
PLGA/chitosan–heparin composite microparticles prepared with microfluidics for the construction of hMSC aggregates†
Min
Ge
a,
Yaqi
Sheng
a,
Shuyue
Qi
a,
Lei
Cao
a,
Yan
Zhang
*ab and
Jun
Yang
 *a
*a
aThe Key Laboratory of Bioactive Materials, Ministry of Education, College of Life Science, Nankai University, Tianjin, 300071, China. E-mail: yangjun106@nankai.edu.cn; Fax: +86 22 23498038; Tel: +86 22 23498038
bState Key Laboratory of Medicinal Chemical Biology, Nankai University, Tianjin, 300350, China. E-mail: zhangyan2016@nankai.edu.cn; Tel: +86 22 85358618
First published on 28th September 2020
Abstract
Incorporating poly(lactic-co-glycolic) acid (PLGA) microparticles into human mesenchymal stem cells (hMSC) aggregates has shown promising application prospects. However, the acidic degradation products and burst release of PLGA microparticles still need to be ameliorated. In this study, the PLGA/chitosan–heparin (P/C–h) composite microparticles were successfully fabricated by integrating the double emulsion and microfluidic technology through the precise manipulation of the emulsion composition and flow rate of the two-phase in a flow-focusing chip. The P/C–h microparticles were highly monodispersed with a diameter of 23.45 ± 0.25 μm and shell–core structure of the PLGA encapsulated C–h complex, which were suitable for the fabrication of hMSC aggregates. When the mass ratio of PLGA to the C–h complex was optimized to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the pH of the leach liquor of P/C–h microparticles remained neutral. Compared with those of PLGA microparticles, the cytotoxicity and the initial burst release (loaded FGF-2 and VEGF) were both significantly reduced in P/C–h microparticles. Furthermore, the survival, stemness, as well as secretion and migration abilities of cells in hMSC aggregates incorporating P/C–h microparticles were also enhanced. In summary, the P/C–h composite microparticles prepared by the droplet microfluidic technique support the optimal biological and functional profile of the hMSC aggregates, which may facilitate the clinical applications of MSC-based therapy.
:1, the pH of the leach liquor of P/C–h microparticles remained neutral. Compared with those of PLGA microparticles, the cytotoxicity and the initial burst release (loaded FGF-2 and VEGF) were both significantly reduced in P/C–h microparticles. Furthermore, the survival, stemness, as well as secretion and migration abilities of cells in hMSC aggregates incorporating P/C–h microparticles were also enhanced. In summary, the P/C–h composite microparticles prepared by the droplet microfluidic technique support the optimal biological and functional profile of the hMSC aggregates, which may facilitate the clinical applications of MSC-based therapy.
1. Introduction
Mesenchymal stem cells (MSCs) have currently become main candidates for the cell treatment and tissue construction because of their wide source, self-renewal, and paracrine ability.1,2 However, the declining cell viability and biological functions of MSCs after implantation or during in vitro culture has limitations that need to be tackled in their clinical applications.3 Three-dimensional (3D) cell aggregation is one of the promising techniques that was believed to preserve MSCs phenotype and innate properties by replicating the ‘physiological tissues’ microenvironment in a spatially relevant manner.4,5 Moreover, the introduction of polymeric microparticles to form cell aggregates can improve the diffusion of oxygen and nutrients in the aggregates, and further modulate the biochemical and physical properties of the interior microenvironment.6–8Poly(lactic-co-glycolic acid) (PLGA) microparticles, an easily-modified biomaterial certified by the American Food and Drug Administration (FDA) for biosafety, have been widely used in the delivery of growth factors and the preparation of cell aggregates.9–11 However, the acidic degradation products of PLGA caused an initial burst release and chemical degradation of loaded proteins, and even induced inflammatory reactions in vivo.12,13 It is well known that chitosan is a structural basic polysaccharide, and heparin has high affinity with various growth factors.14,15 In this study, in order to mitigate the adverse effects of the acid microenvironment caused by the degradation of PLGA, we compounded chitosan and heparin into PLGA microparticles (P/C–h), and used them to improve the cellular functions of human MSC (hMSC) aggregates.
Emulsion polymerization is a conventional method used for the manufacture of PLGA microparticles. However, the obtained microparticles are usually not uniform in size and have a wide size distribution, which limit their application efficiency in regulating the cellular functions.16 In recent years, with the development of micro–nano processing for generating uniform droplets in high-throughput, a droplet microfluidic system has been utilized as an advanced technology due to remarkable advantages in controlling the chip structure and flow rate of the two-phase of emulsion polymerization.17,18 Herein, we attempted to develop a method that combines microfluidic and double emulsion reaction to efficiently prepare the uniform P/C–h composite microparticles.
In this study, the P/C–h composite microparticles were fabricated using droplet microfluidic technology. First, the composition and flow rate of the continuous and dispersed phases were optimized to form the uniform P/C–h composite microparticles. The basic characterizations and biocompatibility of the P/C–h composite microparticles, including their physical and chemical properties, degradation properties, growth factor-releasing ability, cytotoxicity as well as hemolytic properties, were investigated. Furthermore, the P/C–h composite microparticles were used for the fabrication of hMSC aggregates. The cellular survival, migration and secretion were evaluated to determine the effects of the P/C–h composite microparticles on the biological functions of hMSCs cultured in the aggregates. These P/C–h composite microparticles can be used to support the development of novel 3D stem-cell culture systems in stem-cell-based clinical applications.
2. Materials and methods
2.1 Preparation of the chitosan and heparin complex
The chitosan and heparin (C–h) complex was prepared based on ionic interaction. Briefly, heparin (Aladdin, Shanghai, China) was dissolved in deionized water at a concentration of 5 mg mL−1, and the chitosan (Aladdin, Shanghai, China) solution (5 mg mL−1) was prepared using 1% acetic acid solution. Then, 1 mL heparin solution was titrated individually with 1, 2, 3, 4, and 5 mL of chitosan solution with electromagnetic stirring at 500 rpm under room temperature for 30 minutes. Then, the precipitated C–h complex was harvested by centrifugation at 1000 rpm for 10 minutes.The constitution of the C–h complex was characterized by Fourier-transform infrared spectroscopy (FTIR; TENSOR2, Bruker, German). Dimethylmethylene blue was used for the quantification of the unreacted heparin in the supernatant.19 Briefly, 50 μL supernatant was added to the toluidine blue working solution containing 10.7 μg 1,9-dimethylmethylene blue chloride (Aladdin, Shanghai, China) and 55 mM formic acid (Aladdin, Shanghai, China). The absorbance at 520 nm was determined using a multifunctional microplate reader (Bio-Rad, California, USA). Orange II was used for quantification of the unreacted chitosan in the supernatant.20 Briefly, 50 μL supernatant was added to 20 μL orange II solution (1 mM; Aladdin, Shanghai, China) and gently agitated for 10 minutes. After being centrifuged at 12000 rpm for 5 minutes, the supernatant was harvested and detected using the multifunctional microplate reader under 484 nm.
For zeta potential detection, 100 μL of the heparin solution was titrated individually with 100, 200, 300, 400, and 500 μL of the chitosan solution to form the C–h complex. Subsequently, the C–h complex was dilute to 750 μL with water before the zeta potential measurements using a Zetasizer Nano Z (Malvern, Westborough, MA).
2.2 Preparation of the P/C–h composite microparticles
The obtained C–h complex was suspended in PBS (Gibico, California, USA) and thoroughly homogenized using a high-speed homogenizer (IKA, Staufen, Germany). Then, 0.1 mL of the C–h complex suspension (containing 0 mg, 10 mg, and 20 mg C–h complex, respectively) was added to 1 mL PLGA (Mw = 12000, lactide:glycolide = 50:50, Ji’nan Daigang, Shandong, China)/dichloromethane (DCM; Aladdin, Shanghai, China) solution (40 mg mL−1). The primary emulsion was obtained by undergoing ultrasonic emulsification on ice for 2 minutes, and was used as the dispersed phase in the following steps. 2% (w/v) polyvinyl alcohol (PVA; Aladdin, Shanghai, China) solution was used as the continuous phase. The dispersed phase and the continuous phase were both delivered into the microfluidic device using syringe pumps (Longer, Baoding, China). Before introducing the dispersed phase, flow-focusing microchannels in the chip were wetted with 1 M HCl and 1 M NaOH for 30 minutes. The size of the microparticles was regulated through changing the flow rate of the two-phase. The mono-dispersed droplets were generated continuously at the junction of the flow-focusing microchannel, and harvested in 2% PVA. The droplets were stirred at 300 rpm for 4 hours to volatilize DCM, and then the P/C–h composite microparticles were collected after centrifugation at 1000 rpm and freeze-dried using lyophilization (Biocoll, Beijing, China). Finally, the P/C–h composite microparticles with different compositions were obtained (P/C–h 2:1 referred to the mass ratio of PLGA to the C–h complex at 40 mg:20 mg, and P/C–h 4:1 referred to the mass ratio of PLGA to the C–h complex at 40 mg:10 mg).
PLGA microparticles were prepared using the same method as that used to prepare the P/C–h composite microparticles, except for using PBS instead of the C–h complex. The composite microparticles loaded with growth factors were fabricated with heparin, which was bound with 4.5 μg fibroblast growth factor-basic (FGF-2; R&D Systems, Minneapolis, MN, USA) or vascular endothelial growth factor (VEGF; R&D Systems, Minneapolis, MN, USA) before use.
