
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoporous hybrid core–shell nanoparticles for sequential release†
Mandy
Jahns
*ab,
Dawid Peter
Warwas
a,
Marc Robert
Krey
a,
Katharina
Nolte
a,
Sandra
König
 c,
Michael
Fröba
c and
Peter
Behrens
*ab
c,
Michael
Fröba
c and
Peter
Behrens
*ab
aInstitute for Inorganic Chemistry, Gottfried Wilhelm Leibniz University Hannover, 30167 Hannover, Germany. E-mail: peter.behrens@acb.uni-hannover.de; Fax: +49-511/762-3006; Tel: +49-511/762-3660
bCluster of Excellence Hearing4all, Hannover, Germany
cInstitute of Inorganic and Applied Chemistry, University of Hamburg, 20146 Hamburg, Germany
First published on 9th December 2019
Abstract
In this article, a new type of core–shell nanoparticle is introduced. In contrast to most reported core–shell systems, the particles presented here consist of a porous core as well as a porous shell using only non-metal materials. The core–shell nanoparticles were successfully synthesized using nanoporous silica nanoparticles (NPSNPs) as the starting material, which were coated with nanoporous phenylene-bridged organosilica, resulting in a total particle diameter of about 80 nm. The combination of a hydrophilic nanoporous silica core and a more hydrophobic nanoporous organosilica shell provides regions of different chemical character and slightly different pore sizes within one particle. These different properties combined in one particle enable the selective adsorption of guest molecules at different parts of the particle depending on the molecular charge and polarity. On the other hand, the core–shell make-up of the particles provides a sequential release of guest molecules adsorbed at different parts of the nanoparticles. As a proof of concept, loading and release experiments with dyes were performed using non polar fluorescein and polar and charged methylene blue as model guest molecules. Non polar fluorescein is mostly adsorbed on the hydrophobic organosilica shell and therefore quickly released whereas the polar methylene blue, accumulated in the hydrophilic silica core, is only released subsequently. This occurs in small doses for an extended time corresponding to a sustained release over at least one year, controlled by the organosilica shell which acts as a diffusion barrier. An initial experiment with two drugs — non polar ibuprofen and polar and charged procaine hydrochloride — has been carried out as well and shows that the core–shell nanoparticles presented here can also be used for the sequential release of more relevant combinations of molecules.
Introduction
Core–shell nanoparticles, which have been synthesized since the early 1990s,1,2 are an interesting group of nanoparticles as they combine different kinds of materials and therefore are usually multifunctional. For example, a coating shell offers options for modifications changing the accessibility of the core and therefore its reactivity. Furthermore, it can influence the dispersibility, stability of the core or other features. Core–shell nanoparticles occur in a wide variety based on their size, shape and structure, ranging from single layer nanoparticles to multi-shell types or particles with hollow parts.1,3 Often, such nanoparticles consist of dense metal or metal oxide cores, e.g., gold4–6 or magnetite,7–10 and therefore can act as catalytically active centers or provide magnetic properties. Only a few core–shell systems have been described which contain a dense silica core.11,12 For example, Haffer et al.13 reported a one-pot synthesis producing dense silica coated with periodic mesoporous organosilica (PMO). Here, the core only has a passive function, as in this case the impermeability of dense silica was intended to shorten diffusion paths for HPLC applications.Mesoporous silicas are often used as an outer shell in the construction of core–shell nanoparticles. Then, the large permanent cargo space can be used to store and release substances, and reactive silanol groups on the surface enable modifications via post-grafting methods and therefore allow for an adaptation of the surface chemistry to the intended application.14 For uses in the biomedical field, mesoporous silica shows good biocompatibility and low toxicity, a topic which has been intensively studied and reported.15–19
PMOs were discovered in 199920–22 and offer similar advantages as silica. They are built up from bridged bissilylated organosilanes of the type (RO)3Si–R′–Si(OR)3 providing inherent and homogeneously distributed organic functionalities within the highly porous network.23–25 They are also assumed to be biocompatible26 and offer options for further modifications via post-grafting or via functionalization of the organic bridging units.14
Since organosilicas are very versatile materials, there are several examples of existing core–shell nanoparticles containing this material, mainly originating from ethane-bridged precursors.27–30 However, practically all of these nanoparticles contain dense cores or are hollow. Nevertheless, core–shell nanoparticles with a core which is also porous could show interesting properties and be beneficial for several applications like adsorption or drug delivery.31 In general, apart from the present work, core–shell nanoparticles with a mesoporous silica core and a PMO shell have only been reported by Song et al.32 They synthesized a sulfonic acid-modified porous silica core coated with a porous organosilica shell for catalytic applications. Anwander's group recently reported on a PMO core/mesoporous silica shell material and used it for the controlled release of a single agent, a fungicide.30
The presence of two chemically different pore systems, however, implies that two different substances could also be released and delivered, for example, two drugs.33 Most of the described nanoporous materials release only a single active agent, although the release of a combination of drugs, especially sequentially in time, might be beneficial for many applications, e.g., for the treatment of implant-associated infections. In such cases, healing could be supported by using an anti-inflammatory drug in the initial days to deal with acute inflammations or infections, and later on, by using a healing-promoting drug over several weeks. In the literature, sequential release materials or systems are known, but most of these are based on polymers.34,35 Accordingly, long-term release systems which have been described so far are mainly based on polymers as well.36,37 However, there are some disadvantages with regard to polymer-based systems. The incorporation of drugs can cause drastic changes in the mechanical properties of the polymer, with the incorporated drug molecules acting as plasticizers or disturbing the cross-linking of the polymers. Another disadvantage might be that frequently used polymers in biomedicine, like silicones, are not biodegradable, and so cannot be used for applications where a healing-supporting system is needed only temporarily until the body itself can resume its function.
Herein, we describe the construction of core–shell nanoparticles with a core of mesoporous silica and a shell of mesoporous phenylene-bridged organosilica; we use the different hydrophilicities of the two materials (hydrophilic silica/hydrophobic organosilica) for the autonomous selective adsorption of active agents at different parts of the particles, as demonstrated in Scheme 1.
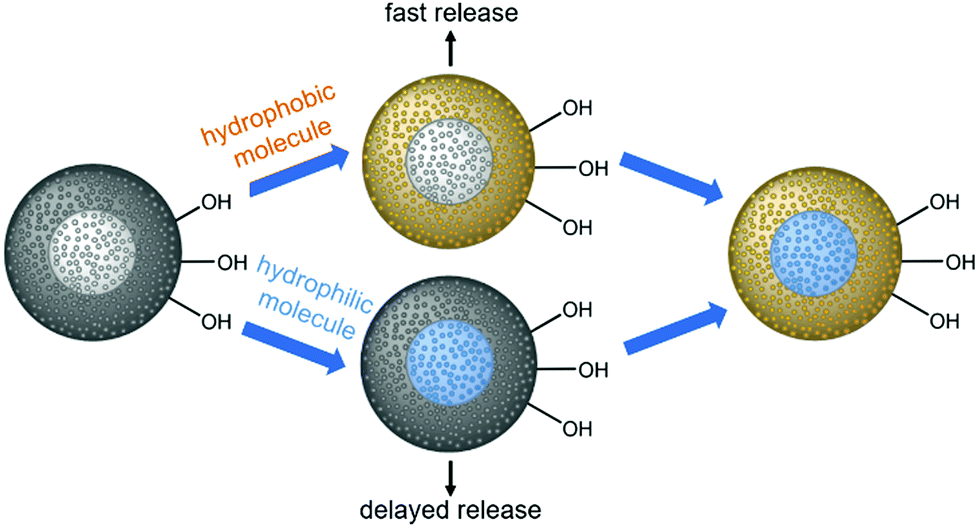 | ||
| Scheme 1 Loading of an entirely porous core–shell nanoparticle consisting of a hydrophilic silica core and a hydrophobic organosilica shell. Hydrophobic (orange) and hydrophilic guest molecules (blue) are selectively adsorbed at different parts of the nanoparticles, leading to different release behaviors depending on the diffusion path length. | ||
It is assumed that hydrophobic guest molecules will preferably be adsorbed in the more hydrophobic shell with relatively large organic bridging units, whereas hydrophilic guest molecules will accumulate in the hydrophilic silica core. Regarding the release of such molecules, we expected a fast release from the shell and a delayed release from the core, due to different diffusion path lengths, but also due to the hydrophobic environment presented by the PMO part through which the hydrophilic guest molecules from the core must travel. We explain the synthesis of the core–shell silica-organosilica nanoparticles and demonstrate sequential delivery using a pair of easily detectable dyes (methylene blue/fluorescein) and a pair of relevant drugs (procaine hydrochloride/ibuprofen).
