
Rh(I)-Catalyzed regioselective arylcarboxylation of acrylamides with arylboronic acids and CO2†
Lei
Cai
ab,
Lei
Fu
a,
Chunlin
Zhou
ab,
Yuzhen
Gao
 a,
Shangda
Li
a and
Gang
Li
*ab
a,
Shangda
Li
a and
Gang
Li
*ab
aKey Laboratory of Coal to Ethylene Glycol and Its Related Technology, Center for Excellence in Molecular Synthesis, Fujian Institute of Research on the Structure of Matter, Fuzhou, Fujian 350002, China
bFujian College, University of Chinese Academy of Sciences, Beijing, 100049, China. E-mail: gangli@fjirsm.ac.cn
First published on 24th September 2020
Abstract
The first Rh(I)-catalyzed regioselective arylcarboxylation of electron-deficient acrylamides with arylboronic acids under atmospheric pressure of CO2 has been developed. A range of acrylamides and arylboronic acids were compatible with this reaction under redox-neutral conditions, leading to a series of malonate derivatives that are versatile building blocks in organic syntheses.
In recent years, numerous transformations for fine chemical synthesis have been achieved through the use of carbon dioxide (CO2), which is nontoxic, low cost, abundant, and sustainable, and is often considered an ideal one-carbon (C1) building block.1 In particular, notable achievements have been made in the functionalization of alkenes with CO2via hydrocarboxylation with transition-metal catalysis and/or photocatalysis,2 including a few examples using Rh(I)-catalysis2d,g,l,r,t (Scheme 1a). Difunctionalization of alkenes with CO2, in contrast, has been less explored,3–5 although several elegant transformations of this kind have been achieved by Xi's,5a,g Popp's,5b,h Martin's,5c Yu's5d,f and Wu's5e groups. To date, however, only a single method of arylcarboxylation of alkenes is available through visible-light-driven reductive arylcarboxylation with CO2 and aryl halides, and has been demonstrated by our group recently [Scheme 1b(i)],6 besides two isolated examples of titanocene-catalyzed arylcarboxylation of dienes reported by Xi's group [Scheme 1b(ii)].5g However, the substrates of this photocatalytic method were limited to styrenes and not applicable to electron-deficient α,β-unsaturated substrates such as acrylamides. Therefore, a complementary method that could overcome this limitation is highly desirable.
 | ||
| Scheme 1 Carboxylation of alkenes with CO2. | ||
As part of our continuing interest in developing Rh-catalyzed carboxylation using CO2,7 we considered whether it is possible to achieve a Rh-catalyzed difunctionalization of α,β-unsaturated compounds with CO2.8 Compared with the Rh-catalyzed 1,4-addition of α,β-unsaturated compounds with organometallic reagents, Rh-catalyzed difunctionalization of these compounds is much less documented and the corresponding arylcarboxylation has not been achieved to date with Rh catalysis.9 Herein, we report the first Rh(I)-catalyzed arylcarboxylation of acrylamides with arylboronic acids and CO2.