2.3 Characterization of the P/C–h composite microparticles
The morphology of the P/C–h composite microparticles was observed using a scanning electron microscope (SEM; JEOL, Tokyo, Japan). The sizes of the microparticles were measured from SEM micrographs using nano measurer granularity analysis software (version 1.2). 100 microparticles were randomly chosen to calculate the average diameter in each group. The chemical constitution of the P/C–h composite microparticles was characterized using FTIR, thermal gravimetric analysis (TGA; TG209, Netzsch, German) and differential scanning calorimetry (DSC; DSC209, Netzsch, German). To present the distribution of PLGA and the C–h complex in the composite microparticles, the PLGA/DCM was labeled with Nile red and the chitosan was labeled with FITC. The fluorescently labeled composite microparticles were observed under a confocal microscope (Leica, Wetzlar, Germany). Meanwhile, the cross-sectioned slices of composite microparticles were prepared using a freezing microtome (Leica, Wetzlar, Germany) and observed using SEM.2.4 The degradation property of the P/C–h composite microparticles
The microparticles were immersed into PBS for 15 days. The pH value change of the PBS was monitored. Meanwhile, the microparticles were taken out from PBS, freeze-dried, weighted and underwent morphological observation using SEM every 3 days. The mass loss was calculated using the following equation:| Mass loss (%) = (M0 − Md)/M0 × 100 |
2.5 The release ability of the P/C–h composite microparticles
To detect the release ability of FGF-2 and VEGF from microparticles, 2 mg microparticles (for individual PLGA, P/C–h 4:1, and P/C–h 2:1) were suspended in 0.5 mL PBSTB (PBS containing 0.02% Tween 20 and 10 mg mL−1 BSA), shaken and incubated at 37 °C for 15 days. The supernatant was collected every 3 days, and fresh PBSTB was supplemented after collection. The collected supernatants were stored at −20 °C. The concentrations of FGF-2 and VEGF in the supernatants were measured using enzyme-linked immunosorbent assay (ELISA) kits (Abcam, Cambridge, UK) according to the instructions. All experiments were performed in triplicate.
2.6 Cell culture
The hMSCs used in this study were isolated from human umbilical cords (HUXUC-01001, Cyagen Biosciences, Guangzhou, China). The cells were confirmed to be positive for CD29, CD44 and CD105 (>70%), and negative for CD14 and CD45 (<5%) by flow cytometry before use. hMSCs were cultured in 100 mm tissue culture dishes (Corning, New York, USA) to a density of 1 × 106 cells per dish with DMEM/F12 (HyClone, Logan, Utah, USA) containing 10% (v/v) fetal bovine serum (FBS; Gibco, California, USA) at 37 °C in humidified air containing 5% CO2. The medium was changed every 3 days. After reaching confluence, the cells were passaged with 0.25% trypsin–EDTA solution (Sigma-Aldrich, St. Louis, MO, USA) and used at passage 5 in all subsequent experiments.2.7 Evaluation of cytocompatibility and the hemolytic analysis
The hMSCs were seeded at 6 × 103 cells per well into 96-well plates, and individually cultured with PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles at equal numbers. After being cultured for 1, 3, and 7 days, the viabilities of hMSCs were quantified by a CCK-8 kit (Dojindo, Kumamoto, Japan) according to the instructions. The absorbance of each well was measured at 450 nm.
For the hemolytic analysis, the PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles were immersed separately in PBS for 14 days, and the supernatants were collected after centrifugation. 2 mL of blood from the ICR mouse was collected and washed with PBS until the supernatant became colorless. Then, 200 μL erythrocyte suspension was mixed with 800 μL collected supernatants. The erythrocytes incubated in 800 μL of H2O and 800 μL of PBS were used as the positive and negative control, respectively. After 4 hours of incubation at 37 °C, the mixture was centrifuged at 10000 rpm for 5 minutes. Then, 100 μL of the supernatant in each group was taken into a 96-well plate, and measured using the multifunctional microplate reader at 577 nm.
2.8 Preparation and characterization of hMSC aggregates
The PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles were coated with collagen by incubating in 0.1% w/v collagen solution at room temperature for 2 hours. Then, the microparticles were collected by centrifugation at 12000 rpm for 5 minutes, and mixed with hMSCs at a cell:microparticle ratio of 3:1. The hMSC aggregates were prepared according to our previous procedures.21 Briefly, the suspension containing hMSCs and microparticles was added to Aggrewell™ 400 plates (STEMCELL Technologies, Los Angeles, CA, USA) at a density of 6 × 105 cells per insert (500 hMSCs per microwell). Afterwards, the plates were centrifuged at 1000 rpm for 5 minutes to force the aggregation of hMSCs and microparticles. The cell aggregates were observed and imaged using an optical microscope (Olympus, Tokyo, Japan). The average diameters of the aggregates were evaluated by Image J software.
After incubation at 37 °C for 12 hours, the aggregates were transferred and cultured in ultra-low attachment 6-well plates (Corning, New York, USA). The aggregates were observed and photographed using the optical microscope (Olympus, Tokyo, Japan). The aggregates without microparticles (MSC aggregates) were prepared and used as the control group for subsequent studies. To investigate the distribution of microparticles in the aggregates, the aggregates were capsulated in alginic acid beads (2% w/v; Sigma-Aldrich, St. Louis, MO, USA) that were generated by crosslinking in CaCl2 solution (0.01 M) for 5 minutes. After washing with PBS three times, the beads were fixed with 4% (v/v) paraformaldehyde for 1 day, embedded in paraffin, sliced using a rotary microtome (Leica, Wetzlar, Germany) and finally stained with the hematoxylin–eosin (H&E) staining kit (Solarbio life sciences, Beijing, China) according to the instructions. The stained slices were observed and photographed using the optical microscope. For F-actin/nuclei staining, the Nile red-labeled microparticles were used to prepare hMSC aggregates. The aggregates were fixed with 4% (v/v) paraformaldehyde for 10 minutes and incubated with 0.1% (v/v) Triton X-100 in PBS for 5 minutes. After being blocked with 1% (w/v) BSA for 30 minutes, the cells were counterstained using FITC-phalloidin and 4′,6-diamidino-2-phenylindole (Sigma-Aldrich, St. Louis, MO, USA) for 15 minutes. The aggregates were observed and photographed using the confocal microscope.
2.9 Viability of hMSCs in the aggregates
The aggregates were stained using a Live/Dead Molecular Probes staining kit (Invitrogen, California, USA) according to the instructions after being cultured for 7 days, and then photographed using the confocal microscope. The cellular viability of the aggregates was quantified using the CCK-8 kit, as described above.2.10 Evaluation of the biological functions of the aggregates in vitro
For reverse transcription-polymerase chain reaction (RT-PCR) analysis, the total RNA of the aggregates was isolated using a TRIzol reagent (Invitrogen, Carlsbad, CA, USA) after being cultured for 1, 3, and 7 days. Then, the cDNA was synthesized using Moloney murine leukemia virus reverse transcriptase and random primers. Afterwards, cDNA samples were subjected to PCR amplification using rTaq polymerase (Takara Bio Inc., Shiga, Japan). Subsequently, the PCR products were separated using 1% agarose gel electrophoresis and stained with ethidium bromide (10 mg mL−1, Sigma-Aldrich, St. Louis, MO, USA). Gene expression levels were normalized to β-actin. The specific primer pairs are listed in Table 1. Five samples per group were included to obtain the statistical results.| Genes | Forward primer (5′–3′) | Reverse primer (5′–3′) | Product length (bp) | T m (°C) |
|---|---|---|---|---|
| Bax | CCCCCGAGAGGTCTTTTTCC | GAGACAGGGACATCAGTCGC | 260 | 60 |
| Bcl-2 | GAACTGGGGGAGGATTGTGG | ACTTCACTTGTGGCCCAGAT | 298 | 60 |
| Nanog | AATGGTGTGACGCAGGGATG | GCATGCAGGACTGCAGAGAT | 223 | 58 |
| Sox-2 | AACCAGCGCATGGACAGTTA | GACTTGACCACCGAACCCAT | 278 | 59 |
| EGF | CTGAATGTCCCCTGTCCCAC | GGTTGCATTGACCCATCTGC | 463 | 60 |
| FGF2 | TCCACCTATAATTGGTCAAAGTGGT | CATCAGTTACCAGCTCCCCC | 121 | 59 |
| TGF-β | AGGGCTACCATGCCAACTTC | GACACAGAGATCCGCAGTCC | 385 | 60 |
| VEGF | GCCATCCAATCGAGACCCTG | ATTAGACAGCAGCGGGCAC | 367 | 60 |
| β-Actin | AGCGAGCATCCCCCAAAGTT | GGGCACGAAGGCTCATCATT | 285 | 60 |
For real-time fluorescence quantitative PCR (qRT-PCR), total RNA was isolated using the TRIzol reagent after culturing for 1, 3, and 7 days. The qRT-PCR was carried out using the SuperScript III Platinum Two-Step qRT-PCR Kit with SYBR Green (Invitrogen, Carlsbad, CA, USA) according to the instructions, and performed on a qRT-PCR instrument (ABI 7900HT, Applied Biosystems, USA). Gene expression levels were normalized to β-actin. Primers used in qRT-PCR are listed in Table 1. Five samples per group were included to obtain the statistical results.
