
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocyclic complexes based on [N⋯I⋯N]+ halogen bonds†
Shilin
Yu
 a,
Elina
Kalenius
a,
Antonio
Frontera
b and
Kari
Rissanen
*a
a,
Elina
Kalenius
a,
Antonio
Frontera
b and
Kari
Rissanen
*a
aUniversity of Jyvaskyla, Department of Chemistry, 40014, Jyväskylä, Finland. E-mail: kari.t.rissanen@jyu.fi
bDepartment of Chemistry, Universitat de les Illes Balears, Crts de Valldemossa km 7.6, 07122, Palma de Mallorca Baleares, Spain
First published on 26th October 2021
Abstract
New 1–2 nm macrocyclic iodine(I) complexes prepared VIA a simple ligand exchange reaction manifest rigid 0.5–1 nm cavities that bind the hexafluorophosphate anion in the gas phase. The size of the cavities and the electrostatic interactions with the iodine(I) cations influence the anion binding properties of these macrocyclic complexes.
The iodine(I) cation (aka iodenium ion) can be considered as an ‘‘ultra-polarized’’ iodine atom that can act as a very strong bis-halogen-bond donor by forming a three-center-four-electron (3c–4e) halogen bond with two Lewis bases (L) resulting in [L–I–L]A (A = anion) complexes1 with the specific bonding character of the iodine(I) cation.2 The 3c–4e halogen bond, viz. [N⋯I⋯N]+, is a robust silver(I)-like building block with enhanced linearity of the N⋯I⋯N bond, and thus has great potential as a construction unit for many intricate supramolecular structures. The iconic example of an iodine(I) complex is Barluenga's reagent,3 bis(pyridine)iodine(I) tetrafluoroborate [(py)2I]BF4, known in synthetic chemistry as an iodinating agent for aromatic electrophilic substitutions and aromatic amines, yet can also be used as an oxidizing agent.4–9 However, probably due to the reactivity as an iodinating reagent, the incorporation of iodine(I) as a component in larger supramolecular systems like molecular capsules or cages,10–13 rotaxanes,14 monomolecular helicates,15 and XOFs,16 has only recently been described. In addition to the “simple” symmetric1 and asymmetric17 [L–I–L]A complexes several monomeric clamp-type iodine(I) complexes have been intensely studied by Erdélyi.18–22 In one case an I+-clamp complex has been suggested to form a dimer, yet it was found to be unstable and experimentally detectable only at low temperatures (−40 °C).23
Inspired by the known three ligands (L1–L3) and their dimeric silver(I) metallacycles,24–26 [(AgL)2]2+, and the previously successfully applied1,10–13,15–17 [N⋯Ag⋯N]+ → [N⋯I⋯N]+ cation exchange reaction, we envisaged that construction of the corresponding halogen-bonded macrocyclic bis-iodine(I) complexes [(IL)2](PF6)2 (1–3, Scheme 1) could be achieved. The initial tries with the silver(I) metallacycles and elemental iodine did not proceed cleanly due to the possibility of the metallocycle to open during the exchange reaction and induction of subsequent side reactions, thus an alternative route, viz. a ligand exchange reaction previous applied by Dutton27 for simpler iodine(I) complexes, was successfully used. Using a stable iodine(I) complex bis(1-methyl-1H-1,2,3-triazole) iodine(I) hexafluoro-phosphate ([I(mtz)2]PF6)28 (see ESI†), and ligands L1–L3, the halogen-bonded iodine(I) macrocyclic complexes 1–3 were obtained with 79–87% yield.
