
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of organic and inorganic gunshot residues from hands using complexing agents and LC-MS/MS†
William
Feeney
 *a,
Korina
Menking-Hoggatt
b,
Courtney
Vander Pyl
b,
Colby E.
Ott
b,
Suzanne
Bell
b,
Luis
Arroyo
b and
Tatiana
Trejos
*b
*a,
Korina
Menking-Hoggatt
b,
Courtney
Vander Pyl
b,
Colby E.
Ott
b,
Suzanne
Bell
b,
Luis
Arroyo
b and
Tatiana
Trejos
*b
aC. Eugene Bennett Department of Chemistry, West Virginia University, 1600 University Ave, Morgantown, WV, USA
bDepartment of Forensic and Investigative Sciences, West Virginia University, USA. E-mail: tatiana.trejos@mail.wvu.edu
First published on 16th June 2021
Abstract
Gunshot residue (GSR) refers to a conglomerate consisting of both organic molecules (OGSR) and inorganic species (IGSR). Historically, forensic examiners have focused only on identifying the IGSR particles by their morphology and elemental composition. Nonetheless, modern ammunition formulations and challenges with the GSR transference (such as secondary and tertiary transfer) have driven research efforts for more comprehensive examinations, requiring alternative analytical techniques. This study proposes the use of LC-MS/MS for chromatographic separation and dual detection of inorganic and organic residues. The detection of both target species in the same sample increases the confidence that chemical profiles came from a gun's discharge instead of non-firearm-related sources. This strategy implements supramolecular molecules that complex with the IGSR species, allowing them to elute from the column towards the mass spectrometer while retaining isotopic ratios for quick and unambiguous identification. The macrocycle (18-crown-6-ether) complexes with lead and barium, while antimony complexes with a chelating agent (tartaric acid). The total analysis time for OGSR and IGSR in one sample is under 20 minutes. This manuscript expands from a previous proof-of-concept publication by improving figures of merit, increasing the target analytes, testing the method's feasibility through a more extensive set of authentic specimens collected from the hands of both shooters and non-shooters, and comparing performance with other analytical techniques such as ICP-MS, electrochemical methods and LIBS. The linear dynamic ranges (LDR) spread across the low ppb range for OGSR (0.3–200 ppb) and low ppm range (0.1–6.0 ppm) for IGSR. The method's accuracy increased overall when both organic and inorganic profiles were combined.
1. Introduction
1.1 Trace analysis disciplines – gunshot residue
Gunshot residue (GSR) comprises two main components, organic (OGSR) and inorganic (IGSR), which arise from different locations within the ammunition. The OGSR compounds originate from the propellant and lubricant, whereas IGSR particulates emanate from the primer, bullet, and cartridge casing. After a deflagration event, those analytes can be dispersed and spread onto various surrounding surfaces, including hair, clothing, and hands. Due to the constituents' nature and various environmental factors, proficient collection and storage of the samples are essential to preserve the GSR compounds and increase the likelihood of detection. Typical indicators for IGSR are Pb, Ba, and Sb which are formed from the initial products lead styphnate (C6HN3O8Pb), barium nitrate (Ba(NO3)2), and antimony trisulfide (Sb2S3). Some of the more common OGSR analytes are diphenylamine (DPA), nitroglycerin (NG), ethyl centralite (EC), and 2,4-dinitrotoluene (2,4-DNT).2,3 Other compounds monitored, primarily formed by the combustion event and degradation of DPA, include 2-dinitrodiphenylamine (2-NDPA), 4-nitrodiphenylamine (4-NDPA), and N-nitrosodiphenylamine (N-NDPA). These compounds' functional roles vary from detonation or blasting agents (explosives, oxidizers, fuel) to binding and performance materials (stabilizers and plasticizers).2,4
Recently, manufacturers have introduced variants of ammunition labeled as “green”, non-toxic, heavy-metal-free, or lead-free. These products incorporate different starting materials to achieve similar results to traditional ammunition while reducing exposure of the shooter and environment to harmful heavy metals. Although this type of ammunition is not widely observed in casework yet, its emergence has required researchers to characterize and adapt interpretation criteria for non-toxic primers.5–8
Even though SEM/EDS is efficient in characterizing micron-sized inorganic particles, the method is not compatible with further sequential examination for OGSR, as factors such as high vacuum conditions, operating parameters, and compound volatility can cause substantial analyte loss. Hence if SEM/EDS analysis is to be used in conjunction with another analysis technique, the OGSR constituents must be collected first. Another disadvantage is that the analysis time for SEM/EDS typically takes 2–8 hours per sample, depending on the sample's nature and instrumental configurations. Additionally, hand residues associated with occupations such as electricians, welders, and mechanics can lead to false-positives and higher error rates.10 A recent study has proposed a solution to OGSR loss by first analyzing the OGSR from the stub using mild solvent extraction with UHPLC-ESI-MS/MS followed by IGSR examination from the same SEM collection stub.11 These authors demonstrated that by using gentle mechanistic motions from a pipette, the IGSR particulates are not significantly disturbed.
Current advancements in instrumentation techniques have shown great promise for evaluating GSR evidence. Instruments such as LIBS, LA-ICP-MS, and TOF-SIMS offer non- or minimally destructive analysis, small particle detection, and the capacity to produce high-quality images with multielement or isotopic composition information.12–14 High-resolution instruments such as ICP-MS can provide additional isotopic and elemental information in the low part-per-billion (ppb) range.15,16
LIBS, electrochemistry, and Raman spectroscopy facilitate sample screenings for quick characterizations with minimal sample preparation, and the sample remains almost unaltered. Additionally, the GSR stub used in these methods is compatible with the confirmatory (SEM/EDS) analysis, allowing further sample manipulation.
Extraction for OGSR constituents use common solvents (methanol and acetonitrile), whereas IGSR elemental identification requires a more rigorous heavy metal digestion (nitric acid and hydrochloric acid). Therefore, methods like capillary electrophoresis and LC-MS/MS are classified as destructive methods.35,36 However, if these methods were incorporated in a comprehensive workflow, the morphology analysis required can be conducted after organic extractions and instrumental analysis (via capillary electrophoresis, DESI, or LC-MS/MS) and before the losing OGSR due to vacuum conditions via SEM/EDS.11,37–40
Due to the structure of IGSR particles, further sample manipulations are required to identify and quantify samples properly via LC-MS/MS. Ideally, these chemical analyses should provide unique spectral signatures without altering the core information of the analyte of interest. This consideration led to the investigation of complexation chemistry. Host–guest chemistry applies larger organic molecules to self-assemble and form complexes called metal–ligand (M–L) complexes. Macrocycles like 18-crown-6-ether (18C6) encapsulate metal ions through noncovalent, electrostatic interactions.35,41,42 Several benefits from this interaction include transportation through both the column and mass analyzer, an extensive range of metal analytes, and retaining of natural isotopic abundance patterns. Other molecules like EDTA and tartaric acid employ a different strategy for cation binding, known as chelation. Chelation involves the formation of physical coordinate bonds between a ligand and a single central atom. Although this can provide similar benefits as host–guest chemistry, more extensive factors like pH must be considered.
The goal of this work addresses key points which include: (1) validate our previous proof-of-concept study by enhancing figures of merit such as detection limits (LOD), quantitation limits (LOQ), bias, and expanding the evaluation of performance rates with authentic specimens,35 (2) investigate interactions and expand upon antimony detection, and (3) establish an identification criterion for a “positive” GSR sample based on baseline authentic samples. This work also evaluates two substrates, including tesa® Tack and the traditional carbon adhesive tape. Furthermore, we investigate various sample types, including samples collected from shooters, non-shooter skin backgrounds, and post-shooting activity.
2. Materials and methods
2.1. Consumables
Optima® LC/MS grade methanol (MeOH), acetonitrile (ACN), and water (H2O), all containing 0.1% formic acid (FA), were obtained from Thermo-Fisher Scientific (Watham, MA) and used either as extraction solvents or mobile phases. Standard (neat) organic constituents used in this study included: akardite II (AK2), ethyl centralite (EC), methyl centralite (MC), diphenylamine (DPA), N-nitrosodiphenylamine (N-NDPA), 2-nitrodiphenylamine (2-NDPA), and 4-nitrodiphenylamine (4-NDPA), all obtained from Sigma-Aldrich and Accustandard® (New Haven, CT). Calibration curves were generated from working stock solutions and diluted in MeOH (≥95%). Complexing agents 18-crown-6-ether (18C6) and tartaric acid (TT) were purchased from Sigma-Aldrich (St. Louis, MO 99% purity) and diluted using MeOH and water, respectively. Metal IGSR standards used to form complexes utilized ICP-MS metal standards (VHG Laboratories, Manchester, NH) and methanol. Micro-bulk digestions utilized nitric (HNO3) and hydrochloric (HCl) acids (ultrapure grade, Thermo-Fisher Scientific, Waltham, MA). Two internal standards were used throughout the study including deuterated diphenylamine-d10 (D10-DPA) which was purchased from CDN Isotopes (Quebec, Canada) and used as the internal standard for the organic molecules and thallium (Tl) for the IGSR (VHG Laboratories, Manchester, NH) and complexing agents.2.2. Firearms, ammunition, and protocols
Hands from the hands of volunteers, both from shooters and non-shooter background individuals were collected under our institutional IRB protocol #1506706336 using carbon adhesive. Aluminum SEM stubs were prepared with two layers of carbon adhesive tape and covered with plastic stub storage containers (Ted Pella, Inc.).In this study, three different firearms were used for the collection of authentic shooter samples: 9 mm Springfield XD9 semi-automatic pistol, 40 caliber Springfield XD40 semi-automatic pistol, and a Taurus 357 Magnum revolver. The ammunition was either loaded by the manufacturer (Remington®, Blazer®, Federal®) or in-house (Winchester®, CCI®). Each specialty ammunition consisted of brass Starline™ cartridge cases loaded with either Winchester® or CCI® small pistol primers, Winchester® 231 propellant, and Speer® 9 mm total metal jacketed bullets. All firing events were performed at the indoor WVU ballistics laboratory and the respective analysis conducted at the Oglebay Hall research building.