After extensive investigation of reaction conditions (see also the ESI†), a set of promising reaction conditions were revealed by using N-methyl-N-phenylacrylamide 1a and phenylboronic acid 2a as the model substrates with [Rh(cod)Cl]2 as the catalyst in the presence of Cs2CO3 under CO2 (1 atm) in DMA. Pleasingly, 40% of the desired phenylcarboxylation product 3a was detected, while 25% of Heck-type product 4 through a β-H elimination process and 7% of 1,4-addition product 5 were also identified as the major side products (Table 1, entry 1). It should be noted that traces of hydrocarboxylation and alkene reduction products could also be detected. Moreover, for the ease of reaction result analysis, the initial carboxylic acid products were converted into their ester counterparts after the phenylcarboxylation reaction ceased. In order to suppress the formation of side products 4 and 5 and increase the desired product, ligands such as dppe and IPr·HCl were added to tune the reactivity of the Rh catalyst (entries 2 and 3, see also the ESI† for more ligands used). Unfortunately, no significant improvement was observed. To our delight, however, the yield of the desired product was slightly increased when an organic base TMEDA (B1) or PMDETA (B2) was introduced into the reaction (entries 4 and 5). Subsequently, different silver salts, which are often used to increase the reactivity of Rh catalysts, were extensively investigated as additives for the reaction (entries 6–9; see also the ESI†), and it was found that AgOTf was the best choice to produce the best yield of 3a, leading to the least yield of 4 and 5 (entry 9). Comparing the results of entry 5 and entries 6–8, it appears that the silver salt mainly inhibited the formation of side product 4. However, the exact role of the silver salt is not clear at present.10 Notably, the amine additive (PMDETA) might also be a potential tridentate ligand to form a silver complex in the solution to increase the solubility of the silver salts such as AgCl.11,10c Interestingly, CuCl could also slightly improve the reaction (entry 10, see also the ESI†). Other bases or solvents were also evaluated, but no increase in the yield of the desired product was observed (entries 11–14). Other Rh catalysts were then tested (entries 15–18), and only [Rh(cod)OH]2 could lead to rather high yield of the product (entry 17). Importantly, the efficiency of the reaction was reduced evidently with a lower loading of the [Rh(cod)Cl]2 catalyst (entry 19) or without B2 (entry 20). Moreover, no product was received without the Rh catalyst (entry 21). Finally, when the reaction was run under argon instead of CO2, no desired product was found, indicating that CO2 was not generated in situ from caesium carbonate (entry 22).