
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of analytical assay performance of antibody-gated indicator-releasing mesoporous silica particles†
Elena
Costa
 ,
Estela
Climent
,
Kornelia
Gawlitza
,
Wei
Wan‡
,
Michael G.
Weller
and
Knut
Rurack
*
,
Estela
Climent
,
Kornelia
Gawlitza
,
Wei
Wan‡
,
Michael G.
Weller
and
Knut
Rurack
*
Bundesanstalt für Materialforschung und prüfung (BAM), Richard-Willstätter-Str. 11, D-12489 Berlin, Germany. E-mail: knut.rurack@bam.de
First published on 29th May 2020
Abstract
Antibody-gated indicator delivery (gAID) systems based on mesoporous silica nano- and microparticle scaffolds are a promising class of materials for the sensitive chemical detection of small-molecule analytes in simple test formats such as lateral flow assays (LFAs) or microfluidic chips. Their architecture is reminiscent of drug delivery systems, only that reporter molecules instead of drugs are stored in the voids of a porous host particle. In addition, the pores are closed with macromolecular “caps” through a tailored “gatekeeping” recognition chemistry so that the caps are opened when an analyte has reacted with a “gatekeeper”. The subsequent uncapping leads to a release of a large number of indicator molecules, endowing the system with signal amplification features. Particular benefits of such systems are their modularity and adaptability. With the example of the immunochemical detection of type-I pyrethroids by fluorescent dye-releasing gAID systems, the influence of several tuning modes on the optimisation of such hybrid sensory materials is introduced here. In particular, different mesoporous silica supports (from nano- and microparticles to platelets and short fibres), different functionalisation routes and different loading sequences were assessed. The materials’ performances were evaluated by studying their temporal response behaviour and detection sensitivity, including the tightness of pore closure (through the amount of blank release in the absence of analyte) and the release kinetics. Our results indicate that the better the paratope-accommodating Fab region of the antibody “cap” fits into the host material's pore opening, the better the closing/opening mechanism can be controlled. Because such materials are well-suited for LFAs, performance assessment included a test-strip format besides conventional assays in suspension. In combination with dyes as indicators and smartphones for read-out, simple analytical tests for use by untrained personnel directly at a point-of-need such as an aeroplane cabin can be devised, allowing for sensitivities down to the μg kg−1 range in <5 min with case-required selectivities.
Introduction
The empowerment of interested individuals and employees or co-workers of civil services, commercial enterprises or non-governmental organisations to carry out reliable and relevant analytical measurements directly at a point-of-need is one of the current drivers for the development of miniaturised analytical systems, quick tests and wearables.1–7 As in laboratory-based analysis, the quality and acceptance of a test or device are governed by its sensitivity, selectivity, immunity against false positives and false negatives as well as robustness and repeatability. In recent years, gated indicator-releasing micro- and nanoparticles have become a promising approach in the field because they allow reaching rather low limits of detection already in comparatively simple assay formats due to their inherent features of signal amplification.8,9 Conceptually, gated indicator-releasing materials are closely related to drug delivery systems.10–13 Highly porous carrier particles are loaded with cargo molecules that are released in the designated operational action. However, in contrast to drug delivery systems, indicator release systems contain indicators or reporters instead of drugs, and their pores are usually closed with macromolecular “caps”. The latter are attached to the outer surface of the carrier particle through a dedicated “gatekeeping” recognition chemistry. One of the partners of this gatekeeping chemistry reacts with the analyte upon its arrival, the cap is displaced, the pore opened, and a large number of indicator molecules are released. Besides architectural similarities, the key difference between the two types of functional materials is that many drug delivery systems should deliver their cargo over a longer period, often many hours. In contrast, their biosensor counterparts should show fast release kinetics, on the order of <5 min. Also, the blank or non-specific release of indicators in the absence of an analyte should be optimally zero, avoiding false positives or signal drifts. It is evident that optimisation procedures and system fine-tuning are supposedly different for both types of materials. However, most examples on gated indicator-releasing particles reported in the literature so far show rather slow kinetics of ≥20 min until a stable signal/plateau is reached.9,14A particularly attractive approach in this field is the use of an immunochemical gating mechanism as it combines high modularity and a generic design with the intrinsic features of immunoassays, i.e., high sensitivity and selectivity. Regarding antibody-gated indicator delivery (gAID) systems, antibodies have been used by others and us in both roles, i.e. as caps and as gatekeepers.15–21 Aiming for simplicity, the use of antibodies as caps seems more appealing because they do not have to be chemically modified for covalent grafting to a particle surface. If one considers the size of antibodies and their structural subunits22 as well as the pore sizes of porous silica materials,23 it seems evident that the combination will determine the performance of the system. However, comprehensive studies in this direction are lacking. In this work, several gAID materials for small-molecule sensing were thus prepared, altering system parameters such as pore diameter, localisation of grafting/functionalisation and loading sequence. The detection of permethrin, a type-I pyrethroid, and 3-phenoxybenzoic acid (3-PBA, Scheme 1), the hapten used for antibody generation, served as the analytical model applications because type-I pyrethroids play an essential role in aeroplane disinfection,24,25 their measurement via simple and rapid tests by flight staff during stopover times of aeroplanes becoming increasingly important. Whereas the best performing system has been assessed by us recently for its intended purpose,26 the present contribution discusses a first in-depth study of the various chemical tuning options of such gAID systems with the final aim of obtaining a potent lateral flow assay for pesticide detection. Whenever working with commercial antibodies like in the present case in which a monoclonal antibody against type-I pyrethroids is commercially available,27–29—and thus not having access to different clones, i.e., to antibodies with different affinities21—chemical system tuning is the most efficient and best possible way to optimize such hybrid materials regarding their analytical performance.
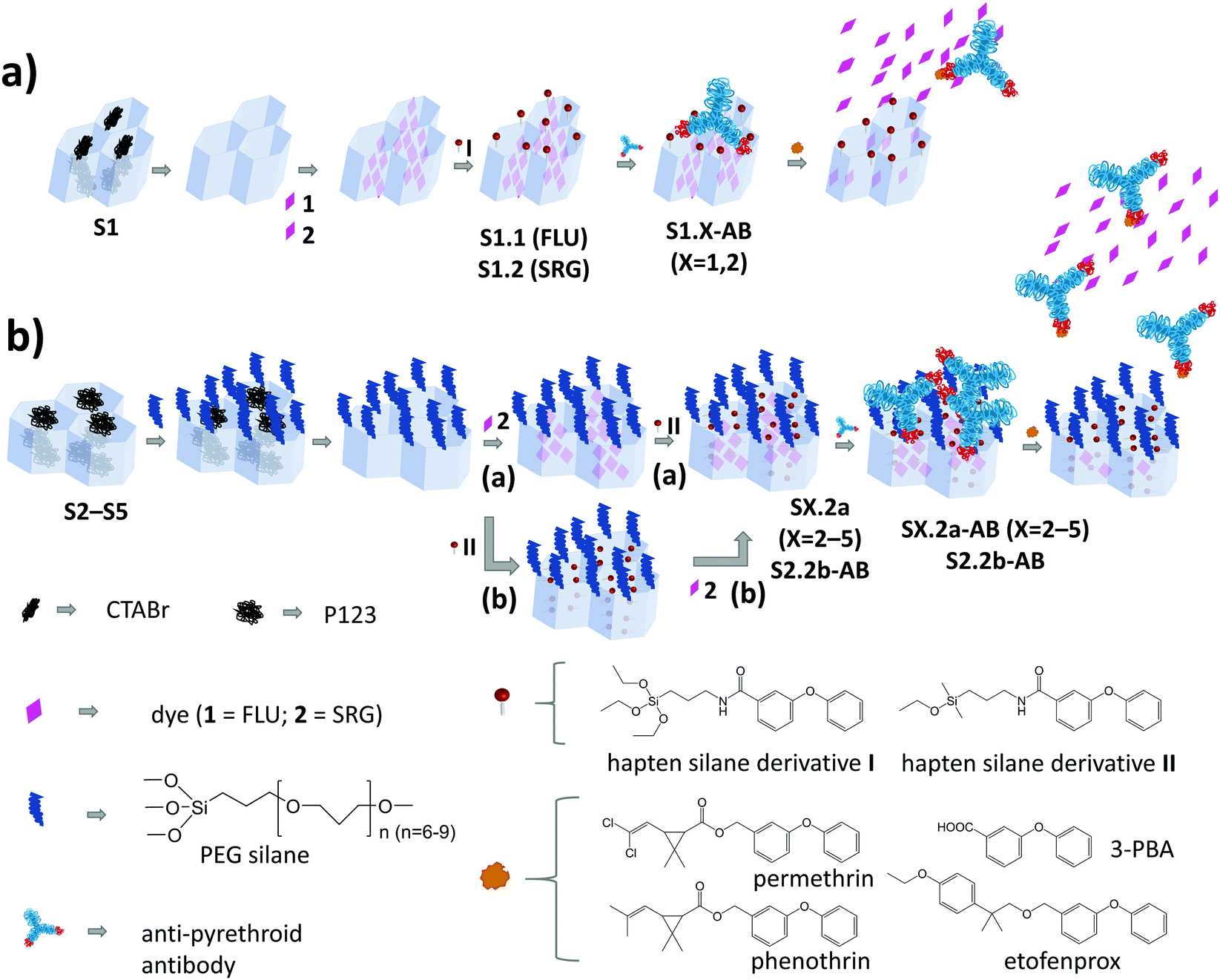 | ||
| Scheme 1 Architecture and synthesis routes of the antibody-gated mesoporous materials (a) containing MCM-41 (S1) as mesoporous scaffold and (b) S2–S5 containing different types of SBA scaffolds (S2: SBA-15, conventional long fibres; S3: SBA-15 platelets; S4: SBA-15 long fibres with enlarged pores; S5: SBA-16 nanoparticles). | ||
Experimental
Synthesis of 3-phenoxybenzoic acid silane derivatives I and II
Hapten derivative I was obtained through an amidation reaction of 3-PBA and (3-aminopropyl)triethoxysilane (APTES) using a modification of the active ester method following described procedures.30 Synthetic details are given in the ESI.† Hapten derivative II was obtained along with a similar procedure but using 3-(ethoxydimethylsilyl)propylamine instead of APTES.26Synthesis of mesoporous silica materials
MCM-41 (Mobil Composition of Matter No. 41) nanoparticles (S1),31 long SBA-15 (Santa Barbara Amorphous type material no. 15) fibres with narrow pores (S2),26,32 SBA-15 platelets (S3),33 long SBA-15 fibres with larger pores (S4)34 and SBA-16 microparticles (S5)35 were prepared according to the respective reports in the literature. Whereas S1 is a calcinated material, S2–S5 still contain the templating surfactants in their pores. A fraction of the latter materials was also calcinated to facilitate characterisation by transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM) and nitrogen adsorption/desorption measurements. Synthetic details are given in the ESI.†Synthesis of S1.1 and S1.2
To load the dyes into the pores of the carriers, 185 mg of the respective particles were suspended in an acetonitrile (MeCN) solution containing either 2,7-dichlorofluorescein (FLU) for S1.1 or sulphorhodamine G (SRG) for S1.2, both at 800 μmol dye g−1 particles and the suspensions were stirred overnight at 45 °C. Thereafter, 63 μL (37.5 μmol) of the hapten derivative I in tetrahydrofuran (THF) solution were added, and stirring was continued for 5.5 h. After centrifugation (5 min, 9000 rpm), washing three times with MeCN and drying for 1 h in a vacuum, S1.1 and S1.2 were obtained as orangish and pinkish coloured solid materials.General optimisation of the capping step