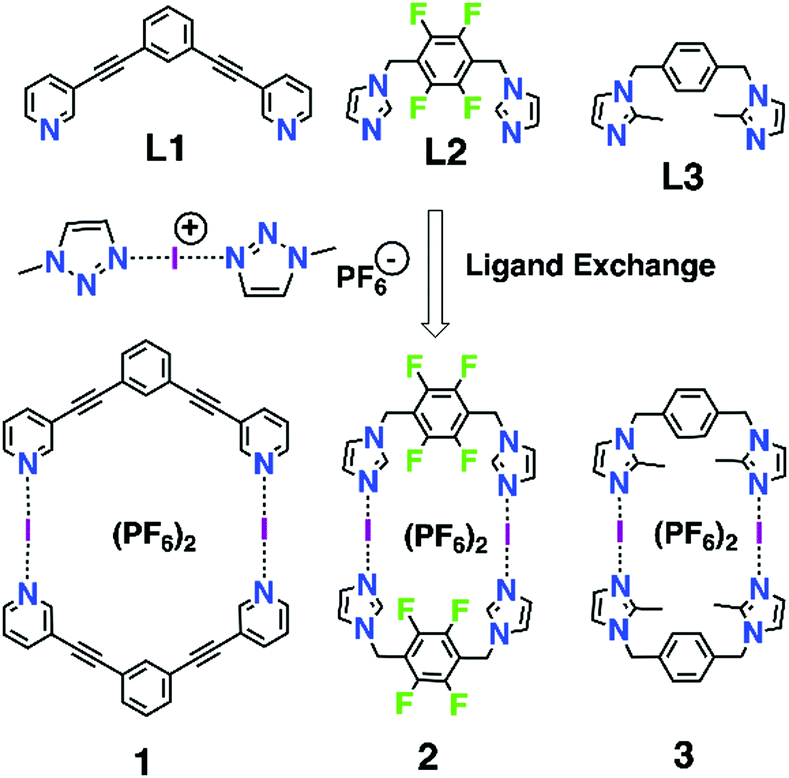 | ||
| Scheme 1 Ligands L1–L3 used and the target macrocycles 1–3. | ||
The 1H NMR analyses of 1 and 2 (Fig. 1) suggested, as expected, the formation of one highly symmetrical complex. The complexes (1 or 2) are kinetically stable and symmetric; the integrals for each of the protons on the ligand remain unchanged, and each peak remains sharp, indicating the formation of only one complex. The downfield shift of the signals (HA, HD and HC) and the upfield shift of signal HB for complex 1 as compared to those for L1 (Δδ = 0.05, 0.25, 0.13 and −0.06 ppm, respectively), and the downfield shift of the signals for complex 2 as compared to those for L2 (Δδ = 0.30, 0.27, 0.28 and 0.12 ppm, respectively), are characteristic of the iodine–ligand complexation.17–19,29 The methyl groups in relation with the macrocyclic core result in three district conformations (up-down-up-down; up-up-down-down and all-up) for the methylimidazole complex 3. The 1H NMR spectrum of 3 suggested there are two distinct complexes (Fig. S20, ESI†). The X-ray structures26,30 of the silver(I) metallacycle from ligand 3 shows only the up-up-down-down conformation. Computational studies (see below and ESI†) of the three conformations indicate that the all-up is least stable, thus the 1H NMR of 3 is interpreted to contain a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 ratio of up-up-down-down (3a) and up-down-up-down (3b) conformations (see ESI†).
:40 ratio of up-up-down-down (3a) and up-down-up-down (3b) conformations (see ESI†).
