
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclobutene based macrocycles†‡
Pan
Wang§
 ab,
Ruqiang
Lu§
a,
Arthur
France-Lanord
c,
Yanming
Wang
c,
Jingjing
Zhou
b,
Jeffrey C.
Grossman
*c and
Timothy M.
Swager
*a
ab,
Ruqiang
Lu§
a,
Arthur
France-Lanord
c,
Yanming
Wang
c,
Jingjing
Zhou
b,
Jeffrey C.
Grossman
*c and
Timothy M.
Swager
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Massachusetts 02139, USA. E-mail: tswager@mit.edu
bSchool of Science, Westlake University, Hangzhou 310024, Zhejiang Province, China
cDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Massachusetts 02139, USA. E-mail: jcg@mit.edu
First published on 19th November 2020
Abstract
Nanoscopic macrocycles could have unusual magnetic, optical, and electronic properties when compared to their linear counterparts. Conjugated π-systems in unsaturated macrocycles are particularly interesting as they have no end groups that limit electronic delocalization in equivalent linear oligomers. The rigid four-membered ring structure in 3,4-bis(methylene)cyclobutene with vicinal connections provides a vertex with an angle slightly less than 90°, which promotes macrocycle formation. We report herein a facile high-yielding synthesis of a series of 3,4-bis(methylene)cyclobutene-base π-conjugation macrocycles. The structure–property studies reveal that the smaller macrocycles are rigid crystalline frameworks and display symmetrical conformations in solution. The electrochemical, photophysical and magnetic properties of these macrocycles were also studied with a framework of characterization methods, revealing their size- and linkage-dependent properties. Density functional theory (DFT) calculations and molecular dynamics (MD) simulations at the molecular level suggest that several possible configurations are possible for macrocycles with larger ring sizes.
Introduction
The concepts of supramolecular chemistry provide a natural translation of molecule constructions to create functional materials.1–3 Supramolecular assemblies containing extended π-conjugated macrocycles are particularly attractive for applications in organic electronics/devices,4,5 examples of note include cyclic porphyrins,6,7 annulenes,8–10 phenylenes11–15 and oligothiophenes.16–18 The most common building blocks of π-extended macrocycles are phenylenes, ethynylenes, thiophenes and pyrroles. Methods to achieve high yields of macrocycles include the use of templates,19 and post cyclization reactions are needed to release the macrocyclic products from the templates. Most macrocyclic precursors are rigid structures that create vertices with angular orientations larger than 90° at their reacting sites. An advantage of the cyclobutene structure is that it can create a vertex angle of about 90° which will be more favorable for cyclic structures, but has rarely been explored in macrocycle synthesis.As a highly strained and air-sensitive isomer of benzene, 3,4-bis(methylene)cyclobutene has drawn much attention since it was first synthesized in the 1960s.20 The air-stable analogues were developed by Toda et al., by the thermal conversion of diallenes in the crystalline state.21–23 The introduction of aromatic substituents on the exocyclic methylenes and electron-withdrawing groups into the cyclobutene skeleton provides improved stability. Four-membered ring compounds that contain a bis(methylene)cyclobutene base structure have been extensively studied,23–26 and we have been interested to extend this unique structure as a vertex in macrocyclic compounds with different conjugation linkages to produce a series of fully conjugated macrocycles with persistent defined shapes.
