 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-nuclear, high-pressure, operando FlowNMR spectroscopic study of Rh/PPh3 – catalysed hydroformylation of 1-hexene†
Alejandro
Bara-Estaún
 ab,
Catherine L.
Lyall
ab,
John P.
Lowe
ab,
Paul G.
Pringle
d,
Paul C. J.
Kamer
e,
Robert
Franke
f and
Ulrich
Hintermair
*abc
ab,
Catherine L.
Lyall
ab,
John P.
Lowe
ab,
Paul G.
Pringle
d,
Paul C. J.
Kamer
e,
Robert
Franke
f and
Ulrich
Hintermair
*abc
aDepartment of Chemistry, University of Bath, Claverton Down, BA2 7AY Bath, UK. E-mail: u.hintermair@bath.ac.uk
bDynamic Reaction Monitoring Facility, University of Bath, Claverton Down, BA2 7AY Bath, UK
cCentre for Sustainable & Circular Technologies, University of Bath, Bath BA2 7AY, UK
dSchool of Chemistry, University of Bristol, Cantock’s Close, Bristol BS8 1TS, UK
eLeibniz Institute for Catalysis, Albert-Einstein-Straße 29A, 18059 Rostock, Germany
fEvonik Performance Materials GmbH, Paul-Baumann-Straße 1, 45772 Marl, Germany
First published on 26th March 2020
Abstract
The hydroformylation of 1-hexene with 12 bar of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2/CO in the presence of the catalytic system [Rh(acac)(CO)2]/PPh3 was successfully studied by real-time multinuclear high-resolution FlowNMR spectroscopy at 50 °C. Quantitative reaction progress curves that yield rates as well as chemo- and regioselectivities have been obtained with varying P/Rh loadings. Dissolved H2 can be monitored in solution to ensure true operando conditions without gas limitation. 31P{1H} and selective excitation 1H pulse sequences have been periodically interleaved with 1H FlowNMR measurements to detect Rh–phosphine intermediates during the catalysis. Stopped-flow experiments in combination with diffusion measurements and 2D heteronuclear correlation experiments showed the known tris-phosphine complex [RhH(CO)(PPh3)3] to generate rapidly exchanging isomers of the bis-phosphine complex [Rh(CO)2(PPh3)2] under CO pressure that directly enter the catalytic cycle. A new mono-phosphine acyl complex has been identified as an in-cycle reaction intermediate.
:1 H2/CO in the presence of the catalytic system [Rh(acac)(CO)2]/PPh3 was successfully studied by real-time multinuclear high-resolution FlowNMR spectroscopy at 50 °C. Quantitative reaction progress curves that yield rates as well as chemo- and regioselectivities have been obtained with varying P/Rh loadings. Dissolved H2 can be monitored in solution to ensure true operando conditions without gas limitation. 31P{1H} and selective excitation 1H pulse sequences have been periodically interleaved with 1H FlowNMR measurements to detect Rh–phosphine intermediates during the catalysis. Stopped-flow experiments in combination with diffusion measurements and 2D heteronuclear correlation experiments showed the known tris-phosphine complex [RhH(CO)(PPh3)3] to generate rapidly exchanging isomers of the bis-phosphine complex [Rh(CO)2(PPh3)2] under CO pressure that directly enter the catalytic cycle. A new mono-phosphine acyl complex has been identified as an in-cycle reaction intermediate.
Introduction
Hydroformylation, also known as the “oxo process”, reacts olefins with a mixture of CO and H2 (“syngas”) to yield aldehydes in the presence of a suitable catalyst.1 With terminal alkenes (α-olefins), the proportions of linear n-aldehydes and branched iso-aldehydes formed are a measure of the regioselectivity of the catalyst (Fig. 1).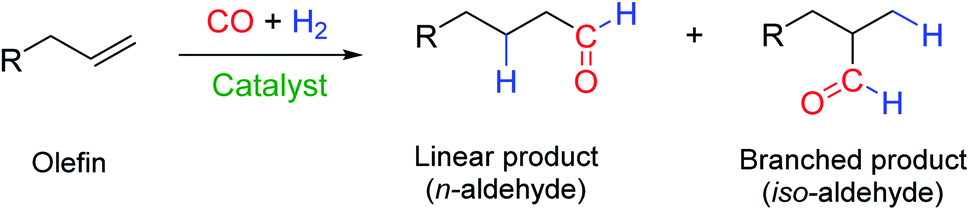 | ||
| Fig. 1 General chemical equation of a hydroformylation reaction. | ||
Chemoselectivity towards aldehyde formation is also important in the process as many catalysts also produce internal olefins (isomerization of α-olefins), paraffins (hydrogenation of olefins) and alcohols (hydrogenation of aldehydes) to various degrees.2
Otto (Roelen) discovered the hydroformylation of olefins in 1938 following its initial observation as a side reaction in Fischer–Tropsch chemistry.3 Hydroformylation has since undergone rapid development to become one of the largest industrial homogeneously catalysed processes, as well as an important academic research field with a large number of papers and patents from all over the world.2 Although rarely used for chemical synthesis on small scale,4 hydroformylation is frequently employed as a benchmark reaction for new ligands due the intricate interplay of chemo-, regio- and enantio-selectivity of the reaction.5 Although Ir, Ru and Fe based hydroformylation catalysts are known, Rh and Co complexes still stand out due to their superior activity, especially on an industrial scale.6 Co-based catalysts are tolerant toward trace impurities in the alkene feedstock, but require relatively harsh reaction conditions (>150 °C and >100 bar syngas) and display significant hydrogenation activity. The latter is often exploited to directly produce alcohols,7 albeit accompanied by significant paraffin co-production and formation of heavy aldol condensation side-products. Rh-based catalysts are generally more active and more selective than Co catalysts and operate under milder conditions (<100 °C and <50 bar syngas) but Rh is decisively more expensive due its rarity and widespread use in the automotive industry.8 Nevertheless, its higher efficiency explains why over 80% of all hydroformylation plants built after 1985 utilize Rh catalysts.9 In 2008, the global production of oxo compounds was about 10.4 million metric tons, and many large-scale plants manufacture several hundred thousand metric tons per year.10 Linear aldehydes are the main product as they are target intermediates for subsequent bulk chemical production of esters, alcohols and amines, while branched aldehydes are mostly desired for fine chemical production, partly due to the generation of a stereogenic centre at the formylated secondary carbon (Fig. 2).11,12 Long chain α-olefin substrates are usually synthesised by oligomerization of ethylene,13 whereas styrene and related molecules are preferred for asymmetric hydroformylation for fine chemical production.14,15
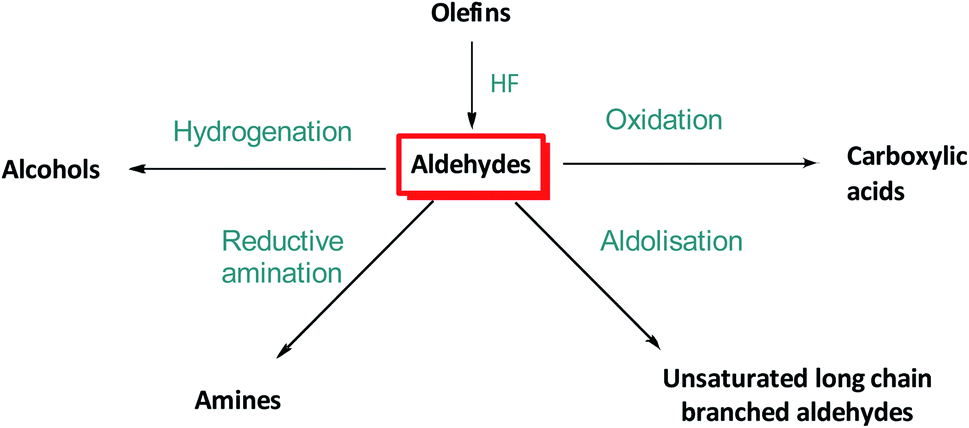 | ||
| Fig. 2 Industrial uses of aldehydes generated by olefin hydroformylation.2 | ||
Many organometallic Rh species have been investigated with a large variety of mono- and bidentate phosphorus ligands such as phosphites and phosphines.16–19 The identity of the ligand is crucial as they can tune, through their electronic and steric properties, the activity, regioselectivity, chemoselectivity and enantioselectivity of the hydroformylation reaction.20,21 The first mechanistic study of this reaction using [RhH(CO)(PPh3)3] as the precursor was carried out over 50 years ago by Wilkinson22 who proposed a mechanism for the olefin hydroformylation that is still accepted as conceptually correct. Since then many studies with a variety of reaction conditions, ligands and metals have been performed, both experimentally and computationally.16,23–26 Despite the large number of publications in which different mechanisms are proposed, several of the suggested intermediate species have never been observed. The difficulty of studying this reaction under working conditions is due to the high syngas pressure and temperatures employed, in addition to the O2 sensitivity of the Rh catalyst.27 A combination of kinetic studies, in situ IR investigations and high-pressure 31P NMR studies in static sapphire tubes have contributed much to this field.28,29 However, very few operando studies (in situ measurements during catalytic turnover) have been performed to verify mechanistic hypotheses and understand the electronic and steric parameters of the ligands and any other conditions that may affect the catalytic performance. Van Leeuwen, Kamer and co-workers have developed bespoke high-pressure infrared spectroscopy (HP-IR) autoclaves that allow operando IR studies of fast catalytic reactions under realistic conditions,24 and other groups have used similar setups to follow Rh-catalysed hydroformylation on different scales.30,31 IR spectroscopy offers high sensitivity and fast measurements compared to NMR spectroscopy, but it requires calibration to be quantitative and provides limited structural information on the intermediates observed.32 On-line high resolution FlowNMR spectroscopy has recently been shown to be a powerful operando reaction monitoring technique for homogeneous catalytic systems under realistic conditions due to its quantitative nature and high specificity in complex reaction mixtures.33 A recent report includes an example of an operando investigation of Rh/phosphite-catalysed hydroformylation using FlowNMR but with a limited amount of data and experimental details.34 Landis recently reported a detailed kinetic study of Rh-catalysed hydroformylation using a static 10 mm reactor within a high-field magnet connected to liquid dosing and gas recirculation.35
Here, we investigate the hydroformylation of 1-hexene in the presence of [Rh(acac)(CO)2] and PPh3 by operando high-resolution FlowNMR spectroscopy from an actively mixed pressure autoclave using periodic cycles of interleaved 1H NMR, 31P{1H} NMR and selective excitation 1H NMR measurements to gain insight into the speciation of the Rh/PPh3 catalyst under reaction conditions.
Results and discussion
The hydroformylation of 1-hexene in the presence of [Rh(acac)(CO)2] and PPh3 has been studied by FlowNMR spectroscopy under 12 bar of H2/CO at 50 °C. The experiments have been carried out as described in the ESI,† with [Rh(acac)(CO)2] catalyst loadings of 2.5 mM and 1-hexene concentration of 500 mM corresponding to [S]/[Rh] = 200 (or 0.5 mol% cat.) throughout all experiments. The concentration of PPh3 has been varied from 0–50 mM resulting in a ratio of [PPh3]/[Rh] = 0–20. The autoclave was charged with 22.4 mL of non-deuterated toluene containing 100 mM of 1,3,5-trimethoxybenzene as internal standard. H2 and CO were added separately in a 1:1 ratio.