Before each session, the working areas were cleaned and covered with butcher paper, and samples manipulated with disposable nitrile gloves. To minimize cross-contamination, our research team established a workflow within five separate laboratory areas. One room is dedicated to preparing the sampling stubs before collection, which is located in the building within our trace laboratory. After collection, the samples are stored in a refrigerator until extraction, which is conducted in a separate laboratory room equipped with dedicated hoods and benchtops. Finally, instrumental analysis is performed on the third floor for the LC-MS/MS, ICP-MS, LIBS, on the second floor for EC, and on the ground level for the SEM-EDS examinations.
Moreover, the shooting range is located in another building a few miles apart from the research laboratory, and firearms and ammunition are stored in dedicated safety rooms away from research spaces. Within the ballistics lab, firearm discharge occurs inside the shooting range, while collection from the individual's hands is done on the annexed laboratory. Also, working areas are cleaned daily before any sample preparation or analysis. The team members have specific scheduled roles to avoid that an individual who has recently fired or manipulated a firearm enters the laboratory areas. The collection team members and shooters dressed in Tyvek® suits for additional protection from carryover.
Our standard operating procedure includes several reagent blanks and negative control samples to monitor any potential contamination. Reagent blanks are analyzed between each sample to monitor any carryover or unexpected contamination. Negative controls are prepared from clean carbon stubs that had not been used for hands collection, but that undergo the same extraction protocol as the respective sample batch. In addition, negative controls from the hands of the collecting individuals undergo the whole analytical process from collection to extraction to analysis. Negative control samples are obtained from the hands of the collection team at the beginning and end of each collection session.
The shooters discharged five consecutive shots inside the WVU ballistics range. Although the number of shots fired per shooter varies on a case-to-case basis, the number of discharges were chosen in this study based on previously reported literature as well as the casework experience of colleagues, who reported that typically a shooter fires 3–5 shots when using revolvers and pistols in criminal activities.10,43 After firing 5 shots, the firearm was cleared and placed with the range officer. The shooters then proceeded to the collection laboratory, where both left- and right hands (palm and back) were stubbed 15 times in the areas from the index finger to the thumb. Each firing event generated four samples, which is outlined in Fig. 1. After sample collection, the shooters washed their hands and repeated the process. After the firing session concluded, the collection team obtained additional negative control samples to test potential secondary transfers or unintended contamination. Once all samples were stored, the Tyvek® suits, parchment paper, and gloves were discarded.
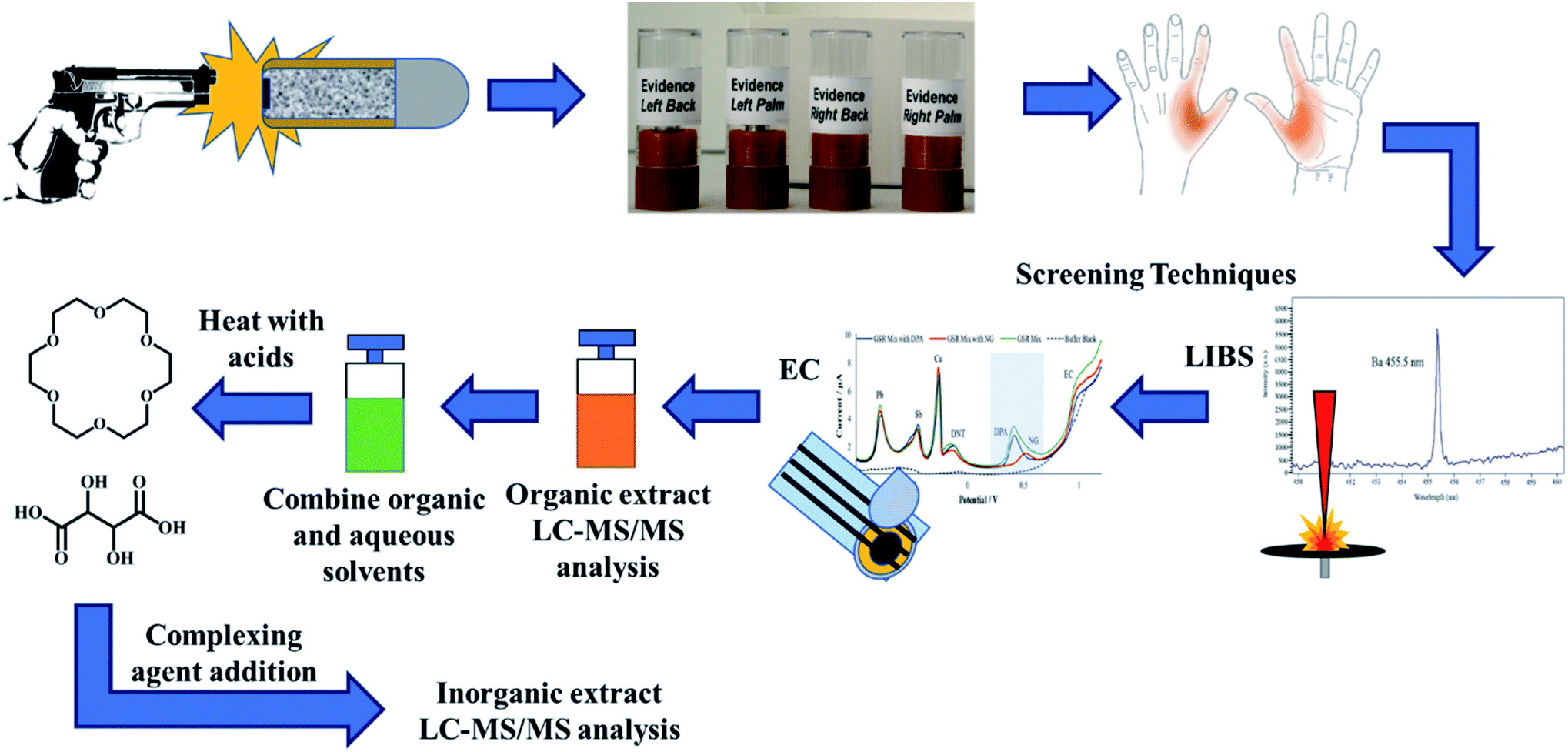 | ||
| Fig. 1 Breakdown of the extraction and analysis process, including cross-validation from screening techniques using a single sample set from a discharge event. From one firing session, each hand is stubbed in the areas highlighted in orange to generate four samples. LIBS first characterizes all four stubs using elemental analysis obtained from the IGSR particulates. The same samples are then followed by electrochemical methods where both IGSR and OGSR are both monitored. The organic extracts from those tests are then tested via LC-MS/MS for OGSR separation and then micro-bulk digestion and complexation for IGSR characterization. | ||
For the activity sessions, three different activities were performed after 5 successive discharges including hand rubbing, hand sanitizer application, and running. For the first activity, the subjects clasped both hands together and vigorously rubbed for approximately 30 seconds. After that time, the collectors proceeded to stub the subjects index and thumb areas. The second activity followed the similar motion as the first activity except for hand sanitizer application. For the final activity, the subjects exited the range and proceeded to run for approximately 60 seconds outside of the range. The collection team then proceeded to stub theirs hands after they entered the ballistics laboratory.
2.3. LC-MS/MS methods
For the inorganics, a Hamilton PRP-X100 cation exchange guard column (10 μm 2.1 × 33 mm) was added to the LC system. The crown ether complexes eluted from the column using an isocratic flow at 90% A/10% B in positive electrospray (ESI) conditions. At 4 minutes, the source polarity switched to negative ESI (ESI−) mode, and the composition of mobile phases switched to 98% A/2% B for the tartaric acid complexes. The injection volume for the IGSR method was 10 μL.
To further address the bias of this method, a primer-only standard was used to evaluate the percent recovery of the micro-bulk digestion method. This was compared to the validated ICP-MS method45 to further compare the concentrations observed in the LC-MS/MS (see Section 2.4.4). The parameters of the ICP-MS parameters and compared digestion methodology are further detailed by Menking-Hoggatt et al.45
2.4. Comparisons with various analytical methods
Our research group has published various innovative methods that can complement current practice. The techniques have been selected to be compatible and complementary to SEM-EDS while providing faster and informative data for case triaging. These include LIBS and electrochemistry, which have demonstrated overall accuracy greater than 90% in several population sets of authentic hand specimens. A goal of the methods, or a combination of them, is to become adoptable in the laboratory and on-site crime scene settings for more effective and streamline decision-making processes. In this study, LC-MS/MS is presented as a powerful tool to provide confirmatory information by dual detection of OGSR and IGSR components. To compare the method's performance, the results from authentic sets are compared to practically non-destructive LIBS and ED methods, and the feasibility of using LC-MS/MS alone or in combination with fast pre-screening tools is evaluated. Detailed information on the methods is described below. On the other hand, this study aims to compare the performance of tesa® Tack polymer versus traditional carbon stubs for the collection of IGSR and OGSR. The LC-MS/MS results were compared to ICP-MS, as detailed below, to evaluate the recovery efficiency.For LC-MS/MS analysis, six consecutive washes of methanol are taken directly onto the surface substrate (6 × 50 μL) for a total of 300 μL. The aliquots were transferred to a 0.2 μm filtration vial and centrifuged for three minutes. The washings were aliquoted into a second filter apparatus (0.45 μm) to ensure no residual polymer or adhesive surfactants could suppress analyte signals. For OGSR analysis, 100 μL of the filtered solution was aliquoted into a separate vial, and D10-DPA was spiked to yield a final concentration of 150 ppb. The remaining 200 μL was dried under a steady stream of N2 until ∼50 μL remained in the centrifuge vial. A 500 μL acid mixture of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 concentrated acid solution (HCl:HNO3) was applied to the substrate surface and gently pipetted on the carbon adhesive surface to remove any remaining particulates. This acid mixture was added to the 50 μL remnant of the organic wash and heated at 85 °C for 10 minutes inside a sterile, plastic centrifuge tube for micro-bulk digestion. After 10 minutes, the solution cooled, and a high concentration of the complexing agents (1 ppm) was added to that mixture to ensure pairings of the solubilized particulates with host agents could self-assemble and form complexes.
:1 concentrated acid solution (HCl:HNO3) was applied to the substrate surface and gently pipetted on the carbon adhesive surface to remove any remaining particulates. This acid mixture was added to the 50 μL remnant of the organic wash and heated at 85 °C for 10 minutes inside a sterile, plastic centrifuge tube for micro-bulk digestion. After 10 minutes, the solution cooled, and a high concentration of the complexing agents (1 ppm) was added to that mixture to ensure pairings of the solubilized particulates with host agents could self-assemble and form complexes.