Experimental section
Chemicals
All chemicals were used as received without further purification. Ethanol (EMPLURA, absolute) was purchased from Merck (Darmstadt, Germany). The remaining chemicals stated in the following were purchased from Sigma-Aldrich (München, Germany): cetyltrimethylammonium bromide (≥96%; CTAB), diethanolamine (≥99.5%; DEA), tetraethylorthosilicate (≥99%; TEOS), hydrogen peroxide (30%), ammonia (25% in water), hydrochloric acid (2 M), 1,4-bis(triethoxysilyl)benzene (≥96%; BTEB), methylene blue hydrate (≥97%; MB), fluorescein (Reag. Ph. Eur.; FC) and phosphate buffered saline (PBS). For all synthesis and modification steps, ultrapure water (18.2 MΩ cm) was used.Synthesis of NPSNPs
Nanoporous silica nanoparticles serving as the starting material for core–shell nanoparticles were synthesized according to an adapted literature procedure of Qiao et al.38 as previously presented by Neumann et al.39 6.32 g CTAB and 0.46 g DEA were dissolved in 26.8 mL ethanol and 150 mL water for 30 minutes at 40 °C, and subsequently 17.12 g of TEOS were added. This mixture was stirred for 2 h at 40 °C and then centrifuged at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g. The white product was washed twice in water and once in ethanol by re-dispersing it in the solvent using ultrasonication and a subsequent centrifugation (18000 g). The particles were dried at 60 °C and calcined at 550 °C for 5 h with a heating rate of 1 °C min−1 to remove the surfactant. These particles are denoted as NPSNP_ca.
000g. The white product was washed twice in water and once in ethanol by re-dispersing it in the solvent using ultrasonication and a subsequent centrifugation (18000 g). The particles were dried at 60 °C and calcined at 550 °C for 5 h with a heating rate of 1 °C min−1 to remove the surfactant. These particles are denoted as NPSNP_ca.
Pretreatment or extraction of NPSNPs
In the preparation of the core–shell nanoparticles, it was noted that freshly calcined nanoparticles lead to larger core–shell nanoparticles due to the agglomeration of silica nanoparticles before the growth of the organosilica shell. Nevertheless, a successful synthesis with calcined nanoparticles as the starting material is possible, if they are stored for 3 months or longer. For better reproducibility of the size of core–shell nanoparticles and a more time-efficient synthesis, a pretreatment or an extraction of the silica cores is recommended.For the pretreatment,40 2.00 g of calcined NPSNPs were stirred in a solution of 50 mL water, 10 mL ammonia (25%) and 10 mL hydrogen peroxide (30%) for 20 minutes. Afterwards, the particles were filtered, washed with water several times and dried at 60 °C. These particles are denoted as NPSNP_pt.
As an alternative to the pretreatment, an extraction instead of a calcination can be performed. For this, 5.00 g of as-synthesized and dried NPSNPs were stirred in a solution of 150 mL ethanol and 6 mL hydrochloric acid (2 M) in a round-bottomed flask under reflux at 100 °C for 24 h. Afterwards, the nanoparticles were filtered, washed with ethanol several times and dried at 60 °C. These particles are denoted as NPSNP_ex.
Synthesis of the organosilica shell
For the synthesis of the organosilica shell, the silica nanoparticles acting as the starting material were sieved with a mesh made of stainless steel (Retsch, Haan, Germany; mesh width 200 μm). The coating of the silica spheres was performed using a synthesis procedure adapted from ref. 13. 1.00 g of sieved NPSNPs was dispersed in a mixture of 90 mL ethanol and 1.13 mL water via ultrasonication for at least 2 h. To the well-dispersed silica nanoparticles, 10 mL ammonia (25%), 200 mL water and 30 mL of a CTAB solution (0.11 M in ethanol/water mixture VEtOH:VH2O = 1:2) were added. This mixture was stirred for 4 h at ambient temperature (25 °C), then 2.2 mL of BTEB were added and further stirred overnight. Afterwards, the reaction mixture was transferred to screw lid glasses and stored at 60 °C for 24 h. The white precipitate was then filtered, washed with water and ethanol several times and dried at 60 °C. To remove the surfactant, an extraction analogous to the previously described procedure was performed. The core–shell nanoparticles are denoted as CSNPs.
Loading procedure
The two dyes methylene blue (MB) and fluorescein (FC) were loaded into the CSNPs simultaneously and were thus allowed to choose their preferred environment in the core or in the shell. For the loading, 100 mg of the resulting nanoparticles were stored in 10 mL of an ethanolic solution containing methylene blue (25 mM) and fluorescein (20 mM) for 2 days at 37 °C. As a blind sample, nanoparticles were stored in a pure solvent. As references, nanoparticles were stored in solutions containing only one of the dyes at the same concentration. Afterwards, the particles were centrifuged (6000 rpm), washed once with pure solvent and dried at 60 °C. In the following, nanoparticles loaded with both dyes are denoted as CSNP_FCMB, the blind sample as CSNP_BL and the references as CSNP_MB or CSNP_FC, marking their loading with methylene blue or fluorescein, respectively.The loading with two drugs was also performed simultaneously, using an ethanol–water mixture (1:1) as the solvent and active agents at concentrations of 10 mM. Loading was carried out at 37 °C for 3 d. In addition to the sample containing both drugs, reference samples with only one of the two drugs were prepared. The ratio of particles to solvent as well as the processing after the loading were as mentioned before. In the following, nanoparticles loaded with both drugs are denoted as CSNP_IBUPRO and the references as CSNP_PRO or CSNP_IBU, marking their loading with procaine hydrochloride or ibuprofen.
Release experiments
The release experiments were performed in a three-fold determination in polypropylene tubes. To 5 mg of nanoparticles, 2 mL of pre-heated PBS were added and the mixture was stored at 37 °C. After certain times, the samples were centrifuged (15000 rpm). The supernatants were separated and stored for UV/Vis analysis and fresh medium was added to the nanoparticles and the mixture was vortexed. This was repeated over several months.
Measurement and characterization
For investigations of particle size and morphology, transmission electron microscopy (TEM) was performed using an FEI Tecnai G2 F20 TMP instrument with a 200 kV field emission gun in bright-field mode. For the preparation of the samples, the nanoparticles were dispersed in ethanol via ultrasonication, dropped on a grid (400-mesh carbon-coated copper grids purchased from Quantifoil) and dried.To characterize the organic components of the nanoparticles and to confirm the loading with dyes, Fourier-transform infrared spectroscopy (FT-IR) was conducted using a Bruker Tensor 27 instrument in transmission mode after the preparation of KBr pellets (Sigma Aldrich). The spectra were recorded in the range of 4000–400 cm−1 with a resolution of 2 cm−1, baseline-corrected and normalized.
Thermogravimetric analysis (TG) and differential scanning calorimetry (DSC) were performed using a Netzsch STA 409 PC/PG instrument using an Ar–O2-atmosphere (80:20) in the range of 40–1000 °C with a heating rate of 5 K min−1.
For zeta potential and dynamic light scattering (DLS) measurements, a Malvern Instruments Zetasizer Nano NS instrument equipped with an autotitrator was used. The sample was dispersed in water (zeta potential) or ethanol (DLS). For the adjustment of the pH value, hydrochloric acid (0.2 M) and sodium hydroxide (0.2 M) were used.
Nitrogen physisorption measurements were performed on empty and loaded samples using a Quantachrome Autosorb-3 instrument. Samples were outgassed in a vacuum for 24 h at 100 °C or 60 °C (loaded samples). For calculation of surface areas and pore size distributions, the software ASiQwin (version 2.0) from Quantachrome was used applying the Brunauer–Emmett–Teller (BET) equation and density functional theory (DFT). The experimental data were fitted to Quantachrome Kernel “N2 at 77 K on silica (cylinder./sphere pore, NLDFT ads. Model)”. The single point method at p/p0 = 0.95 was applied to determine the total pore volumes.
Water physisorption measurements were performed using a Quantachrome Aquadyne DVS-2 instrument at 25 °C in the range of 5–95% RH in 2% steps.
Hysteresis scans using desorption scanning were done with a 3Flex instrument from Micromeritics Instrument Corp. with nitrogen at 77 K.
Supernatants from the release studies of the dyes were measured via UV/Vis-spectroscopy carried out using a Tecan Spark 10 M instrument at the corresponding absorption maxima of the dyes at 37 °C. Supernatants from the release studies of the drugs were analysed via HPLC measurements at 35 °C using an Agilent instrument of the 1100 series with a Sciencix DAD-detector and a deuterium lamp. As the column, a Raptor™ C18 5 μm with dimensions of 150 × 4.6 mm and a pore width of 90 Å was used. As the eluent, a mixture of water and acetonitrile starting with a ratio of water:acetonitrile = 95:5 and ending at a ratio of 5:95 over a period of 15 min was used.
