
Thermal enhancement of upconversion emission in nanocrystals: a comprehensive summary
Rui
Shi
 *a,
Eduardo D.
Martinez
b,
Carlos D. S.
Brites
a and
Luís D.
Carlos
*a
*a,
Eduardo D.
Martinez
b,
Carlos D. S.
Brites
a and
Luís D.
Carlos
*a
aPhantom-g, CICECO-Aveiro Institute of Materials, Physics Department, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: ruishi@ua.pt; lcarlos@ua.pt
bInstituto de Nanociencia y Nanotecnología (INN), Centro Atómico Bariloche, Comisión Nacional de Energía Atómica (CNEA), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Av. E. Bustillo 9500, R8402AGP San Carlos de Bariloche, Río Negro, Argentina
First published on 16th November 2020
Abstract
Luminescence thermal stability is a major figure of merit of lanthanide-doped nanoparticles playing an essential role in determining their potential applications in advanced optics. Unfortunately, considering the intensification of multiple electron-vibration interactions as temperature increases, luminescence thermal quenching of lanthanide-doped materials is generally considered to be inevitable. Recently, the emergence of thermally enhanced upconversion luminescence in lanthanide-doped nanoparticles seemed to challenge this stereotype, and the research on this topic rapidly aroused wide attention. While considerable efforts have been made to explore the origin of this phenomenon, the key mechanism of luminescence enhancement is still under debate. Here, to sort out the context of this intriguing finding, the reported results on this exciting topic are reviewed, and the corresponding enhancement mechanisms as proposed by different researchers are summarized. Detailed analyses are provided to evaluate the contribution of the most believed “surface-attached moisture desorption” process on the overall luminescence enhancement of lanthanide-doped nanoparticles at elevated temperatures. The impacts of other surface-related processes and shell passivation on the luminescence behaviour of the lanthanide-doped materials are also elaborated. Lack of standardization in the reported data and the absence of important experimental information, which greatly hinders the cross-checking and reanalysis of the results, is emphasized as well. On the foundation of these discussions, it is realized that the thermal-induced luminescence enhancement is a form of recovery process against the strong luminescence quenching in the system, and the enhancement degree is closely associated with the extent of luminescence loss induced by various quenching effects beforehand.
1. Introduction
Photon upconversion, involving the sequential absorption of two or more low-energy photons leading to the emission of light at a shorter wavelength, has attracted great attention in the last few decades.1–5 Different from early reported upconversion phenomena, such as second-harmonic generation6 and two-photon absorption,7 which could be only triggered by high-power excitation sources, nowadays photon upconversion is easily achieved in lanthanide-doped inorganic materials by employing a commercial low power-density laser or even a near-infrared light-emitting diode lamp.8 Due to the ladder-like 4f level structures of trivalent lanthanide ions (Ln3+) and the relatively long lifetimes of their excited states, two or more low-energy excitation photons are absorbed successively, leading to the stepwise pumping of electrons into the upper excited states, resulting in high-energy radiative emission. Besides, the creative concept of “energy transfer (ET) upconversion” as proposed by Auzel9,10 introduced the critical role of sensitizer ions (such as Yb3+ and Nd3+) possessing the efficient absorption of excitation photons into the system. This strategy for surpassing the constraint of insufficient light absorptivity of the activator ions (such as Er3+, Tm3+, and Ho3+) greatly enhances the upconversion luminescence (UCL) intensity of materials.11 Starting from the beginning of this century, the rapid progress of nanotechnology has brought new opportunities and challenges to develop nanosized materials with high UCL efficiency. The control of the spatial distributions of different dopants in the nanoparticles (NPs) has been realized as an effective way to modulate the involved ET pathway and further boost the luminescence intensity of Ln3+-doped materials.12–14 One illustrative example is the successful development of core–shell Ln3+-doped upconversion NPs (UCNPs) with the activator and sensitizer ions localized in different regions (i.e. in the core or shell of the NPs).15 Because of these remarkable improvements, Ln3+-doped UCNPs have been widely applied in various fields, such as bioimaging,16 anti-counterfeiting,17 holography,18 near-field microscopies,19 and local temperature monitoring.20Despite these great achievements, the fairly low UCL quantum yield (QY) of Ln3+-doped UCNPs is still one of the main drawbacks restricting their practical applications.21,22 For example, Yb3+/Er3+ co-doped β-NaYF4, one of the most used Ln3+-doped UCNPs, presents an upconversion QY typically less than 1% upon 980 nm commercial laser excitation,23 which is far from the QY value (∼10%) of its bulk counterpart.24 Given the identical crystal structure, site occupancy, and doping concentration of these two forms, surface-related processes are believed to be the major factor behind this huge difference in the luminescence performance (the relaxation of transition selection rules, which plays a vital role in controlling the 4f–4f luminescence properties, is also similar in both cases). As one of the unique features of NPs, the large surface-to-volume ratio (SVR) makes the luminescence properties more sensitive to the particle surface. Considering the nonlinearity of the upconversion process, a minor effect of the surface-related quenching on the primary transition of the sensitizer ions will have a great impact on the subsequently high-order UCL properties of the activators in the NP.
Recently, the topic of thermally enhanced UCL has attracted considerable attention25,26 (although no monograph review has yet been published). Several groups reported the UCL enhancement of Ln3+-doped NPs as temperature increased from room temperature (RT) to high temperatures (420–470 K),27–29 which contradicted the well-known and conventional luminescence thermal quenching.30 Although substantial experimental results have been reported in the past 15 years, the mechanism underpinning this phenomenon is still under debate. Heat is commonly considered to have a negative effect on the luminescence performance of optical materials and de-excitation processes (e.g., thermal-induced ionization,31 multiphonon relaxation,32 and electron transfer33) are efficiently promoted at high temperatures leading to a strong luminescence thermal quenching of individual centres. For the multiple ET-based processes, such as photon upconversion, this quenching effect should be even amplified because all of the involved transitions are affected.34 Moreover, subtle changes in the surface-related effects at the nanoscale may induce drastic changes in the luminescence of the NPs, being potentiated as temperature rises, resulting, thus, in an intricate emission temperature dependence.35 Understanding the temperature-dependent luminescence quenching mechanism of UCNPs can not only present an in-depth comprehension of the structure–property relationship at the nanoscale but also benefit the development of novel nanophotonic materials with particular and intriguing features. In this perspective, we will firstly review the relevant experimental findings on thermally enhanced UCL in Ln3+-doped UCNPs, emphasising the current understandings and the existing puzzles. Lastly, we will provide our viewpoints on this stimulating phenomenon.
2. Historical perspective on thermally enhanced UCL in Ln3+-doped UCNPs
2.1. Early observations
Before getting into the review and discussion, a summary of the main observations and corresponding interpretations on the thermally enhanced UCL phenomena in Ln3+-doped UCNPs by distinct research groups in recent years is tabulated in Table 1, which facilitates readers for comparison.| Year | Observations | Interpretation | Schematic illustration | Ref. |
|---|---|---|---|---|
| 2005 | • Increase of integrated photon flux of the Er3+ emissions in Yb3+/Er3+ co-doped β-NaYF4 powder as temperature increased from 10 to 100 K | The preferable population of the slightly high energy 2F5/2 multiplet of Yb3+ at high temperatures, resulting in a more efficient Yb3+-to-Er3+ ET | Fig. 1c | 36 |
| 2014–2015 | • Thermal enhancement of UCL above-RT in small-sized Yb3+/Ln3+ (Ln = Er3+, Ho3+, Tm3+) co-doped β-NaYF4 NPs | The phenomena were caused by overcoming the restricted phonon bottleneck effect at high temperatures | 29 and 44 | |
| • Excluded the contributions of laser-induced recrystallization and thermal-enhanced excitation light absorptivity to the phenomena | ||||
| • Detected a more significant UCL enhancement as the particle size decreased | ||||
| 2016 | • Found a “decrease-first increase-later” tendency of UCL intensity of Er3+ as temperature increased in 75 nm Yb3+/Er3+ co-doped α-NaYF4 NPs, and a similar but rather weaker emission variation reappeared upon subsequent heating | The increase in UCL intensity was induced by the “adsorption–desorption” process of a small amount of H2O molecules and other organic solvent residuals on the particle surface | 45 | |
| 2017 | • Observed the luminescence enhancement and the increase of decay time of Yb3+ 2F5/2–2F7/2 transition as temperature increased in 10 nm Yb3+/Ln3+ (Ln3+ = Ho3+, Tm3+) co-doped β-NaGdF4 NPs | The suppression of the OH vibration-induced (originated in the H2O molecules around NPs) de-excitation of the Yb3+ 2F5/2 state at high temperatures was the main factor inducing the thermally enhanced UCL | 46 | |
| • Recorded the weak thermal quenching of Tm3+ UCL in the core–shell Yb3+/Tm3+ co-doped UCNPs with a 3.5 nm thick inert-shell | ||||
| • Observed the weak thermal quenching of Tm3+ UCL of the core-only Yb3+/Tm3+ sample when measuring in argon atmosphere or dispersing the NPs in 1-octadecene | ||||
| • Observed the thermally enhanced UCL of Tb3+ 5D3 and 5D4 states in Tb3+-doped LiYbF4 UCNPs complying with “cooperative upconversion” mechanisms | The increasing of the population of the high-energy Stark level of the Yb3+ 2F5/2 state, favoured the phonon-assisted cooperative upconversion process and resulted in a noteworthy UCL enhancement | Fig. 6c | 63 | |
| • Observed a reversible thermally enhanced UCL in Yb3+/Eu3+ co-doped β-NaGdF4 UCNPs following the “cooperative upconversion” mechanisms | The thermal-induced lattice expansion deactivated the energy migration efficiency and suppressed the surface-related quenching effect, leading to the more favourable Yb3+-to-activators ET and resulting in the thermally enhanced UCL | Fig. 6e | 59 | |
| • Emphasized the stronger absolute UCL intensity of large-sized NPs than that of small-sized NPs even at high temperatures | ||||
| • Detected a slight thermal-induced lattice expansion as temperature increased. | ||||
| 2018 | • Found the significant thermal enhancement of Tm3+ UCL in the core-only Yb3+/Tm3+ UCNPs with high Yb3+ concentration, while the thermal quenching of Tm3+ UCL were detected in the samples either with core–shell geometry, or after annealing at high temperature or with μm-size | An efficient ET process was achieved with the participation of “surface phonons” generated by the [Yb⋯O] chelation on the surface of the UCNPs, and more “surface phonons” were created at high temperature and immediately coupled with Yb3+, then transferred the trapped energy to the Tm3+ excited state producing brighter UCL | Fig. 3a | 28 |
• Detected a more pronounced UCL enhancement when the size of the NPs decreased, and a 2000-fold increase in Tm3+ 1G4 UCL was recorded in the 9.7 nm-sized Yb3+/Tm3+ co-doped UCNPs at 453![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K K |
||||
| The essence of “surface phonon”-assisted enhancement mechanism was the level-broadening of the Yb3+ 2F5/2 state at high temperature, which reduced the energy mismatch between the 4f–4f transitions of Yb3+ and activators | Fig. 3d | 49 | ||
| • Observed a reversible thermally enhanced UCL of Ln3+ (Ln3+ = Tm3+, Ho3+, Er3+) in a new-type fluoride system (20 nm-sized Yb3+/Ln3+ co-doped Na3ZrF7 UCNPs) | The thermal-induced trapped electron release was the dominant process inducing the thermally enhanced UCL | 50 | ||
| • Detected the UCL enhancement in the ligand-free and inert-shell coating samples | ||||
| • Presented an increase in both the luminescence intensities and the Yb3+ 2F5/2 decay times accompany with the significant thermally enhanced UCL in small-sized Yb3+/Ln3+ (Ln3+ = Tm3+, Ho3+, Er3+) co-doped UCNPs as temperature increased, which could not be explained by “surface phonon”-assisted enhancement mechanism | The work provided more evidence supporting their previous understandings on this topic in 2017 and raised some questions on the “surface phonon”-assisted enhancement mechanism | Fig. 4c | 53 | |
| • Showed that the temperature dependence of UCL intensity of sample was closely correlated to the measurement atmosphere and the thermally enhanced UCL was only observed in the atmosphere containing H2O molecules | ||||
| • Observed the luminescence thermal quenching of OA ligand-stabilized Yb3+/Ln3+ co-doped UCNPs in dry Ar, which could not be explained by “surface phonon”-assisted enhancement mechanism | ||||
| • Emphasized that the entirely different temperature dependence of UCL of core–shell sample could be obtained by employing different shell-coating methods | ||||
| • Obtained a more significant UCL enhancement at elevated temperature in the core–shell system by introducing the structural defect | The UCL enhancement could be strengthened exploiting the defects as excitation energy reservoirs through the inequivalence substitution | 54 | ||
| 2019 | • Observed the thermally enhanced UCL in Yb3+/Ln3+ (Ln3+ = Tm3+, Er3+) co-doped UCNPs with 2 nm inert-shell coating, and a larger UCL enhancement was detected by doping 20%Yb3+ into the shell | The luminescence enhancement was associated with the incomplete core shielding and Yb3+-to-Yb3+ energy migration in the system | 55 | |
| • Detected a non-reversible thermally enhanced UCL in core-only Yb3+/Tm3+ β-NaGdF4 UCNPs | The nanoparticle sintering process at high temperatures (>500 K) caused the irreversible thermal enhancement of UCL | 56 | ||
| • Reported the thermally enhanced UCL in a non-fluoride host, Yb3+/Ln3+ (Ln3+ = Tm3+, Ho3+, Er3+) co-doped NaY(WO4)2 | The strong thermally enhanced UCL was ascribed to the removal of surface moisture as temperature increased | 27 | ||
| • Excluded the contributions of thermal-induced phase transition and particle coalescence to the phenomena | ||||
| • Showed that the temperature dependence of UCL intensity of sample was closely correlated to the measurement atmosphere | ||||
| • Found a slight increase of Yb3+ 2F5/2 decay rate of 49%Yb3+ doped NPs in N2 and the significant decrease of that in air as temperature increased | ||||
| • Detected a tiny mass gain (0.5%) of NPs in the cooling phase when temperature dropped back to 370 K from high temperature by employing the thermogravimetric analysis, which was attributed to the moisture re-adsorption | ||||
| • Reported the thermally enhanced UCL in OA-capped 2%Er3+ doped NaYbF4 @25%Yb3+ doped NaLuF4 UCNP powder, while the general thermal quenching of Er3+ UCL was observed by removing the oleate ligand or by using an inert NaLuF4 shell | The thermal-induced lattice expansion reduced the energy migration efficiency in the system, causing the suppression of energy dissipation by surface quenchers and finally resulting in the thermally enhanced UCL | 58 | ||
| • Found that the thermally enhanced UCL in core–shell Yb3+/Er3+ doped β-NaGdF4 NPs was only observed in the H2O containing atmospheres | The direct coupling of Yb3+ 2F5/2 state to the OH overtone vibration of the H2O molecule was mainly responsible for the significant UCL quenching in the system, and this effect was largely eliminated at high temperatures, resulting in the thermally enhanced UCL | 57 | ||
| • Observed the shell thickness dependent UCL enhancement of core–shell UCNPs at elevated temperature | ||||
| • Derived the maximum coupling distance (∼11 nm) of excited Yb3+ and the OH vibration of surface H2O | ||||
| 2020 | • Detected a thermally enhanced UCL of Er3+ 4F9/2 state in a new-type fluoride system (27 nm Yb3+/Er3+ co-doped K3ZrF7 UCNPs) when temperature increased from 273 K to 453 K, while a drastically UCL decrease was observed when temperature further increased | 60 | ||
