 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIs reducing new particle formation a plausible solution to mitigate particulate air pollution in Beijing and other Chinese megacities?†
Markku
Kulmala
 *ab,
Lubna
Dada
b,
Kaspar R.
Daellenbach
b,
Chao
Yan
ab,
Dominik
Stolzenburg
b,
Jenni
Kontkanen
b,
Ekaterina
Ezhova
b,
Simo
Hakala
b,
Saana
Tuovinen
b,
Tom V.
Kokkonen
b,
Mona
Kurppa
b,
Runlong
Cai
b,
Ying
Zhou
a,
Rujing
Yin
c,
Rima
Baalbaki
b,
Tommy
Chan
ab,
Biwu
Chu
bd,
Chenjuan
Deng
c,
Yueyun
Fu
c,
Maofa
Ge
e,
Hong
He
d,
Liine
Heikkinen
b,
Heikki
Junninen
f,
Yiliang
Liu
g,
Yiqun
Lu
g,
Wei
Nie
h,
Anton
Rusanen
b,
Ville
Vakkari
ij,
Yonghong
Wang
b,
Gan
Yang
g,
Lei
Yao
b,
Jun
Zheng
k,
Joni
Kujansuu
ab,
Juha
Kangasluoma
ab,
Tuukka
Petäjä
abh,
Pauli
Paasonen
b,
Leena
Järvi
bl,
Douglas
Worsnop
bm,
Aijun
Ding
h,
Yongchun
Liu
a,
Lin
Wang
g,
Jingkun
Jiang
c,
Federico
Bianchi
ab and
Veli-Matti
Kerminen
b
*ab,
Lubna
Dada
b,
Kaspar R.
Daellenbach
b,
Chao
Yan
ab,
Dominik
Stolzenburg
b,
Jenni
Kontkanen
b,
Ekaterina
Ezhova
b,
Simo
Hakala
b,
Saana
Tuovinen
b,
Tom V.
Kokkonen
b,
Mona
Kurppa
b,
Runlong
Cai
b,
Ying
Zhou
a,
Rujing
Yin
c,
Rima
Baalbaki
b,
Tommy
Chan
ab,
Biwu
Chu
bd,
Chenjuan
Deng
c,
Yueyun
Fu
c,
Maofa
Ge
e,
Hong
He
d,
Liine
Heikkinen
b,
Heikki
Junninen
f,
Yiliang
Liu
g,
Yiqun
Lu
g,
Wei
Nie
h,
Anton
Rusanen
b,
Ville
Vakkari
ij,
Yonghong
Wang
b,
Gan
Yang
g,
Lei
Yao
b,
Jun
Zheng
k,
Joni
Kujansuu
ab,
Juha
Kangasluoma
ab,
Tuukka
Petäjä
abh,
Pauli
Paasonen
b,
Leena
Järvi
bl,
Douglas
Worsnop
bm,
Aijun
Ding
h,
Yongchun
Liu
a,
Lin
Wang
g,
Jingkun
Jiang
c,
Federico
Bianchi
ab and
Veli-Matti
Kerminen
b
aAerosol and Haze Laboratory, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing, China. E-mail: markku.kulmala@helsinki.fi
bInstitute for Atmospheric and Earth System Research/Physics, Faculty of Science, University of Helsinki, Finland
cState Key Joint Laboratory of Environment Simulation and Pollution Control, State Environmental Protection Key Laboratory of Sources and Control of Air Pollution Complex, School of Environment, Tsinghua University, Beijing, China
dState Key Joint Laboratory of Environment Simulation and Pollution Control, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing, China
eInstitute of Chemistry, Chinese Academy of Sciences, Beijing, China
fInstitute of Physics, University of Tartu, Ülikooli 18, EE-50090 Tartu, Estonia
gShanghai Key Laboratory of Atmospheric Particle Pollution and Prevention (LAP3), Department of Environmental Science & Engineering, Fudan University, Shanghai 200438, China
hJoint International Research Laboratory of Atmospheric and Earth System Sciences, School of Atmospheric Sciences, Nanjing University, 210023 Nanjing, China
iFinnish Meteorological Institute, 00560 Helsinki, Finland
jAtmospheric Chemistry Research Group, Chemical Resource Beneficiation, North-West University, Potchefstroom, South Africa
kJiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Nanjing University of Information Science & Technology, Nanjing 210044, China
lHelsinki Institute of Sustainability Science, University of Helsinki, Finland
mAerodyne Research Inc., Billerica, Massachusetts 01821, USA
First published on 26th August 2020
Abstract
Atmospheric gas-to-particle conversion is a crucial or even dominant contributor to haze formation in Chinese megacities in terms of aerosol number, surface area and mass. Based on our comprehensive observations in Beijing during 15 January 2018–31 March 2019, we are able to show that 80–90% of the aerosol mass (PM2.5) was formed via atmospheric reactions during the haze days and over 65% of the number concentration of haze particles resulted from new particle formation (NPF). Furthermore, the haze formation was faster when the subsequent growth of newly formed particles was enhanced. Our findings suggest that in practice almost all present-day haze episodes originate from NPF, mainly since the direct emission of primary particles in Beijing has considerably decreased during recent years. We also show that reducing the subsequent growth rate of freshly formed particles by a factor of 3–5 would delay the buildup of haze episodes by 1–3 days. Actually, this delay would decrease the length of each haze episode, so that the number of annual haze days could be approximately halved. Such improvement in air quality can be achieved with targeted reduction of gas-phase precursors for NPF, mainly dimethyl amine and ammonia, and further reductions of SO2 emissions. Furthermore, reduction of anthropogenic organic and inorganic precursor emissions would slow down the growth rate of newly-formed particles and consequently reduce the haze formation.