Also, to test the capability of the LC-MS/MS method to be applied as a confirmatory tool after fast screening. A subset of samples was analyzed by a newly developed approach in our group by LIBS and electrochemistry, followed by LC-MS/MS confirmation on the same stub. LIBS first characterized the four stubs for the IGSR elemental information. Micro-spatial information was obtained about the samples, and multiple wavelengths were monitored GSR elements of interest using a ∼15 mJ pulse laser as per a previously validated method.45,47 After LIBS analysis, the samples were extracted using organic solvents and aqueous buffers for electrochemical methods. First, 100 μL of acetonitrile with gentle mechanical pipette washings was applied to the ablated area of the stub. This extract was split into 50 μL aliquots in 650 μL microcentrifuge tubes. One aliquot was dried under a steady stream of N2 gas and the other was saved for LC-MS/MS analysis. While the drying was occurring, 50 μL of a 0.1 M acetate buffer (pH 4.0) was distributed on the stub's surface with gentle mechanical washings. This aliquot was used to reconstitute the dried down organic fraction. After vortexing, the combined 50 μL drop, encompassing the organic and inorganic fractions, was placed on a carbon electrode for electrochemical analysis by square-wave voltammetry and monitored for both OGSR and IGSR simultaneously.12,46,47
After electrochemistry, the organic extract was run on LC-MS/MS to confirm and detect any OGSR. After analyzing the organic samples, the combined acetonitrile and buffer samples was subjected to the micro-bulk method mentioned above. The complexing agents were added to the mixture after cooling. Each extract was injected once for each hand area, for four replicates per individual (left, right, palm and back). Fig. 1 illustrates the entire combined extraction and analysis process from a single firing event and the same sample.
An ICP-MS instrument (Agilent 7800, Santa Clara, CA) with a MicroMist nebulizer and double pass quartz Scott-type spray chamber was used for the analysis using the parameters reported by Menking-Hoggatt et al.45 After these digestions, both of the acid mixtures were diluted to 2% nitric acid mixtures where they would be compared to a calibration curve ranging from 0–300 ppb within the same matrix of 2% nitric acid. For the ICP standards and the WIN p-GSR standard, we performed three replicates across three days (n = 9). We spiked indium at 150 ppb for the internal standard and performed quality control (QC) runs at two concentrations (25 and 50 ppb) to monitor any loss of response for the instrument. Additionally, we performed method blanks where no analytes were spiked onto the substrate surfaces. We applied the acids to the bare substrates to monitored any potential enhancements or suppression of signal.
3. Results and discussion
3.1 Identification, characterization, and optimization of OGSR and IGSR
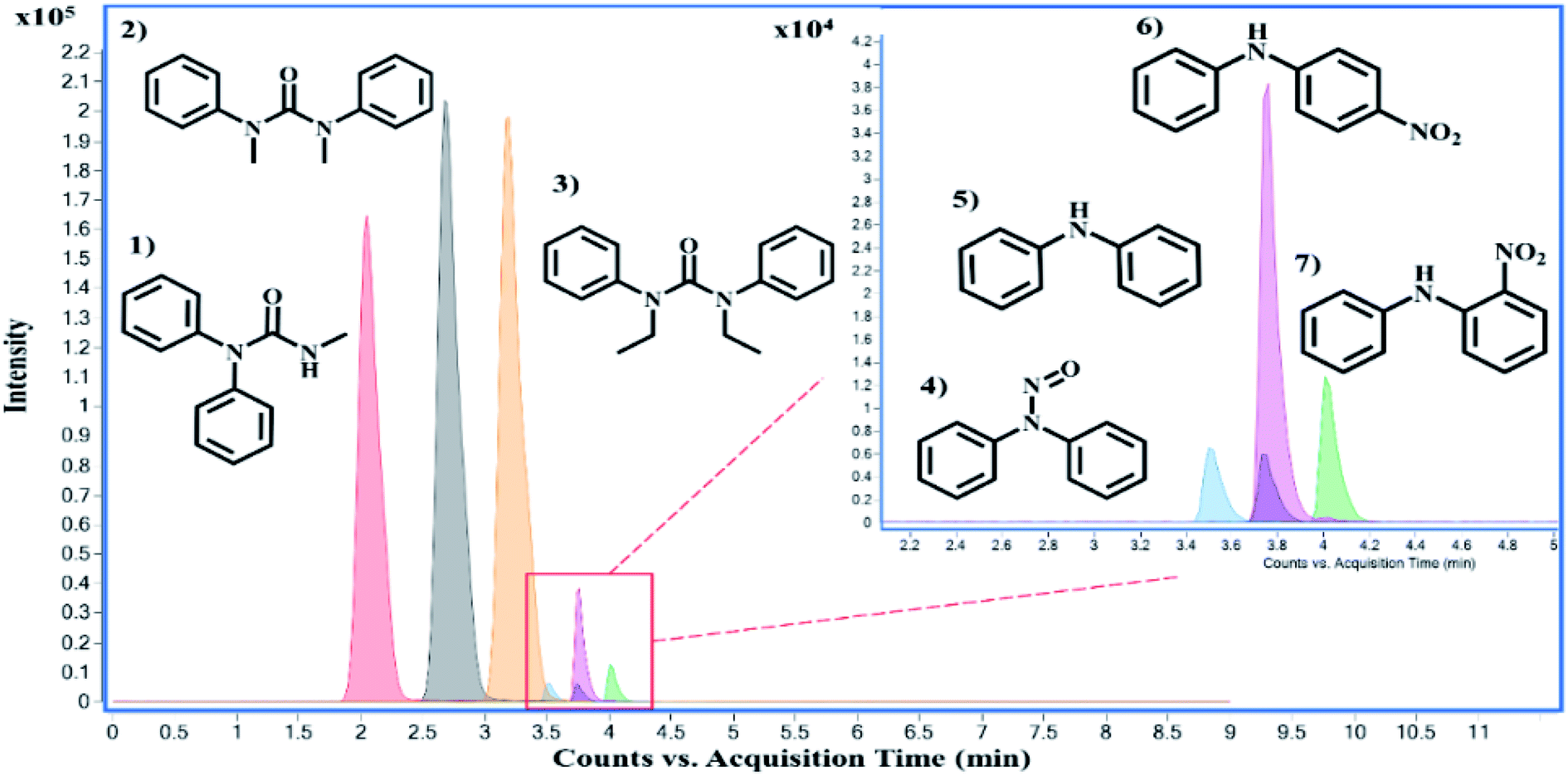 | ||
| Fig. 2 Chromatography separation of the seven OGSR compounds using the pentafluorophenyl column. Standards of OGSR are eluted (minutes) based on their polarities and interactions with the pentafluorophenyl column in the following order: (1) akardite II (2.00 min), (2) methyl centralite (2.67 min), (3) ethyl centralite (3.19 min), (4) N-nitrosodiphenylamine (3.49 min), (5) diphenylamine (3.75 min), (6) 4-nitrodiphenylamine (3.76 min), and (7) 2-nitrodiphenylamine (4.00 min). Each OGSR compound was measured with an electrospray source in (+) mode. | ||
There are over one hundred OGSR compounds that have been reported in different studies. However, we selected seven major compounds (Fig. 2) that are more indicative of gunshot residue as they are not common in the environment nor prevalent in non-shooter populations.10 One advantage of LC-MS methods is they can be easily expanded to additional GSR analytes as long as they are compatible with the ionization requirements. Amongst the constituent list is AK2, which differs from DPA by an amide addition, and 2-NDPA, a positional isomer to 4-NDPA. These OGSR additions to our method were made due to shifts in modern propellant formulations and potential degradation and deflagration entropy of more prominent indicators such as DPA. Interestingly, compounds like MC and EC or 4-NDPA and 2-NDPA, which only differ by methyl groups or the nitro-group position, can be identified by elution time alone.
Source conditions were also optimized for improved sensitivities of the OGSR compounds. These conditions were modeled after Ali et al., who utilized a similar LC-MS/MS system.33 However, the discernable difference between the two methodologies is the increased transitions for all compounds in our method, barring 2-NDPA, and the lowered injection volume (1 μL vs. 5 μL). As expected, most OGSR analytes were suitable to ESI(+). However, it is important to note that two typical OGSR markers, nitroglycerin (NG) and 2,4-dinitrotoluene (2,4-DNT), were not observed using electrospray ionization in positive mode. Several technical publications documented this observation using the same instrument source setup: the Agilent Jet Stream® ESI (AJS-ESI).50 The limitation in ionization is potentially due to the chemical environment conditions of the analyte, which showed very low efficiency in positive ionization. Although ESI(−) mode can detect these compounds, it requires the assistance of signal enhancers' to promote adduct formation (salt forms).51 These anionic additives include acetates, chlorides, iodides, and other nitrate sources.51 Varying sources like atmospheric pressure chemical ionization (APCI−), UV detectors, and recently direct sample analysis (DSA) can circumvent signal problems and observe these compounds easily.52–55 The addition of Na or K to the medium may be useful in some instances, but it was not considered a viable option in our study since these two elements are expected to be widely distributed in the environment and in the skin surfaces sampled for GSR. Also, Na and K are easy to ionize and may cause in-source ionization competition that can affect the efficacy of the metal clusters response. Unfortunately, these options require either dopants or other sources (e.g., APPI) which may lead to additional costs and complications to a forensic laboratory. Therefore, a decision was made to maintain the method's simplicity by using ESI in positive mode while monitoring seven key OGSR components. Alternatively, our approach can incorporate rapid screening with electrochemical methods, one can identify and characterize these two troublesome compounds (NG and 2,4-DNT).12
A Q-Exactive® orbitrap was utilized to corroborate the findings and confirm the M–L complexes' identity from the QQQ full scan. Flow injection analysis (FIA) and manual manipulations of the source conditions revealed the presence of 1:1 as well as 1:2 ratios metal to ligand complexes. Additionally, water adducts could also be observed but were less abundant than the nitrated species. A full fragmentation breakdown and isotopic ratio observation are illustrated in Fig. 3 and Table 1 for barium and lead species. Unfortunately, an Sb complex could not be formed with the chosen macrocycle; however, a different mechanism and complexing agent unlocked the identification and will be discussed later.