The expected products from the reaction of 1-hexene with H2/CO in the presence of [Rh(acac)(CO)2] and PPh3 resulting from hydrogenation, isomerization and hydroformylation are shown in Scheme 1. From previous investigations the predominance of hydroformylation can be expected under these conditions, along with some isomerization and possibly traces of hydrogenation.36
 | ||
| Scheme 1 Possible primary reaction products from 1-hexene under hydroformylation conditions. | ||
All reactions were carried out under batch conditions in a 100 mL thick-walled glass autoclave, with an aliquot of the reaction mixture continuously recirculating through the temperature-controlled FlowNMR tubing in a closed-loop (Fig. 3). This setup ensures the sample within the spectrometer is continually refreshed with access to the reagent gases in the headspace, making every solution fraction representative of the mixture in the reaction vessel with a ca. 30 s delay to the FlowNMR tube under typical reaction conditions (flow rate of 4 mL min−1). Our modified setup enables working at temperatures of up to 140 °C and pressures of up to 20 bar (for details see the ESI†).
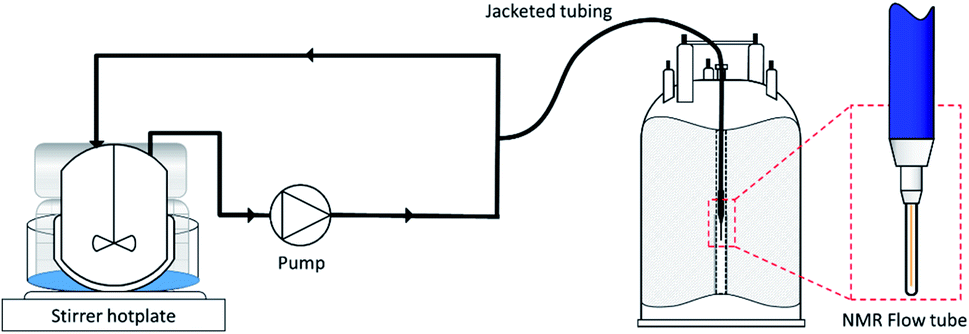 | ||
| Fig. 3 Schematic of the FlowNMR apparatus (not to scale). | ||
The first reaction was performed at 60 °C with 10 bar of H2/CO (1:1), where the autoclave was initially pressurised with H2 for 1 h before adding CO to trigger the hydroformylation reaction. After consideration of significant flow correction factors (see ESI†), reaction progress and chemoselectivity trends may be extracted from the NMR data (Fig. 4).
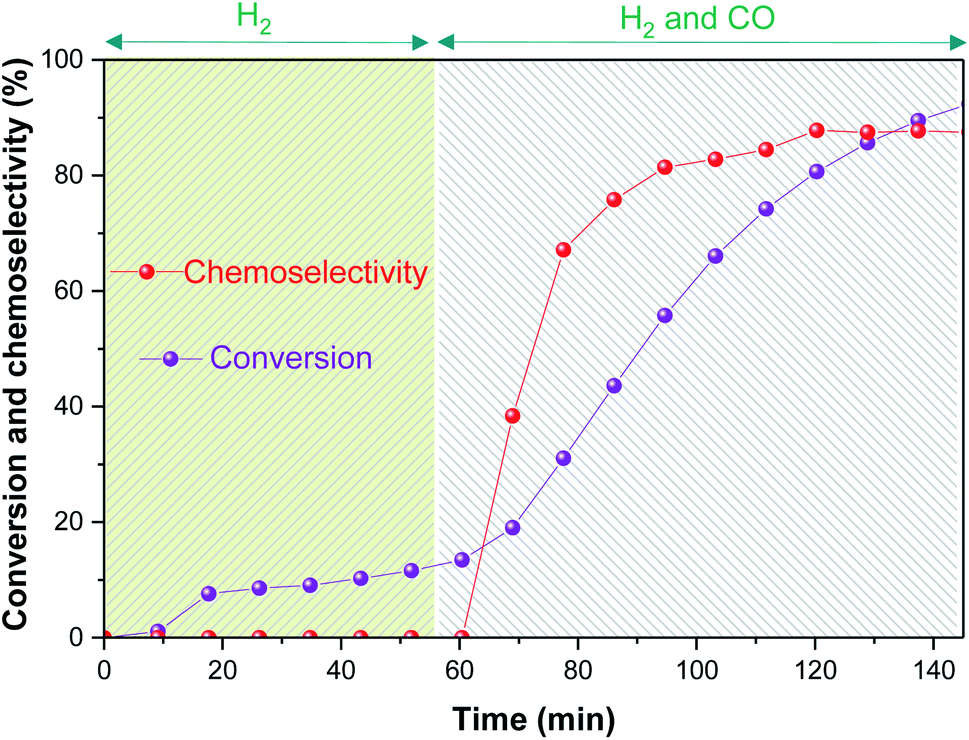 | ||
| Fig. 4 Substrate conversion and chemoselectivity to aldehydes of the [Rh(CO)2(acac)] + 6PPh3 catalysed hydroformylation of 1-hexene in toluene at 60 °C and 10 bar of H2/CO (1:1) monitored using operando1H FlowNMR spectroscopy. [Rh(CO)2(acac)] = 2.5 mM, [PPh3] = 15 mM, [1-hexene] = 500 mM. From t = 5–55 min only 5 bar H2 were present, after which another 5 bar CO were added. | ||
This initial experiment showed that high-resolution FlowNMR with an air-sensitive catalyst system is possible with a fully temperature-controlled and pressurized setup, allowed the identification of relevant NMR signals, and gave an indication of the reaction rate under the conditions applied. The identity of all of the organic products observed by 1H NMR (n- and iso-heptanal, 2- and 3-hexene) were confirmed by GC-MS (see ESI†), which also ruled out formation of any significant quantities of other side products under the conditions used. Thus, quantitative product distribution profiles may be plotted from the 1H FlowNMR data (Fig. 5).
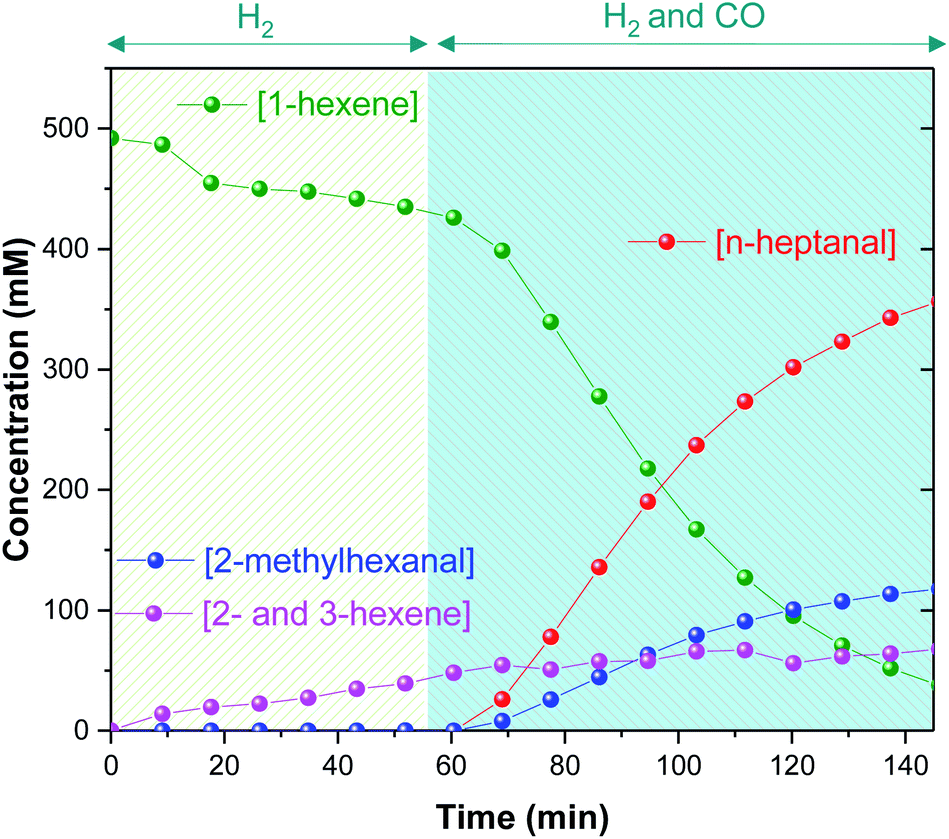 | ||
| Fig. 5 Product formation and substrate consumption profiles of [Rh(CO)2(acac)] + 6PPh3 catalysed hydroformylation of 1-hexene in toluene at 60 °C and 10 bar of H2/CO (1:1) monitored using operando1H FlowNMR spectroscopy. [Rh(CO)2(acac)] = 2.5 mM, [PPh3] = 15 mM, [1-hexene] = 500 mM. | ||
The observation of only isomerization of 1-hexene slowly occurring with kobs = 0.90 mM min−1 immediately after adding H2 suggests catalyst activation to a hydride complex to be fast, but without any appreciable hydrogenation activity (at 60 °C with 6 equiv. of PPh3). Consumption of CO proceeded without any significant lag phase and hydroformylation smoothly progressed with a stable 3:1 linear/branched selectivity over the course of the reaction, alongside some continued isomerisation at kobs = 0.36 mM min−1 until the end of the reaction. Maximum reaction rates were found in the first 40 min after the pressurisation with CO, with substrate consumption k1-hexene(obs) = 8.76 mM min−1, product formation kn-heptanal(obs) = 6.43 mM min−1 and k2-methylhexanal(obs) = 2.15 mM min−1. These values as well as the observed aldehyde regioselectivity align well with previously reported data16,36–40 and are consistent with previous kinetic studies.22
In the following studies the temperature was decreased to 50 °C and data acquisition optimised to increase temporal resolution. The reactor was pressurized with both gases (12 bar 1:1 H2/CO) at the same time to immediately start the hydroformylation. A series of experiments were conducted where the amount of PPh3 was varied under otherwise identical conditions (Fig. 6).