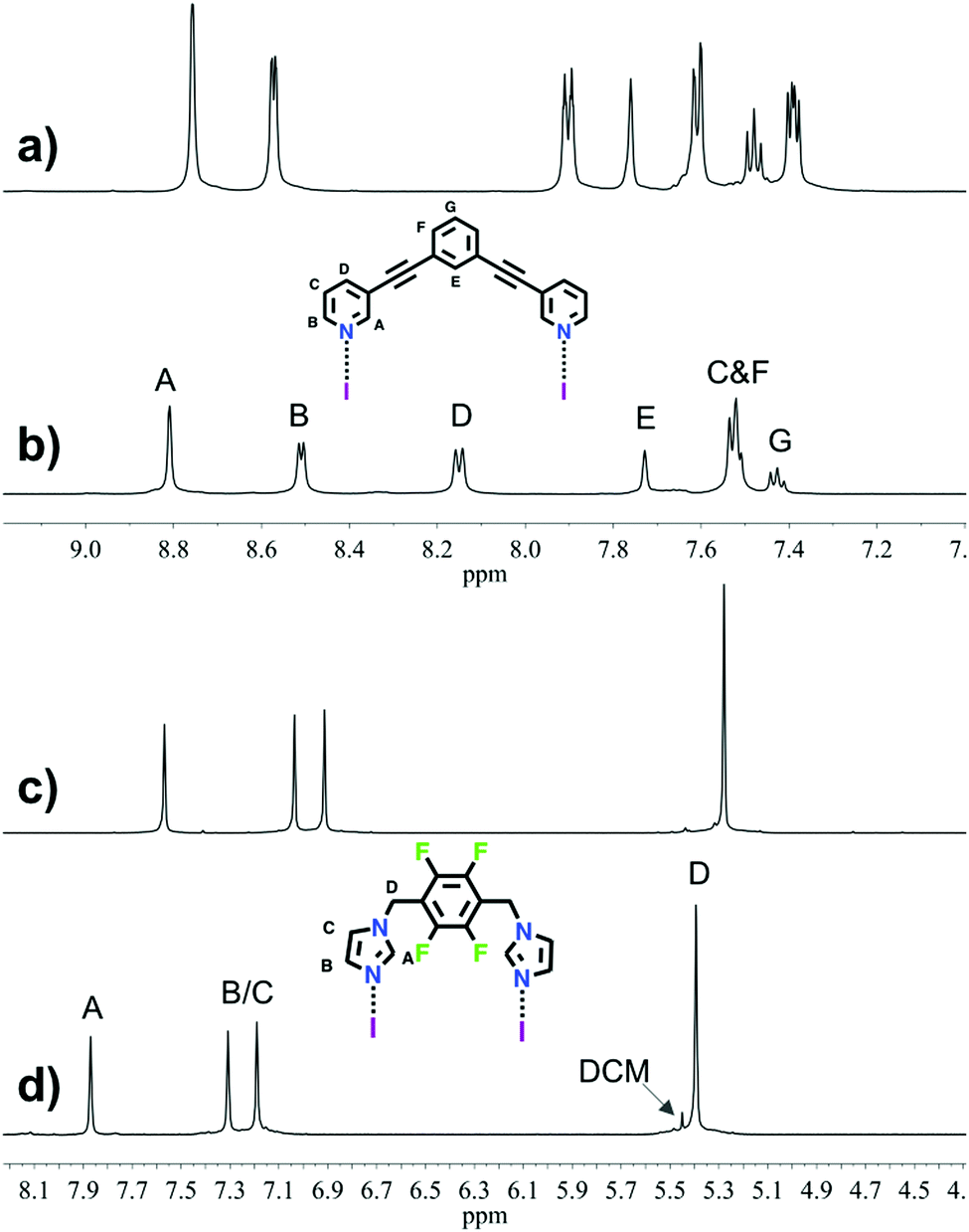 | ||
| Fig. 1 Selected region of 1H NMR spectra (500 MHz, CD3CN, 298 K) for ligand L1 (a), macrocycle 1 (b), the ligand L2 (c) and macrocycle 2 (d). | ||
The formation of stable iodine(I) complexes with N⋯I⋯N 3c–4e halogen bonds was also confirmed by the N-atom coordination shifts (Δδ15Ncoord) of −90 to −110 ppm (Table 1) in the 1H–15N HMBC NMR spectra. The two nitrogen atoms are directly involved in the N⋯I⋯N 3c–4e bond, which, in combination, results in large chemical shift changes.31 The values are comparable to the coordination shifts observed in similar iodine(I) complexes (Fig. S25, ESI†).12,17,18
| L1/1 | L2/2 | L3/3a/3b | |
|---|---|---|---|
| δ15 | −62.7/−173.3 | −117.3/216.1 | −120.1/−210.1/212.1 |
| Δδ15Ncoord | −110.6 | −98.8 | −90.5(3a)/−92.0(3b) |
In order to probe the size of the macrocycles in solution 1H DOSY NMR spectra was measured in CD3CN. The diffusion constants for complexes 1–3 are ca. two times smaller, evidencing discrete macrocycles two times larger in size than the ligands L1–L3 (Table S1, ESI†), confirming that these two complexes are dimeric, not monomeric or polymeric.