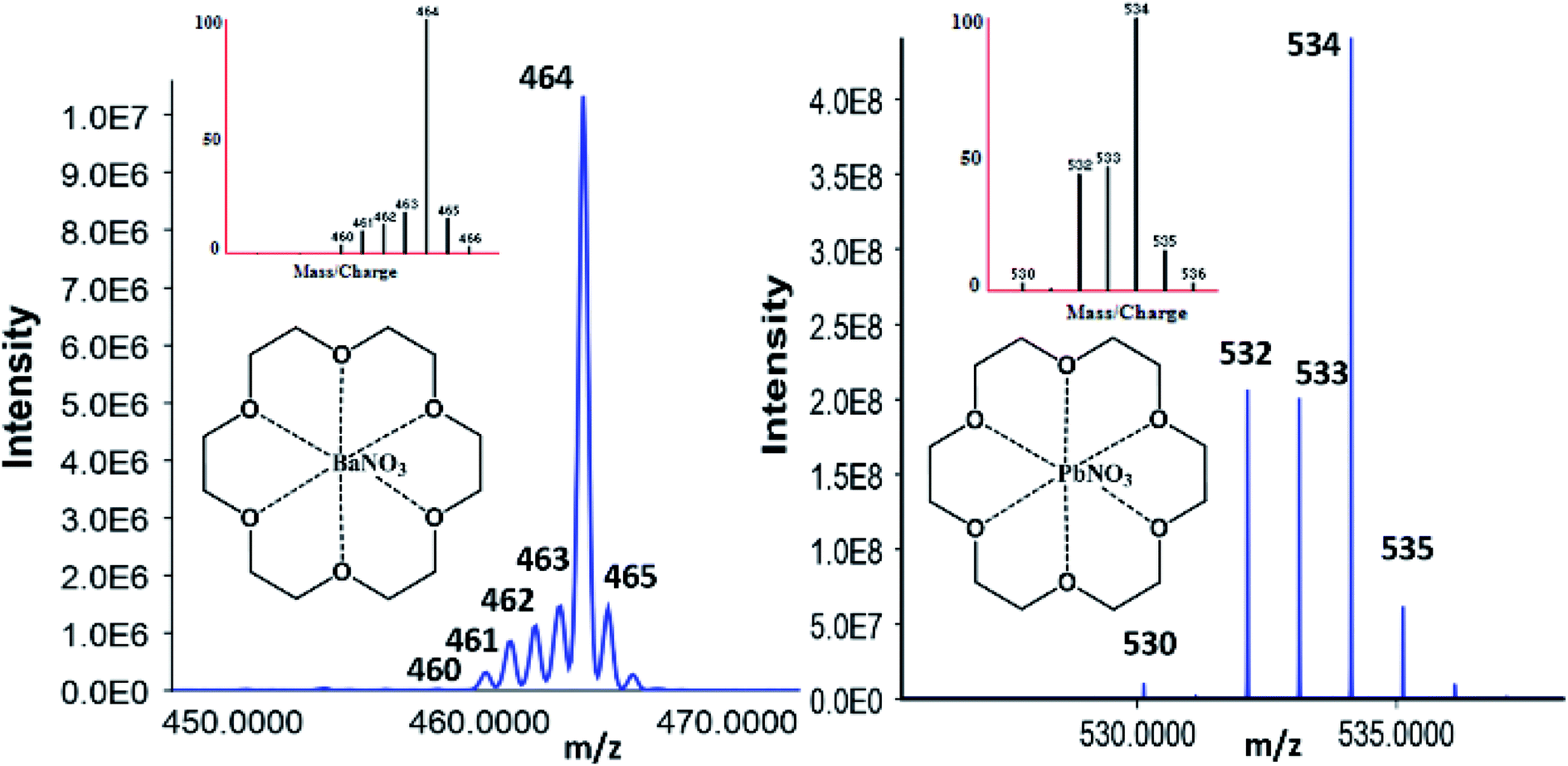 | ||
| Fig. 3 Orbitrap confirmation of 18C6 [Ba–NO3] with Ba natural abundance (left) and [Pb–NO3] with Pb natural abundances (right) patterns correlating to the simulated M–L isotopic distribution. | ||
| Identification | Ions (m/z) observed | Identification | MS1 (nitrated forms) | MSn (lone metals) |
|---|---|---|---|---|
| Barium, BaNO3 (200 Da), 18C6 (264 Da), C12H24O6 | 464.0498 | [L + 138Ba + NO3]+ | 200.9003 [138Ba + NO3]+ | 137.9003 [138Ba]+ |
| 463.0504 | [L + 137Ba + NO3]+ | 199.9009 [137Ba + NO3]+ | 136.9009 [137Ba]+ | |
| 462.0491 | [L + 136Ba + NO3]+ | 198.8997 [136Ba + NO3]+ | 135.8997 [136Ba]+ | |
| 461.0502 | [L + 135Ba + NO3]+ | 197.9008 [135Ba + NO3]+ | 134.9008 [135Ba]+ | |
| 460.0491 | [L + 134Ba + NO3]+ | 196.8972 [134Ba + NO3]+ | 133.8972 [134Ba]+ | |
| Lead, PbNO3 (270 Da), 18C6 (264 Da), C12H24O6 | 534.0498 | [L + 208Pb + NO3]+ | 270.1003 [208Pb + NO3]+ | 208.1003 [208Pb]+ |
| 533.0504 | [L + 207Pb + NO3]+ | 269.0109 [207Pb + NO3]+ | 207.0109 [207Pb]+ | |
| 532.0491 | [L + 206Pb + NO3]+ | 268.0097 [206Pb + NO3]+ | 206.0097 [206Pb]+ | |
| 530.0491 | [L + 204Pb + NO3]+ | 266.0012 [204Pb + NO3]+ | 204.0012 [204Pb]+ |
After orbitrap verification, source conditions and optimizations were performed on the QQQ using the Optimizer® software. Fragmentor and collision energy voltages were monitored from 10–100 V and 10–250 V with 10 V increments to yield the highest signal possible, respectively. The most abundant product ions for the barium-complex were the nitrated species (200.1 Da) and the lone metal species (208.1 Da) for the lead complex. Because of the mechanism and factors for complex formation, the decreasing m/z values must be included when building the acquisition method. For instance, the barium-complex forms various precursor ion m/z values consistent with its isotope values at 464.1 (138), 463.1 (137), 462.1 (136), 461.1 (135), and 460.1 (134). Therefore, the acquisition method must account for these values to gain confidence in the observed structure, which is not required for OGSR analysis.
3.2 Choices of complexing agents
Our goal was to select a single complexing agent to capture the three critical elemental constituents of GSR (barium, lead, and antimony). However, extensive literature review and experimentation demonstrated that this was not possible. Antimony (a metalloid like arsenic and bismuth) demonstrated different complexation characteristics that necessitated selection of a separate chelating agent for this element. While this adds another ingredient to the complexing solution, we demonstrated that it is still possible to detect Ba, Pb, and Sb via LC-MS/MS.Crown ethers employ various binding mechanisms, including electrostatic interactions, hydrogen bonding, and/or van der Waals forces, depending on the guest analyte. Like other host compounds, these molecules have shown their effectiveness in various fields, including forensics for explosive and gunshot residue analysis to toxic metal removal in wastewater.35,57,58 The strength of complexation is dependent on various factors, including the internal cavity size of the crown ether, the atomic radii, and the charge of analytes. The appealing aspect of these interactions involves identifying metal ions and their robust nature in forming metal–ligand complexes. The creation of these complexes increases an analyte's molecular weight while maintaining natural isotopic abundances. Additionally, the host macrocycles provide transportation and interactions through a column for MS/MS detection.
Characterizing complexes relies heavily on the instrumentation parameters. Previous experiments performed in our group focused on mobilities via ion mobility spectrometry CID experimentation via LC-MS/MS.35,59 For instance, when inspecting Pb, the addition of 18C6 increases the molecular weight by 264.1 Da yielding 472.1 Da for the lone metal and 534.1 Da when nitrated. When the 534.1 Da complex is fragmented, the Pb–NO3 (270 Da) product ion is predominant and displays similar isotopic distributions to the lead ion. When the 270 Da is fragmented further, the 208.1 (52%), 207.1 (22%), 206.1 (24%), and 204.1 (2%) ions are observed, thus solidifying high confidence for the precursor complex in question.
Other aspects like charge state and size of ions are essential when forming complexes. Additionally, competition for the macrocycle's internal cavity can ultimately affect the response of the metal detection. Because of validation protocols, internal standards should be similar in structure and exhibit comparable ionization energies to measure analyte concentrations accurately. For the OGSR method, D10-diphenylamine was coeluted with DPA but possessed unique transitions for simple identification. The IGSR method used thallium (Tl) as the internal standard since it is typically not found in GSR and is similar in size (150 pm) to both Ba2+ (135 pm) and Pb2+ (119 pm). Without Tl+ present, 18C6 showed preferential binding for Ba versus Pb, illustrated in Fig. S1.† Thallium lowered the intensities for both IGSR constituents yielding the selectivity trend of Tl > Ba > Pb. In contrast, antimony (76 pm) showed no efficient binding affinity with 18C6. This observation shows that the size and charge of the guest play a crucial role in binding selectivity.
Additionally, tartaric acid can yield enantiomeric complexes (D- and L-) and be separated by HPLC and capillary electrophoresis.65 For adequate separations, the tartrate complexes can be dissolved in water and salt solvents and further yet, diluted in various working stock solutions with specific solvents such as methanol. Tartaric acid is readily dissolvable in water but not in methanol, which is why more water adducts are observed versus other salts.
However, the adducts may also be attributed to altering voltages of an ESI source. This can cause the isotope ratios to differ from recorded natural abundances 57% (121Sb) and 43% (123Sb), outlined by Schug et al. and other groups (Fig. 4).61,62,66–69 For instance, by altering the nozzle voltage from 500 V to 2500 V, the complex at 537 m/z appears and disappears, respectively. This investigation showed that the structure of the tartrate complexes can shift in not only the adducts but also in the charge states. This finding has been outlined by Schug and others where altering the voltages of both the fragmentor and within the collision chamber.62,64 The tartrate complexes can undergo homolytic cleavages as the complex reorients throughout the mass analyzer. Thus, resulting in the different losses of peroxides and water adducts. From these observations, the ligand trends does favor the complex with 2− charge (267 Da) than the 1− charged complexes (415, 433, 450, and 537 Da) illustrated in Fig. 4.