1. Introduction
The rapid, large-scale urbanization and industrialization of China are unique in history. Consequently, China’s air pollution situation has worsened dramatically during the last 2–3 decades, as emissions from industrial activities, energy production and traffic have increased.1 However, the clean air policies implemented in China after 2010 have reduced the emissions of some major pollutants (e.g. SO2, CO, NOx, primary particulate matter) by up to several tens of percent, while having little effect on ammonia and volatile organic compound (VOC) emissions.2 Highly non-linear processes involving radiative transfer, atmospheric boundary layer (ABL) development, atmospheric chemistry and aerosol dynamics create an urban pollution cocktail and generate secondary atmospheric pollutants, including ultrafine particles, fine particulate matter and ozone.3–7 Among the multiple negative environmental and economic consequences of these pollutants,8 their adverse effects on human health are of the greatest concern in China.9–11Beijing and its vicinity is one of the most polluted areas in China, where high particle mass concentrations causing haze are most frequent in winter.12–16 The Beijing winter haze tends to occur in 5 to 7 day cycles, during which cleaner periods are followed by haze episodes that typically last for 3 days.3,17,18 The haze occurs preferably under southerly winds which bring warm and humid air along with air pollutants from the industrial areas located south of Beijing.3,12,13,16,19–23 The local meteorological conditions found to be associated with haze formation in Beijing include low wind speeds, high humidity of air and low intensity of solar radiation.18,23–25 Climate warming arguably influences weather conditions in such a way that the likelihood of winter haze episodes in Beijing has already increased, and is likely to increase further in the future.12,26–29
The development and ultimate severity of a haze episode depend on the interplay between air pollutant emissions, their atmospheric transport, and the processes responsible for accumulating and transforming these pollutants in the ABL.7 In the case of Beijing haze, prior research has focused mainly on the processes and feedbacks causing the highest particle mass concentrations, typically encountered during the final stages of haze development.23 In 2014, Guo et al.3 reported that atmospheric new particle formation (NPF) tends to precede winter haze episodes in Beijing. However, it has remained unclear whether NPF and haze formation are causally connected and, in case they are, to which extent NPF might affect the formation and properties of haze. Here, we investigate how NPF and subsequent particle growth affect the initial steps of haze formation in Beijing and how reduced NPF and/or subsequent growth can affect haze reduction.
2. Materials and methods
The measurement site is at the campus building of the Beijing University of Chemical Technology (BUCT) in the western part of Beijing (39°56′31′′N, 116°17′50′′E). BUCT is located near the 3rd Ring Road of Beijing, and therefore, close to fresh traffic emissions and is also surrounded by residential areas. The particle number size distribution of 1 nm to 10 μm was measured with a homemade Diethylene Glycol Scanning Mobility Particle Sizer (DEG-SMPS) and with a Particle Size Magnifier (PSM; Airmodus A11) and a Neutral cluster and Air Ion Spectrometer (NAIS; Airel Ltd) in parallel. Additionally, ions in the mobility diameter range of 0.8–42 nm were measured with the ion mode of the NAIS. More details on the measurements of the particle number size distributions and the parameters derived from the acquired data can be found in the ESI.† Additionally, meteorological variables were measured by the Vaisala automatic weather station AWS310 at the BUCT site. Trace gases including NOx, CO, O3 and SO2 were also measured at BUCT (see ESI† for more details). To measure the composition of PM2.5, an online Time-of-Flight Aerosol Chemical Speciation Monitor (ToF-ACSM) equipped with a PM2.5 lens was utilized at the BUCT station from February 21 to April 7, 2018. The Black Carbon (BC) mass concentration and absorption coefficient were measured by a dual spot aethalometer at BUCT. Details of the measurement, chemical composition and source apportionment of PM2.5 are provided in the ESI.†3. From new particle formation to haze
In large urban areas, like Eastern China where megacities are tied to each other, the sub-30 nm particle population has several sources and formation pathways. First, atmospheric chemical reactions produce vapors that participate in atmospheric clustering and subsequent growth of these clusters to form nucleation mode particles.30 Second, there are direct emissions of particles down to 5 nm diameter or even below, especially related to traffic emissions.31–33 Since various gas-to-particle conversion processes are heavily interlinked, it is difficult to resolve which fraction of sub-30 nm particles originates from direct (primary) emissions and which fraction results from atmospheric reactions (secondary formation).We performed comprehensive measurements in Beijing between 15 January 2018 and 31 March 2019. The measurement site is located on a rooftop of the campus building at the Beijing University of Chemical Technology (BUCT) in the western part of Beijing (Fig. S1†). A full description of the instrumentation used is given in the ESI, Section 1.† During the measurements, we observed 189 NPF event days, of which over 50 were followed by a haze episode (Fig. S2–S4†). While none of these haze episodes were extremely severe, most of them lasted for more than two days so that the total number of haze days was 158 (haze lasting at least 8 hours). Interestingly, there was an NPF event before almost every haze episode, whereas practically no NPF was detected during any of the haze episodes. Typically, within 15–20 hours, particles associated with the observed NPF events grew to sizes large enough (>100 nm diameter) to be considered as haze particles (Fig. S5†). Both atmospheric clustering and direct emissions, especially from traffic, contributed to the sub-30 nm particle population. The observed particle formation and growth rates (Table S1†) indicate that atmospheric clustering contributes significantly to NPF. Traffic and other sources contributed to the emissions of precursor compounds participating in clustering, particle growth and secondary aerosol mass formation during and before the haze periods.