In general, the amounts of antibody and bovine serum albumin (BSA) were optimised to yield maximum capping while using a minimum amount of substance. While this resulted in slightly different amounts to be used for the single materials, the optimal amount of BSA was the same for all the materials loaded with SRG and only different for S1.1-AB.Synthesis and optimisation of S1.1-AB
The initial gated materials S1.1-AB were obtained by suspending fractions of 1 mg S1.1 in 0.45 mL phosphate buffered saline (PBS, 10 mM) containing 500 ppm FLU (to avoid losses of FLU from the pores) and adding 2.8 μL of a solution of monoclonal anti-pyrethroids antibody (clone number PY-1) in 10 mM PBS (2.85 mg mL−1). The suspensions were mixed in a carousel at room temperature for 30 min before 50 μL of BSA in 10 mM PBS (5 mg mL−1) or only PBS were added to each suspension and stirring was continued for 1.5 h. Afterwards, the materials were centrifuged and washed twice with 1 mL PBS. Subsequently, 1 mL PBS was added to the material, and the suspension was divided into three fractions each containing ca. 333 μg of the material. The fractions were centrifuged, and the materials obtained were dried for 1 h in a vacuum concentrator at room temperature before transfer to a refrigerator for storage, yielding S1.1-AB. These two batches of S1.1-AB capped in presence or absence of BSA served as the starting point for the optimisations reported below.Synthesis of S1.2-AB
Hybrid material S1.2-AB was obtained in a similar way as S1.1-AB using 1 μL of the antibody (2.85 mg mL−1 in PBS), and BSA as well as PBS at concentrations of 50 mg mL−1 as well as 80 mM. Both materials S1.1-AB and S1.2-AB showed stable performance for a storage period of ca. 3 months (in 80 mM PBS at 4 °C in a refrigerator). After this time, less release of dye was observed.Synthesis of S2.2a, S3.2a, S4.2a and S5.2a
185 μL of (0.4 mmol) 3-{methoxy[poly(ethyleneoxy)]propyl}trimethoxysilane (PEGS) were added to 200 mg of each material (S2, S3, S4 and S5), suspended in 6 mL absolute ethanol (EtOH (abs)) and left stirring overnight at 40 °C. Afterwards, the materials were washed with EtOH, centrifuged three times and dried in a vacuum for 5 h. After the PEG coating, the template was removed from the pores of the carrier materials by extraction. For this purpose, 200 mg of the modified materials were suspended in 25 mL EtOH (abs) containing 0.2 mL of HCl (37%) and stirred at 100 °C for 17 h. Thereafter, the materials were washed with water and EtOH until neutral pH and dried at 50 °C overnight in a vacuum oven, yielding the corresponding PEGylated materials. To load the dye into the pores of the carriers, 100 mg of the PEGylated materials were suspended in an acetonitrile solution containing SRG (800 μmol dye g−1 particles) before stirring the solution overnight at 45 °C. Subsequently, 100 μL (80 μmol) of the hapten derivative II (in THF) were added, and stirring was continued for 5.5 h. After centrifugation (5 min, 9000 rpm) and washing three times with MeCN, the materials S2.2a, S3.2a, S4.2a and S5.2a were dried for 1 h in a vacuum before transfer to a refrigerator.Synthesis of S2.2b
Material S2.2b was obtained following a similar procedure as for S2.2a, only that hapten derivative II was added before the loading of SRG. For that purpose, 185 μL (0.4 mmol) of PEGS were added to 200 mg S2, suspended in 6 mL EtOH (abs) and left stirring overnight at 40 °C as before. Afterwards, the material was washed with EtOH and dried in a vacuum. The template was removed by extraction after suspending the PEGylated material in 25 mL EtOH (abs) containing 0.2 mL HCl (37%) and stirring at 100 °C for 17 h. The material was washed with water and EtOH until neutral pH and dried at 50 °C overnight in a vacuum oven. In a second step, 100 mg of the PEGylated materials were suspended in 2.5 mL of MeCN, and 100 μL (80 μmol) of the hapten derivative II (in THF solution) were added before stirring for 5.5 h. The suspension was centrifuged (5 min, 9000 rpm), washed three times with MeCN and the solid was dried for 24 h in a vacuum. Finally, SRG was loaded into the pores by suspending the material functionalised with both, the hapten derivative II and the PEG moieties, in an acetonitrile solution containing SRG (800 μmol dye g−1 particles) and stirring overnight at 45 °C. After drying for 1 h in a vacuum, S2.2b was transferred to a refrigerator.All the fully prepared but uncapped materials are stable for more than 1 year as assessed by regularly capping small portions of them and running assays for comparison alongside.
Synthesis of S2.2a-AB, S2.2b-AB, S3.2a-AB, S4.2a-AB, and S5.2a-AB
To cap the materials, 5 μL of the antibody were suspended in 450 μL PBS (80 mM) solution before the addition of 1 mg of S2.2a, S2.2b or S3.2a. In the case of S4.2a and S5.2a, 2 μL of antibody were used. Subsequently, the solutions were kept rotating for 2 h. After 30 min, 50 μL of BSA solution (50 mg mL−1 in 80 mM PBS) were added. Finally, the particles were centrifuged and washed once with PBS (80 mM), which was sufficient as monitored via UV/vis absorption measurements. The obtained materials (S2.2a-AB, S2.2b-AB, S3.2a-AB, S4.2a-AB and S5.2a-AB) were re-suspended in 1 mL PBS and transferred to the refrigerator for storage as stock solutions of 1 mg mL−1, to avoid partial degradation of the antibody during the drying process. The stock solutions were stable for ca. 1 month, where after release of dye also in the absence of an analyte was observed.Kinetic release experiments under optimised conditions
Two fractions of 200 μL of a stock solution (1 mg mL−1 PBS) of a material were diluted in 900 μL PBS buffer (80 mM). Whereas 120 μL i-propanol (iPrOH) were added to one fraction, serving as control, 120 μL of 3-PBA in iPrOH were added to the other fraction, arriving at a concentration of 500 μg kg−1 3-PBA in the final solution also containing 10% of iPrOH as co-solvent. Fractions of 150 μL were centrifuged in both cases immediately after addition and after certain time intervals, before transferring 100 μL of the corresponding supernatants into a black quartz cell with an optically active volume of 3 × 15 × 10 mm and measuring the fluorescence intensity at 522 nm with 490 nm excitation for FLU and at 550 nm with excitation at 532 nm for SRG.Concentration-dependent studies under optimised conditions
330 μL of a stock solution (1 mg mL−1, in PBS) of the corresponding materials were diluted in 2200 μL PBS buffer (80 mM). The suspension was split into fractions of 140 μL, and 16 μL of an iPrOH solution containing different amounts of permethrin were added to each fraction. The suspensions were stirred for 5 min, and after centrifugation, the fluorescence of the supernatants was recorded as described above.Strips and lateral flow assay assembly
In analogy to our findings, that PEGylation of glass fibre strips improves the robustness of the assay in the lateral flow assay (LFA) format,26 the same strategy was employed here. The strips were prepared as described previously,26 using native glass fibre membranes as base and sample pad, between which a PEGylated glass fibre membrane is sandwiched as a conjugate pad. The gated materials S1.2-AB, S2a-AB, S2.2b-AB, S3a-AB, S4a-AB or S5a-AB were deposited onto the conjugate pad before sandwiching. In a typical experiment, 2.5 μL of a solution of SX.2y-AB (X = 1–5; y = a, b) in PBS (1 mg mL−1) were deposited with a pipette onto the conjugate pad. As reported before, the strips used here contained a focussing line in the detection zone.26 With respect to stability, the fully prepared strips showed unchanged performance over a storage period of ca. 3 months (stored in a sealed vial at 4 °C in a refrigerator) with an acceptable error of ca. 5% between replicates. After this time, the blank release of dye in the absence of the analyte was more pronounced.Results and discussion
Design of the gAID system
A schematic representation of the architecture and assembly routes of antibody-gated indicator-delivering materials is depicted in Scheme 1.The first strategy relied on the use of approximately spherical MCM-41 silica nanoparticles as carriers, with a particle diameter of ca. 100 nm and a mesopore diameter of 2–3 nm (Scheme 1a).31,36 After silica particle synthesis and removal of the surfactant cetyltrimethylammonium bromide (CTABr) by calcination, the material S1 was suspended in a solution containing fluorescein (FLU; dye 1) as the indicator to load the mesopores with. Afterwards, the particles’ external surface was functionalised with hapten derivative I, leading to solid S1.1. In the last step, the mesopores were capped by suspending S1.1 in a solution containing a monoclonal anti-pyrethroid antibody. The antibodies bind to the haptens located on the external surface of the material, eventually capping the pores (S1.1-AB, Scheme 1a). Considering the dimensions, in a more realistic version of this simplified sketch, a Fab (fragment antigen binding) part that contains the paratope region of the antibody (ca. 7 × 5 nm)22 should cover 2–3 pores, most likely in a random fashion, resembling a larger (ill-fitting) lid covering several smaller pots. Because of the rather low loading efficiency of FLU into S1.1 (Table 2) and the protolytic equilibria as well as pH-dependent fluorescence quantum yield of the dye, stretching from the acidic to the mildly basic pH range,37,38 an analogous material employing sulforhodamine G (SRG) as a dye (S1.2) was prepared.