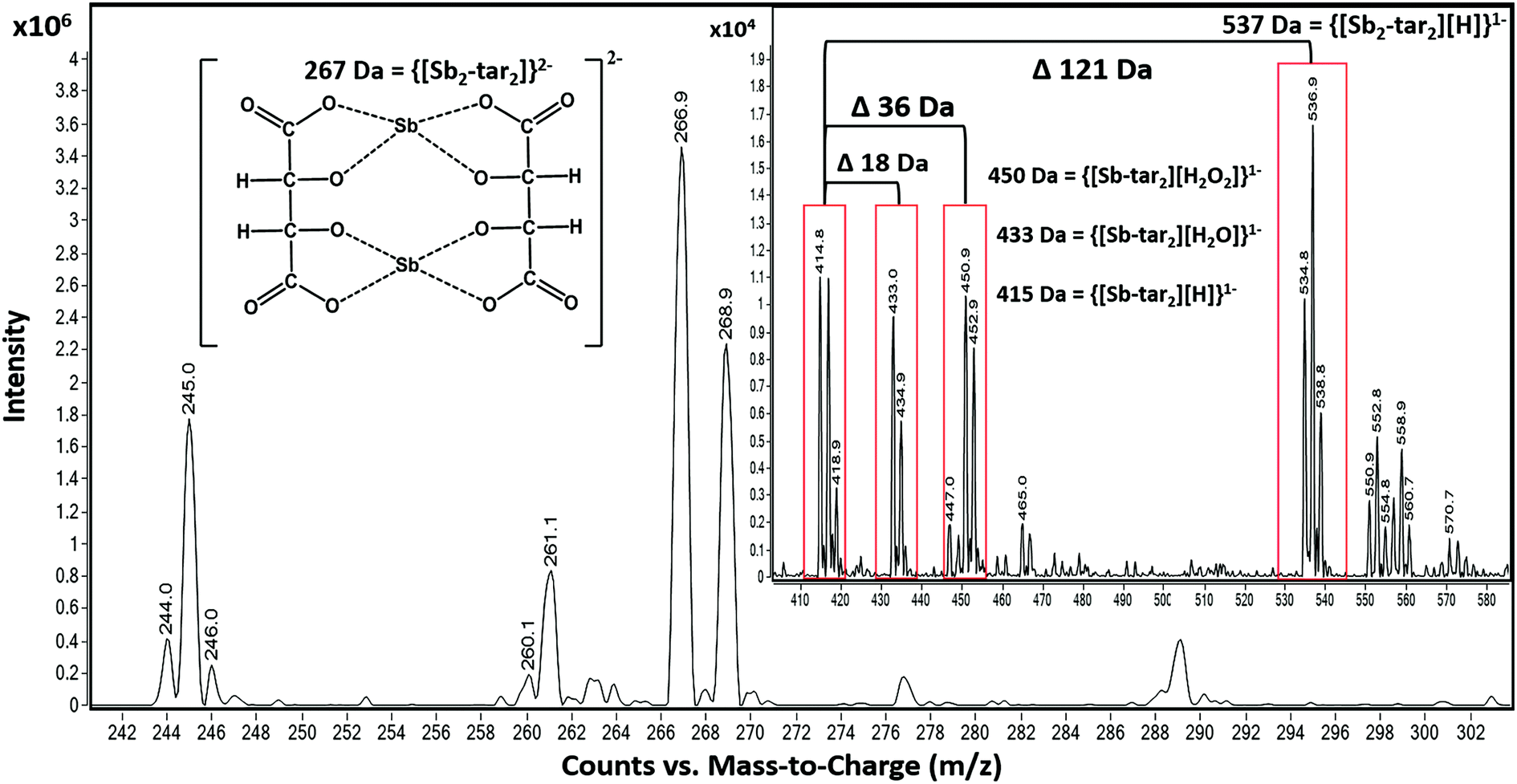 | ||
| Fig. 4 Flow injection analysis of the various tartrate complexes with Sb in ESI− mode. Adducts formation is still prevalent, and because of the nature of the ligand, multiple Sb ions can bind to multiple ligands. For instance, the difference between 415 Da and 433 Da is an m/z value of 18, a water molecule. However, from 415 Da to 537 Da is the addition of an Sb ion. | ||
Prior knowledge of complexation illustrated that 18C6 was not as sensitive to Sb versus Ba and Pb; therefore, tartaric acid was tested as the possible “one host” solution. Although both Ba and Pb formed complexes with the tartaric acid, their signals were vastly smaller than Sb. Ultimately, we decided to incorporate both complexing agents, tartaric acid and 18C6, in equal parts for the final solution.
3.3 Analytical validation and figures of merit
:1 IGSR complexes 100 ppb to 25 ppm (1.0–250 ng). The mean, standard deviation, and 95% confidence intervals are measured against the D10-DPA and Tl quantifier ion ratios for the OGSR and IGSR, respectively. The Agilent MassHunter® Quantitative Analysis software was used to calculate the bias, LDR, and other figures of merit. The absolute LOD and LOQ (ppb and ppm) were calculated for a 1 μL injection and 10 μL injection for OGSR and IGSR, respectively. The estimated LOD and LOQ (pg and ng) were calculated and adjusted for the injections previously mentioned.
| Group | Constituent | LOD (ppb) | LOD (pg) | LOQ (ppb) | LOQ (pg) |
m/z MRM [M + H]+ precursor:product |
Ion ratio % (n = 15) | R 2 | %RSD intra | %RSD inter | LDR (pg) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| OGSR | AK 2 | 0.3 | 0.3 | 0.90 | 0.90 | 227:170:93 |
70.7 | 0.999 | 1.3 | 4.8 | 1–200 |
| DPA | 3.4 | 3.4 | 10.1 | 10.1 | 170:93:65 |
20.4 | 0.998 | 3.7 | 10.4 | 10–200 | |
| EC | 1.0 | 1.0 | 3.0 | 3.0 | 269:148:120 |
91.6 | 0.999 | 1.1 | 9.3 | 5–200 | |
| MC | 0.30 | 0.30 | 0.90 | 0.90 | 241:134:106 |
56.0 | 0.999 | 2.7 | 11.0 | 1–200 | |
| N-NDPA | 4.6 | 4.6 | 13.9 | 13.9 | 199:169:65 |
12.7 | 0.997 | 4.8 | 6.0 | 25–200 | |
| 4-NDPA | 3.0 | 3.0 | 9.0 | 9.0 | 215:198:167 |
92.0 | 0.999 | 7.9 | 6.4 | 10–200 | |
| 2-NDPA | 2.7 | 2.7 | 8.2 | 8.2 | 215:180 |
100.0 | 0.997 | 4.6 | 4.0 | 10–200 |
| Group | Constituent | LOD (ppm) | LOD (ng) | LOQ (ppm) | LOQ (ng) |
m/z MRM precursor:product |
Ion ratio % (n = 15) | R 2 | %RSD intra | %RSD inter | LDR (ng) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IGSR | Ba | 0.10 | 1 | 0.30 | 3.0 | 464:200:138 |
71.70 (138) | 0.998 | 8.4 | 4.6 | 1–60 |
| 463:199:137 |
11.23 (137) | ||||||||||
| 462:198:136 |
7.85 (136) | ||||||||||
| 461:197:135 |
6.59 (135) | ||||||||||
| Pb | 0.20 | 2 | 0.60 | 6.0 | 534:270:208 |
52.40 (208) | 0.982 | 6.4 | 5.7 | 2–60 | |
| 533:269:207 |
22.10 (207) | ||||||||||
| 532:268:206 |
24.10 (206) | ||||||||||
| Sb | 5.0 | 50 | 15.0 | 150 | 267:121 |
57.21 (121) | 0.974 | 2.7 | 11.0 | 50–250 | |
| 269:123 |
42.79 (123) |
As mentioned before, the LOD and LOQ values were improved from the previous proof-of-concept study for both IGSR and OGSR. For OGSR, we included an additional qualifier ion barring 2-NDPA, while Ali et al. served as a guide for source parameters due to similar instrumentation conditions.33 Improvement of the IGSR constituents was contributed to manual optimization and understanding of the complexation process. If more agent is present, more guest analyte can be encapsulated for lower detection. The LDR for Sb is higher due to its formation nature and a greater ionization potential and energies required to dissociate the complex. It is important to note that the 50–250 ng range for Sb is on the cusp of the range when referencing authentic samples. Therefore, we continue to monitor its presence but will need further investigations to circumvent the complex's ionization energy.
The linearity of each compound was assessed through calibration curves and intra- and interday studies. All compounds and elements showed a linear response with an R2 value ≥0.974. The working calibration ranges were 1 ppb to 200 ppb (OGSR) and 100 ppb to 25 ppm (IGSR). Further statistical analysis evaluated an accurate quantitation, including R2 coefficients and residual plots with less than 10% RSD. Residuals plots assess the nature of the samples collected using several considerations: (1) randomly distributed, (2) the variances are equal, and (3) values are normally distributed across predicted values. The residuals' variance did not increase with concentration until after 200 ppb and 6 ppm (Ba and Pb) and 25 ppm (Sb). This observation confirms homoscedasticity across data points, thus ensuring randomness. The relationship between the independent and dependent variables and their variances is displayed in Fig. S2.†
3.3.1.1. Background and collection substrate considerations: recovery study and comparison to ICP-MS digestion method. A critical aspect of the method validation was to consider the performance and practicality of the substrate used for the collection and retention of the inorganic and organic components of interest from the hands of an individual. For this purpose, we compared the recovery of IGSR and OGSR from tesa® Tack polymer and the more universal sampling media carbon adhesive. Our ultimate goal was to evaluate if the LC-MS/MS method can incorporate the use of carbon stubs to be more compatible with current practice and avoid the change of sampling protocols widely used in the GSR community by both law enforcement and laboratory personnel.
The surface extraction of each substrate was evaluated for any possible suppressants. In part, this concern was due to the substrates possessing both polymeric and adhesive qualities that could potentially interfere with the column performance. We were less concerned with the selectivity against the substrates due to choosing specific m/z values and MRM transitions unique to elements and compounds.
