
Introducing borane clusters into polymeric frameworks: architecture, synthesis, and applications
Jing
Yan
,
Weihong
Yang
,
Qiuyu
Zhang
 * and
Yi
Yan
*
* and
Yi
Yan
*
Department of Chemistry, School of Chemistry and Chemical Engineering, Key Laboratory of Special Functional and Smart Polymer Materials of Ministry of Industry and Information Technology, MOE Key Laboratory of Material Physics and Chemistry under Extraordinary Conditions, Northwestern Polytechnical University, Xi’an, 710129, China. E-mail: yanyi@nwpu.edu.cn; qyzhang@nwpu.edu.cn
First published on 2nd September 2020
Abstract
Borane clusters represent a unique class of nano-objects not only because of their special coordination and 3D structure but also due to their broad applications ranging from heat resistance coating to cancer therapy agent. Borane cluster-containing polymers (BCCPs) can effectively integrate the merits of both borane clusters and polymers. During the last two decades, with the progress of boron chemistry and the development of advanced polymerization techniques, BCCPs with different architectures and properties have been developed. The introduction of borane clusters into polymeric frameworks not only improves the chemical and thermal stability of traditional polymers but also endows BCCPs with many specific properties, such as photoluminescence, chemical sensing, heat resistance, and boron neutron capture therapy. This feature article gives an overview of the preparation of BCCPs, especially focusing on the design and synthetic methodology. We expect that this review will be helpful to researchers working in the fields of polymer chemistry and materials science.
 Jing Yan | Jing Yan studied applied chemistry at the College of Chemistry in Shandong Normal University and obtained her bachelor's degree in 2007. She obtained her PhD degree from the College of Chemistry, Jilin University in 2012 under the guidance of Dr Hongyan Wang. From 2012 to 2015, she worked as a postdoctoral researcher at Queen's University, Canada and as a research assistant in Dr Qian Wang's group at University of South Carolina, USA. She joined the faculty at the School of Chemistry and Chemical Engineering of Northwestern Polytechnical University in July 2015. Her research interests focus on biomedical polymers. |
 Weihong Yang | Weihong Yang was born in Shaanxi China in 1991. She obtained her bachelor's and master's degree from Beijing University of Chemical Technology. She is now pursuing her PhD degree under the guidance of Dr Qiuyu Zhang and Dr Yi Yan at the School of Chemistry and Chemical Engineering in Northwestern Polytechnical University. Her research focuses on the design and preparation of polyelectrolytes and anion exchange membranes in fuel cells. |
 Qiuyu Zhang | Qiuyu Zhang studied analytical chemistry at Beijing University of Chemical Technology and obtained her bachelor's degree in 1987 and master's degree at the Institute of Chemistry, Chinese Academy of Sciences. She obtained her PhD degree in materials science in 1999 under the guidance of Dr Mokuang Kang. From 2002 to 2004, she was supported by RSC and carried out scientific research in Loughborough University. Since 2000, she has been appointed as a full professor in the School of Chemistry and Chemical Engineering. Her research interests focus on applied surface science, porous polymers, and high-performance polymers. |
 Yi Yan | Yi Yan studied chemistry at the College of Chemistry, Jilin University and obtained his bachelor's degree in 2006 and PhD degree from the State Key Laboratory of Supramolecular Structure and Materials in 2011 under the guidance of Dr Lixin Wu. Then, he did his postdoctoral research with Prof. Anne Petitjean at Queen's University, Canada and Prof. Chuanbing Tang at University of South Carolina, USA. He joined the faculty of School of Chemistry and Chemical Engineering at Northwestern Polytechnical University in May 2015. His research interests focus on supramolecular self-assembly and the applications of metal-containing polymers. |
1. Introduction
Polymer chemistry has gained great momentum1 during the last 100 years by integrating the progresses of inorganic,2,3 organic,4 as well as supramolecular chemistry.5 On the one hand, new polymerization techniques,6 such as atom transfer radical polymerization (ATRP),7 reversible addition–fragmentation chain transfer (RAFT) polymerization,8 nitroxide-mediated polymerization (NMP), and ring-opening metathesis polymerization (ROMP),9 have been broadly used to achieve good control over the polymeric architecture; on the other hand, different functional building blocks, such as carbon nanotubes,10 fullerenes,11 polyhedral oligomeric silsesquioxanes (POSS),12 and polyoxometalates (POMs)13,14 have been introduced into polymeric frameworks in order to achieve more functionalities. To prepare more functional polymeric materials, the development of new building blocks is still one of the main strategies.The electron-deficient nature of boron element leads to a number of interesting characteristics,15,16 such as special coordination as well as the tendency to form a π-conjugated system with strong luminescence.17 More importantly, due to the large neutron absorption cross-section of its isotope 10B, the boron-containing compounds have become potential candidates for cancer therapy and the corresponding boron neutron capture therapy (BNCT) has been developed.18 As a result, boron-containing polymers with different architectures, such as linear, dendritic, as well as cross-linked networks, have been developed during the last fifty years.17,19–21
Borane clusters represent unique kinds of boron-containing compounds not only because of their special coordination and unusual 3-dimensional structure22,23 but also due to their broad applications ranging from the precursor of ceramics24 to BNCT agents.25 From the view of practical application, on the one hand, the clustered structure increases the concentration of boron and makes them superior candidates for BNCT agents; on the other hand, such a compact structure also improves the chemical and thermal stability, thus enabling their applications in aerospace coatings and so on (Fig. 1). Therefore, borane clusters have been widely used as building blocks for the construction of functional polymeric materials with tunable architectures, amphiphilicity, as well as biocompatibility.26 With the recent progress of controlled polymerization techniques, a number of borane cluster-containing polymers (BCCPs) have been developed during the last two decades. Although some reviews have summarized the progress of boron-containing polymers, there are only few summaries focused on BCCPs.27–29 Herein, this feature article will mainly summarize the most recent progress on BCCPs with a special focus on the synthetic methodology. The architectures of BCCPs are divided into the following kinds: (1) linear, (2) dendrimer-like and dendron, (3) macrocycles, and (4) metal–organic frameworks (MOFs). With regard to the preparation methods, both routine approaches such as stepwise polymerization and some special approaches such as electropolymerization and controlled polymerization will be described. Meanwhile, some borane-cluster containing supramolecular structures will also be discussed. Furthermore, some promising applications of BCCPs will also be introduced.