Thermally enhanced UCL in Ln3+-doped materials was reported back in 2005. Suyver et al. reported that the integrated photon flux of Er3+ emissions arising from the 4I13/2, 4S3/2, and 2H9/2 excited states in Yb3+/Er3+ co-doped β-NaYF4 powder showed a step increase as temperature increased from 10 to 100 K, keeping almost constant when the temperature further increased to 200 K.36 Based on high-resolution excitation spectra (Fig. 1a and b) and Yb3+ and Er3+ energy level structures, the authors explained these observations by a preferable population of the slightly high energy 2F5/2|1〉 multiplet of Yb3+ at high temperatures, which was more energy-resonant with the Er3+ 4I11/2 state, thus resulting in a more efficient Yb3+–Er3+ ET (Fig. 1c). Later in 2013, Yu et al. observed an anomalous luminescence enhancement in Yb3+/Er3+ co-doped β-NaYF4 UCNPs with sizes of around 25 and 45 nm and its bulk form in the 10–150 K range.37 However, these enhancements only occurred at cryogenic temperatures, whereas the thermal quenching behaviour of UCL was generally reported above RT.38–40
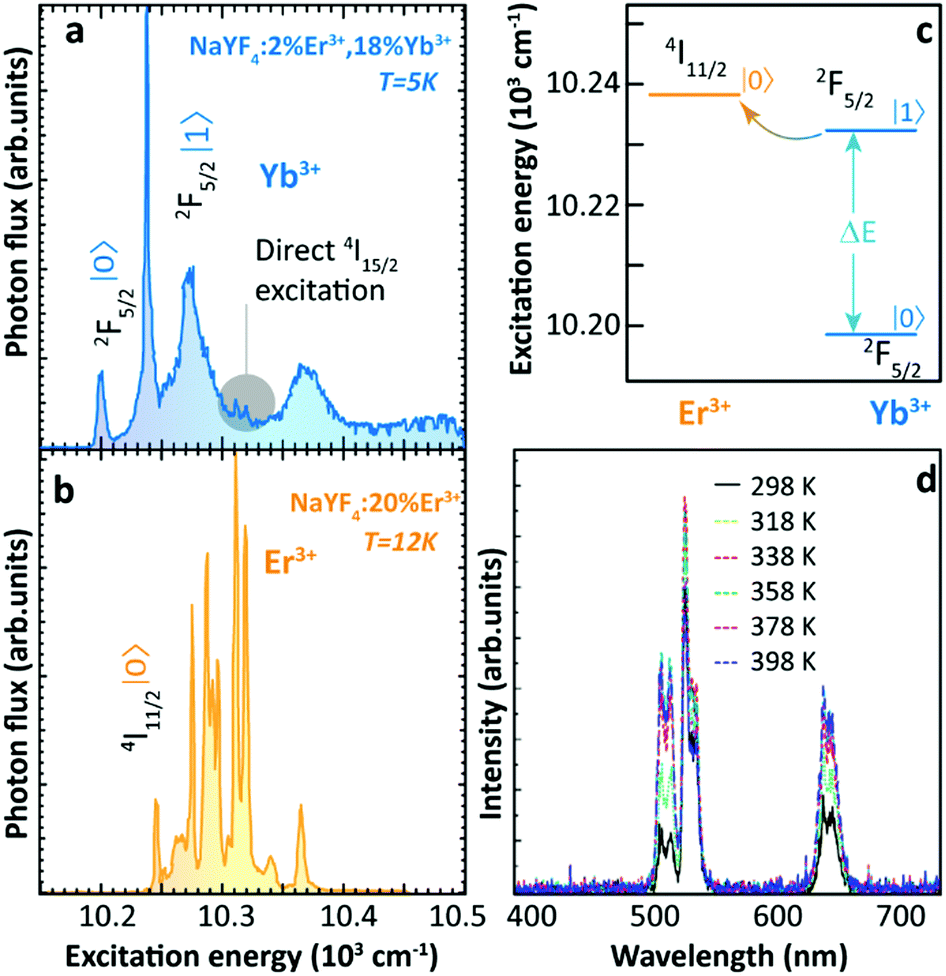 | ||
| Fig. 1 High-resolution excitation spectra of (a) Yb3+/Er3+ co-doped and (b) Er3+ single-doped β-NaYF4 bulk samples at low temperatures by monitoring the Er3+ 4S3/2 → 4I15/2 transition at 550 nm. (c) Schematic energy level diagram of Yb3+ 2F5/2 and Er3+ 4I11/2 excited multiplets. Adapted from ref. 36 with permission from Elsevier. (d) Temperature-dependent UCL spectra of 24 nm-sized Yb3+/Er3+ co-doped β-NaYF4 UCNPs. Adapted with permission from ref. 29. Copyright (2014) American Chemical Society. | ||
In 2014, Shao's group reported the above-RT thermally enhanced UCL in small-sized Yb3+/Er3+ co-doped β-NaYF4 NPs for the first time.29 Upon 975 nm excitation, the UCL intensity of Er3+ in the 24 nm-sized UCNPs increased with temperature from 298 to 358 K (Fig. 1d). Preliminary attempts to explore the origin of this result excluded a possible laser-induced recrystallization effect41 because the observed phenomenon was completely reversible. Also, the increase in excitation light absorptivity was ruled out because no variation was detected in the diffuse reflection spectra of the sample at increased temperatures. Emission spectra of Er3+ under 378 nm direct excitation were recorded at different temperatures and the luminescence thermal quenching was observed, which foreboded that the sensitizer Yb3+ ions played a crucial role in triggering the thermally enhanced UCL. To evaluate the contribution of surface-related effects on the enhancement, the temperature-dependent UCL of core-only and inert-shell coating samples with similar diameters was recorded. The UCL enhancement tendency was observed in both cases. Therefore, the authors claimed no direct relationship between thermally enhanced UCL and surface-related processes. Based on their observations and integrating them with the interpretation of Suyver et al.36 and others,42 the authors concluded that the thermally enhanced UCL was caused by overcoming the restricted phonon bottleneck effect at high temperatures. Because of the small energy differences (40–90 cm−1) between Yb3+ 2F7/2 → 2F5/2 and Er3+ 4I15/2 → 4I11/2 and 4I11/2 → 4F7/2 transitions, the Yb3+–Er3+ ET process was considered to be active only with the participation of low-energy phonons. As the particle size was reduced to the nanoscale, the phonon density of states of the material became discrete, and thus these low-energy acoustic phonon modes were cutoff.43 As the temperature increased, this phonon confinement effect was weakened, leading to more efficient Yb3+–Er3+ ET. In addition, the authors noticed that the luminescence enhancement became more significant with the decrease of particle size. For instance, the UCL intensity increased about 3 times as temperature increased from 298 to 338 K for 7 nm β-NaGdF4 nanospheres, while a luminescence thermal quenching was observed when the particle size was larger than 32 nm.29
2.2. Deeper understandings of the thermal enhancement mechanism
In 2015, Shao's group observed the thermally enhanced UCL in UCNPs with distinct upconversion couples (Yb3+–Ho3+ and Yb3+–Tm3+)44 (Fig. 2a and b). Upon 975 nm excitation, the UCL intensities of large-sized Yb3+/Ho3+ (or Yb3+/Tm3+) co-doped β-NaYF4 nanowires quenched as temperature increased. In contrast, a significant UCL enhancement was observed in all small-sized β-NaGdF4 UCNPs (∼8 nm), and a weaker enhancement was reported for larger UCNPs. Besides, it was observed that the UCL enhancement depended on the activator ion: 52.1 times for Yb3+–Ho3+, 6.2 times for Yb3+–Tm3+, and 3.3 times for Yb3+–Er3+, increasing the temperature from 298 to 398 K. This was tentatively attributed to the different energy matching degrees between the 4f–4f transitions of Yb3+ and the activators.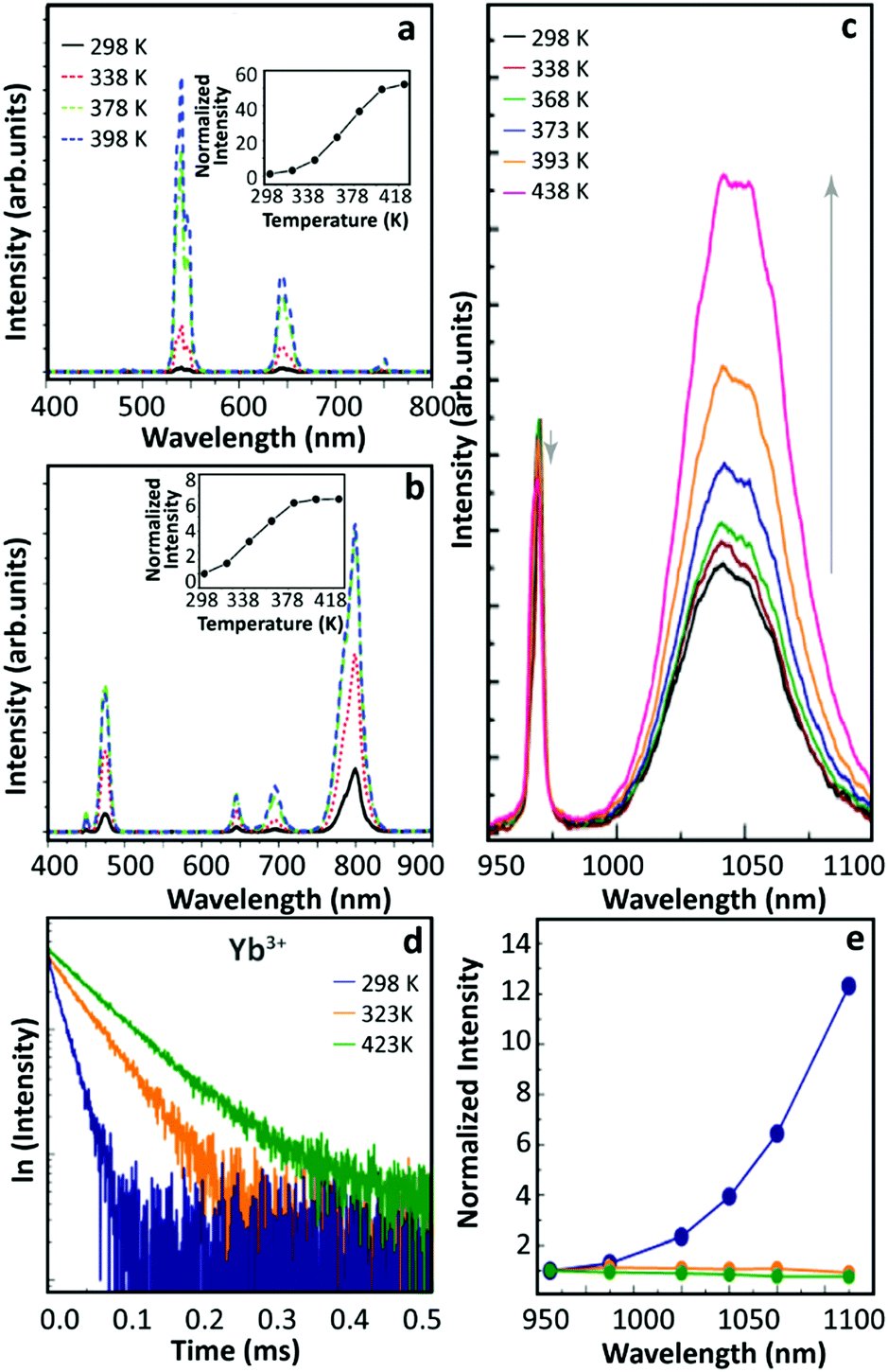 | ||
| Fig. 2 Temperature-dependent UCL spectra of (a) Yb3+/Ho3+ and (b) Yb3+/Tm3+ co-doped β-NaGdF4 UCNPs and the corresponding temperature-dependent integrated intensities. Adapted from ref. 44 with permission from Wiley-VCH. (c) Emission spectra of Yb3+/Ho3+ co-doped β-NaGdF4 UCNPs with 970 nm excitation and (d) 2F5/2 decay curves (λex = 980 nm, λem = 1050 nm) at different temperatures. (e) Temperature-dependent normalized UCL intensities of core-only Yb3+/Tm3+ co-doped β-NaGdF4 UCNPs in air (blue), in Ar (green), and Yb3+/Tm3+ co-doped β-NaGdF4@β-NaGdF4 core–shell UCNPs in air (orange). Adapted from ref. 46 with permission from The Royal Society of Chemistry. | ||
In 2016, Tong et al. reported an interesting temperature-dependent UCL variation of Yb3+/Er3+ co-doped α-NaYF4 NPs with a size of around 75 nm.45 As the temperature increased, the UCL intensity of Er3+ gradually decreased to a minimum at 483 K and then it increased as temperature further increased to 573 K. Upon cooling, the UCL intensity was recovered (as usual), while a similar but rather weaker “decrease-increase” emission intensity variation reappeared in the subsequent heating phase. The authors suggested that this anomaly was induced by the “adsorption–desorption” process of a small amount of H2O molecules and other organic solvent residuals on the particle surface at different temperatures.
Shao's group revisited thermally enhanced UCL later in 2017.46 Upon 975 nm excitation, they observed that the overall UCL intensities of ultra-small Yb3+/Ho3+ (or Yb3+/Tm3+) co-doped β-NaGdF4 UCNPs (<10 nm) increased as the temperature increased from 298 to 423 K. The luminescence spectra of Yb3+ in the co-doped sample were then recorded at increased temperatures (Fig. 2c), and the luminescence enhancement was detected. Recording the temperature-dependent luminescence decay of the Yb3+ 2F5/2 state, all the curves followed a mono-exponential trend and the decay times of the Yb3+ 2F5/2 state increased on heating (Fig. 2d), pointing out a gradual weakening of the de-excitation of the Yb3+ 2F5/2 state. The temperature-dependent UCL intensities of Tm3+ were recorded in the core–shell Yb3+/Tm3+ co-doped UCNPs with a 3.5 nm thick inert-shell and no UCL enhancement was observed, which was in contradiction with their previous result on the 2 nm thick shell-coating Yb3+/Er3+ sample.29 Besides, they measured the UCL intensity of a core-only Yb3+/Tm3+ sample in an argon (Ar) atmosphere at increased temperatures which was pre-heated in Ar at 423 K, and a weak thermal quenching was observed (Fig. 2e). Besides, the temperature-dependent emission spectra of this sample were collected dispersing the NPs in 1-octadecene, and the Tm3+ UCL intensity decreased monotonously as the solution temperature increased. Based on these results, and inspired by the analysis of other researchers,47 the authors suggested that the suppression of OH vibration-induced de-excitation of the Yb3+ 2F5/2 state at high temperatures was the main factor inducing the thermally enhanced UCL, instead of the phonon confinement effect as proposed before.29 According to this rationale, the OH vibrations of H2O molecules around NPs could induce the luminescence quenching of Yb3+ ions situated near the particle surface with a certain probability, even when the surface was fulfilled by oleic acid (OA) ligands. A feeble de-excitation of the Yb3+ 2F5/2 state would cause a severe reduction in the subsequent excited state population of the activator, resulting in a significant UCL quenching in the system. As the temperature increased, these H2O molecules gradually desorbed from the surface weakening the de-excitation of the Yb3+ 2F5/2 state and resulting in the UCL enhancement of the material. Moreover, when the temperature decreased some air moisture was again adsorbed on the particle surface and, thus, reversibility was achieved in cycling experiments.
2.3. Recent advances and the role of the surface for the thermal enhancement of upconversion luminescence
In 2018, Zhou et al. provided a new explanation about the thermally enhanced UCL in UCNPs.28 They suggested that an efficient sensitizer-to-activator ET was achieved with the participation of surface phonons generated by the chelating between the Yb3+ ions and oxygen atoms on the surface of UCNPs. As the temperature increased, more surface phonons were created by the [Yb⋯O] complexes and immediately coupled with Yb3+, then transferred the trapped energy to the Tm3+ excited state producing brighter UCL (Fig. 3a). Combining theoretical calculation and Raman spectra, the vibration energies of these surface phonons were estimated to be in the 510–560 cm−1 range. Significant enhancement of the Tm3+ 1G4 luminescence was detected in the core-only Yb3+/Tm3+ UCNPs with high Yb3+ concentration. The UCL intensities of the Yb3+/Ho3+ and Yb3+/Er3+ samples were recorded at increased temperatures, and the largest enhancement was detected in the Yb3+/Tm3+ system (Fig. 3b), in clear contradiction with the observations of Shao's group.44 In this work, Zhou et al. claimed that the contribution of the phonon confinement effect29 on the thermally enhanced UCL was excluded because the enhancement was also detected in NPs larger than 40 nm (in diameter). Recently, the role of the phonon confinement effect in controlling the relaxation between close-spaced Ln3+ Stark levels in ultra-small UCNPs was revisited.48 The UCL intensities of Tm3+ were also recorded at increased temperatures in core–shell Yb3+/Tm3+ co-doped NPs, co-doped NPs after annealing at 773 K, and co-doped μm-sized rods. As the temperature increased, the thermal quenching of UCL was observed in all the samples, which foreboded that a surface phonon was only created by direct chelation between Yb3+ and O2− of the surface ligand. Moreover, the authors reported that a decrease in the size of the NPs resulted in a more pronounced UCL enhancement, and a 30-fold enhancement of Tm3+ luminescence was achieved in the 29 nm-sized co-doped samples. Following this strategy, a record value of a 2000-fold increase in Tm3+ 1G4 UCL intensity was registered in the 9.7 nm-sized Yb3+/Tm3+ co-doped UCNPs at 453K (Fig. 3c). In a consequent work, Liang et al. pointed out that this surface phonon essentially resulted in the level-broadening of the Yb3+ 2F5/2 state that reduced the energy mismatch between the 4f–4f transitions of the Yb3+ ions and the activators49 (Fig. 3d).
 | ||
| Fig. 3 (a) Schematic illustration of the surface phonon-assisted enhancement mechanism, (b) temperature-dependent normalized UCL intensities of Tm3+: 1G4 → 3H6, Ho3+: 5S2(5F4) → 5I8 and Er3+: 4F9/2 → 4I15/2 emissions in Yb3+/Ln3+ (Ln3+ = Tm3+, Ho3+, Er3+) co-doped β-NaYF4 UCNPs with 980 nm excitation, and (c) Tm3+ blue emission spectra of 9.7 nm sized Yb3+/Tm3+ co-doped β-NaYF4 UCNPs at 453 and 303 K upon 980 nm excitation. Adapted from ref. 28 with permission from the Nature Publishing Group. (d) Level-broadening of the Yb3+ 2F5/2 state at high temperature. Adapted from ref. 49 with permission from the Nature Publishing Group. (e) Temperature-dependent normalized UCL intensities of Yb3+/Er3+ co-doped Na3ZrF7 UCNPs upon 980 nm excitation in the heating and reheating phases. Adapted from ref. 50 with permission from The Royal Society of Chemistry. | ||
In the same year, Lei et al. noticed a reversible thermal enhancement of UCL in the 20 nm-sized Yb3+/Ln3+ (Ln3+ = Tm3+, Ho3+, Er3+) co-doped Na3ZrF7 UCNPs50 (Fig. 3e). This enhancement was detected both in the ligand-free sample and in a sample coated with an inert-shell. It was also observed that the luminescence enhancement was sensitive to the doping concentration, being a weak thermal quenching detected in the core-only 5%Yb3+/20%Lu3+/2%Er3+ sample. The initial thermally enhanced UCL was recovered after a 15%Yb3+-doped active-shell coating. The temperature-dependent UCL of core-only 20%Yb3+/2%Er3+ UCNPs was measured under different pumping powers, and the enhancement was much larger under high than under low-power excitation. Moreover, the UCL intensity of the sample recorded in a cryogenic temperature range (20–290 K) presented a thermal quenching. Based on first-principles calculations, the authors rationalized their experimental results and stated that the thermal-induced trapped electron release was the dominant process inducing the thermally enhanced UCL. A similar mechanism of trapped electron release was adopted independently to elucidate the luminescence enhancement in some bulk optical materials.51,52
Also in 2018, Shao's group raised some questions on the surface phonon-assisted enhancement mechanism28 and presented new evidence supporting their previous understanding on this topic.53 Three distinct Yb3+/Ln3+ (Ln3+ = Tm3+, Ho3+, Er3+) co-doped UCNPs with different diameters, and one co-doped micron-sized rod-like sample were studied (Fig. 4a). The Ho3+-activated UCNPs showed the most significant thermal enhancement of UCL, in accordance with their previous result.44 Besides, the work presented an intriguing increase in both the luminescence intensities and the 2F5/2 decay times with increasing temperature. According to the surface phonon-assisted enhancement mechanism, the UCL enhancement of the activator was achieved at the expense of emission loss of Yb3+ because the assistance of surface phonons facilitated the Yb3+-to-activator ET causing the decrease of the Yb3+ 2F5/2 luminescence and the increase of its decay rate, in disagreement with the current results, as pointed out by the authors. The UCL intensities of Yb3+/Ln3+ co-doped UCNPs were recorded at elevated temperatures in different atmospheres (Fig. 4b), and the thermally enhanced UCL was found to be closely correlated to the suppression of the H2O molecule-induced quenching effect at high temperatures (Fig. 4c). In the same work, the authors stated that the surface phonon-assisted enhancement mechanism also failed to explain the luminescence thermal quenching behaviour of OA ligand-stabilized Yb3+/Ln3+ co-doped UCNPs as observed in dry Ar while the surface phonon was certainly formed due to the interaction between the OA ligand and the Yb3+ ions.
 | ||
| Fig. 4 (a) Morphologies of Yb3+/Ln3+ (Ln3+ = Tm3+, Ho3+, Er3+) co-doped UCNPs, (b) the normalized UCL intensities of Yb3+/Ln3+ co-doped UCNPs at different temperatures in Ar and Ar/H2O atmospheres, (c) schematic diagram of thermally enhanced UCL in core-only UCNPs, and (d) temperature-dependent normalized UCL intensities of Yb3+/Tm3+ co-doped β-NaGdF4@β-NaGdF4 core–shell UCNPs with different shell thicknesses synthesized via different shell coating methods. Adapted with permission from ref. 53. Copyright (2018) American Chemical Society. | ||
The authors also noticed that the thermally enhanced UCL was sensitive to other factors, and the effect of the shell coating was especially emphasized. In line with their previous report,46 the inert-shell as formed by the successive layer-by-layer (LBL) shell-growth method, strongly suppressed the impact of surface moisture on the core luminescence, and the so-called “critical thickness” of the inert-shell was estimated to be 3.5 nm (Fig. 4d). This “critical thickness” refers to the shell thickness value for which the UCL enhancement caused by the decoupling of surface OH to the core compensates exactly the intrinsic quenching of core luminescence at increased temperatures for a given core–shell UCNP, resulting in a temperature-independent UCL. Besides, they found that the thermally enhanced UCL was regenerated in the LBL method shell coating UCNPs if 20%Yb3+ were introduced into the shell (active-shell), which implied that the excitation energy of core Yb3+ could be delivered through Yb3+-to-Yb3+ energy migration to the particle surface and quenched by OH vibration of moisture. Intriguingly, different conclusions were drawn by employing an alternative shell coating method (Fig. 4d). Adopting the one-pot heating-up (OPH) shell-growth method, the significant thermally enhanced UCL was detected in the core-(inert)shell NPs, even for shell thickness reaching 3.5 nm, suggesting an incomplete shielding of the core from the surface moisture.