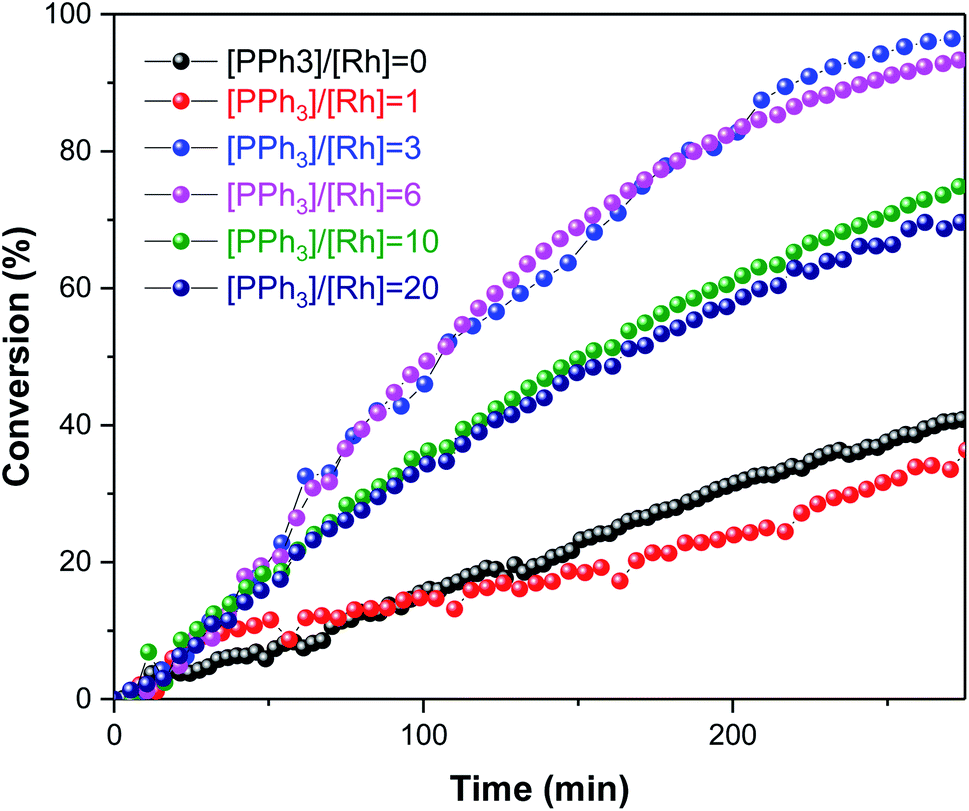 | ||
| Fig. 6 Substrate conversion profiles of [Rh(CO)2(acac)] + PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1) monitored using operando1H FlowNMR spectroscopy. [Rh(CO)2(acac)] = 2.5 mM, [1-hexene] = 500 mM, [PPh3] varied as indicated. | ||
As expected, the amount of PPh3 added had a non-linear influence on the activity. With no phosphine present the Rh catalyst yielded some aldehyde production with a moderate L/B ratio of 2.4:1 in the first 30 min of the reaction, but mostly produced 3-hexene that did not undergo hydroformylation (Fig. 7); this behaviour is known for [RhH(CO)4] catalysed hydroformylation.39 With one equivalent of PPh3 added the system afforded a similar rate (Fig. 6), but with much higher chemoselectivity for aldehyde production with increased L/B ratios (Fig. 7). Under these conditions some isomerisation still occurred alongside hydroformylation, leading to decreasing L/B ratios over time as the 2-hexene formed underwent hydroformylation to 2-methylhexanal (Fig. 7). With 3–6 equivalents of PPh3/Rh the rate of 1-hexene consumption was about four times higher, with >95% chemoselectivity to aldehyde formation at stable L/B ratios of 3:1 at 50 °C. When 10–20 equivalents of PPh3 were added high chemo- and regio-selectivities were maintained, but at reduced overall reaction rates due to PPh3 competing with the olefin substrates for coordination to the Rh centre. Again, this behaviour is well documented in the literature,41 and the overall rates observed in our FlowNMR studies (Table S4†) are in line with previous reports.37
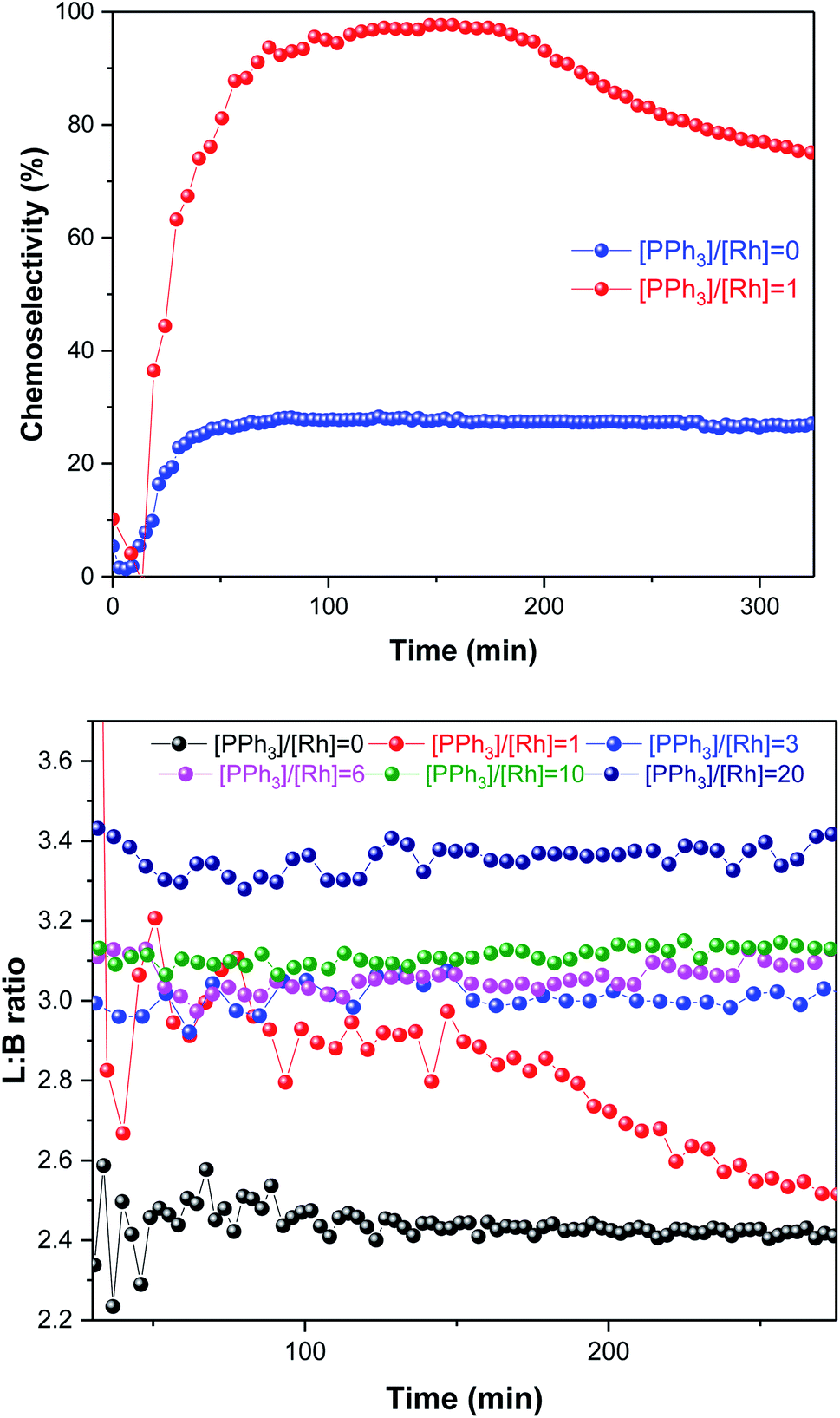 | ||
| Fig. 7 Aldehyde chemoselectivity (upper) and regioselectivity profiles (lower) of [Rh(CO)2(acac)] + PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1) monitored using operando1H FlowNMR spectroscopy. [Rh(CO)2(acac)] = 2.5 mM, [1-hexene] = 500 mM, [PPh3] varied as indicated. | ||
As in some of our previous work,42 it was also possible to track the amount of H2 dissolved in solution by 1H FlowNMR during the hydroformylation reaction (Fig. 8) after considering flow effects and the ortho/para ratio of H2 at 50 °C.43 Ensuring that the volume element analysed in the NMR (temporarily disconnected from the autoclave headspace) is still under turnover conditions and not deprived of any reagent is important to ascertain that the results obtained are truly operando and not just in situ. From the concentration plots shown in Fig. 9 it can be seen that dissolution of H2 into solution was rapid under the conditions applied, and because the reactor had been sealed after the initial pressurisation, its decreasing concentration over time tracked the amount of H2 consumption by the reaction. Although we have not quantified the gas–liquid distribution of the system, the lower H2 uptake by the reaction without PPh3 concurs with the observation of mainly olefin isomerisation occurring. Even for the faster reactions with 3–6 equivalents of PPh3 that did consume H2/CO in producing aldehydes, the system never entered a gas-limited regime showing effective mixing.
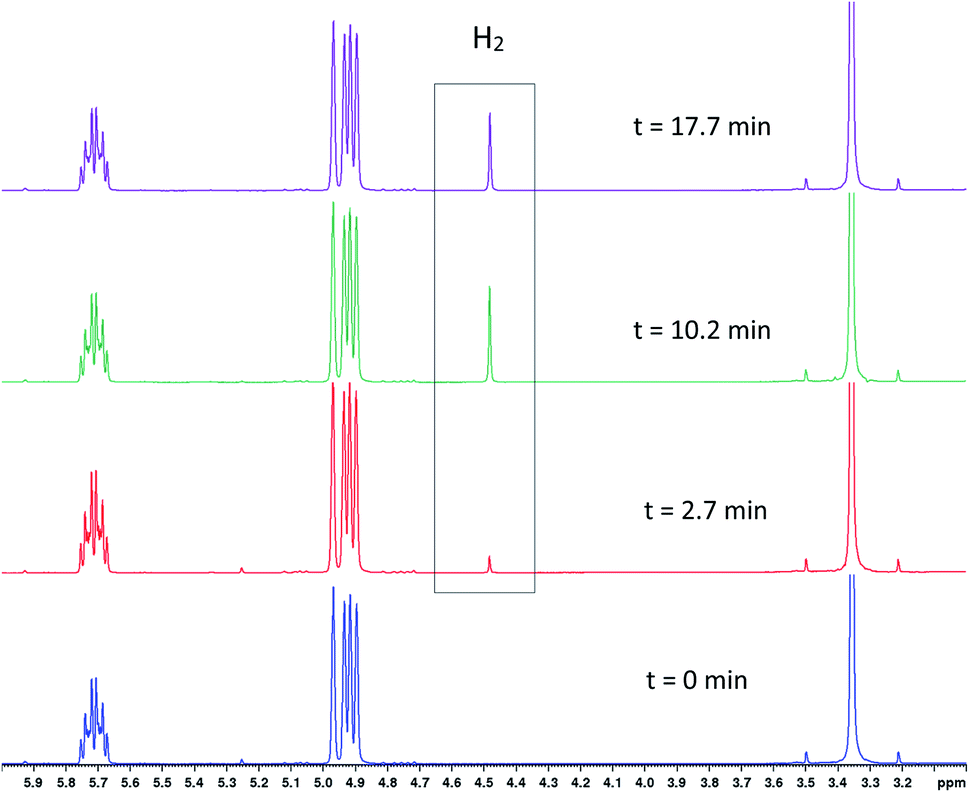 | ||
| Fig. 8
1H NMR spectra showing the appearance of dissolved H2 during [Rh(CO)2(acac)] + 6PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1) from operando1H FlowNMR spectroscopy. [Rh(CO)2(acac)] = 2.5 mM, [PPh3] = 15 mM, [1-hexene] = 500 mM. | ||