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Co-Base | Additive | Yieldb (%) (3a/4/5/1a) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), CO2 (1 atm, closed), catalyst (2.5 mol%), ligand (5 mol%), Cs2CO3 (0.2 mmol), additive (5 mol%), Co-base (50 mol%), and DMA (1 mL), 60 °C, 24 h. b Yield was determined by 1H NMR with CH2Br2 as the internal standard. c dppe (5 mol% was added as a ligand, dppe: 1,2-bis(diphenylphosphino)ethane). d IPr·HCl (5 mol% was added, IPr·HCl: 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride. e CsF (0.2 mmol) as a base. f KOtBu (0.2 mmol) as a base. g DMF as solvent. h NMP as solvent. i [Rh(cod)Cl]2 (1 mol%) as a catalyst. j Ar (1 atm, closed) was used instead of CO2. B1: N,N,N′,N′-tetramethylethylenediamine (TMEDA). B2: N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA). | ||||
| 1 | [Rh(cod)Cl]2 | — | — | 40/25/7/— |
| 2c | [Rh(cod)Cl]2 | — | — | 43/18/16/— |
| 3d | [Rh(cod)Cl]2 | — | — | 36/23/5/— |
| 4 | [Rh(cod)Cl]2 | B1 | — | 47/27/6/— |
| 5 | [Rh(cod)Cl]2 | B2 | — | 49/27/3/— |
| 6 | [Rh(cod)Cl]2 | B2 | AgF | 73/5/14/— |
| 7 | [Rh(cod)Cl]2 | B2 | Ag2O | 75/6/13/— |
| 8 | [Rh(cod)Cl]2 | B2 | AgCl | 77/6/13/— |
| 9 | [Rh(cod)Cl] 2 | B2 | AgOTf | 83/6/5/— |
| 10 | [Rh(cod)Cl]2 | B2 | CuCl | 58/18/7/— |
| 11e | [Rh(cod)Cl]2 | B2 | AgOTf | 13/14/57/2 |
| 12f | [Rh(cod)Cl]2 | B2 | AgOTf | 43/14/12/— |
| 13g | [Rh(cod)Cl]2 | B2 | AgOTf | 19/9/52/— |
| 14h | [Rh(cod)Cl]2 | B2 | AgOTf | 15/9/73/— |
| 15 | Rh(cod)2OTf | B2 | AgOTf | 59/16/8/— |
| 16 | [Rh(CO)2Cl]2 | B2 | AgOTf | —/10/—/81 |
| 17 | [Rh(cod)OH]2 | B2 | AgOTf | 80/9/11/— |
| 18 | [Rh(coe)2Cl]2 | B2 | AgOTf | —/—/—/84 |
| 19i | [Rh(cod)Cl]2 | B2 | AgOTf | 46/14/3/18 |
| 20 | [Rh(cod)Cl]2 | — | AgOTf | 46/17/13/— |
| 21 | — | B2 | AgOTf | —/—/—/53 |
| 22j | [Rh(cod)Cl]2 | B2 | AgOTf | —/8/70/— |
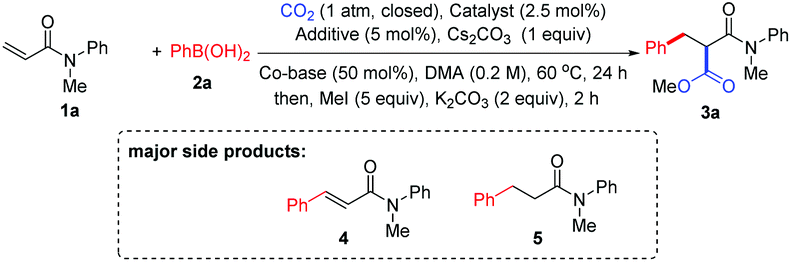
With the optimized reaction conditions in hand, the method was then tested with a range of arylboronic acids (Table 2). First, good isolated yields were obtained with model phenylboronic acid through esterification with different aryl halides (3a′ and 3a′′). To our delight, arylboronic acids with electron-rich or electron-deficient substituents at the ortho- or meta-position of the phenyl group were tolerated, providing moderate to good yields of malonates (3b–3f). It was also found that several types of substituents at the para-position were suitable for the reaction (3g–3k). It is worth noting that multisubstituted phenylboronic acids could also react with 1a to produce the desired products in acceptable yields (3l–3o). It should also be mentioned that the formation of the side products from Heck-type coupling or 1,4-addition could not be suppressed completely for the above examples (see the ESI† for details).
| Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol, 1.5 equiv.), CO2 (1 atm, closed), [Rh(cod)Cl]2 (2.5 mol%), Cs2CO3 (0.2 mmol), AgOTf (5 mol%), PMDETA (50 mol%), and DMA (1 mL), 60 °C, 24 h; then MeI, or BuBr, or BnBr (1 mmol), and K2CO3 (0.4 mmol). Isolated yields; the yield of 3a in parentheses is that of 1 mmol (1a) scale reaction.a Ag2O (5 mol%) as an additive.b KOtBu (0.6 mmol) as a base.c Cs2CO3 (0.6 mmol) as a base.d 2 (0.4 mmol) was used.e AgCl (5 mol%) as an additive.f AgF (5 mol%) as an additive.g [Rh(cod)Cl]2 (5 mol%) as a catalyst. |
|---|
|
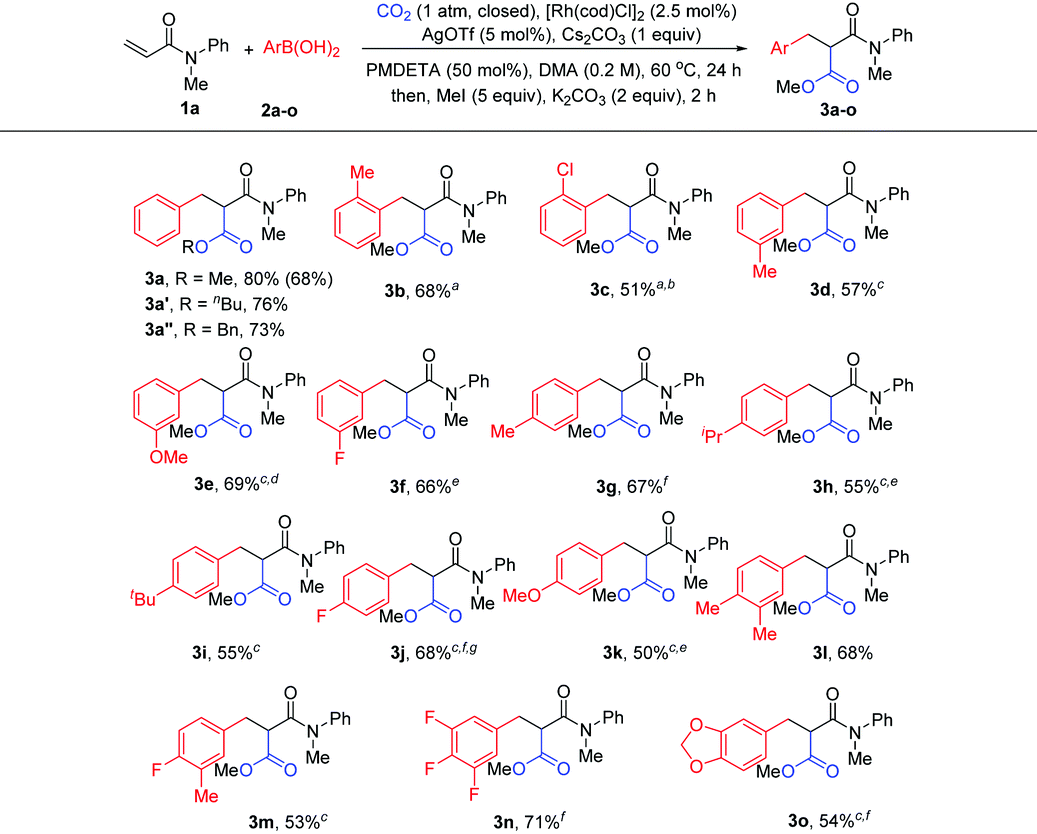
The scope of acrylamides was then examined. As shown in Table 3, acrylamides with electron-donating and electron-withdrawing groups at the meta- or para-position of the aryl group of acrylamides were well tolerated (3ab–3ak), affording the desired products smoothly. Unfortunately, acrylamides bearing substituents at the ortho-position of the acrylamide's aryl group exhibited low reactivity, which is possibly due to the increased steric hindrance. Moreover, a di-substituted acrylamide was also a viable substrate for this reaction (3al). When replacing the N-protecting group Me with Ph and Bn (3am and 3an), the corresponding products were still generated. Notably, indoline- and tetrahydroquinoline-derived acrylamides were compatible with the reaction, yielding the desired products in good yields (3ao and 3ap). In addition, it is noteworthy that the side products from Heck-type coupling or 1,4-addition were also present and were hard to suppress for these examples (see the ESI† for details).
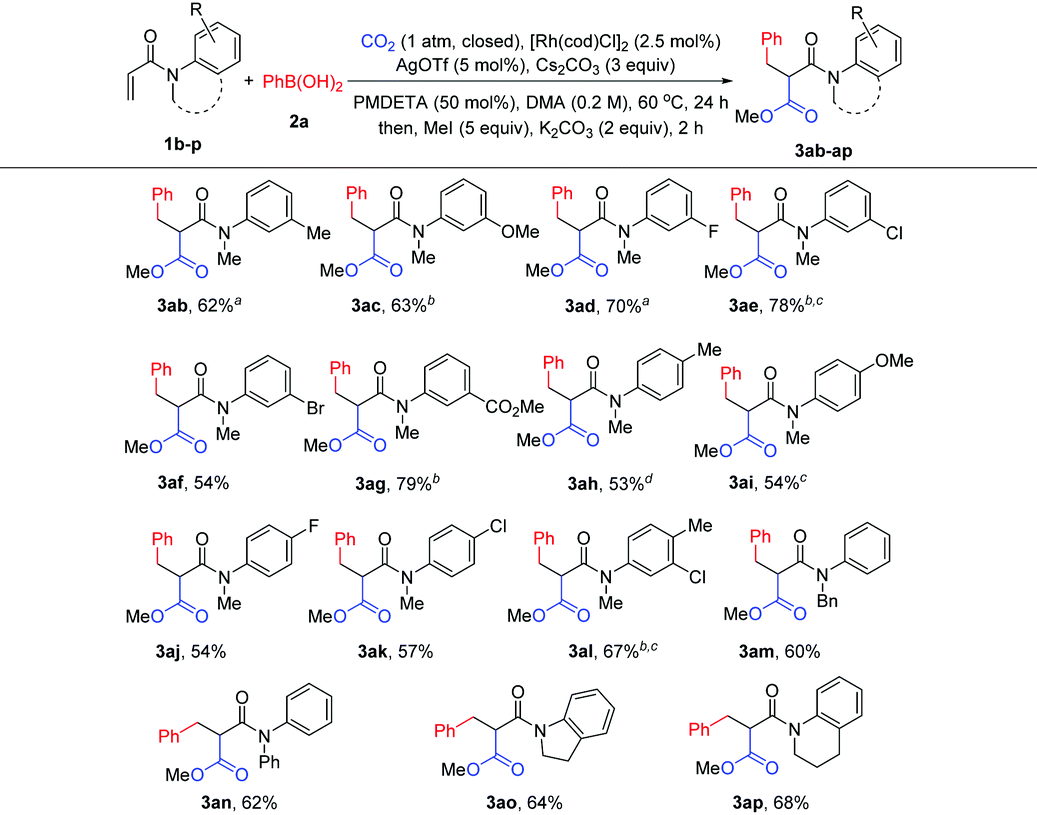
To gain insight into this novel arylcarboxylation method, some control experiments were performed. As shown in Scheme 2, subjecting side product 5 to standard conditions could not lead to the desired product 3a (Scheme 2a). No 3a could be formed without adding the Rh catalyst under standard conditions and with AgCl as the additive which could be formed from [Rh(cod)Cl]2 and AgOTf in the reaction (Scheme 2b). These two experiments indicated that the conjugate addition and the nucleophilic addition of CO2 by the rhodium enolate should occur sequentially without the hydrolysis of the rhodium enolate,9d and that the weakly basic conditions of the reaction were not sufficient to induce carboxylation of the α-position of the amide 5. Further experiments also indicated the presence of a rhodium enolate (Scheme 2c) and its importance for the second functionalization after conjugate addition (Scheme 2d). Finally, the product could be elaborated to generate oxindole-type compounds which are versatile building blocks for organic synthesis and are often found as the core structure of natural products and molecules of medicinal importance (Scheme 2e).12
 | ||