The architecture of S1.X (X = 1, 2), which is the most commonly used in the literature, might not be optimal in two aspects. On one hand, the dimensions of the cap and the pore diameter are not well matching. On the other hand, because of the unidirectional pore system of MCM-41, a significant amount of haptens are supposedly grafted to non-porous parts of the external surface. The antibodies bound to those haptens are not capping any pore and binding of analytes by them would not lead to a pore opening and visible response of released indicators. Another strategy for capping was thus developed, consisting of the choice of materials with larger pore sizes and the separate functionalisation of external and internal surfaces (Scheme 1b). SBA-15 particles of different morphology and SBA-16 microparticles were therefore synthesised (S2–S5).32–35,39–41 In comparison to the previously mentioned MCM-41, these particles were larger in size (500 nm–2 μm) and possessed larger pores (7–17 nm). The two-step functionalisation process included two different silane derivatives, PEG and hapten. Methodologically, the external surface was blocked through PEG silane (PEGS) grafting of the as-prepared silica scaffolds, still containing the surfactant template in the pores.32 After the extraction of the template, the indicator dyes were loaded into the pores, before the hapten derivatives were grafted in a second functionalisation step, yielding S2.2a, S3.2a, S4.2a and S5.2a. This strategy was expected to localise the haptens preferentially at the outer rims of the pore openings, which should allow the Fab fragments of the antibodies to enter the larger pores and close them better. In contrast to the first “lid-on-pots” approach, the second one is more reminiscent of a cork closing a bottle. To avoid self-condensation of triethoxy silanes when functionalising only a very confined area, which could potentially block the pore openings, monoethoxy silane hapten derivative II was used instead of triethoxy silane I. For the assessment of the importance of the synthesis sequence, material S2.2b was prepared, for which SRG loading was done after hapten grafting. Such a grafting of hapten after template extraction and before dye loading would (putatively) coat the entire inner pore walls with hapten. In a final step, the closing procedure with the antibodies was applied for all the SBA-based materials, resulting in SX.2y-AB (X = 2–5; y = a, b).
Materials syntheses and characterisation
The mesoporous carrier particles were prepared according to literature procedures, obtaining MCM-41 nanoparticles S1 under alkaline conditions and the different SBA-type particles S2–S5 under acidic conditions (see ESI†).31,33–35,41S1–S5, as all the other functionalised materials, were characterised using standard techniques. The structure and morphology of the materials were assessed by TEM, STEM and N2 adsorption/desorption studies.TEM images confirmed the typical hexagonal porosity and channels of calcinated materials S1–S431,33,34,41 and the typical cubic array of SBA-16 microparticles S5.35Fig. 1 gives an overview of the morphologies and the differences between the more-ordered and smaller particles of S1 and the more complicated structure of the larger SBA carriers (S2–S5).
 | ||
| Fig. 1 Transmission electron micrographs of calcinated S1–S5. | ||
Nitrogen adsorption/desorption studies showed a type-IV isotherm typical for such materials. Values of specific surface areas (in m2 g−1) were estimated using Brunauer–Emmett–Teller (BET) theory as an evaluation method in a 0–1 range of (P/P0). Pore size distributions were calculated using the desorption branch of the isotherms and the Barrett–Joyner–Halenda (BJH) method (Fig. 2). The pore sizes were derived from the maximum of the peak in the distribution curves. The results are collected in Table 1. The calcinated MCM-41 material S1 shows two well-distinguished adsorption steps in the adsorption/desorption isotherms, ascribed to the nitrogen condensation inside the mesopores by capillarity, and the nitrogen adsorption on the outer particle surface (Fig. 2, blue curve). The absence of a hysteresis loop for the MCM-41 scaffold in this interval supports the existence of uniform cylindrical mesopores. Calcinated SBA-type carriers (S2–S5) show hysteresis loops in the region of P/P0 = 0.4–0.9, indicating gas retention inside the pores and slower desorption through the more complicated morphology of the voids such as, for instance, vesicle- and worm-like pores (Fig. 2, orange, green, red and grey curves).
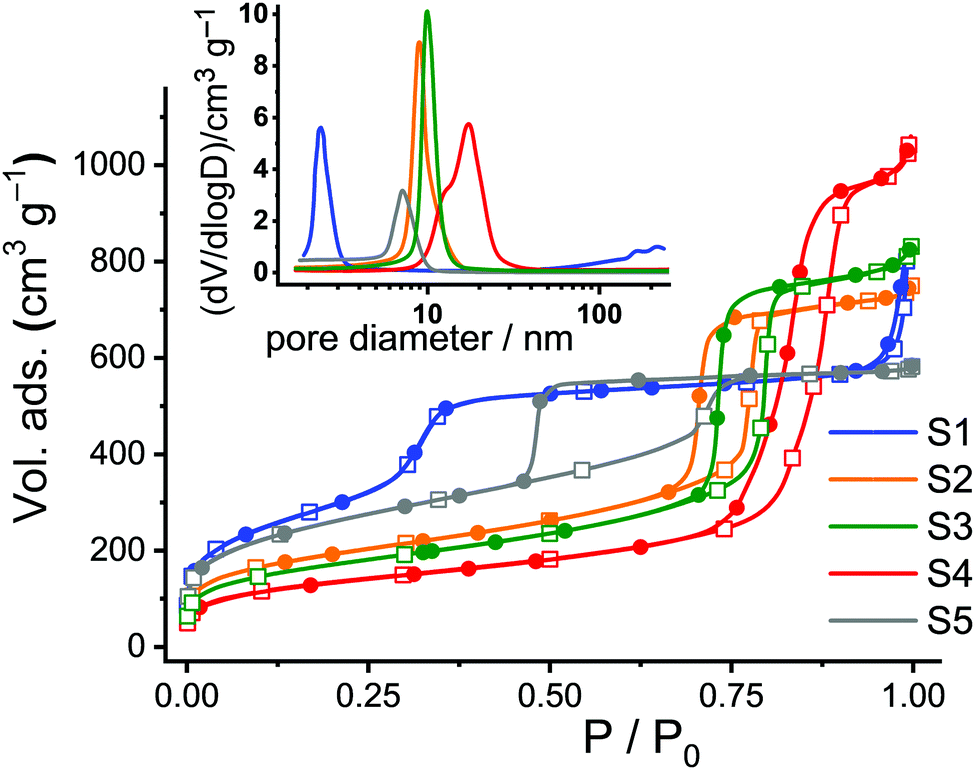 | ||
| Fig. 2 Adsorption (hollow squares) and desorption (solid circles) isotherms for calcinated S1–S5. Inset: Corresponding pore size distributions. | ||
| BET surface/m2 g−1 | Total pore volume/cm3 g−1 | Pore diameter/nm | N pores g−1/×1016 | Total internal area/% | Dimension (diameter or size)/nm | |
|---|---|---|---|---|---|---|
| S1 | 950 | 1.06 | 2.15 ± 0.05 | 84 | 55 | 100 ± 30 |
| S2 | 700 | 1.01 | 8.85 ± 0.05 | 0.43 | 20 | 920 ± 240 × 520 ± 90 |
| S3 | 620 | 1.15 | 9.80 ± 0.10 | 0.41 | 19 | 580 ± 70 × 270 ± 50 |
| S4 | 490 | 1.50 | 16.65 ± 0.05 | 0.35 | 18 | 770 ± 80 × 420 ± 20 |
| S5 | 940 | 0.63 | 7.00 ± 0.10 | 0.74 | 13 | 770 ± 130 |
SBA-15 platelets S3 were prepared by adding small amounts of Zr(IV) ions into the conventional SBA-15 synthesis solutions, resulting in objects of smaller particle size and slightly larger pore diameter compared to the long fibres of S2. The size effect is attributed to the so-called “salting out” effect in which the presence of Zr(IV) reduces the critical micelle concentration (CMC) of the P123 triblock copolymer, further acting as a catalyst to accelerate the hydrolysis and condensation of tetraethylorthosilicate (TEOS) on P123 micelles in the synthesis solution.33,41 On the other hand, the slight pore expansion of S3 compared to S2 can be attributed to the fact that Zr(IV) species may interact with the hydrophilic poly(ethylene oxide) chains of P123 most likely through coordinate or dative bonds, leading to an expansion of the micelles. Concerning S4, it is known that pore sizes can be further enlarged by the addition of 1,3,5-trimethylbenzene (TMB) as a swelling agent for P123 and by pre-hydrolysis of TEOS.34 Thus, S4 possesses a broad pore size distribution with a centre at 16.6 nm.34 Finally, Fig. 2 reveals a larger hysteresis loop for S5 compared to S2–S4 because of its cubic structure.35
The total surface areas (BET surface), total pore volumes, and the pore diameters were estimated from the analysis of the adsorption/desorption isotherms. To get a better idea of the ratio of the inner to the outer surface area, the hexagonal or cubic pores of the single carriers were taken as cylinders and the total number of pores per gram of solid as well as the total internal surface area were calculated (Table 1). The remaining percentage of the surface is thus attributed to the external area, corresponding to the (enveloping) external surface of the materials.