For evaluating the adhesion and migration abilities of hMSCs, the matrigel (Conning, New York, USA) was added to a 48-well plate (100 μL per well) and incubated at 37 °C for 30 minutes. The aggregates were cultured in ultra-low attachment 6-well plates for 7 days, and then transferred onto the matrigel and cultured for another 3 days. The cellular spreading and migrating from the aggregates were observed and photographed using an optical microscope. The F-actin/nuclei staining was performed using FITC-phalloidin and 4′,6-diamidino-2-phenylindole, as described above.
For the microarray assay of the aggregates, the total RNA of the MSC aggregates or the aggregates incorporated with the P/C–h 2:1 composite microparticles (MSC/P/C–h 2:1) were isolated using the TRIzol reagent as described above after being cultured for 36 hours. 0.5 μg of cellular RNA was used for labeling the reactions. Microarray experiments were performed using Agilent 8 × 60 K oligonucleotide arrays, which cover the entire statistical analysis of the human genome. Labeling and Gene Ontology analyses were performed by Capital Bio Technology (Beijing, China), as per the protocol of the manufacturer.
2.11 Statistical analysis
GraphPad Prism Version 5.0 software for Windows (GraphPad, San Diego, CA) was used for statistical analysis. Data analysis was performed by the one-way analysis of variance (ANOVA). The minimum significance level was set at *p < 0.05, **p < 0.01 and ***p < 0.001.3. Results
3.1 Preparation and characterization of the P/C–h composite microparticles
First, the ratio of the C–h complex for fabricating the P/C–h composite microparticles was optimized. According to the results of FTIR spectra (Fig. S1, ESI†) and the residual contents of chitosan and heparin in the supernatant (Fig. S2, ESI†), as well as the zeta potential of synthetic products (Fig. S3, ESI†), heparin was completely titrated by chitosan when the chitosan to heparin ratio was 3:1. Therefore, we chose this ratio to fabricate the P/C–h composite microparticles.
The steps to prepare the P/C–h composite microparticles are shown in Fig. 1A. The surface hydrophilicity of the flow-focusing channels was increased after hydrophilic treatment by HCl and NaOH. The size of the microparticles can be adjusted by changing the flow rate of the two-phase, ranging between 0.5–1.1 mL h−1 for the continuous phase (2% PVA) and 0.2–0.8 mL h−1 for the disperse phase (the primary emulsion) (Fig. 1B). In addition, the sizes of the microparticles were tuned by adjusting the concentration of two-phase (Fig. 1C). In the case of preparing microparticles at 23.5 μm, the flow rates of the continuous phase and dispersed phase were held at 1.1 and 0.4 mL h−1, respectively. The cell-scale (average diameters of 10–20 μm) microparticles of 23.5 μm were used for hMSC aggregates construction and the following biological function experiments.
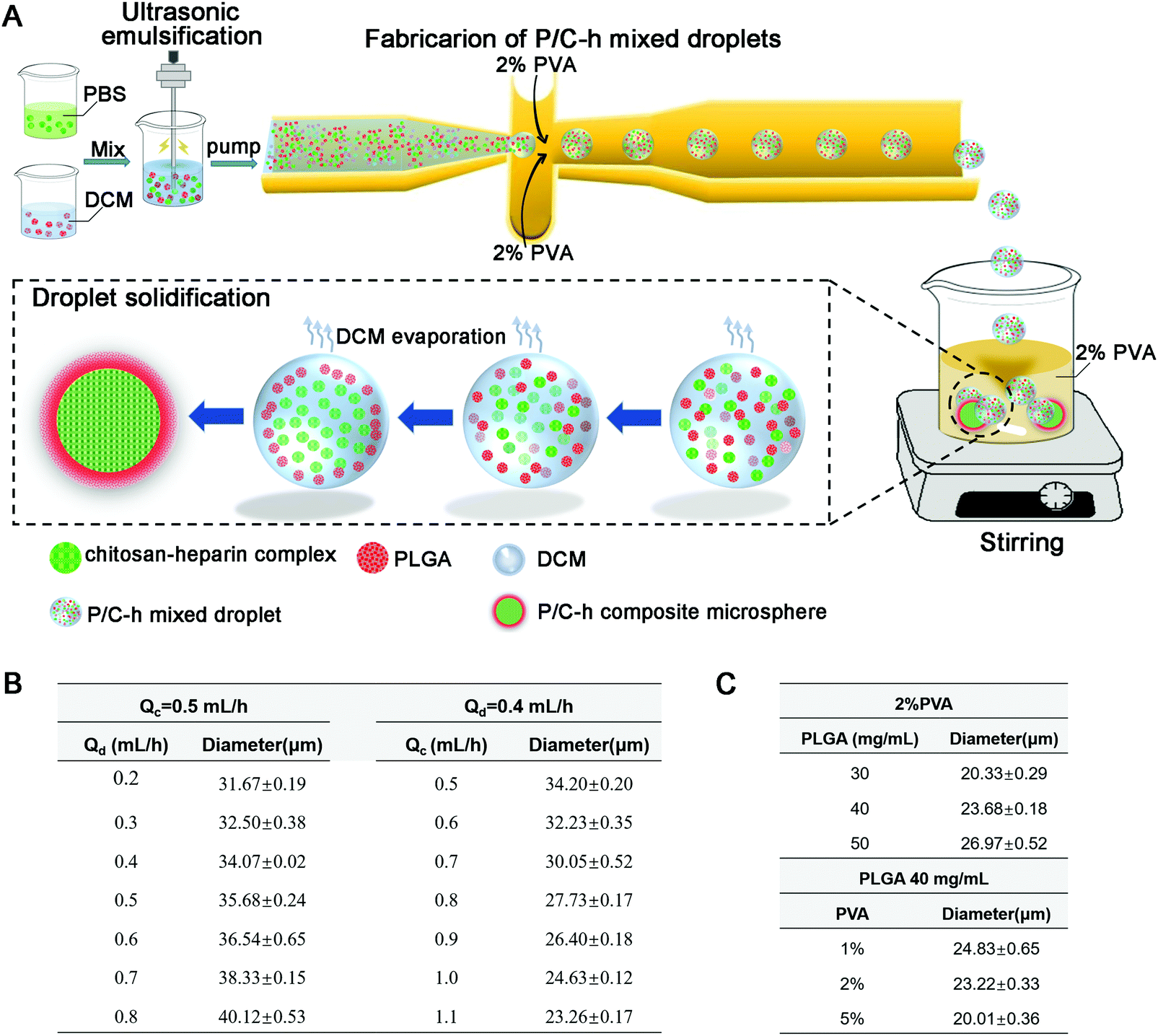 | ||
| Fig. 1 (A) Schematic illustration of P/C–h composite microparticles prepared by droplet microfluidic technology. (B) Optimization of microparticle size by regulating the flow rate of the two-phase. (C) Optimization of microparticle size by regulating the composition of the two-phase. | ||
The SEM images showed that the composite microparticles had uniform sizes, smooth surfaces and a shell-core structure (Fig. 2A and B). To visualize the distribution of PLGA and the C–h complex, the Nile red-labeled PLGA and FITC-labeled C–h complex were used to fabricate the composite microparticles. As shown in Fig. 2C, the shell of the composite microparticles was composed of PLGA, while the core was composed of the C–h complex.