Herein, we report the efficient template-free synthesis of a series of cyclobutene-based π-conjugation macrocycles with different ring sizes in high yields benefiting from the 3,4-bis(methylene)cyclobutene core structure bearing angular orientations less than 90°. Two-dimensional nuclear magnetic resonance (2D) NMR and variable temperature (VT)-NMR analysis demonstrated that these macrocycles are highly symmetrical in solution and single crystal X-ray crystallography revealed the packing mode and the planarity of the main-chain carbon atoms. The synthesized macrocycles can be chemically oxidized and the formation of radical cations (that can provide polarons in the solid state) was monitored by electron paramagnetic resonance (EPR) spectroscopy. SEM images revealed the relatively rigid crystalline macrocyclic framework. The optical and electrochemical properties were investigated, revealing the size- and linkage-dependent properties of these macrocycles (Fig. 1).
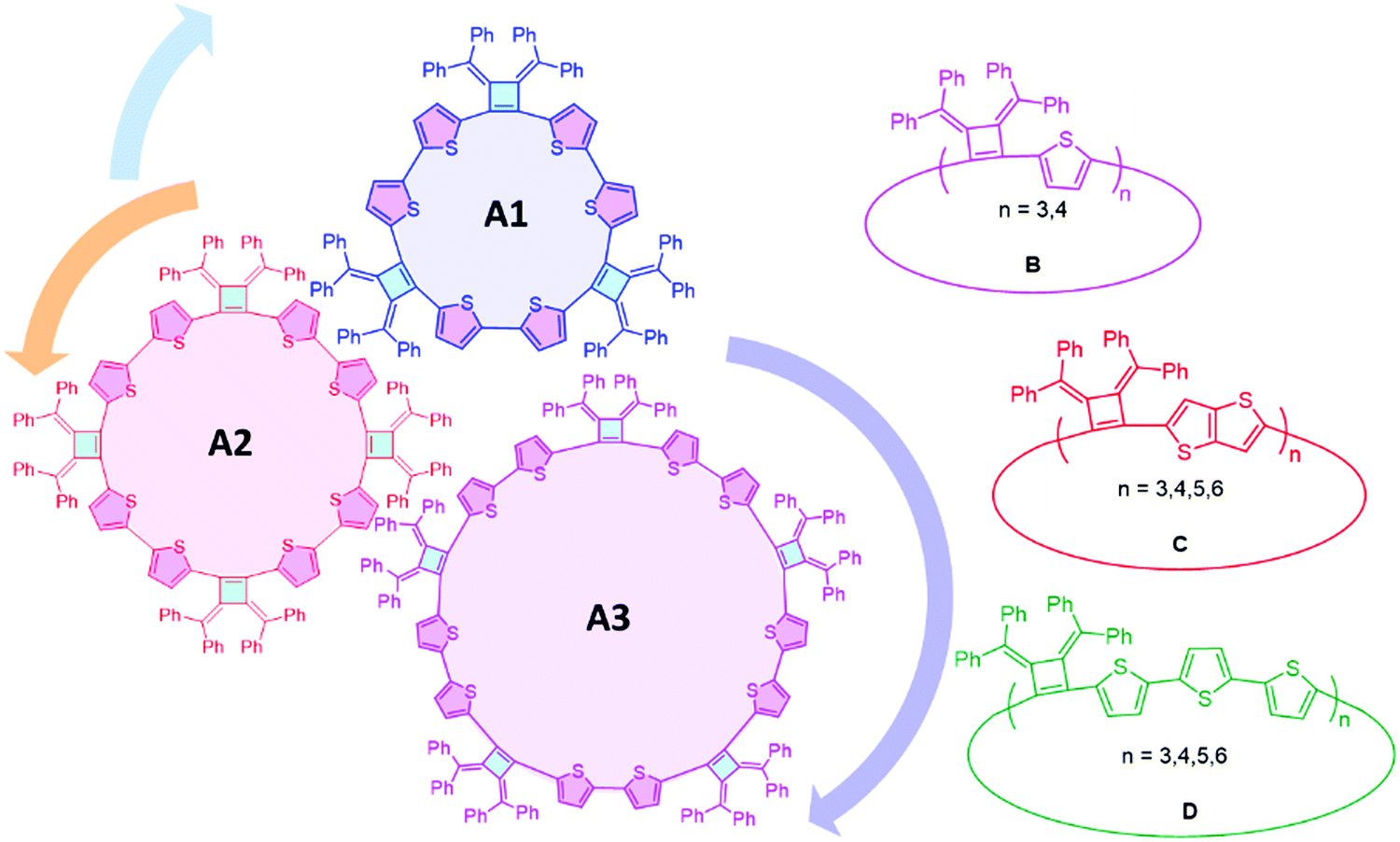 | ||
| Fig. 1 Schematic structures of synthesized cyclobutene based macrocycles. | ||
Materials design and synthesis
Our interest in the target macrocycles were guided by the computed geometries of 1,2-dibromo-3,4-bis(diphenylmethylene)cyclobutene 1, using DFT (ωB97X-D/6-31G*). The optimized geometry displays notable intramolecular π–π interactions between the two inner phenyl rings on the two double bonds. The distance of C3–C4 is 1.52 Å, and the distance of C1–C2 is 1.35 Å (Scheme 1). The angles between C1–Br bond and C2–Br bond is computed to be 82°, a value similar to that obtained in a single crystal structure of an analogue.22 The rigidly fixed molecular geometry of 1 is poised to bias away from the formation of linear oligomers in cross-coupling sequences. This geometric relationship provides a rational and practical synthesis of macrocycles via cross-coupling reactions of 1,2-dibromo-3,4-bis(diphenylmethylene)cyclobutene with mono-thiophene, fused thiophene, bi-thiophene or tri-thiophene comonomers.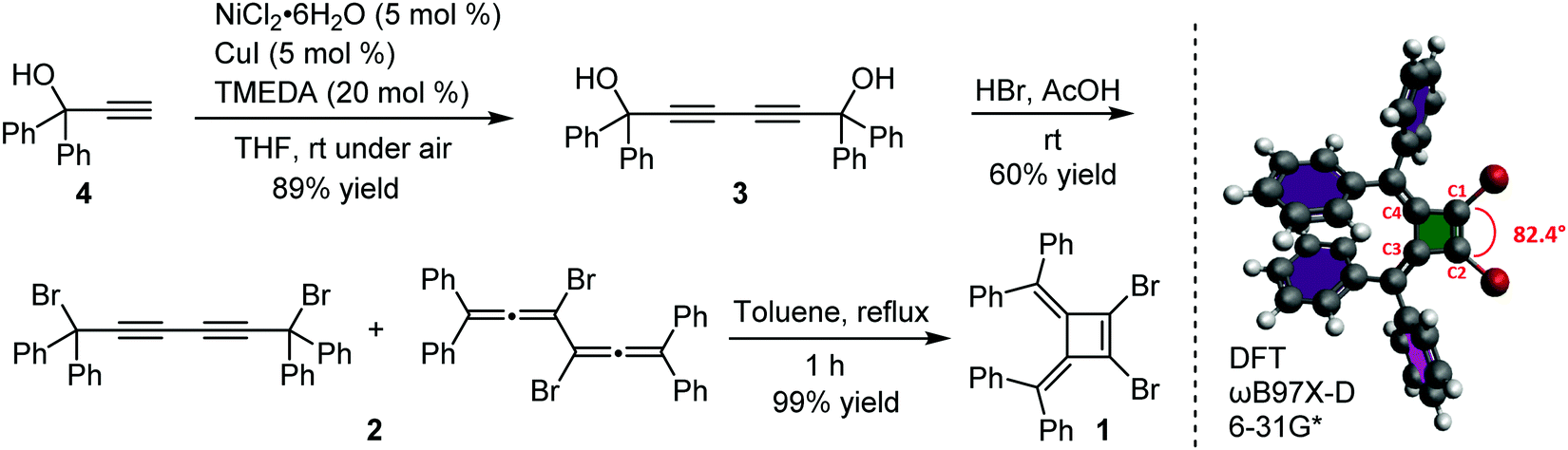 | ||
| Scheme 1 Synthesis and optimized structure of 1,2-dibromo-3,4-bis(diphenylmethylene)cyclobutene. | ||
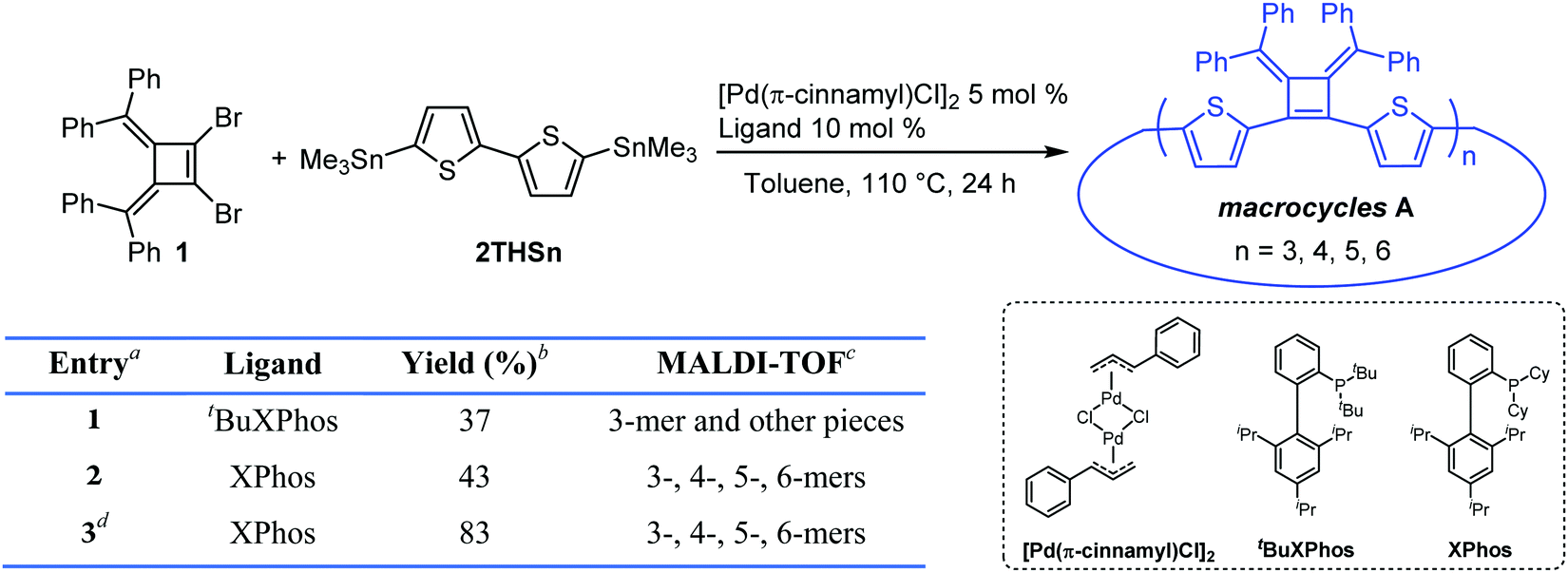
We synthesized 1,2-dibromo-3,4-bis(diphenylmethylene)cyclobutene 1via a three-step synthetic route as shown in Scheme 1. Glaser-Hay oxidative coupling reaction of the corresponding terminal alkyne 4 gave compound 3 in 89% yield. Following Toda's synthetic method,22 upon treatment of 3 with hydrobromic acid in acetic acid provided the brominated products 2 as a mixture. Thermal rearrangement in refluxing toluene afforded pure cyclobutene 1 in quantitative yield within 1 h. We explored cyclization reactions using Stille-coupling of cyclobutene 1 with 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (2THSn). As shown in Table 1, using [Pd(π-cinnamyl)Cl]2 as the precatalyst and tBuXPhos as the ligand, the Stille-coupling reaction in toluene after 24 h gave a 34% yield of soluble red solids and side products that are not soluble in common organic solvents (Table 1, entry 1). MALDI-TOF mass spectrometry analysis reveals that the soluble portion are primarily macrocycles with 3 repeating units. Changing the ligand from tBuXPhos to XPhos increased the yield of the soluble portion to 43% (Table 1, entry 2). Microwave reaction conditions with a shortened reaction time to 4 h, increased the yield of soluble products to 83% (Table 1, entry 3). The MALDI-TOF mass spectrum analysis of macrocycle mixtures indicate the presence of four different ring-size macrocycles, namely the 3-mer A1, 4-mer A2, 5-mer A3 and a trace of the 6-mer A4 (From MALDI-TOF mass spectrometry: A1 (m/z = 1632.223, n = 3), A2 (m/z = 2176.527, n = 4), A3 (m/z = 2720.659, n = 5), A4 (m/z =3265.815, n = 6). The observed isotope distributions of all the different ring-size macrocycles from MALDI-TOF mass spectrometry are in agreement with the calculated values.