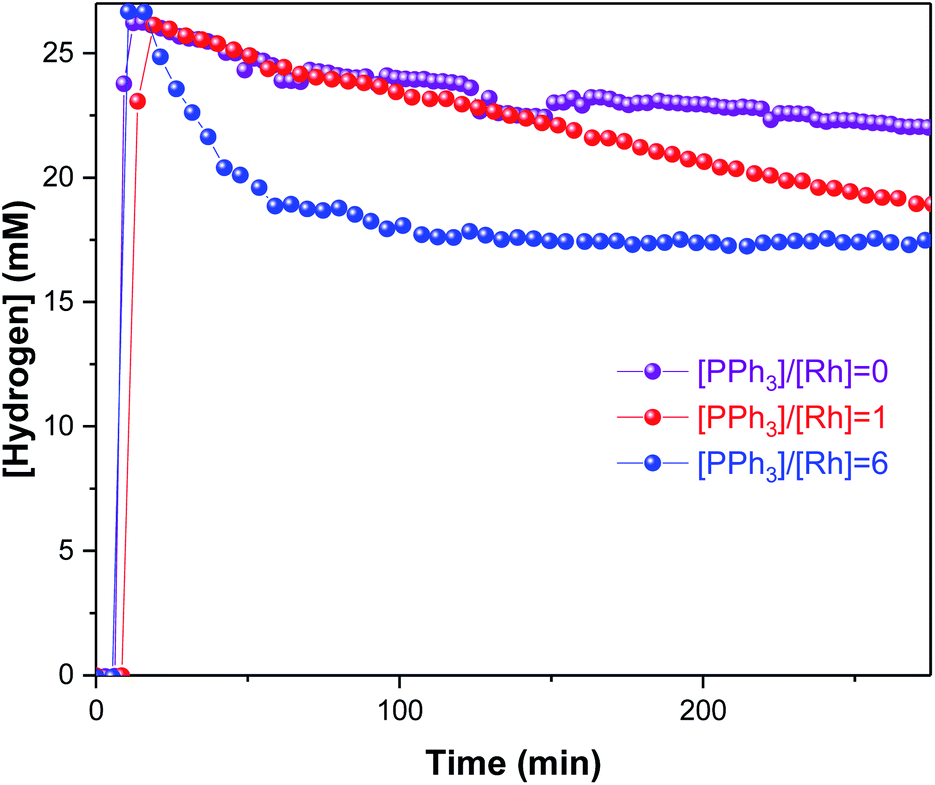 | ||
| Fig. 9 Hydrogen concentration profiles during [Rh(CO)2(acac)] + PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1) monitored using operando1H FlowNMR spectroscopy. [Rh(CO)2(acac)] = 2.5 mM, [1-hexene] = 500 mM, [PPh3] varied as indicated. | ||
Under the conditions applied, the reaction is believed to proceed via a dissociative mechanism (Scheme 2)27,44,45 initially proposed by Wilkinson for ethylene as the substrate,22 analogous to Heck’s mechanism for Co-catalysed hydroformylation.46
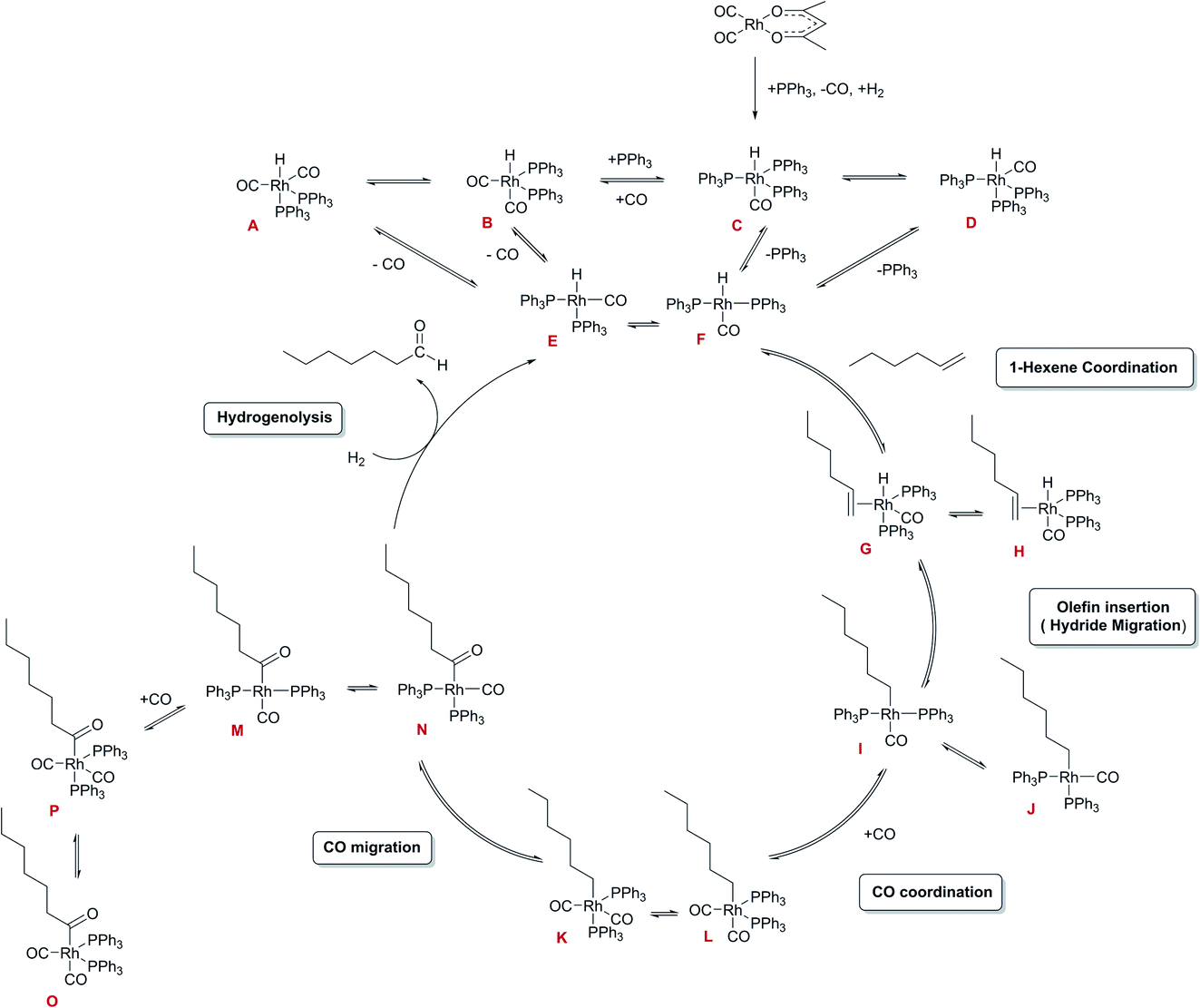 | ||
| Scheme 2 Simplified dissociative mechanism for the Rh-catalysed hydroformylation of hexene (only heptanal formation shown). All in-cycle reaction intermediates are believed to contain two PPh3 ligands per Rh. | ||
The mechanism starts with the formation of unsaturated complexes E and F by dissociation of CO or ligand from stable five-coordinate RhI hydrido-carbonyl complexes. Thereafter, the olefin coordinates to give the olefin-hydrido-carbonyl complexes G and H that can undergo intramolecular hydride migration to form the alkyl complexes I and J. Coordination of CO to I and J results in the alkyl-carbonyl complexes K and L that can undergo a migratory CO insertion to form acyl complexes M and N which may coordinate additional CO to form the more stable five-coordinate acyl complexes O or P. The last step to complete the cycle is the hydrogenolysis to release the aldehyde and recover complexes E and F. The latter is generally believed to be the only irreversible process and constitutes the turnover limiting step in the cycle.22 Among the various intermediates proposed throughout the catalytic cycle, only the penta-coordinated complex [RhH(CO)(PPh3)3] C has been fully characterized by NMR and IR spectroscopy as well as X-ray crystallography.44,47,48 The related [RhH(CO)2(PPh3)2] complexes A and B have been characterized by NMR and IR spectroscopy in solution due to their fluxional behaviour caused by the rapid exchange of PPh3 ligands from equatorial to axial positions.49,50 The in-cycle acyl rhodium complex [Rh{CO(CH2)5(CH3)}(CO)2(PPh3)2] P has been identified by NMR spectroscopy, in addition to some off-cycle dinuclear Rh species such as [Rh2(CO)4(PPh3)4], [Rh2(CO)5(PPh3)3] and [Rh2(CO)6(PPh3)2].27,51 For the PCy3 ligand the tetra-coordinated complex [RhH(CO)(PCy3)2] has been synthesised and monitored spectroscopically under hydroformylation conditions,52 although this catalyst is very slow due to the bulky, strongly binding trialkylphosphine.
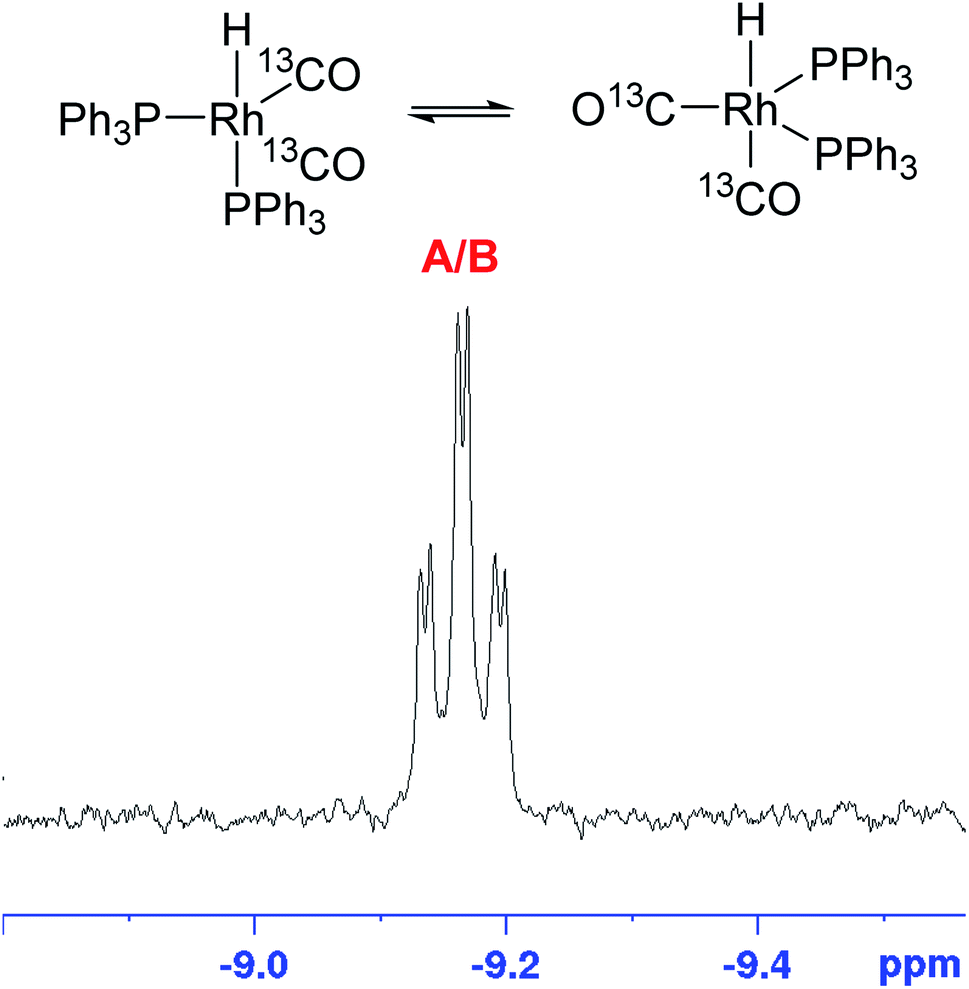
In order to gain some insight into the speciation of the [Rh(acac)(CO)2]/PPh3 catalyst system during catalysis we measured selectively excited 1H NMR spectra53 focussing on the negative shift region where Rh–H resonances are expected, and also acquired 31P{1H} NMR spectra during the reaction in flow. With optimised acquisition parameters using a 500 MHz spectrometer fitted with a cryoprobe (see ESI for details†) we were able to acquire these alongside the standard 1H FlowNMR for monitoring reaction progress in a sequential manner throughout the reaction in a single experiment at 2.5 mM [Rh] concentration in non-deuterated solvent. The hydride FlowNMR spectra thus acquired at 50 °C showed the smooth growth of a broad singlet peak centred at −9.18 ppm (12 Hz FWHM) over the course of the reaction (Fig. 10).
