
Band gap engineering of donor–acceptor co-crystals by complementary two-point hydrogen bonding†‡
Nathan
Yee
 ,
Afshin
Dadvand
and
Dmitrii F.
Perepichka
*
,
Afshin
Dadvand
and
Dmitrii F.
Perepichka
*
Department of Chemistry, McGill University, Montreal, Qc H3A 0B8, Canada. E-mail: dmitrii.perepichka@mcgill.ca
First published on 14th October 2020
Abstract
We report a detailed investigation of a series of new charge-transfer (CT) complexes assembled via a two-point complementary hydrogen bonding (H-bonding) of diindolopyrrole (DIP) electron donors with o-quinone and diazafluorenone acceptors. Unidirectional polarization through the DD⋯AA type H-bonding leads to a dramatic perturbation of electronic levels of the donor and the acceptor. π-Stacking of the H-bonded pairs results in strong charge-transfer (HOMO–LUMO) interactions in their ground state, manifested in low energy optical absorption. Density functional theory (DFT) calculations predict a H-bonding induced rise of the HOMOD (by up to 0.5 eV) and lowering of the LUMOA (by up to 0.7 eV). As a result, the complexes of relatively weak electron donors and acceptor ability exhibit remarkably low optical energy gaps (down to <0.8 eV), that can be tuned by varying the ionization potential and electron affinity of the individual components. Single crystal X-ray analysis for 6 complexes displayed H-bond lengths between 1.9 and 2.3 Å and short π-stacking distances (≥3.2 Å), in line with strong donor–acceptor interactions. Thin-film transistors of such a H-bonded complex, fabricated by vacuum co-sublimation of PhDIP and pyrenetetraone, showed ambipolar charge transport with unusual ‘double dip’ characteristics.
1. Introduction
Organic semiconductors (OSCs) have attracted considerable interest because of their applications in optoelectronic devices such as OFETs, OPVs, and OLEDs.1–5 The supramolecular arrangement of OSCs in the solid state is a critical parameter in determining device performance.6 Several intermolecular interactions have been used to control their self-assembly including van der Waals, ionic and π–π interactions, and hydrogen bonding.7,8 Among these, H-bonding is perhaps the most versatile and reliable tool allowing for the engineering of well-defined supramolecular structures because of its high fidelity and directionality.9–14The industrial pigments indigo, quinacridone, and epindolidione are among the earliest known examples of π-conjugated materials with H-bonding functionality.15 Their utility in OFET devices was recently uncovered, bringing H-bonding to light as a tool for controlling the solid state structure of OSCs.16–18 H-bonding has also been used to modulate the redox properties of tetrathiafulvalene (TTF) based organic metals.19–21
Recent years have seen a renewal of interest in molecular co-crystals of donor and acceptor OSCs as optoelectronic materials with applications as ambipolar transistors22–26 and bicomponent ferroelectrics.27–29 However, controlling the self-assembly of bicomponent OSCs is far more challenging than single component OSCs. The strategies used for the rational control of D/A pairs include co-crystallization of strong D/A pairs,30–33 shape complementarity34,35 and halogen bonding.36,37
Previously, our lab reported the co-assembly of a weak donor/acceptor pair (P2P and NDI) through complementary 3-point H-bonding into well-defined supramolecular structures22 (Scheme 1a). Single crystal FETs of the H-bonded co-assemblies exhibited ambipolar transport with modest mobilities. We have also predicted that H-bonding between the D and A components could lead to polarization effectively reducing the band-gap of the complex comparing to that expected based on the energy off-set between the donor (HOMOD) and acceptor (LUMOA).38 However the observed effect was relatively weak (∼0.1–0.2 eV) because each component has both H-bond donor and acceptor (Scheme 1a). We hypothesized that such polarization would be greatly enhanced if all H-bond donors were placed on an electron-donor and all H-bond acceptors were located on the π-acceptor.39
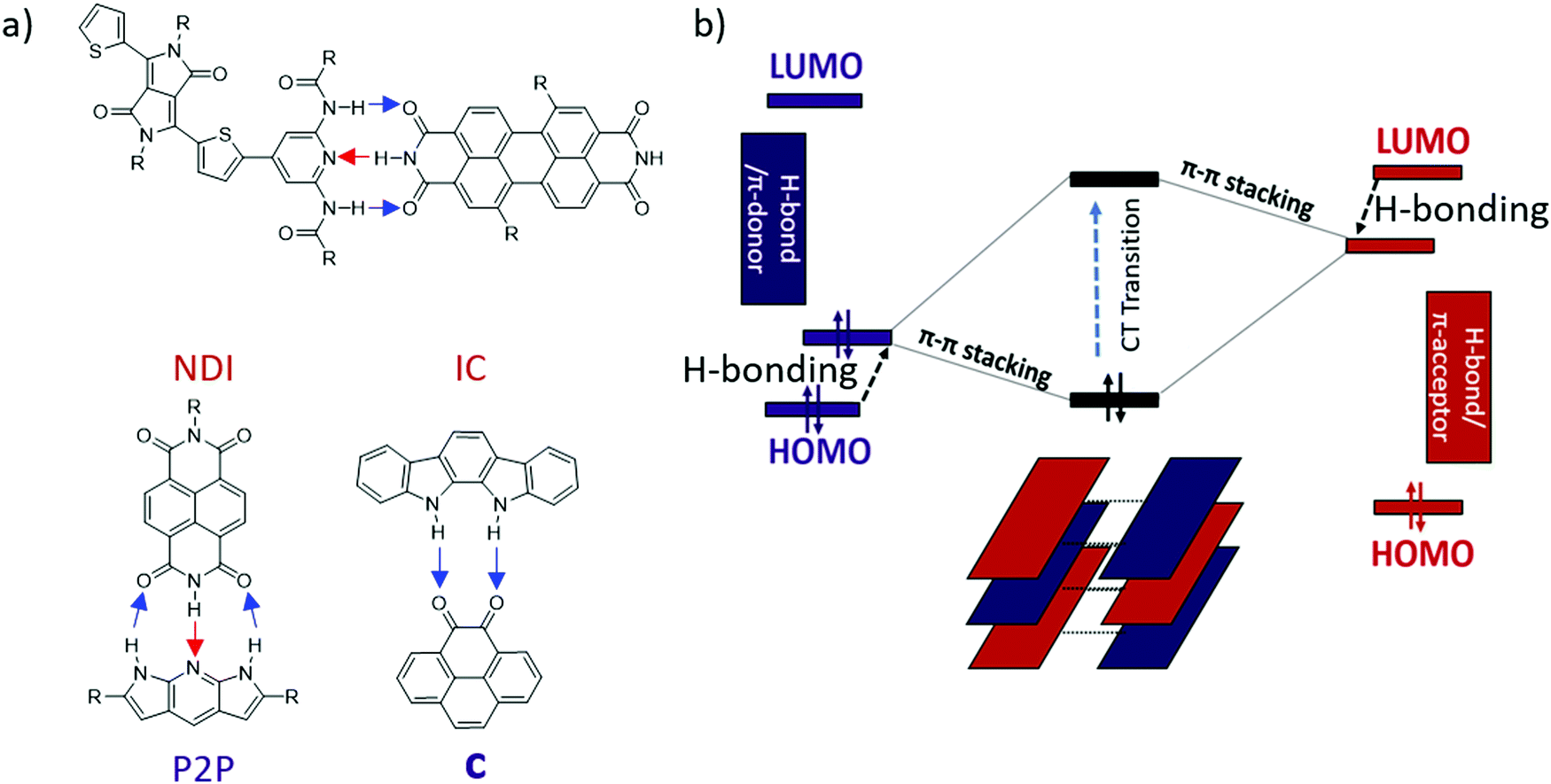 | ||
| Scheme 1 (a) Previously reported H-bonded multicomponent semiconductors22,39,40 (blue arrows show proton displacement from electron donor to acceptor and red arrows show proton displacement from electron acceptor to donor), (b) Energy diagram of donor (left), acceptor (right), and CT state (middle). Black arrows show change in the HOMO/LUMO energy level upon H-bonding. | ||
In this work we present a new series of DD⋯AA H-bonded bicomponent semiconductors composed of strong π-electron donors derived from diindolopyrrole (DIP) and a series of π-electron acceptors a–f (Scheme 2) and demonstrate how H-bonding can be employed to control their co-crystallization and their electronic properties. The high HOMO of DIP comparing to other donors used in this context39 leads to stronger donor–acceptors interactions and allows us to control the bandgap in the 0.8–1.7 eV range by varying the acceptor counterparts. The effect of H-bonding on the HOMO of the complexes is now directly probed by photoelectron spectroscopy providing experimental evidence for orbital polarization via H-bonding. The generality of the effect and fidelity of the two-point H-bonding interactions was confirmed for six complexes by single crystal XRD, illustrating the versatility of this concept as a tool for supramolecular design of materials, across a broad space of structural variations. Thin film organic field effect transistors (OFETs) fabricated with sublimed complex of DIP 1 with pyrenetetraone e exhibited ambipolar transport, demonstrating that such vacuum processing is viable for the fabrication of H-bonded thin film devices based on multicomponent materials.
