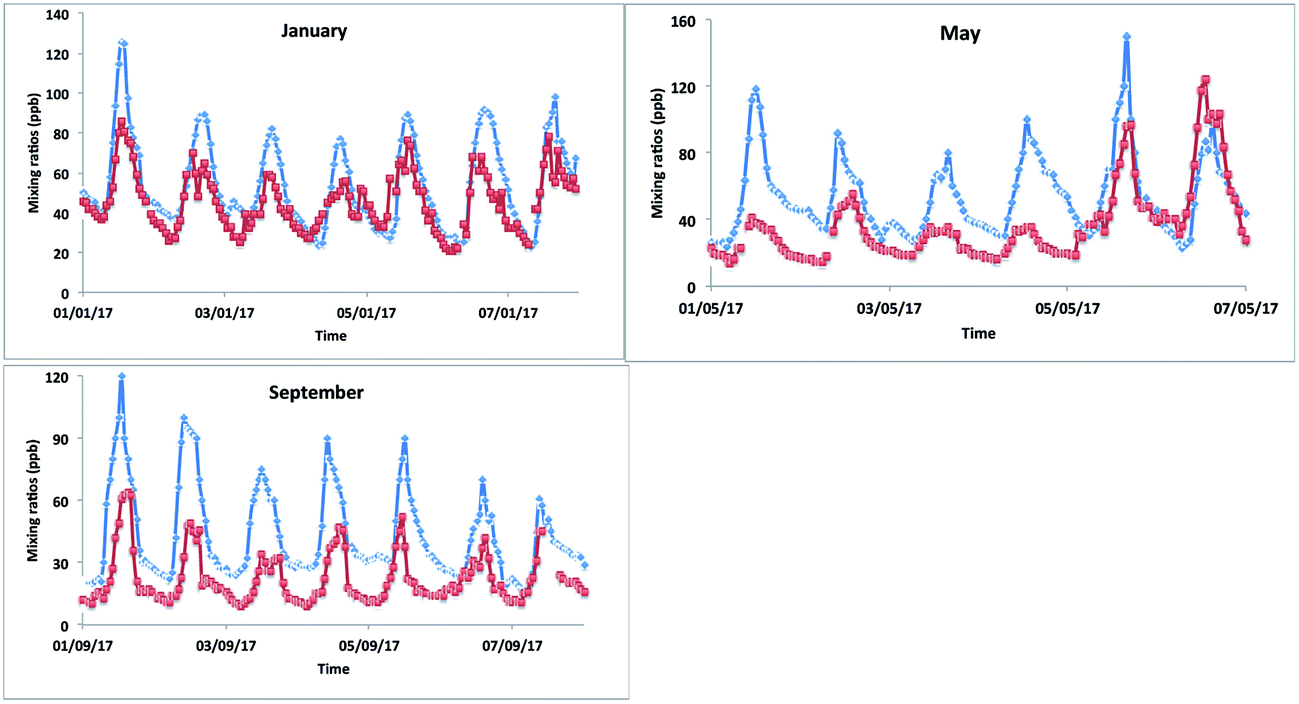 | ||
| Fig. 10 The model–measurement comparison of [OX] levels in the Bangkok roadside site Thonburi for January, May, and September 2017. The red and blue lines represent measurement and modelled data, respectively. | ||
| Jan | May | Sep | ||||||
|---|---|---|---|---|---|---|---|---|
| Bias (ppb) | RMSE (ppb) | Pear. corr. | Bias (ppb) | RMSE (ppb) | Pear. corr. | Bias (ppb) | RMSE (ppb) | Pear. corr. |
| 9.4 | 15.3 | 0.86 | 16.8 | 27.4 | 0.55 | 21.4 | 25.8 | 0.79 |
Conclusions
The background and local oxidant contributions at London and Bangkok roadside sites to the total oxidant levels are investigated and found to be 32 ppb [OX]B and 0.21[NOx] ppb [OX]L at Marylebone Road, 32 ppb [OX]B and 0.12[NOx] ppb [OX]L at Din Daeng, 29 ppb [OX]B and 0.26[NOx] ppb [OX]L at Thonburi and 31 ppb [OX]B and 0.18[NOx] ppb [OX]L at Chokchai. The seasonal variation of [OX]B shows a maximum in spring season for London and dry season for Bangkok. The diurnal variation of both [OX]L and [OX]B for both London and Bangkok shows a peak during the afternoon. Decreasing trends of [OX]L levels at London and Bangkok roadside sites are found for the period of 2001–2019 and 2005–2015, respectively. There is also a decreasing trend of [OX]B levels at Marylebone Road and Chokchai with no significant changes at Din Daeng and Thonburi. Overall, the adopted emission controls in both the U.K. and Thailand have been successful in the decrease of [OX]B and [OX]L levels. The [OX]L at the London roadside site is higher than that at the Bangkok roadside sites. However, a large number of policy initiatives have been taken following the London Mayor’s Air Quality Strategy. This includes the roll out of zero-emission buses, implementing the Ultra Low Emission Zone legislation and including the Congestion Charge Zone across London to improve the oxidant levels in central London. To assess the London Mayor’s policy, this type of analysis must be carried out periodically for checking the [OX] levels, which will be beneficial in terms of improved quality of life, for both people and ecosystems. The model, WRF-Chem-CRI was able to reproduce the temporal trends and mixing ratios of [OX]B in London well, highlighting its capability as a regional air quality model which could be