Fig. 1 shows the median time evolution of the total particle number concentration (N), condensation sink (CS) and volume concentration (V), along with the corresponding size distributions, when considering all the 2 day periods during which an NPF event day was followed by a day with no apparent NPF (see Fig. S4† for this subset of days). The following day was in many cases a haze day. NPF starts to increase the sub-3 nm and total particle number concentrations at around 08:00 during the first day. As a result, N peaks at around noon and then declines gradually to a level that still considerably exceeds the total particle number concentration observed before the NPF event. Sub-3 nm particles contribute little to N after about 16:00–17:00 during the first day, suggesting that atmospheric clustering and particularly the growth of clusters to the nucleation mode become inactive after the late afternoon of the first day. Traffic emissions cause increases in N (Fig. 1) during both morning and evening rush hours, and contribute to the observed increases in CS and V during the following hours. Besides traffic, there are no detectable changes in N caused by other primary particle sources during the median 2 day period. Both CS and V increase in the afternoon of the first day, with no decline thereafter, indicating a continuous production of (secondary) particulate surface and mass until the end of the 2 day period. The observed increases are clearly faster during the daytime than at night, which suggests that photochemical reactions enhance the formation of secondary particulate matter. The temporal behavior of the daytime CS and V is very different between the two days: during the first day no increase in CS or V can be observed until a few hours after the start of NPF, while during the second day both increase notably in the early morning. This means either that the initiation of haze formation is connected causally with NPF, or that both of these phenomena are caused by a common set of processes or atmospheric conditions.
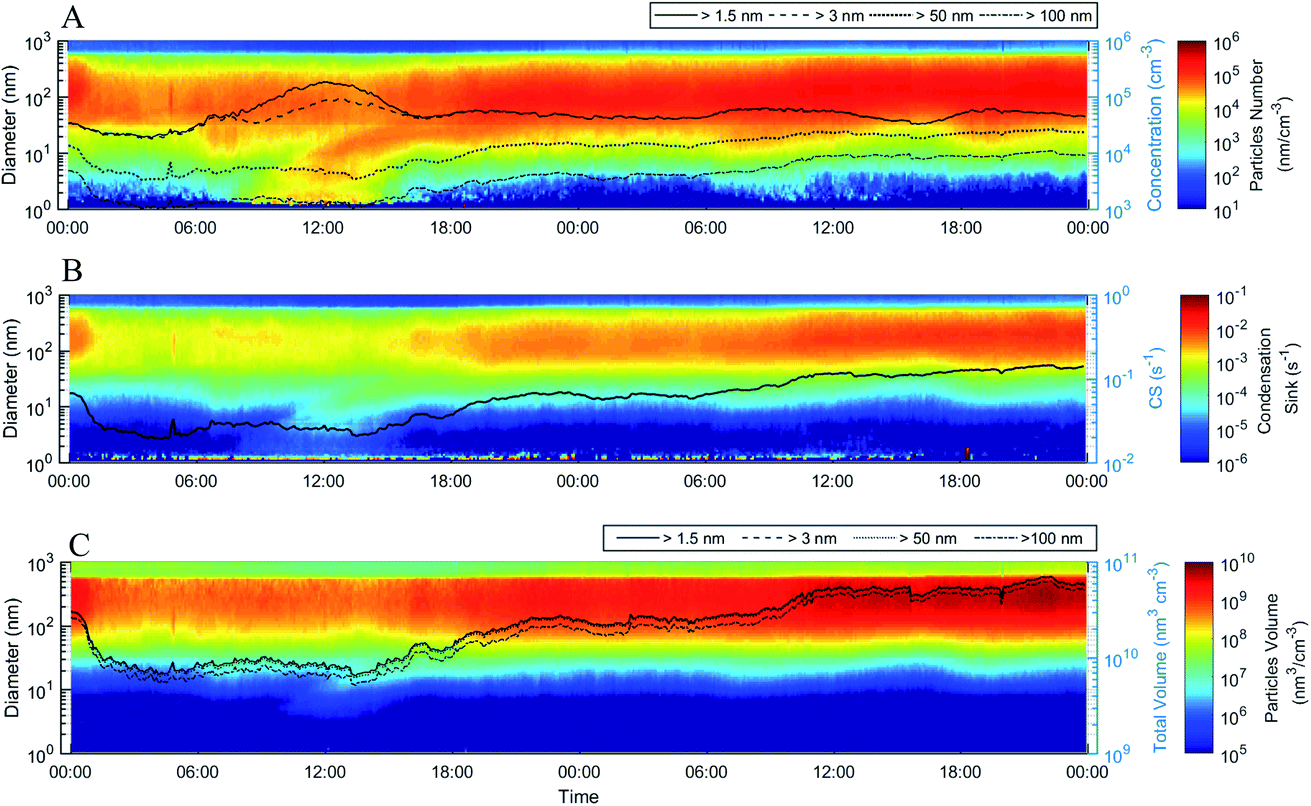 | ||
| Fig. 1 Time evolution (local time) of the (A) particle number size distribution and total number concentrations of particles larger than 1.5 nm, 3 nm, 50 nm and 100 nm in diameter, (B) condensation sink size distribution and total condensation sink (solid line), and (C) particle volume size distribution and total volume concentrations of particles larger than 1.5 nm, 3 nm, 50 nm and 100 nm in diameter. The surface plots are calculated as a median of 15 cases (Table S2†), in which an NPF event day was followed by a non-event day (this could be a haze day or a day in the process of forming haze). In (A), right after the start of NPF, we observe a major increase in the number concentrations of particles with diameters larger than 1.5 nm and 3 nm, as demonstrated by the solid and broad-dashed lines. Around 6 hours later, we observe an increase in the number concentrations of 50 nm and 100 nm particles. At the onset of NPF, we observe a small contribution of the recently-formed particles to the total condensation sink (solid line in (B)), which later systematically increases concurrent with the increase in larger particle numbers (>50 nm and 100 nm) observed in (A). Finally, similar to the CS, we observe a contribution to the total volume of particles, even though the volume of particles is dominated by larger particles, easily visible from the fact that particles with diameters larger than 1.5 nm, 3 nm, 50 nm and 100 nm have almost the same total volume. A drop in visibility is observed around midnight on the second day (Fig. S18†). | ||
Beijing is located not very far from the northern clean mountain regions. As a result, if the measured air mass continued to arrive from the north, the particles formed by atmospheric clustering grew slowly in size, making it difficult to separate them from particles emitted by traffic (Fig. S6†). However, when the measured air mass started to circle over more populated areas (Fig. S7†), the particles formed by atmospheric clustering grew rapidly beyond 30 nm (Fig. S8†) so that they became distinguishable from traffic emissions.