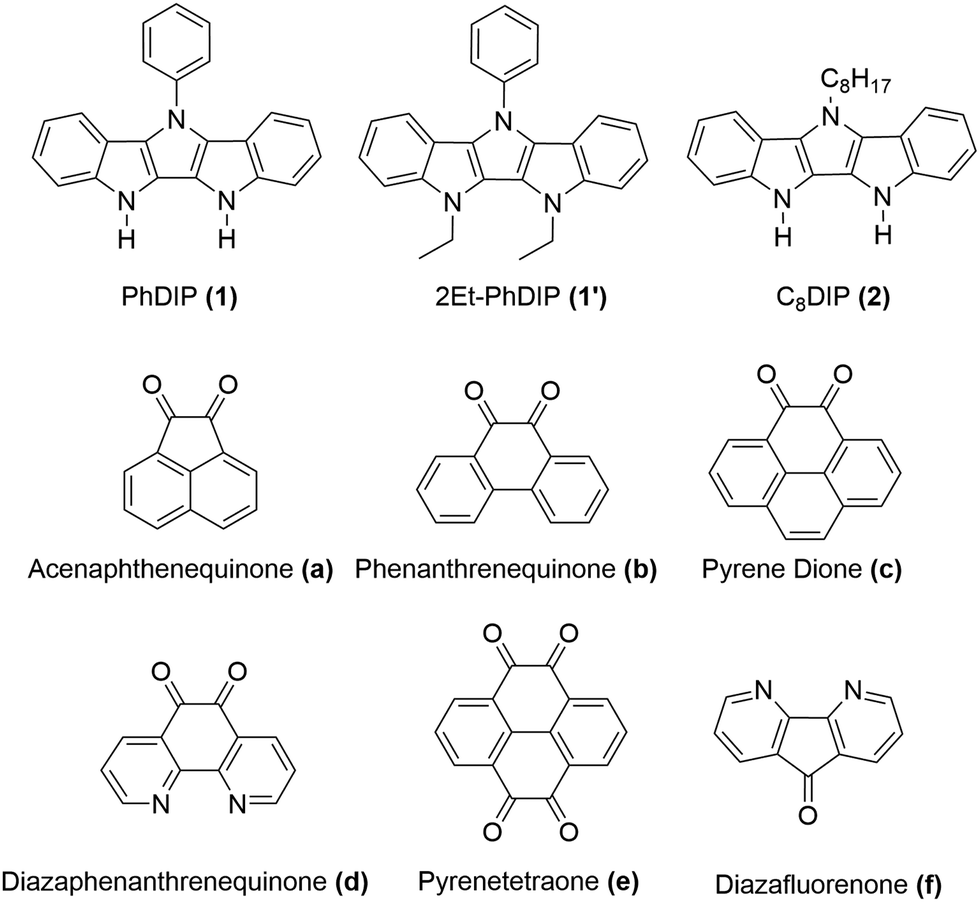 | ||
| Scheme 2 Structures of donors (1, 1′, 2) and acceptors (a–f). | ||
2. Results and discussion
Molecular design
In choosing appropriate molecular components for DD⋯AA type H-bonding, we looked for electron-donors and acceptors with H-bonding functionality integrated with their π-conjugated system. The selected donors and acceptors should possess complementary H-bonding motifs such that proton transfer is unidirectional, i.e. all H-bond donor groups are located on an electron-donor molecule and all H-bond accepting groups on the electron-acceptor molecule. Based on these considerations, two diindolopyrroles 1 and 2 (Scheme 2) were synthesized as H-bonding π-electron donors, following a published procedure.41N-Alkylated derivative 1′ was also synthesized as a control compound lacking H-bonding functionality. Cyclic voltammograms of 1, 2, and 1′ exhibit reversible oxidations at 0.00 V, −0.10 V and −0.19 V ferrocene (Fc) (Fig. 1), from which the HOMO levels of −4.80, −4.70, and −4.61 eV, respectively, were estimated. The lower oxidation potentials of 2 and 1′ comparing to 1 is a result of the electron-donating alkyl substituents. The DFT predicted HOMOs for 1, 2 and 1′ were −4.66, −4.55 and −4.50 eV, respectively, in agreement with the electrochemically determined values. All three donors have wide optical gaps, with the absorption onsets at <400 nm (>3.1 eV, Fig. S2, ESI‡).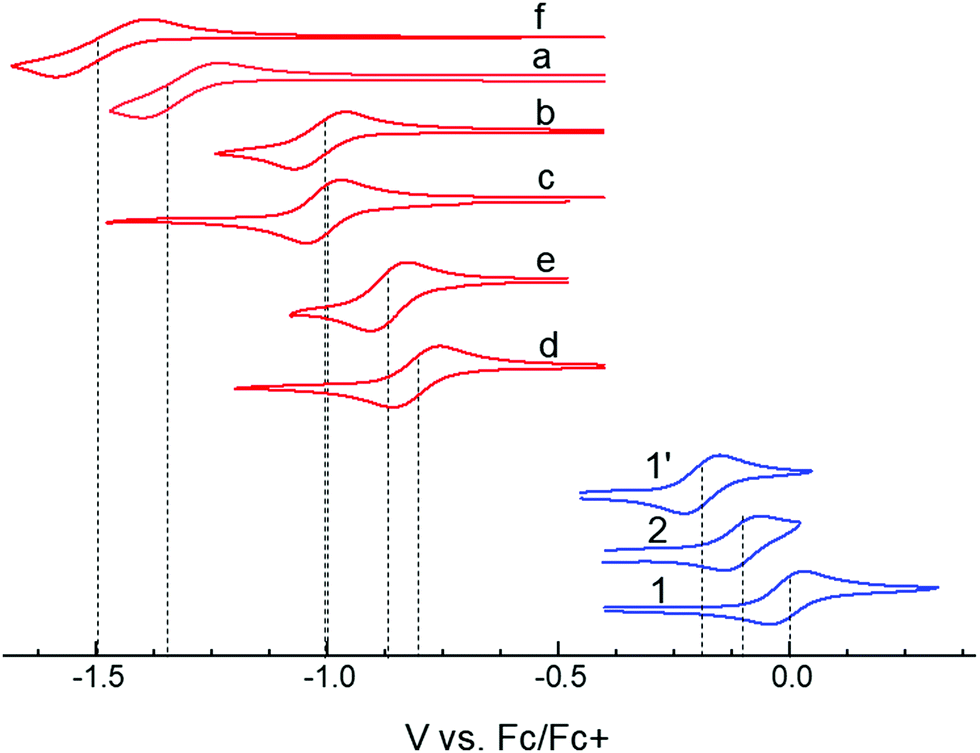 | ||
| Fig. 1 Cyclic voltammograms of studied donors and acceptors in MeCN. HOMO/LUMO energies were estimated from the oxidation and reduction potentials vs. Fc/Fc+, respectively, as −(eE1/2 + 4.8 eV). | ||
For the H-bonding electron acceptors, we selected a series of o-quinones (a–e) and diazafluorenone (f) (Scheme 2). The acceptors had reversible reductions ranging from −1.48 V (f) to −0.81 V (d) vs. Fc (Fig. 1), corresponding to LUMO energies of −3.32 and −3.99 eV, respectively. These values are supported by the LUMO energies calculated by DFT (−3.21 and −4.15 eV, B3LYP/-31G(d)).