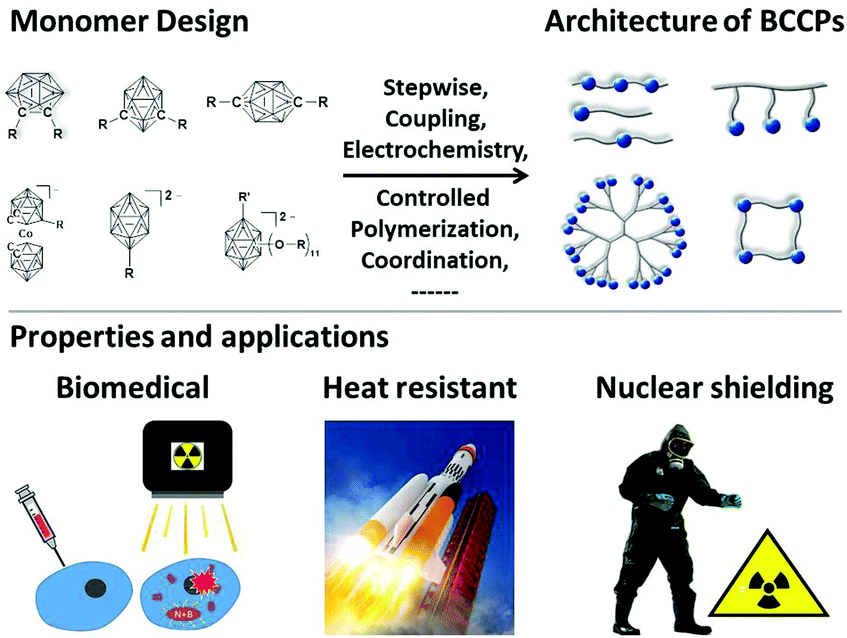 | ||
| Fig. 1 Schematic of the monomer design and the architectures of BCCPs, and the representative properties and applications. | ||
The first step for incorporating borane clusters into polymeric frameworks is to synthesize functional group-modified borane cluster derivatives. Generally, neutral carborane clusters with mono- and bis-substitutions (o-, m-, p-), negatively-charged mono-substituted cobaltabis(dicarbollide) carborane cluster, and negatively-charged borane clusters, as well as multiply-substituted borane clusters were used as the building blocks (Fig. 1). Starting from these building blocks, a variety of polymeric architectures can be prepared through different strategies, such as polycondensation, electropolymerization, coupling polymerization, and controlled polymerization.
2. Linear borane cluster-containing polymers
In principle, linear structured BCCPs are usually prepared through the reaction between bis-substituted borane clusters and the corresponding reactants (Fig. 2). Routine polycondensation method, electropolymerization, and coupling polymerization have been developed. To achieve good control over the polymer architecture, tunable molecular weight, and narrow distribution, controlled polymerization strategies have also been used.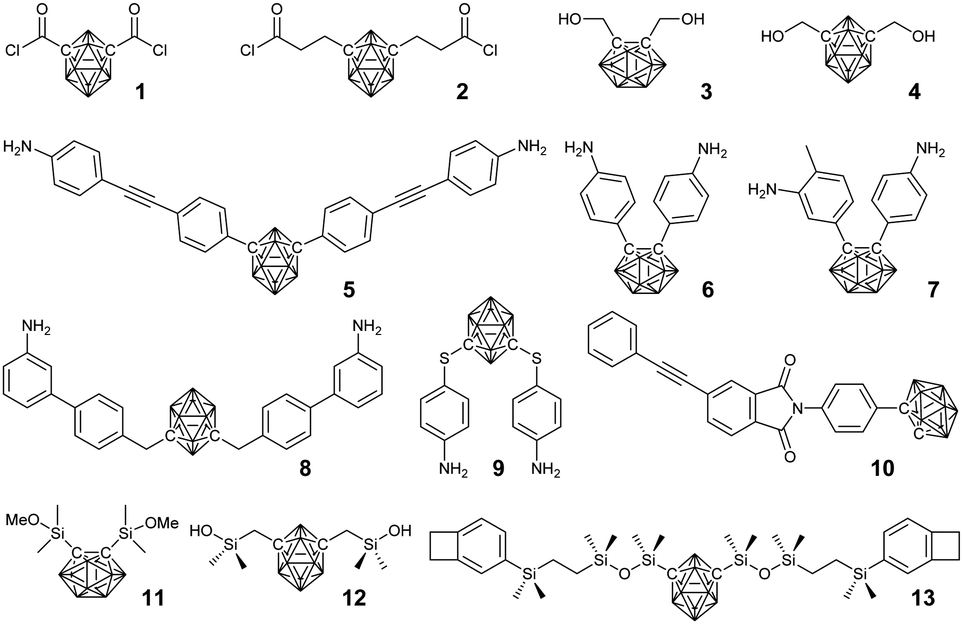 | ||
| Fig. 2 Borane cluster-containing monomers used in the preparation of the corresponding PIs, epoxy resins, and polysiloxanes. | ||
2.1 Linear BCCPs prepared via routine stepwise polymerization
The facile symmetrical modification of carborane provided the easiest way to BCCPs via polycondensation. For example, m-carboranedicarboxylic acyl chlorides 1 and 2 can undergo routine polycondensation with different diols (such as carborane-containing 3 and 4) and diamines to afford the corresponding polyesters30 and polyamides31 with low molecular weight. A suitable hydroxyl-terminated polyester can also be further used as a pre-polymer to afford segmented borane cluster-containing polyurethanes.32 As a result of the incorporation of the borane cluster, the thermal stability and antioxidation property of the resultant BCCPs is greatly improved. Therefore, borane clusters are usually integrated with engineering polymers via stepwise polymerization, resulting in the corresponding polyimides (PIs), epoxy resins, and polysiloxanes. In principle, borane cluster-containing PIs can be prepared from polycondensation between borane cluster-containing diamines (monomer 5–9)33,34 and commercially available dianhydrides. The resultant BCCPs showed good solubility in various organic solvents, thus enabling solution processing. More importantly, the thermal property and thermal-oxidative stability can be tuned by varying the ratio of borane cluster-containing amines.35,36 Meanwhile, carborane-containing phenylethynyl-terminated imide compound (10) can also be used as a modifier to improve the stability of phenylethynyl-terminated imide oligomers.37–39 These amine-terminated monomers can also be used as curing agents for the preparation of different epoxy resins with improved thermal stability and excellent thermal-oxidative stability.40–42In the case of borane cluster-containing polysiloxane, poly(carborane-siloxane) is widely used in the fields of medicine, aerospace, and automobiles. Generally, there are two different synthetic routes: one is the polycondensation between dialkoxysilane (11) and dichlorosilane,43 and the other one is the polycondensation between silicon hydroxyl (12) and diethylamino.44,45 The symmetrical grafting of benzocyclobutene groups on carborane (13) enabled the preparation of the corresponding BCCPs via ring-opening polymerization.46
2.2 Linear BCCPs prepared via electropolymerization
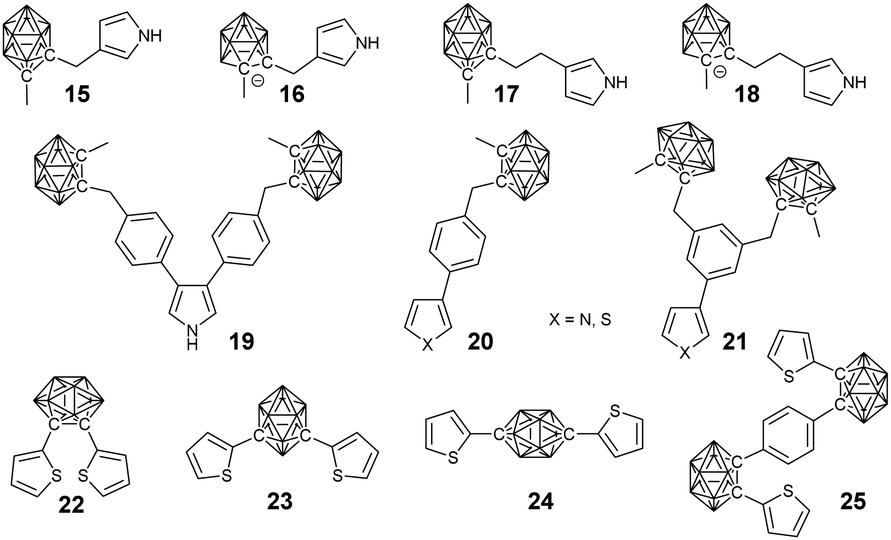
To the best of our knowledge, Teixidor's group reported the first example of BCCPs prepared via electropolymerization (Fig. 3).47 More importantly, this work implied the possibility that the covalent modification of the carborane cluster with electropolymerizable groups, such as thiophene and pyrrole, enabled the preparation of carborane-containing polymers through electrochemistry. Different borane cluster-containing monomers were designed and successfully polymerized or copolymerized with other monomers via electrochemistry. | ||
| Fig. 3 Electropolymerization of monomer 14 to prepare BCCP P1. | ||
For example, the electropolymerization of the negatively charged monomer 14 gave a self-doped semi-conductive polypyrrole (PPy). Meanwhile, it can also be copolymerized with pyrrole to afford different degrees of self-doping.48 Indeed, the introduction of borane cluster into PPy greatly improved the overoxidation resistance and may broaden the application of these materials. Vicente's group carried out a systematic study on the electrosynthesis of BCCPs. Normally, borane cluster-containing monomers with substitution at the 3-position of pyrrole is preferred due to their easier electropolymerization in comparison with the N-substituted ones.49 To study the effects of spacer length and charge of the boron cluster on the electropolymerization efficiency, monomers 15–18 were synthesized. It was found that monomer 17 with an ethyl spacer could be more efficiently and easily electropolymerized than monomer 15 with a methyl spacer.50 In contrast to the feasible electropolymerization of neutral carboranes, their corresponding anionic derivatives 16 and 18 were found to be inert under different electropolymerization conditions. It may because the short spacer is not able to overcome the electronic and steric effects of the anionic carborane compared with the ease of electropolymerization of the anionic monomer 14.48