Despite the underlying uncertainties mentioned above, we attempted to estimate the fraction of accumulation mode particles (>100 nm diameter) originating from direct emissions compared to secondary formation through different approaches.34 Our results suggest that 65–80% of the accumulation mode particles originate from secondary formation (NPF) in the atmosphere during the observed haze episodes (Fig. 2A, B and S9, S10†), and that this fraction was already clearly above 50% before haze formation. Analyses of the particle phase composition combined with advanced source apportionment techniques (Fig. S11–S14†) showed that about 80–90% of the fine particle mass was secondary during the haze episodes (Fig. 2B), with secondary inorganic matter aerosols (SIAs, mostly NH4NO3 and (NH4)2SO4) being about twice as abundant as secondary organic aerosols (SOAs).
 | ||
| Fig. 2 (A) The fraction of primary particles in the accumulation mode (>100 nm) estimated using the NOx tracer method (see ESI, Section 3.1), for the percentile line of 1% in Fig. S9 and S10.† (B) Relative contributions of black carbon (BC), primary organic aerosols (POAs) from traffic, cooking, coal and biomass burning, secondary organic aerosols (SOAs), ammonium (NH4), nitrate (NO3), sulphate (SO4), and chloride (Cl) to the total PM2.5 mass concentration as a function of the non-refractory PM2.5 mass concentration. Our observations mean that most of the accumulation mode particles are secondary, which confirms our hypothesis that secondary sources also contribute to the number concentration and not only to the mass concentration. With increasing PM2.5, the relative contribution of secondary PM2.5 (SOAs, NH4, NO3, SO4, Cl) increases up to 90% of the total PM2.5 mass concentration. | ||
4. The estimated time it takes from NPF to haze formation
We investigated the time between NPF and subsequent formation of haze. These results (Fig. 3) show that faster haze formation (reflected by smaller time difference Δt on the y-axis) was associated primarily with a larger number of particles produced by the NPF event preceding the haze, and secondarily with a higher growth rate (GR) of the newly formed particles. Since the eventual clearing of haze is dictated by larger-scale weather conditions, the length of a haze episode is strongly related to how quickly haze forms. If the underlying cause governing the speed of haze formation is NPF, then our findings suggest that NPF has the potential to considerably increase the total number of haze days, typically by 1–2 days for each haze episode. On the other hand, since during our observation period we observed 57 haze episodes and since the normal cycle from haze formation to clearance lasts up to 7 days,3,17,18 we could cut the number of haze days to almost half by reducing the vapors responsible for the growth of nucleation mode particles. | ||
| Fig. 3 The time difference between the end of the NPF event and the time when the PM2.5 concentration reaches 100 μg m−3 as a function of the (left panel) total number of 3 nm particles produced during the NPF event and (right panel) total number of 10 nm particles produced during the NPF event. The points are divided into points with low growth rate (GR < 6 nm h−1) shown in blue and points with high growth rate (GR > 6 nm h−1) shown in yellow. The data points are binned based on the number concentration (x-axis). A linear fit for the points with low GR is shown as a dashed line and one for the points with high GR is shown as a solid line. Our results show that an increase in the total number of formed 3 nm or 10 nm particles results in a shorter time needed to form haze. Accordingly, a decrease in the intensity of NPF could result in delaying the haze formation. The data set with larger values of GR shows faster haze development. More specifically, decreasing the total number of formed 3 nm particles by a factor of 2 results in delaying the haze formation by up to 40 and 60 hours for cases with low and high GR, respectively. Reducing particle growth rates by 50% (a shift from yellow to blue points), e.g. by reducing anthropogenic VOC concentrations, will delay haze formation by between about 20 and 50 hours. | ||
After establishing the strong linkage between NPF and haze formation, we explore whether faster haze formation could be causally connected to NPF in the following sections.