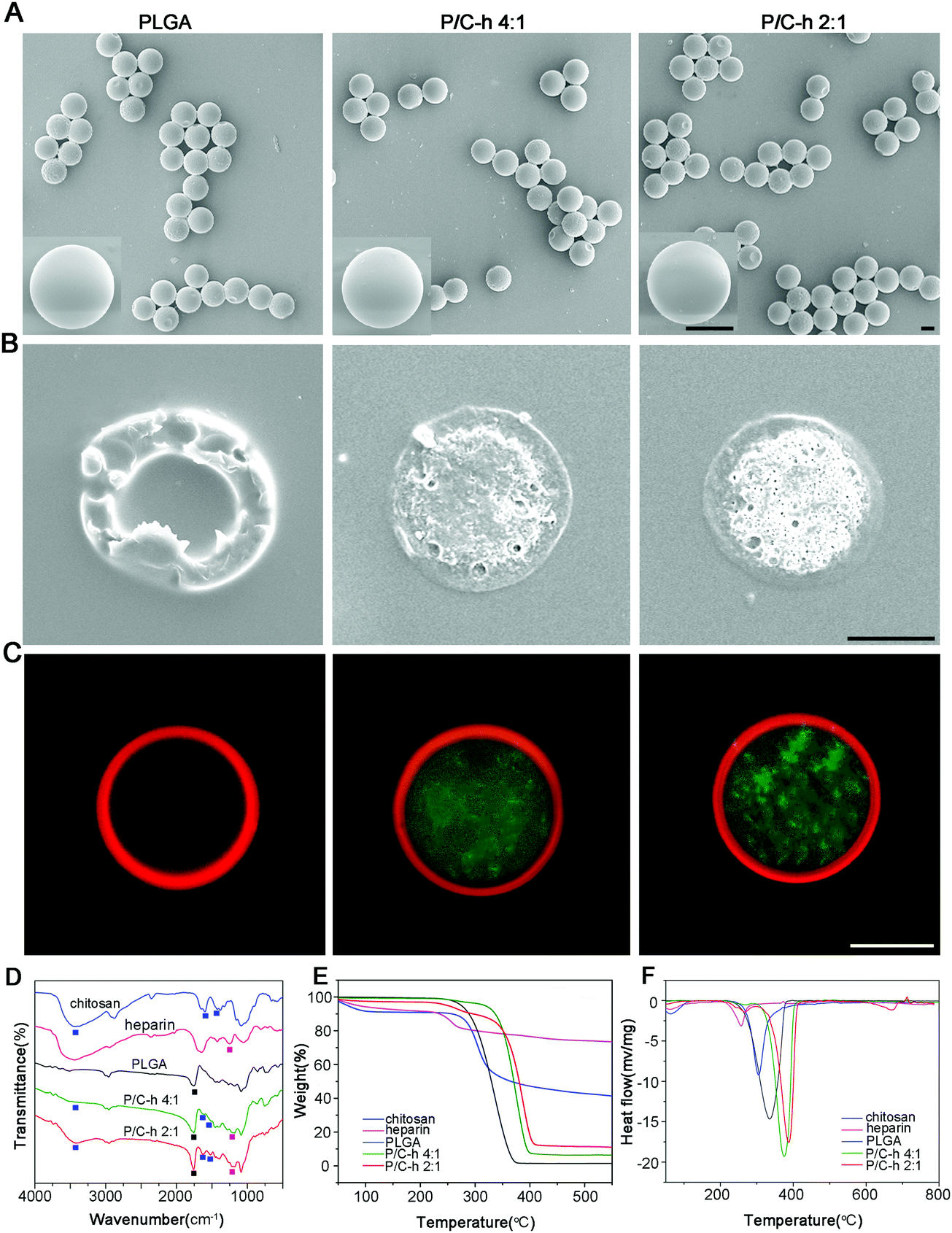 | ||
| Fig. 2 Characterization of PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles. (A) SEM images of the microparticles. (B) SEM images of the cross-sections of microparticles. (C) Confocal microscope images of the cross-sections of microparticles (red: Nile red labeled PLGA, green: FITC labeled chitosan) scale bars = 10 μm. (D) FTIR spectra. (E) TGA curves. (F) DSC analysis. | ||
The chemical compositions of the microparticles (PLGA, P/C–h 4:1, and P/C–h 2:1) were analyzed by FTIR spectra (Fig. 2D). To be specific, the peaks at 1597, 1422 and 1318 cm−1 correspond to the –NH stretching vibrations of chitosan,22 peaks at 1257 and 815 cm−1 correspond to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O–S stretching vibration of the sulfate group in heparin.23 Besides, the peak at 1755 cm−1 corresponds to the stretching vibration of the CO bond in PLGA.24 There were typical peaks of PLGA, chitosan, and heparin observed in the spectra of the P/C–h 4:1 and P/C–h 2:1 composite microparticles. In the FTIR spectrum of the P/C–h 2:1 composite microparticles, the peak at around 3500–3100 cm−1 became broader. This indicated the strong ionic interaction between chitosan and heparin.
O and C–O–S stretching vibration of the sulfate group in heparin.23 Besides, the peak at 1755 cm−1 corresponds to the stretching vibration of the CO bond in PLGA.24 There were typical peaks of PLGA, chitosan, and heparin observed in the spectra of the P/C–h 4:1 and P/C–h 2:1 composite microparticles. In the FTIR spectrum of the P/C–h 2:1 composite microparticles, the peak at around 3500–3100 cm−1 became broader. This indicated the strong ionic interaction between chitosan and heparin.
Fig. 2E showed the typical TGA curves of PLGA, chitosan and heparin. All samples exhibited a two-stage weight loss. Accordingly, two endothermic peaks were visible in the DSC analysis (Fig. 2F). During the first stage from 25 °C to 100 °C, all samples showed only slight weight loss due to the evaporation of adsorbed water from polymers. During the second stage, all samples showed considerable weight loss, which was attributed to the decomposition of the polymers. Additionally, the percentages of the weight loss in the P/C–h 2:1 composite microparticles (88.72%) were lower than that in the P/C–h 4:1 composite microparticles (93.55%) and PLGA microparticles (100%), indicating a higher content of C–h complex in the P/C–h 2:1 composite microparticles.
3.2 Degradation properties of the P/C–h composite microparticles
The SEM images of the degraded microparticles are shown in Fig. 3A. The surface of the P/C–h 2:1 composite microparticles became rough, but its shape was still basically spherical after 12 days of incubation. In contrast, the structure of the PLGA microparticles appeared fragmented and the initial spherical shape was disintegrated. After 15 days of incubation, the PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles lost 40%, 23%, and 19% of their initial weight (Fig. 3B), respectively, and the weight loss of the PLGA microparticles was significantly greater than that in the other groups. As shown in Fig. 3C, the pH value of degradation of PLGA microparticles was reduced rapidly from 7.4 to 6.2 during the 15 days. As expected, the pH values of the degradation products of the P/C–h 2:1 composite microparticles were always higher than that of the PLGA microparticles, which only decreased to 7.2 after 15 days of incubation.
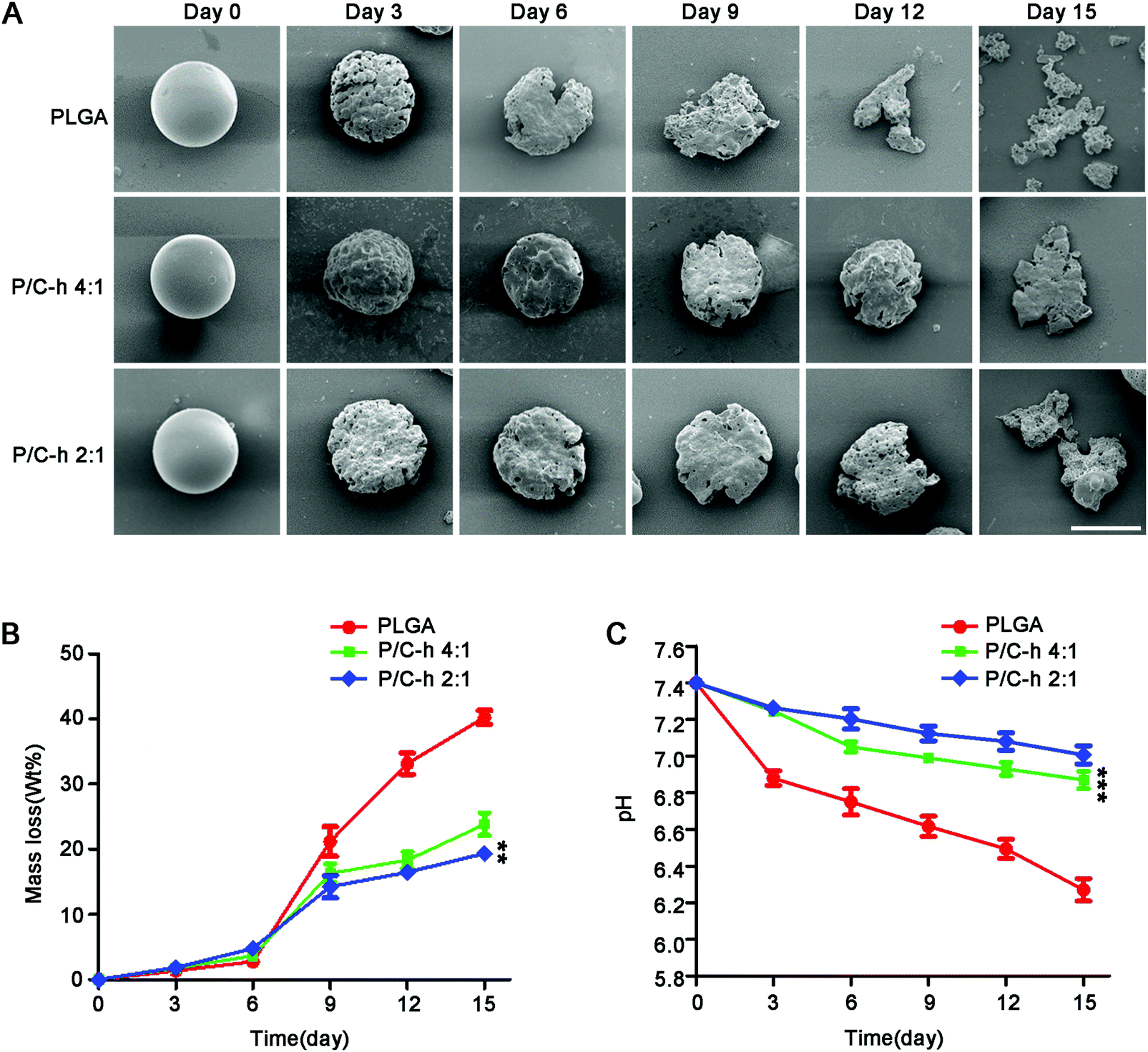 | ||
| Fig. 3 Degradation properties of PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles. (A) SEM images of the microparticles. Scale bars = 20 μm. (B) Weight loss of microparticles immersed in PBS for 0, 3, 6, 9, 12 and 15 days. (C) The pH value changes of the solutions immerging the microparticles for 0, 3, 6, 9, 12 and 15 days. The data were reported as the mean ± SD, n = 5. *** significant difference, p < 0.001. ** significant difference, p < 0.01. | ||
3.3 Cytocompatibility of the P/C–h composite microparticles
As shown in Fig. 4A, the cell activity of the PLGA group was lower than that of the TCPS group, especially on the 7th day, indicating that the degradation products of PLGA inhibited the activity of hMSCs. However, the cell activity in the P/C–h 2:1 group was considerably higher than that in the PLGA group. Consistent with the CCK-8 results, the hemolytic test results indicated that the degradation products of the PLGA microparticles caused slight hemolysis, while those of the P/C–h 2:1 composite microparticles were anhemolytic (Fig. 4B).