a useful tool for predicting long-term changes in [OX]B by accounting for changes in the emissions. Periodic overestimations of model [OX] mixing ratios in the Bangkok roadside site suggest errors in the EDGAR emission inventory, which is based on population rather than emission control technology. Further analysis with scaling of the emission inventory would be required to determine the extent of this overestimation, and if there are other factors driving this observation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Department for Environment, Food and Rural Affairs (Defra) for supporting UK monitoring network data and Pollution Control Department (PCD) for providing Bangkok air pollution data. This work is the collaboration between UK and Thai scientists facilitated by funding from the GCRF Global Impact Accelerator Account 2018 (Grant code-EP/S515899/1). DES, JCM, PN, RH, and MAHK also thank Newton NERC Fund (grant code-NE/P014674/1), Thailand Research Fund (grant code-RDG6030008), Bristol ChemLabS and the Primary Science Teaching Trust under whose auspices various aspects of this work were supported. AF thanks EPSRC for her PhD studentship. CJP’s work was carried out at Jet Propulsion Laboratory, California Institute of Technology, under contract with the National Aeronautics and Space Administration (NASA), and was supported by the Upper Atmosphere Research and Tropospheric Chemistry Programs. © 2020 all rights reserved.References
- WHO, Ambient Air Pollution. A Global Assessment of Exposure and Burden of Disease, World Health Organisation, Geneva, Switzerland, 2016 Search PubMed.
- P. A. Leighton, Photochemistry of Air Pollution, Academic Press, New York, 1961 Search PubMed.
- J. H. Seinfeld and S. N. Pandis, in Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, New Jersey, United States, 2nd edn, 2006 Search PubMed.
- D. Kley, H. Geiss and V. A. Mohnen, Atmos. Environ., 1994, 28, 149–158 CrossRef CAS.
- L. J. Clapp and M. E. Jenkin, Atmos. Environ., 2001, 35, 6391–6405 CrossRef CAS.
- M. E. Jenkin, Atmos. Environ., 2004, 38, 5117–5129 CrossRef CAS.
- M. E. Jenkin, Atmos. Environ., 2004, 38, 5131–5138 CrossRef CAS.
- A. Notario, I. Bravo, J. A. Adame, Y. Díaz-de-Mera, A. Aranda, A. Rodríguez and D. Rodríguez, Atmos. Res., 2012, 104–105, 217–226 CrossRef CAS.