First, we started from the unresolved puzzle of why and by which mechanisms NPF is possible in a polluted environment like Beijing.30 Based on our recent observations that clusters composed of sulfuric acid (H2SO4) and dimethyl amine (DMA) are crucial in producing new aerosol particles in both Shanghai and Beijing,35,36 we estimated the effective condensation sink (CSeff) of such clusters using heterogeneous nucleation theory37 (Fig. 4A, see details in the ESI, Section 3.3†). We found that, depending on the contact angle of heterogeneous nucleation and the properties of the nucleating vapor, CSeff can be substantially lower, by a factor of more than 100, than CS obtained using the traditional assumption that clusters are scavenged by larger (mainly accumulation mode) particles upon every collision with them. Low values of CSeff/CS for H2SO4–DMA clusters can explain why such clusters are able to survive in polluted air to produce growing nanoparticles. Additionally, this observation indicates that vapors with a low heterogeneous nucleation probability on accumulation mode particles might prefer condensing onto the nucleation mode over the accumulation mode. Investigating the contact angles of vapours under atmospherically relevant conditions remains essential, especially since the contact angles of nucleation of atmospheric vapors are still unknown.
 | ||
| Fig. 4 (A) The ratio between effective and total condensation sink, CSeff/CS, for sulfuric acid–DMA clusters as a function of condensing cluster mass was estimated using heterogeneous nucleation theory with a constant contact angle of 45° (see details in the ESI, Section 3.4†). (B) Measured and modeled effect of the growth rate of freshly formed particles by NPF on the time needed for formation of haze. The measured data in the figure covers the 15 months in Beijing between Jan 1 2018 and Mar 31 2019; the Shanghai data (see the ESI, Section 1.9† for details) spans the dates from 25 February 2014 to 25 February 2016. The curve shows the results of model calculations. The model simulations are described in the ESI (Section 3.3).† | ||
Second, regardless of their exact origin and formation pathway, fresh particles produced by NPF are likely to have a chemical composition very different from that of accumulation mode particles. The chemically distinct aerosol surface and volume of these particles facilitate heterogeneous reactions that contribute to their growth38 and, as a side product, produce secondary aerosol mass. We found that the growth rates of aerosol particles produced by NPF (measured as a function of time and size, Fig. S7†) appear not to depend on the particle size for particles larger than 25 nm. This is consistent with surface reactions (reactive uptake) being the dominant growth mechanism39 for these particles.
Third, we investigated the effect of enhanced aerosol mass growth (dM/dt = ΔM/Δt) due to surface reactions on haze formation by assuming that the increased reactive surface area is due to NPF. We used a simple conceptual model and set the initial (before NPF) aerosol mass concentration and growth rate equal to 10 μg m−3 and 1 μg m−3 per hour, respectively. The results (Table 1) show that even small enhancements in the mass growth rate due to new reactive aerosol surfaces will lead to significant increases in the accumulated aerosol mass, and that a mass growth rate enhancement of 10% is sufficient for haze (100 μg m−3) to form within 24 hours. This supports our observations that without NPF the time needed for haze formation is significantly longer.
| Enhanced mass growth rate (times the original value) | Additional aerosol mass after 10 hours | Additional aerosol mass after 24 hours | The time it takes for the aerosol mass concentration to reach 100 μg m−3 |
|---|---|---|---|
| 1.01 | 1 μg m−3 | 6.5 μg m−3 | 57 hours |
| 1.04 | 4.8 μg m−3 | 37.5 μg m−3 | 31 hours |
| 1.10 | 16 μg m−3 | 212.4 μg m−3 | 18 hours |
How would preferential heterogeneous nucleation and reactive uptake on the nucleation mode compared with the accumulation mode affect how fast haze forms? We performed simulations using a simple but realistic model (see ESI, Section 3.4, Fig. S15†) that has two modes, a growing mode (originating from NPF) and an accumulation mode, and two different vapors. We assume that one of these vapors condenses irreversibly onto both modes, representing all low-volatility vapors, whereas the other vapor can be taken up only by the growing mode, representing vapors that show preferential heterogeneous nucleation or reactive uptake with nucleation mode particles. By repeating these simulations for a large number of cases with different initial conditions, we obtained essentially the same result as that observed in the atmosphere (see Fig. 4B): higher growth rates of the nucleation particle mode following NPF lead to faster haze formation.