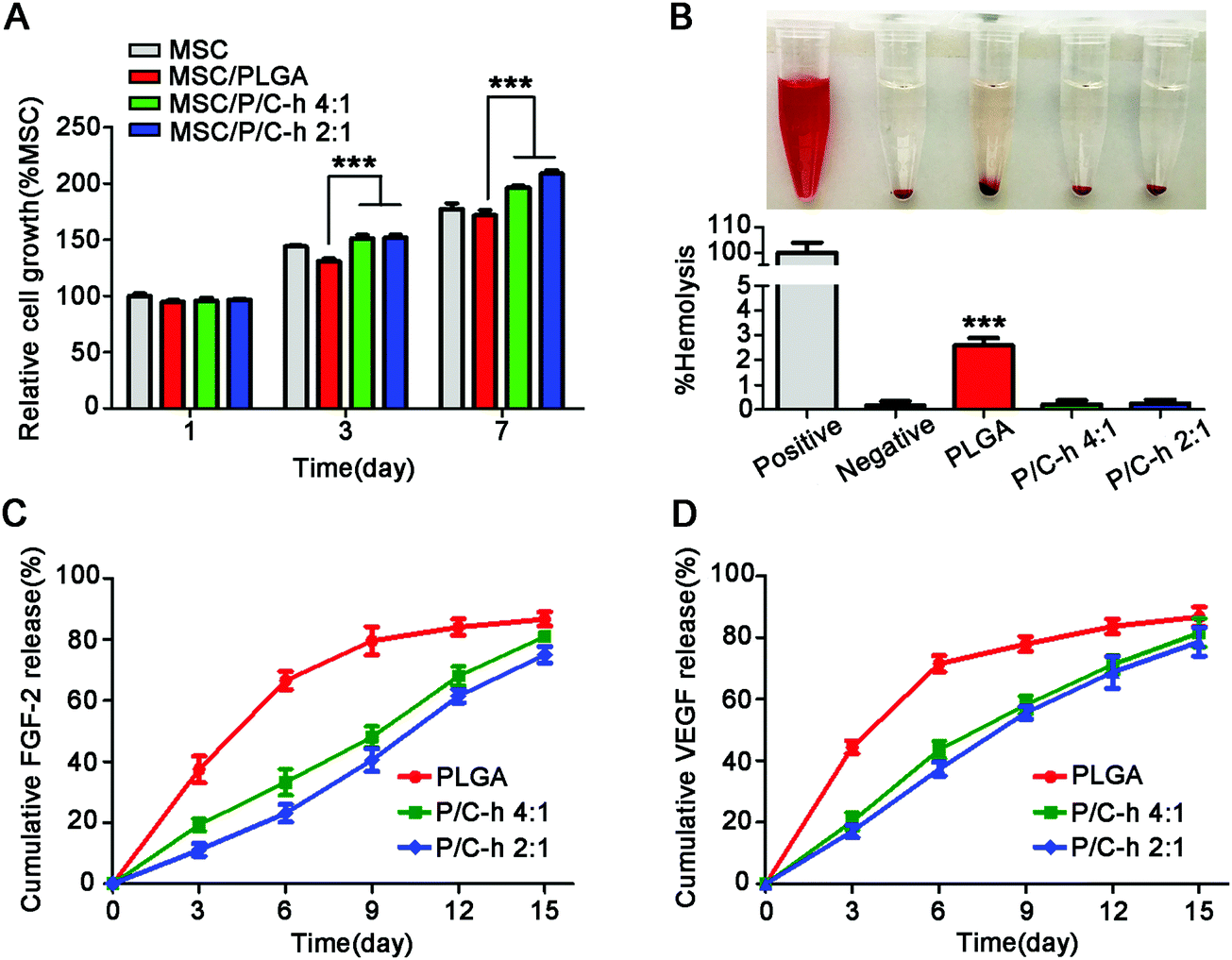 | ||
| Fig. 4 Cytocompatibility and controlled release ability of PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles. (A) CCK-8 assay of hMSCs cultured with microparticles for 1, 3, 7 days. (B) The hemolysis analysis of the degradation products. (C) The controlled release of FGF-2 from PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles. (D) The controlled release of VEGF from PLGA, P/C–h 4:1, and P/C–h 2:1 microparticles. The data were reported as the mean ± SD, n = 5. *** significant difference, p < 0.001. ** significant difference, p < 0.01 and * significant difference, p < 0.05. | ||
3.4 The release of growth factors by the P/C–h composite microparticles
Fig. 4C and D displayed the cumulative release profiles of the growth factors from microparticles over 15 days. Apparently, a high initial burst release of FGF-2 and VEGF appeared in the PLGA microparticles, i.e., approximately 37.4 ± 3.1% of FGF-2 and 44.5 ± 0.9% of VEGF were released during the first 3 days. Over 84.6 ± 2.5% of FGF-2 and 86.8 ± 1.5% of VEGF were released from the PLGA microparticles within 15 days. The growth factors were released slowly and continuously from the P/C–h 2:1 composite microparticles during the 15 days period, and finally release up to 74.9 ± 1.7% of FGF-2 or 78.6 ± 1.9% VEGF, respectively. In particular, the P/C–h 2:1 composite microparticles only released 11.2 ± 1.9% of FGF-2 and 17.5 ± 1.5% of VEGF during the first 3 days, which was significantly lower than that of the PLGA microparticles.
3.5 Effect of composite microparticles on hMSCs in the aggregates
The preparation procedure of the aggregates is shown in Fig. 5A. The PLGA microparticles and cells were initially mixed at ratios of 3:1, 1:1, and 1:3. It was found that more stable aggregates were formed when the initial addition ratio of microparticles to cells was 1:3, which was then used for the preparation of the hMSC aggregates. Under this condition, the ratio of microparticles to cells in the formed aggregates was 1:4.16 (Fig. 5B). After incubation for 12 hours, the aggregates in all groups showed a spherical structure with a dense cell layer on the surface (Fig. 5C). The diameter of the MSC aggregates was 98.6 ± 10.5 μm, whereas the diameter of the aggregates incorporated with microparticles including MSC/PLGA, MSC/P/C–h 4:1 and MSC/P/C–h 2:1, were 115.3 ± 18.8 μm, 117.2 ± 15.3 μm and 116.3 ± 18.5 μm, respectively. There was no significant difference between the diameter of the MSC aggregates with or without microparticles (p > 0.05). The H&E and F-actin/nuclei staining images of the aggregates’ sections showed that the microparticles were distributed throughout the aggregates, and were surrounded by cells (Fig. 5D and E).