Despite rigorous attempts X-ray quality crystals of 1–3 were not obtained. Large basis set DFT calculations have been applied for the macrocycles 1–3 (Fig. 3 and Fig. S32, ESI†). The geometries of the optimized complexes have been compared with the X-ray structures of the corresponding silver(I) metallacycles retrieved from the CSD database. In general, the optimized geometries of the macrocyclic XB complexes (1–3) are very similar to the X-ray geometries of the corresponding silver(I) metallacycles.
The main structural difference is that the N⋯I⋯N angles in 1–3 are more linear than the N⋯Ag⋯N analogues. Moreover, the intra-complex I+···I+ distances are shorter than the Ag+···Ag+ distances in complexes 1 and 2 and longer in complex 3 (see Fig. 2), their variations are very likely caused by the different packing and anion–cation interactions in the solid-state of the silver(I) metallacycles.
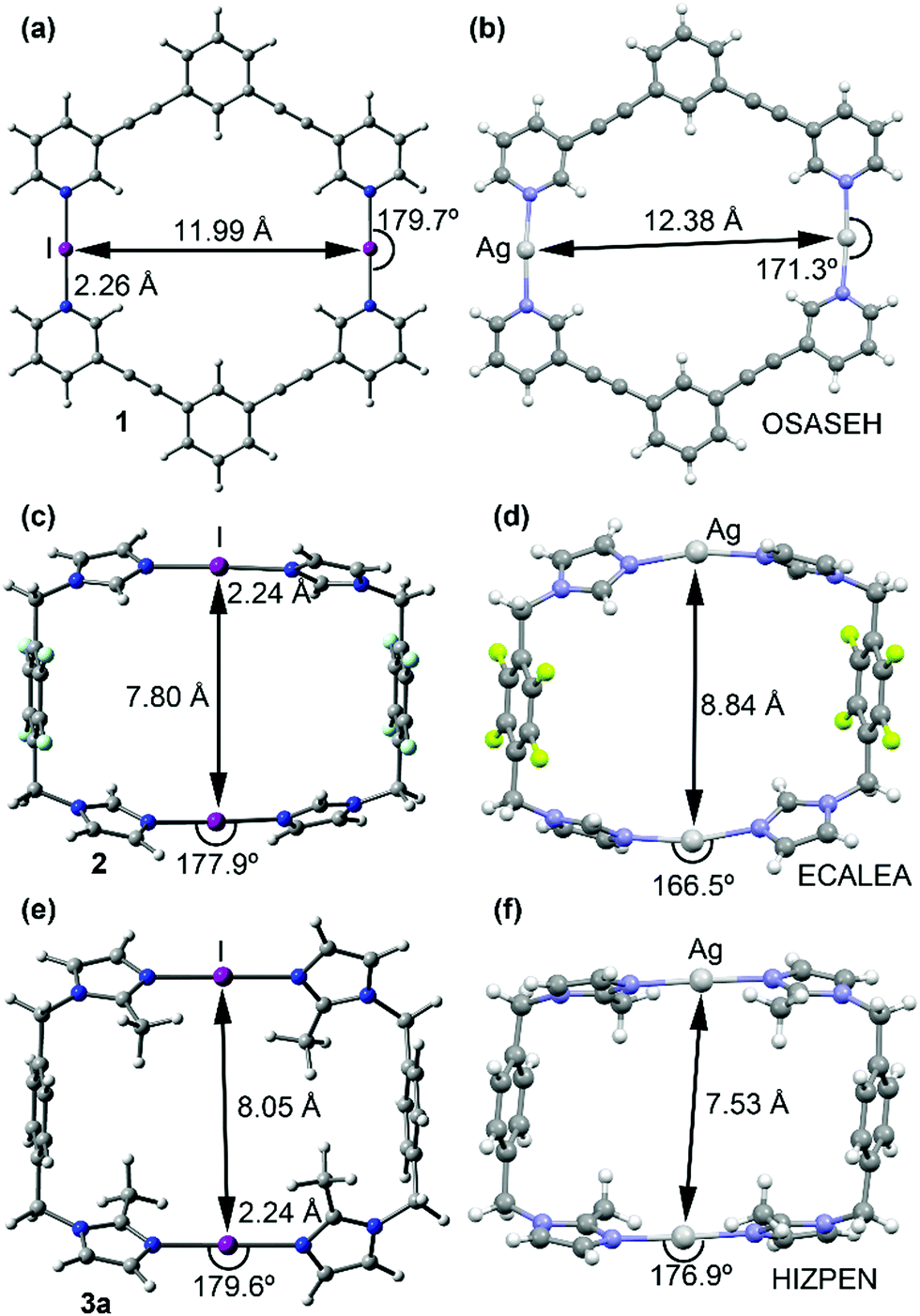 | ||
| Fig. 2 Optimized geometries of compounds 1 (a), 2 (b) and 3a (c) at the M06-2X/def2TZVp level of theory. The X-ray structures of the corresponding Ag(I) metallacycles (CSD refcodes OSASEH (b) and ECALEA (d) and HIZPEN (f)). | ||
The stoichiometries and structures of the complexes were further verified using electrospray ionization ion mobility mass spectrometry (ESI-IMMS). In gas phase, the complexes were observed to complex the PF6− anion into the cavity of the macrocycle and ions [1-PF6]+, [2-PF6]+ and [3-PF6]+ appeared in spectra with high abundance (ESI,† Fig. S28–S30 and Fig. 3.) The MS/MS analysis also confirmed the composition as [2L + 2I + PF6]+ and showed structure-related dissociation of macrocycles through consecutive eliminations of IPF6 or HPF6 and ligands L1, L2 or L3.
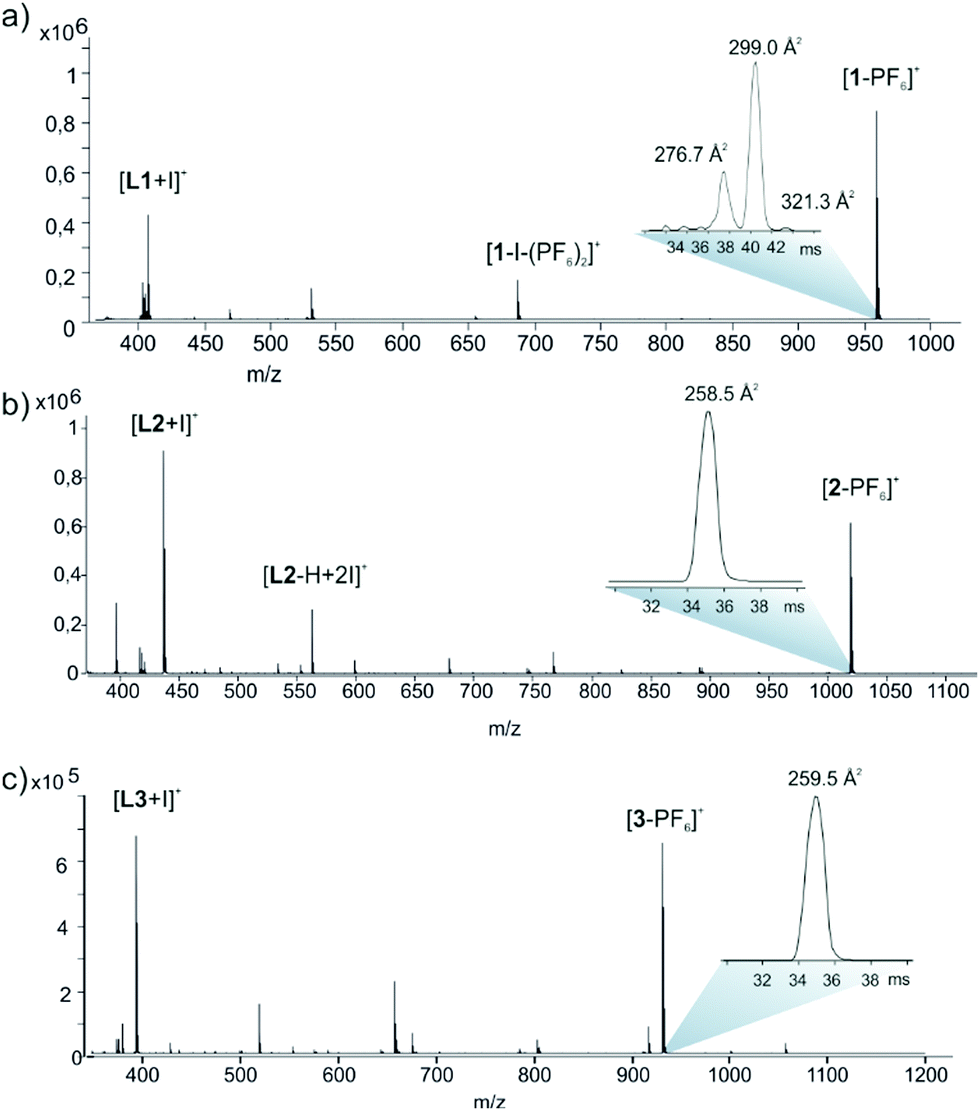 | ||
| Fig. 3 (+)ESI-QTOF mass spectrum (a) 1, (b) 2 and (c) 3 in MeCN. Insets show IM-TOF arrival time distributions for ions [C-PF6]+. | ||
The correlation between theoretical and experimental structures was further analyzed by ion-mobility mass spectrometry experiments.32 To visualize the IM-MS data, [1,2,3-PF6]+ complexes were DFT-optimized (Fig. 4). The calculations show that the cavity in 2 or 3 just suitable for the PF6− anion which fits very well inside the cavity, as evidenced by the optimized geometries of the host–guest complexes. An interesting combination of non-covalent interactions is formed. The complex [2-PF6]+ manifests two anion–π interactions, due to the presence of two electron-deficient tetrafluorophenyl rings in the macrocycle (marked as red dashed lines in Fig. 4a). In fact, two fluorine atoms of the anion point to the center of the electron-deficient rings (distance 3.16 Å). Additional Csp2–H⋯F HBs (2.37 Å) and electrostatic forces I+⋯PF6− firmly hold the guest inside the cavity. The orientation of the anionic guest is different in [3-PF6]+ because