Our previous study successfully used tesa® Tack polymer for GSR collection by fully submerging and exposing it to organic solvents and harsh acidic conditions.35 This polymer was very attractive in our initial study because it was commercially sold (http://www.tesa.com) as a non-greasy, putty and suggested by BKA colleagues based on their experiences (Dr Ludwieg Niewoehner) and adapted for the previous project.35 However, because we want this method to comply with GSR cases, without changing current sampling protocols, the universal method of carbon tape stubs was evaluated. Thus, a newly developed extraction method was optimized for both OGSR and IGSR components for the carbon tape by utilizing surface washings. These washings displayed suppression of analytical signals, which led to the incorporation of filters. The OGSR extraction used 100% methanol, where the IGSR analysis used a micro-bulk method that needed heat and concentrated acids as microliter volumes.
One substantial difference between this work and our previous proof-of-concept study is the OGSR and IGSR recovery assessment from the substrates. Following the Eurachem validation guideline, we conducted a recovery comparison experiment using ESI involving both carbon tape and tesa® Tack.35 Bias or percent recovery is not only crucial to indicate potential trends or underlying characteristics/properties present but addresses the effectiveness of a method by measuring the “closeness” of extraction results versus the reference or “true” spiked value onto substrates.
There are two critical differences between the tesa® Tack and carbon tape. First, the polymer exhibits porous properties similar to the skin's epidermal layer, allowing for significant absorption of OGSR. Secondly, the thickness is vastly different compared to the carbon tape. Since tesa® Tack is thicker than the carbon tape, the OGSR components can adsorb into the Tack's inner layers, creating a more significant challenge for extraction. Three different concentration levels (low, medium, high) assessed the repeatability and reproducibility and determined the variability in the results; 25 ppb, 50 ppb, and 165 ppb for OGSR (Table 4) and 1 ppm, 3 ppm, and 5 ppm for IGSR (Table 5). The low, medium, and high values were chosen to represent concentrations that can be fully quantified from the validated method and represent concentrations observed in authentic items.
| Expected conc. | Substrate | MC % recovery | EC % recovery | DPA % recovery | N-NDPA % recovery | 4-NDPA % recovery | 2-NDPA % recovery |
|---|---|---|---|---|---|---|---|
| 25 ppb | Carbon tape | 41.2 ± 0.74 | 42.1 ± 2.81 | 59.9 ± 3.29 | 85.0 ± 2.34 | 44.8 ± 5.87 | 83.4 ± 5.33 |
| tesa® Tack | 28.8 ± 1.49 | 17.3 ± 1.59 | 36.9 ± 1.69 | 55.2 ± 3.83 | 17.8 ± 8.45 | 52.9 ± 11.2 | |
| 50 ppb | Carbon tape | 77.5 ± 1.47 | 67.8 ± 1.36 | 74.2 ± 1.71 | 83.5 ± 2.43 | 40.4 ± 1.38 | 86.2 ± 1.60 |
| tesa® Tack | 38.6 ± 0.40 | 27.1 ± 0.95 | 55.6 ± 0.51 | 48.5 ± 9.03 | 23.4 ± 1.15 | 57.7 ± 1.49 | |
| 165 ppb | Carbon tape | 61.7 ± 2.59 | 80.5 ± 1.58 | 57.6 ± 1.25 | 54.9 ± 1.77 | 35.7 ± 0.77 | 53.5 ± 1.00 |
| tesa® Tack | 30.5 ± 2.52 | 27.1 ± 1.86 | 35.5 ± 1.24 | 24.7 ± 1.70 | 20.6 ± 1.29 | 23.5 ± 0.78 |
| Expected conc. | Substrate | Ba % recovery | Pb % recovery |
|---|---|---|---|
| 1 ppm | Carbon tape | 67.1 ± 0.97 | 57.8 ± 3.28 |
| tesa® Tack | 57.9 ± 1.67 | 45.5 ± 2.98 | |
| 3 ppm | Carbon tape | 74.7 ± 2.44 | 44.4 ± 3.13 |
| tesa® Tack | 83.3 ± 4.53 | 44.6 ± 1.39 | |
| 5 ppm | Carbon tape | 84.7 ± 3.12 | 44.3 ± 1.27 |
| tesa® Tack | 86.1 ± 1.14 | 43.5 ± 1.05 |
From the recovery experiment, the OGSR constituents' results indicated that all components had less than 10% RSD, barring 2-NDPA at 25 ppb for the tesa® Tack, 11.2%. The percent recovery values ranged from 35.7–86.2% for carbon tape and 17.3–57.7% for tesa® Tack. A one-tailed t-test (at p = 0.05) showed the carbon tape has significantly higher recoveries than the tesa® Tack at all concentration ranges tested.
It is worth mentioning the extraction methods are not exhaustive as we designed the method for surface washing rather than a complete submersion extraction. The rationale for this decision was two-fold: (1) it minimizes potential undesired contribution from the adhesive that can be detrimental for LC-MS, and (2) gentle to mild washings further increase the possibility of not displacing particles for further analysis, as demonstrated by Bonnar et al.11
Conversely, the IGSR percent recoveries were comparable for both substrates, showing no significant differences between Ba and Pb observed using a one-tail t-test. The recovery range for the carbon tape was 44–85%, and the tesa® Tack ranged from 43–86%, which is illustrated by Table 5. As a result, the carbon tape was chosen for the remaining of the study as it improved OGSR recovery while not having a detriment effect in the IGSR, as compared to the tesa® Tack.
This procedure utilized ICP-MS standards directly spiked onto each substrate and allowed to dry completely. After drying, surface extractions followed, which used concentrated acids and the micro-bulk procedure mentioned previously. However, this only answers one part of the recovery process as those standards do not truly represent a particulates' nature because they are already solubilized. Therefore, a standard that corresponds to the morphological characteristics and composition relating to ASTM E1588-20 would better represent authentic samples and monitor the behavior of IGSR in concentrated acid washings and digestion, as discussed below.
3.3.1.1.1. Cross-validation of recovery of IGSR via ICP-MS. With OGSR compounds, variables like lipophilicity and evaporation rates play a significant role in the recovery from exposed skin and other sampling areas. Because the environment heavily influences IGSR formation and placement, sufficient analysis is more dependent on factors like extraction procedures and substrate surfaces versus skin absorption. Additionally, OGSR standards correlate more with authentic samples than ICP-MS standards spiked onto substrate surfaces. Therefore, intact IGSR particulates offer a greater evaluation of factors like extraction protocols and interactions.
Tailor-made primer-only GSR (p-GSR) standards originating from a Winchester primer was previously developed and characterized in our group, and was used to evaluate the extraction protocol's effectiveness.45 These standards are made of p-GSR obtained from the actual discharge of a primer in a firearm, therefore providing similar conditions and concentrations that an analyst would expect from an authentic sample. These “matrix-matched” microparticle standards were characterized via ICP-MS, SEM/EDS, and LIBS methods to confirm the concentration, stability, and elemental composition and morphology of the particles. Results showed that this standard correlates to the expected concentration found in the primers.45
Since IGSR is more susceptible to secondary and tertiary transfer, factors like substrate composition and extraction protocol can affect practitioners' analysis. We address both by substrate exposure to heavy acids with multiple washings to a final volume of 500 μL. Therefore, both the extraction and substrate considerations were compared via digestion methods; 85° for 10 minutes in concentrated acid (LC-MS/MS) versus 60 minutes with dilution to 10% nitric acid (ICP-MS). Our findings demonstrated that the percent recoveries were higher with the 60 minute digestion protocol but with less than a 10% difference. Furthermore, there was no statistically significant difference between tesa® Tack and carbon tape regarding the recovery of Pb and Ba. This difference explains more of the interaction with the 18-crown-6-ether, where the binding energy is dependent on the size and charge of the metallic species present in a solution.
After the collection via carbon-adhesive stub, LIBS was used as the first instrument for analysis. LIBS has several appealing aspects, including ambient conditions, quick analysis times (<2 minutes per stub), and compositional information comparable to SEM/EDS.47 Because of the ambient and laser conditions, the OGSR constituents are not substantially lost, and the overall IGSR morphology remains practically uncompromised. The preservation of both constituents allows for further analysis, such as electrochemical methods. By washing a portion of the sample's surface with two solvents (acetonitrile and aqueous buffer), OGSR and IGSR are effectively extracted. With an analysis time comparable to LIBS (<5–10 minutes), the advantage of electrochemical methods is the capability to simultaneously detecting both constituents.46 Furthermore, combining these screening methods has demonstrated their effectiveness in increasing confidence in GSR samples with accuracies superior to 95%, with the advantage that analysis can be followed by SEM/EDS confirmation.12 Because electrochemical methods generate extracts compatible with LC-MS/MS, this study explored if these washings could be used with the micro-bulk digestion and complexing agents.
Because LC-MS/MS offers superior LOD and LOQ and consumes a small sample volume (1 μL), the remaining aliquot can be used for further confirmation methodologies, if needed. After running the organic extract from electrochemistry, the aqueous buffer aliquot with the organic extract can be combined and pushed through with the micro-bulk digestion and complexing agent addition allowing OGSR and IGSR identification by LC-MS/MS. Additionally, we tested the electrochemical extraction collection efficiency by running subsequent washings from an extracted stub. Our findings showed that the subsequent washings show either no signal or signal below the LOQ, which confirms that the electrochemical extractions are very efficient in collecting both constituents. These results illustrate the potential of utilizing various instrumentation techniques to gain more informative data from a single sample without compromising the entire stub.