2.4. Understanding the possible underpinning mechanisms explaining the thermal enhancement
Additional efforts were made to unravel the origin of this phenomenon. Lei et al. found that the UCL enhancement could be further strengthened exploiting the defects as excitation energy reservoirs through the inequivalence substitution.54 In 2019, Martínez et al. reported the thermally enhanced UCL in Yb3+/Tm3+ (or Yb3+/Er3+) co-doped UCNPs with 2 nm inert-shell coating55 (Fig. 5a and b). By doping 20%Yb3+ into the shell, a slightly larger UCL enhancement was observed. These studies suggested that the luminescence enhancement was associated with the incomplete core shielding by a thin shell and Yb3+-to-Yb3+ energy migration in the system. Meanwhile, Qiu's group reported the thermally enhanced UCL in core-only Yb3+/Tm3+ β-NaGdF4 UCNPs. It was found that the particle sintering process proceeded at high temperatures (>500 K) and caused the irreversible thermal enhancement of UCL56 (Fig. 5c and d). Later, Meijerink's group reported the thermally enhanced UCL in a non-fluoride host, Yb3+/Ln3+ (Ln3+ = Tm3+, Ho3+, Er3+) co-doped NaY(WO4)2.27 As the temperature increased from 300 to 600 K in air, the UCL intensities of the activators increased gradually up to the maxima, and then decreased in all of these co-doped UCNPs with different Yb3+ concentrations. The decay dynamics of the Er3+ 4S3/2 and 4F9/2 states were recorded at increased temperatures in the co-doped sample, and the luminescence rise and decay times became longer as the temperature increased, indicating the weakening of nonradiative relaxations in the intermediate and emitting states of Yb3+ and Er3+. Similar results were obtained for the Er3+ single-doped sample. The results of in situ temperature-dependent XRD and TEM measurements (Fig. 5e) pointed out that the thermally enhanced UCL was not caused by the thermal-induced phase transition nor by particle coalescence. The temperature-dependent UCL measurement of co-doped UCNPs was also conducted in dry N2, that presented a UCL intensity with reversible thermal quenching/recovering performances in the heating–cooling processes after the first heating–cooling cycle (Fig. 5f). Additionally, the 2F5/2 decays in 49%Yb3+ doped NPs were measured at elevated temperatures, both in air and dry N2. With the increase of the temperature, the 2F5/2 decay rate slightly increased in N2 (from 3.17 × 104 s−1 (300 K) to 4.11 × 104 s−1 (500 K)) and significantly decreased in the air (from 1.10 × 105 s−1 (300 K) to 4.46 × 104 s−1 (500 K)). Based on these results, the authors proposed that the strong thermally enhanced UCL was ascribed to the removal of surface moisture as the temperature increased. To further confirm this hypothesis, thermogravimetric analysis (TGA) and Fourier Transform Infrared (FT-IR) measurements were carried out. After the mass loss in the initial TGA heating process, a tiny mass gain (0.5%) of NPs was detected in the cooling phase when the temperature was below 370 K (Fig. 5g), which was attributed to the moisture re-adsorption. Moreover, the characteristic OH stretching vibration band was observed in the FT-IR spectrum of the sample even after heating treatment at 570 K. | ||
| Fig. 5 (a) Temperature-dependent UCL spectra upon 980 nm excitation of the heterogeneous system containing two different kinds of Yb3+/Ln3+ (Ln3+ = Er3+, Tm) co-doped β-NaGdF4 UCNPs deposited onto an AgNWs/PMMA film and (b) the corresponding integrated intensities of overall Er3+ (or Tm3+) 4f–4f emissions in the whole spectra at different temperatures. Adapted from ref. 55 with permission from the Wiley-VCH. (c) Tm3+ 1G4 → 3H6 UCL intensities of Yb3+/Tm3+ co-doped β-NaGdF4 UCNPs in one “heating–cooling” cycle and (d) the corresponding sample morphology changes before and after heating to 565 K. Adapted from ref. 56 with permission from The Royal Society of Chemistry. (e) Temperature-dependent XRD patterns and morphology of Yb3+/Er3+ co-doped NaY(WO4)2 UCNPs at 300 and 600 K, (f) temperature-dependent UCL intensities of Yb3+/Er3+ co-doped NPs within a continuous “heating–cooling” cycle in air or N2 upon 980 nm excitation, and (g) TGA of the sample in the first cooling and reheating phases in air. Adapted from ref. 27 with permission from The Royal Society of Chemistry. (h) Yb3+ 2F5/2 emission spectrum of Yb3+/Er3+ co-doped β-NaGdF4@β-NaGdF4 core–shell UCNPs and the absorption spectra of H2O and toluene. Adapted with permission from ref. 57. Copyright (2019) American Chemical Society. | ||
Later, Wang's group observed a thermally enhanced UCL of Er3+ in OA-capped 2%Er3+-doped NaYbF4@25%Yb3+-doped NaLuF4 UCNPs, while the thermal quenching of Er3+ UCL was observed by removing the oleate ligand or by using an inert NaLuF4 shell.58 The authors considered that the thermal-induced lattice expansion reduced the energy migration efficiency in the system,59 causing the suppression of energy dissipation by surface quenchers and finally resulting in the luminescence enhancement. In the same year, Shao's group gave an additional contribution to this discussion investigating the UCNP luminescence loss mechanisms.57 A series of core–shell Yb3+/Er3+ doped β-NaGdF4 NPs with an identical core diameter (5.7 nm) and different shell thicknesses (from 1.1 to 17.7 nm) were studied. The thinnest shell coating co-doped UCNPs were used to record the UCL intensity at increased temperatures in several atmospheres (air, Ar, Ar/H2O, and Ar/D2O). The thermal enhancement of UCL was observed in the H2O containing atmospheres, and the thermal quenching was observed in the other atmospheres, ratifying the importance of the moisture-induced effect. Distinctly from what they reported before, the authors argued that the direct coupling of Yb3+ 2F5/2 excited state to the OH overtone vibration of the H2O molecule (instead of the OH fundamental vibration-involved multiphonon relaxation), was the main pathway inducing the strong de-excitation of the 2F5/2 state, leading to significant UCL quenching. This was supported by the comparison between the emission spectrum of Yb3+ and the absorption spectrum of H2O (Fig. 5h). Additionally, the temperature dependence of the UCL intensities was recorded for these samples. A “critical thickness” of 5.4 nm was obtained, a value 1.5 times larger than that reported before.53 The 2F5/2 luminescence decay in these samples permitted the authors to conclude that the maximum coupling distance of excited Yb3+ and the OH vibration of surface H2O was of the order of 11 nm.
In 2020, Hong's group reported the thermally enhanced UCL of the Er3+ 4F9/2 state in a new-type fluoride system, 27 nm-sized Yb3+/Er3+ co-doped K3ZrF7 UCNPs.60 As the temperature increased from 273 to 453 K, a more than 10-fold increase of Er3+ red UCL intensity was detected. This result further proved the generalization of thermally enhanced UCL phenomena in various Ln3+-doped UCNP systems. Recently, some researchers showed that the thermally enhanced UCL was detected in Ln3+-doped inorganic bulk compounds, in which their crystal structures exhibited the unusual negative thermal expansion properties. Zou et al. reported a unique thermally induced lattice contraction in the Er3+-doped Yb2W3O12 material.61 As the temperature increased from 303 to 573 K, a 29-fold enhancement of Er3+ green UCL was observed. Later in 2020, they found a 5-fold UCL enhancement in the analogous Yb3+/Ho3+ co-doped Sc2Mo3O12 bulk material as the temperature increased.62 The authors demonstrated that the gradual decrease of interionic distance between Yb3+ and the activators at increased temperatures played an essential role in enhancing the Yb3+ → activators ET efficiency in the system, finally resulting in the thermally enhanced UCL of the activators.
2.5. Thermal enhancement in NPs with distinct upconversion mechanisms
Despite the observation of thermally enhanced UCL in the systems displaying “ET upconversion” couples, some researchers found a similar phenomenon in other types of upconversion pairs, as complying with “cooperative upconversion” mechanisms, such as Yb3+–Tb3+ and Yb3+–Eu3+. In 2017, Chen's group reported the multiphoton UCL of the Tb3+ 5D3 and 5D4 states in Tb3+-doped LiYbF4 NPs upon high-density 980 nm laser excitation.63 As the temperature increased from 10 to 300 K, the overall UCL intensity increased almost an order of magnitude with the gradual decrease of the Yb3+ luminescence in 30%Tb3+-doped UCNPs (Fig. 6a and b). The authors attributed their observations to the temperature-induced increase of the population of the high-energy Stark level of the Yb3+ 2F5/2 state. This increase favoured the phonon-assisted upconversion process, resulting in a noteworthy UCL enhancement (Fig. 6c). | ||
| Fig. 6 (a) Temperature-dependent UCL spectra of LiYbF4:30%Tb3+@LiYF4 core–shell UCNPs upon 980 nm excitation and (b) the corresponding temperature-dependent normalized Tb3+ and Yb3+ emission intensities, and (c) the schematic illustration of the phonon-assisted “cooperative upconversion” mechanism. Adapted from ref. 63 with permission from The Royal Society of Chemistry. (d) Temperature-dependent UCL spectra of Yb3+/Eu3+ co-doped β-NaGdF4 UCNPs upon 980 nm excitation and the inset showing the corresponding normalized Eu3+ 5D0 → 7F2 emission intensities as a function of temperature, and (e) a schematic illustration of the lattice thermal expansion-induced quenching suppression mechanism. Adapted from ref. 59 with permission from The Royal Society of Chemistry. | ||
In the same year, Wang's group reported a reversible thermally enhanced UCL in Yb3+/Eu3+ co-doped β-NaGdF4 UCNPs.59 Heating the particles from 300 to 423 K, the UCL of Eu3+ showed a 16-fold enhancement in 20%Yb3+/10%Eu3+ co-doped β-NaGdF4 upon 980 nm excitation (Fig. 6d). Moreover, this enhancement exhibited a dependence on the particle size, which was more significant for the smaller UCNPs. It is noteworthy that the authors emphasized that the absolute UCL intensity of large-sized NPs was always stronger than that of small-sized NPs (even at high temperatures), and thus the thermally enhanced UCL was associated with the surface-related effects. The Yb3+-to-Yb3+ energy migration was regarded as the main non-radiative deactivation pathway64 which was further supported by the observation of a gradual decrease of the 2F5/2 decay rate in the co-doped sample as the temperature increased. Additionally, a slight thermal-induced lattice expansion was detected. Besides that, the UCL intensity of Eu3+ in the core–shell co-doped UCNPs was recorded at increased temperatures, and a weak luminescence enhancement was also discerned. Based on these observations, a different enhancement mechanism was suggested (Fig. 6e). As the temperature increased, the averaged Yb3+–Yb3+ distance becomes larger due to the lattice expansion, leading to the deactivation of Yb3+-to-Yb3+ energy migration, therefore suppressing the surface-related quenching effect. Consequently, the Yb3+-to-activators ET was favoured which resulted in the thermally enhanced UCL.
3. Emerging applications for thermally enhanced UCNPs
Apart from the basic comprehension of the thermally enhanced UCL, it soon becomes clear that this phenomenon can be used for conceiving innovative solutions in luminescence thermometry, thermochromism, and lithography.3.1. Luminescence thermometry
Luminescence thermometry is a technique using the thermal dependence of the photophysical properties of a given material to determine the temperature of its surroundings. One of the most interesting classes of phosphors for luminescence thermometry is Ln3+-doped materials. The applications of Ln3+ ions in luminescence thermometry were recently reviewed by some of us,65,66 however, several other reviews not circumscribed to Ln3+ ions were also published.67–69 Ladder-like energy levels of Ln3+ ions make them unique probes for thermometry applications. As the separation of the 4f energy levels is on the order of hundreds of cm−1, it can be easily bridged near RT (the so-called thermally coupled energy levels) by thermal redistribution, which is governed by the Boltzmann law. For the illustrative example of two thermally coupled emitting levels whose energy difference between the barycentres of the 2 → 0 and 1 → 0 emission bands is ΔE, the 2 → 0 (I2) and 1 → 0 (I1) intensity ratio is given by: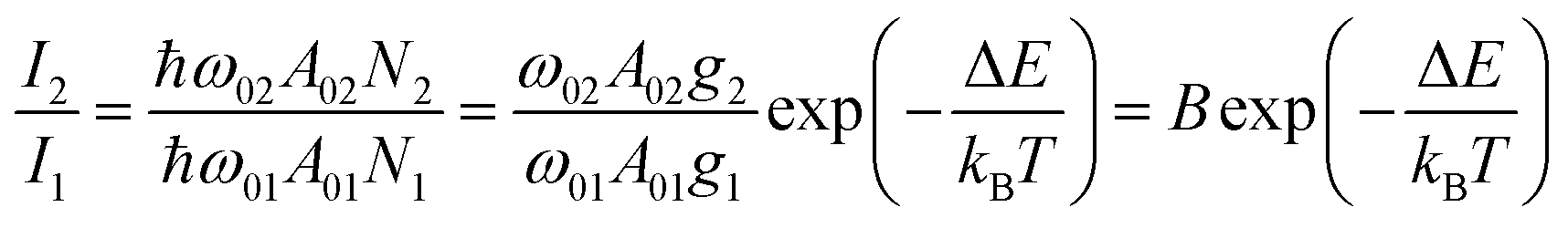 | (1) |
The UCNP-based thermometers exploit the intensity ratio of two emissions originated from thermally coupled energy levels and are intrinsically primary thermometers (i.e. the temperature can be calculated avoiding any calibration process). In contrast, the thermometers that are referred to an external temperature reference requiring a calibration procedure are termed as secondary thermometers. The restriction to the energy difference ΔE in the examples involving thermally coupled levels (to ensure the strong coupling, typically ΔE < 1000 cm−1) precludes relative thermal sensitivity (Sr) values near RT higher than 1.5% K−1.70 Larger ΔE between the thermally coupled levels decreases the thermalization of the upper |2〉 level, resulting in lower luminescence intensity.
To overcome this bottleneck, strategies for designing novel luminescence intensity ratio-based thermometers to further improve Sr should be considered other than the thermally coupled strategy, besides playing with the size of UCNPs or with the phonon energy of the hosts. The wisest approaches consist of using two distinct (and thermally decoupled) emission lines of the same Ln3+ ion or two emitting levels of distinct centres. A recent review by Cheng et al.70 discussed in detail these strategies for improving the thermometric performance based on the “fully-decoupled” or “moderately-coupled” emitting levels or emitting levels in which ET was mediated or thermally assisted by host or ligand energy levels.
The conventional thermal quenching is responsible for narrowing the operating temperature range of the luminescent thermometers because when the intensity of the transitions reaches the baseline fluctuations the device luminescent thermometer falls out of its operating range. Therefore, the thermally enhanced UCL offers the possibility of overcoming this limitation, and thus the strong luminescence at high temperatures is easily recorded even using cost-effective portable detectors. Regarding the thermal dependence of the Sr, the well-known inverse proportionality with temperature squared precludes Sr values beyond 1% K−1 for temperature above 400 K. However, this rule is relaxed when the value of temperature is derived from the ratio between non-thermally coupled transitions, and higher Sr are achievable.
There are still a few studies exploiting the thermally enhanced UCL for producing highly sensitive nanothermometers. Following the pioneering work of Shao's group in 2014,29 the utility of thermally enhanced UCL was harnessed almost immediately. In 2015, Shao's group reported the application of UCNPs with different sizes as a strategy for the development of materials for luminescence thermometry.44 They formed dry nanopowders combining large-sized Yb3+/Ho3+ (or Yb3+/Tm3+) NaYF4 nanowires with Yb3+/Tm3+ (or Yb3+/Ho3+) NaGdF4 NPs (∼8 nm). In 2019 Martínez et al. used nanocomposite transparent films combining a poly(methyl methacrylate) (PMMA) matrix and a percolating network of silver nanowires (AgNWs) to control the local temperature and therefore to fine-tune the emission intensity of UCNPs55 (Fig. 7a and b). The concept was to use nanocomposites formed by two types of UCNPs with opposite luminescence thermal responses, one containing Er3+ and the other containing Tm3+ ions. The UCL intensity of the smaller of the particles increased with the increase in temperature while that of the larger ones decreased, making the value of Sr considerably higher.