For the respective functionalised materials, N2 adsorption/desorption studies were not performed because of the comparatively high amount of material that is required for this method in the format available to us. However, the structure of the materials was corroborated via STEM studies. As an example, materials S2.2a and S2.2a-AB showed the typical porosity in a similar way to their calcinated precursor material S2 (Fig. 3a). The content of the different organic moieties of the hybrid materials was estimated through elemental analysis (EA), thermogravimetric analysis (TGA) and energy-dispersive X-ray spectroscopy (EDX) analysis. Contents of H, C, S and N estimated through EA allowed to calculate the amount of PEG, dye and hapten present on or inside the functionalised carriers (Table 2). The EA results were compared with TGA, showing isothermal traces with a steep weight loss between 150–600 °C, which is attributed to the decomposition of organic matter (Fig. S1, ESI†). Both analyses agreed well, yielding a total organic content of 30–35% for the solids prepared with SRG, yet of only 5–10% for S1.1 prepared with FLU, the lower loading efficiency with FLU being presumably due to the reasons mentioned in the section on system design above.
 | ||
| Fig. 3 (a) Scanning transmission electron micrographs of calcinated S2, S2.2a, and S2.2a-AB. (b)–(d) STEM images and corresponding EDX element maps of Si, S and C for (b) S2, (c) S2.2a and (d) S2.2a-AB. | ||
| EA | TGA | |||||
|---|---|---|---|---|---|---|
| FLU % | SRG % | HAPTEN % | PEG % | TOTAL % | TOTAL % | |
| S1.1 | 6.6 | — | 2.0 | — | 8.6 | 9.4 |
| S1.2 | — | 29.4 | 2.0 | — | 31.3 | 29.6 |
| S2.2a | — | 15.0 | 1.7 | 15.2 | 31.9 | 30.8 |
| S2.2b | — | 17.0 | 3.2 | 14.8 | 35.0 | 34.3 |
| S3.2a | — | 12.3 | 1.6 | 17.6 | 30.2 | 30.0 |
| S4.2a | — | 21.4 | 2.2 | 11.1 | 34.7 | 31.3 |
| S5.2a | — | 16.7 | 3.5 | 10.2 | 30.4 | 29.3 |
For the larger carriers, the first step of silane grafting yields PEG amounts in the final materials that vary between ca. 10–20%. As expected, subsequent functionalisation by hapten grafting was more efficient when the hapten silane derivative was added to a material with empty pores (S2.2b) than to one with pores already loaded with SRG (S2.2a). Although we cannot spatially resolve the true location of haptens in a single channel, these results suggest that loading of a dye prior to hapten grafting leads to a preferential condensation of the haptens closer to the pore openings. As the external surface was already largely coated by PEG moieties during the first functionalisation step, less hapten could be covalently anchored to S2.2a than to S2.2b.
EDX analysis from TEM and STEM was used to determine the average amount of Na, P, Si, and C for the different steps of materials preparation (see Fig. 3b–d, Fig. S2 and Table S1, ESI† exemplarily for materials S2 (calcinated), S2.2a and S2.2a-AB). As can be seen, the modification of S2 with SRG and hapten (S2.2a) leads to an increase of Na, S and C, all atoms being present in the molecules used (SRG as sodium salt). After the incorporation of the antibody, a significant increase of the carbon content (due to the presence of IgG proteins and BSA) and Na/P (due to PBS buffer) was observed. In addition, a decrease of the content of S in S2.2a-AB in comparison with S2.2a can be observed which can be related to a loss of dye during the washing steps after the antibody capping procedure, see also the discussion in ESI.† Despite the trends found, it is essential to note that the values in Table S1 (ESI†) are just estimations and cannot be considered as quantitative, considering that the uncertainty of measurements is considerably high (ca. 25%) and the materials are partially damaged during scanning because of the high energy of the beam.
Optimisation and performance evaluation of the systems in suspension
At first, the responses of the materials prepared according to the conventional approach (S1.1-AB and S1.2-AB, Scheme 1a), in which the hapten is located on the outer surface, have been investigated. During these initial experiments, the amounts of antibody and BSA, employed as a stabilising agent, the ionic strength and the possibility of use of an organic co-solvent (commonly required when aiming to analyse target molecules that are as hydrophobic as the pyrethroids here) have been established, to find the best conditions under which capping is most efficient while being economical in the use of a substance (see Experimental above). The optimisation of the capping step is crucial to achieving maximum sensitivity while at the same time keeping the leaching of the dye in the absence of the analyte at a minimum. Release experiments were then generally carried out by comparison of the amount of liberated dye in the presence or the absence of an analyte. Only in the presence of the analyte should uncapping of the pores lead to a significant increase of fluorescence in the supernatant after the solids have been separated by centrifugation.As the antibody requires neutral pH, phosphate-buffered saline (PBS) solution of pH 7.5 was used. For the study of the release kinetics, fractions of 333 μg of S1.1-AB were suspended in 400 μL of PBS buffer (10 mM). 200 μL of that suspension were diluted with 900 μL PBS (10 mM) and 30 μL of iPrOH, whereas the other 200 μL were diluted with 900 μL PBS and 30 μL of permethrin in iPrOH (250 ppm), arriving at a final concentration of 6.6 mg kg−1 of permethrin and 2.65% of iPrOH in the solution, small amounts of iPrOH facilitating the ready dissolution of the hydrophobic analyte.
The results reported in Fig. 4 demonstrate that the assay is only working efficiently if BSA is present during the antibody capping of the functionalised MCM-41 scaffold. Presumably, during the capping of the pores, the BSA stabilizes the antibody after it bound to the hapten on the silica scaffold, leading to enhanced interaction.
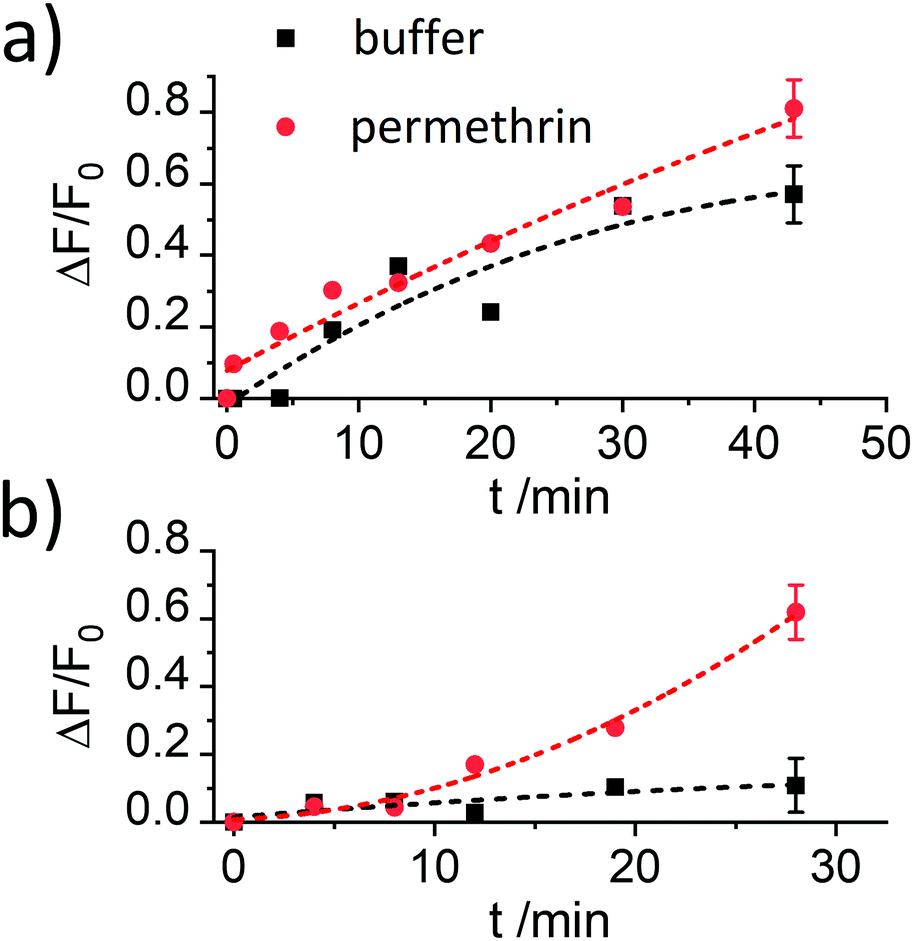 | ||
| Fig. 4 Increase in fluorescence registered at 522 nm (λexc = 490 nm) in the presence (red) and the absence (black) of permethrin (6.6 mg kg−1) for the supernatants of suspensions of two batches of materials S1.1-AB capped with antibody in (a) the absence and (b) the presence of BSA as blocking agent as a function of time between analyte addition to the suspension and centrifugation of the suspension. The lines are included only as a guide to the eye for better illustration. | ||
Despite showing already a favourable distinction between blank and analyte, the overall kinetics in Fig. 4b are too slow. As mentioned in the Introduction, response times on the order of <5 min until signal saturation are required for rapid tests. The next step was thus to accelerate the assay by modifying the ionic strength and the amount of organic co-solvent. Therefore, experiments in the presence of 5% of iPrOH in 10 mM and 80 mM PBS buffer were performed. As can be seen in Fig. 5a, the speed of dye release increased with the amount of organic co-solvent. We tentatively assume that both, increased solubility and better diffusion of the pyrethroid in this mixture led to this favourable effect.