Synthesis of hydrogen-bonded complexes
The H-bonded complexes can be readily obtained by mechanical mixing (grinding) of the individual components,42 co-crystallization from solution, or vacuum co-sublimation. Single crystals of the H-bonded complexes of 1 were grown by slow evaporation of the solvent (MeCN or 1,2-dichloroethane). We note that crystallization of complexes of 2 is significantly more challenging due to the presence of the long alkyl (octyl) chain. Thus, in characterizing the effect of H-bonding interactions on solid state properties, we mainly focused on characterization of H-bonded complexes of acceptor 1. Complexation leads to a dramatic color change (from colorless and light yellow/orange to dark green, Fig. 3b inset) indicative of a ground-state CT interaction. The infrared spectra of the complexes exhibited a significant red-shift (70–105 cm−1) in the N–H stretching vibrations and a smaller red-shift in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration (3–23 cm−1) vs. that of the individual diones a–e, which are indicative of a reduction in bond order due to H-bonding (Table S1, ESI‡).
O vibration (3–23 cm−1) vs. that of the individual diones a–e, which are indicative of a reduction in bond order due to H-bonding (Table S1, ESI‡).
DFT calculations
The H-bonding interactions and the resulting perturbation of the electronic properties of the donors and the acceptors were explored via DFT calculations using the B3LYP hybrid functional commonly used to describe the donor–acceptor43,44 and H-bonded complexes.45–47 In the gas phase, complexes of diones (a–e) and diazafluorenone (f) acceptors all exhibited similar binding energies of ca. 12 kcal mol−1 (for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding, Table 1). This is lower than the binding energies of P2P⋯NDI (Eb ∼ 20 kcal mol−1),38 and can be explained by fewer H-bonds as well as slight mismatch of the relative position and orientation of H-bond donating and accepting pairs in the interacting molecules (Fig. 2). Gas phase calculations of the H-bonded complexes show no overlap between HOMO and LUMO orbitals of the H-bonded complexes; instead, they remain almost fully localized on the donor and acceptor molecules, respectively (Fig. 2 and Fig. S1, ESI‡).
:1 binding, Table 1). This is lower than the binding energies of P2P⋯NDI (Eb ∼ 20 kcal mol−1),38 and can be explained by fewer H-bonds as well as slight mismatch of the relative position and orientation of H-bond donating and accepting pairs in the interacting molecules (Fig. 2). Gas phase calculations of the H-bonded complexes show no overlap between HOMO and LUMO orbitals of the H-bonded complexes; instead, they remain almost fully localized on the donor and acceptor molecules, respectively (Fig. 2 and Fig. S1, ESI‡).
| Acceptor | a | b | c | d | e | f |
|---|---|---|---|---|---|---|
|
a Shift vs. HOMOD (or LUMOA).
b Difference between HLG of a complex and the HOMOD–LUMOA off-set of the individual components.
c Energy is for 2:1 binding (12e).
|
||||||
| E b (kcal mol−1) | 11.6 | 12.4 | 12.6 | 11.5 | 22.5c | 11.2 |
| HOMOD | −4.66 | −4.66 | −4.66 | −4.66 | −4.66 | −4.66 |
| HOMOD–A (eV) | −4.17 | −4.23 | −4.14 | −4.42 | −4.58 | −4.34 |
| ΔHOMOa (eV) | 0.49 | 0.43 | 0.52 | 0.24 | 0.08 | 0.32 |
| LUMOA | −2.61 | −2.98 | −2.96 | −3.32 | −3.35 | −2.66 |
| LUMOD–A (eV) | −3.39 | −3.64 | −3.56 | −3.86 | −3.97 | −3.24 |
| ΔLUMOa (eV) | −0.78 | −0.66 | −0.60 | −0.54 | −0.62 | −0.58 |
| HLGD–A | 0.78 | 0.59 | 0.58 | 0.56 | 0.61 | 1.10 |
| ΔHLGb | 1.27 | 1.09 | 1.12 | 0.83 | 0.70 | 0.90 |
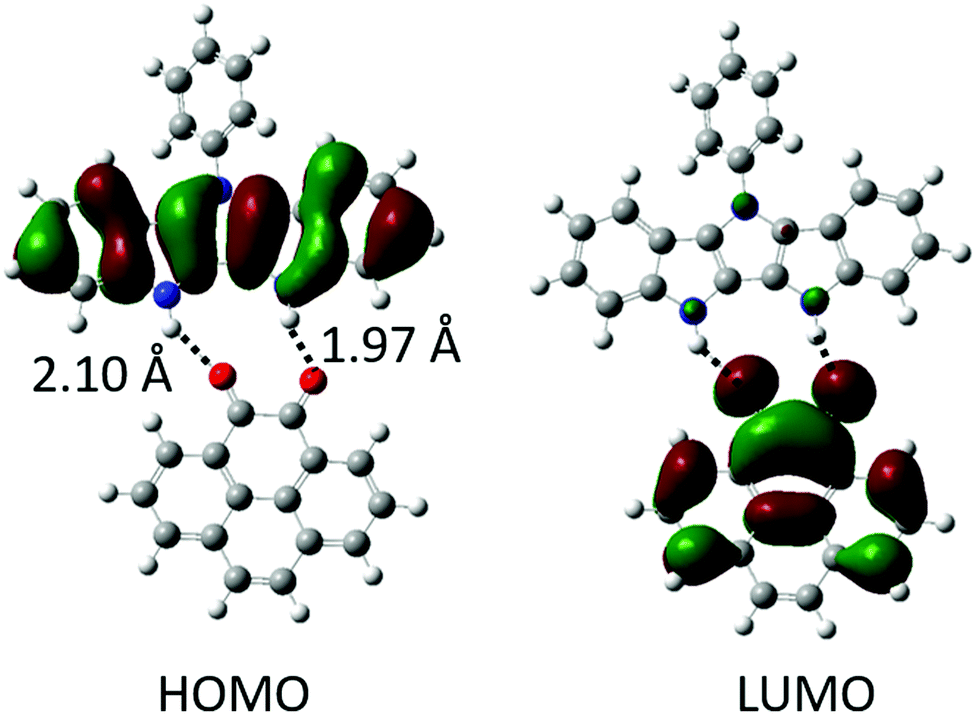 | ||
| Fig. 2 Surface topologies of H-bonded complexes 1c with calculated H-bond lengths shown. | ||
As expected (Scheme 1b), the H-bonding results in the raising of HOMOD and lowering of LUMOA upon complexation. The calculated HOMOD and LUMOA of the individual and H-bonded donors/acceptors along with the resulting HOMO–LUMO gaps (HLGs) of the complexes are summarized in Table 1 (for 1) and S2 (for 2). The perturbation of the molecular orbitals upon H-bonding increases with decreasing acceptor strength, i.e. the weakest π-donor/acceptor pairs exhibit the greatest perturbation of MOs. This is attributed to orbital (charge transfer) interactions which is more significant for stronger π-donors/acceptors. This trend is reflected in the ΔHLGs (i.e. the change in LUMOA–HOMOD offset of the donor–acceptor pair upon complexation), where the weakest acceptor 1a exhibits the largest ΔHLG (1.24 eV), while the strongest acceptors 1d and 12e (2:1 complex of donor 1 with acceptor e) exhibit the smallest ΔHLGs (∼0.70 eV) (Table 1). However, the resulting HLGs of the complexes follow the opposite trend, i.e. the complexes with the strongest acceptor have the narrowest band gaps. Thus, the HLG is largely determined by acceptor strength, but weaker acceptors display a larger orbital perturbation (ΔHLG) upon H-bonding. For comparison, smaller H-bonding induced ΔHLGs (0.1–0.3 eV) have been reported for nucleobases and some D/A complexes,13,38,48–50 confirming that the effect of polarization is much greater in unidirectional DD⋯AA than bidirectional (e.g. DAD⋯ADA) H-bonding. We also note that, the ΔHLG in the DIP complexes system is slightly lower compared to our previously studied indolo[2,3-a]carbazole (weaker donor (ΔHLG ≤ 1.5 eV)),39 suggesting that using a stronger donor results in less pronounced modulation of the ΔHLG, but still allows achieving a significantly narrower band gaps in the H-bonded complexes.