|
|
|||
|---|---|---|---|
| Entrya | Ligand | Yieldb (%) | MALDI-TOFc |
| a Reaction conditions: 0.15 mmol scale, 0.05 M, under N2 atmosphere. b Yield of the soluble mixture of macrocycle portion. Macrocycles were purified by passing through a pad of Celite and precipitated from MeOH. c 2-[(2E)-3-(4-tert-Butylphenyl)-2-methylprop-2-enylidene]malononitrile (DCTB) was used as the matrix in MALDI-TOF mass spectrometry with a PEG3000 calibration standard. d The reaction was carried out in a microwave reactor at 110 °C for 4 h. | |||
| 1 | t BuXPhos | 37 | 3-mer and other pieces |
| 2 | XPhos | 43 | 3-, 4-, 5-, 6-mers |
| 3d | XPhos | 83 | 3-, 4-, 5-, 6-mers |
The relative constitutions of macrocycles A1, A2 and A3 were quantified and isolated using high-performance liquid chromatography (HPLC) analysis (Fig. 2c). A4 was present only in trace quantities and wasn’t studied in detail. UV-vis spectroscopic analysis (Fig. 2d) shows that A1 has the maximum absorption band at 426 nm, whereas A2 is red shifted to 440 nm as a result of its longer effective conjugation length. A3 shows an absorption band at 427 nm similar with A1, suggesting a twisted configuration of the larger ring. Our observations suggest that there is no full delocalization of the whole π-system as the ring size increases, thus limiting the conjugation length in the ring. The molar absorption coefficients of the three macrocycles are around 7.0 × 104 M−1 cm−1 to 8.2 × 104 M−1 cm−1 at 427 nm, and the relative concentrations in the soluble reaction products were roughly estimated based on the integrated peak areas from HPLC analysis. As can be seen in Fig. 2c, the HPLC trace area with monitoring at 430 nm indicates the relative percentages of A1, A2, A3 to be 55.2%, 26.4% and 18.4% respectively. All of the macrocycles are emissive when dissolved in tetrahydrofuran (THF) and show a similar emission profile around 630 nm (Fig. 2e and f).
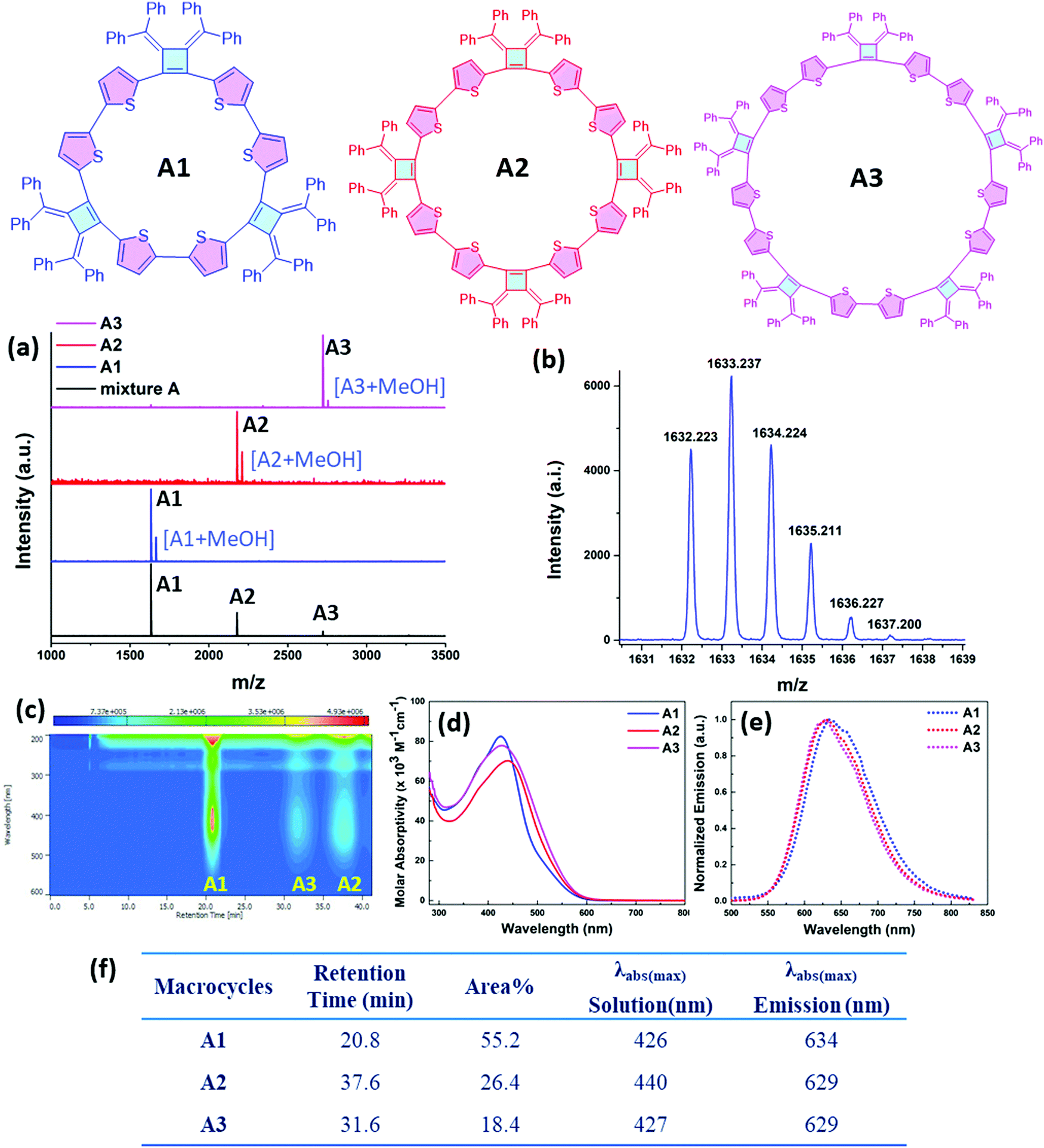 | ||
| Fig. 2 (a) MALDI-TOF mass spectrometry data of macrocycle mixtures A and macrocycles A1, A2 and A3 after HPLC separation. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as the matrix with PEG3000 as a calibration standard. (b) Isotope distribution of A1 in the MALDI-TOF mass spectrometry. (c) 2D contour map UV-vis spectrum for the HPLC separation in heptane/THF using a ZORBAX-CN prep-column. (d) UV-vis spectra and (e) normalized emission spectra of the isolated macrocycle A1, A2 and A3 in THF solutions. The emission spectra were measured after excitation at the maximum of each absorption wavelength. (f) A summary table of the HPLC separation results, and photophysical properties of the compositions in macrocycle A mixtures. | ||
1H NMR spectra of the three isolated macrocycles A1–A3 indicate these macrocycles are highly symmetric in solution that only one set of proton signals of the repeating unit was observed. The combined information from 2D NMR heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond correlation (HMBC) analysis, allows for the assignment of all the proton signals (Fig. S1–S3, ESI‡). The protons on thiophene ring display different chemical shifts for each ring-size macrocycle (Fig. 3). For example, as shown for A1, the proton Ha, which is the closest to the cyclobutene ring, is shielded with a chemical shift at 5.67 ppm, whereas the proton Hb is de-shielded and resonates at 6.58 ppm. The different chemical shifts of the different ring-size macrocycles are indicative of the differences in their electronic and conformational structures.