We tested this criterion against two types of authentic sample data sets and compare them with LIBS and electrochemical methods. The two data sets consisted of 95 baseline shooter samples (Table 6) and 78 activity samples (Table 7). The baseline samples involved shooting in the WVU ballistics range and immediately collecting after a shooting event. These samples were extracted using the LC-MS/MS methodology involving the filters and concentrated acid surface washings. The baseline results are represented in Table 6 and include success rates and detection for LC-MS/MS. The collection methodology involves generating four separate carbon stub samples labelled as such, left-palm (LP), left-back (LB), right-palm (RP), and right-back (RB). The first shooting session used a single type of ammunition (Winchester), while the second shooting session utilized a mixture of ammunition and firearms (Federal, Blazer, 9 mm and 40 caliber pistols). Moreover, a subset of 30 non-shooter background samples was monitored to assess potential false-positives and establish critical thresholds. All background samples resulted in 100% true-negative rate, where all potential peaks were below LOD and LOQ.
| Session | Analyte | Range (ppb) | Mean (ppb) | Median (ppb) | Positive set | Combined constituent data | |
|---|---|---|---|---|---|---|---|
| 1 | OGSR | AK2 | 0.50–296 | 15.4 | 6.5 | 18/20, 90% | 19/20, 95% |
| MC | ND | ||||||
| EC | 1.1–350 | 13.8 | 2.8 | ||||
| DPA | 4.5–498 | 102 | 74 | ||||
| N-NDPA | 6.8–271 | 58 | 36 | ||||
| 4-NDPA | 3.7–30 | 10.9 | 8.2 | ||||
| 2-NDPA | 6.2–58 | 16.7 | 9.7 | ||||
| Range (ppm) | Mean (ppm) | Median (ppm) | |||||
| IGSR | Ba | 1.1–8.9 | 4.07 | 3.5 | 20/20, 100% | ||
| Pb | 1.1–5.2 | 1.83 | 1.4 | ||||
| Sb | 6.1–10.1 | 5.6 | 6.1 | ||||
| 2 | OGSR | AK2 | 28–55 | 42.8 | 42.1 | 33/75, 44% | 75/75, 100% |
| MC | ND | ||||||
| EC | 15.4–81.5 | 42.1 | 40.3 | ||||
| DPA | 11.2–65.3 | 30.1 | 25.1 | ||||
| N-NDPA | ND | ||||||
| 4-NDPA | 9.3–38.2 | 20.4 | 16.8 | ||||
| 2-NDPA | 9.1–57.9 | 25.5 | 20.8 | ||||
| Range (ppm) | Mean (ppm) | Median (ppm) | |||||
| IGSR | Ba | 1.7–4.5 | 2.9 | 2.8 | 50/75, 66.7% | ||
| Pb | 0.75–2.5 | 1.5 | 1.2 | ||||
| Sb | ND | ND | ND | ||||
| Session | Activity | LC-MS/MS | LIBS | LIBS + EC | |
|---|---|---|---|---|---|
| OGSR (%) | OGSR + IGSR (%) | IGSR (%) | OGSR + IGSR (%) | ||
| 1 | Rub (n = 13) | 15.4 | 84.6 | 0 | 23.1 |
| Hand sanitizer (n = 13) | 7.7 | 53.8 | 15.4 | 30.8 | |
| Running (n = 13) | 15.4 | 23.1 | 15.4 | 15.4 | |
| Baseline (no activity) (n = 5) | 100 | 100 | 60 | 100 | |
| Background (non-shooters) (n = 4) | 0 | 0 | 0 | 0 | |
| 2 | Rub (n = 13) | 30.8 | 84.6 | 46.2 | 92.3 |
| Hand sanitizer (n = 13) | 69.2 | 92.3 | 38.5 | 84.6 | |
| Running (n = 13) | 23.1 | 38.5 | 61.5 | 92.3 | |
| Baseline (no activity) (n = 5) | 100 | 100 | 60 | 100 | |
| Background (non-shooters) (n = 4) | 0 | 0 | 0 | 0 | |
Interestingly, the second session decreased overall OGSR and IGSR separate performance rates but demonstrated the GSR detection potential when combined (100% true positive rate). These differences may be attributed a myriad of factors including the cartridge ejection port locations, composition of the powder and primers, or the randomness of each firing event. By using various ammunition formulae, the amounts and types of IGSR and OGSR deposited in hands was variable and at some extent, representative of casework.
The 78 activity samples were utilized to monitor the behavior of GSR for more realistic scenarios. These activities included vigorous hand rubbing, the application of hand sanitizer, and running for one minute after discharge events. These samples were cross-validated with LIBS and electrochemical methods and report the success rates and detection for LC-MS/MS compared to LIBS and electrochemistry, represented in Table 7. These results were compared to 10 baseline (no-activity) shooting samples and eight negative control (non-shooter) samples. The negative control samples were collected from the collection team before and after the shooting session to monitor any potential contamination.
Additionally, we saw the effect of weather and its role in the recovery of these analytes. The first shooting session involved inclement weather in the form of rain. The shooter's hands showed significant amounts of water when they entered the lab after the running activity. The results indicated the OGSR compounds were more affected versus the IGSR particulates ranging from 7.7–15.4% for LC-MS/MS. This consideration presents another point when researching more “real-world” scenarios where weather could be impactful. For all samples, methyl centralite was not detected in any samples, which may be largely attributed to a shift in propellant formulation. On the other hand, we observed an increase in AK2 detection in these items.
Observing the analyte response versus activity under clearer weather conditions, the OGSR responses for the LC-MS/MS were more significant for the hand sanitizer versus hand rubbing and running. This may be primarily due to a “pseudo” organic extraction using isopropyl alcohol in direct contact with the hands. Conversely, running for ∼one minute produced the lowest response for OGSR, which may be due to sweat formation allowing for the potential of increased absorption into the epidermal layer. Looking at the LIBS elemental responses, we observed an inverse relationship for the IGSR particulates where running was the greatest response followed by rubbing then hand sanitizer. This may indicate that the IGSR particulates are more influenced by physical interactions with hands in the forms of washing or sanitizing. Although these remarks and comments are speculative, the results illustrate the need to expand research efforts and interest in the persistence and fate of GSR analytes after a discharge event. Also, the preliminary post-shooting activity data demonstrates that the detection of OGSR and IGSR by LC-MS/MS was feasible even for samples exposed to factors that decrease the chances for detection.
4. Conclusions
Herein, we demonstrate the use of host–guest chemistry to reach application in another discipline. Crown ethers are incredibly versatile not only for their transportation properties but also for their ability to retain isotopic ratios of the metals.35,41,70,71 The method proved efficient for the detection of Pb and Ba. On the other hand, the inability to detect antimony in authentic samples is primarily due to the chelated metal complex's detection limitations and ionization. Current research efforts focus on investigating optimal complexing agents to encompass IGSR analytes to lower the ionization potentials. Finding host molecules suitable for complexing all IGSR metals is inherently difficult because of the wide variety of complexing agents available and the intricacies associated with the interactions between the metal species and host molecules.More importantly, we investigated and validated an LC-MS/MS method to integrate into a broader workflow of GSR analysis. LC-MS/MS is a widely found technique in crime laboratories that primarily analyze drugs of abuse or toxicology samples. The figures of merit established represent this technique's capability but does not explicitly envelop all instrumentation models. This methodology's appealing aspect is the ability to use one sample for various screening tests like LIBS or electrochemistry followed by confirmatory techniques such as LC-MS/MS.12,30,33 Following the ASTM E1588-20 guideline for GSR analysis allows this research to apply a universal collection substrate, compositional considerations, and an established standard instrument for reference.
However, without considering both components, the evidentiary value and understanding of GSR behavior may remain stagnant. The key to this method's successful deployment in the forensics field is to adopt LC-MS/MS approaches to increase confidence in the identification of GSR on a sample with both IGSR and OGSR information. The figures of merit presented demonstrate the ability of the method to identify and quantitate both OGSR and IGSR. With a further understanding and additional research concerning elemental and isotopic data when bound to host molecules, an instrumental technique like LC-MS/MS can provide additional quantitation for practitioners. Not only does this method increase the accuracy in GSR analysis and interpretation, but it yields an alternative avenue and resolves the need to choose between IGSR or OGSR. Finally, the approaches proposed in this study can be applied in other fields, monitoring exposure and environmental compartments near ammunition manufacturing or shooting facilities.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We would like to acknowledge both Dr Keith Morris and James Hamilton for their support during the collection of authentic samples and access to the WVU ballistics laboratory. We would also like to acknowledge Kourtney Dalzell for her assistance in collection and analysis of the electrochemical results. Additionally, we would like to acknowledge Dr Callee Walsh and Dr Sandra Majuta for assistance and access to the orbitrap instrument in the BNRF. This project is sponsored by Award No. #2019-R2-CX-044 from the National Institute of Justice to West Virginia University. The opinions, findings, and conclusions are those of the authors and do not necessarily reflect those of the Department of Justice.References
- T. Trejos, S. Koch and A. Mehltretter, Forensic Chem., 2020, 18, 100223 CrossRef CAS.
- L. S. Blakey, G. P. Sharples, K. Chana and J. W. Birkett, J. Forensic Sci., 2018, 63, 9–19 CrossRef PubMed.
- Z. Brozek-Mucha, Anal. Bioanal. Chem., 2017, 409, 5803–5811 CrossRef CAS PubMed.
- O. Dalby, D. Butler and J. W. Birkett, J. Forensic Sci., 2010, 55, 924–943 CrossRef PubMed.
- M. A. de Carvalho, M. Talhavini, M. F. Pimentel, J. M. Amigo, C. Pasquini, S. Alves, I. T. Weber, M. Albino de Carvalho, M. Talhavini, M. F. Pimentel, J. M. Amigo, C. Pasquini, S. A. Junior and I. T. Weber, Anal. Methods, 2018, 10, 4711–4717 RSC.
- F. S. Romolo, M. J. Bailey, J. de Jesus, L. Manna and M. Donghi, Sci. Justice, 2019, 59, 181–189 CrossRef PubMed.
- M. Donghi, K. Mason and F. S. Romolo, J. Forensic Sci., 2019, 64, 1658–1667 CrossRef CAS PubMed.
- K. Mason and R. Wuhrer, Microsc. Microanal., 2017, 23, 1074–1075 CrossRef.
- ASTM International, E1588-20 Standard Practice for Gunshot Residue Analysis by Scanning Electron Microscopy/Energy Dispersive X-Ray Spectrometry, ASTM International, West Conshohocken, PA, 2020, DOI:10.1520/E1588-20.
- W. Feeney, C. Vander Pyl, S. Bell and T. Trejos, Forensic Chem., 2020, 19, 100250 CrossRef CAS.
- C. Bonnar, E. C. Moule, N. Lucas, K. E. Seyfang, R. P. Dunsmore, R. S. Popelka-Filcoff, K. Redman and K. Paul Kirkbride, Forensic Sci. Int., 2020, 314, 110389 CrossRef CAS PubMed.
- T. Trejos, C. V Pyl, K. Menking-Hoggatt, A. L. Alvarado and L. E. Arroyo, Forensic Chem., 2018, 8, 146–156 CrossRef CAS.
- M. Lopez-Lopez, C. Alvarez-Llamas, J. Pisonero, C. Garcia-Ruiz and N. Bordel, Forensic Sci. Int., 2017, 273, 124–131 CrossRef CAS PubMed.