Compared with the pyrrole monomers, the electropolymerization of the thiophene monomers was more challenging due to the possible overoxidation at higher oxidation potentials required by the generation of radical cation intermediates. To study the effect of thiophene groups, a series of thiophene monomers (20, 21) with borane cluster substitution were developed.51 For the pyrrole-based mono-substituted monomer 20-N and bi-substituted monomer 19, electropolymerization yielded the corresponding conducting polymers. However, monomer 21-N cannot be electropolymerized, possibly due to the high steric hindrance of its 3-dicarboranylmethylphenyl substituent. In comparison, electropolymerization of the thiophene monomers with the same substituent can be feasible at high potential, which was very close to the mono-substituted monomer (20).
To control the geometry and conductivity of the resulting polymer, three isomers bearing o- (22), m- (23) and p-substituted carboranes (24) were designed.52,53 These monomers were readily electropolymerized with the oxidation potentials decreasing in the order 22 < 23 < 24. The lower oxidation potential required for monomer 22 facilitated the electropolymerization process by overcoming the so-called “polythiophene paradox” and achieving high conductivity, which can be ascribed to the conjugated conformation resulting from the intramolecular β–β′ cyclization reaction of monomer 24. On the other hand, the electropolymerization of the Z-shaped monomer 25 was unsuccessful due to the strong electron-withdrawing character of the bridge between the two thienyl units.53
The copolymerization of monomer 23 and 3,4-ethylenedioxythiophene (EDOT) with different monomer ratio gave a series of electrochromic films.54 The absorbance of these films can be tuned between the range of 522 and 565 nm, which was dependent on the feed ratio. During the redox process, the color of these films can be reversibly changed between purple and blue, which can be achieved within 2 s.
2.3 Linear BCCPs prepared via coupling polymerization
Based on the facile formation of the C–C bond, different cross coupling reactions were also developed to prepare the corresponding conjugated BCCPs. To the best of our knowledge, Carter's group reported the first π-conjugated BCCPs through Yamamoto coupling between the silylcarborane-containing fluorene monomer 26 with the aid of Ni(COD)2.55 The molecular weight of the as-prepared BCCPs was constrained by steric hindrance and can be increased by copolymerization with alkyl substituted fluorene monomers.To prepare main-chain type BCCPs, monomer 27 was designed and synthesized from the reaction of the adduct of decaborane and dimethylsulfide with a suitable acetylene. Following the classical Yamamoto condition, main-chain BCCPs can be prepared with reasonable yield.56 The molecular weight of the as-prepared BCCPs was ca. 2.4–10 kg mol−1, which may be ascribed to the possible intramolecular effects associated with the kinked, hindered conformation of the o-carborane monomer. As a result, BCCPs with higher molecular weight could be prepared from the Yamamoto coupling of the corresponding p-carborane monomer.57 Meanwhile, the copolymerization of the o-carborane monomer with 2,7-dibromo-9,9-dihexylfluorene can also be used to achieve BCCPs with higher molecular weight. More importantly, increasing the feed ratio of 2,7-dibromo-9,9-dihexylfluorene can gradually increase the local excited state and therefore tune the photo emission property in both the solid state and the solution state.58 The photoluminescence property of the o-carborane-based BCCPs can also be modulated through interactions with different small molecules, which can be used as vapochromatic photoluminescent sensors towards volatile organic molecules, such as tetrahydrofuran, ethyl acetate, methylene chloride, acetone, methanol, toluene, and hexanes.59
Besides Yamamoto coupling, Sonogashira–Hagihara coupling between monomer 28 and different diynes was also developed to prepare π-conjugated BCCPs with the aid of Ph(PPh3)4/CuI. The molecular weight of the as-prepared BCCPs can be as high as 4100, with Đ as large as 1.7.60 Interestingly, these BCCPs displayed aggregation-induced emission property. To increase the molecular weight, m-carborane monomer was designed.61 Owing to the wide-angle of such a monomer, the molecular weight of the resulting BCCPs as high as 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 can be achieved. Coupling between monomer 29 and the axially chiral binaphthyl unit can also endow the resulting BCCPs with a chiral structure, as indicated by the corresponding circular dichroism spectrum.62 In order to achieve a higher molecular weight, p-substituted monomer was designed and synthesized by Heeney and co-workers, which can be polymerized through Stille coupling. Tributylstannyl-substituted monomer 31 was synthesized from commercially available o-carborane with reasonable yield. The Stille copolymerization of 31 and 2,6-dibromo-4,4-bis-(dodecyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CDT) and 3,6-bis(2-bromothien-5-yl)2,5-bis(2-octyldodecyl)pyrrolo[3,4-c]-pyrole-1,4(2H,5H)-dione (DPP) was performed under microwave conditions, catalyzed by Pd2(dba)3/P(o-tol)3.63 The resulting BCCPs were soluble in chlorobenzene and chloroform. Interestingly, the Suzuki coupling of bis(boronic ester)-substituted monomer 30 and CDT failed due to the competing deboronation. To extend the monomer structure in Stille coupling and to prepare the corresponding BCCPs, carborane was connected to thiophene (32) via a double bond through the classic Wittig strategy.64 These monomers can undergo subsequent Stille polymerization with 2,5-bis(trimethylstannyl)thiophene to give BCCPs with a molecular weight of ca. 12 kg mol−1. The introduction of o-carborane into the polythiophene system significantly improved the polymer ionization potential and influenced the optoelectronic, photovoltaic, and charge-transporting properties (Fig. 4 and 5).