D–A complexation in solution
Mixing the solutions of donor and acceptor in CH2Cl2 results in characteristic dark color due to the low energy charge-transfer absorption of corresponding complexes. Since the CT absorption band is dependent on the HOMO–LUMO offset of the D/A pairs, its energy can be tuned by varying both the donor and the acceptor components (Scheme 1b). The optical band gap (Eg) in solution was estimated from the absorption edge and ranges from 1.90 eV (645 nm) for 1a to 1.36 eV (910 nm) for 1d and 1e (Fig. 3a and Fig. S3, ESI,‡Table 2). The onset of absorption bands for the complexes of octyl-substituted diindolopyrrole 2 were consistently red-shifted by 0.10–0.20 eV compared to complexes of phenyl-substituted 1 (Fig. S4, ESI‡), in line with the higher HOMO of 2 (Fig. 1).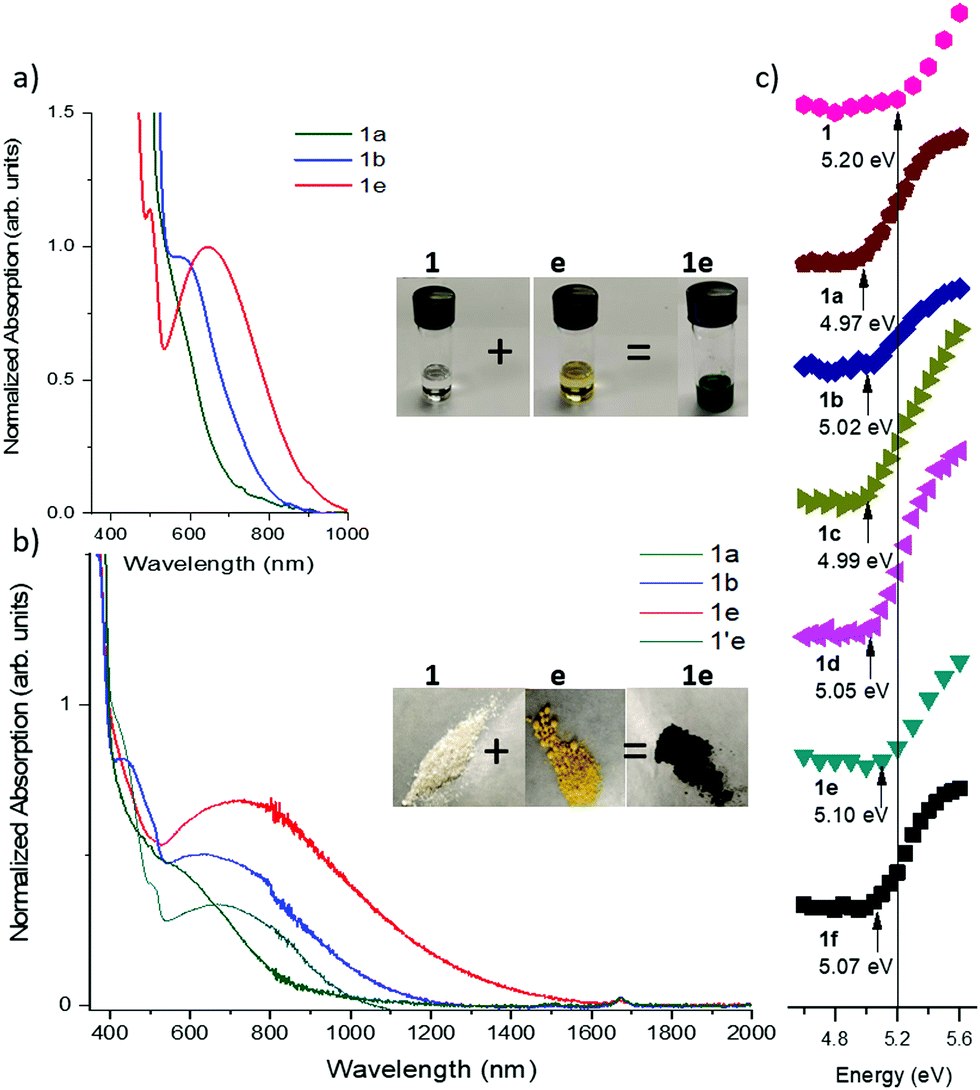 | ||
| Fig. 3 (a) Solution absorption spectra of complexes of 1a, 1b, and 1e (8 mM). (b) Solid state diffuse reflectance spectra of 1a, 1b, 1e and 1′e. (c) PESA characterization of 1 and its complexes with acceptors (solid state). Ionization energies are depicted by black arrows; black line shows the ionization energy of 1. | ||
| Acceptor | 1 λ onset/nm (HLG/eV) | 2 λ onset/nm (HLG/eV) | 1′ λ onset/nm (HLG/eV) | IC λ onset/nm |
|---|---|---|---|---|
| a | 930 (1.51) | — | 820 (1.77) | 760 |
| b | 1160 (1.17) | 1160 (1.15) | 910 (1.48) | 945 |
| c | 1160(1.20) | 1180 (1.10) | 990 (1.34) | 945 |
| d | 1250(1.08) | 1380 (0.96) | 1030 (1.30) | 945 |
| e | 1360 (0.96) | 1610 (0.78) | 1060 (1.27) | 1201 |
| f | 790 (1.73) | — | 675 (2.00) | — |
To probe the role of H-bonding, we also studied complexation of N,N′-diethyl-PhDIP 1′ (with no H-bond donor sites). While the absorption spectra in concentrated solutions of 1′ with acceptors e and c also exhibited CT transitions, the corresponding bands were much less intense than those complexes of 1 (Fig. S5, ESI‡), despite 1′ being a stronger donor than 1. Thus, while CT interactions can take place without H-bonding, the complexation in solution is only efficient for strong donor–acceptor pairs, and even then, it requires higher concentrations compared to corresponding H-bonded pairs. We attribute the more intense CT absorption to a higher concentration of the complexes of 1, afforded by the cooperativity effect of H-bonded complexation and hierarchical D/A stacking.38,40,51,52 Indeed, no solution complexation was observed for acceptor e and N,N′-dimethylindolo[2,3-a]carbazole while the corresponding H-bonding donor IC (HOMO = −5.10 eV) exhibited a very CT absorption in the same conditions.39
D–A interactions in the solid state
The H-bonded complexes were prepared by precipitation from MeCN or mechanical mixing of the D/A components,53 and their optical properties were studied via diffuse reflectance UV/vis spectroscopy (Fig. 3b and Fig. S6, S7, ESI‡). Complexation leads to the emergence of a low-energy CT band with onsets ranging from 1360 nm (0.96 eV) (12e) to 790 nm (1.74 eV) (1f). The onset values correlate with the electron affinity of the acceptors, i.e. stronger acceptors exhibit lower band gaps. The band-gaps were extracted by applying the Kubelka–Munk transformation (Fig. S7 and S8, ESI‡) and the data is summarized in Table 2. It was also expected that a stronger donor results in narrower band gaps, and indeed, the band gaps of complexes of 1 were 0.10–0.30 eV lower than corresponding complexes of IC with the same acceptors.39 To further corroborate this point, we synthesized H-bonded complexes with 2, whose HOMO is higher than that of 1 by 0.10 V as determined by CV (0.04 eV by DFT). The complexes of 2 exhibited CT bands ranging between 1160 nm (1.19 eV) to 1380 (0.76 eV), corresponding to band gaps up to 0.21 eV narrower than complexes of 1.To probe the role of H-bonding, we also prepared the complex 1′e consisting of an alkylated donor (1′) and the same acceptor (e). Complex 1e has a lower energy band edge at 1360 nm (0.96 eV) compared to 1′e which has a band edge at 1110 nm (1.12 eV) (Fig. 3b). Considering that HOMO of 1′ is 0.13 eV above that of 1, the observed difference in their band-gaps should correspond to the effect of H-bond induced polarization, which can be estimated about 0.3 eV (0.13 eV + 0.16 eV).