- J. Coumbaros, K. P. Kirkbride, G. Klass and W. Skinner, Forensic Sci. Int., 2001, 119, 72–81 CrossRef CAS PubMed.
- G. Vanini, R. M. Souza, C. A. Destefani, B. B. Merlo, T. M. Piorotti, E. V. R. de Castro, M. Carneiro and W. Romao, Microchem. J., 2014, 115, 106–112 CrossRef CAS.
- M. I. A. Halim, M. F. Safian, E. Elias, S. S. Shazali and IEEE, Identification of Gunshot Residue from Trace Element by Using ICP/OES, 2013 Search PubMed.
- E. Goudsmits, G. P. Sharples and J. W. Birkett, Sci. Justice, 2016, 56, 421–425 CrossRef PubMed.
- S. R. Khandasammy, A. Rzhevskii and I. K. Lednev, Anal. Chem., 2019, 91, 11731–11737 CrossRef CAS PubMed.
- J. Bueno and I. K. Lednev, Anal. Chem., 2014, 86, 3389–3396 CrossRef CAS PubMed.
- D. Muller, A. Levy, A. Vinokurov, M. Ravreby, R. Shelef, E. Wolf, B. Eldar and B. Glattstein, J. Forensic Sci., 2007, 52, 75–78 CrossRef CAS PubMed.
- B. Stevens, S. Bell and K. Adams, Forensic Chem., 2016, 2, 55–62 CrossRef CAS.
- C. Weyermann, V. Belaud, F. Riva and F. S. Romolo, Forensic Sci. Int., 2009, 186, 29–35 CrossRef CAS PubMed.
- S. Benito, Z. Abrego, A. Sanchez, N. Unceta, M. A. Goicolea, R. J. Barrio, A. Sánchez, N. Unceta, M. A. Goicolea and R. J. Barrio, Forensic Sci. Int., 2015, 246, 79–85 CrossRef CAS PubMed.
- A. L. Gassner and C. Weyermann, Forensic Sci. Int., 2016, 264, 47–55 CrossRef CAS PubMed.
- A. M. O'Mahony, J. R. Windmiller, I. A. Samek, A. J. Bandodkar and J. Wang, Electrochem. Commun., 2012, 23, 52–55 CrossRef.
- A. Dona-Fernandez, I. de Andres-Gimeno, P. Santiago-Toribio, E. Valtuille-Fernandez, F. Aller-Sanchez and A. Heras-Gonzalez, Forensic Sci. Int., 2018, 292, 167–175 CrossRef CAS PubMed.
- L. A. Fambro, D. D. Vandenbos, M. B. Rosenberg and C. R. Dockery, Appl. Spectrosc., 2017, 71, 699–708 CrossRef CAS PubMed.
- E. Hondrogiannis, D. Andersen and A. W. Miziolek, inConference on Next-Generation Spectroscopic Technologies VI, Spie-Int Soc Optical Engineering, Bellingham, 2013, vol. 8726, p. 87260P Search PubMed.
- M. B. Rosenberg and C. R. Dockery, Appl. Spectrosc., 2008, 62, 1238–1241 CrossRef CAS PubMed.
- Z. Abrego, N. Grijalba, N. Unceta, M. Maguregui, A. Sanchez, A. Fernández-Isla, M. A. Goicolea, R. J. Barrió, A. Fernandez-Isla, M. A. Goicolea and R. J. Barrio, Analyst, 2014, 139, 6232–6241 RSC.
- M. Gallidabino, C. Weyermann, F. S. Romolo and F. Taroni, Sci. Justice, 2013, 53, 41–48 CrossRef PubMed.
- J. W. Moran and S. Bell, Anal. Chem., 2014, 86, 6071–6079 CrossRef CAS PubMed.
- L. Ali, K. Brown, H. Castellano and S. J. Wetzel, J. Forensic Sci., 2016, 61, 928–938 CrossRef CAS PubMed.
- A. Tarifa and J. R. Almirall, Sci. Justice, 2015, 55, 168–175 CrossRef PubMed.
- S. Bell and W. Feeney, Forensic Sci. Int., 2019, 299, 215–222 CrossRef CAS PubMed.
- W. A. MacCrehan, K. D. Smith and W. F. Rowe, J. Forensic Sci., 1998, 43, 119–124 CAS.
- M. Morelato, A. Beavis, A. Ogle, P. Doble, P. Kirkbride and C. Roux, Forensic Sci. Int., 2012, 217, 101–106 CrossRef CAS PubMed.
- V. Redouté Minzière, D. Werner, D. Schneider, M. Manganelli, B. Jung, C. Weyermann and A. L. Gassner, J. Forensic Sci., 2020, 65(4) DOI:10.1111/1556-4029.14314.
- E. Bernal Morales and A. L. Revilla Vázquez, J. Chromatogr. A, 2004, 1061, 225–233 CrossRef CAS PubMed.
- R. V Taudte, C. Roux, L. Blanes, M. Horder, K. P. Kirkbride and A. Beavis, Anal. Bioanal. Chem., 2016, 408, 2567–2576 CrossRef PubMed.
- S. M. Blair, E. C. Kempen and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 1998, 9, 1049–1059 CrossRef CAS.
- Y. Tao and R. R. Julian, J. Am. Soc. Mass Spectrom., 2013, 24, 1634–1640 CrossRef CAS PubMed.
- D. C. Reedy and C. S. Koper, Inj. Prev., 2003, 9, 151–155 CrossRef CAS PubMed.
- Eurachem, The Fitness for Purpose of Analytical Methods, 1998 Search PubMed.
- K. Menking-Hoggatt, C. Martinez, C. Vander, E. Heller, E. Chip, L. Arroyo, T. Trejos, C. Vander Pyl, E. Heller, E. “Chip” Pollock, L. Arroyo and T. Trejos, Talanta, 2021, 225, 121984 CrossRef CAS PubMed.
- C. E. Ott, K. A. Dalzell, P. J. Calderón-Arce, A. L. Alvarado-Gámez, T. Trejos and L. E. Arroyo, J. Forensic Sci., 2020, 65, 1935–1944 CrossRef CAS PubMed.
- K. Menking-Hoggatt, L. Arroyo, J. Curran and T. Trejos, J. Chemom., 2021, 35, 1–13 CrossRef.
- M. Maitre, M. Horder, K. P. Kirkbride, A. L. Gassner, C. Weyermann, C. Roux and A. Beavis, Forensic Sci. Int., 2018, 292, 1–10 CrossRef CAS PubMed.
- D. Laza, B. Nys, J. De Kinder, A. K. D. Mesmaeker and C. Moucheron, J. Forensic Sci., 2007, 52, 842–850 CrossRef CAS PubMed.
- E. M. Thurman and I. Ferrer, Agilent Technologies Technical Note, 2012, 1–8 Search PubMed.
- G. Gaiffe, M. C. Bridoux, C. Costanza and R. B. Cole, J. Mass Spectrom., 2018, 53, 21–29 CrossRef CAS PubMed.
- C. Bonnar, R. Popelka-Filcoff and K. P. Kirkbride, J. Am. Soc. Mass Spectrom., 2020, 31, 1943–1956 CrossRef CAS PubMed.
- M. Roberts, N. Petraco and M. Gittings, Sci. Justice, 2015, 55, 467–471 CrossRef PubMed.
- S. Park, J. Lee, S. G. Cho, E. M. Goh, S. Lee, S. S. Koh and J. Kim, Bull. Korean Chem. Soc., 2013, 34, 3659–3664 CrossRef CAS.
- D. Perret, S. Marchese, A. Gentili, R. Curini, A. Terracciano, E. Bafile and F. Romolo, Chromatographia, 2008, 68, 517–524 CrossRef CAS.
- I. V. Kolesnichenko and E. V. Anslyn, Chem. Soc. Rev., 2017, 46, 2385–2390 RSC.
- S. Kafashi, M. Yaftian and A. Zamani, J. Solution Chem., 2015, 44, 1798–1811 CrossRef CAS.
- N. Islam and S. S. Chimni, J. Mol. Struct., 2017, 1130, 781–790 CrossRef CAS.
- B. Yeager, K. Bustin, J. Stewart, R. Dross and S. Bell, Anal. Methods, 2015, 7, 9683–9691 RSC.
- M. Tella and G. S. Pokrovski, Geochim. Cosmochim. Acta, 2009, 73, 268–290 CrossRef CAS.
- A. B. Wijeratne, S. E. Spencer, J. Gracia, D. W. Armstrong and K. A. Schug, J. Am. Soc. Mass Spectrom., 2009, 20, 2100–2105 CrossRef CAS PubMed.
- A. B. Wijeratne and K. A. Schug, J. Sep. Sci., 2009, 32, 1537–1547 CrossRef CAS PubMed.
- J. M. Casas, G. Crisóstomo and L. Cifuentes, Can. J. Chem. Eng., 2008, 82, 175–183 CrossRef.
- A. B. Wijeratne, S. H. Yang, D. W. Armstrong and K. A. Schug, Anal. Chem., 2010, 82, 5141–5146 CrossRef CAS.
- H. F. Lau, N. M. Quek, W. S. Law, J. H. Zhao, P. C. Hauser and S. F. Y. Li, Electrophoresis, 2011, 32, 1190–1194 CrossRef CAS PubMed.
- A. B. Wijeratne, J. Gracia, S. H. Yang, P. Kroll, D. W. Armstrong and K. A. Schug, Inorg. Chem. Commun., 2010, 13, 1504–1508 CrossRef CAS.
- D. Marcovich and R. E. Tapscott, J. Am. Chem. Soc., 1980, 102, 5712–5717 CrossRef CAS.
- R. C. Palenik, K. A. Abboud and G. J. Palenik, Inorg. Chim. Acta, 2005, 358, 1034–1040 CrossRef CAS.
- X. Li, T. Reich, M. Kersten and C. Jing, Environ. Sci. Technol., 2019, 53, 5221–5229 CrossRef CAS.
- Y. Inokuchi, O. V. Boyarkin, R. Kusaka, T. Haino, T. Ebata and T. R. Rizzo, J. Phys. Chem. A, 2012, 116, 4057–4068 CrossRef CAS PubMed.
- V. Rudiger, H. J. Schneider, V. P. Solov'ev, V. P. Kazachenko and O. A. Raevsky, Eur. J. Org. Chem., 1999, 1847–1856 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ay00778e |
| This journal is © The Royal Society of Chemistry 2021 |