- N. Mazzeo, L. Venegas and H. Choren, Atmos. Environ., 2005, 39, 3055–3068 CrossRef CAS.
- S. Beirle, K. F. Boersma, U. Platt, M. G. Lawrence and T. Wagner, Science, 2011, 333, 1737–1739 CrossRef CAS.
- J. Cyrys, M. Eeftens, J. Heinrich, C. Ampe, A. Armengaud, R. Beelen, T. Bellander, T. Beregszaszi, M. Birk, G. Cesaroni, M. Cirach, K. de Hoogh, A. De Nazelle, F. de Vocht, C. Declercq, A. Dedele, K. Dimakopoulou, K. Eriksen, C. Galassir, R. Grauleviciene, G. Grivas, O. Gruzieva, A. H. Gustafsson, B. Hoffmann, M. Iakovides, A. Ineichen, U. Kramer, T. Lanki, P. Lozano, C. Madsen, K. Meliefste, L. Modig, A. Moelter, G. Mosler, M. Nieuwenhuijsen, M. Nonnemacher, M. Oldenwening, A. Peters, S. Pontet, N. Probst-Hensch, U. Quass, O. Raaschou-Nielsen, A. Ranzi, D. Sugiri, E. G. Stephanou, P. Taimisto, M. Y. Tsai, E. Vaskovi, S. Villani, M. Wang, B. Brunekreef and G. Hoek, Atmos. Environ., 2012, 62, 374–390 CrossRef CAS.
- T. Wang, P.-C. Wang, F. Hendrick, H. Yu and M. Van Roozendael, Atmos. Oceanic Sci. Lett., 2015, 8, 392–396 Search PubMed.
- L. Jaeglé, L. Steinberger, R. V. Martin and K. Chance, Faraday Discuss., 2005, 130, 407–423 RSC.
- R. J. van der A, H. J. Eskes, K. F. Boersma, T. P. C. van Noije, M. Van Roozendael, I. De Smedt, D. H. M. U. Peters and E. W. Meijer, J. Geophys. Res., 2008, 113, D04302 CrossRef.
- F. Liu, S. Beirle, Q. Zhang, R. J. van der A, B. Zheng, D. Tong and K. He, Atmos. Chem. Phys., 2017, 17, 9261–9275 CrossRef CAS.
- M. L. Williams, R. W. Atkinson, H. R. Anderson and F. J. Kelly, Air Qual., Atmos. Health, 2014, 7, 407–414 CrossRef CAS.
- C. Yang, H. Li, R. Chen, W. Xu, C. Wang, L. A. Tse, Z. Zhao and H. Kan, Environ. Res. Lett., 2016, 11, 074014 CrossRef.
- S. Weichenthal, L. L. Pinault and R. T. Burnett, Sci. Rep., 2017, 7, 16401 CrossRef.
- United Nations, The World’s cities in 2018, Data Booklet, New York, United States, 2018 Search PubMed.
- R. Brook and K. King, Updated analysis of air pollution exposure in London, Report to Greater London Authority, Aether, 2017, https://www.london.gov.uk/sites/default/files/aether_updated_london_air_pollution_exposure_final_20-2-17.pdf, accessed June 2020 Search PubMed.
- City of London Air Quality Strategy, Delivering healthy air in the City of London, The Air Quality Team, London, UK, 2019–2024 Search PubMed.
- UNECE, The 1999 Gothenburg Protocol to Abate Acidification, Eutrophication and Ground-Level Ozone, https://www.unece.org/env/lrtap/multi_h1.htm, accessed June 2020 Search PubMed.
- B. Richmond, A. Misra, M. Broomfield, P. Brown, E. Karagianni, T. Murrells, Y. Pang, N. Passant, B. Pearson, R. Stewart, G. Thistlethwaite, D. Wakeling, C. Walker, J. Wiltshire, M. Hobson, M. Gibbs, T. Misselbrook, U. Dragosits and S. Tomlinson, UK informative inventory report (1990 to 2017), Ricardo Energy & Environment, Defra, https://uk-air.defra.gov.uk/assets/documents/reports/cat09/1904121008_GB_IIR_2019_v2.0.pdf, accessed June 2020 Search PubMed.