Photoelectron yield spectroscopy in air (PESA) was conducted to probe the effect of solid-state complexation on the ionization potential (IP) of H-bonded complexes of 1. The measured IP of the pure crystalline 1 is 5.20 eV, and it reduced by 0.10–0.20 eV upon complexation with acceptors, with weaker acceptors exhibiting a greater ΔIP, corroborating the DFT-predicted trend.
Crystal structure of H-bonded DA complexes
X-ray crystallographic structures of 1 with all six acceptors were obtained, demonstrating that the two-point H-bonding provides a robust platform for D/A co-assembly. For acceptor e, we were able to grow both the 2:1 (12e) and 1:1 (1e) H-bonded complex (Fig. S11, ESI‡). The hydrogen bond length of complexes ranged from 1.95–2.30 Å (Fig. 4, top), consistent with DFT results (2.00–2.20 Å) and our previously published IC complexes (2.00–2.11 Å). The individual donor and acceptor molecules are almost completely planar (except for 36–44° out-of-plane twist of the Ph group in 1).
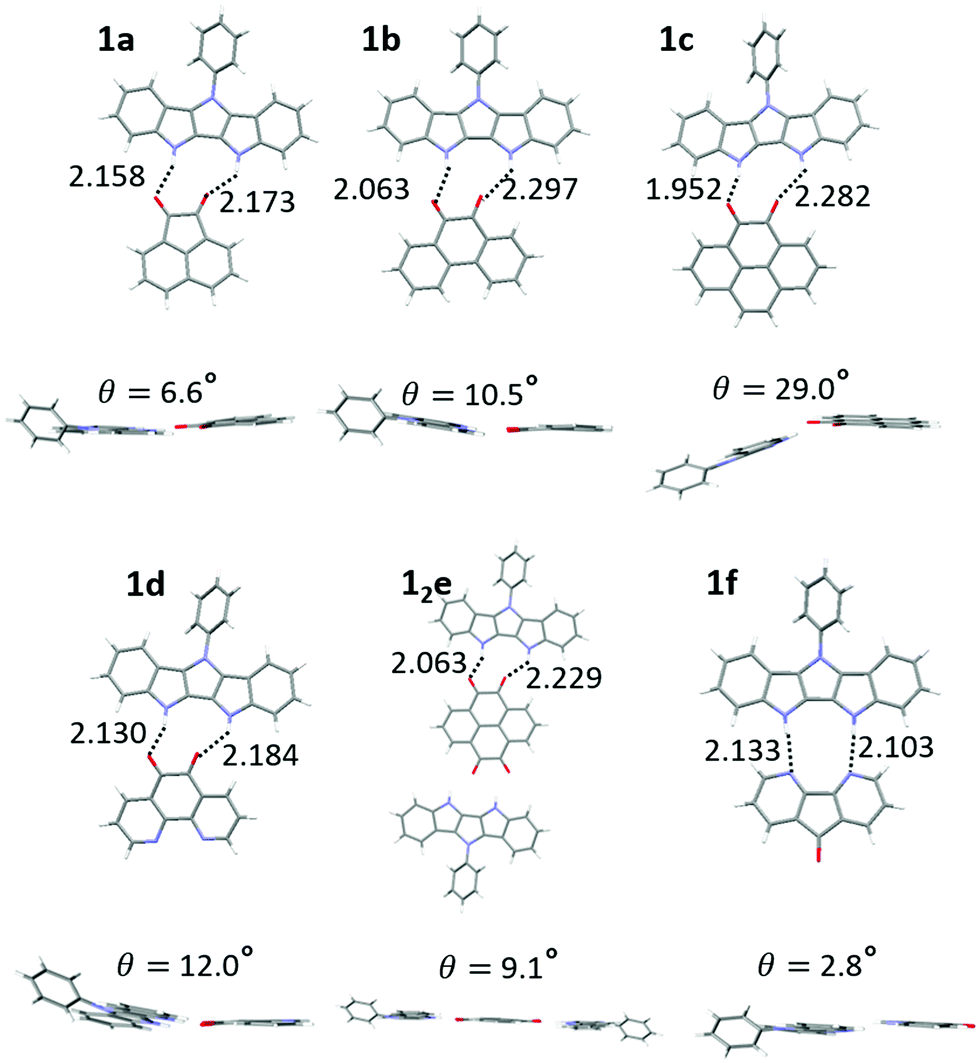 | ||
| Fig. 4 Crystal structures of complexes of 1a–1f displaying H bond lengths and dihedral angles between the donor and acceptor planes. | ||
However, larger geometric distortions are observed in complexes of 1 compared to IC, with one of oxygen atoms of the quinone acceptor located in between the two NH groups on the donor. In some complexes the donor and acceptor components are not coplanar, with an interplanar angle up to 29° (1c, Fig. 4). We attribute these distortions to a combination of the crystal packing and the mismatch between the orientations of the H-bond donor groups (NH) and H-bond acceptor groups (CO), that cannot achieve their ideal binding geometry (180° for NH⋯O and 120° for CO⋯H) in the DIP scaffold. The better match between the orientations of the H-bond donor (NH) and H-bond acceptor (R–N:) in diazafluorenone complex 1f leads to a more favorable binding geometry, reflected in very little distortion and the binding energy comparable to that of quinone complexes despite N being a weaker H-bond acceptor than O (Table 1). We note that H-bonding of 1 and d could in principle occur via either the carbonyl groups or the nitrogen atoms. Only the former interaction was observed in the crystals of 1d, in line with the gas phase calculations predicting a 0.8 kcal mol−1 stronger binding via the carbonyl groups (Fig. S10, ESI‡).
All crystal structures exhibit some variation of a mixed-stack arrangement which maximizes D/A π-interactions.38 The corresponding π⋯π contacts between donor and acceptor molecules are very short (C⋯C distances 3.04–3.30 Å), with stronger D/A pairs tending to have shorter stacking distances (Fig. 5). 1a, 1c, 1d, and 1f display a “typical” mixed stack (DADA) where a donor sits directly atop an acceptor and little to no DD or AA π-contacts (Fig. 5). For pyrenetetraone (e) acceptor, the expected 1:2 stoichiometry of H-bonded complex was confirmed crystallographically. The adjacent D⋯A⋯D H-bonded triads within the π-stacks are rotated by 90° vs. each other, which allows for both D⋯A (shortest C⋯C contact 3.04 Å) and D⋯D (shortest C⋯C contact 3.15 Å) π-contacts (Fig. 5e). The crystal structure of 1b (Fig. 5b) exhibits a DDAA π-stacking motif, also possessing both DA (3.25 Å) and DD (3.31 Å) interactions, but the dimeric nature of this stacking does not provide for continuous p- (or n-) transport channel. Thus, among all 6 complexes, 12e appears to have the most suitable packing and the lowest band-gap justifying its study as an OSC material.