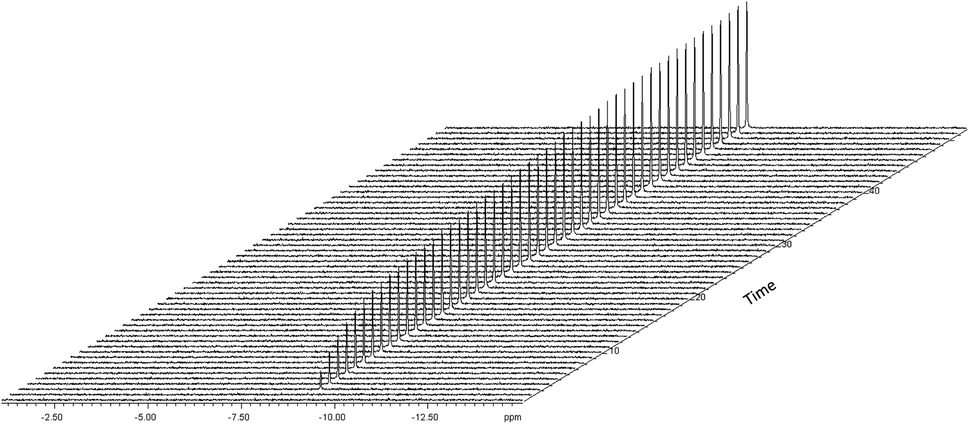 | ||
| Fig. 10 Rhodium–hydride formation during [Rh(CO)2(acac)] + 6PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1) monitored using selective excitation 1H FlowNMR spectroscopy. [Rh(CO)2(acac)] = 2.5 mM, [1-hexene] = 500 mM, [PPh3] = 15 mM. | ||
This hydride signal appeared immediately after adding syngas to the reaction mixture, but not with H2 alone. No other hydride resonances were detected, and the same signal was observed for any P/Rh ratios >1. For 1 and 0 equivalents of PPh3, no hydride signals were detected. The absence of any defined Rh–H coupling patterns in the broad singlet at −9.18 ppm is expected due to the known low magnitude of 1JRhH of <10 Hz.54
The corresponding 31P{1H} FlowNMR spectra of mixtures of [Rh(acac)(CO)2] and PPh3 did not show any signals in the absence of syngas, likely due to rapid chemical exchange between a number of unknown species (bubbling indicative of CO release can be observed visually upon combining [Rh(acac)(CO)2] and PPh3). Immediately after pressurising the mixture with syngas the 31P{1H} FlowNMR spectra regained shape and showed three major resonances: a large, broad singlet peak at −4.9 ppm (68.5 Hz FWHM) originating from free PPh3, a small sharp singlet at 25.1 ppm from traces of PPh3 oxide (as verified with an authentic sample), and a well-defined doublet with a coupling constant of 139.2 Hz centred at 37.3 ppm (Fig. 11).
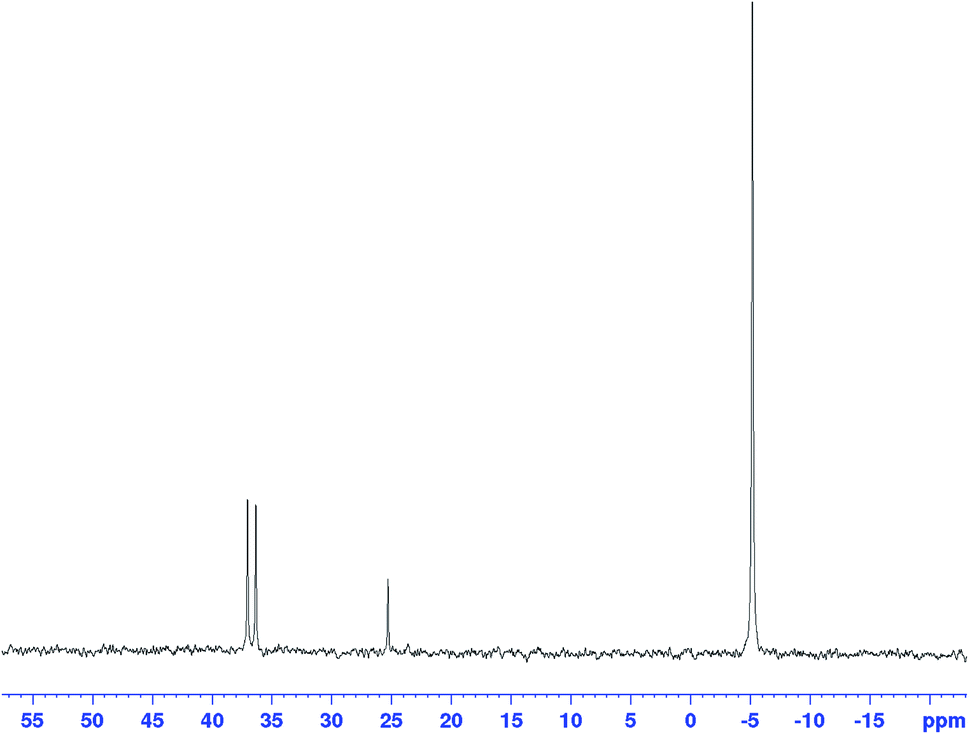 | ||
| Fig. 11
31P{1H} FlowNMR spectrum recorded during [Rh(CO)2(acac)] + 6PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1) at t = 395 min. [Rh(CO)2(acac)] = 2.5 mM, [1-hexene] = 500 mM, [PPh3] = 15 mM. | ||
The growth of the 31P{1H} doublet at 37.3 ppm tracked the appearance of the broad 1H signal at −9.18 ppm. 31P DOSY and 1H DOSY spectra showed very similar diffusion coefficients of 7.7 × 10−10 m2 s−1 and 7.1 × 10−10 m2 s−1 for both (see Fig. S15 and S16†), but the apparent absence of any 2JPH coupling in the 1H signal initially seemed surprising. A 1H–31P HMBC spectrum did however clearly establish both resonances to originate from the interchanging coordination isomers A ↔ B of [RhH(CO)2(PPh3)2] (Fig. 12). The rapid intramolecular exchange of the PPh3 between equatorial and apical positions, known to be facile in trigonal-bipyramidal Rh(I) complexes by way of Berry pseudo-rotation, obscures the observation of any 2JPH couplings in this complex under syngas at 50 °C.49,50 Additional intermolecular exchange with free PPh3 (A ↔ B ↔ C ↔ D as indicated by the slightly broadened 31P{1H} signal at −4.9 ppm) may contribute further to the fluxionality of this complex.
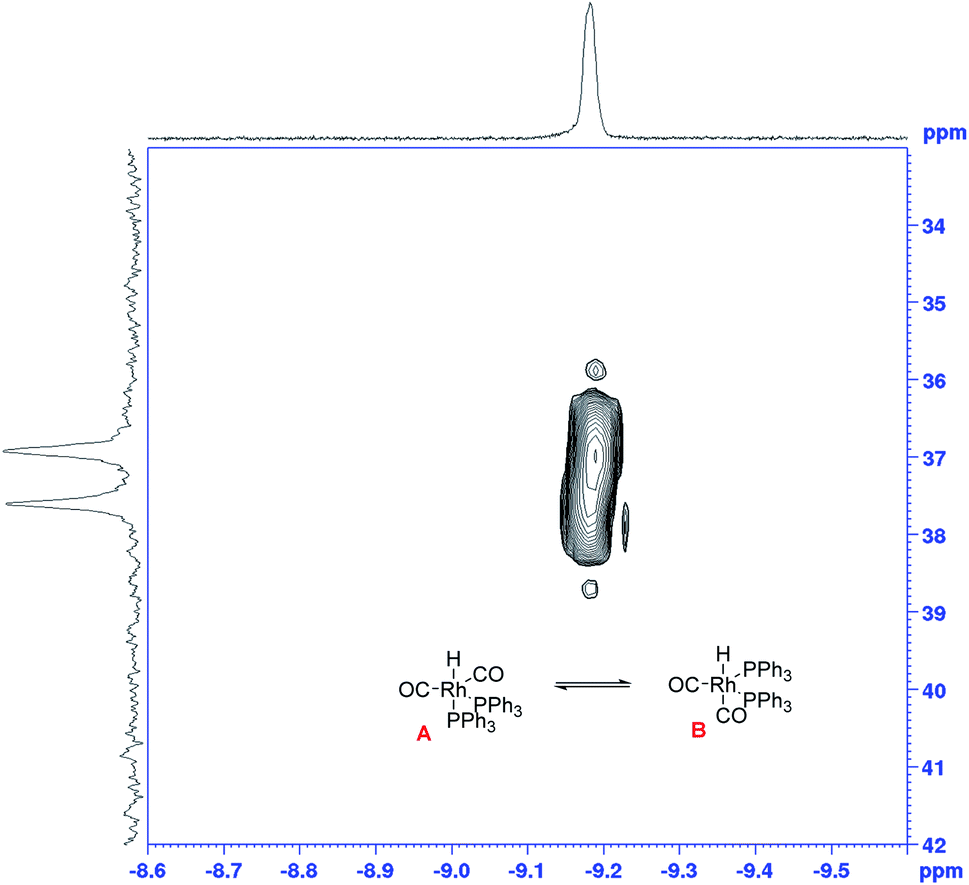 | ||
| Fig. 12
1H–31P HMBC NMR spectrum recorded during [Rh(CO)2(acac)] + 6PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1) at t = 325 min. [Rh(CO)2(acac)] = 2.5 mM, [1-hexene] = 500 mM, [PPh3] = 15 mM. | ||
To test the connectivity of A/B to the catalytic cycle, one flow run was deliberately interrupted by stopping the pump to temporarily isolate the sample in the magnet from the gas supply in the reactor headspace. From the 1H FlowNMR spectra it could be seen that the reaction progressed for about 2 min until the H2 (and presumably CO) dissolved in solution had been consumed (Fig. 13). Once the sample in the tip of the flow tube had run out of syngas, both the 31P and 1H signals of the hydrido-carbonyl complexes A/B started to fade away from the spectra (Fig. 13). As soon as flow was resumed and the sample fed back into the reactor where it was brought into contact with syngas again, hydroformylation activity resumed accompanied by re-appearance of [RhH(CO)2(PPh3)2]. This experiment demonstrated this complex to be of direct relevance to the catalytic cycle, representing the major reaction intermediate under the conditions applied.