5. Potential solutions for mitigating winter haze
In Fig. 5 we summarize our main findings from clustering to haze formation. The particulate matter formed during haze episodes in Beijing is mainly secondary, in terms of both particle number and mass concentration (Fig. 2). This, together with our other findings listed above, suggests that a reduction in the strength of NPF and subsequent particle growth would delay the starting times of haze episodes and reduce particle mass concentrations during these episodes. By reducing the number of particles formed during an NPF event by a factor of 4, we might be able to delay the initiation of haze by up to 40 hours, particularly if we can decrease their growth rate at the same time. The most effective way to reduce NPF strength is to reduce DMA emissions together with H2SO4 concentrations originating predominantly from gas-phase SO2 oxidation, since H2SO4–DMA clustering has been shown to be an effective pathway in megacities.35 Reductions in anthropogenic inorganic and organic gas-phase precursor emission would decrease the growth rates of newly-formed particles and further delay haze formation. A potential delay of 1–2 days per haze period would mean 16 to 32 fewer haze days each winter in Beijing. | ||
| Fig. 5 The main mechanisms of NPF and subsequent particle growth. The initial stages of NPF are dominated by formation of sulfuric acid–DMA clusters, with additional contributions by ammonia. Growth of particles larger than 3 nm in diameter is mainly due to anthropogenic low-volatility organic and inorganic compounds. Heterogeneous chemistry, including reactive uptake of aerosol precursors, is crucial for the production of secondary aerosol mass. | ||
Major clean air policies have already been implemented to reduce SO2 emissions in China,2 especially in the North China Plain surrounding Beijing. As a result, SO2 concentrations in the surface air have decreased considerably during the recent years.40 Though the sulfate aerosol mass decreased, the observed reduction in the PM2.5 concentration was smaller,40 partly due to compensating effects of more efficient nitrate formation.41 Other contributors to PM2.5 (SOAs, NH4+ and NO3−) will likely be targeted in future emission reductions. When it comes to particle number concentrations, practically no change in the NPF event frequency in Beijing was observed in the past four years (Fig. S3†). This indicates that SO2 concentrations have probably not yet reached levels low enough to make them a limiting factor for NPF in Beijing. The apparent lack of major emission changes for the other precursors for NPF and subsequent particle growth, especially NH3 and VOCs2 and probably also amines, during recent years is consistent with this picture.
During our research period of about 400 days in Beijing, we had 189 NPF event days and more than 50 of them were followed by a haze episode. In practice almost no haze episodes were recorded without a preceding NPF event. Based on our results, we identify potential solutions to reduce aerosol formation and growth rates that will reduce the strength and frequency of NPF, leading to less haze formation: (1) further reducing SO2 emissions to decrease H2SO4 concentrations and weaken both NPF and formation of particulate sulfate; (2) reducing amine and ammonia emissions to decrease the strength of NPF; and (3) reducing anthropogenic organic and inorganic gas-phase precursor emissions to decrease the rate at which particles formed by NPF grow into haze particles. Using our current emission estimates for the Beijing–Tianjing–Hebei area (see ESI†), the regional SO2 emissions are split mainly between industrial combustion (60%), residential combustion (20%) and power production (10%), with more than 70% of these emissions being related to coal combustion, whereas approximately 90% of the NH3 emissions originate from agriculture. No official emission data for amines in this region exists, but our measurements in Beijing indicate a large contribution from local traffic. Based on Fig. 3 and 4, a reduction by a factor of 3–5 in the particle growth rate will delay the onset of haze by 1–3 days and consequently provide more time for the synoptic meteorological situation to change. This has the potential to cut the total number of haze days by more than half. In order to verify the efficiency of imposed control mechanisms, and to understand how the mutual effects of aerosol precursor emissions and changes in condensation sink affect NPF and ultimately haze, we need continuous and comprehensive observations in Chinese megacities, as suggested by Kulmala.6
Author contributions
All co-authors contributed to revising and commenting on the manuscript.Scientific discussions and interpretations
MK, LD, KRD, CY, DS, JKon, EE, TP, PP, DW, AD, LW, JJ, FB, VMK.Data analysis
MK, LD, KRD, CY, JKon, EE, SH, ST, TK, MoK, RC, YZ, RY, RB.Modeling
EE, ST.Writing
MK, LD, KRD, CY, DS, JKon, VMK.Measurements
KRD, CY, TK, YZ, RY, TC, BC, CD, YF, LH, HJ, YiLiu, YLu, AR, VV, YW, GY, LY, JZ, JKan, JKuj, YoLiu.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Maija Peltola for providing the growth rate code. The work is supported by the Academy of Finland via the Center of Excellence in Atmospheric Sciences (project no. 307331) and the European Research Council via ATM-GTP (742206). This research has also received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no. 895875 (“NPF-PANDA”), the Academy of Finland (project no. 316114 & 315203) as well as the Doctoral Programme in Atmospheric Sciences at the University of Helsinki. Partial support from the National Key R&D Program of China (2017YFC0209503 & 2017YFC0209505), the National Natural Science Foundation of China (21876094 & 91644213 & 41730106), and Samsung PM2.5 SRP is acknowledged. Moreover, this research received support from a Swiss National Science postdoc mobility grant (P2EZP2_181599).References
- R. Han, et al., Spatial and temporal variation of haze in China from 1961 to 2012, J. Environ. Sci., 2016, 46, 134–146, DOI:10.1016/j.jes.2015.12.033.