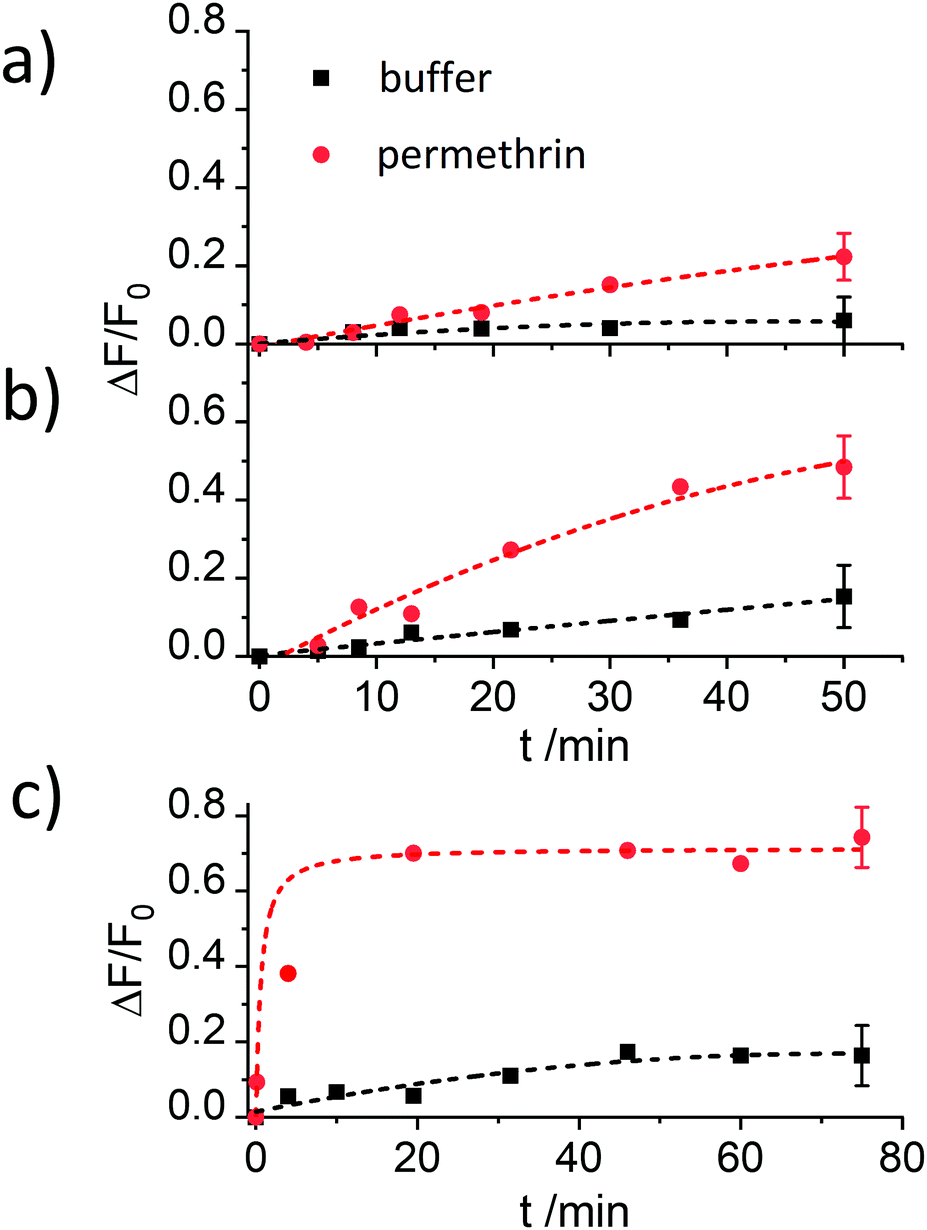 | ||
| Fig. 5 Increase in fluorescence registered at 522 nm (λexc = 490 nm) in the presence (red) and the absence (black) of permethrin (6.6 mg kg−1) for the supernatants of suspensions of two batches of materials S1.1-AB capped with antibody in the presence of BSA in (a) 10 mM PBS containing 5% of iPrOH, (b) 80 mM PBS containing 5% of iPrOH and (c) 80 mM PBS containing 10% of iPrOH as a function of time between analyte addition to the suspension and centrifugation of the suspension. The lines are included only as a guide to the eye for better illustration. | ||
Regarding the ionic strength of the buffer, Fig. 5b shows that a higher ionic strength entails a higher release of dye. 80 mM PBS was found to guarantee a proper capping and an efficient response. In the next series, the amount of co-solvent was further adjusted by carrying out release experiments in the presence of 5, 10, 15 and 20% of iPrOH (in 80 mM PBS). The presence of 10% of iPrOH showed the fastest release and most pronounced difference between blank and analyte (Fig. 5c). Higher amounts of iPrOH produced a strong release already in the absence of the analyte, ascribed to the partial denaturation of the antibody (Fig. S3, ESI†).
After having optimised the conditions, concentration-dependent studies were carried out to assess the sensitivity of the system. For this purpose, 333 μg of the corresponding S1.1-AB material were suspended in 2200 μL of PBS buffer (80 mM), and the procedure as detailed in the respective section of the Experimental was followed. As Fig. 6 illustrates, an increase of dye released as a function of the concentration of permethrin was observed, arriving at a limit of detection (LOD) of 30 μg kg−1. In comparison to similar systems described in the literature, this sensitivity is only moderate.19,20 A plausible reason can be the low loading efficiency of FLU (Table 2). To verify this, the analogously prepared material containing sulphorhodamine G (SRG, as sodium salt) instead of FLU, S1.2-AB, was tested under the optimised assay conditions. As reported above, elemental and thermogravimetric analysis revealed much higher loading rates for S1.2-AB compared to S1.1-AB. Indeed, concentration-dependent studies employing this material showed an increase in sensitivity of more than one order of magnitude, realizing a LOD < 1 μg kg−1 (Fig. 6). A comparison of the sensitivity of materials S1.1-AB and S1.2-AB is also helpful in understanding why the response behaviour of such gAID systems is more complex than only an antibody binding to an antigen in an ELISA. Although both materials contained the same amount of hapten (see Table 2) and were capped with the same antibody along with the same protocol, the EC50 values that result from fitting the datasets in Fig. 6 to a four-parametric logistic function are distinctly different, i.e., 142 nM for S1.1-AB yet only 5.1 nM for S1.2-AB. These EC50 values thus do not represent a binding constant but rather response constants, see also ESI.†
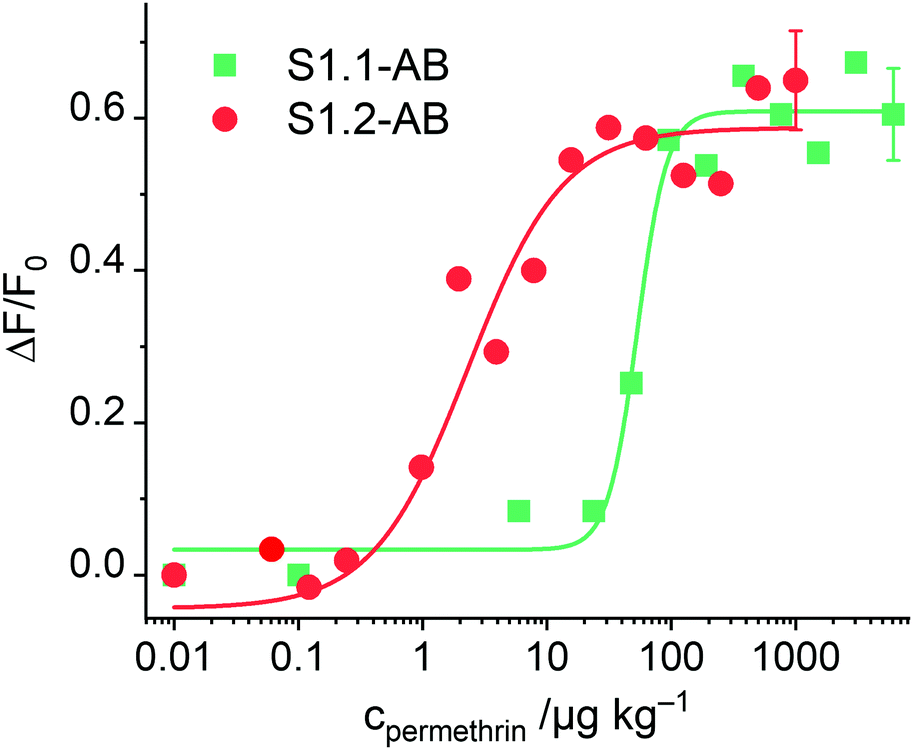 | ||
| Fig. 6 Increase in fluorescence of FLU and SRG released from S1.1-AB and S1.2-AB as a function of permethrin concentration after 5 min of reaction; the lines exemplify four-parametric logistic fits (for instrumental conditions used for S1.1-AB, see Fig. 4; λexc = 532 nm and λem = 550 nm used for S1.2-AB). | ||
Once the conditions were optimised, our aim was to verify the design considerations outlined above, i.e., whether assay performance improves when materials parameters and functionalisation sequences are fine-tuned. For this purpose, all the materials described in Scheme 1b were loaded with SRG. As already mentioned in the Experimental, the amount of antibody had to be optimised for SX.2y-AB (X = 2–5; y = a, b), see Fig. S4, ESI.†SX.2y-AB (X = 2–5; y = a, b) were stored in a stock solution (1 mg mL−1 PBS) in the refrigerator. Kinetic studies were performed to assess the tightness of pore closure and release kinetics, in analogy to the description for S1.1-AB above (with 80 mM PBS and 10% of iPrOH). As a model analyte, 3-phenoxybenzoic acid (3-PBA) was used, as 3-PBA has been used for antibody generation and resembles well type-I pyrethroids. Whereas S2.2a-AB and S3.2a-AB with a pore size of ca. 10 nm produced the best OFF/ON response when having been capped with 5 μL of antibody solution, for S4.2a with a larger pore diameter of ca. 15 nm, 2 μL performed best. S5.2a, in contrast, was unable to produce any response with all the amounts tested.