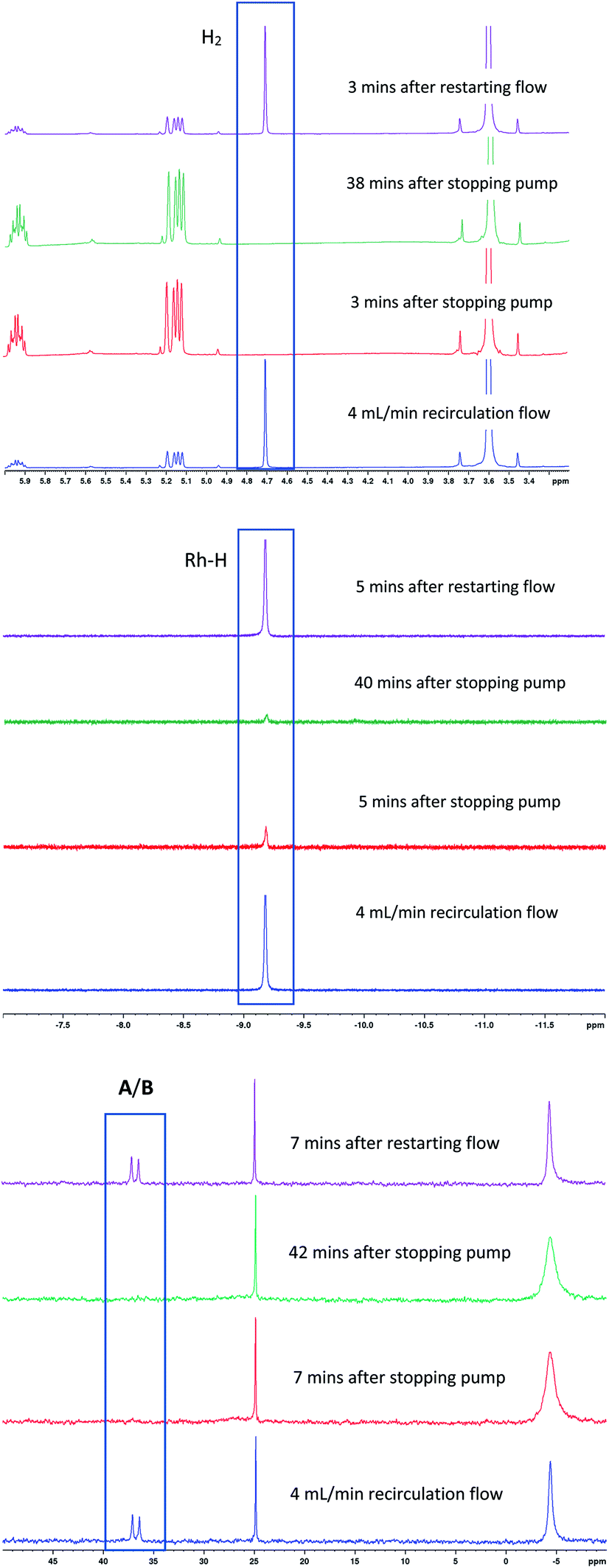 | ||
| Fig. 13
1H and 31P{1H} FlowNMR spectra of [Rh(CO)2(acac)] + 6PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1) during stop/start cycling of flow. [Rh(CO)2(acac)] = 2.5 mM, [1-hexene] = 500 mM, [PPh3] = 15 mM. | ||
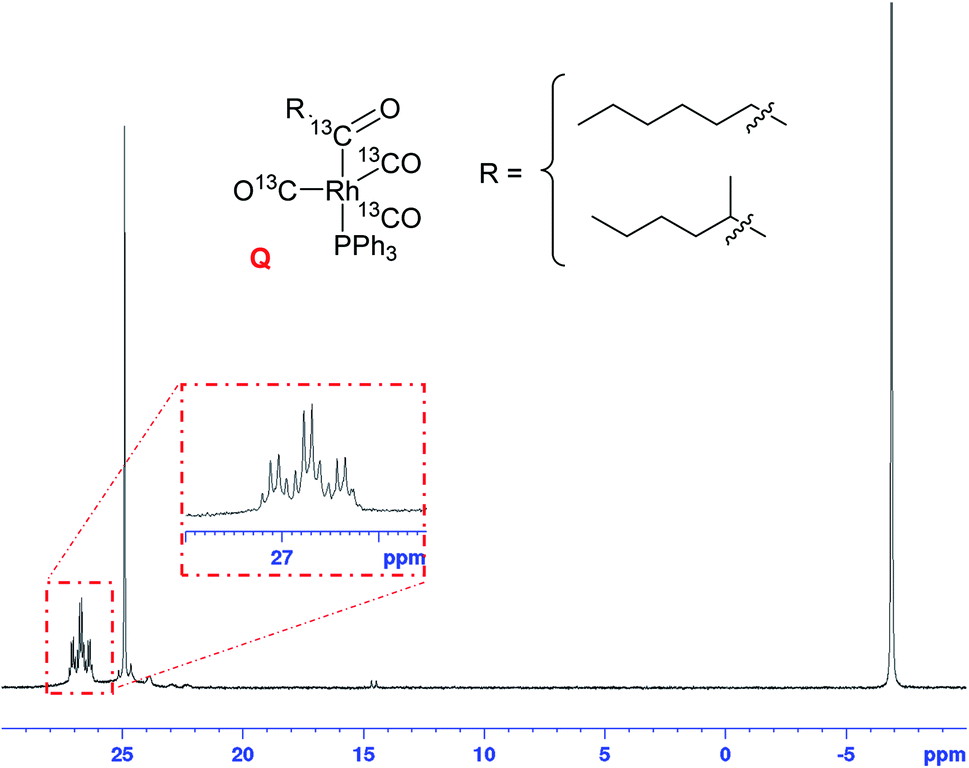
When the excess H2/CO was vented at the end of the reaction (after complete consumption of hexene) and the sample analysed under an atmosphere of argon, the only Rh–phosphine species detected by NMR was the known [RhH(CO)(PPh3)3] complex C resulting from substitution of one CO by PPh3. However, when the catalyst was deprived of syngas mid-reaction (i.e. in the presence of hexene substrate) as described above, no hydride resonance could be observed at all in the selectively excited 1H NMR, but a new signal was detected in the 31P{1H} NMR. Interestingly, it was the same species that was the only Rh–phosphine complex detected by FlowNMR throughout the reaction when PPh3/Rh = 1 (Fig. 14). The doublet peak centred at 27.7 ppm had a relatively low 1JRhP of 71.8 Hz and no associated hydride signal.
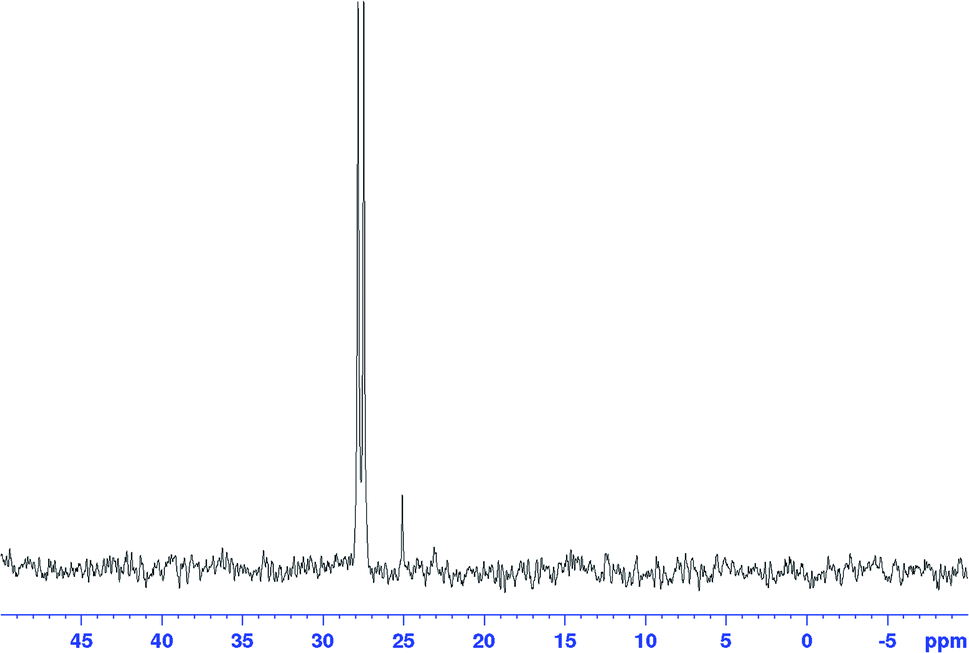 | ||
| Fig. 14
31P{1H} FlowNMR spectrum recorded during [Rh(CO)2(acac)] + 1PPh3 catalysed hydroformylation of 1-hexene in toluene at 50 °C and 12 bar of H2/CO (1:1). The same doublet at 27.7 ppm was also observed from mixtures with higher PPh3/Rh ratios when deprived of syngas in the presence of 1-hexene. [Rh(CO)2(acac)] = 2.5 mM, [1-hexene] = 500 mM, [PPh3] = 2.5 mM. | ||
The conditions under which this complex was observed together with its spectroscopic signatures suggested that it may be an acyl intermediate prior to release of product via hydrogenolysis (O–N, Scheme 2). Indeed, a very similar 31P{1H} NMR signal (doublet at 27.5 ppm with 1JRhP = 78.0 Hz) had previously been assigned to [Rh(CO(CH2)5CH3)(CO)2(PPh3)2] but not fully characterised.27 A 31P DOSY spectrum gave a diffusion coefficient of 7.35 × 10−10 m2 s−1 for our signal at 27.7 ppm (Fig. S17†), very similar to the monomeric [RhH(CO)2(PPh3)2] complexes A/B analysed under the same conditions.
When the stable complex [RhH(CO)(PPh3)3] C was pressurised with CO in the presence of 1-hexene but without H2 under otherwise identical reaction conditions, C disappeared from the 1H spectra with a transient trace of A/B before all hydride signals vanished (Fig. 15). The corresponding 31P{1H} NMR spectra showed the same sequence of transformation, eventually yielding the doublet at 27.7 ppm along with one equivalent of OPPh3 at 24 ppm and one equivalent of PPh3 at −4.9 ppm (Fig. 15). From this observation we can deduce that only one PPh3 is coordinated to the Rh centre in this compound thus identified as the acyl complex [Rh(CO(CH2)5CH3)(CO)3(PPh3)] (Q). As expected, no aldehyde formation was observed in this experiment. Pressurising the mixture with CO and H2 immediately led to hydroformylation activity with the acyl complex Q disappearing and A/B dominating the phosphorus and hydride FlowNMR spectra just as when starting from [Rh(acac)(CO)2] + PPh3 (see Fig. 10 and 11).
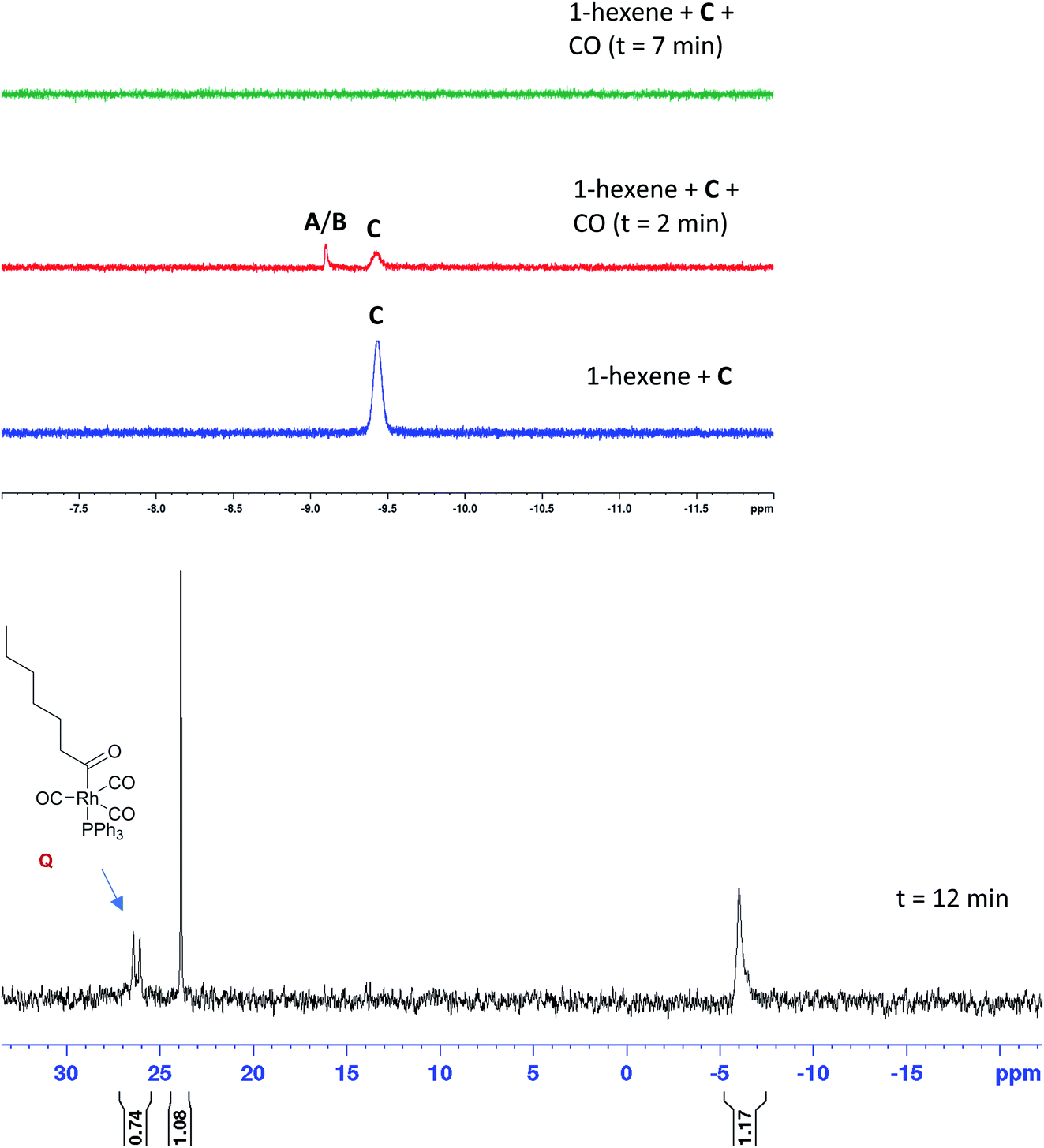 | ||
| Fig. 15 Selective excitation 1H and 31P{1H} FlowNMR spectra of [RhH(CO)(PPh3)3] (2.5 mM) and 1-hexene (500 mM) under 6 bar of CO in toluene at 50 °C. | ||
The observation of a mono-phosphine acyl complex as the major catalyst species prior to rate-limiting hydrogenolysis is surprising given the long-accepted view that all in-cycle intermediates in Wilkinson's dissociative mechanism would be bis-phosphine complexes (Scheme 2).27,45 In support of this notion, Brown reported the NMR spectroscopic characterisation of bis-phosphine acyl complex P obtained from [RhHCO(PPh3)3] (C) and 1-octene under atmospheric pressure of 13CO.44 However, the facile displacement of PPh3 by CO observed in the generation of A/B from C under 5 bar of CO (Fig. 15) suggests that a similar equilibrium may generate mono-phosphine complexes from Brown's bis-phosphine complex under catalytic reaction conditions of >1 bar CO.