- B. Zheng, et al., Trends in China’s anthropogenic emissions since 2010 as the consequence of clean air actions, Atmos. Chem. Phys., 2018, 18, 14095–14111, DOI:10.5194/acp-18-14095-2018.
- S. Guo, et al., Elucidating severe urban haze formation in China, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17373–17378, DOI:10.1073/pnas.1419604111.
- R.-J. Huang, et al., High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 2014, 514, 218, DOI:10.1038/nature13774.
- M. Kulmala, Atmospheric chemistry: China’s choking cocktail, Nature, 2015, 526, 497, DOI:10.1038/526497a.
- M. Kulmala, Build a global Earth observatory, Nature, 2018, 553, 21–23, DOI:10.1038/d41586-017-08967-y.
- Z. Li, et al., Aerosol and boundary-layer interactions and impact on air quality, Natl. Sci. Rev., 2017, 4, 810–833, DOI:10.1093/nsr/nwx117.
- Y. Gu, et al., Impacts of sectoral emissions in China and the implications: air quality, public health, crop production, and economic costs, Environ. Res. Lett., 2018, 13, 084008, DOI:10.1088/1748-9326/aad138.
- R. Burnett, et al., Global estimates of mortality associated with long-term exposure to outdoor fine particulate matter, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9592–9597, DOI:10.1073/pnas.1803222115.
- J. Hu, et al., Premature Mortality Attributable to Particulate Matter in China: Source Contributions and Responses to Reductions, Environ. Sci. Technol., 2017, 51, 9950–9959, DOI:10.1021/acs.est.7b03193.
- J. Lelieveld, J. S. Evans, M. Fnais, D. Giannadaki and A. Pozzer, The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 2015, 525, 367, DOI:10.1038/nature15371.
- W. Cai, K. Li, H. Liao, H. Wang and L. Wu, Weather conditions conducive to Beijing severe haze more frequent under climate change, Nat. Clim. Change, 2017, 7, 257, DOI:10.1038/nclimate3249.
- H. Chen and H. Wang, Haze Days in North China and the associated atmospheric circulations based on daily visibility data from 1960 to 2012, J. Geophys. Res.: Atmos., 2015, 120, 5895–5909, DOI:10.1002/2015jd023225.
- J. He, et al., Analyses of winter circulation types and their impacts on haze pollution in Beijing, Atmos. Environ., 2018, 192, 94–103, DOI:10.1016/j.atmosenv.2018.08.060.
- B. Lv, B. Zhang and Y. Bai, A systematic analysis of PM2.5 in Beijing and its sources from 2000 to 2012, Atmos. Environ., 2016, 124, 98–108, DOI:10.1016/j.atmosenv.2015.09.031.
- X. Wang, et al., Characteristics and classification of PM2.5 pollution episodes in Beijing from 2013 to 2015, Sci. Total Environ., 2018, 612, 170–179, DOI:10.1016/j.scitotenv.2017.08.206.
- Y. Jia, K. A. Rahn, K. He, T. Wen and Y. Wang, A novel technique for quantifying the regional component of urban aerosol solely from its sawtooth cycles, J. Geophys. Res.: Atmos., 2008, 113, D21309, DOI:10.1029/2008jd010389.
- G. Zheng, et al., Episode-Based Evolution Pattern Analysis of Haze Pollution: Method Development and Results from Beijing, China, Environ. Sci. Technol., 2016, 50, 4632–4641, DOI:10.1021/acs.est.5b05593.
- C. Jiang, H. Wang, T. Zhao, T. Li and H. Che, Modeling study of PM2.5 pollutant transport across cities in China’s Jing–Jin–Ji region during a severe haze episode in December 2013, Atmos. Chem. Phys., 2015, 15, 5803–5814, DOI:10.5194/acp-15-5803-2015.
- X. Tie, et al., A budget analysis of the formation of haze in Beijing, Atmos. Environ., 2015, 100, 25–36, DOI:10.1016/j.atmosenv.2014.10.038.
- P. Wu, Y. Ding and Y. Liu, Atmospheric circulation and dynamic mechanism for persistent haze events in the Beijing–Tianjin–Hebei region, Adv. Atmos. Sci., 2017, 34, 429–440, DOI:10.1007/s00376-016-6158-z.