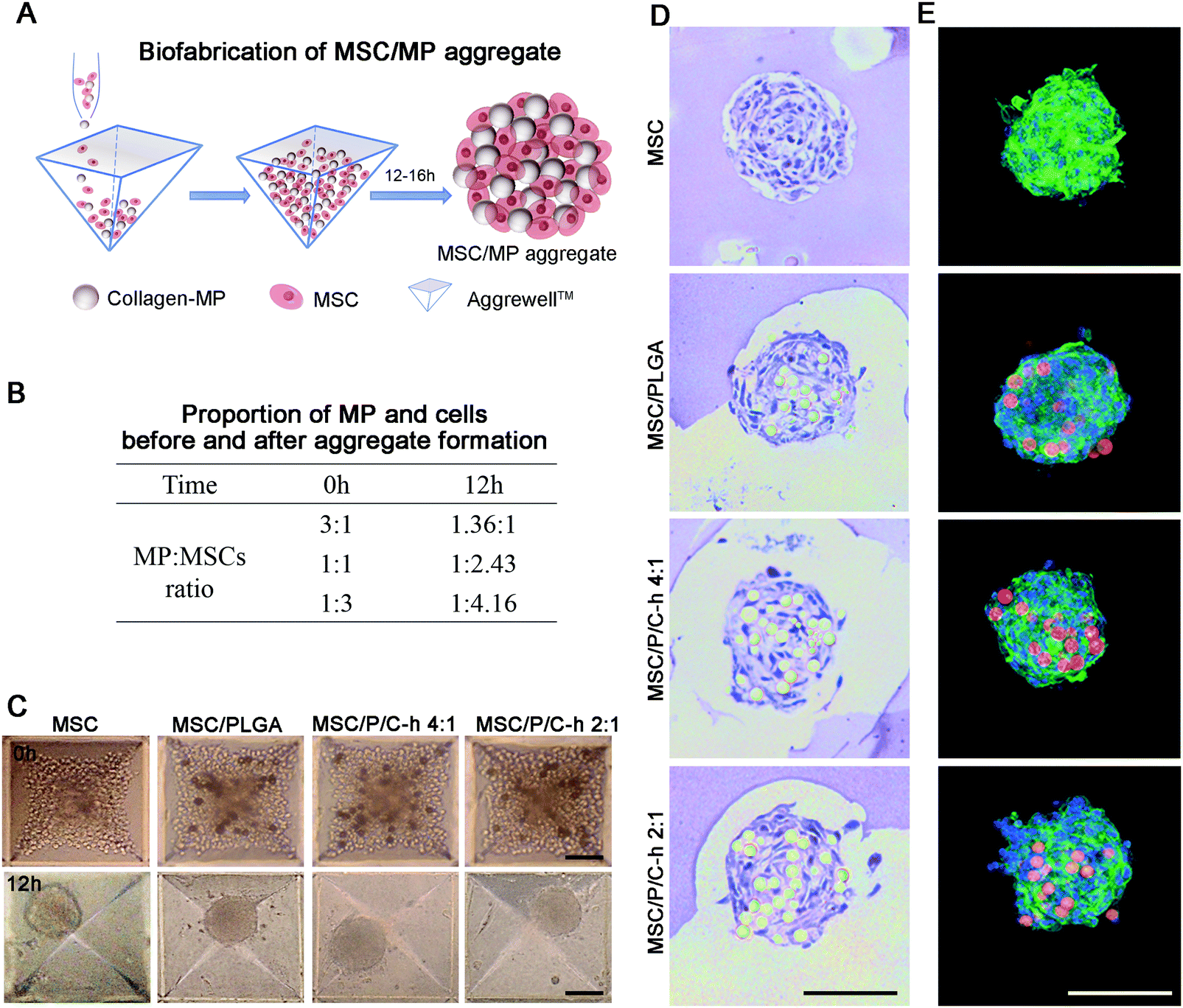 | ||
| Fig. 5 Preparation and characterization of the aggregates. (A) Schematic representation of the stepwise protocol of hMSC aggregates fabrication. (B) The quantitative statistics of the ratio of microparticles to hMSCs at different microparticles addition ratios. (C) Optical microscopy images of hMSC aggregates constructed with different microparticles (D) H&E staining and (E) F-actin/nuclei staining images of MSC, MSC/PLGA, MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates. Scale bars = 100 μm. | ||
3.6 Survival of hMSCs in the aggregates
The results of the live/dead staining study showed that there were no significant dead cells (red) detected in the MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates after culturing for 7 days (Fig. 6A). However, on day 3, several dead cells appeared in the MSC aggregates and the MSC/PLGA aggregates. As shown in Fig. 6B, the cellular viability showed the most obvious sustained increase in the MSC/P/C–h 2:1 group. In contrast, it was slightly decreased in the MSC and MSC/PLGA groups, indicating that apoptosis may have occurred in these aggregates during cultivation. Moreover, the cells in the MSC/P/C–h 2:1 group showed lower expression of apoptosis markers Bax and higher expression of anti-apoptosis marker Bcl-2 compared with other groups (Fig. 6C).
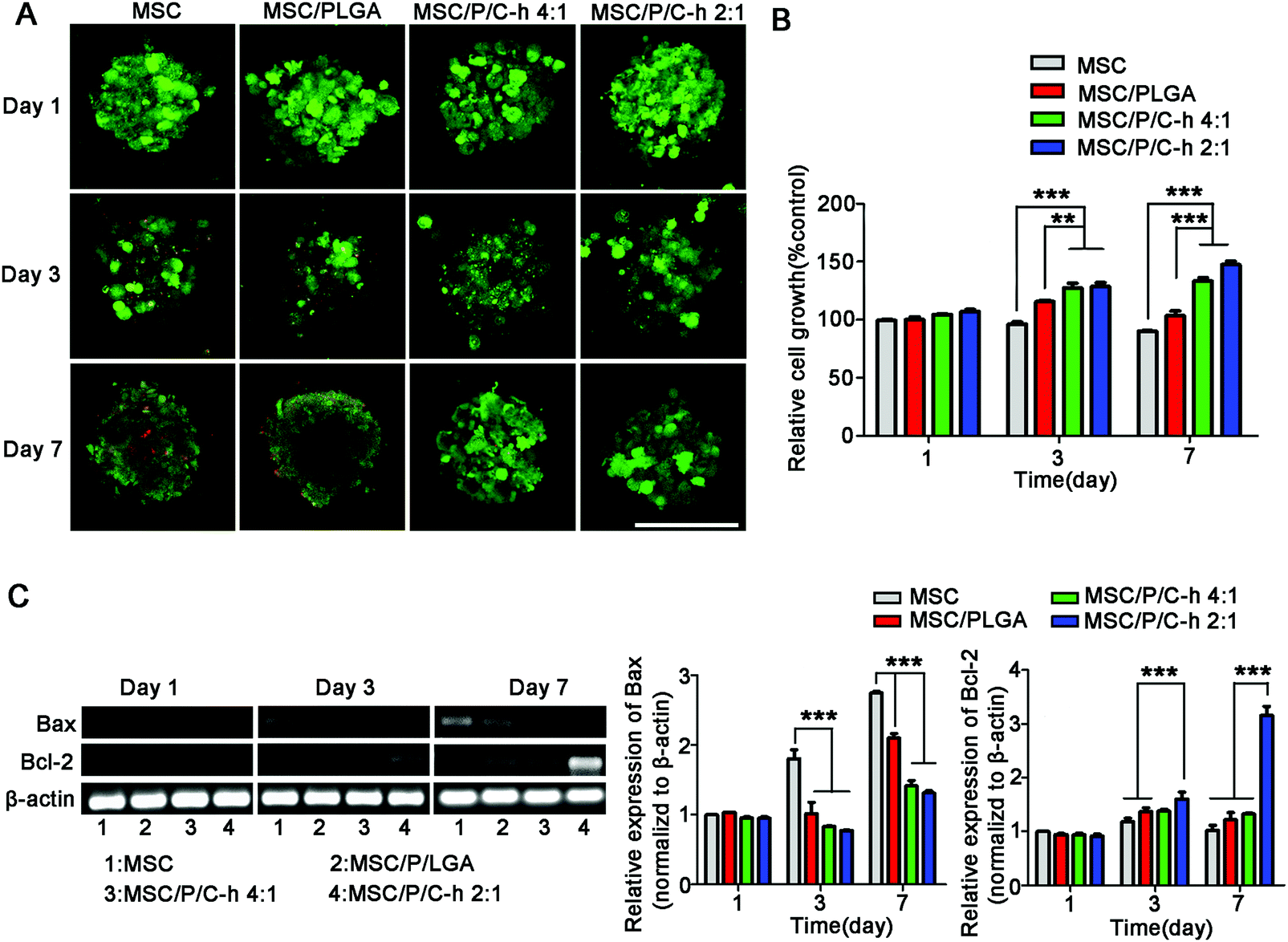 | ||
| Fig. 6 The viabilities of hMSCs in the aggregates. (A) Live/dead staining of the MSC, MSC/PLGA, MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates after cultured for 1, 3 and 7 days. Scale bars = 150 μm. (B) CCK-8 assay of the MSC, MSC/PLGA, MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates after cultured for 1, 3 and 7 days. (C) Expressions of Bax and Bcl-2 of MSC, MSC/PLGA, MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates were evaluated by PCR on day 1, 3 and 7. The expression values were normalized to β-actin. The data were reported as the mean ± SD, n = 5. *** significant difference, p < 0.001. ** significant difference, p < 0.01 and * significant difference, p < 0.05. | ||
3.7 Biological functions maintenance of hMSCs in the aggregates
As shown in Fig. 7A, the expressions of the stemness markers, including Nanog and Sox-2, of the cells in the MSC/P/C–h 2:1 aggregates were steadily maintained, while those in the MSC and MSC/PLGA aggregates were significantly decreased.