Results and discussion
Characterization of core–shell nanoparticles
A detailed characterization of the NPSNPs is not given here since porous silica nanoparticles are well-known and have been extensively reported in the literature.39,41 Nevertheless, some characteristics are mentioned in comparison with the final core–shell product, primarily to describe the changes occurring due to the shell synthesis.FT-IR spectra (Fig. 1A; differences marked by grey boxes) show characteristic vibrational bands at 525 cm−1 (Si−C stretching mode) and between 1491 cm−1 and 1387 cm−1 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching and C–H vibrations) for the organic bridging units of the organosilica. These occur in addition to the dominant bands for siloxane at 1076 cm−1 and OH valence vibrations (from silanol groups as well as water) at 3400 cm−1. Note that these bands are weaker in the CSNP material, as is the bending vibration of H2O (1630 cm−1). The bands which appear newly upon formation of the PMO shell match with known data from the literature42 and confirm the formation of organosilica and its stability during the extraction at elevated temperatures (100 °C).
C stretching and C–H vibrations) for the organic bridging units of the organosilica. These occur in addition to the dominant bands for siloxane at 1076 cm−1 and OH valence vibrations (from silanol groups as well as water) at 3400 cm−1. Note that these bands are weaker in the CSNP material, as is the bending vibration of H2O (1630 cm−1). The bands which appear newly upon formation of the PMO shell match with known data from the literature42 and confirm the formation of organosilica and its stability during the extraction at elevated temperatures (100 °C).
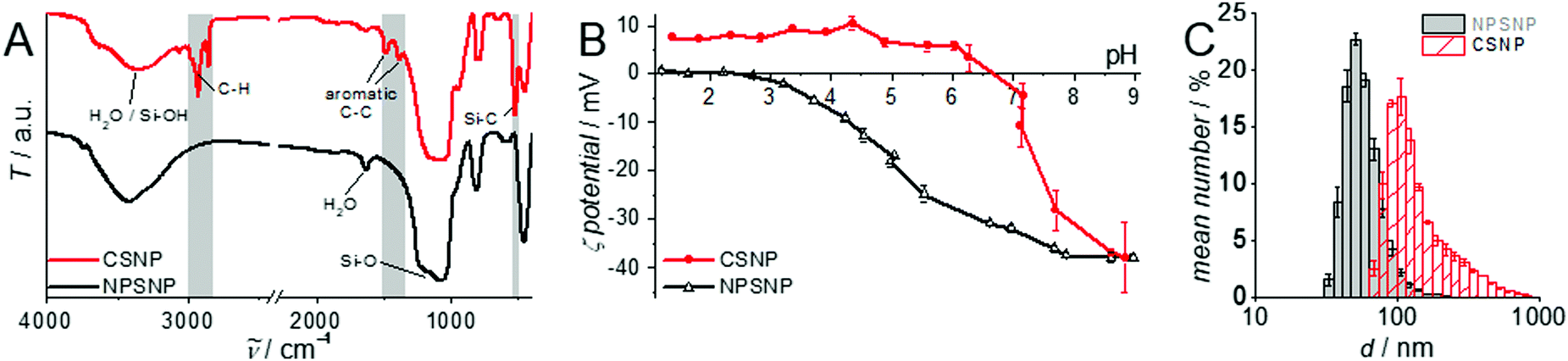 | ||
| Fig. 1 Characterization of CSNPs (red) in comparison to the core NPSNPs (black). (A) FT-IR spectroscopy; additional bands appearing after the addition of the PMO shell are marked in grey. (B) pH-Dependent zeta potential measurement showing a shift of the isoelectric point after the shell growth. (C) DLS investigations show an increasing particle size comparing the starting material NPSNP (black/grey) and CSNPs (red) equipped with an organosilica shell. | ||
The successful shell growth can also be confirmed by comparison of pH-dependent zeta potential measurements (Fig. 1B) performed on particle suspensions before and after shell formation. Most characteristic is the change of the isoelectric point from pH 2.4 (NPSNP) to 6.6 (CSNP). Noteworthy, the zeta potential at pH 9 is practically the same (−37 mV) for both types of nanoparticles; this is caused by deprotonated and therefore negatively charged silanolate groups, which are present in silica as well as in organosilica. These curves reveal the stability of nanoparticle dispersions under basic conditions due to the highly negative zeta potential. At the pH value of PBS (7.4), the particles are negatively charged, so a good electrostatic interaction with positively charged methylene blue or procaine hydrochloride in the following loading and release experiments is expected.
An important feature of the CSNPs presented here is that they are entirely porous. Nitrogen physisorption measurements showed that the NPSNPs and the core–shell nanoparticles exhibit high porosities with BET surface areas of 1000 m2 g−1 and 600 m2 g−1, respectively. This is also confirmed by the determined total pore volumes of 1.0 cm3 g−1 for the NPSNPs and 0.6 cm3 g−1 for the CSNPs. In addition, the pore size distribution (Fig. 2B) for the core–shell nanoparticles shows two maxima at 2.6 nm and 3.8 nm for the pore diameter. The assumption that one of both pore diameters can be assigned to either the silica or the organosilica is confirmed by comparison of the pore size distributions of pure phenylene PMO and nanoporous silica nanoparticles which were stored in a reaction solution for shell growth without adding the organosilica precursor. These diameters show a maximum at 2.6 nm for the PMO material and a maximum of 4.1 nm for the silica core material. Both these maxima are combined in the bimodal pore size distribution of CSNPs. The physisorption isotherm of the CSNPs with type IV behavior (Fig. 2A) also shows a characteristic drop of the desorption branch at a relative pressure of 0.47 indicating a cavitation process.43 This often occurs in combination with ink-bottle44 pores which have small pore necks in comparison to larger pore cavities. Spontaneously formed gas bubbles lead to a sudden emptying of the pore system resulting in a sharp drop at the hysteresis closure. Such larger cavities might have built up at the interface between the core and shell when one takes into account that during the synthesis of the PMO shell, the nanoporous silica core might have partially dissolved as a consequence of the basic conditions present during this final synthesis step. A similar behavior has already been reported by Haffer et al.13 for similar core–shell nanoparticles, which, however, contain a dense nonporous silica core. To further characterize this special feature, a hysteresis scan measurement was performed (see the ESI,† Section S1 and Fig. S1) which confirms the cavitation mechanism for the desorption.
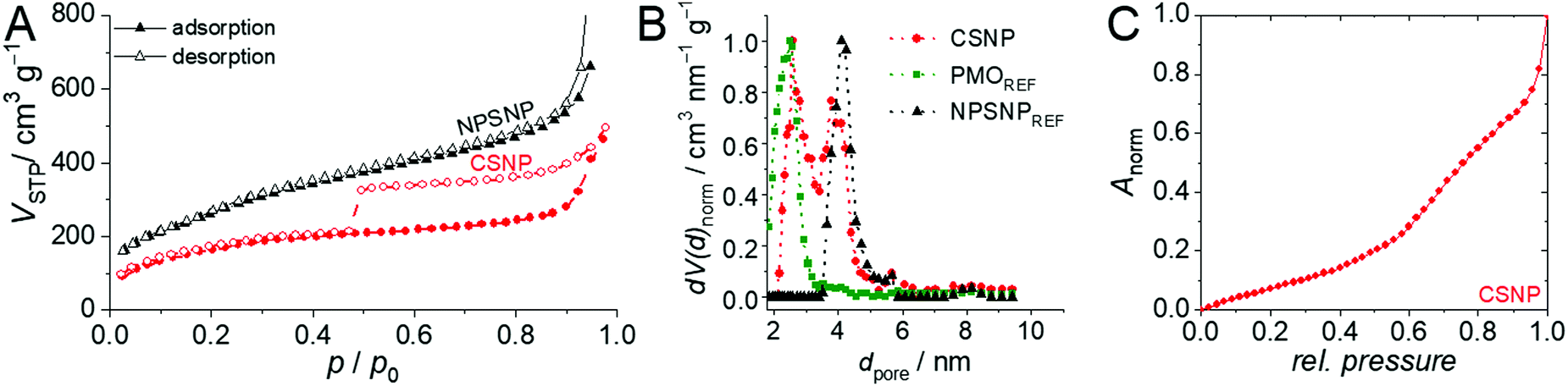 | ||
| Fig. 2 Results of physisorption measurements. (A) Nitrogen physisorption isotherms for NPSNPs (black) and CSNPs (red). (B) Normalized pore size distributions; the pore size distributions of a pure PMO material (red) and a pure NPSNP material (black) are shown for reference; note that the CSNP sample shows two maxima which can be assigned to the NPSNP core and the PMO shell. (C) Normalized water adsorption isotherm of CSNPs at 25 °C with an increasing slope from a relative pressure of 0.6. | ||
To investigate the hydrophilicity and the surface polarity of the materials, water physisorption measurements were performed (see Fig. 2C). Water adsorption on the CSNP material occurs in two steps. The first one starts at very low pressures, as is typical of strongly hydrophilic mesoporous silica. From a relative pressure of about 0.6 onwards, water uptake increases. Mietner et al.45 reported water sorption isotherms for pure mesoporous silica and phenylene-bridged PMO materials. The sorption curve measured on CSNPs can in principle be rationalized as a superposition of these isotherms, however, with the strong increases due to capillary condensation missing.