400 can be achieved. Coupling between monomer 29 and the axially chiral binaphthyl unit can also endow the resulting BCCPs with a chiral structure, as indicated by the corresponding circular dichroism spectrum.62 In order to achieve a higher molecular weight, p-substituted monomer was designed and synthesized by Heeney and co-workers, which can be polymerized through Stille coupling. Tributylstannyl-substituted monomer 31 was synthesized from commercially available o-carborane with reasonable yield. The Stille copolymerization of 31 and 2,6-dibromo-4,4-bis-(dodecyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CDT) and 3,6-bis(2-bromothien-5-yl)2,5-bis(2-octyldodecyl)pyrrolo[3,4-c]-pyrole-1,4(2H,5H)-dione (DPP) was performed under microwave conditions, catalyzed by Pd2(dba)3/P(o-tol)3.63 The resulting BCCPs were soluble in chlorobenzene and chloroform. Interestingly, the Suzuki coupling of bis(boronic ester)-substituted monomer 30 and CDT failed due to the competing deboronation. To extend the monomer structure in Stille coupling and to prepare the corresponding BCCPs, carborane was connected to thiophene (32) via a double bond through the classic Wittig strategy.64 These monomers can undergo subsequent Stille polymerization with 2,5-bis(trimethylstannyl)thiophene to give BCCPs with a molecular weight of ca. 12 kg mol−1. The introduction of o-carborane into the polythiophene system significantly improved the polymer ionization potential and influenced the optoelectronic, photovoltaic, and charge-transporting properties (Fig. 4 and 5).
 | ||
| Fig. 4 Different carborane-containing monomers used in the study of the preparation of BCCPs via electropolymerization. | ||
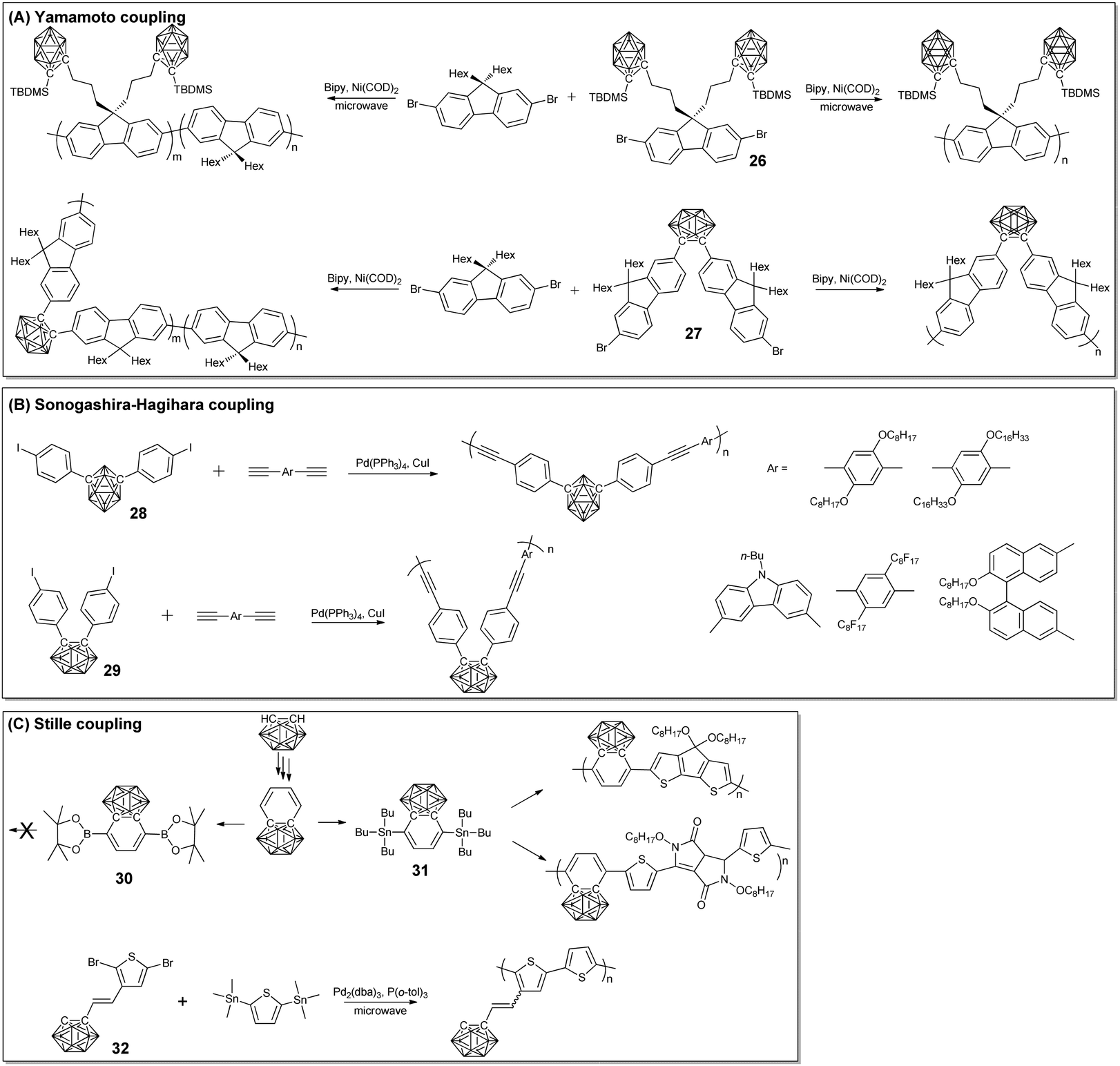 | ||
| Fig. 5 Linear BCCPs synthesized from different coupling methods: (A) Yamamoto coupling, (B) Sonogashira–Hagihara coupling, and (C) Stille coupling. | ||
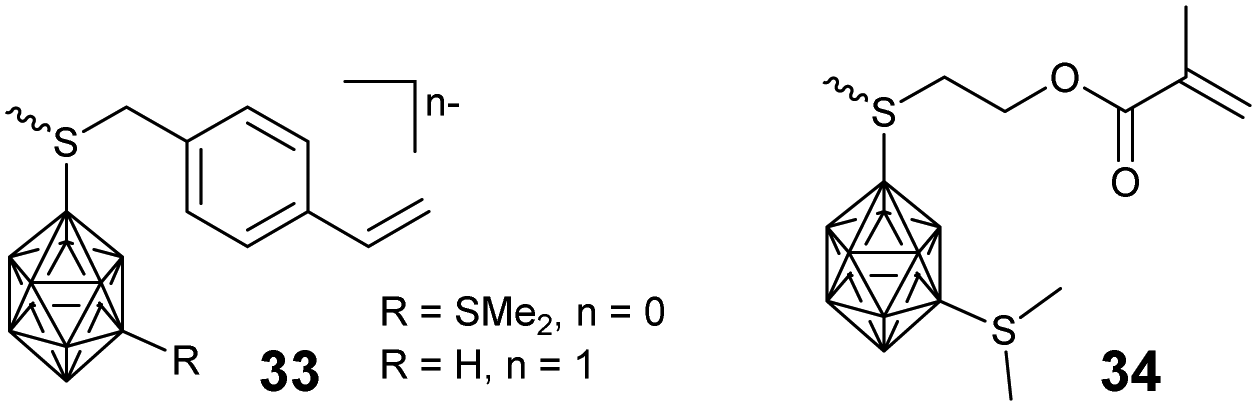 | ||
| Fig. 6 Borane cluster-containing monomers used in free radical polymerization. | ||
 | ||
| Fig. 7 Linear BCCPs prepared through the ATRP approach. | ||
 | ||
| Fig. 8 Linear BCCPs prepared from the RAFT polymerization approach. | ||
Alternately, the one-pot coupling reaction of dichloro-oligosilanes with dilithiated m-carborane was also developed to prepare poly(carborane-silane).65 Due to the disadvantage of such a strategy, the molecular weight of the target BCCPs was relatively low, irrespective of the substitution position of the carborane.66
2.4 Linear BCCPs prepared via controlled polymerization
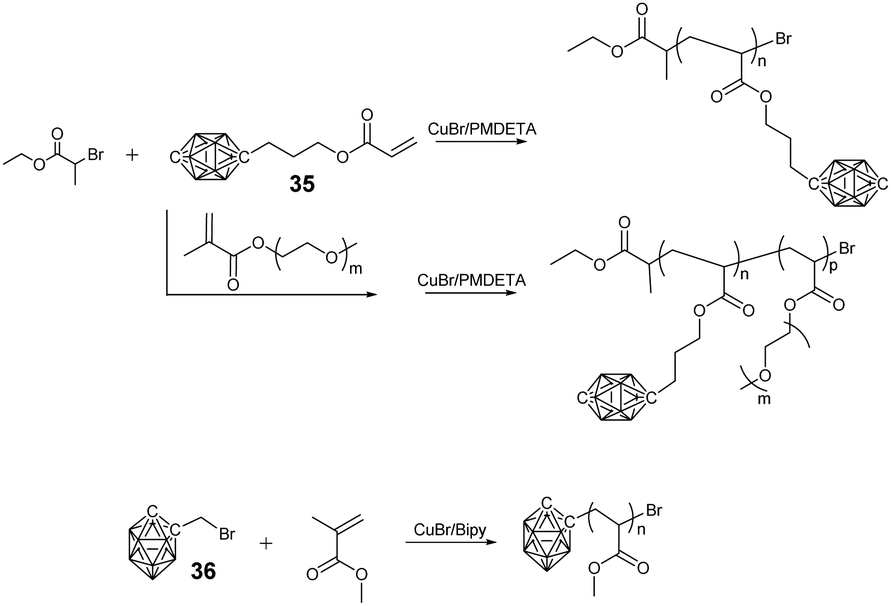