The release kinetics of the materials are displayed in Fig. 7. These results show that better control of the closing/opening mechanism is achieved when the Fab region of the antibody (with a size of ca. 7 × 5 nm) containing the paratope region matches better the pore size of the carriers. For carriers with pores that are either distinctly larger than those of MCM-41 yet almost the same size as a Fab region (S5.2a-AB) or that are much larger than a Fab region (S4.2a-AB), pore closure with antibodies does not seem to be very controlled so that dye is released in both, the presence and the absence of the analyte. When increasing the pore size from S5.2a-AB to S2.2a-AB or decreasing the pore size from S4.2a-AB to S3.2a-AB, performance improves, with the 8.8 nm-pore diameter material (S2.2a-AB) showing the best features of kinetics, relative signal increase, and achievable signal intensity. Regarding the latter, the order of the absolute achievable signal in terms of the maximum amount of dye released (after allowing the system to fully release its cargo in the presence of an excess of the analyte) is S5.2a-AB > S2.2a-AB > S4.2a-AB > S3.2a-AB > S1.2a-AB. A comparison of S2.2a-AB and S2.2b-AB finally provided insight into the role of the functionalisation sequence. For S2.2b-AB, grafting of the hapten was performed before dye loading (Scheme 1b). Even though the grafting efficiency of the hapten was higher for S2.2b-AB, the kinetic release experiments showed slower and inferior response compared with S2.2a-AB (Fig. 7, top panels). These results support the assumption that a preferential localisation of the hapten in the vicinity of the entrance to the pores is more decisive than a high degree of coating of the inner pore walls.
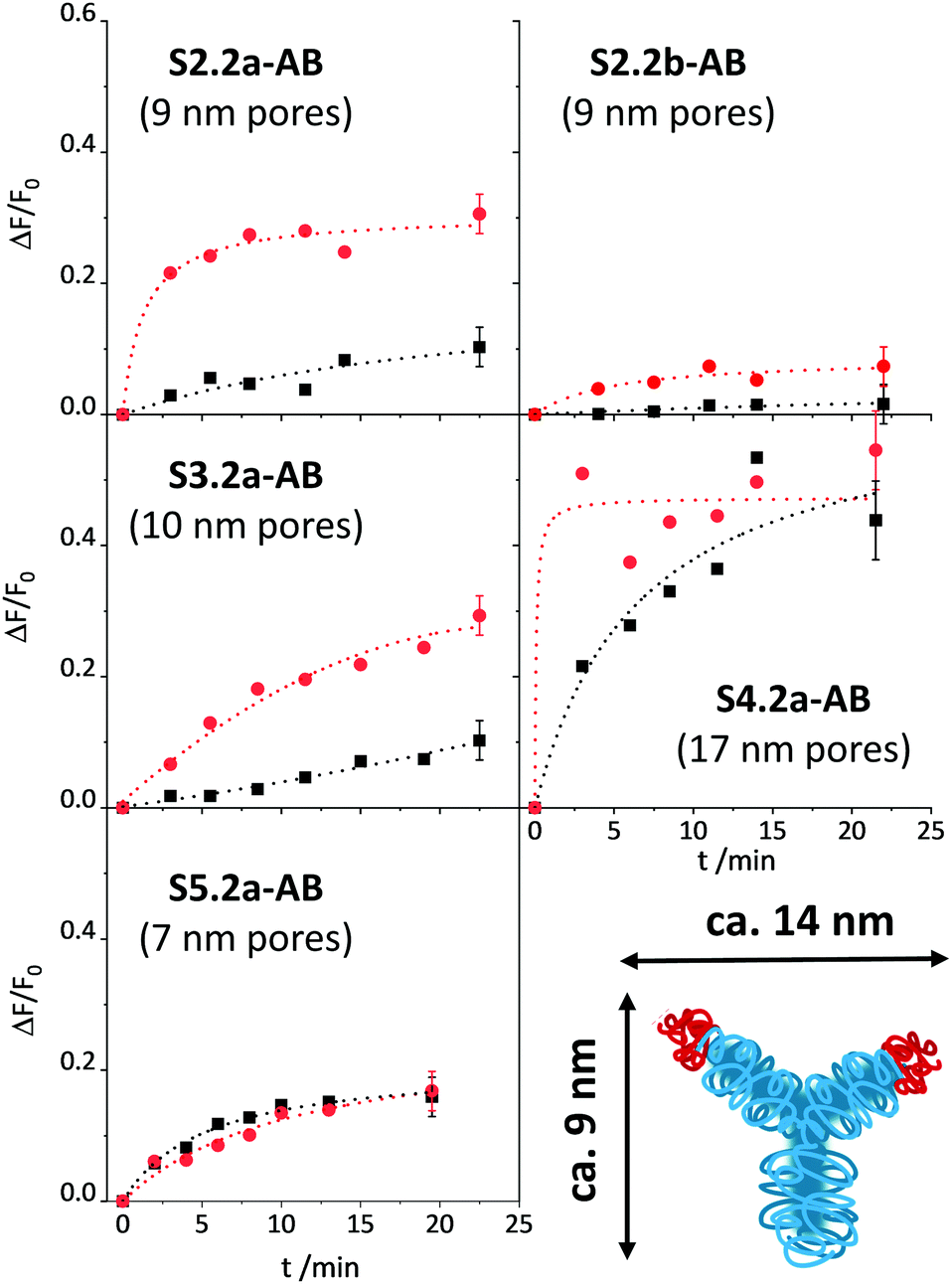 | ||
| Fig. 7 Increase in fluorescence (λexc = 532 nm; λem = 550 nm) in the absence (black) and the presence (red) of 500 μg kg−1 3-PBA for the supernatants of suspensions of the respective SBA materials as indicated in PBS buffer (80 mM, pH = 7.5, 10% iPrOH) as a function of time between analyte addition to the suspension and centrifugation of the suspension. The lines are included only as a guide to the eye for better illustration. | ||
In classic immunoanalytical chemistry, the ratio of the hapten to the antibody is also a factor that governs performance. If one approximates these values from the anchored hapten data in Table 2 and the amounts of antibody employed for capping, the materials of this small library can be ordered according to their hapten-antibody ratio as S3.2a-AB (0.3) ≈ S2.2a-AB (0.3) < S2.2b-AB (0.6) < S4.2a-AB (1.1) < S5.2a-AB (1.7) < S1.2-AB (2.0). The lowest values are thus found for the best performing materials S2.2a-AB and S3.2a-AB, which suggests that also for such gAID systems, low hapten coatings lead to more efficient antibody binding. However, it has to be taken into account that for such systems as discussed here, morphological aspects also have a considerable impact and putative molecular interaction stoichiometries have to be considered on that background. Because it is also essentially difficult to determine the true amount of bound antibody, the ratios given here are only crude approximations.
As the most promising material, S2.2a-AB was then subjected to titrations with both, 3-PBA and permethrin. As can be seen in Fig. 8a, the expected correlation of increasing dye release with increasing analyte concentration was observed, in agreement with a “displacement” of the antibody from the solid surface upon successful competition of the analyte with the binding sites and escape of the dye, estimating LODs of 2.4 ± 1.2 ppb and 0.6 ± 0.4 μg kg−1 as well as a dynamic range between 2.5 and 54.5 μg kg−1 and 0.7 and 3.5 μg kg−1 for permethrin and 3-PBA, respectively. Moreover, the data yield an EC50response = 28 nM for S2.2a-AB and permethrin.