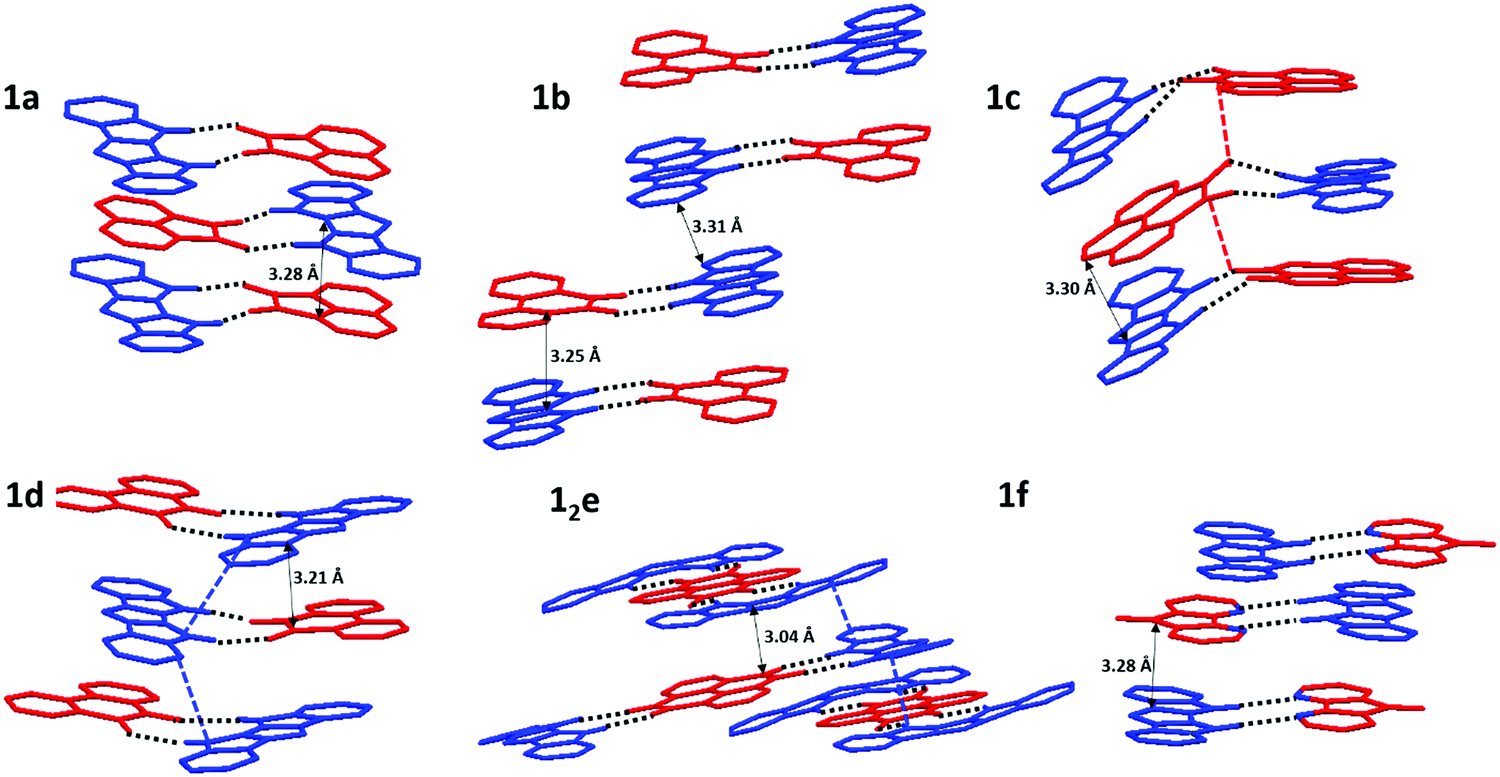 | ||
| Fig. 5 Crystal structures of complexes 1a–1f. Donor (1) is displayed in blue, acceptors (a–f) are displayed in red. Closest C–C distances are shown by black arrows. Continuous donor (blue) and acceptor (red) channels (possible p-/n-transport pathways) are indicated by dotted lines. Ph substituent and MeCN solvent molecules in 12e are omitted for clarity. | ||
Charge transport properties of donor molecules and H-bonded DA complexes
By itself, DIP 1 is a p-type OSC which shows a modest hole mobility of 10−5 cm2 V−1 s−1 in vacuum deposited films.54 To our knowledge, there has been no report of OFETs based on compound e. We were able to prepare thin-film OFETs by co-sublimation of the H-bonded complex 12e powder, although efforts to co-sublime complexes of 1 with more volatile acceptors (c and d) led to deposition of the individual acceptor component. The film of 12e exhibited red-shifted CO and N–H vibrations compared to the individual donor/acceptor components which is indicative of a reduction in bond order due to H-bonding, and displayed a low energy CT absorption resembling that of the solid 12e (Fig. S9, ESI‡). The OFETs based on 12e exhibited ambipolar charge transport with average hole and electron mobilities of 10−5 cm2 V−1 s−1 and 10−6 cm2 V−1 s−1, respectively (Fig. 6). The hole-dominated ambipolar behavior is clearly seen in both output and transfer characteristics of 12e. This was expected based on the crystal packing of 12e that shows continuous π-stacks of donor 1 (with overlapping HOMOs providing a channel for hole conduction), but lack of contacts between acceptor molecules e leave the superexchange interactions as the only possibility for the electron conduction. In addition, the HOMO of 12e crystals (−5.1 eV based on PESA measurements) is aligned with the work function of Au electrodes (−5.3 eV) while there is a significant barrier for electron injection (LUMO = −4.15 eV).
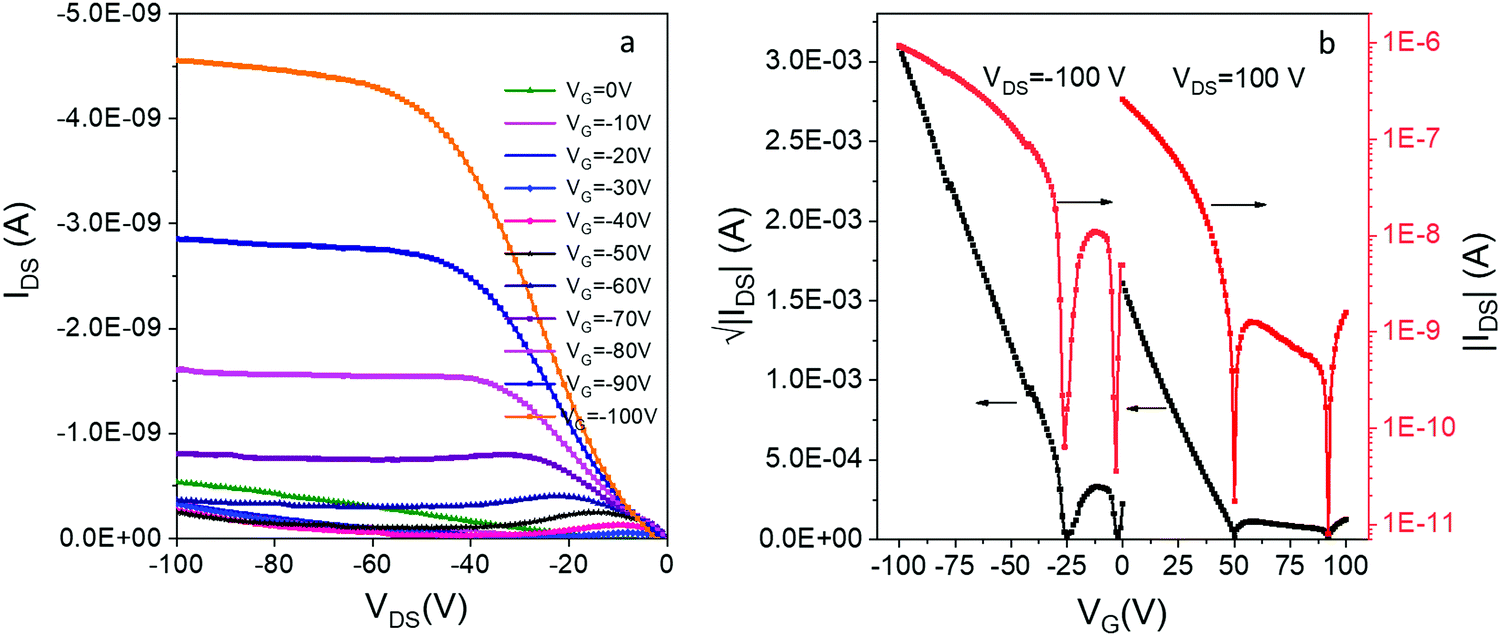 | ||
| Fig. 6 (a) The output and (b) transfer characteristics of bottom-contact bottom-gate OFETs based on thin film of 12e. | ||
We have also observed an unusual ‘double-dip’ (two minima in conductance) in the transfer curves, for both positively and negatively biased devices. Similar behavior has been previously been observed in inorganic ambipolar FETs, most notably those based on graphene.55,56 The effect likely originates from the charge transfer at the semiconductor/metal interface that leads to different neutrality points in the channel and at the contacts. It has been used for building memory devices with graphene-based FET,55 but no such applications have been yet reported for OFETs.