DLS measurements (Fig. 1C) and TEM investigations (Fig. 3) were used to characterize nanoparticles based on their sizes and morphologies (SEM images are shown in the ESI,† Section S2 and Fig. S2). Particle size distributions derived from DLS measurements show that the NPSNPs possess a hydrodynamic diameter of about 50 nm, which increases to 100 nm after the growth of the organosilica shell. However, the measurement also shows that the particles tend to agglomerate which was expected with regard to the pH-dependent zeta potential measurement, showing a value of −3.3 mV at the neutral pH of water. The increasing particle size also confirms the presence of the coating of the cores. TEM images show consistent results with particle diameters of 40 nm for the NPSNPs (Fig. 3A and B) and a total diameter of 80 nm for the CSNPs with a shell thickness of about 20 nm (Fig. 3C and D). In addition, TEM investigations could also confirm a certain agglomeration tendency for the CSNPs suggested by DLS analysis. Despite this, the spherical shape of both types of particles as well as their porosity is clearly revealed. It is also shown that the core–shell nanoparticles mostly include only a single core resulting overall in a monodisperse product.
 | ||
| Fig. 3 TEM images of NPSNPs (A and B) and CSNPs (C and D). (A) Non-agglomerated spherical NPSNPs with a particle diameter of ca. 40 nm; (B) close-up revealing the porous nature. (C) Spherical CSNPs with a particle size of ca. 80 nm containing in most of cases only one core; (D) close-up revealing the core–shell character of the CSNPs; the shell has a thickness of ca. 20 nm. | ||
Importance of silanol group density of NPSNPs
Multiple syntheses to reproduce the core–shell nanoparticles with a size of 80 nm revealed the importance of pretreatment of the NPSNP surface. In all syntheses, a complete organosilica-shell around the NPSNPs was obtained but the reproduction of the particle size failed whenever NPSNPs were used directly after the calcination (Fig. 4A and B). The particles were mostly built up from multiple cores, which had agglomerated before the coating with organosilica took place, resulting in much larger nanoparticles with a total size of approximately 350 nm. A few single core nanoparticles were larger with a size of 160 nm due to a thicker shell since the – also present – agglomerated NPSNPs consumed less of the organosilica species than individual particles would have done. However, NPSNP samples, which had been stored under ambient conditions for longer times (e.g., three months), consistently produced small CSNPs of similar size, with each CSNP containing only one NPSNP core. Concluding from these observations, we assumed that the density of silanol groups on the NPSNP surface plays a key role in the successful preparation of such CSNPs with favorable characteristics. This can be rationalized when the silanol groups act as anchor points for the attachment of the organosilica shell. Directly after calcination, the silanol surface density is strongly reduced due to temperature-induced condensation reactions of silanol groups to siloxane groups. During extended storage under typical laboratory conditions, including air humidity, new silanol groups are formed by the hydrolysis of siloxane bonds.46 To prove this hypothesis, additional silanol groups on the surface of the NPSNPs were generated using a mixture of hydrogen peroxide and ammonia to obtain pretreated silica nanoparticles (sample NPSNP_pt). With the NPSNP_pt, regular small CSNPs with one core per particle were obtained (Fig. 4C and E). Another option to adjust an appropriate amount of silanol groups on the surface of the NPSNPs is to use an extraction procedure instead of calcination for the removal of the surfactant. As extraction occurs at lower temperatures than calcination, condensation reactions are disfavored and NPSNP_ex with a high silanol surface density is obtained. In fact, NPSNP_ex samples deliver regular small CSNPs, each containing one core per particle (Fig. 4D and F). This line of reasoning is substantiated by FT-IR spectroscopy where the intensity of the signals related to silanol groups and water is higher for NPSNP_pt and NPSNP_ex samples than for freshly calcined NPSNPs (see ESI,† Section S3 and Fig. S3). Thus, both precursor nanoparticles, NPSNP_pt and NPSNP_ex, offer the possibility to synthesize regular small core–shell particles with time-efficiency. However, physisorption measurements revealed a strongly reduced surface area of 375 m2 g−1 for NPSNP_pt samples in contrast to NPSNP_ex with a surface area of 725 m2 g−1. The strongly decreased surface area for NPSNP_pt results from partial degradation of the silica during the pretreatment due to the slightly basic conditions. The value for NPSNP_ex is still smaller in comparison to the BET surface area of NPSNP_ca (1000 m2 g−1). This can probably be ascribed to residual surfactant molecules still present in the pores after extraction; weak FT-IR bands appearing at 2850 cm−1 and 2925 cm−1 for this sample support this interpretation (see ESI,† Section S2 and Fig. 2). Accordingly, with regard to applications requiring high porosity, the extracted nanoparticles NPSNP_ex or the calcined ones, NPSNP_ca, should be preferred. On the other hand, when traces of residual surfactant are causing concern (as in biomedical applications due to the cytotoxicity properties of the surfactant), NPSNP_pt or NPSNP_ca are the materials of choice. In any case, the NPSNP_ca have to be aged before use.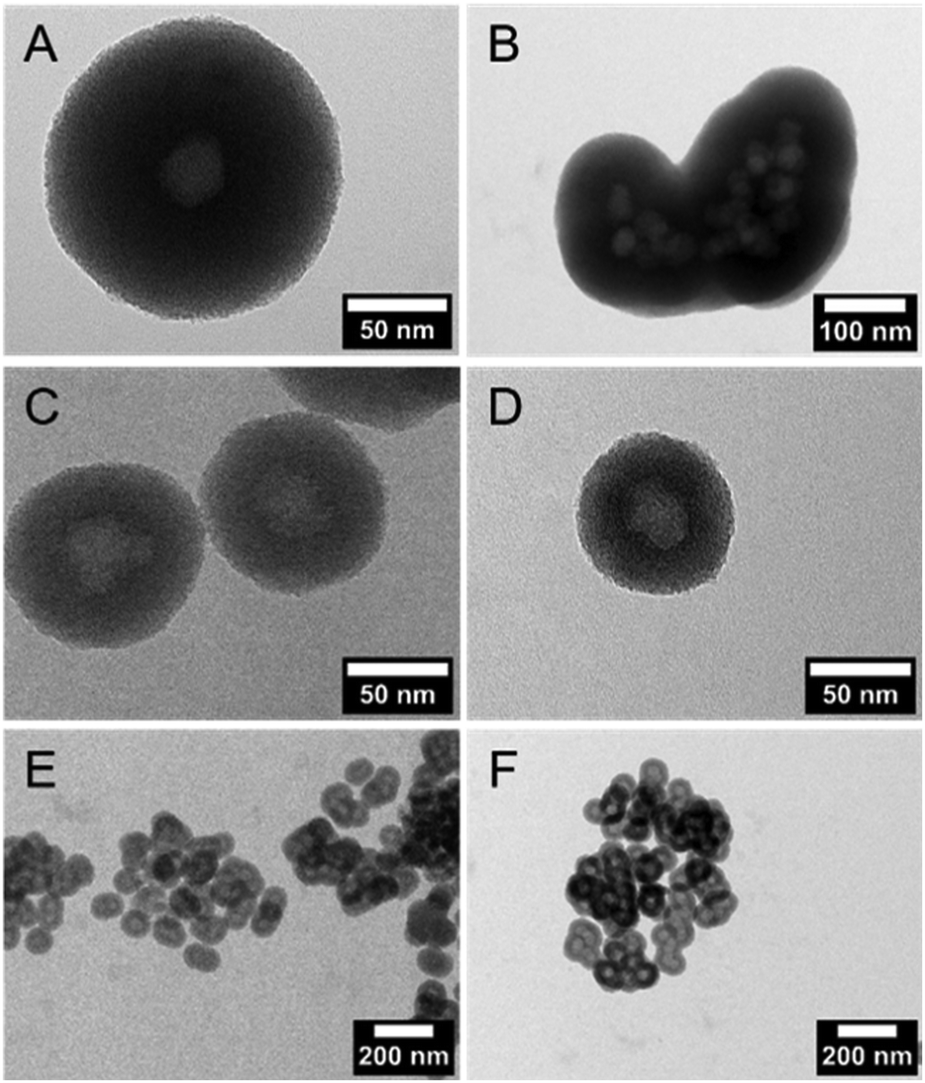 | ||
| Fig. 4 TEM images of CSNP resulting from freshly calcined NPSNPs either yield large spherical particles containing one core and a thick organosilica shell (A) or very large dysmorphic particles containing multiple agglomerated cores in the center of the organosilica shell (B). Syntheses based on NPSNP_pt (C and E) and NPSNP_ex (D and F) result in core–shell nanoparticles containing a single (or very few) core particle and possessing a shell with a thickness of ca. 20 nm, resulting in a total size of about 80 nm. | ||
Loading with dyes
The successful loading of the nanoparticles (specifically, CSNPs using NPSNP_ex as the core) with dyes was confirmed qualitatively by a change of color from white to blue, turquoise or yellow for CSNP_MB, CSNP_FCMB or CSNP_FC, respectively. Besides, there are additional bands in the FT-IR spectra (Fig. 5) assignable to the incorporated dyes. There are vibrations at 1600 cm−1 and from 1400–1340 cm−1 which can be attributed to vibrations of carboxylate groups or CN bonds as well as aromatic carbon–carbon bonds, which are part of the dyes. In addition, the incorporation of the dyes was indicated via TG-DSC measurements (see the ESI,† Section S4 and Fig. S4) showing a higher mass loss for loaded samples than for the blind sample CSNP_BL. Taking into account the mass loss due to adsorbed solvents, the amount of loaded dye was calculated. 5 mass% (m%) of the dye were loaded into CSNP_MB and 2 m% were loaded into CSNP_FCMB; for CSNP_FC, a mass loss due to the incorporated dye compared to CSNP_BL could not be detected.