In principle, a facile method to prepare BCCPs with a large molecular weight is free radical polymerization (FRP). For example, negatively-charged borane cluster-containing styrene monomer 33 and the corresponding methacrylate monomer 34 were designed to prepare the side chain BCCPs through FRP (Fig. 6). Theoretically, these monomers can also be copolymerized with other classical monomers, such as methylmethacrylate (MMA), 2-hydroxyethylmethacrylate (HEMA), 2-hydroxyethylacrylate (HEA), acrylamide (AA), and styrene.67 However, one disadvantage of FRP is the poor control of the polymerization process. To get better control over the molecular weight and its distribution as well as the block structure of the resulting BCCPs, controlled polymerization techniques such as ATRP, RAFT, NMP, as well as ROMP were employed. Adronov proposed and reported the first example of BCCPs prepared by the ATRP of acrylate monomer 35 in DMF (Fig. 7).68The resulting linear BCCP was a white powder with Đ as low as 1.14, which can be dissolved in a polar organic solvent such as acetonitrile and DMF but is insoluble in water. To improve the water solubility and biocompatibility required in biomedical application, the copolymerization of monomer 35 and hydrophilic monomer such as poly(ethylene glycol) monomethyl ether methacrylate (PEGMA) was carried out.69 The direct copolymerization of monomer 35 and PEGMA via ATRP gave a water-soluble copolymer with a Đ of 1.3, indicating the “living” characteristic of the polymerization process. As an alternative approach, the block copolymerization of monomer 35 and PEGMA was not very successful, as indicated by the shift of Đ from 1.14 for the macroinitiator to 2.9 for the resulting block copolymer, which may be attributed to the possible termination induced by radical coupling during the aggregation of the amphiphilic block copolymer. Interestingly, the chain extension from PCL-PGALPA-r-POEGMA to the methacrylate monomer 35 was demonstrated to be very successful.70
Besides the side chain BCCPs, the borane cluster can also be modified as an ATRP initiator to prepare borane cluster-labeled BCCPs. 1-Bromomethyl-carborane 36 can be used to initiate the ATRP of MMA with the aid of Cu(I)Cl and 2,2′-bipyridine. The good control of such an ATRP process can be proved by the corresponding linear relationship between ln([M]0/[M]) and the reaction time in the kinetic plot as well as the narrow Đ.71
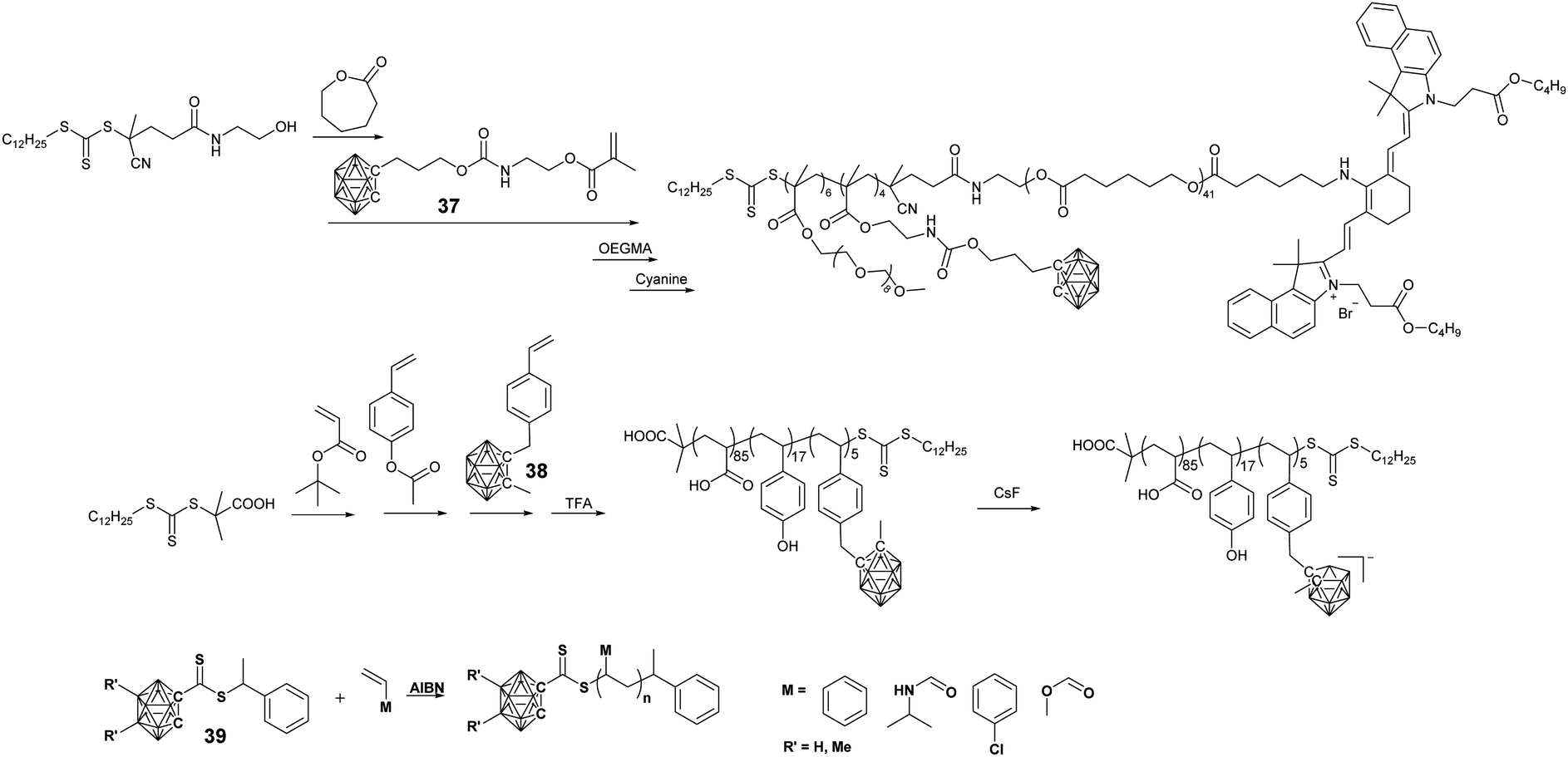