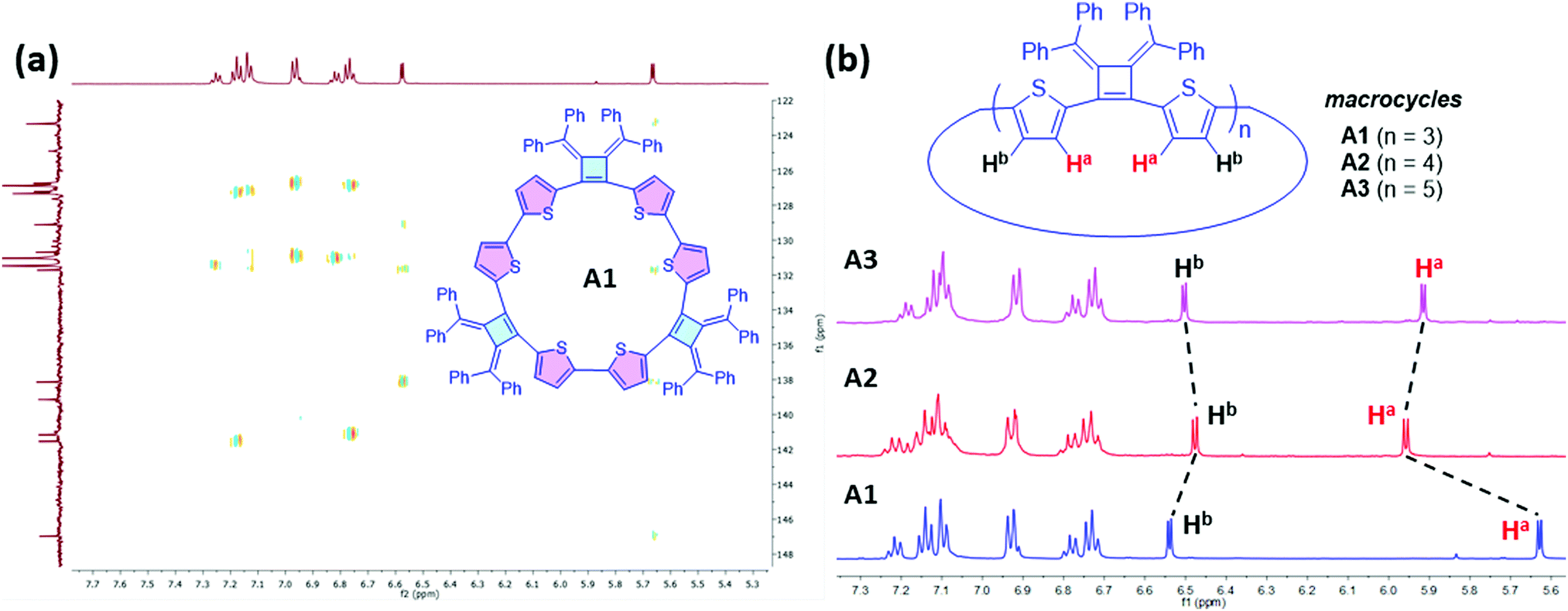 | ||
| Fig. 3 Structural elucidation of Macrocycles A1, A2 and A3 by NMR spectroscopy. (a) 2D NMR HMBC (500 MHz, 25 °C, THF-d8) spectrum of A1. (b) 1H NMR (500 MHz, 25 °C, THF-d8) spectra of A1, A2 and A3. | ||
X-ray crystal structure and self-assembly
The structure of macrocycle A1 was elucidated by X-ray crystallography. The single crystal was prepared by vapor diffusion of methanol into a chlorobenzene solution of A1. The single crystal structure (Fig. 4a) reveals a macrocycle in an isosceles triangular shape with cyclobutene rings are located at each vertex and thiophene rings on sides. The internuclear distances of C13–C2 and C14–C7 are 9.152 Å and 9.162 Å respectively, while the distance of C1–C8 is 8.894 Å. The structure produces a large inner cavity with potential applications in host–guest chemistry.2,27 The bithiophene connector favors a trans configuration on the two longer sides of the isosceles triangle, with a cis configuration providing the reduced length on the other side. It is noteworthy that we do not observe 1H NMR resonances for the different conformations in solution even under low temperatures (0 °C to −90 °C in VT-NMR, Fig. S4, ESI‡), indicating that either there is one single conformer in solution, or there is a very rapid interconversion of the two isomers. The conformations also prevent the main-chain atoms of the macrocyclic structure from being in plane of the isosceles triangle. The root-mean-square deviation (RMSD) of the main-chain carbon atoms from the calculated plane is 0.172 Å. The phenyl rings on the cyclobutenes are twisted out of plane to reduce steric repulsions to each other. The two inner phenyl rings twist to promote face-to-face π-stacking with a distance of 3.419 Å between the planes of the phenyls (the distance between C615 and the mean plane of C511, C513 and C515) (Fig. 4a and Fig. S12a, ESI‡). A1 forms stacked dimers with a distance of 4.223 Å between the planes of the isosceles triangles (Fig. 4c). These dimers further organize in a 1-D ‘slipped-stacked’ fashion with a distance of 9.454 Å along a axis (Fig. S12b, ESI‡).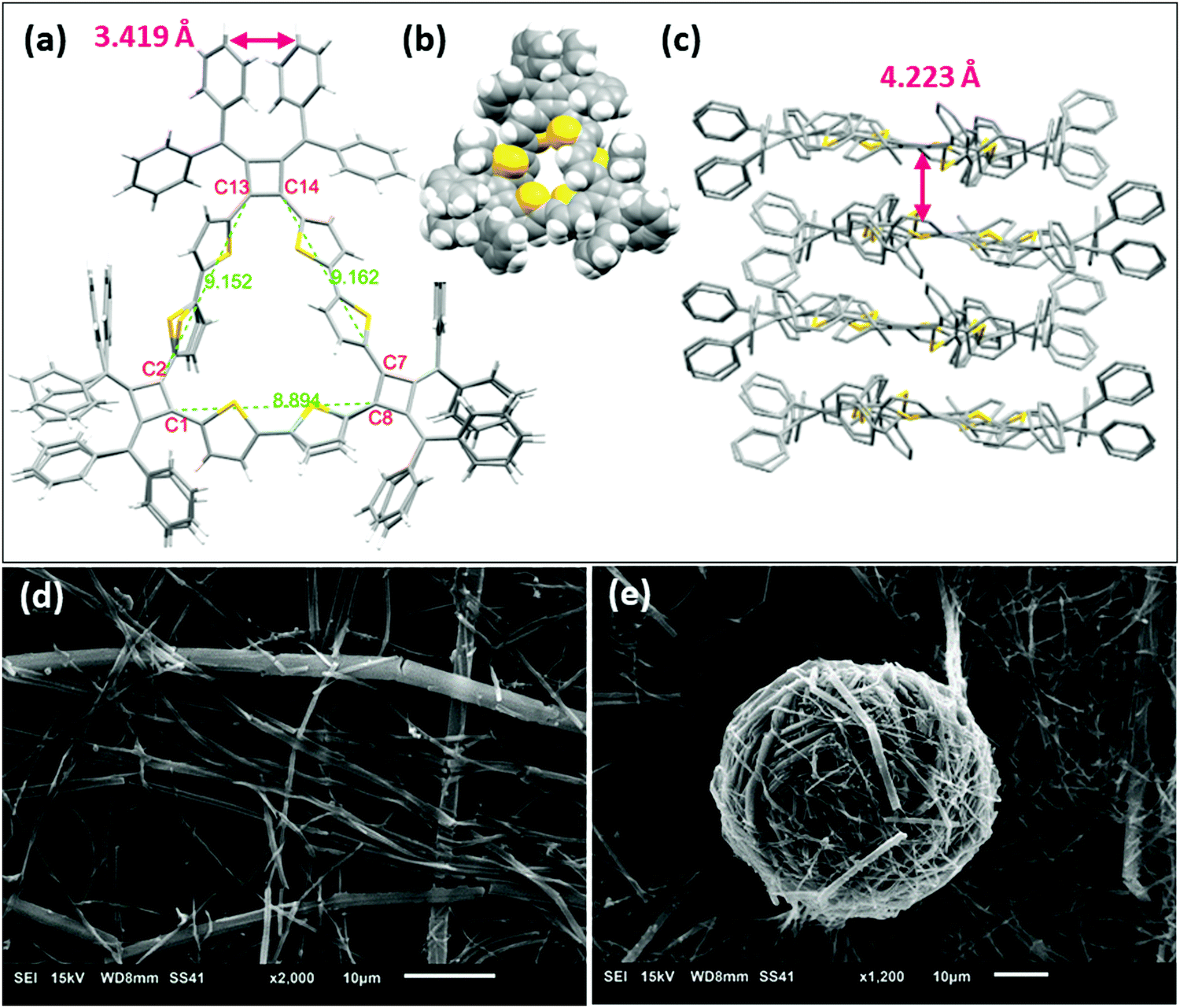 | ||
| Fig. 4 X-ray crystal structure of macrocycle A1, (a) top view, stick model; (b) space filling model; (c) side view of the stacked model; (d) SEM images of crystals of A1 grown in chlorobenzene and (e) crystal fibrils of A1 grown in THF. | ||
Macrocycle A1 crystallizes more readily than the other macrocycles and displays interesting self-assembled morphologies when grown from different solvents.28 As visualized by scanning electron microscopy (SEM), crystals of A1 tend to form cylinders in various diameters when grown in chlorobenzene (Fig. 4d) and form fibrous structures when grown in THF (Fig. 4e). These fibrils have a smaller diameter that assemble into balls.