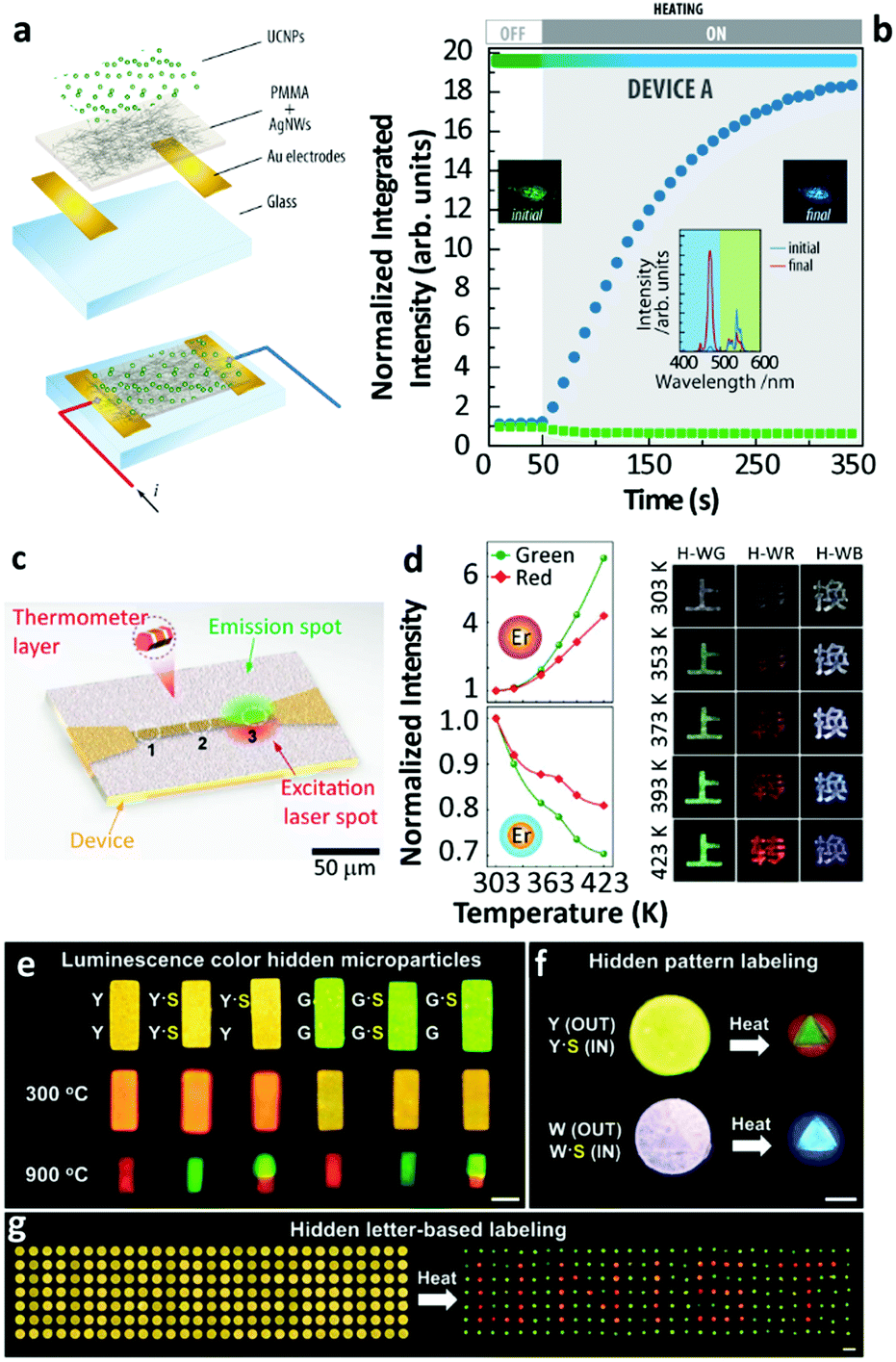 | ||
| Fig. 7 (a) Exploded view and schematic of an assembled electrothermal device and (b) the UCL intensity and overall luminescence colour change in the device upon electrothermal control. Reproduced from ref. 55 with permission from Wiley-VCH. (c) The illustration of real-time sensing of local temperature changes in a microelectronic device coated with nanothermometers. Reprinted with permission from ref. 71. Copyright (2019) American Chemical Society. (d) Temperature-dependent UCL of Yb3+/Er3+ NaGdF4 UCNPs with an inert-shell or Yb3+-doped active-shell coating, and temperature-responsive colour changes of Chinese characters printed with the corresponding hybrid inks. Reproduced from ref. 72 with permission from Wiley-VCH. (e) Temperature-sensitive luminescence colour hidden microparticles, (f) hidden pattern- and (g) hidden letter-based labelling system. Reproduced from ref. 73 with permission from the Wiley-VCH. | ||
Using the ratio of intensities between the 1G4 → 3H6 (originated in small-sized Tm3+ doped NPs) and the 4S3/2 → 4I15/2 (originated in large-sized Er3+ doped NPs) a maximum value of Sr (5.88% K−1 at 339 K) was reported. This constituted a more than 6-fold improvement relative to the value calculated at the same temperature using the commonly reported ratio between the Er3+ 4S3/2 → 4I15/2/2H11/2 → 4I15/2 transitions in the green spectral range. As an added benefit the intensity ratio involving the 4S3/2 and 2H11/2 levels was found to be independent of the size of the NPs, and the morphology and phase of the host lattice, constituting an inner primary thermometer that was very useful for the system calibration.
In a further development of the same strategy, the same authors extended the concept of reporting how Yb3+/Er3+ and Yb3+/Tm3+ co-doped UCNPs of distinct sizes embedding PMMA films could be used to fabricate self-calibrated double luminescent thermometers.74 Moreover, this report assessed the figures-of-merit of thermometers by combining mixtures of UCNPs with distinct sizes (e.g., large-sized Er3+- and small-sized Tm3+-doped UCNPs and small-sized Er3+- and large-sized Tm3+-doped UCNPs). As the nanocomposites contained a primary thermometer operating based on the Er3+ 4S3/2 → 4I15/2/2H11/2 → 4I15/2 transitions and a secondary thermometer that used the intensity ratio of Tm3+ and Er3+ transitions, the primary thermometer was used to calibrate the secondary one (that displayed a higher Sr and a lower temperature uncertainty), avoiding recurrent and time-consuming calibration procedures whenever the system operated under new experimental conditions.
A recent work of Jin's group explored the same strategy to boost the value of Sr in UCNPs.71 The concept of opposite thermally affected luminescence behaviour was exploited by constructing heterogeneous Ln3+-doped UCNPs for dual-emitting centre-based thermometry. By multistep synthesis, the authors fabricated UCNPs with selected regions co-doped with Yb3+/Nd3+ or Yb3+/Er3+ displaying thermally enhanced or quenching performances, respectively. Following this strategy, a record-breaking Sr (9.6% K−1 at RT) was achieved. The nanoparticles were then successfully used to probe the local temperature in an electrical track dissipating energy by the Joule effect (Fig. 7c).
3.2. Thermochromism
The opposite temperature-dependent UCL features of each set of particles resulted in a thermochromic luminescence of the mixed powder, whereby the emission colour shifted from green to blue depending exclusively on temperature. Shao's group applied this concept for the development of anti-counterfeiting technologies.46,75 This idea was then followed by Lei and collaborators who worked on the engineering of crystal defects in UCNPs to improve the thermally enhanced luminescence properties. They confirmed the usefulness of Yb3+/Ln3+ co-doped Na3ZrF7 UCNPs for thermochromic inks.50 Soon after, the same authors proposed the addition of low valence dopant ions (mainly Ca2+) in the composition of NaGdF4-based UCNPs to further improve the thermally enhanced UCL and the thermochromic performance of composite inks.54Through rational design and controlled synthesis, Hu et al. recently presented thermochromic nanocomposite inks based on the combination of small-sized UCNPs with inert-shell or Yb3+-doped active-shell coatings.72 Because of different thermally induced luminescence variation tendencies of the adopted shell coating UCNPs, the emission colour of the nanocomposite inks could shift throughout the chromaticity diagram following the increase in temperature (Fig. 7d). Recent works also showed that the thermochromic performance was obtained in other systems by employing the thermally enhanced UCL behaviour of the materials. Zou et al. developed a thermochromism system by combining two distinct Er3+-doped upconversion phosphors (Yb2W3O12 and Yb2WO6) together, where their UCL intensities possessed the opposite temperature dependences.61 Continuing this work, they found a 5-fold UCL enhancement in the analogous Yb3+/Ho3+ co-doped Sc2Mo3O12 bulk material as the temperature increased, and a significant luminescence colour shift was achieved, accordingly.62
A step forward in thermochromic nanocomposites is the integration of UCNPs with heating elements that can control electrically the local temperature around the luminescence emitters. With this idea, Martínez et al. developed bilayer systems formed by a semi-transparent conductive bottom layer and a top layer formed by UCNPs55 (Fig. 7a). The temperature increase was controlled by the bottom layer and resulted in the luminescence enhancement or quenching of small-sized or large-sized UCNPs, respectively, which allowed electrical control of the emission colour throughout the chromatic scale. An important feature was that the structure could be formed on flexible and transparent substrates opening the door for optoelectronic flexible devices.25
3.3. Lithography
The ability to include UCNPs in optical and optoelectronic devices in a localized and selective manner is one of the objectives sought. In this regard, several authors have shown the possibility of applying different lithography techniques to pattern UCNP deposits.76–78 Although most works did not harvest the phenomenon of thermally enhanced UCL, recent developments have taken this feature into account. Baek and co-workers developed a maskless flow lithography technique dispersing Ln3+-doped NaYF4 UCNPs and SiO2 NPs in a photocurable resin (polyurethane acrylate)73 (Fig. 7e–g). The polymer solution was flowed into a polydimethylsiloxane (PDMS) microfluidic channel and polymerized with patterned UV light. The subsequent thermal treatment resulted in the UCL enhancement of the material, which demonstrated the application potential in the multiple colour encryption field. Martínez and co-workers developed a maskless lithography method based on the photothermal action of gold nanostars deposited over a thermoplastic nanocomposite containing small-sized UCNPs.79 The thermally enhanced UCL in Er3+-doped UCNPs was analysed to probe the local temperature in the laser spot used for writing, and luminescence patterns on rigid and flexible substrates were produced.4. Discussion
4.1. Contention
Based on the above extensive revision, we can easily realize that many contradictory experimental findings have been reported and some incompatible explanations have been proposed to address the origin of thermally enhanced UCL in Ln3+-doped UCNPs. The rapid desorption of surface-attached H2O molecules at high temperatures is regarded as one of the most likely inducements. It has been reported that the massive luminescence quenching of Ln3+-doped UCNPs was observed when dispersing in aqueous solution,80–82 and the OH vibration was regarded as the most deleterious cause. Because of large SVR, NPs exposed to air have a certain probability of adsorbing the moisture in the air and forming a hydration layer around the particle surface. Therefore, the luminescence properties of UCNPs even in the powder form will be affected by the H2O molecule-induced quenching effect. If so, the thermal-induced UCL enhancement can be roughly regarded as a recovery process through which the sample regains the emission features missed due to the quenching effects. In order to deeply comprehend the nature of thermally enhanced UCL in UCNPs, a clear and aforehand understanding of the detailed luminescence quenching mechanism of the involved Ln3+ in the system is essential. In this section, this issue will be explicitly discussed.Luminescence properties of the optical material are strongly associated with the ambient medium. For Ln3+-doped UCNPs, the medium-dominated effect can not only affect the intrinsic radiative transition rate of the centre but also have a great impact on the nonradiative relaxation probabilities of their excited states.83 The former can be well-described by the local-field effect,84,85 and the latter is largely controlled by multiple processes. One of the dominant processes is the solvent quenching effect by vibration coupling. Provided that UCNPs are dispersed in H2O, the nonradiative relaxation between two closely spaced states will be triggered with the participation of suitable vibrations of the H2O molecule, resulting in a noticeable de-excitation of the upper energy state. In 2015, Arppe et al. studied the H2O molecule-induced quenching effect on the luminescence of core-only Yb3+/Er3+ (or Yb3+/Tm3+) co-doped NPs.47 Upon 380 nm excitation, the luminescence decay of the Er3+ 4S3/2 state in the co-doped sample was faster in H2O than in deuterated water (D2O, with much lower OD vibrational energy) as shown in Fig. 8a, while the luminescence decay of the Er3+ 4F9/2 state was almost identical in these two solutions. Supplemental experiments further revealed that the intensity of the 4F9/2 → 4I15/2 transition in this sample remained constant on increasing the proportion of H2O in D2O (Fig. 8b). These results indicated that the OH vibration contributed differently to the quenching behaviour of Er3+ visible luminescence from different excited levels. Besides, it showed that the decay rates of Yb3+ 2F5/2 luminescence in H2O were always faster than those in D2O (Fig. 8c). Consequently, the authors concluded that the direct de-excitation of the Yb3+ 2F5/2 state through the coupling interaction with the OH vibration of H2O was the main factor causing the UCL quenching in the system. Moreover, the authors suggested that the long-range Yb3+-to-Yb3+ energy migration would let all Yb3+ ions in the particle being strongly susceptible to the quenching effect of OH vibration because of the high concentration of Yb3+ in the general-studied UCNPs.86
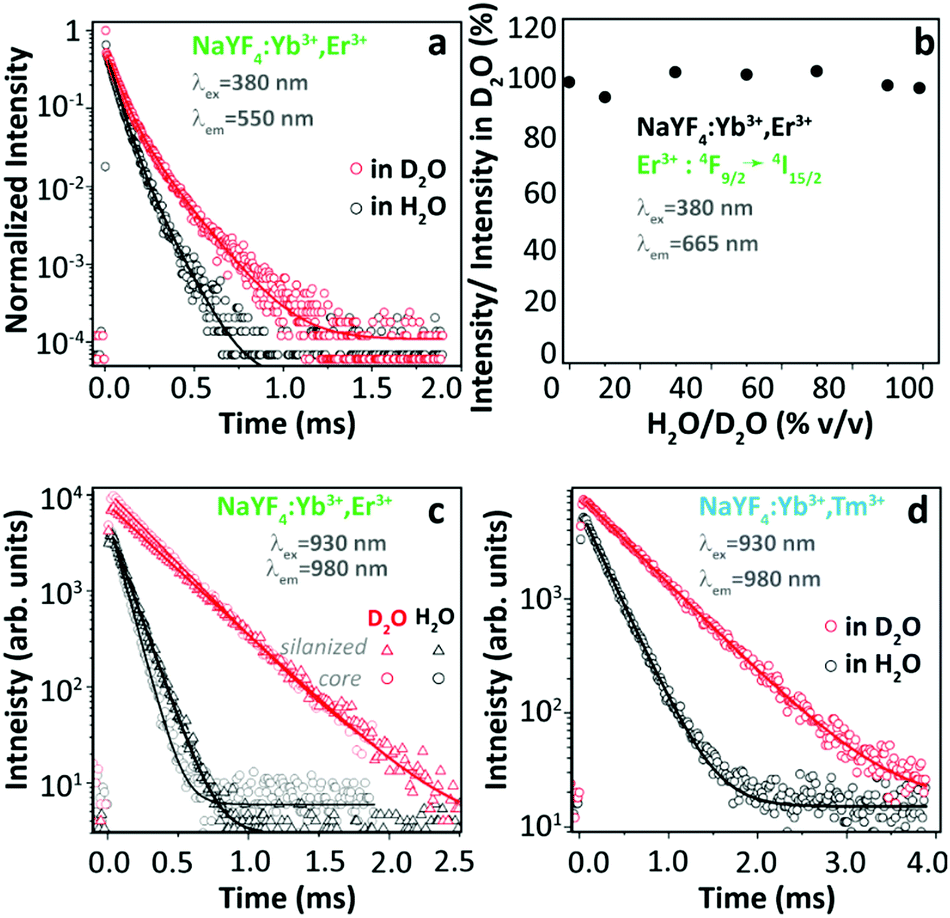 | ||
| Fig. 8 (a) Decay curves of Er3+ 4S3/2 → 4I15/2 luminescence (λex = 380 nm, λem = 550 nm) in Yb3+/Er3+ co-doped β-NaYF4 UCNPs in H2O and in D2O, and (b) normalized emission intensities of Er3+ 4F9/2 → 4I15/2 luminescence upon 380 nm excitation in D2O with increasing the proportion of H2O. (c and d) Decay curves of Yb3+ 2F5/2 → 2F7/2 luminescence (λex = 930 nm, λem = 980 nm) in Yb3+/Er3+ and Yb3+/Tm3+ co-doped UCNPs with or without a silica shell in H2O and in D2O. Reproduced from ref. 47 with permission from The Royal Society of Chemistry. | ||
Although experiments have shown that the presence of H2O certainly induced the strong luminescence loss of the material, few studies were published exploring the underlying mechanism behind the phenomena.87–90 A full understanding of this issue requires not only the clear comprehension of the UCL mechanism91–95 but also the knowledge of the effect of OH vibration on the excited electron population88,96 and subsequent luminescence dynamics of each excited state involved in the upconversion process.97 In addition, attention should be paid to considering the synergistic effects imposed by the other variables on the luminescence properties of the material,98 such as laser-induced local heating and the delocalization of dopants within the structure. Fortunately, some attempts have been made to reveal the solvent molecule-induced luminescence quenching mechanism of Ln3+ and the results are helpful to comprehend the nature of thermally enhanced UCL in Ln3+-doped UCNPs.