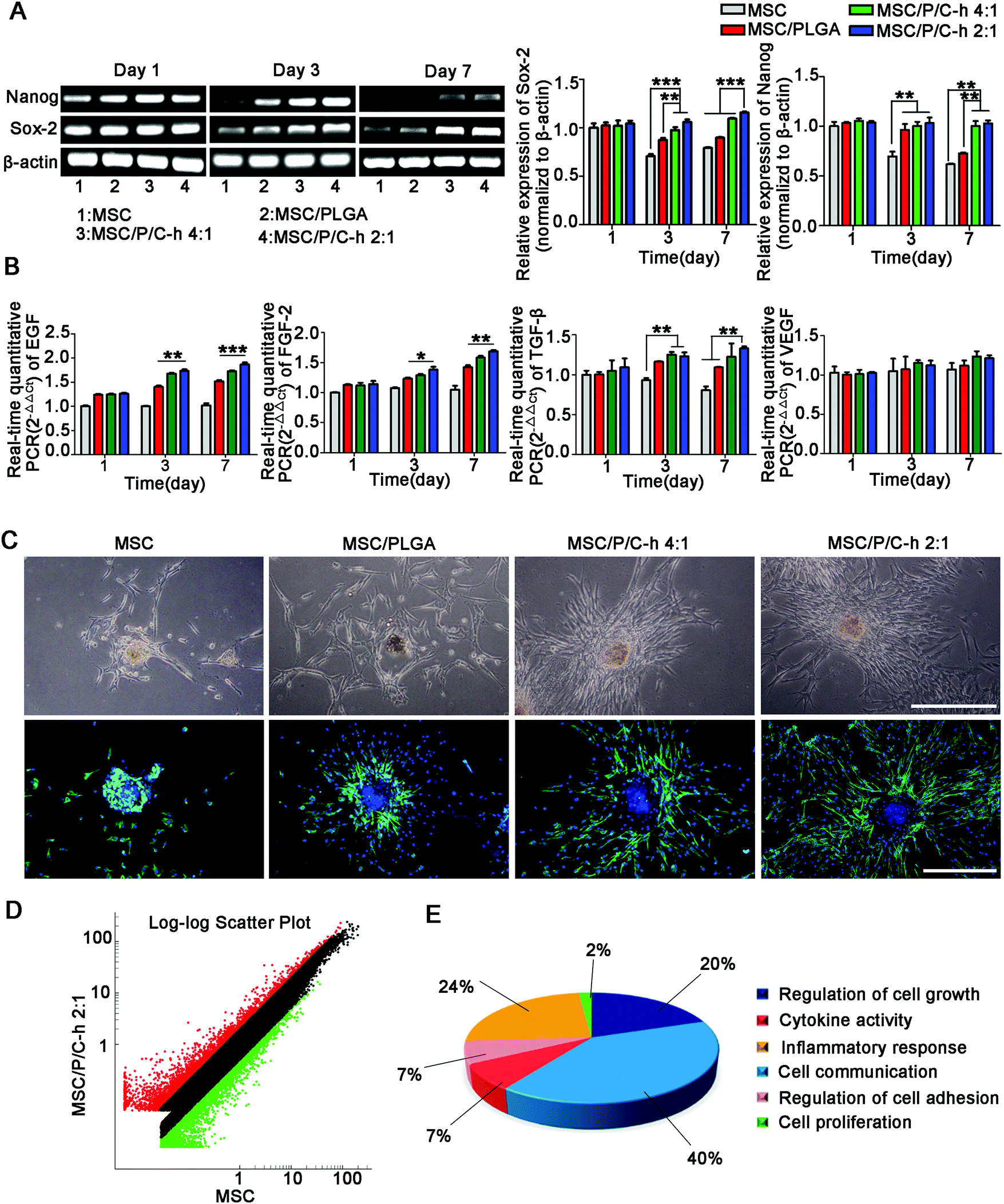 | ||
| Fig. 7 The biological activities of hMSCs in the aggregates. (A) Expression of Nanog and Sox-2 of MSC, MSC/PLGA, MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates on day 1, 3, and 7 were analysis by PCR. (B) Expression of EGF, FGF-2, TGF-β and VEGF of MSC, MSC/PLGA, MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates on day 1, 3, and 7 were analysis by RT-PCR. The data were reported as the mean ± SD, n = 5. *** significant difference, p < 0.001. ** significant difference, p < 0.01. * significant difference, p < 0.05. (C) Optical microscopy and F-actin/nuclei staining images of hMSCs migrated from the MSC, MSC/PLGA, MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates after cultured for 3 days. Scale bars = 200 μm. (D) Volcano plot of the differently expressed genes between MSC and MSC/P/C–h 2:1 aggregates. (E) Statistical distribution of the enriched gene ontology terms. | ||
The secretion of various cytokines is an extremely important function of MSCs for the regulation of trauma recovery and tissue regeneration.25,26 Here, the mRNA expressions of epidermal growth factor (EGF), FGF-2, transforming growth factor (TGF-β) and VEGF were selected to evaluate the biological functions of hMSCs in the aggregates. The expressions of EGF, FGF-2 and TGF-β detected in the aggregates incorporating the P/C–h composite microparticles were significantly higher than the MSC and MSC/PLGA aggregates from day 3 to day 7 (Fig. 7B).
After being cultured on the matrigel for 3 days, the hMSCs migrated out from the aggregates incorporating microparticles, while the MSC aggregates still maintained a spherical shape (Fig. 7C). Compared to the MSC/PLGA aggregates, more fibroblast-like cells were detected in the MSC/P/C–h 4:1 and MSC/P/C–h 2:1 aggregates, whereas the migration area of the cells in the MSC/P/C–h 2:1 group was significantly higher than that in the MSC/P/C–h 4:1 group (Fig. S2, ESI†). These results indicated that the hMSCs migrating from the MSC/P/C–h 2:1 aggregates had the highest cellular activity among these groups.
To further evaluate the effect of the P/C–h 2:1 composite microparticles on the biological functions of hMSCs in the aggregates, the transcriptional profiling was analyzed using the Agilent Gene Chip. The results showed that 161 genes were upregulated and 55 genes were downregulated in the MSC/P/C–h 2:1 aggregates compared with the MSC aggregates (Fig. 7D and Table S1, ESI†). The gene ontology enrichment analysis identified 6 biological functions, including the regulation of cell growth and adhesion, cytokine activity, cell communication and inflammatory response (the lowest p-value = 9.05 × 10−3) (Fig. 7E and Table S2, ESI†).
4. Discussion
The incorporation of microparticles into cell aggregates has allowed the further development of the 3D cell culture. The cell-scaled polymer microparticles as ‘synthetic neighbors’ incorporated into cell aggregates have become a powerful tool to direct the cellular response by maintaining the diffusion of nutrients, cytokines, and small molecules.27 The chemical/physical and architectural features of the microparticles are closely correlated with the cellular response.4 The PLGA microparticles that are integrated within the cell aggregates enable the localized and continuous delivery of soluble diffusing cues, such as proteins and small molecules, which can better bio-mimic the reconstruction of the ‘physiological tissues’ microenvironment in a spatially and temporally relevant manner.28 The design of these microparticles should take the influence of the physicochemical properties of the biomaterials on cellular biological functions into consideration, but also the effect on the local microenvironment.29The shell–core structure of the microparticles has been widely used in tissue engineering for encapsulating the ‘targets’ into two distinct components.30 Here, the composite microparticles with a shell–core structure of PLGA-encapsulated C–h complex (P/C–h) were prepared for the construction of the hMSC aggregates. The C–h complex was distributed (aqueous solution) in emulsion droplets (PLGA in DCM) at first and subsequently converged to the inside of the microparticles. This led to the formation of a shell–core structure (Fig. 1A) due to the different interfacial tension between the PLGA in DCM solution and the C–h complex in aqueous solution.31 Along with the evaporation of DCM, the internal phase separation process was accelerated by the external fusion of PLGA and the internal fusion of the C–h complex. Thus, the monodispersed uniform P/C–h composite microparticles with shell–core structure were generated (Fig. 2B). This core–shell structure would allow the loading of two distinct components, such as model drugs and growth factors, and release at a controlled rate.31 The PLGA shell can protect the inner components from dissolution or hydrolysis,32 as well as provide active sites for the conjugation with the extracellular matrix (ECM) via Schiff's base substitution or Michael addition reactions,33 and for the immobilization of the fusion protein via hydrophobic bond.19
It was reported that chitosan can neutralize the acidic degradation products of PLGA by its NH2 base, while heparin can control the release of various growth factors due to its high affinity with them.15,34 As shown in Fig. 3, the acidic microenvironment generated by the degradation of PLGA has been mitigated by the addition of the C–h complex, and the biocompatibility was also significantly enhanced compared with that of the PLGA microparticles. Furthermore, the release of the growth factors by the P/C–h composite microparticles was also more stable compared with PLGA microparticles (Fig. 4). These results indicated that the C–h complex in the core of the composite microparticles significantly improved the expected functions of the PLGA microparticles for stem cell applications.
The size distribution of the microparticles is crucial for biomedical applications because the uniform microparticles ensure the consistency in the degradation rate of microparticles and the release profiles of the loaded cytokines.35 In droplet microfluidic technology, the design of the microchannels is important to the structure and stability of the generated microparticles.36 Here, we applied a flow-focusing microchannel for the preparation of the P/C–h composite microparticles. The uniform P/C–h composite microparticles were fabricated due to the large shear stress of the microchannel (Fig. 2A and B), which was beneficial to the homogeneous distribution of the loaded cytokines in the cell aggregates.37,38 The size of these microparticles in this system could be controlled by regulating the flow rate between the two-phase in the microfluidic microchannel (Fig. 1B). Otherwise, the size of the microparticles can also be adjusted by tuning the concentrations of the two-phase (Fig. 1C). Reducing the concentration of the dispersed phase could lead to decreased viscous stress. This makes the dispersed phase flow break up more easily, and the smaller sizes of the microparticles were ultimately generated. Similarly, increasing the concentration of the continuous phase may result in an increase in the shear stress, as more PVA is added to produce a greater viscosity.39 Through adjusting the parameters above, the size, structure, and composition of the microparticles can be tailored in a highly controllable manner to accomplish specific functions for biomedical applications.
In recent years, 3D cell aggregates have become a promising strategy to replace the well-established two-dimensional cell culture approaches.40 However, the acidic degradation products of PLGA would lead to the removal of certain ionic species, such as calcium and iron ions.41,42 This causes a dysfunction in the divalent metallic cation-dependent cell adhesion molecule, such as the cadherins of adhesion junctions, which is a core molecule that mediates cell aggregation.43,44 Besides, the iron chelation would affect a variety of cellular biochemical pathways, including the Kreb's cycle and oxidative phosphorylation in the mitochondria, and have the greatest effect on the cell cycle.45 In short, even though the degradation products of PLGA are known to be largely noncytotoxic, the altered pH value of the microenvironment would still decrease the cell proliferation and accelerated cell differentiation. This is not conducive to the precise regulation of the cell behavior in stem cell aggregates and the full realization of cell functions.