Macrocycles using linkers with different conjugation lengths
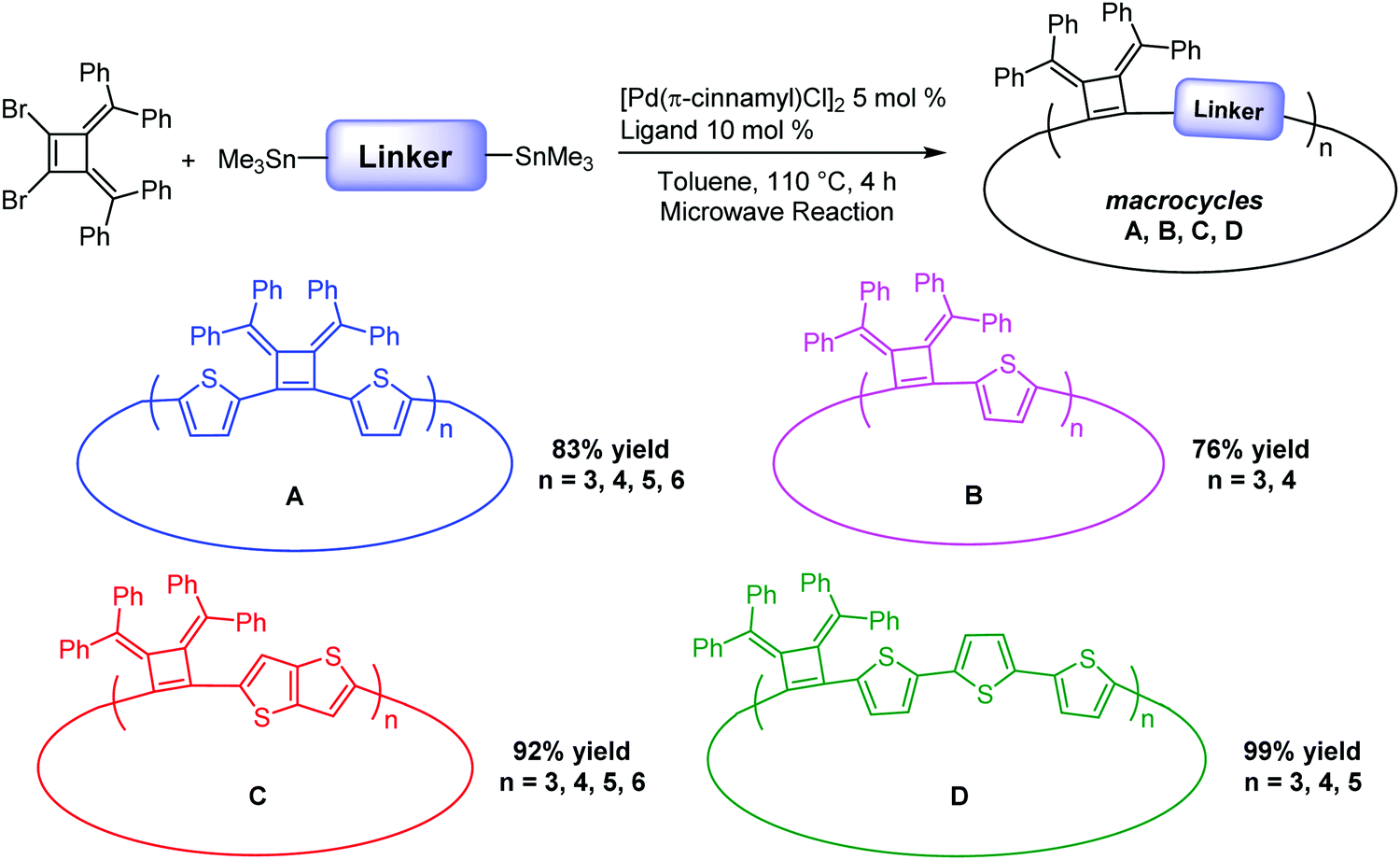
Linkers adjoining the cyclobutenes play an important role in controlling structure, effective conjugation length and photophysical properties. Our synthetic method affords modular access to macrocycles with monothiophene, bithiophene, fused thiophenes and terthiophene linkages in good to excellent yields (Table 2). Single thiophene linkages produce macrocycles B with 3 to 4 repeating units; fused thiophene rings give macrocycle C with 3 to 6 repeating units; and terthiophenes produce the largest macrocycle D with 3 to 5 repeating units. Their compositions contain 3- to 6-mers as confirmed by MALDI-TOF mass spectrometry (Fig. S5–S7, ESI‡).|
|
||||||
|---|---|---|---|---|---|---|
| Macrocyclesa | Yieldb (%) | Tdc (°C) | λ abs(max) solutiond (nm) | λ em(max) emissione (nm) | λ abs(max) filmf (nm) | Quantum yieldg (%) |
| a Standard reaction conditions: 0.15 mmol scale, 0.05 M, under N2 atmosphere. b Yield of the soluble products. The macrocycles were purified by passing through a pad of Celite and precipitated from MeOH. c Initial decomposition temperature, onset temperature in TGA analysis. TGA analysis was conducted in N2 atmosphere with a ramping rate 20 °C min−1 to 900 °C. d In THF solution. e The excitation is at the maximum absorption wavelength. f Spin-coated films on glass slides with macrocycles dissolved in THF solution. g Quantum yields were measured in THF solutions with 1,4-bis(5-phenyloxazole-2-yl)benzene (POPOP) as the standard. | ||||||
| A | 83 | 374 | 422 | 628 | 445 | 5.9 |
| B | 76 | 339 | 352 | 688 | 380 | 6.5 |
| C | 92 | 328 | 387 | 660 | 383 | 7.6 |
| D | 99 | 339 | 440 | 603 | 440 | 7.3 |
As summarized in Table 1, thermal gravimetric analysis (TGA) reveals that the macrocycles display high thermal stability with initial decomposition temperatures ranging from 328–374 °C (Fig. S9, ESI‡). Differential scanning calorimetry (DSC) analysis of the macrocycle mixtures shows no obvious thermal transitions between 100 °C to 350 °C as expected as a result of the mixture of macrocycles and conformations, thereof appears to produce an amorphous state.