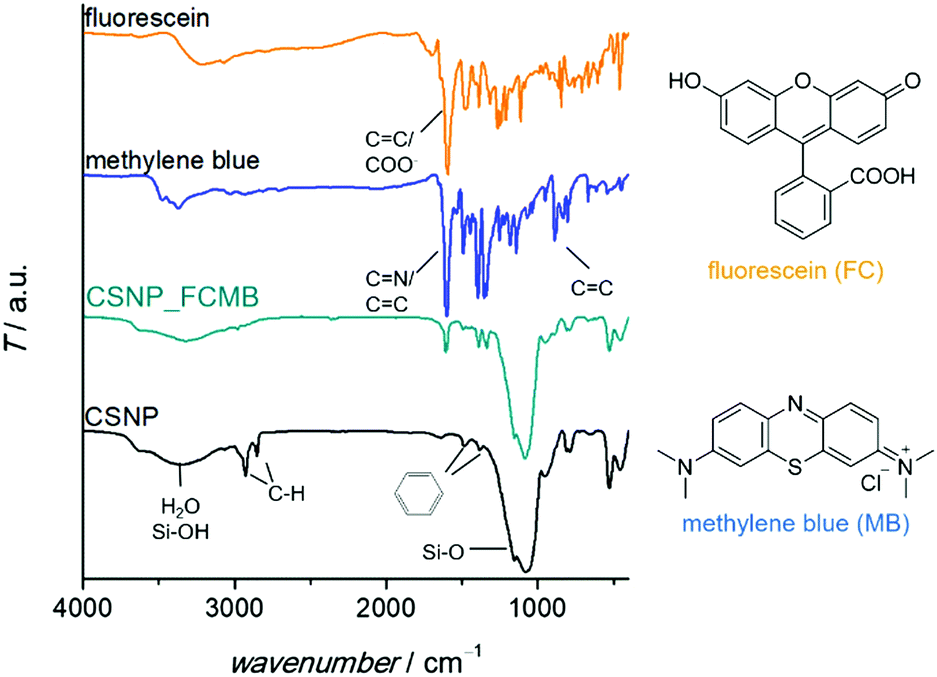 | ||
| Fig. 5 FT-IR spectra. Black trace: CSNP (based on NPSNP_ex core particles) before loading showing the characteristic bands for silica and for phenylene-bridged PMO. Green trace: CSNP_FCMB (CSNP after loading with both dyes): the presence of both dyes is confirmed due to additional bands resulting from the incorporated dyes. This mainly results from CN-bonds of MB and from carboxylate groups of FC and can also be found in the spectra of methylene blue (blue trace) and fluorescein (orange trace). The molecular structures of the dyes are depicted on the right. | ||
Loading with drugs
For the drugs, the loading was confirmed via HPLC measurements. The drug solutions were analyzed before and after the loading assuming that the difference between both concentrations had been adsorbed by the nanoparticles. With this method, for the nanoparticles loaded with a single drug, an amount of 51.8 μg mg−1 of ibuprofen and 163 μg mg−1 of procaine hydrochloride was determined. For the CSNPs containing both drugs, the values were slightly higher with 59.9 μg mg−1 of hydrophobic ibuprofen and 178.4 μg mg−1 of the hydrophilic drug.Release experiments with dyes
Release experiments were carried out in PBS at 37 °C for CSNPs loaded with either fluorescein or methylene blue or with both dyes simultaneously. As shown in Fig. S5 (see the ESI,† Section S5), the supernatants for the reference sample CSNP_FC have a weak yellow color and exhibit a strong green fluorescence under UV at the very beginning of the release, both of which fade rapidly. Correspondingly, the release curve in Fig. 6 (orange trace) shows an initial burst release quickly reaching a plateau with an amount of 3 μg mg−1 released after 35 days. According to the quick release, the diffusion path is assumed to be short and therefore the fluorescein is likely to be adsorbed on the surface and in the PMO shell of the particle.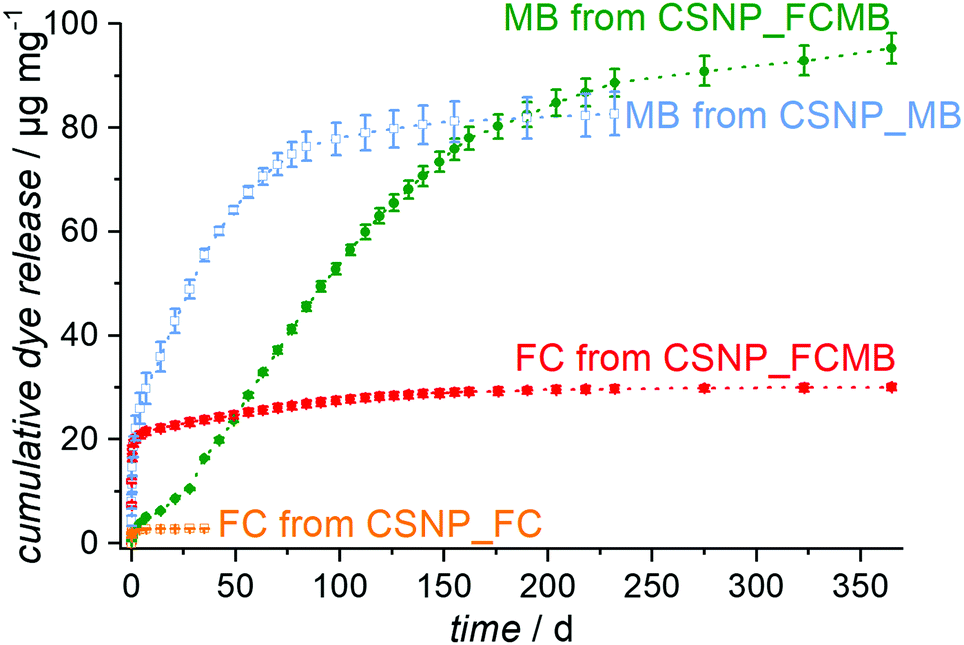 | ||
| Fig. 6 Cumulative release of dye release systems. CSNP_FC shows a fast burst release of fluorescein (orange curve); CSNP_MB shows (after an initial burst) an extended and nearly linear release of MB (blue curve). From CSNP_FCMB, a sequential release is observed. First, fluorescein is released quickly, again in a burst release fashion (red curve). The liberation of methylene blue is clearly delayed and shows a strong increase only after 28 days (green curve). Then, there is a continuous delivery of MB extending up to more than 200 days. | ||
In contrast, the reference sample CSNP_MB (supernatants are shown in the ESI,† Section S5 and Fig. S5) releases the hydrophilic methylene blue, after an initial rise, in a nearly linear fashion over several weeks (Fig. 6; blue curve). This initial release probably results from dye molecules adsorbed at the particle surface or located in the shell; however, the delayed release occurring later on indicates that most of the dye is accumulated in the hydrophilic core. The delayed release results from the longer diffusion path and the linearity continues until the dye reservoir inside the particle is consumed; then, also for CSNP_MB, a plateau is reached with a total amount of 83 μg mg−1 of released methylene blue after 232 days. It might be expected that the dual release from CSNP_FCMB nanoparticles loaded with both dyes should result in a mere combination of the individual release curves observed on CSNP_FC and CSNP_MB. This is not the case, however. Fig. 7 presents the supernatants obtained from the dual release experiment under visible and UV light. Under visible light, the samples show a yellow color in the beginning based on the fast release of fluorescein. Afterwards, the color changes to light green (after 1 d) and then to blue; thereafter, the intensity of the blue color increases before it starts to fade after 100 d, corresponding to a later start of the release of methylene blue and a highly extended liberation. Under UV light, only the release of fluorescein can be followed. The observations substantiate those made for the fast appearance of the yellow color under visible light, and its fast fading. However, as the fluorescence signal is much more sensitive, the liberation of very small amounts of fluorescein can still be detected even after more than 100 d.