- P. Watcharavitoon, C.-P. Chio and C.-C. Chan, Aerosol Air Qual. Res., 2013, 13, 1741–1754 CrossRef CAS.
- P. Uttamang, V. P. Aneja and A. F. Hanna, Atmos. Chem. Phys., 2018, 18, 12581–12593 CrossRef CAS.
- M. G. Lawrence, T. M. Butler, J. Steinkamp, B. R. Gurjar and J. Lelieveld, Atmos. Chem. Phys., 2007, 7, 3969–3987 CrossRef CAS.
- N. Castell-Balaguer, L. Téllez and E. Mantilla, Environ. Sci. Pollut. Res., 2012, 19, 3461–3480 CrossRef CAS.
- M. A. H. Khan, J. Clements, D. Lowe, G. McFiggans, C. J. Percival and D. E. Shallcross, Atmos. Res., 2019, 229, 145–156 CrossRef CAS.
- A. Charron and R. M. Harrison, Environ. Sci. Technol., 2005, 39, 7768–7776 CrossRef CAS.
- AQEG, Nitrogen dioxide in the United Kingdom. Report of the UK Air Quality Expert Group (AQEG), Prepared for the Department for Environment, Food and Rural Affairs, the Scottish Executive, the Welsh Assembly and the Department of the Environment in Northern Ireland, Defra Publications, London, 2004 Search PubMed.
- E. J. Dunlea, S. C. Herndon, D. D. Nelson, R. M. Volkamer, F. San Martini, P. M. Sheehy, M. S. Zahniser, J. H. Shorter, J. C. Wormhoudt, B. K. Lamb, E. J. Allwine, J. S. Gaffney, N. A. Marley, M. Grutter, C. Marquez, S. Blanco, B. Cardenas, A. Retama, C. R. Ramos Villegas, C. E. Kolb, L. T. Molina and M. J. Molina, Atmos. Chem. Phys., 2007, 7, 2691–2704 CrossRef CAS.
- A. M. Winer, J. W. Peters, J. P. Smith and J. N. Pitts, Environ. Sci. Technol., 1974, 8, 1118–1121 CrossRef CAS.
- AEA, QA/QC procedures for the UK automatic urban and rural air quality monitoring network (AURN), Report to Defra and the Developed Administrations, (AEAT/ENV/R/2837), 2009 Search PubMed.
- G. A. Grell, S. E. Peckkam, R. Schmitz, S. A. McKeen, G. Frost, W. C. Skamarock and B. Eder, Atmos. Environ., 2005, 39, 6957–6975 CrossRef CAS.
- S. Archer-Nicholls, D. Lowe, S. Utembe, J. Allan, R. A. Zaveri, J. D. Fast, Ø. Hodnebrog, H. D. van der Gon and G. McFiggans, Geosci. Model Dev., 2014, 7, 2557–2579 CrossRef.
- D. P. Dee, S. M. Uppala, A. J. Simmons, P. Berrisford, P. Poli, S. Kobayashi, U. Andrae, M. A. Balmaseda, G. Balsamo, P. Bauer, P. Bechtold, A. C. M. Beljaars, L. van de Berg, J. Bidlot, N. Bormann, C. Delsol, R. Dragani, M. Fuentes, A. J. Geer, L. Haimberger, S. B. Healy, H. Hersbach, E. V. Holm, L. Isaksen, P. Kallberg, M. Kohler, M. Matricardi, A. P. McNally, B. M. Monge-Sanz, J.-J. Morcrette, B.-K. Park, C. Peubey, P. de Rosnay, C. Tavolato, J.-N. Thepaut and F. Vitart, Q. J. R. Meteorol. Soc., 2011, 137, 553–597 CrossRef.