| Scheme 2 Control experiments and product elaboration. | ||
Based on the above results, a plausible mechanism is proposed (Scheme 3). First, ligand exchange converts the [Rh(cod)Cl]2 catalyst to an active rhodium species A, which undergoes transmetalation with arylboronic acids 2, leading to a [Rh]–Ar species B. Coordination of B onto 1a, followed by a regioselective addition, produces a rhodium enolate D. This key intermediate D will attack CO2 to afford the carboxylate intermediate E, which ultimately leads to the desired product 3a. On the other hand, rhodium enolate D can also undergo either β-H elimination to produce side product 4 or direct protonation to produce side product 5, respectively.
 | ||
| Scheme 3 Proposed catalytic cycle. | ||
Conclusions
In summary, we have developed the first Rh(I)-catalyzed regioselective arylcarboxylation of electron-deficient acrylamides with CO2 under redox-neutral conditions. A range of acrylamides and arylboronic acids were suitable for this reaction, leading to a series of malonate derivatives that could be converted into other important structures such as oxindoles. Preliminary mechanistic studies indicated the importance of the rhodium enolate. Further investigation of the Rh-catalyzed carboxylation strategy with CO2 is underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the financial support from NSFC (Grant No. 21871257 and 21801240) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000).Notes and references
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS; (b) I. I. F. Boogaerts and S. P. Nolan, Chem. Commun., 2011, 47, 3021 RSC; (c) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (d) Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602 RSC; (e) F. Manjolinho, M. Arndt, K. Gooßen and L. J. Gooßen, ACS Catal., 2012, 2, 2014 CrossRef CAS; (f) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS; (g) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC; (h) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS; (i) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef; (j) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353 CrossRef; (k) M. Börjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739 CrossRef; (l) Z. Zhang, T. Ju, J.-H. Ye and D.-G. Yu, Synlett, 2017, 28, 741 CrossRef CAS; (m) T.-S. Mei, Y.-G. Chen, X.-T. Xu, K. Zhang, Y.-Q. Li, L.-P. Zhang and P. Fang, Synthesis, 2017, 50, 35 CrossRef; (n) J. Luo and I. Larrosa, ChemSusChem, 2017, 10, 3317 CrossRef CAS; (o) W. Zhang, N. Zhang, C. Guo and X. Lü, Chin. J. Org. Chem., 2017, 37, 1309 CrossRef CAS; (p) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948 CrossRef CAS; (q) Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644 CrossRef CAS; (r) F. Tan and G. Yin, Chin. J. Chem., 2018, 36, 545 CrossRef CAS; (s) S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382 RSC.