 | ||
| Fig. 7 Supernatants of the release of CSNP_FCMB starting at 0.5 h up to a sample taken after 1 year. The yellow color and the strong fluorescence under UV light in the beginning show a fast release of FC. A change of color to blue occurs within the first week, afterwards the supernatants are blue and only slightly fluorescent due to the release of most of the FC and the sequential release of MB. | ||
The colorations were quantified and the release curves “FC from CSNP_FCMB” (red curve) and “MB from CSNP_FCMB” (green curve) are shown in Fig. 7. For fluorescein, on a first glance, the general shape of the release curves is similar, irrespective of whether we consider the release from CSNPs which were loaded with FC only or which were loaded with both dyes, both corresponding to a burst release. We therefore rationalize that the non polar fluorescein molecules are predominantly adsorbed on the outer hydrophobic PMO shell. However, for CSNP_FCMB, the total amount of released hydrophobic dye is 30 μg mg−1 and thus ten times higher than that for the single dye system. Besides, there is also a range (from day 1 to day 120) where the red curve shows a linear increase, corresponding to constant albeit small amounts of fluorescein liberation. This indicates that some fluorescein molecules are located deeper inside the particle leading to a longer diffusion path during the release experiments. Both these peculiarities could be explained when it is assumed that methylene blue and fluorescein molecules interact with one another via π–π interactions between their aromatic moieties. Although fluorescein–methylene blue complexes have not been described in the literature, it is well-known that especially methylene blue is prone to form such complexes with other aromatic entities (e.g., nucleo-bases, carbon nanotubes)47,48 and, specifically, also with itself, forming dimers.49,50 Here, we presume that methylene blue interacts with fluorescein, forming MB-FC complexes. Consequently, during loading, part of the fluorescein is carried deeper into the particles, since the methylene blue also interacts electrostatically with the hydrophilic silica in the core. Another difference can be found between CSNP_FCMB (Fig. 6 green curve) and CSNP_MB (blue curve). Methylene blue is also released from the CSNP_FCMB material at the beginning to a minor degree, possibly due to some dye molecules located in the outer PMO shell. However, the released amount is small and only increases after 28 d when most of the fluorescein has left the nanoparticles, indicating that the methylene blue molecules reside in the core of the particles. The shape of the release curve for MB from CSNP_FCMB is very similar to that of MB from CSNP_MB, apart from the shift of about 28 d. In total, 95 μg mg−1 of methylene blue are released from the CSNP_FCMB, extending over a period of at least one year.
Our interpretation of these results is as follows: during loading, the charged and polar MB is preferentially adsorbed in the inner hydrophilic core of the CSNPs. On their travel to the core, MB molecules may carry more FC molecules deeper into the nanoparticles. Nevertheless, the non polar FC molecules predominantly reside in the hydrophobic outer core. It is worth noting that the loading into the different parts of the nanoparticles occurs autonomously and self-selectively from one solution, i.e., a time-consuming individual loading is not necessary. When the release starts, FC is quickly liberated from the shell of the nanoparticles. Only when the pores of the PMO shell have been liberated to a large degree from the FC molecules, the release of MB molecules becomes significant. From then on, similar amounts of MB are released within a time unit in a regular fashion. Here, the PMO shell acts as a diffusion barrier which passively controls the release of MB molecules, firstly due to the long diffusion pathways the molecules have to travel, but secondly also due to the hydrophobic environment which they have to traverse. In this way, a controlled release lasting for a long time is established.
We investigated the particles at the end of the release (after one year) by TEM (see the ESI,† Section S6 and Fig. S6). The particles are basically still intact, at least the shell giving the particles their outer shape is still present. A more thorough time-dependent investigation of the stability of the particles will be given elsewhere.51
Release experiments with drugs
The release behavior of CSNPs as observed in the experiments with the dyes is also of considerable interest for the sequential release of drugs, for example when an antibiotic is initially released quickly and in high doses, followed by, e.g., an anti-inflammatory agent, which is provided for an extended time period. To test for the transferability of the sequential release system, we chose the ibuprofen and procaine hydrochloride system. This dual drug system was used in the testing of a sequential release system based on copolymer nanoparticles with a hydrophobic core and a hydrophilic shell.52 Ibuprofen is a hydrophobic pain-relieving and anti-inflammatory drug and procaine is a local anesthetic drug, made highly water soluble by the formation of the ammonium chloride salt through the addition of HCl (procaine hydrochloride).The resulting release curves, determined by HPLC on supernatants, are presented in Fig. 8. As expected, the hydrophobic drug ibuprofen is liberated with a burst release from the CSNP_IBU sample as well as from the dual drug system CSNP_IBUPRO. Almost all ibuprofen is liberated during the first hours, so that these curves soon reach a plateau with a maximum release of 4.0 μg mg−1 for the reference sample loaded only with ibuprofen and 6.5 μg mg−1 for the amount of ibuprofen from the dual drug system. Interestingly, the higher amount of ibuprofen released from the dual drug CSNPs in comparison to the single drug CSNPs corresponds to the results from the dye experiments. It may be hypothesized that a similar mechanism like the one proposed above is at work here, too, i.e., the introduction of the more hydrophobic substance to the hydrophilic core through interactions between the two guest molecules.
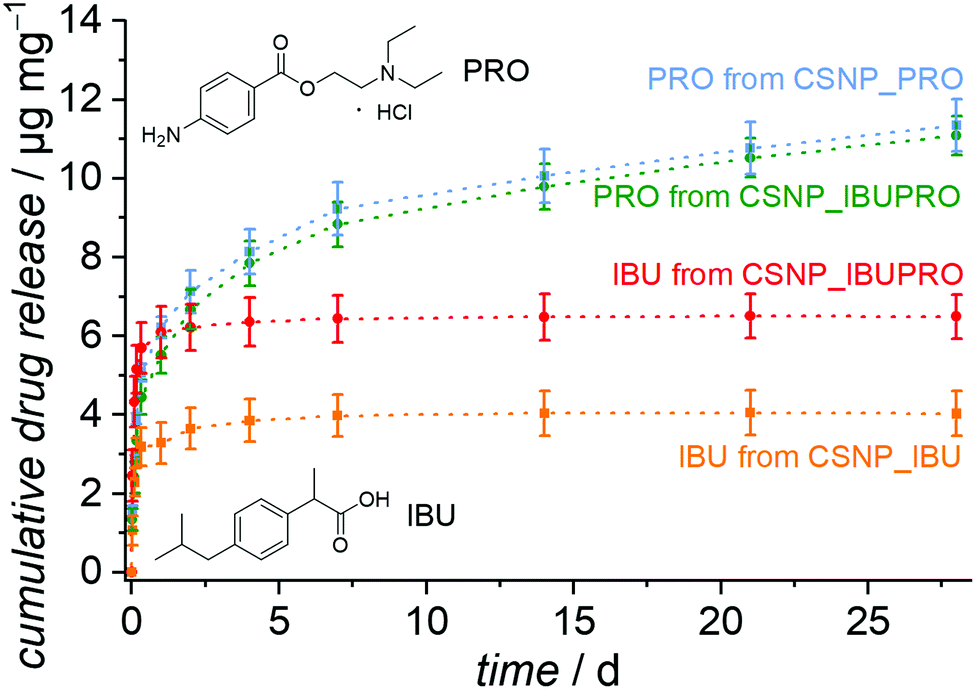 | ||
| Fig. 8 Cumulative release of drug release systems. CSNP_IBU shows a fast burst release of ibuprofen (orange curve); CSNP_PRO shows after an initial burst an extended and nearly linear release of procaine (blue curve). From CSNP_IBUPRO, a sequential release is observed. First, ibuprofen is released quickly (red curve), accompanied, however, by a starting release of procaine (green curve). After the early saturation of the ibuprofen release, the liberation of procaine attains a linear progression, extending for more than 28 days. | ||
For the hydrophilic drug procaine hydrochloride, there is also a strong increase of the amount released in the beginning of the experiment. The release curve then flattens out and from day 7 gives way to a regular continuous linear release, which would possibly still continue for longer than the actual observation time of 28 d. The released amounts after 28 d are 11.4 μg mg−1 for PRO from CSNP_PRO and 11.1 μg mg−1 for PRO from CSNP_IBUPRO.