4. Conclusions
This study shows that two-center DD⋯AA hydrogen bonding is a robust tool for co-assembly of a wide range of p- and n-type semiconducting molecules that significantly affects and enables tuning of their electronic properties. The perturbation of the frontier orbital energies via unidirectional H-bonding transforms weak electron donors and acceptors into very strong donor/acceptor pairs. DFT analysis shows how this enhancement of π-donating and accepting abilities arises from polarization induced by unidirectional H-bonding, which then drives the formation of a CT states through π-stacking, as shown by X-ray crystallography of the full series of complexes 1a–1f. Diffuse reflectance spectroscopy of the H-bonded complexes revealed narrow band gaps (0.78 to 1.73 eV) that can be fine tuned by varying the ionization potential/electron affinity of the H-bonding components. Photoelectron spectroscopy in air (PESA) confirmed the increase of the ionization potential of the donor upon H-bonding complexation. Thin film OFETs of the H-bonded complex 1e exhibited ambipolar charge transport with an unusual “double dip” characteristic.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERC of Canada. We thank Dr T. Maris (University of Montreal) and A. Jonderian (McGill University) for X-ray analyses.References
- S. R. Forrest and M. E. Thompson, Introduction: organic electronics and optoelectronics, Chem. Rev., 2007, 107, 923–925 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Semiconducting π-conjugated systems in field-effect transistors: a material odyssey of organic electronics, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, All-Polymer Solar Cells: Recent Progress, Challenges, and Prospects, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Material insights and challenges for non-fullerene organic solar cells based on small molecular acceptors, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- N. T. Kalyani and S. Dhoble, Organic light emitting diodes: energy saving lighting technology—A review, Renewable Sustainable Energy Rev., 2012, 16, 2696–2723 CrossRef.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, Functionalized pentacene: improved electronic properties from control of solid-state order, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS.
- J. E. Anthony, Functionalized acenes and heteroacenes for organic electronics, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- J. E. Anthony, The larger acenes: versatile organic semiconductors, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS.
- F. J. Hoeben, P. Jonkheijm, E. Meijer and A. P. Schenning, About supramolecular assemblies of π-conjugated systems, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS.
- D. González-Rodríguez and A. P. Schenning, Hydrogen-bonded supramolecular π-functional materials, Chem. Mater., 2011, 23, 310–325 CrossRef.
- O. Ivasenko and D. F. Perepichka, Mastering fundamentals of supramolecular design with carboxylic acids. Common lessons from X-ray crystallography and scanning tunneling microscopy, Chem. Soc. Rev., 2011, 40, 191–206 RSC.
- K. Aratsu, R. Takeya, B. R. Pauw, M. J. Hollamby, Y. Kitamoto, N. Shimizu, H. Takagi, R. Haruki, S.-I. Adachi and S. Yagai, Supramolecular copolymerization driven by integrative self-sorting of hydrogen-bonded rosettes, Nat. Commun., 2020, 11, 1–12 Search PubMed.
- M. Gray, A. O. Cuello, G. Cooke and V. M. Rotello, Hydrogen bonding in redox-modulated molecular recognition. An experimental and theoretical investigation, J. Am. Chem. Soc., 2003, 125, 7882–7888 CrossRef CAS.
- V. Nandwana, I. Samuel, G. Cooke and V. M. Rotello, Aromatic stacking interactions in flavin model systems, Acc. Chem. Res., 2013, 46, 1000–1009 CrossRef CAS.
- E. D. Głowacki, M. Irimia-Vladu, M. Kaltenbrunner, J. Gsiorowski, M. S. White, U. Monkowius, G. Romanazzi, G. P. Suranna, P. Mastrorilli, T. Sekitani, S. Bauer, T. Someya, L. Torsi and N. S. Sariciftci, Hydrogen-bonded semiconducting pigments for air-stable field-effect transistors, Adv. Mater., 2013, 25, 1563–1569 CrossRef.
- F. Zhang, V. Lemaur, W. Choi, P. Kafle, S. Seki, J. Cornil, D. Beljonne and Y. Diao, Repurposing DNA-binding agents as H-bonded organic semiconductors, Nat. Commun., 2019, 19, 4217 CrossRef.
- M. Gsänger, J. H. Oh, M. Könemann, H. W. Höffken, A. M. Krause, Z. Bao and F. Würthner, A crystal-engineered hydrogen-bonded octachloroperylene diimide with a twisted core: an n-channel organic semiconductor, Angew. Chem., Int. Ed., 2010, 49, 740–743 CrossRef.
- Z. He, D. Liu, R. Mao, Q. Tang and Q. Miao, Hydrogen-bonded dihydrotetraazapentacenes, Org. Lett., 2012, 14, 1050–1053 CrossRef CAS.
- T. Murata, Y. Morita, Y. Yakiyama, K. Fukui, H. Yamochi, G. Saito and K. Nakasuji, Hydrogen-bond interaction in organic conductors: redox activation, molecular recognition, structural regulation, and proton transfer in donor–acceptor charge-transfer complexes of TTF-imidazole, J. Am. Chem. Soc., 2007, 129, 10837–10846 CrossRef CAS.
- T. Isono, H. Kamo, A. Ueda, K. Takahashi, A. Nakao, R. Kumai, H. Nakao, K. Kobayashi, Y. Murakami and H. Mori, Hydrogen bond-promoted metallic state in a purely organic single-component conductor under pressure, Nat. Commun., 2013, 4, 1344 CrossRef.
- Y. Morita, E. Miyazaki, K. Fukui, S. Maki and K. Nakasuji, Introduction of amino groups into the dibenzo-TTF π-system: enhanced electron-donating ability and intermolecular hydrogen bonding, Bull. Chem. Soc. Jpn., 2005, 78, 2014–2018 CrossRef CAS.
- H. T. Black and D. F. Perepichka, Crystal engineering of dual channel p/n organic semiconductors by complementary hydrogen bonding, Angew. Chem., Int. Ed., 2014, 126, 2170–2174 CrossRef.
- H. T. Black, H. Lin, F. Bélanger-Gariépy and D. F. Perepichka, Supramolecular control of organic p/n-heterojunctions by complementary hydrogen bonding, Faraday Discuss., 2014, 174, 297–312 RSC.
- Y. Qin, C. Cheng, H. Geng, C. Wang, W. Hu, W. Xu, Z. Shuai and D. Zhu, Efficient ambipolar transport properties in alternate stacking donor–acceptor complexes: from experiment to theory, Phys. Chem. Chem. Phys., 2016, 18, 14094–14103 RSC.
- Y. Qin, J. Zhang, X. Zheng, H. Geng, G. Zhao, W. Xu, W. Hu, Z. Shuai and D. Zhu, Charge-Transfer Complex Crystal Based on Extended-π-Conjugated Acceptor and Sulfur-Bridged Annulene: Charge-Transfer Interaction and Remarkable High Ambipolar Transport Characteristics, Adv. Mater., 2014, 26, 4093–4099 CrossRef CAS.
- A. Mandal, A. Choudhury, S. Sau, P. K. Iyer and P. Mal, Exploring Ambipolar Semiconductor Nature of Binary and Ternary Charge-Transfer Cocrystals of Triphenylene, Pyrene, and TCNQ, J. Phys. Chem. C, 2020, 124, 6544–6553 CrossRef CAS.
- S. Horiuchi, F. Ishii, R. Kumai, Y. Okimoto, H. Tachibana, N. Nagaosa and Y. Tokura, Ferroelectricity near room temperature in co-crystals of nonpolar organic molecules, Nat. Mater., 2005, 4, 163 CrossRef CAS.
- A. S. Tayi, A. Kaeser, M. Matsumoto, T. Aida and S. I. Stupp, Supramolecular ferroelectrics, Nat. Chem., 2015, 7, 281 CrossRef CAS.