In order to confirm this hypothesis and ascertain the structural identity of Q as a mono-phosphine acyl complex we reacted [RhHCO(PPh3)3] (C) and 1-hexene with 5 bar of 99.9% enriched 13C carbon monoxide in toluene. As seen before (Fig. 15), the hydride signal of C vanished within minutes after pressurising C with 13CO in the presence of olefin, transiently showing the formation of A/B (Fig. 16) before all hydride signals disappeared after 60 min at 25 °C. With two 13CO ligands the 1H resonance of A/B was a triplet of doublets with a 2JCH = 30.2 Hz and 1JRhH = 3.9 Hz without any apparent 2JPH due to the fluxionality of this complex (as discussed above).
 | ||
| Fig. 16 Selective excitation 1H NMR spectrum of [RhH(CO)(PPh3)3] (25 mM) and 1-hexene (500 mM) under 5 bar of 13CO in 0.5 mL of toluene at 0 °C showing the transient formation of A/B from C with 13CO. | ||
The corresponding 31P{1H} NMR spectra showed the same sequence of transformations, eventually yielding a virtual triplet of quartets centred at 26.5 ppm along with some OPPh3 at 25 ppm and free PPh3 at −4 ppm (Fig. 17) as seen before for Q (Fig. 15). The 31P resonance associated with the acyl complex coincidentally showed identical 1JRhP = 70.7 Hz and 2JPC = 70.7 Hz couplings to the trans acyl carbon, and a distinct cis2JPC = 17.6 Hz from coupling to three equivalent 13CO ligands. This multiplicity is only possible with a pentacoordinate mono-phosphine tris-carbonyl acyl complex as drawn for Q.
 | ||
| Fig. 17 31P{1H} NMR spectrum of [RhH(CO)(PPh3)3] (25 mM) and 1-hexene (500 mM) under 5 bar of 13CO in toluene at −40 °C, showing the formation of Q from C with 13CO. | ||
The corresponding 13C{1H} NMR spectra showed a variety of distinct carbonyl species in the range of 240–180 ppm (Fig. 18). Free 13CO dissolved in toluene was observed at 184.5 ppm (confirming excess conditions), and the quaternary carbonyls of heptanal and 2-methylhexanal originating from stoichiometric 1-hexene hydroformylation were observed at 200.5 ppm and 203.2 ppm in a 3:1 ratio, respectively. The terminal 13CO ligands of mono-phosphine complex Q were observed as two overlapping doublets of doublets at 192.1 ppm with 1JRhC = 74.5 Hz and 2JCP = 17.5 Hz as well as 1JRhC = 74.7 Hz and 2JCP = 17.3 Hz, confirming the cis2JPC observed in the 31P{1H} NMR spectra (Fig. 17). Interestingly, while only one version of Q was detectable in the 31P{1H} NMR spectra, the two different acyl regio-isomers QL and QB could just be distinguished in the 13C{1H} NMR spectra in the expected 3:1 ratio (with similar chemical shifts and cis2JPC couplings), consistent with the formation of linear and branched aldehydes.
 | ||
| Fig. 18 13C{1H} NMR spectrum of [RhH(CO)(PPh3)3] (25 mM) and 1-hexene (500 mM) under 5 bar of 13CO in toluene at −40 °C showing the formation of Q and O/P from C with 13CO. | ||
Although not visible in the corresponding 31P{1H} NMR spectra due to very fast exchange between axial and equatorial positions, the 13C{1H} doublet at 198.36 ppm with a 1JRhC = 76.7 Hz could be assigned to the terminal 13CO ligands of the bis-phosphine complex O/P previously reported by Brown.44 Upon cooling the sample to −90 °C, coupling of 13CO with 31P could be resolved as a doublet of triplets with a cis2JCP = 20.9 Hz (Fig. S29†) but the corresponding 31P{1H} signal of O/P could still not be detected. The ratio of Q to O/P in the 13C{1H} NMR spectra was about 1.3:1 under 5 bar of 13CO in the presence of 3 equiv. of PPh3 per Rh, similar to the formation of A/B from C in the presence of excess CO (Fig. 15). Consistent with our assignments, the 13C resonances of the terminal acyls in both regio-isomers of Q and O/P could be detected in in the range of 225–240 ppm (Fig. 18), showing characteristic C–Rh and C–P coupling constants despite partial signal overlap (Fig. S30†). Similar to the difficulty of resolving P–H couplings in the rapidly interconverting isomers of A/B (Fig. 11), no C–C couplings between the acyl and terminal carbonyls could be resolved in the 13C{1H} NMR spectra of Q and O/P down to −40 °C due to their highly fluxional nature (all 13C{1H} NMR signals including free 13CO were very broad at room temperature; Fig. S31†). Further confirmation of the assignment of Q as a mixture of two regio-isomers of a mono-phosphine acyl complex and O/P as a mixture of isomers of bis-phosphine acyl complexes came from 1H–13C HMBC and 1H–31P HMBC correlation experiments (Fig. S32 and S33†) as well as a quantitative 13C{1H} 1D NMR measurement showing a 2:1 CO/acyl ratio for O/P and 3:1 CO/acyl ratio for Q in their relative peak integrals (Fig. S35†).
The dynamic equilibrium between Q and O/P was shown by a change in their distribution from 1.3:1 under 5 bar CO to 1:2.5 after the excess CO was vented from the mixture (Fig. S34†). The different conditions used in Brown's experiments44 and our own thus explain the different complexes detected. At higher PPh3 loadings of >3 equiv. to Rh (as typically used in applied hydroformylation) the excess phosphine might overcompensate for the higher CO pressures and shift the equilibrium to the bis-phosphine acyl complex again. When a total of 6 equivalents of PPh3 to Rh were used under 5 bar of 13CO, the ratio of Q to O/P in the 13C{1H} NMR spectra was found to be 1:3 as opposed to 1.3:1 with 3 equiv. PPh3, clearly showing the coordination competition of CO and PPh3 in Qvs.O/P just as in Cvs.A/B (Scheme 3).
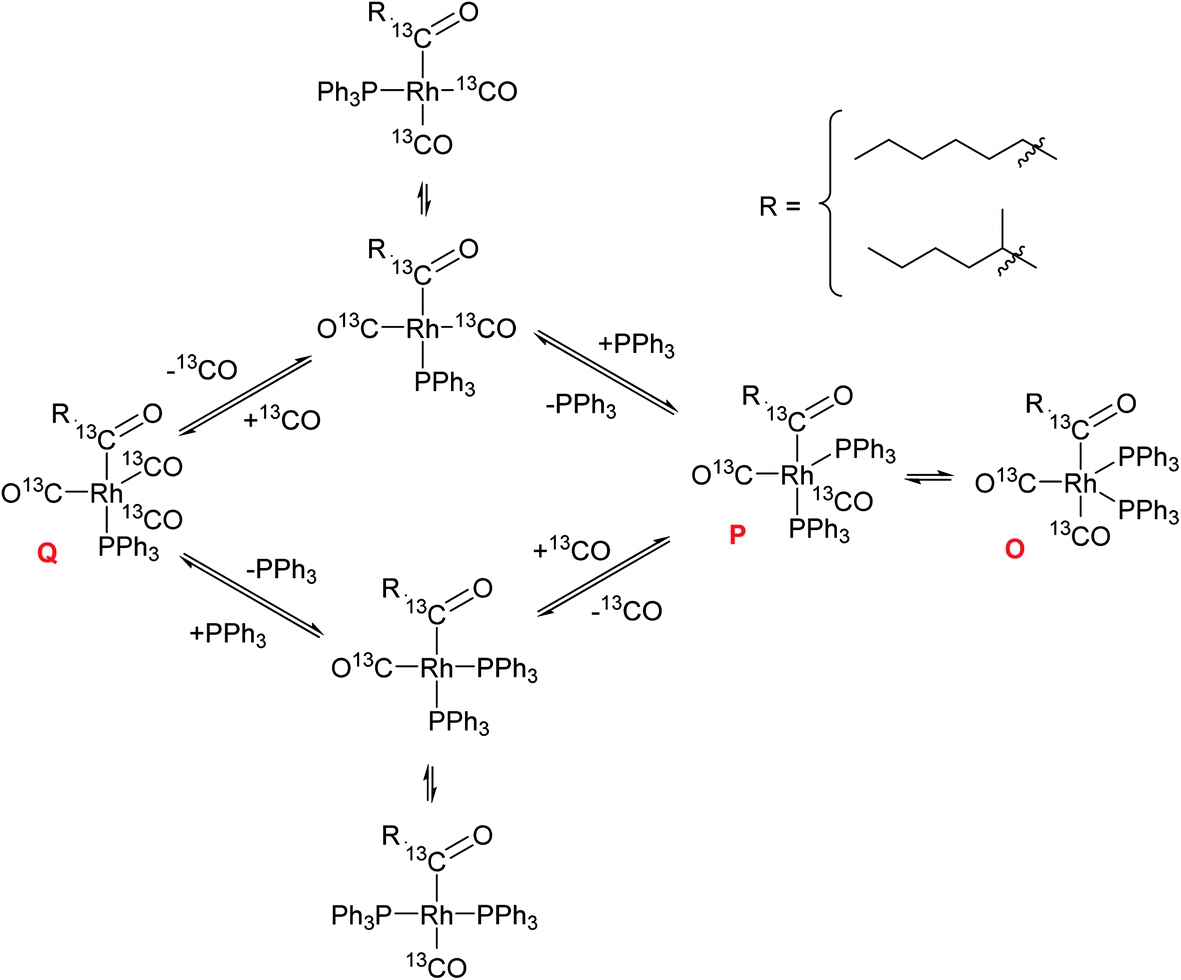 | ||
| Scheme 3 Coordination equilibria between the mono- (Q) and bis-phosphine (O/P) acyl complexes through dynamic PPh3 and CO exchange (unlabelled four-coordinate intermediates not detected but believed to be the species reacting further with H2, see Scheme 2). | ||
These observations provide evidence for several conclusions: (i) olefin coordination does not occur with the off-cycle tris-PPh3 complex C but requires dissociation of at least one PPh3, a process that is facilitated by excess CO; (ii) once bis-phosphine hydrido–carbonyl complexes A/B have been generated they lead into the catalytic cycle, where the sequence of olefin coordination – hydride migration – CO insertion proceeds rapidly through either bis- or mono-phosphine intermediates; (iii) both mono- and bis-phosphine acyl complexes Q and O/P are observable as in-cycle intermediates prior to rate-limiting hydrogenolysis, with their ratios depending on the relative amounts of CO and PPh3 present; (iv) the observation that hydroformylation rates are highest under conditions that see Q dominating over O/P (PPh3/Rh = 3 at 5 bar CO) at unchanged L/B selectivity suggests that hydrogenolysis proceeds predominantly through mono-phosphine acyl intermediates.