In general, a similar interpretation holds for the dual drug release system as for the dual dye release system: the non polar substance ibuprofen resides in the outer PMO shell and becomes initially released in a burst release fashion. The charged and polar substance procaine hydrochloride is to a large extent adsorbed on the hydrophilic core, resulting in an extended linear release where the PMO shell serves as a diffusion barrier. The fact that the sequential character of the drug release is less obvious than that of the dye release may be attributed to several influences. First, the loading concentration was much lower due to the poor solubility of the drugs, meaning that the pores are not fully occupied, allowing for early “leakage” of the core guest substance PRO. Secondly, the fact that the drug molecules are smaller allows for a generally higher diffusivity, allowing for intermixing of the substances. Thirdly, the differences in polarity or hydrophilicity/hydrophobicity are probably less pronounced in the drug molecules than in the dye molecules. Correspondingly, the sequential character of the dual release of ibuprofen and procaine hydrochloride from copolymer core–shell nanoparticles is also less pronounced.52
Finally, we note that for a therapeutic application, the probably more suitable sequence for the release of PRO and IBU would be the other way round, with the procaine acting quickly as a local anaesthetic, followed by the anti-inflammatory drug ibuprofen, as was the case in the work on copolymer nanoparticles. In this respect, it is of interest that recently nanoporous core–shell nanoparticles with an “inverted” construction compared to ours (PMO core/nanoporous silica shell) were described.30
Conclusion
Core–shell nanoparticles consisting of a silica core and an organosilica-shell based on phenylene bridging units, which are both completely nanoporous, were successfully synthesized. For the synthesis, a sufficient density of surface silanol groups on the NPSNPs is necessary, which can be achieved by (i) storing calcined NPSNPs under ambient conditions for extended time periods; (ii) by a pretreatment of calcined NPSNPs with a mixture of hydrogen peroxide and ammonia; (iii) or by using extraction instead of calcination for surfactant removal. An extraction should be preferred due to economy of time and since the pretreatment leads to loss of porosity. The porosity was confirmed using physisorption measurements. The particles showed a bimodal pore size distribution and a physisorption isotherm indicating cavitation. By carrying out DLS and TEM investigations, a particle diameter of 80 nm and a spherical shape were revealed. The core particles have a diameter of ca. 40 nm, and the shell has a thickness of around 20 nm.To test the CSNPs as a sequential delivery system, proof-of-concept experiments were performed with dyes due to simpler analytical determination. The release experiments showed fast releases for the non polar model dye fluorescein indicating a shorter diffusion path and therefore its location in the hydrophobic PMO shell. Then, sequentially, polar methylene blue is released steadily over several months and for a total time of at least one year, proving its preferred accumulation in the hydrophilic silica core, which is supported by electrostatic interactions between the negatively charged silica at a pH of 7.4 and the positively charged dye. The long-term controlled release is ascribed to the PMO shell acting as a diffusion barrier.
After the successful proof of concept, the CSNPs were tested for sequential drug release applications using ibuprofen as an non polar drug molecule and procaine hydrochloride as a hydrophilic active agent. Also, in this case, a sequential release can be observed, although less well differentiated than in the case of the dyes, possibly due to the insufficient differences in polarity or to the simpler interdiffusion of the smaller molecules.
It is noteworthy that the selectivity of the two pore systems versus the adsorption of the polar/non polar drugs/dyes is strong enough so that the guest molecules can be loaded simultaneously in one procedure, avoiding the application of time-consuming individual loading procedures for each drug/dye.
In conclusion, our results prove that the continuously porous CSNPs are promising candidates for the sequential delivery of drugs. The pore systems of the nanoporous silica and the periodic mesoporous organosilica could in principle be further adapted to this task by chemical modification, e.g., by silanization or reactions at the organic bridges. Although the biocompatibility of the individual material components does not give rise to concern, cell culture experiments and animal tests with clinically relevant combinations of drugs should follow to further develop continuously nanoporous CSNPs for sequential drug release.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
This work was supported by the DFG Cluster of Excellence EXC 1077/1 “Hearing4all”, project number 390895286.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Laboratory of Nano and Quantum Engineering (LNQE) for access to the instrument for TEM measurements.References
- R. Ghosh Chaudhuri and S. Paria, Core/Shell Nanoparticles: Classes, Properties, Synthesis Mechanisms, Characterization, and Applications, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- T. Zhao, N.-T. Nguyen, Y. Xie, X. Sun, Q. Li and X. Li, Inorganic Nanocrystals Functionalized Mesoporous Silica Nanoparticles: Fabrication and Enhanced Bio-Applications, Front. Chem., 2017, 5, 1–8 Search PubMed.
- Y. Yang, X. Liu, X. Li, J. Zhao, S. Bai, J. Liu and Q. Yang, A Yolk – Shell Nanoreactor with a Basic Core and an Acidic Shell for Cascade Reactions, Angew. Chem., Int. Ed., 2012, 51, 9164–9168 CrossRef CAS.
- Y. Yang, W. Zhang, Y. Zhang, A. Zheng, H. Sun, X. Li, S. Liu, P. Zhang and X. Zhang, A Single Au Nanoparticle Anchored inside the Porous Shell of Periodic Mesoporous Organosilica Hollow Spheres, Nano Res., 2015, 8, 3404–3411 CrossRef CAS.
- D. Gontero, A. V. Veglia, A. G. Bracamonte and D. Boudreau, Synthesis of Ultraluminescent Gold Core–shell Nanoparticles as Nanoimaging Platforms for Biosensing Applications Based on Metal-Enhanced Fluorescence, RSC Adv., 2017, 7, 10252–10258 RSC.
- P. D. Nallathamby, J. Hopf, L. E. Irimata, T. L. McGinnity and R. K. Roeder, Preparation of Fluorescent Au–SiO2 Core–shell Nanoparticles and Nanorods with Tunable Silica Shell Thickness and Surface Modification for Immunotargeting, J. Mater. Chem. B, 2016, 4, 5418–5428 RSC.
- B. Chang, J. Guo, C. Liu, J. Qian and W. Yang, Surface Functionalization of Magnetic Mesoporous Silica Nanoparticles for Controlled Drug Release, J. Mater. Chem., 2010, 20, 9941–9947 RSC.
- D. Kwon, B. G. Cha, Y. Cho, J. Min, E.-B. Park, S.-J. Kang and J. Kim, Extra-Large Pore Mesoporous Silica Nanoparticles for Directing in Vivo M2 Macrophage Polarization by Delivering IL-4, Nano Lett., 2017, 17, 2747–2756 CrossRef CAS PubMed.
- A. Figuerola, R. Di Corato, L. Manna and T. Pellegrino, From Iron Oxide Nanoparticles towards Advanced Iron-Based Inorganic Materials Designed for Biomedical Applications, Pharm. Res., 2010, 62, 126–143 CrossRef CAS PubMed.
- H. C. Janßen, D. P. Warwas, D. Dahlhaus, J. Meißner, P. Taptimthong, M. Kietzmann, P. Behrens, J. Reifenrath and N. Angrisani, In Vitro and in Vivo Accumulation of Magnetic Nanoporous Silica Nanoparticles on Implant Materials with Different Magnetic Properties, J. Nanobiotechnol., 2018, 16, 96 CrossRef PubMed.
- K. Zhang, H. Chen, X. Chen, Z. Chen, Z. Cui and B. Yang, Monodisperse Silica-Polymer Core-Shell Microspheres via Surface Grafting and Emulsion Polymerization, Macromol. Mater. Eng., 2003, 288, 380–385 CrossRef CAS.
- K. Zhang, L. Zheng, X. Zhang, X. Chen and B. Yang, Silica-PMMA Core-Shell and Hollow Nanospheres, Colloids Surf., A, 2006, 277, 145–150 CrossRef CAS.
- S. Haffer, M. Tiemann and M. Fröba, Periodic Mesoporous Organosilica (PMO) Materials with Uniform Spherical Core–Shell Structure, Chem. – Eur. J., 2010, 16, 10447–10452 CrossRef CAS PubMed.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Silica-Based Mesoporous Organic-Inorganic Hybrid Materials, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- F. Tang, L. Li and D. Chen, Mesoporous Silica Nanoparticles: Synthesis, Biocompatibility and Drug Delivery, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- N. Ehlert, P. P. Mueller, M. Stieve, T. Lenarz and P. Behrens, Mesoporous Silica Films as a Novel Biomaterial: Applications in the Middle Ear, Chem. Soc. Rev., 2013, 42, 3847–3861 RSC.
- S. Williams, A. Neumann, I. Bremer, Y. Su, G. Dräger, C. Kasper and P. Behrens, Nanoporous Silica Nanoparticles as Biomaterials: Evaluation of Different Strategies for the Functionalization with Polysialic Acid by Step-by-Step Cytocompatibility Testing, J. Mater. Sci.: Mater. Med., 2015, 26, 125 CrossRef PubMed.
- K. Möller and T. Bein, Talented Mesoporous Silica Nanoparticles, Chem. Mater., 2017, 29, 371–388 CrossRef.
- Y. Zhou, G. Quan, Q. Wu, X. Zhang, B. Niu, B. Wu, Y. Huang, X. Pan and C. Wu, Mesoporous Silica Nanoparticles for Drug and Gene Delivery, Acta Pharm. Sin. B, 2018, 8, 165–177 CrossRef.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, Novel Mesoporous Materials with a Uniform Distribution of Organic Groups and Inorganic Oxide in Their Frameworks, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Mesoporous Sieves with Unified Hybrid Inorganic/Organic Frameworks, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Periodic Mesoporous Organosilicas with Organic Groups inside the Channel Walls, Nature, 1999, 402, 867–871 CrossRef CAS.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Periodic Mesoporous Organosilicas: From Simple to Complex Bridges; a Comprehensive Overview of Functions, Morphologies and Applications, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- J. G. Croissant, X. Cattoën, M. C. W. Man, J.-O. Durand and N. M. Khashab, Syntheses and Applications of Periodic Mesoporous Organosilica Nanoparticles, Nanoscale, 2015, 7, 20318–20334 RSC.