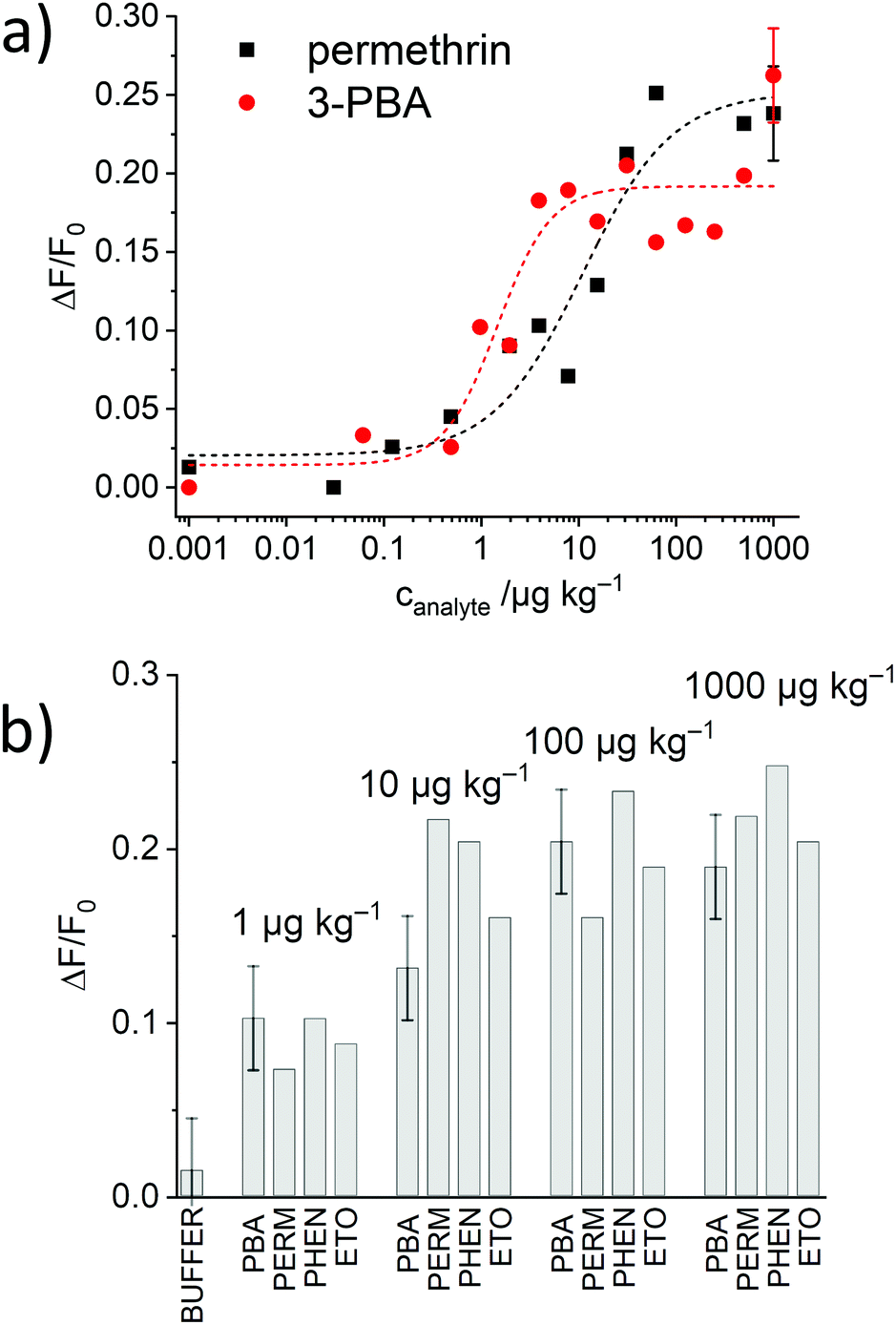 | ||
| Fig. 8 (a) Increase in fluorescence because of SRG released from S2.2a-AB as a function of the concentration of 3-PBA (red) and permethrin (black) after 5 min of reaction; the lines exemplify four-parametric logistic fits. (b) Corresponding data in the presence of 3-PBA, permethrin, phenothrin and etofenprox. | ||
The generic response of the monoclonal anti-pyrethroid antibody was also tested with phenothrin and etofenprox, two other pyrethroids. All of them could be detected with the assay, showing comparable increases in fluorescence and dynamic ranges (Fig. 8b), validating a similar sensitivity for this class of analytes.
Performance evaluation of the systems in LFA format
The last part of the present study was concerned with the question of whether the sequence of performance found in suspension can also be observed when the hybrids are embedded into a test strip and used in a lateral flow assay (LFA). The materials were thus integrated into glass fibre membranes as recently described26 and subjected to LFA tests, aiming at sensitive pyrethroid detection with smartphone read-out.Strip design and architecture were adopted from ref. 26. In essence, the gAID materials were deposited onto a conjugate pad which was sandwiched between a sample pad for sample administration and a base membrane which facilitates the flow of the released dye molecules to a focussing zone ca. 2 cm along the strip for detection. The gAID particles are too large to travel with the flow and remain in the conjugate pad. The ideal response pattern is as follows. In the absence of the analyte, no dye is released, and no fluorescence can be measured in the detection zone. In the presence of the designated target molecules, the dye is released in the conjugate pad and travels along the strip to the detection zone that starts to become fluorescent proportionally to the amounts of indicator dyes released. The smartphone used as a detector and for data analysis was equipped with a 3D printed case, a green LED for excitation and appropriate filters, in analogy to earlier works.26,42–44
The experiments with the materials S1.2-AB, S2.2a-AB, S2.2b-AB, S3.2a-AB, S4.2a-AB and S5.2a-AB on the strip were carried out under optimised conditions, i.e., employing PBS as a buffer (80 mM) containing 1% iPrOH, to avoid non-specific background fluorescence because of remobilisation of fluorescent organic material from the strip assembly. In a typical experiment, the strip was inserted into a 3D-printed polymer cassette to better fix the single pads on the carrier membrane and to enhance the flow. The strips with the different materials were treated with 120 μL of a PBS solution (80 mM, containing 1% iPrOH) with and without permethrin. After 2 min of incubation, the case was opened, and the strip was introduced into the smartphone holder. The fluorescence of the detection zone was registered by taking an image with the smartphone camera, extracting the integrated density (product of mean grey value and area) of the detection zone; data analysis was referenced on a computer with the software ImageJ.45 Concentration-dependent studies showed an increase in the fluorescence signal as a function of the concentration of permethrin in all of the cases. However, the sensitivities found were rather different for the different materials employed (Fig. 9). Interestingly, not only did S2.2a-AB show an excellent sensitivity with a LOD < 1 μg kg−1, but also S1.2-AB. However, the relative signal increase was significantly higher for S2.2a-AB than for S1.2-AB, presumably due to lower background fluorescence. As in the tests in suspension, materials S4.2a-AB and S5.2a-AB, as well as also S3.2a-AB, had lower sensitivities and less signal strength; an inferior performance was also found for S2.2b-AB (Fig. 9). Attractively, the performance pattern found in suspension is retained on the strip, contributing to rational design and fine-tuning.
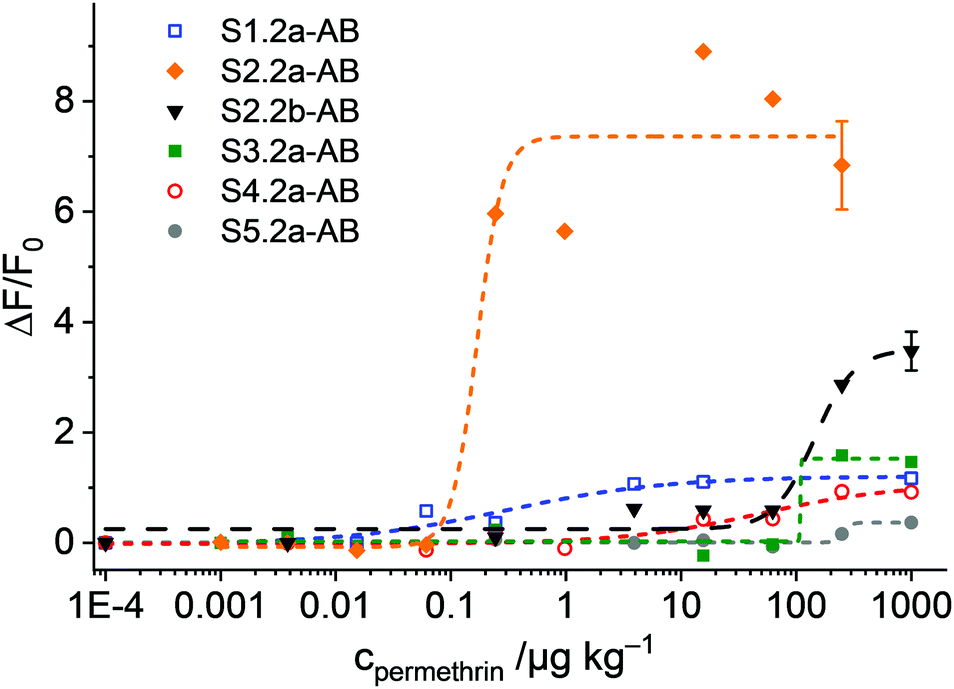 | ||
| Fig. 9 Integrated density enhancement registered for the images taken with the 3D-printed holder and the smartphone as a function of the concentration of permethrin for the materials S1.2-AB, S2.2a-AB, S2.2b-AB, S3.2a-AB, S4.2a-AB, S5.2a-AB and S2.2b-AB; the lines exemplify four-parametric logistic fits. | ||
Amplification features
gAID systems possess intrinsic features of signal amplification by releasing a larger number of indicator molecules upon binding of considerably few analyte molecules. Considering the system design, this chemical amplification depends on concentration, i.e., the amplification factor decreases with an increase of analyte concentration. We thus assessed the amplification factors of the materials, again in suspension, at analyte concentrations that produce a dye release equivalent to 10% of the maximum signal. The order obtained, all at analyte concentrations between ca. 1–10 μg kg−1, S2.2a-AB (1010-fold amplification) > S1.2-AB (860) > S2.2b-AB (197) > S3.2a-AB (12) > S1.1-AB (2.5)—S4.2a-AB and S5.2a-AB did not show a distinct and coherent amplification—also reveals that material S2.2a-AB performs best.Conclusions
This contribution reported the synthesis, characterisation and performance assessment of a series of different antibody-gated indicator-releasing materials, discussing in detail the design considerations and routes of optimisation that allowed to single out a powerful gAID system for the detection of pyrethroids. Whereas the conventional strategy in which the hapten and the antibody are located on the entire outer surface of the carrier particle allows for the fast detection of analytes with moderate sensitivity, the use of scaffolds with larger pore diameters that better match the dimensions of the Fab fragment of an antibody yielded better sensitivity and signal intensities. Different optimisations, such as the presence of BSA as a stabilising agent and the amount of co-solvent employed for better solubility of the hydrophobic analyte, were evaluated and found to contribute to success. Particularly attractive was the sequential grafting of PEG moieties to the particles’ external surface, and of the haptens only to the outer rims of the pores so that the antibody resides partially in the pore openings. S2.2a-AB, having a pore diameter of ca. 9 nm, showed the best control of the closing/opening mechanism, the main driver here presumably being the size match of the Fab and the pore opening. Larger pores (S4.2a-AB) lead to a pronounced release already in the absence of an analyte, whereas cubic particles in combination with slightly too small pores (S5.2a-AB) seem to be a similarly weak-performing approach. Final incorporation of the gAID systems into modified glass fibre membranes for the use of the strips in a lateral flow assay with mobile communication device readout yielded favourable detection limits down to the lower μg kg−1 level in an easy-to-operate manner and an overall assay time of 2–5 min, being well-suited for controlling for residues after for example spraying of pesticides in closed compartments such as aeroplane cabins directly at the point-of-use. This first in-depth study of several tuning strategies for antibody-gated indicator delivery materials did not only yield potent assays for pesticide detection but opens up the way for materials chemists to further advance the architecture of such promising hybrid materials, reaching out beyond sensing applications to diagnostics, theranostics and other areas in which chemical amplification plays a role.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from BAM (MIT-1-17-12, E. Costa) and German Research Foundation (DFG, CL 761/1-1, E. Climent) is gratefully acknowledged. We thank A. Zimathies (BAM) for N2 adsorption/desorption measurements, J. Lisec (BAM) and B. Kobin (Humboldt University Berlin, HUB) for MS analysis, A. Zehl (HUB) for elemental analysis, M. Grüneberg (BAM) for TGA, J. Bell (BAM) for help with the smartphone setup, K. Fast (BAM) for graphic support and S. Ast (AusSI Systems) for fruitful discussions.Notes and references
- D. Erickson and D. Q. Li, Anal. Chim. Acta, 2004, 507, 11–26 CrossRef CAS.