- L. K. Emmons, S. Walters, P. G. Hess, J.-F. Lamarque, G. G. Pfister, D. Fillmore, C. Granier, A. Guenther, D. Kinnison, T. Laepple, J. Orlando, X. Tie, G. Tyndall, C. Wiedinmyer, S. L. Baughcum and S. Kloster, Geosci. Model Dev., 2010, 3, 43–67 CrossRef.
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef CAS.
- J. J. P. Kuenen, A. J. H. Visschedijk, M. Jozwicka and H. A. C. Denier van der Gon, Atmos. Chem. Phys., 2014, 14, 10963–10976 CrossRef.
- D. Simpson, A. Benedictow, H. Berge, R. Bergström, L. D. Emberson, H. Fagerli, C. R. Flechard, G. D. Hayman, M. Gauss, J. E. Jonson, M. E. Jenkin, A. Nyíri, C. Richter, V. S. Semeena, S. Tsyro, J.-P. Tuovinen, Á. Valdebenito and P. Wind, Atmos. Chem. Phys., 2012, 12, 7825–7865 CrossRef CAS.
- M. Crippa, D. Guizzardi, M. Muntean, E. Schaaf, F. Dentener, J. A. van Aardenne, S. Monni, U. Doering, J. G. J. Olivier, V. Pagliari and G. Janssens-Maenhout, Earth Syst. Sci. Data, 2018, 10, 1987–2013 CrossRef.
- G. Huang, R. Brook, M. Crippa, G. Janssens-Maenhout, C. Schieberle, C. Dore, D. Guizzardi, M. Muntean, E. Schaaf and R. Friedrich, Atmos. Chem. Phys., 2017, 17, 7683–7701 CrossRef CAS.
- Y. Chen, O. Wild, E. Ryan, S. K. Sahu, D. Lowe, S. Archer-Nicholls, Y. Wang, G. McFiggans, T. Ansari, V. Singh, R. S. Sokhi, A. Archibald and G. Beig, Atmos. Chem. Phys., 2020, 20, 499–514 CrossRef CAS.
- D. Lowe, WRF-Chem emission tool, The University of Manchester, http://github.com/douglowe/WRF_UoM_EMIT/releases/tag/v1.1, accessed June 2020 Search PubMed.
- S. Tiwari, A. Dahiya and N. Kumar, Atmos. Res., 2015, 157, 119–126 CrossRef CAS.
- I. Nikolova, A. R. MacKenzie, X. Cai, M. S. Alam and R. M. Harrison, Faraday Discuss., 2016, 189, 529–546 RSC.
- P. Takkanon and P. Chantarangul, Arch. Sci. Rev., 2019, 62, 35–46 CrossRef.
- T. A. M. Pugh, A. R. Mackenzie, D. Whyatt and C. N. Hewitt, Environ. Sci. Technol., 2012, 46, 7692–7699 CrossRef CAS.
- D. Carslaw and G. Rhys-Tyler, Atmos. Environ., 2013, 81, 339–347 CrossRef CAS.
- J. B. Heywood, in Internal Combustion Engine Fundamentals, Tata McGraw-Hill, USA, 1988 Search PubMed.
- AQEG, Trends in primary nitrogen dioxide in the UK, Report of the UK Air Quality Expert Group, AQEG, Prepared for the Department for Environment, Food and Rural Affairs, the Scottish Executive, the Welsh Assembly and the Department of the Environment in Northern Ireland, Defra Publications, London, 2007 Search PubMed.
- R. B. Ellison, S. P. Greaves and D. A. Hensher, Transp. Res. D, 2013, 23, 25–33 CrossRef.
- D. C. Carslaw, T. P. Murrells, J. Andersson and M. Keenan, Faraday Discuss., 2016, 189, 439–454 RSC.
- A. Font and G. W. Fuller, Environ. Pollut., 2016, 218, 463–474 CrossRef CAS.