the aromatic rings of the macrocycle are electron-rich. In this case the four fluorine atoms of the anion point towards the methyl groups, establishing multiple Csp3–H⋯F contacts (distances 2.48–2.54 Å). The [3-PF6]+ host–guest complex is further stabilized by I+⋯PF6− ion pair interactions. The IM-MS arrival time distributions (ATDs) showed single drift peaks for the host–guest complexes [2-PF6]+ and [3-PF6]+. The experimentally observed collision cross section values using He as a drift gas (DTCCSHe) for these ions were 184.4 Å2 and 182.3 Å2, respectively. The values show nice correlation (<1.9% difference) with theoretical CCS values (TMLJCCSHe) calculated on basis of DFT-optimized structures (Table S3, ESI†). This comparison between theoretical and experimental results verifies macrocyclic structure, analogues to Ag+ complexes, also in the gas phase. This further suggests that the PF6 anion act as a guest and is located inside the macrocycle. The two conformers observed in 1H NMR spectra for 3 could not be resolved by IM-MS, because the theoretical TMLJCCSHe values for 3a and 3b differ only by 0.98 Å2 (typically ∼1% difference is expected to be resolved with ultra-high resolving power instruments).32 The IM drift peak for [3-PF6]+ has, however, clearly decreased resolution (RFWHM for [3-PF6]+ is only 29.7 whereas for [2-PF6]+ RFWHM = 46.3), which often indicates several closely related, unresolved conformers.