- J. Heikenfeld, A. Jajack, J. Rogers, P. Gutruf, L. Tian, T. Pan, R. Li, M. Khine, J. Kim, J. Wang and J. Kim, Lab Chip, 2018, 18, 217–248 RSC.
- B. B. Gao, X. Li, Y. Q. Yang, J. L. Chu and B. F. He, Analyst, 2019, 144, 6497–6511 RSC.
- C. Dincer, R. Bruch, E. Costa-Rama, M. T. Fernandez-Abedu, A. Merkoci, A. Manz, G. A. Urban and F. Guder, Adv. Mater., 2019, 31, 1806739 CrossRef PubMed.
- R. Pol, F. Cespedes, D. Gabriel and M. Baeza, TrAC, Trends Anal. Chem., 2017, 95, 62–68 CrossRef CAS.
- G. Chen, J. G. Zheng, L. Y. Liu and L. Xu, Small Methods, 2019, 3, 1900688 CrossRef CAS.
- M. Philp and S. L. Fu, Drug Test. Anal., 2018, 10, 95–108 CrossRef CAS PubMed.
- M. Hecht, E. Climent, M. Biyikal, F. Sancenon, R. Martinez-Manez and K. Rurack, Coord. Chem. Rev., 2013, 257, 2589–2606 CrossRef CAS.
- F. Sancenon, L. Pascual, M. Oroval, E. Aznar and R. Martinez-Manez, ChemistryOpen, 2015, 4, 418–437 CrossRef CAS PubMed.
- E. Aznar, M. Oroval, L. Pascual, J. R. Murguia, R. Martinez-Manez and F. Sancenon, Chem. Rev., 2016, 116, 561–718 CrossRef CAS PubMed.
- R. R. Castillo, A. Baeza and M. Vallet-Regi, Biomater. Sci., 2017, 5, 353–377 RSC.
- E. Climent, R. Martinez-Manez, A. Maquieira, F. Sancenon, M. Dolores Marcos, E. M. Brun, J. Soto and P. Amoros, ChemistryOpen, 2012, 1, 251–259 CrossRef CAS PubMed.
- G. K. Xu, X. Q. Feng, B. Li and H. J. Gao, J. Phys. Chem. B, 2012, 116, 6003–6009 CrossRef CAS PubMed.
- Z. Zheng, X. Huang, M. Schenderlein, H. Moehwald, G. K. Xu and D. G. Shchukin, Nanoscale, 2015, 7, 2409–2416 RSC.
- D. P. Tang, B. Q. Liu, R. Niessner, P. W. Li and D. Knopp, Anal. Chem., 2013, 85, 10589–10596 CrossRef CAS PubMed.
- B. Zhang, B. Q. Liu, J. Y. Liao, G. N. Chen and D. P. Tang, Anal. Chem., 2013, 85, 9245–9252 CrossRef CAS PubMed.
- D. P. Tang, Y. X. Lin, Q. Zhou, Y. P. Lin, P. W. Li, R. Niessner and D. Knopp, Anal. Chem., 2014, 86, 11451–11458 Search PubMed.
- J. Gao, Z. K. Guo, S. Q. Yu, F. J. Su, H. M. Ma, B. Du, Q. Wei and X. H. Pang, Biosens. Bioelectron., 2015, 66, 141–145 CrossRef CAS PubMed.
- E. Climent, A. Bernardos, R. Martinez-Manez, A. Maquieira, M. D. Marcos, N. Pastor-Navarro, R. Puchades, F. Sancenon, J. Soto and P. Amoros, J. Am. Chem. Soc., 2009, 131, 14075–14080 CrossRef CAS PubMed.
- E. Climent, R. Martinez-Manez, A. Maquieira, F. Sancenon, M. D. Marcos, E. M. Brun, J. Soto and P. Amoros, ChemistryOpen, 2012, 1, 251–259 CrossRef CAS PubMed.
- E. Climent, D. Groninger, M. Hecht, M. A. Walter, R. Martinez-Manez, M. G. Weller, F. Sancenon, P. Amoros and K. Rurack, Chem. – Eur. J., 2013, 19, 4117–4122 CrossRef CAS PubMed.
- Y. H. Tan, M. Liu, B. Nolting, J. G. Go, J. Gervay-Hague and G.-Y. Liu, ACS Nano, 2008, 2, 2374–2384 CrossRef CAS PubMed.
- D. Zhao, Y. Wan and W. Zhou, Ordered Mesoporous Materials, Wiley-VCH, Weinheim, 2013, ch. 5, pp. 153–217 Search PubMed.
- H. M. Li, Q. Zheng, J. Luo, Z. X. Cheng and J. Q. Xu, Sens. Actuators, B, 2013, 187, 604–610 CrossRef CAS.
- Report of the WHO Ad-hoc Advisory Group on aircraft disinsection for controlling the international spread of vectorborne diseases, Report WHO/HSE/GCR/2016.12, World Health Organization, Geneva, 2016.
- E. Costa, E. Climent, S. Ast, M. G. Weller, J. Canning and K. Rurack, Analyst, 2020 10.1039/D0AN00319K.
- Pyrethroids ELISA Kits PN 500201, PN 500204, Abraxis, Inc., Warminster, PA. The purified monoclonal mouse anti-pyrethroids (PY-1) antibody was kindly provided by Abraxis separately from the kit.
- J. C. Chuang, J. M. Van Emon, M. E. Tefft and N. K. Wilson, J. Environ. Sci. Health, Part B, 2010, 45, 516–523 CrossRef CAS PubMed.
- L. H. Stanker, C. Bigbee, J. Vanemon, B. Watkins, R. H. Jensen, C. Morris and M. Vanderlaan, J. Agric. Food Chem., 1989, 37, 834–839 CrossRef CAS.
- T. Watanabe, G. Shan, D. W. Stoutamire, S. J. Gee and B. D. Hammock, Anal. Chim. Acta, 2001, 444, 119–129 CrossRef CAS.
- Q. Cai, Z.-S. Luo, W.-Q. Pang, Y.-W. Fan, X.-H. Chen and F.-Z. Cui, Chem. Mater., 2001, 13, 258–263 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- S.-Y. Chen, C.-Y. Tang, W.-T. Chuang, J.-J. Lee, Y.-L. Tsai, J. C. C. Chan, C.-Y. Lin, Y.-C. Liu and S. Cheng, Chem. Mater., 2008, 20, 3906–3916 CrossRef CAS.
- S.-Y. Chen, Y.-T. Chen, J.-J. Lee and S. Cheng, J. Mater. Chem., 2011, 21, 5693–5703 RSC.
- G. F. Andrade, D. C. F. Soares, R. G. dos Santos and E. M. B. Sousa, Microporous Mesoporous Mater., 2013, 168, 102–110 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845–8849 CrossRef CAS PubMed.
- R. Sjoback, J. Nygren and M. Kubista, Spectrochim. Acta, Part A, 1995, 51, L7–L21 CrossRef.
- V. R. Batistela, J. D. Cedran, H. P. M. de Oliveira, I. S. Scarminio, L. T. Ueno, A. E. H. Machado and N. Hioka, Dyes Pigm., 2010, 86, 15–24 CrossRef CAS.
- R. Moulin, J. Schmitt, A. Lecchi, J. Degrouard and M. Impéror-Clerc, Soft Matter, 2013, 9, 11085–11092 RSC.
- Q. He, J. Shi, X. Cui, J. Zhao, Y. Chen and J. Zhou, J. Mater. Chem., 2009, 19, 3395–3403 RSC.
- K. Zhuang, G. Yin, X. Pu, X. Chen, X. Liao, Z. Huang and Y. Yao, MRS Commun., 2016, 6, 449–454 CrossRef CAS.
- R. Gotor, P. Ashokkumar, M. Hecht, K. Keil and K. Rurack, Anal. Chem., 2017, 89, 8437–8444 CrossRef CAS PubMed.
- R. Gotor, J. Bell and K. Rurack, J. Mater. Chem. C, 2019, 7, 2250–2256 Search PubMed.
- R. Gotor, C. Tiebe, J. Schlischka, J. Bell and K. Rurack, Energy Fuels, 2017, 31, 11594–11600 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00371a |
| ‡ Present address: Surflay Nanotec GmbH, Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2020 |