- (a) C. M. Williams, J. B. Johnson and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14936 CrossRef CAS; (b) T. Ohishi, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 8114 CrossRef CAS; (c) M. D. Greenhalgh and S. P. Thomas, J. Am. Chem. Soc., 2012, 134, 11900 CrossRef CAS; (d) T. G. Ostapowicz, M. Schmitz, M. Krystof, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 12119 CrossRef CAS; (e) C. Hayashi, T. Hayashi, S. Kikuchi and T. Yamada, Chem. Lett., 2014, 43, 565 CrossRef CAS; (f) L. Wu, Q. Liu, I. Fleischer, R. Jackstell and M. Beller, Nat. Commun., 2014, 5, 3091 CrossRef; (g) S. Kawashima, K. Aikawa and K. Mikami, Eur. J. Org. Chem., 2016, 3166 CrossRef CAS; (h) P. Shao, S. Wang, C. Chen and C. Xi, Org. Lett., 2016, 18, 2050 CrossRef CAS; (i) M. Gaydou, T. Moragas, F. Julia-Hernandez and R. Martin, J. Am. Chem. Soc., 2017, 139, 12161 CrossRef CAS; (j) Y.-Y. Gui, N. Hu, X.-W. Chen, L.-L. Liao, T. Ju, J. H. Ye, Z. Zhang, J. Li and D.-G. Yu, J. Am. Chem. Soc., 2017, 139, 17011 CrossRef CAS; (k) M. Juhl, S. L. R. Laursen, Y. Huang, D. U. Nielsen, K. Daasbjerg and T. Skrydstrup, ACS Catal., 2017, 7, 1392 CrossRef CAS; (l) K. Murata, N. Numasawa, K. Shimomaki, J. Takaya and N. Iwasawa, Chem. Commun., 2017, 53, 3098 RSC; (m) H. Seo, A. Liu and T. F. Jamison, J. Am. Chem. Soc., 2017, 139, 13969 CrossRef CAS; (n) J. Takaya, K. Miyama, C. Zhu and N. Iwasawa, Chem. Commun., 2017, 53, 3982 RSC; (o) T. Ju, Q. Fu, J.-H. Ye, Z. Zhang, L. L. Liao, S. S. Yan, X.-Y. Tian, S. P. Luo, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2018, 57, 13897 CrossRef CAS; (p) Y.-X. Luan and M. Ye, Tetrahedron Lett., 2018, 59, 853 CrossRef CAS; (q) Q.-Y. Meng, S. Wang, G. S. Huff and B. König, J. Am. Chem. Soc., 2018, 140, 3198 CrossRef CAS; (r) L. Pavlovic, J. Vaitla, A. Bayer and K. H. Hopmann, Organometallics, 2018, 37, 941 CrossRef CAS; (s) X. Zhang, C. Shen, C. Xia, X. Tian and L. He, Green Chem., 2018, 20, 5533 RSC; (t) K. Murata, N. Numasawa, K. Shimomaki, J. Takaya and N. Iwasawa, Front. Chem., 2019, 7, 371 CrossRef CAS.
- For reviews on difunctionalization of alkenes, see: (a) Z. Zhang, L. Gong, X.-Y. Zhou, S.-S. Yan, J. Li and D.-G. Yu, Acta Chim. Sin., 2019, 77, 783 CrossRef; (b) L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285 RSC; (c) S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439 CrossRef CAS; (d) T. Koike and M. Akita, Chem, 2018, 4, 409 CrossRef CAS; (e) X. Wu, S. Wu and C. Zhu, Tetrahedron Lett., 2018, 59, 1328 CrossRef CAS; (f) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175 CrossRef CAS; (g) X. Wang and A. Studer, Acc. Chem. Res., 2017, 50, 1712 CrossRef CAS; (h) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS; (i) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef CAS; (j) T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945 CrossRef CAS; (k) M.-Y. Cao, X. Ren and Z. Lu, Tetrahedron Lett., 2015, 56, 3732 CrossRef CAS; (l) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC.
- For intramolecular difunctionalization of alkenes, see: (a) M. Takimoto and M. Mori, J. Am. Chem. Soc., 2002, 124, 10008 CrossRef CAS; (b) M. Takimoto, T. Mizuno, M. Mori and Y. Sato, Tetrahedron, 2006, 62, 7589 CrossRef CAS; (c) A. Nomoto, Y. Kojo, G. Shiino, Y. Tomisaka, I. Mitani, M. Tatsumi and A. Ogawa, Tetrahedron Lett., 2010, 51, 6580 CrossRef CAS; (d) M.-Y. Wang, Y. Cao, X. Liu, N. Wang, L.-N. He and S.-H. Li, Green Chem., 2017, 19, 1240 RSC; (e) J. B. Diccianni, T. Heitmann and T. Diao, J. Org. Chem., 2017, 82, 6895 CrossRef CAS.