- S. S. Park, M. S. Moorthy and C.-S. Ha, Periodic Mesoporous Organosilicas for Advanced Applications, NPG Asia Mater., 2014, 6, e96 CrossRef CAS.
- B. Guan, Y. Cui, Z. Ren, Z. Qiao, L. Wang, Y. Liu and Q. Huo, Highly Ordered Periodic Mesoporous Organosilica Nanoparticles with Controllable Pore Structures, Nanoscale, 2012, 4, 6588 RSC.
- Z. Teng, X. Su, Y. Zheng, J. Zhang, Y. Liu, S. Wang, J. Wu, G. Chen, J. Wang, D. Zhao and G. Lu, A Facile Multi-Interface Transformation Approach to Monodisperse Multiple-Shelled Periodic Mesoporous Organosilica Hollow Spheres, J. Am. Chem. Soc., 2015, 137, 7935–7944 CrossRef CAS PubMed.
- J. Gao, W. Kong, L. Zhou, Y. He, L. Ma, Y. Wang, L. Yin and Y. Jiang, Monodisperse Core-Shell Magnetic Organosilica Nanoflowers with Radial Wrinkle for Lipase Immobilization, Chem. Eng. J., 2017, 309, 70–79 CrossRef CAS.
- J. Liu, H. Q. Yang, F. Kleitz, Z. G. Chen, T. Yang, E. Strounina, G. Q. Lu and S. Z. Qiao, Yolk-Shell Hybrid Materials with a Periodic Mesoporous Organosilica Shell: Ideal Nanoreactors for Selective Alcohol Oxidation, Adv. Funct. Mater., 2012, 22, 591–599 CrossRef CAS.
- L. Luo, Y. Liang, E. S. Erichsen and R. Anwander, Hierarchical Mesoporous Organosilica – Silica Core – Shell Nanoparticles Capable of Controlled Fungicide Release, Chem. – Eur. J., 2018, 24, 7200–7209 CrossRef CAS PubMed.
- Y. Liu, B. Huang, J. Zhu, K. Feng, Y. Yuan and C. Liu, Dual-Generation Dendritic Mesoporous Silica Nanoparticles for Co-Delivery and Kinetically Sequential Drug Release, RSC Adv., 2018, 8, 40598–40610 RSC.
- D. Song, Y. Sun, Q. Zhang, P. Zhang, Y. Guo and J. Leng, Fabrication of Propylsulfonic Acid Functionalized SiO2 Core/PMO Shell Structured PrSO3H–SiO2@Si(R)Si Nanospheres for the Effective Conversion of D-Fructose into Ethyl Levulinate, Appl. Catal., A, 2017, 546, 36–46 CrossRef CAS.
- X. Li, L. Zhou, Y. Wei, A. M. El-Toni, F. Zhang and D. Zhao, Anisotropic Growth-Induced Synthesis of Dual-Compartment Janus Mesoporous Silica Nanoparticles for Bimodal Triggered Drugs Delivery, J. Am. Chem. Soc., 2014, 136, 15086–15092 CrossRef CAS PubMed.
- Z. Zhou, M. Jafari, V. Sriram, J. Kim, J.-Y. Lee, S. J. Ruiz-Torres and S. E. Waltz, Delayed Sequential Co-Delivery of Gefitinib and Doxorubicin for Targeted Combination Chemotherapy, Mol. Pharmaceutics, 2017, 14, 4551–4559 CrossRef CAS PubMed.
- C. Maderuelo, A. Zarzuelo and J. M. Lanao, Critical Factors in the Release of Drugs from Sustained Release Hydrophilic Matrices, J. Controlled Release, 2011, 154, 2–19 CrossRef CAS PubMed.
- W. Guo, J. Zhang, H. Lin, J. Jiang, D. Xiang and F. Qu, Long-Term Controlled Release of Dual Drugs from MBG/PLGA Composite Microspheres, J. Sol–Gel Sci. Technol., 2013, 68, 31–38 CrossRef CAS.
- Y. Liu, C. Jolly, S. Braun, T. Stark, E. Scherer, S. K. Plontke and J. Kiefer, In Vitro and In Vivo Pharmacokinetic Study of a Dexamethasone-Releasing Silicone for Cochlear Implants, Arch. Oto-Rhino-Laryngol., 2016, 273, 1745–1753 CrossRef PubMed.
- Z.-A. Qiao, L. Zhang, M. Guo, Y. Liu and Q. Huo, Synthesis of Mesoporous Silica Nanoparticles via Controlled Hydrolysis and Condensation of Silicon Alkoxide, Chem. Mater., 2009, 21, 3823–3829 CrossRef CAS.
- A. Neumann, A. Christel, C. Kasper and P. Behrens, BMP2-Loaded Nanoporous Silica Nanoparticles Promote Osteogenic Differentiation of Human Mesenchymal Stem Cells, RSC Adv., 2013, 3, 24222–24230 RSC.
- D. M. Knotter, S. de Gendt, P. W. Mertens and M. M. Heyns, Silicon Surface Roughening Mechanisms in Ammonia Hydrogen Peroxide Mixtures, J. Electrochem. Soc., 2000, 147, 736–740 CrossRef CAS.
- H. Fullriede, P. Abendroth, N. Ehlert, K. Doll, J. Schäske, A. Winkel, S. N. Stumpp, M. Stiesch and P. Behrens, pH-Responsive Release of Chlorhexidine from Modified Nanoporous Silica Nanoparticles for Dental Applications, BioNanoMaterials, 2016, 17, 59–72 Search PubMed.
- N. Hao, Y. Yang, H. Wang, P. A. Webley and D. Zhao, Synthesis of Large-Pore Phenyl-Bridged Mesoporous Organosilica with Thick Walls by Evaporation-Induced Self-Assembly for Efficient Benzene Adsorption, J. Colloid Interface Sci., 2010, 346, 429–435 CrossRef CAS PubMed.
- M. Thommes, B. Smarsly, M. Groenewolt, P. I. Ravikovitch and A. V. Neimark, Adsorption Hysteresis of Nitrogen and Argon in Pore Networks and Characterization of Novel Micro- and Mesoporous Silicas, Langmuir, 2006, 22, 756–764 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- J. B. Mietner, F. J. Brieler, Y. J. Lee and M. Fröba, Properties of Water Confined in Periodic Mesoporous Organosilicas: Nanoimprinting the Local Structure, Angew. Chem., Int. Ed., 2017, 56, 12348–12351 CrossRef CAS PubMed.
- S. L. Warring, D. A. Beattie and A. J. McQuillan, Surficial Siloxane-to-Silanol Interconversion during Room-Temperature Hydration/Dehydration of Amorphous Silica Films Observed by ATR-IR and TIR-Raman Spectroscopy, Langmuir, 2016, 32, 1568–1576 CrossRef CAS PubMed.
- E. Tuite and J. M. Kelly, The Interaction of Methylene Blue, Azure B, and Thionine with DNA: Formation of Complexes with Polynucleotides and Mononucleotides as Model Systems, Biopolymers, 1995, 35, 419–433 CrossRef CAS.
- V. V. Chagovets, M. V. Kosevich, S. G. Stepanian, O. A. Boryak, V. S. Shelkovsky, V. V. Orlov, V. S. Leontiev, V. A. Pokrovskiy, L. Adamowicz and V. A. Karachevtsev, Noncovalent Interaction of Methylene Blue with Carbon Nanotubes: Theoretical and Mass Spectrometry Characterization, J. Phys. Chem. C, 2012, 116, 20579–20590 CrossRef CAS.
- O. Yazdani, M. Irandoust, J. B. Ghasemi and S. Hooshmand, Thermodynamic Study of the Dimerization Equilibrium of Methylene Blue, Methylene Green and Thiazole Orange at Various Surfactant Concentrations and Different Ionic Strengths and in Mixed Solvents by Spectral Titration and Chemometric Analysis, Dyes Pigm., 2012, 92, 1031–1041 CrossRef CAS.
- A. Ghanadzadeh Gilani, T. Ghorbanpour and M. Salmanpour, Additive Effect on the Dimer Formation of Thiazine Dyes, J. Mol. Liq., 2013, 177, 273–282 CrossRef CAS.
- M. Jahns, D. P. Warwas, T. R. Jones, K. Nolte, S. Behrens and P. Behrens, Effect of Release Conditions on Porosity and Degradation of Nanoporous Nanoparticles, to be published.
- X. Xu, G. R. Shan and P. Pan, Controlled Co-Delivery of Hydrophilic and Hydrophobic Drugs from Thermosensitive and Crystallizable Copolymer Nanoparticles, J. Appl. Polym. Sci., 2016, 133, 1–10 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sorption experiments (hysteresis scan of CSNPs; SEM images of nanoparticles; IR-measurements of different types of NPSNPs; thermogravimetric measurements of blind and loaded samples; visual photographs of supernatants of release studies on reference samples; TEM images of particles after release). See DOI: 10.1039/c9tb01846h |
| This journal is © The Royal Society of Chemistry 2020 |