4.2. Solvent-induced quenching
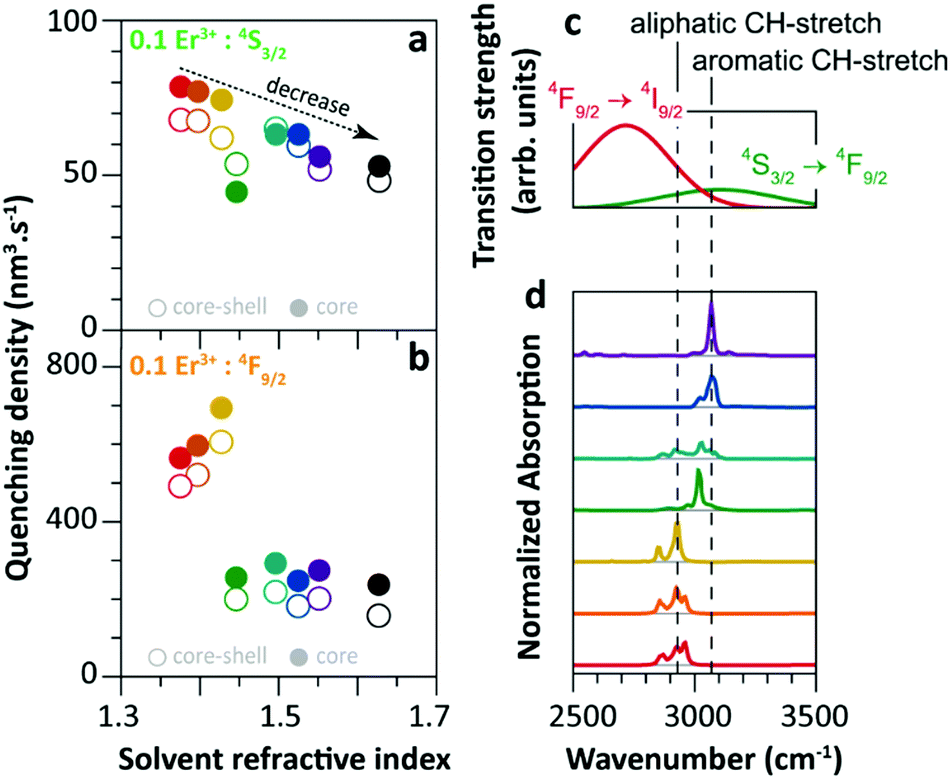
Since 2018 Meijerink's group has focused their attention on understanding the luminescence quenching mechanism in UCNPs. The main motivation of their studies is to attempt to give a quantitative interpretation of the luminescence quenching behaviour of Ln3+ ions. In their first report,35 they developed a solvent quenching model on the basis of the Förster resonant ET (FRET) mechanism, which could simulate the luminescence decay dynamics of Er3+ and Yb3+ excited states when the samples were dispersed in different organic solvents (Fig. 9a).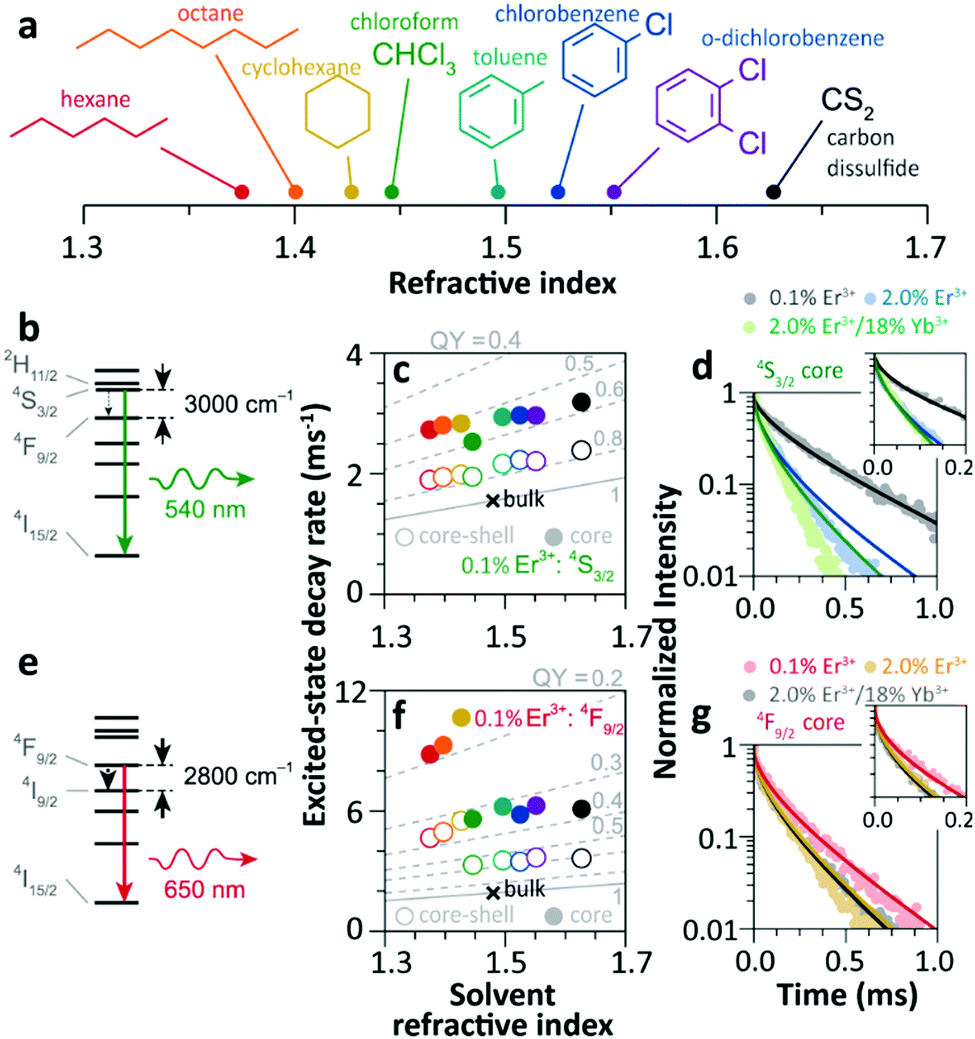 | ||
| Fig. 9 (a) Refractive index of different organic solvents. Please note the color code that will be used along with the work. (b) Energy diagram of Er3+ 4S3/2 state, and (c) averaged decay rates of the 4S3/2 state in 0.1%Er3+-doped β-NaYF4 NPs with core-only and core–shell geometries in the different solvents indicated in (a). The dashed lines represent the simulated QY values (estimated as the ratio of radiative decay rate and the total decay rate of a luminescence centre in a nanocrystal). (d) Decay curves of 4S3/2 luminescence in core-only NPs when dispersing in toluene with different concentrations and the corresponding model simulation results; the inset shows a zoom-in in the initial period. (e) Energy diagram of the Er3+ 4F9/2 state, (f) averaged decay rates of the 4F9/2 state in Er3+-doped β-NaYF4 samples in different solvents, and (g) decay curves of 4F9/2 luminescence in core-only NPs when dispersing in toluene and the corresponding simulation results. Adapted with permission from ref. 35. Copyright (2018) American Chemical Society. | ||
The analyses of the solvent quenching effect on the luminescence decays of the Er3+ 4S3/2 and 4F9/2 states were initially performed in the core-only and core–shell 0.1% Er3+ doped samples (Fig. 9b–g). Considering the small energy gaps (3200 and 2800 cm−1) of the 4S3/2 and 4F9/2 states to their next-lower-energy states (4F9/2 and 4I9/2, respectively), the nonradiative relaxation induced by the CH stretching vibration of solvent molecules (∼3000 cm−1) was significant in the core-only samples. Also, it showed that the solvent quenching effect on the luminescence of the 4S3/2 state was weaker than that of the 4F9/2 one because of the relatively lower oscillator strength of the 4S3/2 → 4F9/2 transition than that of 4F9/2 → 4I9/2.99 Besides, a stronger quenching of Er3+ 4F9/2 luminescence was detected in aliphatic solutions than in aromatic ones (Fig. 9f), and more adaptive energy resonance of the 4F9/2 → 4I9/2 relaxation and the CH stretching vibration in aliphatic solutions served as the main cause. Experimental results also confirmed the strong suppression of the inert-shell coating to the solvent quenching effect in the core–shell NPs.
More significant luminescence quenching was observed in the Er3+-doped NPs with high doping concentration (2%Er3+ and 2%Er3+/18%Yb3+). The solvent-quenching model perfectly described the luminescence decays of the Er3+ 4S3/2 states in these core–shell NPs. However, the simulation only described the initial parts of the 4S3/2 decays in the core-only NPs (blue and green curves in Fig. 9d), and the experimental decay became faster than the simulation in the later period. The authors proposed that it was caused by an efficient Er3+-to-Er3+ energy migration to the particle surface, being rapidly quenched by toluene molecules. In contrast, the model successfully simulated the decays of Er3+ 4F9/2 luminescence in these core-only NPs in toluene (Fig. 9g), and a feeble contribution of energy migration was detected.
The solvent quenching effect on the infrared luminescence of Er3+ 4I11/2 and Yb3+ 2F5/2 states was also studied (Fig. 10). Almost complete quenching (98%) was detected for Er3+ 4I11/2 luminescence in the core-only 2%Er3+ single-doped sample in all solvents (Fig. 10b). Considering the small energy gap of the Er3+ 4I11/2 → 4I13/2 transition (3500 cm−1), the solvent quenching effect was expected to be strong. Surprisingly, the luminescence decay of the Er3+ 4I11/2 state could not be described by the model at all, neither in core-only nor in core–shell NPs (Fig. 10d), and the authors believed that the OH impurity in the crystal structure, introduced during the synthesis, was the main quenching centre, especially in the core–shell samples. In contrast, the solvent quenching effect on the Yb3+ 2F5/2 luminescence was expected to be insignificant considering the large 2F5/2 → 2F7/2 energy gap. While in the core-only 18%Yb3+ doped sample, a strong luminescence quenching of the Yb3+ 2F5/2 state was observed, and the undercoordinated Yb3+ on the particle surface was regarded as the main quenching centre. The shell passivation effect was very efficient, leading to an increase of almost one order of magnitude of the 2F5/2 luminescence in the core–shell 18%Yb3+ single-doped NPs (Fig. 10c). Moreover, the result showed that the decay of 2F5/2 luminescence in the co-doped sample was much faster than that in the single-doped one (Fig. 10e).
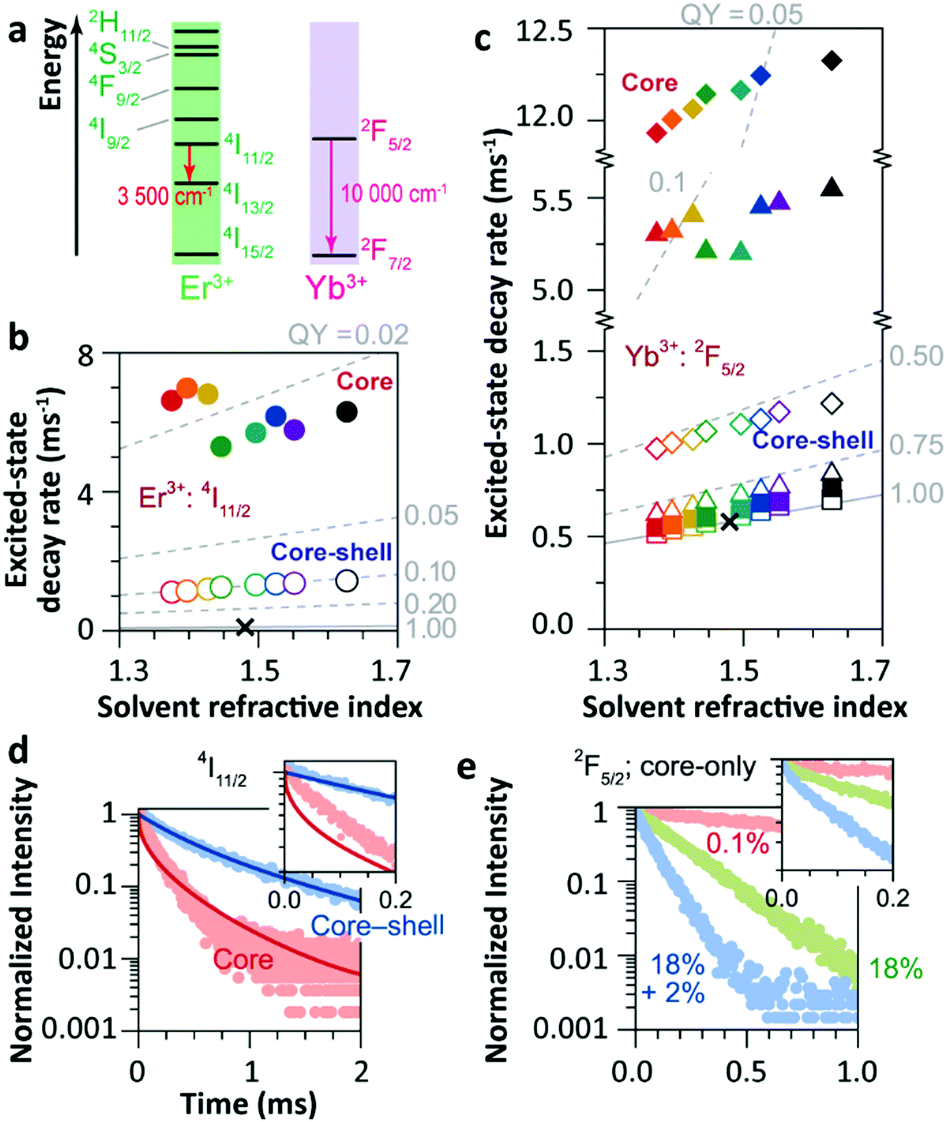 | ||
| Fig. 10 (a) Energy diagram of Er3+ and Yb3+ ions. Averaged decay rates of the (b) 4I11/2 (Er3+) of Er3+ single-doped β-NaYF4 NPs and (c) 2F5/2 (Yb3+) states of Yb3+ single-doped (squares, 0.1%Yb3+; triangles, 18%Yb3+) and Er3+/Yb3+ co-doped (diamonds) β-NaYF4 NPs with core-only and core–shell geometries in the different solvents shown in Fig. 9a (note the previously defined colour code). The dashed lines represent the simulated QY curves. Decay curves of the (d) 4I11/2 and (e) 2F5/2 energy levels in core-only and core–shell UCNPs when dispersing in toluene (points) and the corresponding simulation results (lines). The insets show the zoomed-in view in the first 0.2 ms period. Adapted with permission from ref. 35. Copyright (2018) American Chemical Society. | ||
Later, Huang et al. raised some doubts about the explanations provided by Meijerink's group.100 They suggested that the main quenching pathway of Yb3+ 2F5/2 luminescence was the coupling interaction between the 2F5/2 → 2F7/2 transition of Yb3+ and the overtone vibration of the solvent molecules in the solution,101 instead of the quenching by uncoordinated Yb3+ ions. An overtone transition is defined as the transition between two states separated by more than one vibrational quantum.102,103 The oscillator strength of the 1-overtone transitions is typically two orders of magnitude weaker than that of the fundamental transition, and each successive overtone is roughly one order of magnitude weaker.103 Huang et al. claimed that the quenching effect of the OH overtone vibration on Yb3+ 2F5/2 and Er3+ 4I13/2 states induced the huge luminescence dissipation in β-NaYF4:18%Yb3+ and β-NaYF4:20%Er3+ NPs, respectively. Thus, they speculated that the quenching effect of the CH2 overtone vibration of the organic molecule on the Yb3+ 2F5/2 luminescence should be significant. Meijerink's group made responses to these doubts afterwards104 and claimed that the overtone vibration-induced quenching effect had already been considered in the model. They also confirmed that the quenching effect of the CH2-overtone vibration on the Yb3+ 2F5/2 luminescence was very limited in the diluted-doped (0.1%Yb3+) NPs.
Later, Meijerink's group studied the quenching behaviour of Er3+ and Yb3+ luminescence in UCNPs when dispersing in solutions with different vibration modes.105 Two core–shell samples, β-NaYF4:1%Er3+@β-NaYF4 and β-NaYF4:1%Yb3+@β-NaYF4 with almost identical diameters and three different solvents, cyclohexane, H2O, and D2O were employed. A very strong quenching of the Er3+ 4I11/2 luminescence was observed in H2O (Fig. 11a) because of the resonant OH fundamental vibration with the 4I11/2 → 4I13/2 energy gap. Meanwhile, significant but with almost equal degrees of Er3+ 4I11/2 luminescence quenching was detected in the other two solvents (in D2O, the lifetime of the 4I11/2 state decreases from 10.26 ms (τradiative) to 2.59 ms; in cyclohexane, the lifetime of the 4I11/2 state decreases from 8.54 ms (τradiative) to 2.25 ms), which was expectantly caused by the 2-phonon relaxation in both cases. Huge luminescence quenching of the Er3+ 4I13/2 state was detected in H2O, while no significant quenching was detected in the other two solvents (Fig. 11b). Limited quenching of Yb3+ 2F5/2 luminescence was detected in the three different solvents (Fig. 11c). In addition, concentration-dependent decay dynamics of Yb3+ 2F5/2 luminescence and the shell passivation effect were studied in the system as well (Fig. 11d and e).