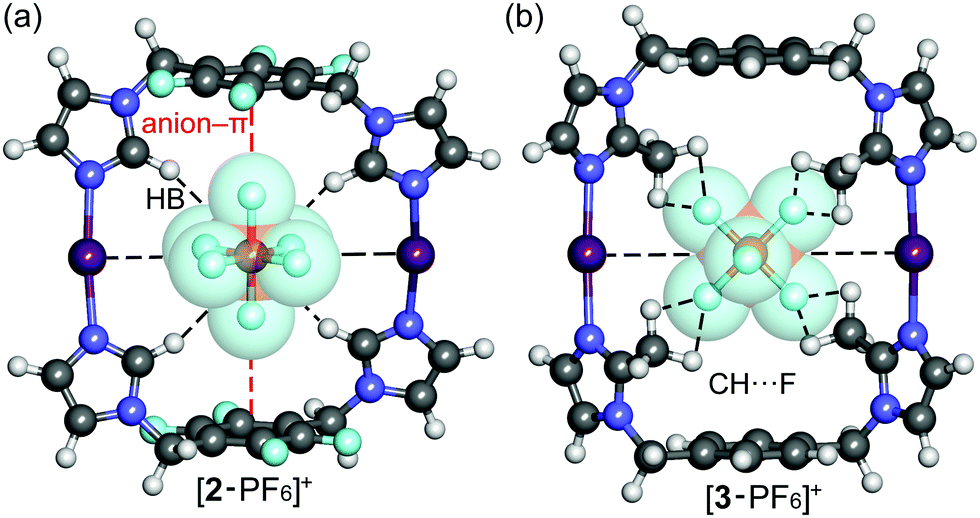 | ||
| Fig. 4 Optimized structures of host–guest complexes of macrocycles 2 (a) and 3 (b) with the PF6− anion at the M06-2X/def2-TZVP level of theory. | ||
The ATD for much larger [1-PF6]+ surprisingly shows distribution of IM drift peaks ranging from 196.6 to 237.8 Å2. The DFT optimization confirm, that the cavity in 1 is much larger than the size of PF6 anion, allowing different locations for a guest within the cavity (see Fig. S32a (ESI†) for one possible host–guest complex). To study this, the structures of endo and exo complexes for [1-PF6]+ were calculated, resulting in TMLJCCSHe values of 228.8 and 231.8 Å2, respectively. Whereas in [2-PF6]+ and [3-PF6]+ the anion has a tight fit, the complexation of PF6− into the larger cavity of [1-PF6]+ results in less specific interactions with the cavity walls and offers larger degrees of freedom for the spatial location of the guest. We conclude that the multiple IM peaks originate from the different locations of loosely bound PF6− counter anion. Also according to the MS measurements the [1-PF6]+ complex is less stable (has lower interaction energy), which is reflected in MS/MS spectra (collision induced dissociation, CID) showing more dissociation and lower relative intensity for [1-PF6]+ precursor ion when compared with the [2-PF6]+ and [3-PF6]+ precursor ions.
In summary, we have demonstrated a simple preparation of three new iodine(I)-based macrocycles via ligand exchange reaction, and studied their structures and host–guest behavior using 1H and 1H–15N HMBC and DOSY NMR, IM-MS and theoretical calculations. Further studies of these systems are underway in our laboratory. The simple preparation route to the previously unreported iodine(I) macrocycles might provide a way to construct iodine(I)-based mechanically interlocked molecules.