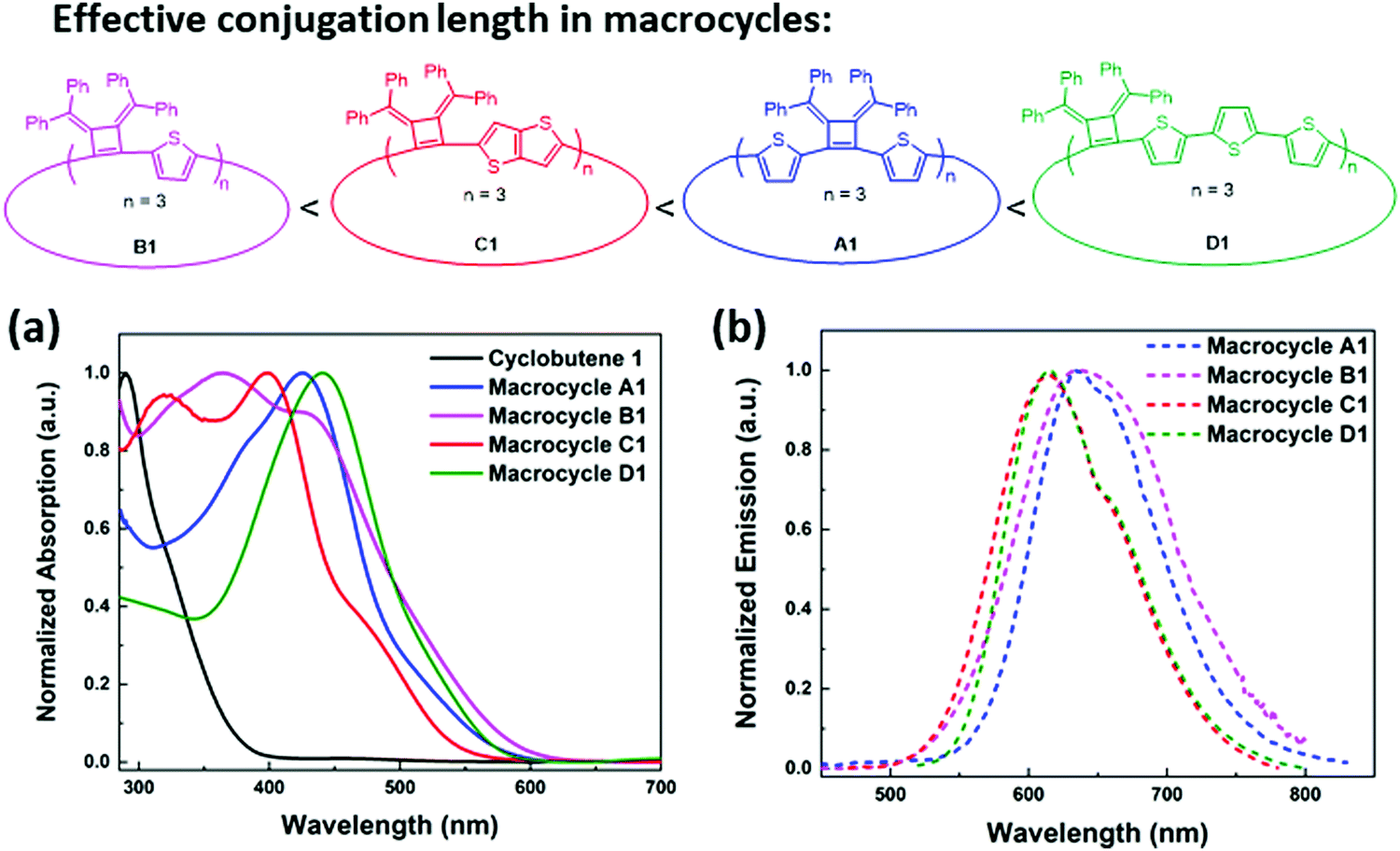
The macrocycle mixtures show different UV-vis absorption behaviors, which are related to their effective conjugation lengths (Fig. S8, ESI‡) and modest emissive quantum yields ranging from 5.9% to 7.6%. Macrocycles with three repeating units have been separated successfully by HPLC prep-column, giving macrocycle A1 (3 repeating units), B1 (3 repeating units), C1(3 repeating units) and D1 (3 repeating units) as single pure products. Their UV-vis and emission spectra were also studied in THF solutions. As shown in Fig. 5, D1 with a terthiophene linker has the longest conjugation length with absorption and emission maximum at λabs(max) of 440 nm and λem(max) of 615 nm respectively, while A1 displays a λabs(max) of 426 nm and a λem(max) of 634 nm. C1 displays two absorption bands at 319 nm and 397 nm, and its emission spectrum shows a λem(max) of 634 nm and a shoulder peak at 658 nm that are related to the absorption and emission of two different transition states. Similarly, B1 presents two absorption bands at 365 nm and 428 nm, and a broad emission band around 637 nm. The difference in conjugation lengths observed in these macrocycles with the same repeating units (three repeating units) could be attributed to the effective conjugation between the cyclobutene monomer and the different linkers. The conformations between the linkers and the cyclobutenes may vary and therefore affect the conformations in macrocycles. Based on the trends in the λmax values, it appears that the length of the linking heterocycles dominates the conformation structures: the terthiophene linker in D1 having the longest conjugation length and the single thiophene of B1 having the lowest degree of conjugation.
 | ||
| Fig. 5 Normalized (a) UV-vis spectra and (b) the emission spectra of the isolated macrocycles A1, B1, C1 and D1 in THF solutions. The emission spectra were measured after excitation at the maximum of each absorption wavelength. | ||
The synthesized macrocycles readily absorb I2 to create “doped” samples. Thin films were prepared by spin coating with macrocycles in THF solution (5 mg mL−1, 1500 rpm) and were then exposed to iodine vapor in a sealed chamber for 60 h at room temperature. After doping, the λabs(max) of the thin film of macrocycles mixtures A shifts from 445 nm to 395 nm, and a new strong absorption band arises at 793 nm that is related to the absorption band of polaronic or bipolaronic states of the macrocycles29 (Fig. 6a). The iodine treated samples also display an induced electron paramagnetic resonance (EPR) signal in solution phase (Fig. 6b). Upon adding excess amount of iodine to the dry toluene solution of macrocycle A1, a broad EPR spectrum was observed with a g-value of 2.0089, indicating the formation of polarons that have localized spin distribution in the macrocycles.30,31 Its spin intensity decreased over time, as the solution undergoes to higher doping levels, creating bipolaronic states (with charges of +4e and +6e) which are spinless.
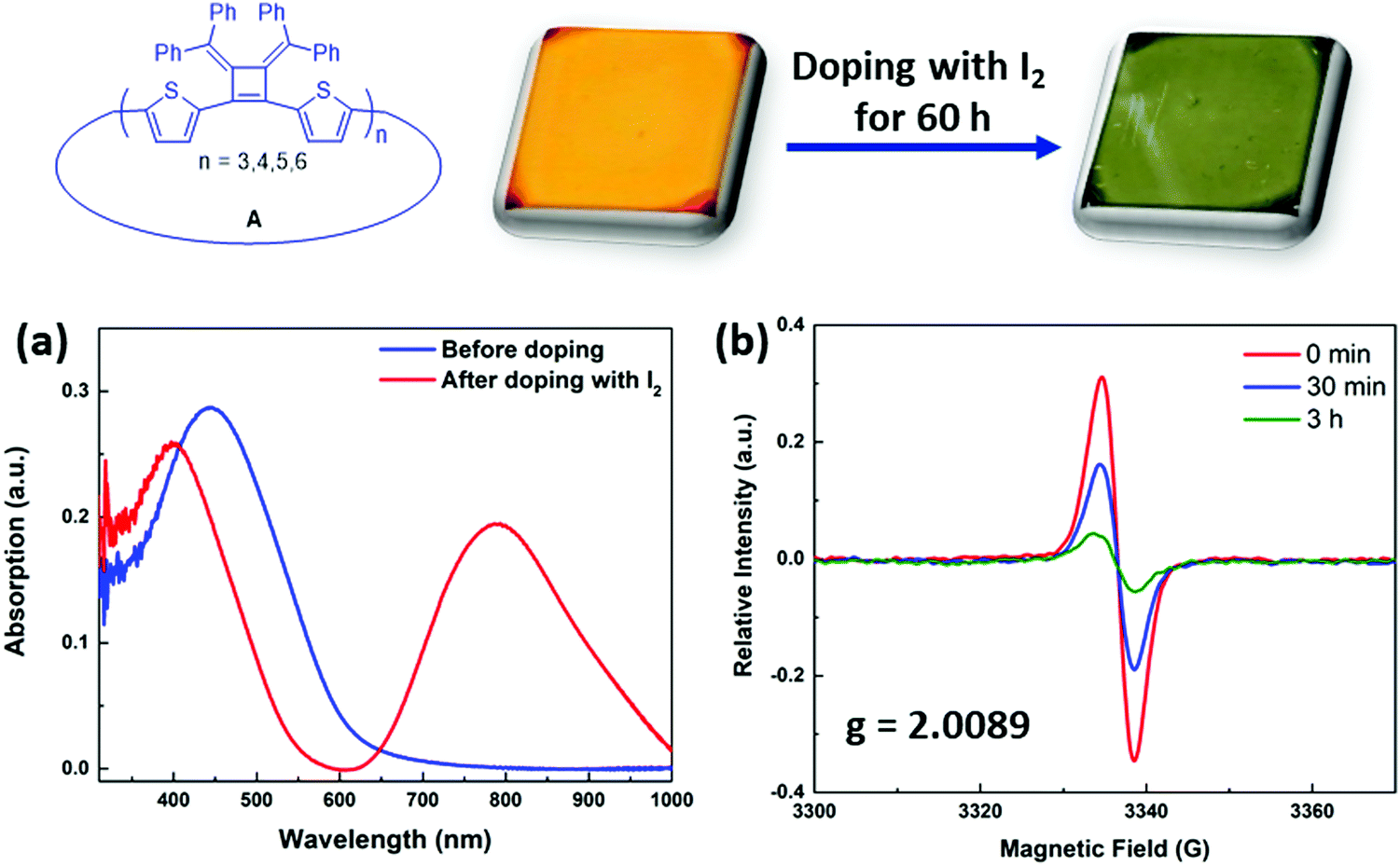 | ||
| Fig. 6 Iodine vapor doping with thin films prepared by spin-coating (5 mg mL−1, 1500 rpm). Macrocycle A was dissolved in THF and spin-coated on glass slides. (a) UV-vis absorption with thin films before and after doping with iodine in a sealed chamber for 60 h. (b) EPR spectra of macrocycle A1 doping with iodine in dry toluene solution. | ||
To further study the redox behavior of these macrocycles, cyclic voltammetry (CV) was conducted on the macrocycle mixtures. A model compound RP1 with two single thiophenes attached to the cyclobutene was synthesized by Suzuki coupling reaction. The CV of RP1 dissolved in dichloromethane (DCM) solution produced that one quasi-reversible redox wave at E1/2 = 0.48 V (vs. Fc/Fc+) (Fig. S10a, ESI‡). The CV of macrocycles A1 spin-coated on the indium tin oxide (ITO) glass displays a single redox wave at E1/2 = 0.40 V (vs. Fc/Fc+) similar to RP1 (Fig. S10b, ESI‡). The CVs of the macrocycle mixtures of A, B, C and D were also studied as thin films on ITO glass and all had quasi-reversible redox waves at slightly lower potentials and thereby establishing the semi-conductive potential of these materials (Fig. S10c, ESI‡).