Reversible addition fragmentation chain transfer (RAFT) polymerization shows better monomer tolerance towards different functional groups and is much more facile than ATRP. Yan's group prepared amphiphilic BCCPs through the sequential RAFT polymerization of monomer 37 and OEGMA (Fig. 8).72,73 Due to the presence of hydrophobic PCL and the polycarborane segment as well as the hydrophilic POEGMA segment, these BCCPs can self-assemble into micelles in an aqueous solution. The conjugation of the pH-responsive polypeptide block can also introduce pH-regulated micelles.73 Furthermore, the terminal hydroxyl group can be further modified with a NIR dye, thus enabling fluorescence imaging-guided BNCT therapy.72
Styrene-derived monomer 38 can also be polymerized through sequential RAFT polymerization to give the terpolymer PAA-b-PHS-b-PSC. Interestingly, in such a BCCP, pH-responsive property and CsF-induced potential deboronation can be used to tune the amphiphilic property of this BCCPs, which thus changes the self-assembled structure as well as their fluorescence property.74
To prepare borane cluster-labeled BCCPs through RAFT polymerization, Spokoyny's group synthesized a carborane-functionalized RAFT agent 39, which could be used in the RAFT polymerization of multiple monomer classes, including NIPAAm, 4-chlorostyrene, and MA.75 RAFT polymerization is well-controlled by using such kinds of chain transfer agents, as indicated by the narrow polydispersity as low as 1.03. The presence of a terminal carborane group enables the corresponding BCCPs with different functions: (i) it showed 2:1 binding with β-CD, as determined by the corresponding isothermal titration calorimetry measurement; (ii) it could be used as a bioorthogonal molecular probe for Raman application due to the B–H vibrational signal in the range of 2350–2600 cm−1.
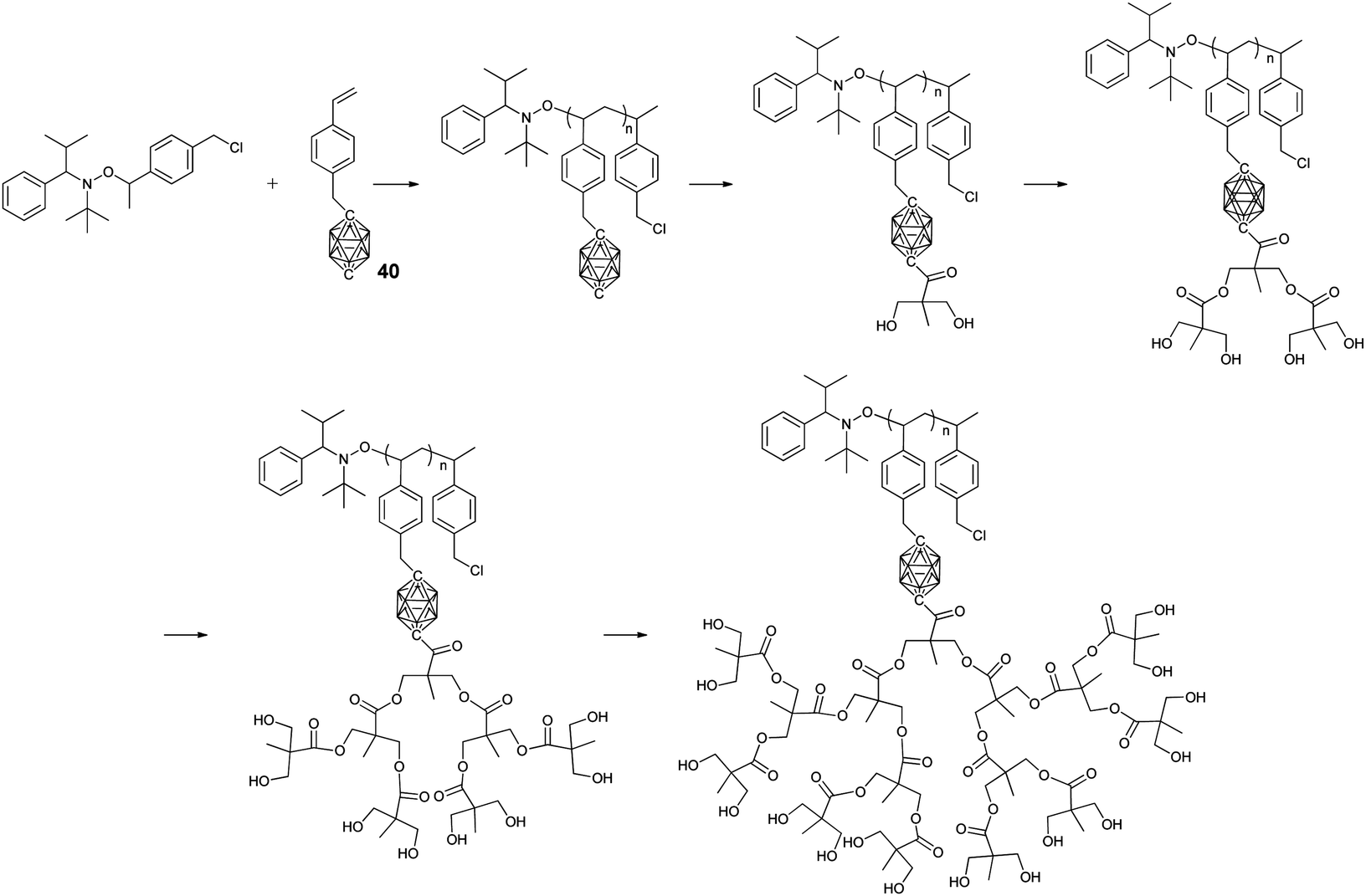
As an effective CRP technique, nitroxide-mediated polymerization (NMP) was also used to prepare BCCPs. As shown in Fig. 9, styrene-based monomer 40 can be successfully polymerized through NMP to give BCCPs with a high molecular weight (up to 22 kg mol−1) and narrow molecular weight distribution (Đ < 1.1).76 The quantitative treatment of the resulting BCCP with n-BuLi and benzylidene-protected anhydride afforded BCCPs with two hydroxyl groups. Following an iterative deprotection and esterification approach, different generations of dendrons can be prepared to give BCCPs with hydroxyl groups as many as 16. Therefore, the water solubility of BCCPs was greatly improved from being insoluble for low generation to 1 mg mL−1 for higher generation. As a result, these water-soluble BCCPs with high boron content were potential candidates for BNCT.