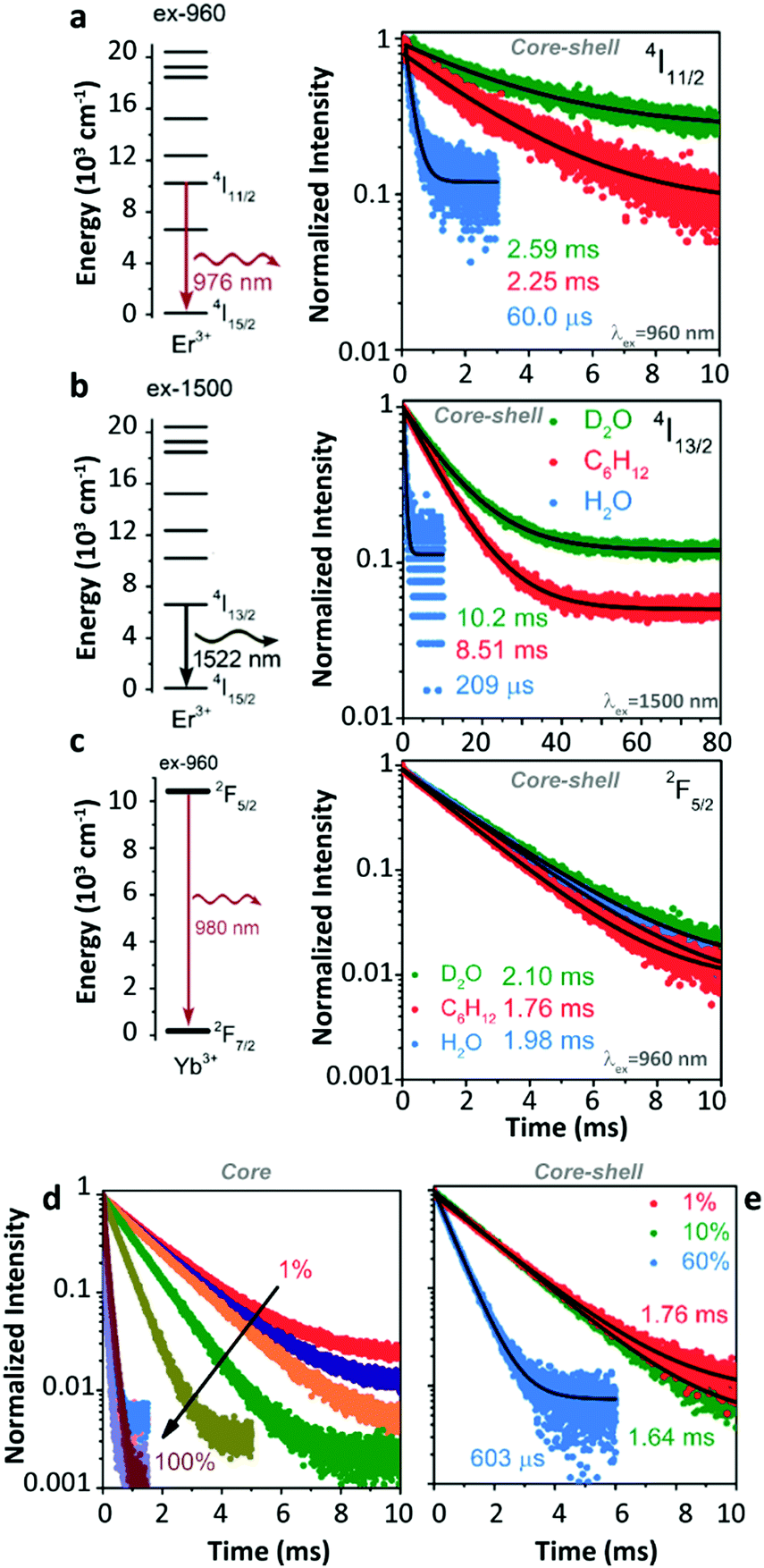 | ||
| Fig. 11 (a and b) Energy diagrams of Er3+ 4I11/2 and 4I13/2 states and the decay curves of 4I11/2 and 4I13/2 luminescence in β-NaYF4:1%Er3+@β-NaYF4 core–shell NPs dispersing in D2O, cyclohexane, and H2O upon 960 nm and 1500 nm excitations, respectively. (c) Energy diagram of the Yb3+ state and decay curves of 2F5/2 luminescence in β-NaYF4:1%Yb3+@β-NaYF4 core–shell NPs dispersing in different solvents. (d and e) Concentration-dependent decay curves of Yb3+ 2F5/2 luminescence in Yb3+ single-doped β-NaYF4 core-only and core–shell NPs as dispersing in cyclohexane upon 960 nm excitation, respectively. Adapted with permission from ref. 105. Copyright (2018) American Chemical Society. | ||
4.3. Interpretation of the solvent quenching effect
Research of Meijerink's group provides valuable guidance to understand the quenching effect of the H2O molecule. An important viewpoint is that the solvent-induced luminescence quenching can be treated as a type of FRET process by dipole–dipole coupling,106 and the quenching efficiency can be quantified by three factors: (1) energy matching between the electronic transition of Ln3+ ions and the vibration of the solvent molecules, (2) the oscillator strengths of the involved transitions and (3) the spatial distance. Similar treatment has been conceptually adopted to describe the quenching behaviours of Ln3+-doped LaF3 and LaPO4 NPs107 and other nanoscale systems.108The proposed model successfully described the quenching behaviour of Er3+ 4F9/2 luminescence in the core-only 2%Er3+-doped sample as considering the strong coupling between the 4F9/2 → 4I9/2 relaxation and the CH vibration (Fig. 9g). However, concerns should be raised to consider the quenching behaviour of Er3+ 4S3/2 luminescence as the doping concentration increased. Experimental results demonstrated that the contribution of cross-relaxation (CR) to the luminescence quenching of the Er3+ 4F9/2 state was negligible in the high-doped samples, while Er3+ 4S3/2 luminescence was greatly suffered because of the suitable CR channels.35 It is noteworthy that the decay curve of 4S3/2 luminescence in the core-only 2%Er3+ doped sample was only simulated in the initial period (Fig. 9d), and a faster decay than the simulation was observed afterward. This decay derivation was largely eliminated after shell coating. Therefore, the authors stated that it was predominantly caused by the Er3+-to-Er3+ energy migration-assisted solvent quenching effect. Yet this explanation needs to be reassessed. Considering the stronger coupling interaction of 4F9/2 → 4I9/2 relaxation to the CH vibration, this additional energy migration-assisted quenching effect on Er3+ 4F9/2 luminescence should be more severe than that of the 4S3/2 luminescence, which did not agree with the results as shown in Fig. 9g. Therefore, we consider the existence of other surface-related quenching processes in the system, which results in the decay deviation as observed above.
The model deviation became more significant when simulating the luminescence decay of Er3+ 4I11/2 and Yb3+ 2F5/2 states. Massive luminescence quenching of the Er3+ 4I11/2 state was observed, while the model could not reproduce the decay dynamics of 4I11/2 luminescence neither in core-only nor in core–shell samples (Fig. 10d), which indicated that the 4I11/2 luminescence was controlled by the other quenching effect. Moreover, the solvent quenching on Yb3+ 2F5/2 luminescence was supposed to be weak even considering the CH 2-overtone vibration (around 9000 cm−1) in the system. The quenching of Yb3+ 2F5/2 luminescence was very limited in the very diluted-doped (0.1%Yb3+) core-only sample,104 while the quenching became noteworthy as the Yb3+ concentration increased to 18% (Fig. 10c). As the solvent quenching effect was almost silent, the author believed that the uncoordinated Yb3+ on the particle surface should serve as the main quenching centre. However, careful consideration should be given to understand the detailed quenching pathway. On the one hand, the large 2F5/2 → 2F7/2 energy gap dismisses the possibility of phonon-assisted nonradiative relaxation in the system, especially in organic solutions without the participation of energetically favoured vibration models. On the other hand, several concentration-related processes can be active in the Yb3+ high-doped samples, especially the Yb3+-to-Yb3+ energy migration.14,86 With the assistance of this process, the averaged distance of excited Yb3+ to the organic solvent molecule is shortened to some extent. Besides, the large number of Yb3+ in the sample directly increases the overall coupling degree of 2F5/2 → 2F7/2 relaxation to the organic solvent vibration in the system. These two factors cooperatively enhance the solvent quenching efficiency of Yb3+ 2F5/2 luminescence even if the energy matching condition is not satisfied. While the solvent quenching effect is still far enough to dominate the overall quenching of Yb3+ luminescence in the core-only high-doped sample dispersed in organic solvents, and the contribution of other surface-related quenching processes should not be ignored.
The solvent quenching effect on the Er3+ 4I13/2 luminescence should be considered as well. Because of the relatively large 4I13/2 → 4I15/2 energy gap (∼6800 cm−1), the CH fundamental vibration-involved quenching effect on the 4I13/2 luminescence was less efficient than that on the luminescence of other Er3+ excited states in the diluted-doped sample (Fig. 11b).109 Hence, some works studied the quenching efficiencies of overtone vibrations on the 4I13/2 luminescence (Fig. 12).110–113 Because of the suitable energy matching of the 4I13/2 → 4I15/2 relaxation to the OH 1-overtone vibration, a stronger quenching effect was expected. As the Er3+ concentration increased, the efficient energy migration between different Er3+ 4I13/2 states was active, resulting in the decay acceleration of the 4I13/2 luminescence in the samples.105
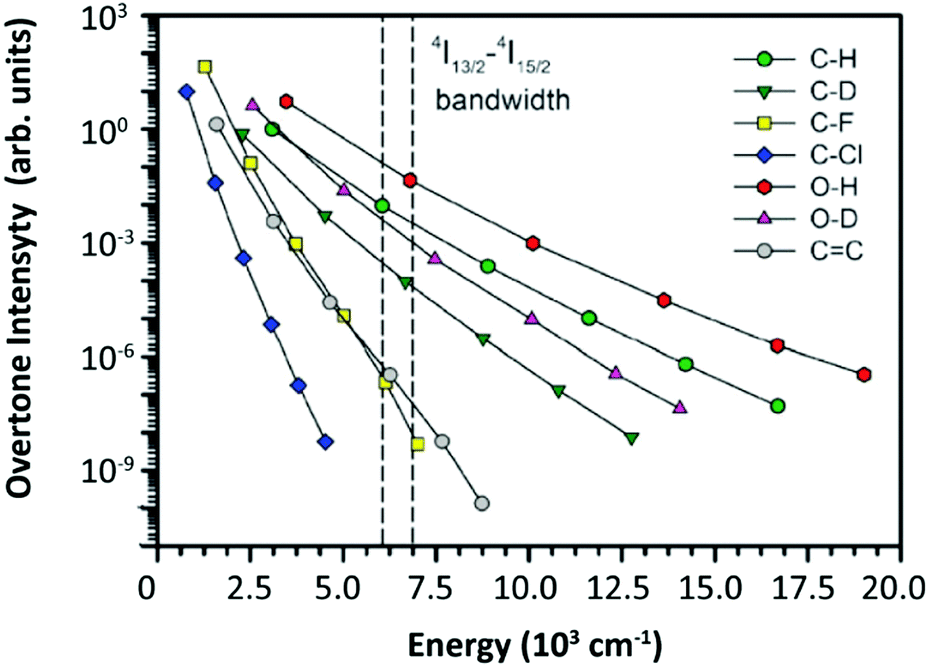 | ||
| Fig. 12 Relative oscillator strength of different types of bonds normalized for the CH fundamental vibration. The 4I13/2–4I15/2 bandwidth of Er3+ was marked by two dashed lines in the figure. Reproduced from ref. 112 with permission from the Centre National de la Recherche Scientifique (CNRS) and The Royal Society of Chemistry. | ||
One thing that should be emphasized is that the above-mentioned quenching effect is only limited to the cases of single-doped samples, and the quenching mechanism becomes more complicated in co-doped samples.114 For example, the solvent quenching effect on Yb3+ 2F5/2 luminescence was very limited in the Yb3+ diluted single-doped sample. While in the Yb3+/Er3+ co-doped system, the excitation energy of Yb3+ can “back-transfer” to Er3+ because of the nearly energy-resonant Er3+ 4I11/2 and Yb3+ 2F5/2 states and then rapidly dissipated.95 This effect was more significant in Yb3+ high-doped samples (Fig. 10e). This synergistic process should be always kept in mind when studying related issues.
A series of interesting results should be noticed. A gradual quenching relief was observed for Er3+ 4S3/2 luminescence when changing the solvent type from aliphatic to aromatic as shown in Fig. 9c and 13a. The corresponding CH fundamental vibration energy increases with this variable sequence of organic solvent molecules and should have gradually diminished the energy mismatching between Er3+ 4S3/2 → 4F9/2 relaxations and CH fundamental vibration (Fig. 13c and d). Similar quenching relief results were detected for Er3+ 4I11/2 and Yb3+ 2F5/2 luminescence as well (Fig. 10). In contrast, no solvent-dependent quenching relief was detected for Er3+ 4S9/2 luminescence (Fig. 13b) which its quenching behaviour was strongly dominated by the solvent quenching effect. Considering that the energy matching is one of the dominant factors inducing the solvent quenching in the system, these quenching relief results indicate that the other quenching process, instead of solvent quenching, plays an essential role in controlling the quenching behaviours of these excited states. This process is active in the situation that the solvent quenching effect is not prominent, and probably raises a “shielding” effect which weakens the solvent quenching effect in the system to some extent.
 | ||
| Fig. 13 (a and b) Solvent quenching densities deduced from model simulation results of the 4S3/2 and 4F9/2 luminescence in Er3+ diluted-doped β-NaYF4 NPs with core-only and core–shell geometries dispersed in the different solvents shown in Fig. 9a (note the already defined colour code). (c) The bandwidth of Er3+ 4F9/2 → 4I9/2 and 4S3/2 → 4F9/2 relaxations and (d) the solvent-dependent oscillator strengths and energies of the CH fundamental vibration modes. Adapted with permission from ref. 35. Copyright (2018) American Chemical Society. | ||
4.4. Other surface-related effects
Most of the above-mentioned pending puzzles can be well solved by introducing other frequently mentioned surface-related quenching processes.115 Because of the nature of the wet synthesis of UCNPs with large SVR, many defect centres are created on the particle surface due to incomplete crystallization.116 Recent research demonstrated that these uncrystallized centres could be transferred to the ordered crystal structure after post-annealing treatment, resulting in several orders of magnitude UCL increase of UCNPs without a change in morphology117,118 (Fig. 14). Different from the random positions of solvent molecules in the solution, these defects are localized on the particle surface generating an electrical dipole because of the imbalance of charge conservation in these regions. Besides the general-proposed irreversible electron–hole recombination-type quenching process induced by surface defects, the coupling interaction between electronic transition of Ln3+ and the assumed surface quenching dipole probably occurs as well, which raises the “surface-dipole quenching effect”. Such type of quenching process probably follows the FRET mechanism, and its quenching efficiency is closely associated with (i) the separated distance between Ln3+ and surface defects, (ii) energy matching of involved transitions, and (iii) the corresponding oscillator strengths. It should be noted in this case that the overall coupling interaction is conceptualized as a point-to-plane ET form (r−4 relationship) provided that the particle surface is abstracted as a spherical plane,119–122 which is different from the common r−6 relationship of point-to-point FRET form. This treatment is still under debate because some researchers believe that the nature of this interaction should be attributed to the electron transfer and the exponential form relationship is more realistic.123 Phenomenologically, the assumed oscillator strength of surface-dipole is predicted to be feeble around 2800 cm−1, moderate at 3200 cm−1 and large at 3500 cm−1 based on the results of the perfect model simulation of Er3+ 4F9/2 luminescence decay, the anomalous decay acceleration of Er3+ 4S3/2 luminescence in the later period, and the total deviation from the model of Er3+ 4I11/2 → 4I13/2 relaxation. As the Er3+ concentration increases, the efficient Er3+-to-Er3+ energy migration shortens the averaged distance between excited Ln3+ and surface defects, thus resulting in a more pronounce surface-dipole quenching effect on the Er3+ luminescence. Based on this hypothesis, the surface-dipole quenching effect, instead of the solvent quenching effect, serves as the dominant role in controlling the luminescence quenching of the Er3+ 4I11/2 state, which well-explains the reason for the significant but almost equal degrees of Er3+ 4I11/2 luminescence quenching as detected in D2O and cyclohexene solutions with entirely different vibration energies (Fig. 11b). Besides, as the assumed surface-dipole gains a gradually increasing oscillator strength from 2800 to 3500 cm−1, partly overlapped with the CH vibration energies of organic molecules, surface-dipoles will possibly couple with the CH vibration of the solvent molecule, which results in the simultaneous weakening of solvent quenching and surface-dipole quenching effects to some extent and leads to the quenching relief of Er3+ 4S3/2 and 4I11/2 luminescence on changing the solvent type from aliphatic to aromatic. In contrast, because of the absence of spectral overlapping between the surface-dipole transition and Er3+ 4F9/2 → 4I9/2 relaxation, no quenching relief is observed for Er3+ 4F9/2 luminescence. Moreover, this surface-dipole quenching effect probably contributes to the significant luminescence quenching of the Yb3+ 2F5/2 state in the highly doped samples as well,124 and a systematic investigation should be done in the future to identify its authenticity. | ||
| Fig. 14 (a and b) HAADF-STEM images, (c and d) intensity profiles as recorded by line scanning (directions of red and green arrows), and (e and f) enlarged crystal edge structure images of Yb3+/Er3+ co-doped KLu2F7 UCNPs before and after heating annealing. (g) UCL spectra of co-doped samples before and after annealing upon 980 nm excitation and the corresponding luminescence photographs. Reprinted with permission from ref. 117. Copyright (2018) American Chemical Society. (h) The photographs of Yb3+/Er3+ co-doped LuF3 UCNPs upon 980 nm excitation before and after laser treatment. Adapted from ref. 118 with permission from Wiley-VCH. | ||
The situation is completely different by changing the solvent from an organic solution to H2O. In consideration of the parity-forbidden nature of Ln3+ 4f–4f transitions, the quenching efficiency of the solvent quenching process largely depends on the oscillator strength of the vibration mode of the solvent molecule which is generally dipole-allowed.35 Compared with the CH vibration of an organic molecule, the OH vibration in H2O gains a larger oscillator strength (Fig. 12), which induces stronger coupling interaction. Besides, most of the above-mentioned energy gaps of Er3+ can be exactly fulfilled by the OH fundamental vibration, such as 4S3/2 → 4F9/2 (3200 cm−1) and 4I11/2 → 4I13/2 (3500 cm−1).88 Even for the large Er3+ 4I13/2 → 4I15/2 energy gap (6800 cm−1), the quenching effect of the OH 1-overtone vibration is still efficient as verified experimentally125 and theoretically.126 One exception is the Er3+ 4F9/2 state. Because of the large energy mismatch between the 4F9/2 → 4I9/2 gap and the OH fundamental vibration, the corresponding coupling interaction is almost negligible, leading to the H2O-insensitivity of 4F9/2 luminescence, as previously observed.47 Despite this, as the overall UCL efficiency depends on the individual electron population of different intermediate states, which all suffered from the OH vibration-induced solvent quenching effect, it is understandable that almost 99.9% UCL decrease has been detected in the core-only UCNPs dispersed in H2O.115 Special consideration should be given to comprehend the OH quenching effect on Yb3+ 2F5/2 luminescence. Different from the CH 2-overtone vibration of the organic molecule, the OH 2-overtone vibration possesses more appropriate energy to fill the Yb3+ 2F5/2 → 2F7/2 energy gap, which results in more efficient nonradiative relaxation of Yb3+ 2F5/2 luminescence in H2O. On the other hand, taking into consideration that the oscillator strength of the OH 2-overtone vibration is far weaker than that of the OH fundamental vibration,127,128 the involved coupling interaction should not be very strong. The trade-off between these two factors largely determines the quenching efficiency of the OH vibration on Yb3+ 2F5/2 luminescence in different diluted-doped systems. Other quenching processes will be active as Yb3+ concentration increases, which further complicates the quenching behaviour of Yb3+ 2F5/2 luminescence. Great efforts should be made in the future to consummate the understanding on this crucial issue.