- (a) P. Shao, S. Wang, C. Chen and C. Xi, Chem. Commun., 2015, 51, 6640 RSC; (b) T. W. Butcher, E. J. McClain, T. G. Hamilton, T. M. Perrone, K. M. Kroner, G. C. Donohoe, N. G. Akhmedov, J. L. Petersen and B. V. Popp, Org. Lett., 2016, 18, 6428 CrossRef CAS; (c) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915 CrossRef CAS; (d) J.-H. Ye, M. Miao, H. Huang, S. S. Yan, Z. B. Yin, W. J. Zhou and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15416 CrossRef CAS; (e) J. Hou, A. Ee, H. Cao, H. W. Ong, J. H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220 CrossRef CAS; (f) Q. Fu, Z.-Y. Bo, J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Nat. Commun., 2019, 10, 3592 CrossRef; (g) W. Hang, S. Zou and C. Xi, ChemCatChem, 2019, 11, 3814 CrossRef CAS; (h) T. M. Perrone, A. S. Gregory, S. W. Knowlden, N. R. Ziemer, R. N. Alsulami, J. L. Petersen and B. V. Popp, ChemCatChem, 2019, 11, 5814 CrossRef CAS; (i) W. J. Yoo, J. Kondo, J. A. Rodriguez-Santamaria, T. V. Q. Nguyen and S. Kobayashi, Angew. Chem., Int. Ed., 2019, 58, 6772 CrossRef CAS; (j) A. Gevorgyan, K. H. Hopmann and A. Bayer, ChemSusChem, 2020, 13, 2080 CrossRef CAS.
- H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122 CrossRef CAS.
- (a) L. Fu, S. Li, Z. Cai, Y. Ding, X.-Q. Guo, L.-P. Zhou, D. Yuan, Q.-F. Sun and G. Li, Nat. Catal., 2018, 1, 469 CrossRef CAS; (b) Z. Cai, S. Li, Y. Gao and G. Li, Adv. Synth. Catal., 2018, 360, 4005 CrossRef CAS; (c) R. Huang, S. Li, L. Fu and G. Li, Asian J. Org. Chem., 2018, 7, 1376 CrossRef CAS; (d) Y. Gao, Z. Cai, S. Li and G. Li, Org. Lett., 2019, 21, 3663 CrossRef CAS.
- (a) H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251 CrossRef CAS; (b) T. Suga, H. Mizuno, J. Takaya and N. Iwasawa, Chem. Commun., 2014, 50, 14360 RSC; (c) T. Suga, T. Saitou, J. Takaya and N. Iwasawa, Chem. Sci., 2017, 8, 1454 RSC; (d) T. Saitou, Y. Jin, K. Isobe, T. Suga, J. Takaya and N. Iwasawa, Chem. – Asian J., 2020, 15, 1941 CrossRef CAS.
- (a) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS; (b) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS; (c) N. Miyaura, Bull. Chem. Soc. Jpn., 2008, 81, 1535 CrossRef CAS; (d) Y. J. Jang, H. Yoon and M. Lautens, Org. Lett., 2015, 17, 3895 CrossRef CAS.
- (a) Q.-W. Song, W. Q. Chen, R. Ma, A. Yu, Q. Y. Li, Y. Chang and L.-N. He, ChemSusChem, 2015, 8, 821 CrossRef CAS; (b) K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524 RSC; (c) K. Sekine, Y. Sadamitsu and T. Yamada, Org. Lett., 2015, 17, 5706 CrossRef CAS; (d) N. Eghbali, J. Eddy and P. T. Anastas, J. Org. Chem., 2008, 73, 6932 CrossRef CAS; (e) X. Zhang, W.-Z. Zhang, X. Ren, L.-L. Zhang and X.-B. Lu, Org. Lett., 2011, 13, 2402 CrossRef CAS.
- I.-S. Chun, J.-A. Kwon, M.-N. Bae, S.-S. Lee and O.-S. Jung, Bull. Korean Chem. Soc., 2006, 27, 1005 CrossRef CAS.
- (a) A. Perry and R. J. Taylor, Chem. Commun., 2009, 45, 3249 RSC; (b) Z. J. Wu, S. R. Li, H. Long and H. C. Xu, Chem. Commun., 2018, 54, 4601 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and data for new compounds. See DOI: 10.1039/d0gc02667k |
| This journal is © The Royal Society of Chemistry 2020 |