Conclusion
The results of this investigation of one of the most widely used homogeneous catalytic systems extend multi-nuclear high-resolution FlowNMR spectroscopy to high temperature systems operating under gas pressure. While not as sensitive as gas chromatography and mass spectrometry or as fast as IR spectroscopy, FlowNMR is inherently quantitative, non-invasive and highly informative and thus powerful for following similar reaction intermediates with high specificity. Being able to quantify dissolved H2 during the analysis is uniquely useful in ensuring true operando conditions, and may be exploited to map out gas-limitation regimes to guide process development and upscaling. The ability to temporarily isolate a sample from the headspace by interruption of flow allows probing the behaviour of the system under gas-limiting conditions and following associated changes in catalyst speciation. Heteronuclear correlation experiments and diffusion measurements may be used to characterise fleeting reaction intermediates and gain insights into their dynamics. In the case of Rh/PPh3 catalysed hydroformylation of 1-hexene, this approach has led to the successful characterisation of [RhH(CO)(PPh3)3], [RhH(CO)2(PPh3)2] as well as bis- and mono-phosphine acyl intermediates under reaction conditions for the first time. Work to further extend the utility of high-resolution FlowNMR spectroscopy to study hydroformylation and other catalytic systems is currently ongoing in our laboratories.Conflicts of interest
RF is an employee of Evonik GmbH who operate industrial hydroformylation plants. The other authors declare no conflicts of interest.Note after first publication
The data in Fig. 16–18 and Scheme 3, and the related discussion, was presented at the Faraday Discussions meeting and added to the manuscript post-meeting.Acknowledgements
This work was supported by the Royal Society (UF160458; fellowship to UH), the EPSRC Dynamic Reaction Monitoring Facility (EP/P001475/1), and the EPSRC Centre for Doctoral Training in Catalysis (EP/L016443; studentship to ABE). The authors thank Dr Christopher Kubis (LIKAT, Germany) for useful discussions and help with pump configurations.References
- H. Adkins and G. Krsek, J. Am. Chem. Soc., 1949, 71, 3051–3055 CrossRef CAS.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- O. Roelen, Chemische Verwertungsgesellschaft Oberhausen m.b.H., DE 849548, 1938 & 1952; US Pat. 2327066, 1943; Chem. Abstr., 1944, 38, 3631 Search PubMed.
- L. Stahl, J. Am. Chem. Soc., 2007, 129, 10297–10298 CrossRef CAS.
- P. C. J. Kamer, J. N. H. Reek and P. W. N. M. van Leeuwen, Rhodium Catalyzed Hydroformylation, Springer International Publishing, 2005 Search PubMed.
- G. R. Stephenson, Organometallic chemistry: The transition elements, 1996 Search PubMed.
- G. Morales Torres, R. Frauenlob, R. Franke and A. Börner, Catal. Sci. Technol., 2015, 5, 34–54 RSC.
- G. Süss-Fink, in Homogeneous Transition Metal Catalyzed Reactions, American Chemical Society, 1992, vol. 230, pp. 28–419 Search PubMed.
- M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17–85 CrossRef CAS.
- S. Naqvi, Oxo Alcohols. Process Economics Program Report 21E, 2010 Search PubMed.
- H. K. Reinius, P. Suomalainen, H. Riihimäki, E. Karvinen, J. Pursiainen and A. O. I. Krause, J. Catal., 2001, 199, 302–308 CrossRef CAS.
- A. Nemati Kharat, F. Rajabi Kouchi and B. Tamaddoni Jahromi, J. Coord. Chem., 2016, 69, 12–19 CrossRef CAS.
- P.-A. R. Breuil, L. Magna and H. Olivier-Bourbigou, Catal. Lett., 2015, 145, 173–192 CrossRef CAS.
- I. Ojima, Chem. Rev., 1988, 88, 1011–1030 CrossRef CAS.
- M. Diéguez, O. Pàmies and C. Claver, Tetrahedron: Asymmetry, 2004, 15, 2113–2122 CrossRef.
- A. A. Dabbawala, R. V. Jasra and H. C. Bajaj, Catal. Commun., 2011, 12, 403–407 CrossRef CAS.
- R. Whyman, Appl. Organomet. Chem., 2001, 15, 315–316 CrossRef CAS.
- L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 336–338 CrossRef CAS PubMed.
- Y. Yan, X. Zhang and X. Zhang, J. Am. Chem. Soc., 2006, 128, 16058–16061 CrossRef CAS PubMed.
- A. M. Trzeciak, T. Głowiak, R. Grzybek and J. J. Ziółkowski, J. Chem. Soc., Dalton Trans., 1997, 1831–1838 RSC.
- M. Sparta, K. J. Børve and V. R. Jensen, J. Am. Chem. Soc., 2007, 129, 8487–8499 CrossRef CAS PubMed.
- D. Evans, J. A. Osborn and G. Wilkinson, J. Chem. Soc. A, 1968, 3133–3142 RSC.
- M. R. Axet, S. Castillon and C. Claver, Inorg. Chim. Acta, 2006, 359, 2973–2979 CrossRef CAS.
- P. C. J. Kamer, A. van Rooy, G. C. Schoemaker and P. W. N. M. van Leeuwen, Coord. Chem. Rev., 2004, 248, 2409–2424 CrossRef CAS.
- A. Aghmiz, C. Claver, A. M. Masdeu-Bultó, D. Maillard and D. Sinou, J. Mol. Catal. A: Chem., 2004, 208, 97–101 CrossRef CAS.
- T. Maji, C. H. Mendis, W. H. Thompson and J. A. Tunge, J. Mol. Catal. A: Chem., 2016, 424, 145–152 CrossRef CAS.
- C. Bianchini, H. M. Lee, A. Meli and F. Vizza, Organometallics, 2000, 19, 849–853 CrossRef CAS.
- S.-I. Pien and S. S. C. Chuang, J. Mol. Catal., 1991, 68, 313–330 CrossRef CAS.
- I. T. Horváth, R. V. Kastrup, A. A. Oswald and E. J. Mozeleski, Catal. Lett., 1989, 2, 85–90 CrossRef.
- C. Kubis, M. Sawall, A. Block, K. Neymeyr, R. Ludwig, A. Börner and D. Selent, Chem.–Eur. J., 2014, 20, 11921–11931 CrossRef CAS PubMed.
- J. M. Dreimann, E. Kohls, H. F. W. Warmeling, M. Stein, L. F. Guo, M. Garland, T. N. Dinh and A. J. Vorholt, ACS Catal., 2019, 9, 4308–4319 CrossRef CAS.
- R. Whyman, K. A. Hunt, R. W. Page and S. Rigby, J. Phys. E: Sci. Instrum., 1984, 17, 559–561 CrossRef CAS.
- A. M. R. Hall, J. C. Chouler, A. Codina, P. T. Gierth, J. P. Lowe and U. Hintermair, Catal. Sci. Technol., 2016, 6, 8406–8417 RSC.
- A. Martínez-Carrión, M. G. Howlett, C. Alamillo-Ferrer, A. D. Clayton, R. A. Bourne, A. Codina, A. Vidal-Ferran, R. W. Adams and J. Burés, Angew. Chem., Int. Ed., 2019, 58, 10189–10193 CrossRef PubMed.
- A. C. Brezny and C. R. Landis, ACS Catal., 2019, 9, 2501–2513 CrossRef CAS.
- P. Suomalainen, H. K. Reinius, H. Riihimäki, R. H. Laitinen, S. Jääskeläinen, M. Haukka, J. T. Pursiainen, T. A. Pakkanen and A. O. I. Krause, J. Mol. Catal. A: Chem., 2001, 169, 67–78 CrossRef CAS.
- R. M. Deshpande, Ind. Eng. Chem. Res., 1988, 27, 1996 CrossRef CAS.
- E. Mieczyńska, A. M. Trzeciak and J. J. Ziółkowski, J. Mol. Catal., 1992, 73, 1–8 CrossRef.
- B. E. Hanson and M. E. Davis, J. Chem. Educ., 1987, 64, 928 CrossRef CAS.
- M. Rosales, G. Chacón, A. González, I. Pacheco, P. J. Baricelli and L. G. Melean, J. Mol. Catal. A: Chem., 2008, 287, 110–114 CrossRef CAS.
- P. J. Baricelli, E. Lujano, M. Modroño, A. C. Marrero, Y. M. García, A. Fuentes and R. A. Sánchez-Delgado, J. Organomet. Chem., 2004, 689, 3782–3792 CrossRef CAS.
- D. B. G. Berry, A. Codina, I. Clegg, C. L. Lyall, J. P. Lowe and U. Hintermair, Faraday Discuss., 2019, 220, 45–57 RSC.
- J. Y. Buser and A. D. McFarland, Chem. Commun., 2014, 50, 4234–4237 RSC.
- J. M. Brown and A. G. Kent, J. Chem. Soc., Perkin Trans. 2, 1987, 1597–1607 RSC.
- C. K. Brown and G. Wilkinson, J. Chem. Soc. A, 1970, 2753–2764 RSC.
- R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5518–5526 CrossRef CAS.
- I. S. Babra, L. S. Morley, S. C. Nyburg and A. W. Parkins, J. Crystallogr. Spectrosc. Res., 1993, 23, 997–1000 CrossRef CAS.
- R. V. Kastrup, J. S. Merola and A. A. Oswald, in Catalytic Aspects of Metal Phosphine Complexes, American Chemical Society, 1982, vol. 196, pp. 3–43 Search PubMed.
- L. A. van der Veen, M. D. K. Boele, F. R. Bregman, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616–11626 CrossRef CAS.
- J. M. Brown, L. R. Canning, A. G. Kent and P. J. Sidebottom, J. Chem. Soc., Chem. Commun., 1982, 721–723 RSC.
- D. Evans, G. Yagupsky and G. Wilkinson, J. Chem. Soc. A, 1968, 2660–2665 RSC.
- M. A. Freeman and D. A. Young, Inorg. Chem., 1986, 25, 1556–1560 CrossRef CAS.
- A. M. R. Hall, P. Dong, A. Codina, J. P. Lowe and U. Hintermair, ACS Catal., 2019, 9, 2079–2090 CrossRef CAS.
- G. J. H. Buisman, L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1997, 16, 5681–5687 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fd00145j |
| This journal is © The Royal Society of Chemistry 2021 |