4.5. Inert-shell coating
Inert-shell coating serves as an effective way to weaken the solvent quenching effect in the system as previously described. In fact, inert-shell coating also provides a remarkable passivation effect to suppress the surface quenching effect on the luminescence of the material with a core–shell geometry. Almost one order of magnitude enhancement of Er3+ and Yb3+ infrared luminescence was observed after a 3 nm thick inert-shell coating as well as a large enhancement of Er3+ visible luminescence35 (Fig. 10). Also, the enhancement degree was more significant in highly doped systems, and a two times larger enhancement of Er3+ 4S3/2 luminescence was observed in 2%Er3+ than in 0.1%Er3+ single-doped samples. In fact, several orders of magnitude UCL enhancement were reported in samples after shell coating.129–131 As a representative example, Haase et al.24 reported that the QY value of UCL of Yb3+/Er3+ co-doped UCNPs (23 nm) increased from 0.01% to 9% after 11 nm thick inert-shell coating, which was close to the highest QY in the bulk sample (10.3%) ever reported (Fig. 15a). Such a passivation effect is significant for Yb3+ 2F5/2 luminescence as well. Luminescence quenching of the Yb3+ 2F5/2 state was totally suppressed in the core–shell 10%Yb3+ single-doped NPs with a 5 nm thick β-NaYF4 inert-shell coating when dispersed in cyclohexane105 (Fig. 11e). More impressively, the OH quenching effect on Yb3+ 2F5/2 luminescence was eliminated for the core–shell 1%Yb3+ single-doped samples even dispersing in H2O (Fig. 11c). However, it is very confounded to entirely comprehend the role of inert-shell coating in the process of relieving the surface quenching effect. The surface quenching effect is generally regarded as the generic term for substantial surface defect-related quenching processes complied with different quenching pathways (such as electron–hole recombination, multipole-multipole interaction, even including exchange interaction), and their individual contributions to the overall UCL quenching efficiency of UCNPs is rarely known. In addition, the spatial distance-dependent quenching efficiencies of these quenching processes should be highly diverse. Because of the localization of defects on the particle surface, the separated distance between excited Ln3+ and these quenching centres can be modified by adjusting the inert-shell thickness, which brings about distinct passivation degrees to different luminescence quenching processes of UCNPs. Together with other variables in the system, fully understanding and further controlling the shell passivation effect on UCL properties of the material with core–shell geometry is extremely difficult. Some attempts had been made to study this issue phenomenologically. Based on the results of Alivisatos's group123 (Fig. 15b) and Berry's group121 (Fig. 15c), the UCL quenching of the Yb3+/Er3+ co-doped sample was efficiently suppressed with about 6 nm thick inert-shell coating, while Shao et al. believed that a far thicker inert-shell (∼11 nm) was required57 (Fig. 15d). The validity of these statements is relative depending on specific sample treatments, experimental conditions, and data analysis criteria. Entirely different shell thickness will be required for absolutely passivating the surface quenching effect of different excited states because of the divergent distance dependences of interaction.123 Even under the conditions of identical apparatus and the same type of NPs, the contributions from different quenching pathways to the luminescence quenching behaviour of a certain Ln3+ still needs to be cautiously interpreted.132 | ||
| Fig. 15 (a) Excitation power density-dependent upconversion QY of Yb3+/Er3+ co-doped β-NaYF4 core-only and core–shell UCNPs with different diameters upon 980 nm excitation. Reproduced from ref. 24 with permission from Wiley-VCH. (b) Shell thickness-dependent decay time of Yb3+ 2F5/2 luminescence in Yb3+/Er3+ co-doped β-NaYF4 core–shell UCNPs and the model simulation result (orange dash line). Reprinted with permission from ref. 123. Copyright (2016) American Chemical Society. (c) Shell thickness-dependent upconversion internal quantum efficiency values of Yb3+/Er3+ co-doped β-NaYF4 core–shell UCNPs and the model simulation result. Reprinted with permission from ref. 121. Copyright (2017) American Chemical Society. (d) Shell thickness-dependent decay time of Yb3+ 2F5/2 luminescence in Yb3+/Er3+ co-doped β-NaGdF4 core–shell UCNPs and the model simulation result (red line). Reprinted with permission from ref. 57. Copyright (2019) American Chemical Society. | ||
4.6. Experimental reproducibility
Lack of criterion-normalized experimental data is regarded as the other apparent obstacle for the understanding of luminescence quenching mechanisms. In a typical setup, 808/980/1530 nm lasers are generally selected as the default and exclusive excitation sources to study the UCL properties of Ln3+-doped UCNPs. However, the electron feeding kinetics of the intermediate state will have greatly influenced the luminescence decay dynamics of the activator upon indirect excitation. Because of the relatively long lifetime of the intermediate state of the sensitizer, it is not possible to extract the intrinsic decay properties of excited states of the activator and gain meaningful insight into its special quenching behaviour. Direct excitation by using different sources and wavelengths can be a suitable way to obtain the intrinsic luminescence decay kinetics of the studied state. However, some special cautions should be given to monitoring the output stability of laser power density.94 A intermediate power density should be selected to avoid triggering other electron population processes.133 The spatial distribution of power density on the excitation laser spot should be further optimized,134 especially for nonlinear UCL processes. Beyond that, more uncertainties in data are brought from the synthesis methodology of NPs. Completely different luminescence quenching behaviour of UCNPs can be induced by the fluctuation in any step during the materials synthesis, such as the choice of wet-reaction composition, synthesis procedure setup, and product post-treatment. Shao's group demonstrated that either the thermally enhanced or quenching UCL of UCNPs with identical core–shell geometry was obtained depending on different shell coating methods.53 This could probably explain the reason why thermally enhanced UCL was still detected in the core–shell UCNPs as reported by Martínez et al.55 and Xu's group.50 Particle size and shell thickness of UCNPs, regarded as two of the most controllable variables, still cannot be accomplished as expected from batch-to-batch in practical terms.135–141 Moreover, recent results showed that the cation intermixing in UCNPs was significant,122,142–145 resulting in the strong luminescence quenching through the deleterious CR processes between different Ln3+, supposedly located in the core or shell regions.144,146 In addition, the concern of cation dopant distributions in nanostructures was addressed frequently.145,147–149 These issues not only perplex the detailed analysis of currently available results but also greatly prevent the explicit understanding of the overall quenching mechanism. Moreover, the precise record of experimental variables (such as the type of protective atmosphere using in the experimental chamber, relative humidity, sample pre-treatment method, equipment calibration procedures, and relevant experimental parameters), is very important during the data collection process.133 The absence of this information in the early works aiming at the topic of thermally enhanced UCL largely restricts the possibility of result reanalysis and data cross-check. In addition, reversibility should be carefully assessed. Despite the fact that most researchers claimed that the phenomenon of thermally enhanced UCL was largely reversible in their investigations, some factors indeed resulted in the irreversible thermal enhancement of UCL.56 For instance, local heat accumulation induced by continuous laser excitation has been proven to induce crystallization of the amorphous surface of NPs, which significantly enhanced the UCL intensity of the studied samples.118 This effect is generally neglected during the successive spectral data collection. It is worth making the necessary efforts in the future to evaluate the contribution to the overall UCL enhancement of photothermally-induced surface structure transformation of the sample during the thermal cycling. This, and the careful reporting of experimental conditions, will facilitate the origin revealing of thermally enhanced UCL in the general cases.5. Conclusions
Retrieving the topic of thermally enhanced UCL in UCNPs, it is gradually realized that this phenomenon is attributed to the concurrent effects of multiple factors. As one of the most dominant, the effect of moisture detachment is carefully discussed. Strong luminescence quenching of Ln3+-doped UCNPs has been observed before when dispersing in aqueous solution, however, the specific role of H2O molecules in affecting the luminescence properties of material was rarely explored, which largely prevented the understanding of the experimental findings. In this work, the luminescence quenching of each excited state in the representative Yb3+/Er3+ co-doped UCNPs is systematically interpreted based on the previously proposed solvent quenching model. The interaction between Ln3+ and H2O molecules is carefully explicated. On the one hand, direct luminescence quenching of the Yb3+ 2F5/2 state by the OH vibration is proved to be very limited in the diluted single-doped sample. On the other hand, the electron populations of most excited states of Er3+ are easily affected by the H2O molecule because of its suitable OH vibration energy, which probably results in a significant de-excitation of these states. As the temperature increases, the adsorptive moisture gradually detaches from the particle surface, leading to a relief of the solvent quenching effect on Er3+ luminescence and inducing the UCL enhancement of the sample.Different from the case of UCNPs thoroughly dispersing in aqueous solution, the solvent quenching effect on the UCL generated from the surface-attached hydration layer of powder-formed NPs is relatively weak when placed in the ambient atmosphere. In contrast, the quenching efficiencies of other processes on the luminescence of UCNPs, possibly arising from the surface and intrinsic defects,124 is high and largely moisture-independent, generally leading to a very weak UCL of the sample at RT. On this occasion, the luminescence recovery caused by weakening the solvent quenching effect will be amplified to some extent, resulting in observable UCL enhancement as the temperature increases. Complete inert-shell coating is proven to effectively suppress the surface-related quenching effects and greatly preserve the UCL intensity of core Ln3+. Hence, in passivated core–shell UCNPs the luminescence recovery gained from the moisture detachment is insufficient to offset the intrinsic UCL quenching at high temperatures; therefore, thermal quenching behaviour is observed. More precisely, the thermal enhancement degree of UCL is associated with the extent of luminescence loss induced by various quenching effects beforehand. Therefore, it is understandable that many conflicting results have been reported because of the large uncertainty in data acquisition in different articles. Moreover, other factors, such as phase transition, thermal-induced line broadening, lattice expansion, as well as kinetic electron population of the intermediated state, should not be neglected. All these mechanisms contribute to the temperature-dependent luminescence properties of UCNP.
Many efforts should be further made to uncover the mystery of the thermally enhanced UCL phenomenon. Lack of criterion-normalized experimental setup is regarded as one of the main obstacles for the acquisition of cross-validated experimental results. Besides that, more uncertainties can be brought from the synthesis methodology of NPs. Future studies should be proceeded to deeply understand the luminescence quenching mechanism in small-sized NPs on the foundation that the unity criteria of sample synthesis, experimental setup, and data analysis are regulated. Only with a fundamental understanding of quenching mechanisms will it be possible to develop UCNPs with superior UCL properties, capable of meeting application requirements, for example in optogenetics,150 visual enhancement,151in vivo imaging152 and intracellular temperature monitoring.153
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially developed under the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 & UIDP/50011/2020, financed by Portuguese funds through the FCT/MEC and when appropriate co-financed by FEDER under the PT2020 Partnership Agreement. Financial support from the project NanoHeatControl, POCI-01-0145-FEDER-031469, funded by FEDER, through POCI and by Portuguese funds (OE), through FCT/MCTES, and by the European Union's Horizon 2020 FET Open program under grant agreements no. 801305 are acknowledged. EDM acknowledges funding from the National Agency for the Promotion of Science and Technology (ANPCyT), through grant PICT 2017-0307. R. S. would like to thank Dr Zijun Wang (École Polytechnique) for valuable discussions.References
- F. Auzel, Chem. Rev., 2004, 104, 139–174 CrossRef CAS.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS.
- M. Bettinelli, L. Carlos and X. Liu, Phys. Today, 2015, 68, 38 CrossRef CAS.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS.
- P. A. Franken, A. E. Hill, C. W. Peters and G. Weinreich, Phys. Rev. Lett., 1961, 7, 118–119 CrossRef.
- W. Kaiser and C. G. B. Garrett, Phys. Rev. Lett., 1961, 7, 229–231 CrossRef CAS.
- Y. Zhong, I. Rostami, Z. Wang, H. Dai and Z. Hu, Adv. Mater., 2015, 27, 6418–6422 CrossRef CAS.
- F. Auzel, C. R. Acad. Sci., 1966, 262, 1016–1019 Search PubMed.
- F. Auzel, J. Lumin., 2020, 223, 116900 CrossRef CAS.
- L. M. Wiesholler, F. Frenzel, B. Grauel, C. Würth, U. Resch-Genger and T. Hirsch, Nanoscale, 2019, 11, 13440–13449 RSC.
- G. Chen, H. Ågren, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Soc. Rev., 2015, 44, 1680–1713 RSC.
- X. Chen, D. Peng, Q. Ju and F. Wang, Chem. Soc. Rev., 2015, 44, 1318–1330 RSC.
- Q. Su, S. Han, X. Xie, H. Zhu, H. Chen, C.-K. Chen, R.-S. Liu, X. Chen, F. Wang and X. Liu, J. Am. Chem. Soc., 2012, 134, 20849–20857 CrossRef CAS.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2014, 8, 32–36 CrossRef CAS.
- R. Deng, F. Qin, R. Chen, W. Huang, M. Hong and X. Liu, Nat. Nanotechnol., 2015, 10, 237–242 CrossRef CAS.
- M. B. Prigozhin, P. C. Maurer, A. M. Courtis, N. Liu, M. D. Wisser, C. Siefe, B. Tian, E. Chan, G. Song, S. Fischer, S. Aloni, D. F. Ogletree, E. S. Barnard, L.-M. Joubert, J. Rao, A. P. Alivisatos, R. M. Macfarlane, B. E. Cohen, Y. Cui, J. A. Dionne and S. Chu, Nat. Nanotechnol., 2019, 14, 420–425 CrossRef CAS.
- C. D. S. Brites, X. Xie, M. L. Debasu, X. Qin, R. Chen, W. Huang, J. Rocha, X. Liu and L. D. Carlos, Nat. Nanotechnol., 2016, 11, 851–856 CrossRef CAS.
- D. J. Gargas, E. M. Chan, A. D. Ostrowski, S. Aloni, M. V. P. Altoe, E. S. Barnard, B. Sanii, J. J. Urban, D. J. Milliron, B. E. Cohen and P. J. Schuck, Nat. Nanotechnol., 2014, 9, 300–305 CrossRef CAS.
- M. S. Meijer, P. A. Rojas-Gutierrez, D. Busko, I. A. Howard, F. Frenzel, C. Würth, U. Resch-Genger, B. S. Richards, A. Turshatov, J. A. Capobianco and S. Bonnet, Phys. Chem. Chem. Phys., 2018, 20, 22556–22562 RSC.
- J.-C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417–1419 RSC.
- C. Homann, L. Krukewitt, F. Frenzel, B. Grauel, C. Würth, U. Resch-Genger and M. Haase, Angew. Chem., Int. Ed., 2018, 57, 8765–8769 CrossRef CAS.
- E. D. Martínez, C. D. S. Brites, L. D. Carlos, R. R. Urbano and C. Rettori, Front. Chem., 2019, 7, 83 CrossRef.
- B. Chen and F. Wang, Trends Chem., 2020, 2, 427–439 CrossRef CAS.
- Z. Wang, J. Christiansen, D. Wezendonk, X. Xie, M. A. van Huis and A. Meijerink, Nanoscale, 2019, 11, 12188–12197 RSC.
- J. Zhou, S. Wen, J. Liao, C. Clarke, S. A. Tawfik, W. Ren, C. Mi, F. Wang and D. Jin, Nat. Photonics, 2018, 12, 154–158 CrossRef CAS.
- D. Li, Q. Shao, Y. Dong and J. Jiang, J. Phys. Chem. C, 2014, 118, 22807–22813 CrossRef CAS.
- V. Bachmann, C. Ronda and A. Meijerink, Chem. Mater., 2009, 21, 2077–2084 CrossRef CAS.
- P. Dorenbos, J. Phys.: Condens. Matter, 2005, 17, 8103–8111 CrossRef CAS.
- C. B. Layne, W. H. Lowdermilk and M. J. Weber, Phys. Rev. B: Condens. Matter Mater. Phys., 1977, 16, 10–20 CrossRef CAS.
- A. A. Setlur and J. J. Shiang, J. Phys. Chem. C, 2010, 114, 2792–2798 CrossRef CAS.
- M. T. Berry and P. S. May, J. Phys. Chem. A, 2015, 119, 9805–9811 CrossRef CAS.
- F. T. Rabouw, P. T. Prins, P. Villanueva-Delgado, M. Castelijns, R. G. Geitenbeek and A. Meijerink, ACS Nano, 2018, 12, 4812–4823 CrossRef CAS.
- J. F. Suyver, J. Grimm, K. W. Krämer and H.-U. Güdel, J. Lumin., 2005, 114, 53–59 CrossRef CAS.
- W. Yu, W. Xu, H. Song and S. Zhang, Dalton Trans., 2014, 43, 6139–6147 RSC.
- J. Zhao, H. Li, Q. Zeng, K. Song, X. Wang and X. Kong, Chem. Lett., 2013, 42, 310–312 CrossRef CAS.
- J. Shan, W. Kong, R. Wei, N. Yao and Y. Ju, J. Appl. Phys., 2010, 107, 054901 CrossRef.
- J. Dong and J. I. Zink, ACS Nano, 2014, 8, 5199–5207 CrossRef CAS.
- A. Bednarkiewicz, D. Wawrzynczyk, A. Gagor, L. Kepinski, M. Kurnatowska, L. Krajczyk, M. Nyk, M. Samoc and W. Strek, Nanotechnology, 2012, 23, 145705 CrossRef CAS.
- G. K. Liu, H. Z. Zhuang and X. Y. Chen, Nano Lett., 2002, 2, 535–539 CrossRef CAS.
- G. K. Liu, X. Y. Chen, H. Z. Zhuang, S. Li and R. S. Niedbala, J. Solid State Chem., 2003, 171, 123–132 CrossRef CAS.
- D. D. Li, Q. Y. Shao, Y. Dong, F. Fang and J. Q. Jiang, Part. Part. Syst. Charact., 2015, 32, 728–733 CrossRef CAS.
- L. Tong, X. Li, R. Hua, T. Peng, Y. Wang, X. Zhang and B. Chen, J. Nanosci. Nanotechnol., 2016, 16, 816–821 CrossRef CAS.
- Q. Shao, G. Zhang, L. Ouyang, Y. Hu, Y. Dong and J. Jiang, Nanoscale, 2017, 9, 12132–12141 RSC.
- R. Arppe, I. Hyppänen, N. Perälä, R. Peltomaa, M. Kaiser, C. Würth, S. Christ, U. Resch-Genger, M. Schäferling and T. Soukka, Nanoscale, 2015, 7, 11746–11757 RSC.
- J. J. H. A. van Hest, G. A. Blab, H. C. Gerritsen, C. de Mello Donega and A. Meijerink, J. Phys. Chem. C, 2018, 122, 3985–3993 CrossRef CAS.
- L. Liang and X. Liu, Nat. Photonics, 2018, 12, 124–125 CrossRef CAS.
- L. Lei, D. Chen, C. Li, F. Huang, J. Zhang and S. Xu, J. Mater. Chem. C, 2018, 6, 5427–5433 RSC.