- A. Font, L. Guiseppin, M. Blangiardo and V. Ghersi, Environ. Pollut., 2019, 249, 1–12 CrossRef CAS.
- M. E. Jenkin, Atmos. Environ., 2008, 42, 5434–5445 CrossRef CAS.
- R. G. Derwent, A. J. Manning, P. G. Simmonds, T. G. Spain and S. O’Doherty, Atmos. Environ., 2018, 179, 279–287 CrossRef CAS.
- M. E. Jenkin, S. R. Utembe and R. G. Derwent, Atmos. Environ., 2008, 42, 323–336 CrossRef CAS.
- K. Tørseth, W. Aas, K. Breivik, A. M. Fjaeraa, M. Fiebig, A. G. Hjellbrekke, C. Lund Myhre, S. Solberg and K. E. Yttri, Atmos. Chem. Phys., 2012, 12, 5447–5481 CrossRef.
- P. Cheewaphongphan, A. Junpen, S. Garivait and S. Chatani, Atmosphere, 2017, 8, 167 CrossRef.
- PCD, Vehicle Emission Regulation and Data, Pollution Control Department, Thailand, http://www.pcd.go.th/info_serv/air_diesel.html#s4, accessed June 2020 Search PubMed.
- B. N. Duncan, L. N. Lamsal, A. M. Thompson, Y. Yoshida, M. M. Hurwitz and K. E. Pickering, J. Geophys. Res.: Atmos., 2016, 121, 976–996 CAS.
- F. Dentener, T. Keating and H. Akimoto, in Hemispheric transport of air pollution, Part A: Ozone and Particulate Matter, Air Pollut. Stud. No. 17, U. N., New York, 2010 Search PubMed.
- M. McCarthy, L. Armstrong and N. Armstrong, Weather, 2019, 74, 382–387 CrossRef.
- N. T. K. Oanh, D. A. Permadi, N. H. Phuc and Y. Zhuang, in Air Quality Status and Management Practices in Asian Developing Countries, Integrated Air Quality Management: Asian Case Studies, ed. N. T. K. Oanh, CRC Press, Taylor & Francis Group, New York, 2013 Search PubMed.
- P. Pochanart, J. Kreasuwun, P. Sukasem, W. Geeratithadaniyom, M. S. Tabucanon, J. Hirokawa, Y. J. Kajii and H. Akimoto, Atmos. Environ., 2001, 35, 2657–2668 CrossRef CAS.
- B.-N. Zhang and N. T. Kim Oanh, Atmos. Environ., 2002, 36, 4211–4222 CrossRef CAS.
- T. Sonkaew and R. Macatangay, Environ. Res. Lett., 2015, 10, 065009 CrossRef.
- D. C. Carslaw and S. D. Beevers, Atmos. Environ., 2004, 38, 3585–3594 CrossRef CAS.
- M. Naja and S. Lal, J. Geophys. Res., 2002, 107, 4197 CrossRef.
- A. R. Vaughan, J. D. Lee, P. K. Misztal, S. Metzger, M. D. Shaw, A. C. Lewis, R. M. Purvis, D. C. Carslaw, A. H. Goldstein, C. N. Hewitt, B. Davison, S. D. Beevers and T. G. Karl, Faraday Discuss., 2016, 189, 455–472 RSC.
- R. E. Dunmore, J. R. Hopkins, R. T. Lidster, J. D. Lee, M. J. Evans, A. R. Rickard, A. C. Lewis and J. F. Hamilton, Atmos. Chem. Phys., 2015, 15, 9983–9996 CrossRef CAS.
- T. Amnuaylojaroen, M. C. Barth, L. K. Emmons, G. R. Carmichael, J. Kreasuwun, S. Prasitwattanaseree and S. Chantara, Atmos. Chem. Phys., 2014, 14, 12983–13012 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data (Fig. S1–S5 and Table S1). See DOI: 10.1039/d0fd00086h |
| This journal is © The Royal Society of Chemistry 2021 |