- Y. Huang, Z. Wang, Z. Chen and Q. Zhang, Organic Cocrystals: Beyond Electrical Conductivities and Field-Effect Transistors (FETs), Angew. Chem., Int. Ed., 2019, 58, 9696–9711 CrossRef CAS.
- J. Zhang, H. Geng, T. S. Virk, Y. Zhao, J. Tan, C. A. Di, W. Xu, K. Singh, W. Hu and Z. Shuai, Sulfur-Bridged Annulene-TCNQ Co-Crystal: A Self-Assembled “Molecular Level Heterojunction” with Air Stable Ambipolar Charge Transport Behavior, Adv. Mater., 2012, 24, 2603–2607 CrossRef CAS.
- M. Kumar, K. V. Rao and S. J. George, Supramolecular charge transfer nanostructures, Phys. Chem. Chem. Phys., 2014, 16, 1300–1313 RSC.
- J. Zhang, J. Tan, Z. Ma, W. Xu, G. Zhao, H. Geng, C. A. Di, W. Hu, Z. Shuai and K. Singh, Fullerene/sulfur-bridged annulene cocrystals: two-dimensional segregated heterojunctions with ambipolar transport properties and photoresponsivity, J. Am. Chem. Soc., 2013, 135, 558–561 CrossRef CAS.
- J. Zhang, P. Gu, G. Long, R. Ganguly, Y. Li, N. Aratani, H. Yamada and Q. Zhang, Switching charge-transfer characteristics from p-type to n-type through molecular “doping”(co-crystallization), Chem. Sci., 2016, 7, 3851–3856 RSC.
- L. Zhu, Y. Yi, Y. Li, E.-G. Kim, V. Coropceanu and J.-L. Brédas, Prediction of remarkable ambipolar charge-transport characteristics in organic mixed-stack charge-transfer crystals, J. Am. Chem. Soc., 2012, 134, 2340–2347 CrossRef CAS.
- S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner and S. Y. Park, Tailor-made highly luminescent and ambipolar transporting organic mixed stacked charge-transfer crystals: an isometric donor–acceptor approach, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS.
- W. Zhu, R. Zheng, Y. Zhen, Z. Yu, H. Dong, H. Fu, Q. Shi and W. Hu, Rational design of charge-transfer interactions in halogen-bonded co-crystals toward versatile solid-state optoelectronics, J. Am. Chem. Soc., 2015, 137, 11038–11046 CrossRef CAS.
- L. Sun, Y. Wang, F. Yang, X. Zhang and W. Hu, Cocrystal engineering: a collaborative strategy toward functional materials, Adv. Mater., 2019, 31, 1902328 CrossRef.
- H. T. Black, N. Yee, Y. Zems and D. F. Perepichka, Complementary Hydrogen Bonding Modulates Electronic Properties and Controls Self-Assembly of Donor/Acceptor Semiconductors, Chem. – Eur. J., 2016, 22, 17251–17261 CrossRef CAS.
- C.-H. Liu, R. Niazi and D. F. Perepichka, Extraordinary Enhancement of π-Electron Donor/Acceptor Ability by DD/AA Complementary Hydrogen Bonding, Angew. Chem., Int. Ed., 2019, 131, 17473–17482 CrossRef.
- D. Ley, C. X. Guzman, K. H. Adolfsson, A. M. Scott and A. B. Braunschweig, Cooperatively assembling donor–acceptor superstructures direct energy into an emergent charge separated state, J. Am. Chem. Soc., 2014, 136, 7809–7812 CrossRef CAS.
- M.-J. Jiang, W.-J. Xiao, J.-C. Huang, W.-S. Li and Y.-Q. Mo, Diindole[3,2-b:4,5-b′]pyrrole as a chromophore containing three successively fused pyrroles: synthesis, optoelectronic properties and π-functionalization, Tetrahedron, 2016, 72, 979–984 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett and W. Jones, Mechanochemistry: opportunities for new and cleaner synthesis, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- M.-S. Liao, Y. Lu, V. D. Parker and S. Scheiner, DFT calculations and spectral measurements of charge-transfer complexes formed by aromatic amines and nitrogen heterocycles with tetracyanoethylene and chloranil, J. Phys. Chem. C, 2003, 107, 8939–8948 CrossRef CAS.
- D. F. Perepichka, M. R. Bryce, C. Pearson, M. C. Petty, E. J. McInnes and J. P. Zhao, A Covalent Tetrathiafulvalene–Tetracyanoquinodimethane Diad: Extremely Low HOMO–LUMO Gap, Thermoexcited Electron Transfer, and High-Quality Langmuir–Blodgett Films, Angew. Chem., Int. Ed., 2003, 115, 4784–4787 CrossRef.
- C. Fonseca Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, Hydrogen bonding in DNA base pairs: reconciliation of theory and experiment, J. Am. Chem. Soc., 2000, 122, 4117–4128 CrossRef.
- G. L. Strati, J. L. Willett and F. A. Momany, A DFT/ab initio study of hydrogen bonding and conformational preference in model cellobiose analogs using B3LYP/6-311++G, Carbohydr. Res., 2002, 337, 1851–1859 CrossRef CAS.
- A. Dey, T.-A. Okamura, N. Ueyama, B. Hedman, K. O. Hodgson and E. Solomon, Sulfur K-edge XAS and DFT calculations on P450 model complexes: effects of hydrogen bonding on electronic structure and redox potentials, J. Am. Chem. Soc., 2005, 127, 12046–12053 CrossRef CAS.
- A. K. Turek, D. J. Hardee, A. M. Ullman, D. G. Nocera and E. N. Jacobsen, Activation of Electron-Deficient Quinones through Hydrogen-Bond-Donor-Coupled Electron Transfer, Angew. Chem., Int. Ed., 2016, 55, 539–544 CrossRef CAS.
- T. Caruso, A. Capobianco and A. Peluso, The oxidation potential of adenosine and adenosine–thymidine base pair in chloroform solution, J. Am. Chem. Soc., 2007, 129, 15347–15353 CrossRef CAS.
- Y. Paukku and G. Hill, Theoretical determination of one-electron redox potentials for DNA bases, base pairs, and stacks, J. Phys. Chem. A, 2011, 115, 4804–4810 CrossRef CAS.
- S. Yagai, T. Kinoshita, M. Higashi, K. Kishikawa, T. Nakanishi, T. Karatsu and A. Kitamura, Diversification of self-organized architectures in supramolecular dye assemblies, J. Am. Chem. Soc., 2007, 129, 13277–13287 CrossRef CAS.
- F. Würthner, C. Thalacker, A. Sautter, W. Schärtl, W. Ibach and O. Hollricher, Hierarchical self-organization of perylene bisimide–melamine assemblies to fluorescent mesoscopic superstructures, Chem. – Eur. J., 2000, 6, 3871–3886 CrossRef.
- We note that PXRDs (Fig. S12) of some of the complexes prepared by precipitation reveal different polymorphs from those observed by single crystal XRD. However, the solid state spectra (UV-vis, PESA, IR) of single crystals and powders were found to be the same.
- N. Yee, A. Dadvand and D. F. Perepichka, Serendipitous Formation of Semiconducting Semi-Nindigo Indigoid by the Degradation of Diindolopyrrole, J. Org. Chem., 2020, 85, 5073–5077 CrossRef CAS.
- A. Di Bartolomeo, F. Giubileo, S. Santandrea, F. Romeo, R. Citro, T. Schroeder and G. Lupina, Charge transfer and partial pinning at the contacts as the origin of a double dip in the transfer characteristics of graphene-based field-effect transistors, Nanotechnology, 2011, 22, 275702 CrossRef.
- H.-Y. Chiu, V. Perebeinos, Y.-M. Lin and P. Avouris, Controllable pn junction formation in monolayer graphene using electrostatic substrate engineering, Nano Lett., 2010, 10, 4634–4639 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Fred Wudl on occasion of his 80th Birthday. |
| ‡ Electronic supplementary information (ESI) available: Methods, FMO topologies, UV-vis-NIR and IR spectra, PXRD, synthesis, NMR spectra. CCDC 2014451–2014456 and 2014479. See DOI: 10.1039/d0qm00500b |
| This journal is © the Partner Organisations 2020 |