- J. Qiao, L. Ning, M. S. Molokeev, Y.-C. Chuang, Q. Liu and Z. Xia, J. Am. Chem. Soc., 2018, 140, 9730–9736 CrossRef CAS.
- R. Shi, L. Ning, Z. Wang, J. Chen, T.-K. Sham, Y. Huang, Z. Qi, C. Li, Q. Tang and H. Liang, Adv. Opt. Mater., 2019, 7, 1901187 CrossRef CAS.
- Y. Hu, Q. Shao, P. Zhang, Y. Dong, F. Fang and J. Jiang, J. Phys. Chem. C, 2018, 122, 26142–26152 CrossRef CAS.
- L. Lei, J. Xia, Y. Cheng, Y. Wang, G. Bai, H. Xia and S. Xu, J. Mater. Chem. C, 2018, 6, 11587–11592 RSC.
- E. D. Martínez, C. D. S. Brites, L. D. Carlos, A. F. García-Flores, R. R. Urbano and C. Rettori, Adv. Funct. Mater., 2019, 29, 1807758 CrossRef.
- D. Li, W. Wang, X. Liu, C. Jiang and J. Qiu, J. Mater. Chem. C, 2019, 7, 4336–4343 RSC.
- Y. Hu, Q. Shao, Y. Dong and J. Jiang, J. Phys. Chem. C, 2019, 123, 22674–22679 CrossRef CAS.
- B. Chen, W. Kong, N. Wang, G. Zhu and F. Wang, Chem. Mater., 2019, 31, 4779–4786 CrossRef CAS.
- X. Cui, Y. Cheng, H. Lin, F. Huang, Q. Wu and Y. Wang, Nanoscale, 2017, 9, 13794–13799 RSC.
- P. Peng, N. Wu, L. Ye, F. Jiang, W. Feng, F. Li, Y. Liu and M. Hong, ACS Nano, 2020 DOI:10.1021/acsnano.0c02601.
- H. Zou, X. Yang, B. Chen, Y. Du, B. Ren, X. Sun, X. Qiao, Q. Zhang and F. Wang, Angew. Chem., Int. Ed., 2019, 58, 17255–17259 CrossRef CAS.
- H. Zou, B. Chen, Y. Hu, Q. Zhang, X. Wang and F. Wang, J. Phys. Chem. Lett., 2020, 11, 3020–3024 CrossRef CAS.
- Q. Zou, P. Huang, W. Zheng, W. You, R. Li, D. Tu, J. Xu and X. Chen, Nanoscale, 2017, 9, 6521–6528 RSC.
- J. Zuo, Q. Li, B. Xue, C. Li, Y. Chang, Y. Zhang, X. Liu, L. Tu, H. Zhang and X. Kong, Nanoscale, 2017, 9, 7941–7946 RSC.
- C. D. S. Brites, S. Balabhadra and L. D. Carlos, Adv. Opt. Mater., 2019, 7, 1801239 CrossRef.
- C. D. S. Brites, A. Millán and L. D. Carlos, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier Science, B. V., Amsterdam, 2016, vol. 49, ch. 281, pp. 339–427 Search PubMed.
- L. D. Carlos and F. Palacio, Thermometry at the nanoscale: Techniques and selected applications, Royal Society of Chemistry, Oxfordshire, 2016 Search PubMed.
- M. Dramićanin, Luminescence Thermometry: Methods, Materials, and Applications, Elsevier, Cambridge, 2018 Search PubMed.
- D. Jaque and F. Vetrone, Nanoscale, 2012, 4, 4301–4326 RSC.
- Y. Cheng, Y. Gao, H. Lin, F. Huang and Y. Wang, J. Mater. Chem. C, 2018, 6, 7462–7478 RSC.
- C. Mi, J. Zhou, F. Wang, G. Lin and D. Jin, Chem. Mater., 2019, 31, 9480–9487 CrossRef CAS.
- Y. Hu, Q. Shao, X. Deng, S. Han, D. Song and J. Jiang, Adv. Mater. Technol., 2019, 4, 1800498 CrossRef.
- D. Baek, T. K. Lee, I. Jeon, S. H. Joo, S. Shin, J. Park, S. J. Kang, S. K. Kwak and J. Lee, Adv. Sci., 2020, 2000104, DOI:10.1002/advs.202000104.
- C. D. S. Brites, E. D. Martínez, R. R. Urbano, C. Rettori and L. D. Carlos, Front. Chem., 2019, 7, 267 CrossRef CAS.
- Y. Hu, Q. Shao, X. Deng, D. Song, S. Han, Y. Dong and J. Jiang, J. Mater. Chem. C, 2019, 7, 11770–11775 RSC.
- H. Hauser, B. Herter, C. L. M. Hofmann, O. Höhn, V. Kübler, S. Fischer, S. Wolf, S. Fasold, F. C. J. M. van Veggel, J. C. Goldschmidt and B. Bläsi, Microelectron. Eng., 2018, 187-188, 154–159 CrossRef CAS.
- W. J. Kim, M. Nyk and P. N. Prasad, Nanotechnology, 2009, 20, 185301 CrossRef.
- F. Kaboli, N. Ghazyani, M. Riahi, H. Zare-Behtash, M. H. Majles Ara and E. Heydari, ACS Appl. Nano Mater., 2019, 2, 3590–3596 CrossRef CAS.
- E. D. Martínez, R. R. Urbano and C. Rettori, ACS Appl. Nano Mater., 2019, 2, 6889–6897 CrossRef.
- J.-C. Boyer, M.-P. Manseau, J. I. Murray and F. C. J. M. van Veggel, Langmuir, 2010, 26, 1157–1164 CrossRef CAS.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- S. Guo, X. Xie, L. Huang and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 847–853 CrossRef CAS.
- D. Toptygin, J. Fluoresc., 2003, 13, 201–219 CrossRef CAS.
- T. Senden, F. T. Rabouw and A. Meijerink, ACS Nano, 2015, 9, 1801–1808 CrossRef CAS.
- C. K. Duan and M. F. Reid, Spectrosc. Lett., 2007, 40, 237–246 CrossRef CAS.
- S. Fischer, N. J. J. Johnson, J. Pichaandi, J. C. Goldschmidt and F. C. J. M. van Veggel, J. Appl. Phys., 2015, 118, 193105 CrossRef.
- M. Kraft, C. Würth, V. Muhr, T. Hirsch and U. Resch-Genger, Nano Res., 2018, 11, 6360–6374 CrossRef CAS.
- C. Würth, M. Kaiser, S. Wilhelm, B. Grauel, T. Hirsch and U. Resch-Genger, Nanoscale, 2017, 9, 4283–4294 RSC.
- D. Yuan, M. C. Tan, R. E. Riman and G. M. Chow, J. Phys. Chem. C, 2013, 117, 13297–13304 CrossRef CAS.
- M. C. Tan, G. A. Kumar, R. E. Riman, M. G. Brik, E. Brown and U. Hommerich, J. Appl. Phys., 2009, 106, 063118 CrossRef.
- R. B. Anderson, S. J. Smith, P. S. May and M. T. Berry, J. Phys. Chem. Lett., 2014, 5, 36–42 CrossRef CAS.
- R. Martín-Rodríguez, F. T. Rabouw, M. Trevisani, M. Bettinelli and A. Meijerink, Adv. Opt. Mater., 2015, 3, 558–567 CrossRef.
- H. Dong, L.-D. Sun and C.-H. Yan, Chem. Soc. Rev., 2015, 44, 1608–1634 RSC.
- P. S. May and M. Berry, Methods Appl. Fluoresc., 2019, 7, 023001 CrossRef CAS.
- M. Kaiser, C. Würth, M. Kraft, T. Soukka and U. Resch-Genger, Nano Res., 2019, 12, 1871–1879 CrossRef CAS.
- I. Hyppänen, N. Höysniemi, R. Arppe, M. Schäferling and T. Soukka, J. Phys. Chem. C, 2017, 121, 6924–6929 CrossRef.
- G. Liu, Chem. Soc. Rev., 2015, 44, 1635–1652 RSC.
- J. Zhao, Z. Lu, Y. Yin, C. McRae, J. A. Piper, J. M. Dawes, D. Jin and E. M. Goldys, Nanoscale, 2013, 5, 944–952 RSC.
- P. Villanueva-Delgado, D. Biner and K. W. Krämer, J. Lumin., 2017, 189, 84–90 CrossRef CAS.
- B. Huang, J. Bergstrand, S. Duan, Q. Zhan, J. Widengren, H. Ågren and H. Liu, ACS Nano, 2018, 12, 10572–10575 CrossRef CAS.
- K. Huang, H. Liu, M. Kraft, S. Shikha, X. Zheng, H. Ågren, C. Würth, U. Resch-Genger and Y. Zhang, Nanoscale, 2018, 10, 250–259 RSC.
- E. S. Medvedev, J. Chem. Phys., 2012, 137, 174307 CrossRef.
- K. K. Lehmann and A. M. Smith, J. Chem. Phys., 1990, 93, 6140–6147 CrossRef CAS.
- F. T. Rabouw, P. T. Prins, P. Villanueva-Delgado, M. Castelijns, R. G. Geitenbeek and A. Meijerink, ACS Nano, 2018, 12, 10576–10577 CrossRef CAS.
- Z. Wang and A. Meijerink, J. Phys. Chem. C, 2018, 122, 26298–26306 CrossRef CAS.
- J. M. F. van Dijk and M. F. H. Schuurmans, J. Chem. Phys., 1983, 78, 5317–5323 CrossRef CAS.
- J. W. Stouwdam, G. A. Hebbink, J. Huskens and F. C. J. M. van Veggel, Chem. Mater., 2003, 15, 4604–4616 CrossRef CAS.
- A. Aharoni, D. Oron, U. Banin, E. Rabani and J. Jortner, Phys. Rev. Lett., 2008, 100, 057404 CrossRef.
- E. Kreidt, C. Kruck and M. Seitz, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2018, vol. 53, pp. 35–79 Search PubMed.
- A. Monguzzi, M. I. Trioni, R. Tubino, A. Milani, L. Brambilla and C. Castiglioni, Synth. Met., 2009, 159, 2410–2412 CrossRef CAS.
- J. Scholten, G. A. Rosser, J. Wahsner, N. Alzakhem, C. Bischof, F. Stog, A. Beeby and M. Seitz, J. Am. Chem. Soc., 2012, 134, 13915–13917 CrossRef CAS.
- A. Monguzzi, A. Milani, L. Lodi, M. I. Trioni, R. Tubino and C. Castiglioni, New J. Chem., 2009, 33, 1542–1548 RSC.
- Y. Yan, A. J. Faber and H. de Waal, J. Non-Cryst. Solids, 1995, 181, 283–290 CrossRef CAS.
- N. J. J. Johnson, S. He, S. Diao, E. M. Chan, H. Dai and A. Almutairi, J. Am. Chem. Soc., 2017, 139, 3275–3282 CrossRef CAS.
- G. Tessitore, G. A. Mandl, M. G. Brik, W. Park and J. A. Capobianco, Nanoscale, 2019, 11, 12015–12029 RSC.
- R. Naccache, Q. Yu and J. A. Capobianco, Adv. Opt. Mater., 2015, 3, 482–509 CrossRef CAS.
- W. Bian, Y. Lin, T. Wang, X. Yu, J. Qiu, M. Zhou, H. Luo, S. F. Yu and X. Xu, ACS Nano, 2018, 12, 3623–3628 CrossRef CAS.
- Q. Min, J. Lei, X. Guo, T. Wang, Q. Yang, D. Zhou, X. Yu, S. F. Yu, J. Qiu, Q. Zhan and X. Xu, Adv. Funct. Mater., 2020, 30, 1906137 CrossRef CAS.
- A. L. Rogach, T. A. Klar, J. M. Lupton, A. Meijerink and J. Feldmann, J. Mater. Chem., 2009, 19, 1208–1221 RSC.
- H. Kuhn, J. Chem. Phys., 1970, 53, 101–108 CrossRef CAS.
- M. Y. Hossan, A. Hor, Q. Luu, S. J. Smith, P. S. May and M. T. Berry, J. Phys. Chem. C, 2017, 121, 16592–16606 CrossRef CAS.
- C. Würth, S. Fischer, B. Grauel, A. P. Alivisatos and U. Resch-Genger, J. Am. Chem. Soc., 2018, 140, 4922–4928 CrossRef.
- S. Fischer, N. D. Bronstein, J. K. Swabeck, E. M. Chan and A. P. Alivisatos, Nano Lett., 2016, 16, 7241–7247 CrossRef CAS.
- X. Qin, L. Shen, L. Liang, S. Han, Z. Yi and X. Liu, J. Phys. Chem. C, 2019, 123, 11151–11161 CrossRef CAS.
- Y. Chun-Lei, D. Shi-Xun, Z. Gang, Z. Jun-Jie, H. Li-Li and J. Zhong-Hong, Chin. Phys. Lett., 2005, 22, 2926 CrossRef.
- L. Ning, L. Lodi, M. I. Trioni, R. Tubino, S. Edvardsson and G. P. Brivio, J. Phys.: Condens. Matter, 2006, 19, 016202 CrossRef.
- T. Salmi, H. G. Kjaergaard and L. Halonen, J. Phys. Chem. A, 2009, 113, 9124–9132 CrossRef CAS.
- T. Salmi, V. Hänninen, A. L. Garden, H. G. Kjaergaard, J. Tennyson and L. Halonen, J. Phys. Chem. A, 2012, 116, 796–797 CrossRef CAS.
- F. Wang, J. Wang and X. Liu, Angew. Chem., Int. Ed., 2010, 49, 7456–7460 CrossRef CAS.
- Y. Zhong, G. Tian, Z. Gu, Y. Yang, L. Gu, Y. Zhao, Y. Ma and J. Yao, Adv. Mater., 2014, 26, 2831–2837 CrossRef CAS.
- S. Fischer, R. D. Mehlenbacher, A. Lay, C. Siefe, A. P. Alivisatos and J. A. Dionne, Nano Lett., 2019, 19, 3878–3885 CrossRef CAS.
- A. Skripka, A. Benayas, C. D. S. Brites, I. R. Martín, L. D. Carlos and F. Vetrone, Nano Lett., 2020, 20, 7648–7654 CrossRef CAS.
- M. Kaiser, C. Würth, M. Kraft, I. Hyppänen, T. Soukka and U. Resch-Genger, Nanoscale, 2017, 9, 10051–10058 RSC.
- M. Mousavi, B. Thomasson, M. Li, M. Kraft, C. Würth, U. Resch-Genger and S. Andersson-Engels, Phys. Chem. Chem. Phys., 2017, 19, 22016–22022 RSC.
- T. Rinkel, J. Nordmann, A. N. Raj and M. Haase, Nanoscale, 2014, 6, 14523–14530 RSC.
- S. Dühnen, T. Rinkel and M. Haase, Chem. Mater., 2015, 27, 4033–4039 CrossRef.
- P. B. May, J. D. Suter, P. S. May and M. T. Berry, J. Phys. Chem. C, 2016, 120, 9482–9489 CrossRef CAS.
- H.-X. Mai, Y.-W. Zhang, L.-D. Sun and C.-H. Yan, J. Phys. Chem. C, 2007, 111, 13730–13739 CrossRef CAS.
- N. J. J. Johnson, A. Korinek, C. Dong and F. C. J. M. van Veggel, J. Am. Chem. Soc., 2012, 134, 11068–11071 CrossRef CAS.
- B. Voss and M. Haase, ACS Nano, 2013, 7, 11242–11254 CrossRef CAS.
- B. Amouroux, C. Roux, J.-D. Marty, M. Pasturel, A. Bouchet, M. Sliwa, O. Leroux, F. Gauffre and C. Coudret, Inorg. Chem., 2019, 58, 5082–5088 CrossRef CAS.
- S. Dühnen and M. Haase, Chem. Mater., 2015, 27, 8375–8386 CrossRef.
- L. Liu, X. Li, Y. Fan, C. Wang, A. M. El-Toni, M. S. Alhoshan, D. Zhao and F. Zhang, Chem. Mater., 2019, 31, 5608–5615 CrossRef CAS.
- D. Hudry, I. A. Howard, R. Popescu, D. Gerthsen and B. S. Richards, Adv. Mater., 2019, 31, 1900623 CrossRef.
- C. Dong, J. Pichaandi, T. Regier and F. C. J. M. van Veggel, J. Phys. Chem. C, 2011, 115, 15950–15958 CrossRef CAS.
- D. Hudry, R. Popescu, D. Busko, M. Diaz-Lopez, M. Abeykoon, P. Bordet, D. Gerthsen, I. A. Howard and B. S. Richards, J. Mater. Chem. C, 2019, 7, 1164–1172 RSC.
- J. F. Suyver, R. Meester, J. J. Kelly and A. Meijerink, J. Lumin., 2003, 102–103, 182–188 CrossRef CAS.
- T. C. Droubay, T. C. Kaspar, B. P. Kaspar and S. A. Chambers, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 075324 CrossRef.
- A. Podhorodecki, B. Krajnik, L. W. Golacki, U. Kostiv, G. Pawlik, M. Kaczmarek and D. Horák, Nanoscale, 2018, 10, 21186–21196 RSC.
- S. Chen, A. Z. Weitemier, X. Zeng, L. He, X. Wang, Y. Tao, A. J. Y. Huang, Y. Hashimotodani, M. Kano, H. Iwasaki, L. K. Parajuli, S. Okabe, D. B. L. Teh, A. H. All, I. Tsutsui-Kimura, K. F. Tanaka, X. Liu and T. J. McHugh, Science, 2018, 359, 679 CrossRef CAS.
- Y. Ma, J. Bao, Y. Zhang, Z. Li, X. Zhou, C. Wan, L. Huang, Y. Zhao, G. Han and T. Xue, Cell, 2019, 177, 243–255.e215 CrossRef CAS.
- Y. Fan, P. Wang, Y. Lu, R. Wang, L. Zhou, X. Zheng, X. Li, J. A. Piper and F. Zhang, Nat. Nanotechnol., 2018, 13, 941–946 CrossRef CAS.
- R. Piñol, J. Zeler, C. D. S. Brites, Y. Gu, P. Téllez, A. N. Carneiro Neto, T. E. da Silva, R. Moreno-Loshuertos, P. Fernandez-Silva, A. I. Gallego, L. Martinez-Lostao, A. Martínez, L. D. Carlos and A. Millán, Nano Lett., 2020, 20, 6466–6472 CrossRef.
| This journal is © the Owner Societies 2021 |