Computational studies of macrocycles
In order to further understand the structural and electronic properties of these macrocycles, a series of DFT calculations were performed on A1 (n = 3), B1 (n = 3), C1 (n = 3), and D1 (n = 3). For each macrocycle, starting from an initial estimate of the molecular structure, the geometry was optimized in order to minimize the total energy and atomic forces using the Vienna Ab Initio Simulation Package (VASP),32,33 and the Perdew–Burke–Ernzerhof (PBE) functional.34 In a subsequent step, the electronic structure was determined at a higher level of theory, using the ORCA program35 with the PBE0 hybrid functional.36 All details of the DFT calculations are summarized in the ESI.‡The DFT results are presented in Fig. 7. The structure of the A1 macrocycle differs slightly from the crystal structure, and the computed structure has all thiophene rings oriented towards the center of the molecule. This is not the case for all macrocycles and in D1 the thiophene units show alternating orientations. The macrocycles all share two main geometrical features: (1) the inner phenyl rings stack face-to-face with π–π interactions, and (2) the triangular ring formed by the thiophene groups is corrugated. Computed structures reveal similar orientations of the outer phenyl groups of the X-ray crystal structure. Both the HOMO and LUMO orbitals of the macrocycles are significantly delocalized throughout the π-conjugated network for all macrocycles. Finally, the calculated HOMO–LUMO gaps of the A1, B1, C1, and D1 molecules are 2.40, 2.57, 2.54, and 2.42 eV, respectively. It is important to note that the calculated HOMO–LUMO gap is the difference between the vertical ionization potential and the vertical electron affinity, which is typically larger than the actual band gap in the condensed phase as a result of interactions between π-conjugated molecules.37 The DFT calculations are in agreement with the experimental observation of these macrocycles. MD simulations for larger macrocycles were also performed at the molecular level in the gas phase, which suggest a few possible configurations of the isolated macrocycles exist at room temperature (300 K). Here the calculated configurations for macrocycles with higher repeating units (n > 3) demonstrated a tendency to buckle and fold (Fig. S14, ESI‡).
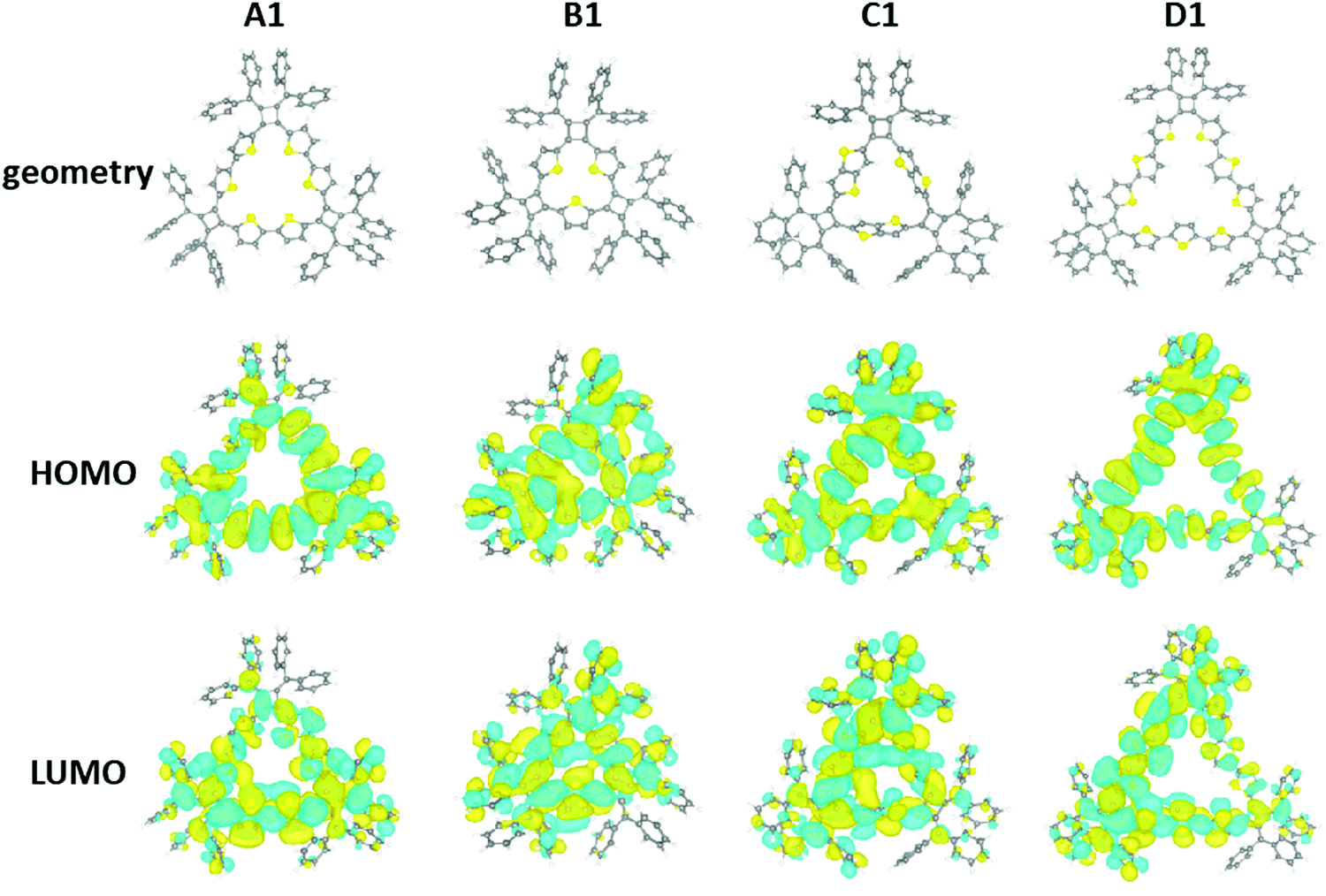 | ||
| Fig. 7 DFT results: structure and frontier orbitals of the A1, B1, C1, and D1 compounds. In the structural models, yellow, grey, and white spheres respectively correspond to sulfur, carbon, and hydrogen atoms. | ||
Conclusion
In summary, a series of cyclobutene-based π-conjugation macrocycles were synthesized by a facile template-free modular method using a cyclobutene building block that provides a vertex angle of less than 90°. The synthesized macrocycles were found to be highly symmetrical in solution by 1H NMR and VT-NMR. X-ray crystallography of one macrocycle ring confirmed the structure and provided insights into the conformations. DFT calculations and MD simulations suggest that several possible configurations are possible for macrocycles with larger ring sizes. The redox activity and nature of the charged species were investigated by cyclic voltammetry and EPR measurements. Our results suggest that the cyclobutene geometry provides facile access to a number of cyclic topologies by incorporating different linkages for the creation of new organic electronic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported the Air Force Office of Scientific Research AFOSR Grant #17RT0904, FA9550-18-1-0341. DFT calculations were supported by Toyota Research Institute (TRI), and computational support was provided by the Extreme Science and Engineering Discovery Environment, supported by the National Science Foundation grant number ACI-1053575.References
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Supramolecular coordination: self-assembly of finite two- and three-dimensional ensembles, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Functional Supramolecular Polymeric Networks: The Marriage of Covalent Polymers and Macrocycle-Based Host-Guest Interactions, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS.