We gratefully acknowledge financial support from the Academy of Finland (K. R. grant no. 317259), the MICIU/AEI of Spain (A. F. project CTQ2017-85821-R FEDER), and the University of Jyvaskyla, Finland.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC.
- L. Turunen and M. Erdélyi, in Halogen Bonding in Solution, ed. S. Huber, Wiley-VCH Verlag GmbH, 2021, pp. 121–151 Search PubMed.
- J. Barluenga, F. González-Bobes, M. C. Murguía, S. R. Ananthoju and J. M. González, Chem. – Eur. J., 2004, 10, 4206–4213 CrossRef CAS PubMed.
- J. A. Creighton, I. Haque and J. L. Wood, Chem. Commun., 1966, 229 RSC.
- J. A. Creighton, I. Haque and J. L. Wood, Chem. Commun., 1966, 892 RSC.
- I. Haque and J. L. Wood, J. Mol. Struct., 1968, 2, 217–238 CrossRef CAS.
- J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC.
- J. Ezquerra, C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, M. A. García-Martín and J. M. González, J. Org. Chem., 1996, 61, 5804–5812 CrossRef CAS.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 1307–1308 RSC.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861–869 CAS.
- U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Chem. Sci., 2018, 9, 8343–8351 RSC.
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714–11718 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- M. Kandrnálová, Z. Kokan, V. Havel, M. Nečas and V. Šindelář, Angew. Chem., Int. Ed., 2019, 58, 18182–18185 CrossRef PubMed.
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdélyi, Angew. Chem., Int. Ed., 2019, 58, 9012–9016 CrossRef CAS PubMed.
- G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831–14835 CrossRef CAS PubMed.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428–8431 RSC.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdélyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- A. A. Neverov, H. X. Feng, K. Hamilton and R. S. Brown, J. Org. Chem., 2003, 68, 3802–3810 CrossRef CAS PubMed.
- L. Turunen, F. B. Németh, D. A. Decato, I. Pápai, O. B. Berryman and M. Erdélyi, Bull. Chem. Soc. Jpn., 2020, 94, 191–196 CrossRef.
- S. Lindblad, F. Boróka Németh, T. Földes, D. von der Heiden, H. G. Vang, Z. L. Driscoll, E. R. Gonnering, I. Pápai, N. Bowling and M. Erdélyi, Chem. – Eur. J., 2021, 27, 13748–13756 CrossRef CAS PubMed.
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Gräfenstein and M. Erdélyi, J. Am. Chem. Soc., 2018, 140, 13503–13513 CrossRef CAS PubMed.
- K. J. Kilpin, M. L. Gower, S. G. Telfer, G. B. Jameson and J. D. Crowley, Inorg. Chem., 2011, 50, 1123–1134 CrossRef CAS PubMed.
- Y. Gao, B. Twamley and J. M. Shreeve, Inorg. Chem., 2006, 45, 1150–1155 CrossRef CAS PubMed.
- L. Dobrzańska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2005, 127, 13134–13135 CrossRef PubMed.
- D. C. Georgiou, P. Butler, E. C. Browne, D. J. D. Wilson and J. L. Dutton, Aust. J. Chem., 2013, 66, 1179–1188 CrossRef CAS.
- S. Yu, P. Kumar, J. S. Ward, A. Frontera and K. Rissanen, Chem, 2021, 7, 948–958 CAS.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topić, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdélyi, Chem. Sci., 2015, 6, 3746–3756 RSC.
- M. du Plessis, V. J. Smith and L. J. Barbour, CrystEngComm, 2014, 16, 4126–4132 RSC.
- S. B. Hakkert, J. Gräfenstein and M. Erdélyi, Faraday Discuss., 2017, 203, 333–346 RSC.
- A. T. Kirk, A. Bohnhorst, C.-R. Raddatz, M. Allers and S. Zimmermann, Anal. Bioanal. Chem., 2019, 411, 6229–6246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compounds characterization, DFT calculations. See DOI: 10.1039/d1cc05616f |
| This journal is © The Royal Society of Chemistry 2021 |