This neutral microenvironment in the aggregates after the degradation of the P/C–h 2:1 composite microparticles was much more beneficial to the survival of hMSCs (Fig. 6A and B) compared with the PLGA microparticles, which was also related to the activation of the Bax/Bcl-2 pathway (Fig. 6C). Moreover, the addition of the C–h complex mitigated the adverse effects of the PLGA microparticle degradation on cellular functions, including significantly preserving the stemness (Fig. 7A), increasing the secretion (Fig. 7B), and elevating the migration capacity (Fig. 7C) of hMSCs in the aggregates. Furthermore, the transcriptional profiling results showed that the P/C–h 2:1 composite microparticles could modulate the microenvironment in the aggregates and induce highly localized responses, such as cell growth and adhesion, cytokine activity, cell communication and inflammatory response (Fig. 7D and E).
Therefore, the P/C–h 2:1 composite microparticles are suitable for the immobilization of ECM-derived proteins to replicate the cell matrix signaling for promoting cell adhesion, or the cellular adhesion molecules to modify them into ‘synthetic cells’ for improving their abilities to direct and modulate the cellular responses and regulate the stem-cell fate.
5. Conclusions
In this work, the P/C–h composite microparticles were fabricated using droplet microfluidic technology and were proved to be able to improve the microenvironment by neutralizing acidic products produced by the degradation of PLGA. Meanwhile, the stemness and cell activity of hMSCs in the aggregates were maintained after incorporation with the P/C–h composite microparticles. Taken together, the P/C–h composite microparticles have the potential to be used as a drug delivery system for the 3D culture of hMSCs in the field of regenerative medicine. Moreover, these P/C–h composite microparticles would be particularly appropriate to support the 3D stem cell aggregate culture, and to integrate more targeted functionality into in vitro organ-on-a-chip models.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant No. 31771067), the Youth Science Fund of the National Nature Science Foundation of China (Grant No. 81703094), and the National Key Research and Development Program of China (No. 2016YFC1101304).Notes and references
- X. Wei, X. Yang, Z. P. Han, F. F. Qu, L. Shao and Y. F. Shi, Acta Pharmacol. Sin., 2013, 34, 747–754 CrossRef CAS.
- Z. Y. Lu, X. Jiang, M. Q. Chen, L. Feng and Y. J. Kang, Biofabrication, 2019, 11, 1–41 Search PubMed.
- S. Sart, T. Ma and Y. Li, BioReserach Open Access, 2014, 3, 137–149 CrossRef CAS.
- S. Sart, A. C. Tsai, Y. Li and T. Ma, Tissue Eng., Part B, 2014, 20, 365–380 CrossRef.
- T. J. Bartosh, J. H. Ylostalo, N. Bazhanov, J. Kuhlman and D. J. Prockop, Stem Cells, 2013, 31, 2443–2456 CrossRef CAS.
- V. W. Ap, I. D. Gates and M. Kallos, Cells Tissues Organs, 2012, 196, 34–47 CrossRef.
- C. C. Ahrens, Z. Dong and W. Li, Acta Biomater., 2017, 62, 64–81 CrossRef CAS.
- G. D. Duraine, W. E. Brown, J. C. Hu and K. A. Athanasiou, Ann. Biomed. Eng., 2015, 43, 543–554 CrossRef.
- J. C. Gomez, J. M. Edgar, A. M. Agbay, E. Bibault, A. Montgomery, N. K. Mohtaram and S. M. Willerth, Cell. Mol. Bioeng., 2015, 8, 307–319 CrossRef CAS.
- L. Y. Jiang, B. Lv and Y. Luo, Biomaterials, 2013, 34, 2665–2673 CrossRef CAS.
- P. Gentile, V. Chiono, I. Carmagnola and P. V. Hatton, Int. J. Mol. Sci., 2014, 15, 3640–3659 CrossRef CAS.
- W. J. Habraken, J. G. Wolke, A. G. Mikos and J. A. Jansen, J. Biomater. Sci., Polym. Ed., 2008, 19, 1171–1188 CrossRef CAS.
- B. S. Zolnik and D. J. Burgess, J. Controlled Release, 2007, 122, 338–344 CrossRef CAS.
- J. A. Edson, D. Ingato, S. Wu, B. Lee and Y. J. Kwon, Biomacromolecules, 2018, 19, 1508–1516 CrossRef CAS.
- S. E. S. Elbert, Acta Biomater., 2014, 10, 1581–1587 CrossRef.
- Y. Y. Yang, H. H. Chia and T. S. Chung, J. Controlled Release, 2000, 69, 81–96 CrossRef CAS.
- I. U. Khan, C. A. Serra, N. Anton and T. F. Vandamme, Expert Opin. Drug Delivery, 2015, 12, 547–562 CrossRef.
- H. T. Liu, H. Wang, W. B. Wei, H. Liu, L. Jiang and J. H. Qin, Small, 2018, 14, 1–9 Search PubMed.
- C. Deblois, M. Cote and C. J. Doillon, Biomaterials, 1994, 15, 665–672 CrossRef CAS.
- A. Drogoz, L. David, C. Rochas, A. Domard and T. Delair, Langmuir, 2007, 23, 10950–10958 CrossRef CAS.
- Y. Zhang, H. L. Mao, C. Gao, S. H. Li, Q. Z. Shuai, J. B. Xu, K. Xu, L. Cao, R. Lang, Z. W. Gu, T. Akaike and J. Yang, Adv. Healthcare Mater., 2016, 5, 1949–1959 CrossRef CAS.
- J. J. Fang, Y. Zhang, S. F. Yan, Z. W. Liu, A. M. He, L. Cui and J. B. Yin, Acta Biomater., 2014, 10, 276–288 CrossRef CAS.
- N. B. Skop, F. Calderon, S. W. Levison, C. D. Gandhi and C. H. Cho, Acta Biomater., 2013, 9, 6834–6843 CrossRef CAS.
- C. T. Tsao, C. H. Chang, Y. Y. Lin, M. F. Wu, J. L. Wang, J. L. Han and K. H. Hsieh, Carbohydr. Res., 2010, 345, 1774–1780 CrossRef CAS.
- F. Barry and M. Murphy, Nat. Rev. Rheumatol., 2013, 9, 584–594 CrossRef CAS.
- Y. F. Shi, Y. Wang, Q. Li, K. Liu, J. Q. Hou, C. S. Shao and Y. Wang, Nat. Rev. Nephrol., 2018, 14, 493–507 CrossRef CAS.
- J. L. Roam, Y. Yan, P. K. Nguyen, I. S. Kinstlinger, M. K. Leuchter, D. A. Hunter, M. D. Wood and D. L. Elbert, Biomaterials, 2015, 72, 112–124 CrossRef CAS.
- Y. J. Li, C. Li, A. Mujeeb, Z. X. Jin and Z. G. Ge, Mater. Lett., 2015, 154, 68–72 CrossRef CAS.
- W. Leong and D. Wang, Trends Biotechnol., 2015, 33, 653–666 CrossRef CAS.
- Z. J. Lin, J. Wu, W. Qiao, Y. Zhao, K. H. Wong, P. K. Chu, L. M. Bian, S. L. Wu, Y. F. Zheng, K. M. Cheung, F. Leung and K. W. Yeung, Biomaterials, 2018, 174, 1–16 CrossRef CAS.
- N. G. Min, M. Ku, J. Yang and S. Kim, Chem. Mater., 2016, 28, 1430–1438 CrossRef CAS.
- M. S. Fleming, T. K. Mandal and D. R. Walt, Chem. Mater., 2001, 13, 2210–2216 CrossRef CAS.
- S. Pathak, S. Regmi, P. Shrestha, I. Choi, K. O. Doh and J. H. Jeong, Small, 2019, 15, 1–13 CrossRef.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 1997, 28, 5–24 CrossRef CAS.
- W. Liu, W. Wu, C. Selomulya and X. D. Chen, Soft Matter, 2011, 7, 3323–3330 RSC.
- J. Li, T. L. Lam, J. P. W. Toh, S. Reuveny, S. K. W. Oh and W. R. Birch, Biomed. Microdevices, 2015, 17, 105.1–105.12 Search PubMed.
- C. Qiu, M. Chen, H. Yan and H. Wu, Adv. Mater., 2007, 19, 1603–1607 CrossRef CAS.
- H. Zhang, E. Tumarkin, R. Peerani, Z. Nie, R. Sullan, G. Walker and E. Kumacheva, J. Am. Chem. Soc., 2006, 128, 12205–12210 CrossRef CAS.
- L. Zhu, Y. Li, Q. Zhang, H. Wang and M. Zhu, Biomed. Microdevices, 2010, 12, 169–177 CrossRef.
- N. T. Elliott and F. Yuan, J. Pharm. Sci., 2011, 100, 59–74 CrossRef CAS.
- F. P. Cavasino, E. D. Dio and C. Sbriziolo, Adv. Mol. Relax. Interact. Processes, 1982, 23, 191–201 CrossRef CAS.
- X. Wang, Med. Hypotheses, 1999, 53, 380–382 CrossRef CAS.
- C. M. Ditre, T. D. Griffin, G. F. Murphy, H. Sueki, B. Telegan, W. C. Johnson, O. R. Yu and E. J. Scott, J. Am. Acad. Dermatol., 1996, 34, 187–195 CrossRef CAS.
- S. M. Burge, Br. J. Dermatol., 2006, 131, 153–159 CrossRef.
- F. Meyer, J. Wardale, S. Best, R. Cameron, N. Rushton and R. Brooks, J. Orthop. Res., 2012, 30, 864–871 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb01593h |
| This journal is © The Royal Society of Chemistry 2020 |