- Y. Zhou, K. Jie, R. Zhao and F. Huang, Supramolecular-Macrocycle-Based Crystalline Organic Materials, Adv. Mater., 2020, 32, 1904824 CrossRef CAS.
- M. Iyoda, J. Yamakawa and M. J. Rahman, Conjugated macrocycles: concepts and applications, Angew. Chem., Int. Ed., 2011, 50, 10522–10553 CrossRef CAS.
- M. Stepien, E. Gonka, M. Zyla and N. Sprutta, Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications, Chem. Rev., 2017, 117, 3479–3716 CrossRef CAS.
- T. Chatterjee, V. S. Shetti, R. Sharma and M. Ravikanth, Heteroatom-Containing Porphyrin Analogues, Chem. Rev., 2017, 117, 3254–3328 CrossRef CAS.
- T. Tanaka and A. Osuka, Chemistry of meso-Aryl-Substituted Expanded Porphyrins: Aromaticity and Molecular Twist, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS.
- F. Sondheimer and R. Wolovsky, Unsaturated Macrocyclic Compounds. XXI.1 The Synthesis of a Series of Fully Conjugated Macrocyclic Polyene-polyynes (Dehydro-annulenes) from 1,5-Hexadiyne, J. Am. Chem. Soc., 1962, 84, 260–269 CrossRef CAS.
- R. Suzuki, H. Tsukuda, N. Watanabe, Y. Kuwatani and I. Ueda, Synthesis, structure and properties of 3,9,15-tri- and 3,6,9,12,15,18-hexasubstituted dodecadehydro[18]annulenes (C18H3R3 and C18R6) with D6h-symmetry, Tetrahedron, 1998, 54, 2477–2496 CrossRef CAS.
- D. Ajami, O. Oeckler, A. Simon and R. Herges, Synthesis of a Möbius aromatic hydrocarbon, Nature, 2003, 426, 819–821 CrossRef CAS.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, Synthesis, Characterization, and Theory of [9]-, [12]-, and [18]Cycloparaphenylene: Carbon Nanohoop Structures, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS.
- S. Yamago, Y. Watanabe and T. Iwamoto, Synthesis of [8]Cycloparaphenylene from a Square-Shaped Tetranuclear Platinum Complex, Angew. Chem., Int. Ed., 2010, 49, 757–759 CrossRef CAS.
- Y. Segawa, D. R. Levine and K. Itami, Topologically Unique Molecular Nanocarbons, Acc. Chem. Res., 2019, 52, 2760–2767 CrossRef CAS.
- Y. Segawa, M. Kuwayama, Y. Hijikata, M. Fushimi, T. Nishihara, J. Pirillo, J. Shirasaki, N. Kubota and K. Itami, Topological molecular nanocarbons: All-benzene catenane and trefoil knot, Science, 2019, 365, 272–276 CrossRef CAS.
- Y. Li, A. Yagi and K. Itami, Synthesis of Highly Twisted, Nonplanar Aromatic Macrocycles Enabled by an Axially Chiral 4,5-Diphenylphenanthrene Building Block, J. Am. Chem. Soc., 2020, 142, 3246–3253 CrossRef CAS.
- T. Yamamoto, M. Hosokawa, M. Nakamura, S.-i. Sato, T. Isono, K. Tajima, T. Satoh, M. Sato, Y. Tezuka, A. Saeki and Y. Kikkawa, Synthesis, Isolation, and Properties of All Head-to-Tail Cyclic Poly(3-hexylthiophene): Fully Delocalized Exciton over the Defect-Free Ring Polymer, Macromolecules, 2018, 51, 9284–9293 CrossRef CAS.
- A. Mishra, C.-Q. Ma and P. Bäuerle, Functional Oligothiophenes: Molecular Design for Multidimensional Nanoarchitectures and Their Applications, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS.
- T. Fujiwara, A. Muranaka, T. Nishinaga, S. Aoyagi, N. Kobayashi, M. Uchiyama, H. Otani and M. Iyoda, Preparation, Spectroscopic Characterization and Theoretical Study of a Three-Dimensional Conjugated 70 pi-Electron Thiophene 6-mer Radical Cation pi-Dimer, J. Am. Chem. Soc., 2020, 142, 5933–5937 CrossRef CAS.
- V. Marti-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization, Chem. Rev., 2015, 115, 8736–8834 CrossRef CAS.
- W. D. Huntsman and H. J. Wristers, 3,4-Dimethylenecyclobutene by Thermal Rearrangement of 1,5-Hexadiyne, J. Am. Chem. Soc., 1963, 85, 3308–3309 CrossRef CAS.
- F. Toda and P. Garratt, Four-membered ring compounds containing bis(methylene)cyclobutene or tetrakis(methylene)cyclobutane moieties. Benzocyclobutadiene, benzodicyclobutadiene, biphenylene, and related compounds, Chem. Rev., 1992, 92, 1685–1707 CrossRef CAS.
- F. Toda, K. Tanaka, T. Tamashima and M. Kato, Stereoselective Thermal Conversion of s-trans-Diallene into Dimethylenecyclobutene via s-cis-Diallene in the Crystalline State, Angew. Chem., Int. Ed., 1998, 37, 2724–2727 CrossRef CAS.
- F. Toda, Naphthocyclobutenes and benzodicyclobutadienes: synthesis in the solid state and anomalies in the bond lengths, Eur. J. Org. Chem., 2000, 1377–1386 CrossRef CAS.
- S. Braverman, E. S. Kumar, M. Cherkinsky, M. Sprecher and I. Goldberg, Electron depleted bis(methylene)cyclobutenes: sulfinyl and sulfonyl substitution, Tetrahedron, 2005, 61, 3547–3557 CrossRef CAS.
- R. R. Parkhurst and T. M. Swager, Synthesis of 3, 4-Bis(benzylidene)cyclobutenes, Synlett, 2011, 1519–1522 CAS.
- R. R. Parkhurst and T. M. Swager, Synthesis and optical properties of phenylene-containing oligoacenes, J. Am. Chem. Soc., 2012, 134, 15351–15356 CrossRef CAS.
- X. Ji, M. Ahmed, L. Long, N. M. Khashab, F. Huang and J. L. Sessler, Adhesive supramolecular polymeric materials constructed from macrocycle-based host–guest interactions, Chem. Soc. Rev., 2019, 48, 2682–2697 RSC.
- H. Chen, C. Huang, Y. Deng, Q. Sun, Q.-L. Zhang, B.-X. Zhu and X.-L. Ni, Solvent-Switched Schiff-Base Macrocycles: Self-Sorting and Self-Assembly-Dependent Unconventional Organic Particles, ACS Nano, 2019, 13, 2840–2848 CrossRef CAS.
- I. Zozoulenko, A. Singh, S. K. Singh, V. Gueskine, X. Crispin and M. Berggren, Polarons, Bipolarons, And Absorption Spectroscopy of PEDOT, ACS Appl. Polym. Mater., 2019, 1, 83–94 CrossRef CAS.
- S. Hayashi, K. Kaneto, K. Yoshino, R. Matsushima and T. Matsuyama, Electrical Conductivity and ESR Studies in Iodine-Doped Polythiophene from Semiconductor to Metallic Regime, J. Phys. Soc. Jpn., 1986, 55, 1971–1980 CrossRef CAS.
- R. P. Kingsborough and T. M. Swager, Polythiophene Hybrids of Transition-Metal Bis(salicylidenimine)s:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Correlation between Structure and Electronic Properties, J. Am. Chem. Soc., 1999, 121, 8825–8834 CrossRef CAS.
Correlation between Structure and Electronic Properties, J. Am. Chem. Soc., 1999, 121, 8825–8834 CrossRef CAS. - G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J.-L. Bredas, Mind the gap!, Mater. Horiz., 2014, 1, 17–19 RSC.
Footnotes |
| † Dedication: To the inspiring example of Fred Wudl and his many pioneering contributions in the design, synthesis and study of organic electronic materials. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2011836. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qm00824a |
| § P. W. and R. L. contributed equally. |
| This journal is © the Partner Organisations 2020 |