- G. J. Zheng, et al., Exploring the severe winter haze in Beijing: the impact of synoptic weather, regional transport and heterogeneous reactions, Atmos. Chem. Phys., 2015, 15, 2969–2983, DOI:10.5194/acp-15-2969-2015.
- J. Zhong, et al., Feedback effects of boundary-layer meteorological factors on cumulative explosive growth of PM2.5 during winter heavy pollution episodes in Beijing from 2013 to 2016, Atmos. Chem. Phys., 2018, 18, 247–258, DOI:10.5194/acp-18-247-2018.
- Z. Chen, et al., Understanding meteorological influences on PM2.5 concentrations across China: a temporal and spatial perspective, Atmos. Chem. Phys., 2018, 18, 5343–5358, DOI:10.5194/acp-18-5343-2018.
- L. Zhang, T. Wang, M. Lv and Q. Zhang, On the severe haze in Beijing during January 2013: unraveling the effects of meteorological anomalies with WRF-Chem, Atmos. Environ., 2015, 104, 11–21, DOI:10.1016/j.atmosenv.2015.01.001.
- H. Chen, H. Wang, J. Sun, Y. Xu and Z. Yin, Anthropogenic fine particulate matter pollution will be exacerbated in eastern China due to 21st century GHG warming, Atmos. Chem. Phys., 2019, 19, 233–243, DOI:10.5194/acp-19-233-2019.
- Z. Han, B. Zhou, Y. Xu, J. Wu and Y. Shi, Projected changes in haze pollution potential in China: an ensemble of regional climate model simulations, Atmos. Chem. Phys., 2017, 17, 10109–10123, DOI:10.5194/acp-17-10109-2017.
- L. Shen, D. J. Jacob, L. J. Mickley, Y. Wang and Q. Zhang, Insignificant effect of climate change on winter haze pollution in Beijing, Atmos. Chem. Phys., 2018, 18, 17489–17496, DOI:10.5194/acp-18-17489-2018.
- X. Zhang, J. Zhong, J. Wang, Y. Wang and Y. Liu, The interdecadal worsening of weather conditions affecting aerosol pollution in the Beijing area in relation to climate warming, Atmos. Chem. Phys., 2018, 18, 5991–5999, DOI:10.5194/acp-18-5991-2018.
- M. Kulmala, V. M. Kerminen, T. Petäjä, A. J. Ding and L. Wang, Atmospheric gas-to-particle conversion: why NPF events are observed in megacities?, Faraday Discuss., 2017, 200, 271–288, 10.1039/c6fd00257a.
- T. Rönkkö, et al., Traffic is a major source of atmospheric nanocluster aerosol, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7549–7554, DOI:10.1073/pnas.1700830114.
- J. Kontkanen, et al., Size-resolved particle number emissions in Beijing determined from measured particle size distributions, Atmos. Chem. Phys., 2020, 20, 11329–11348, DOI:10.5194/acp-20-11329-2020.
- Y. Zhou, et al., Variation of size-segregated particle number concentrations in wintertime Beijing, Atmos. Chem. Phys., 2020, 20, 1201–1216, DOI:10.5194/acp-20-1201-2020.
- M. Kulmala, et al., On the mode-segregated aerosol particle number concentration load: contributions of primary and secondary particles in Hyytiala and Nanjing, Boreal Environ. Res., 2016, 21, 319–331 Search PubMed.
- L. Yao, et al., Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity, Science, 2018, 361, 278–281, DOI:10.1126/science.aao4839.
- C. Deng, et al., Seasonal Characteristics of New Particle Formation and Growth in Urban Beijing, Environ. Sci. Technol., 2020, 54(14), 8547–8557, DOI:10.1021/acs.est.0c00808.
- S. Tuovinen, J. Kontkanen, J. Jiang and M. Kulmala, Investigating the effectiveness of condensation sink based on heterogeneous nucleation theory, J. Aerosol Sci., 2020, 149, 105613, DOI:10.1016/j.jaerosci.2020.105613.
- I. Riipinen, et al., The contribution of organics to atmospheric nanoparticle growth, Nat. Geosci., 2012, 5, 453, DOI:10.1038/ngeo1499.
- S. K. Friedlander, Smoke, Dust, and Haze: Fundamentals of Aerosol Dynamics, Wiley-Interscience, New York, 1977, vol. 333 Search PubMed.
- J. M. Moch, et al., Contribution of Hydroxymethane Sulfonate to Ambient Particulate Matter: A Potential Explanation for High Particulate Sulfur During Severe Winter Haze in Beijing, Geophys. Res. Lett., 2018, 45, 11-969–11-979, DOI:10.1029/2018gl079309.
- X. Fu, et al., Persistent Heavy Winter Nitrate Pollution Driven by Increased Photochemical Oxidants in Northern China, Environ. Sci. Technol., 2020, 54, 3881–3889, DOI:10.1021/acs.est.9b07248.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0fd00078g |
| This journal is © The Royal Society of Chemistry 2021 |