 | ||
| Fig. 9 Linear BCCP with dendritic side chains prepared through the NMP approach. | ||
In comparison with the radical process of ATRP, RAFT, and NMP, ROMP demonstrated advantages in the preparation of block copolymers with good control. Oxonorbornene-based monomer 41 was synthesized through Mitsunobu coupling, which can be easily polymerized with the aid of Grubbs catalyst within minutes to give the corresponding BCCPs with good yields and narrow polydispersity (Đ < 1.1)77 (Fig. 10). More importantly, the living nature of ROMP guaranteed the controlled preparation of the diblock copolymer BCCPs with molecular weight up to 65100 g mol−1 and polydispersity as low as 1.15. Taking advantage of the better monomer tolerance of ROMP, different monomers can be used in the chain extension to prepare the block copolymers. For example, Coughlin and coworkers demonstrated that the ROMP copolymerization of monomer 41 with cyclooctene and subsequent hydrogenation can give polyethylene-like BCCPs.78 In a recent report, Eren and coworkers prepared phosphorous-containing BCCPs. Generally, the phosphaester monomer is used to copolymerize with monomer 41, which can be converted to phosphonic acid-containing BCCPs through reaction with trimethylsilyl bromide and can be potentially used as a heat resistant material.79
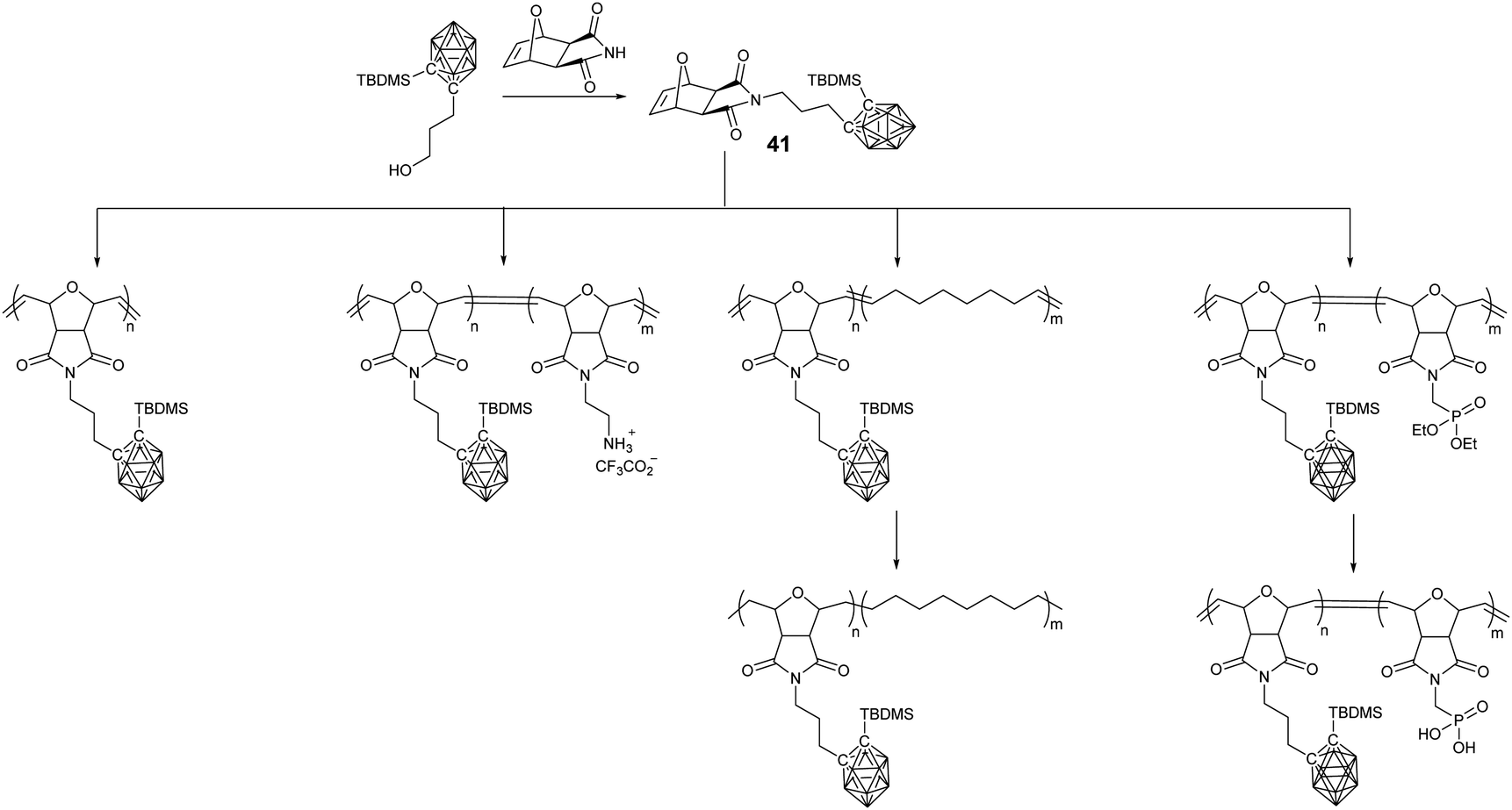 | ||
| Fig. 10 Linear BCCPs prepared through the ROMP approach. | ||
Besides the above controlled polymerization techniques, coordination polymerization can also be used to prepare structure-specific BCCPs. Do and coworkers reported the preparation of polyolefins with pendant carborane clusters by using monomer 42 (Fig. 11) under the catalysis of the [Me2Si(η5-C5Me4) (η1-N-tBu)TiCl2]/methylaluminoxane (MAO) system.80
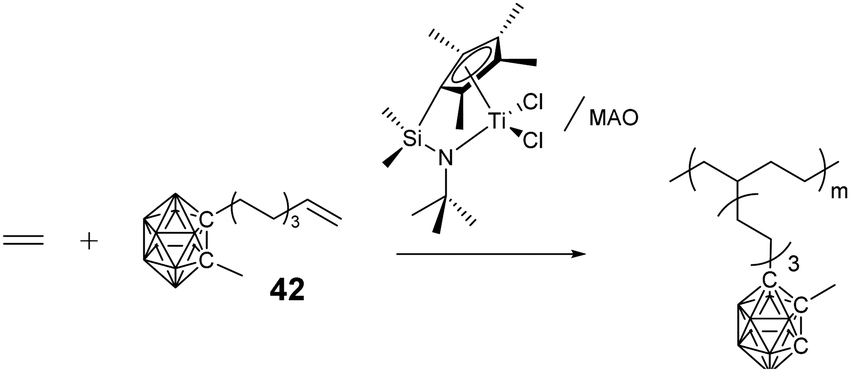 | ||
| Fig. 11 Linear BCCPs prepared through coordination polymerization. | ||
3. Borane cluster-containing dendrimers, macrocycles, and frameworks
Besides the above linear BCCPs, different architectures including dendrimer-like structures, macrocycles, and cross-linked networks have been developed. Furthermore, to make full use of the framework structure and the functions of the metal node, borane cluster-containing metal–organic frameworks have also been developed (Fig. 12–18).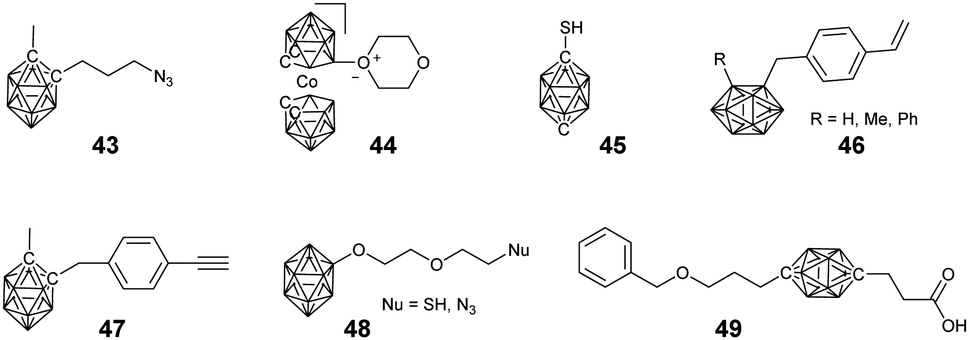 | ||
| Fig. 12 Functional group-modified borane clusters used in the preparation of different dendrimers. | ||
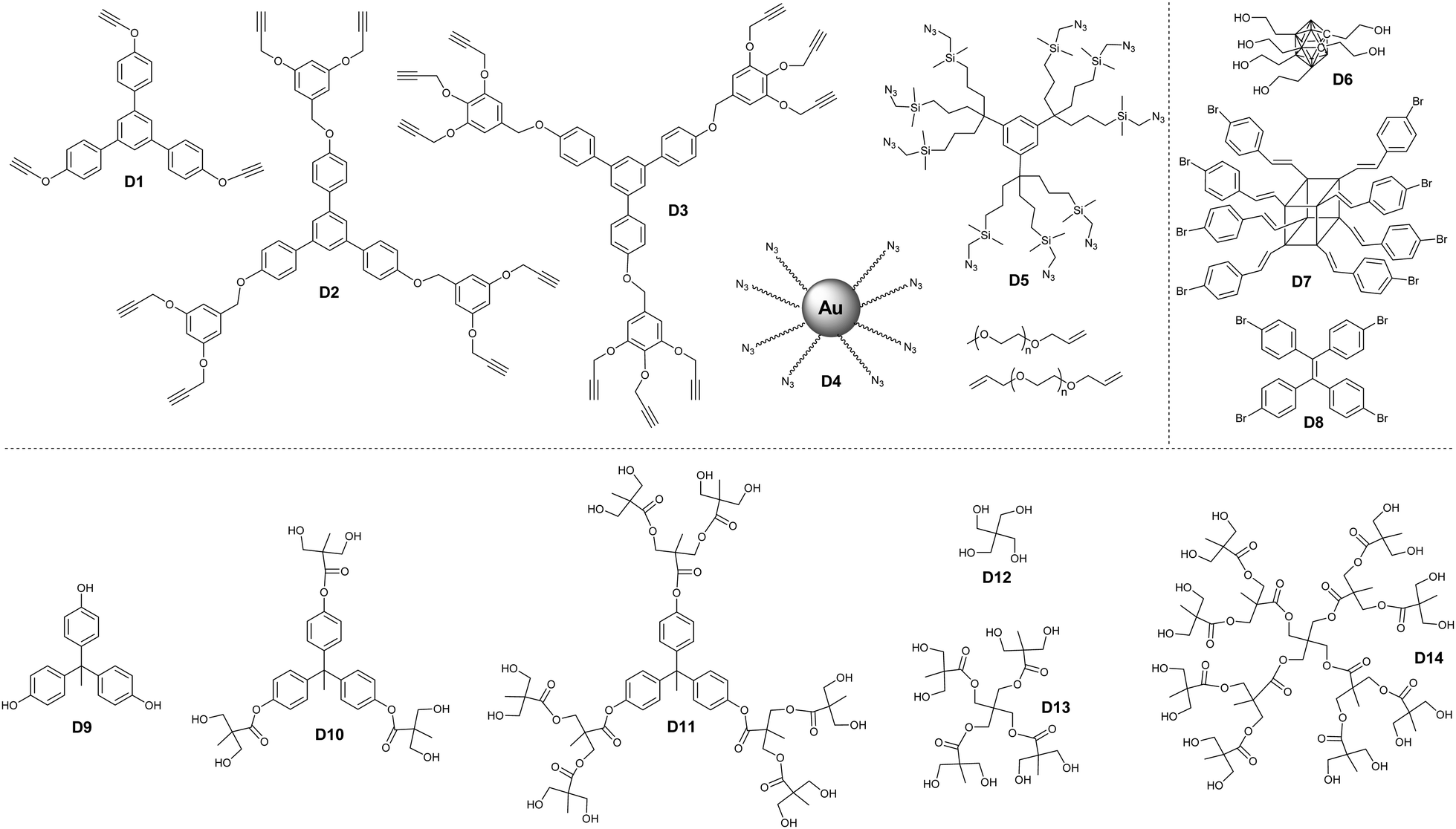 | ||
| Fig. 13 Different dendritic polymers used in the preparation of borane cluster-containing dendrimer-like structures. | ||
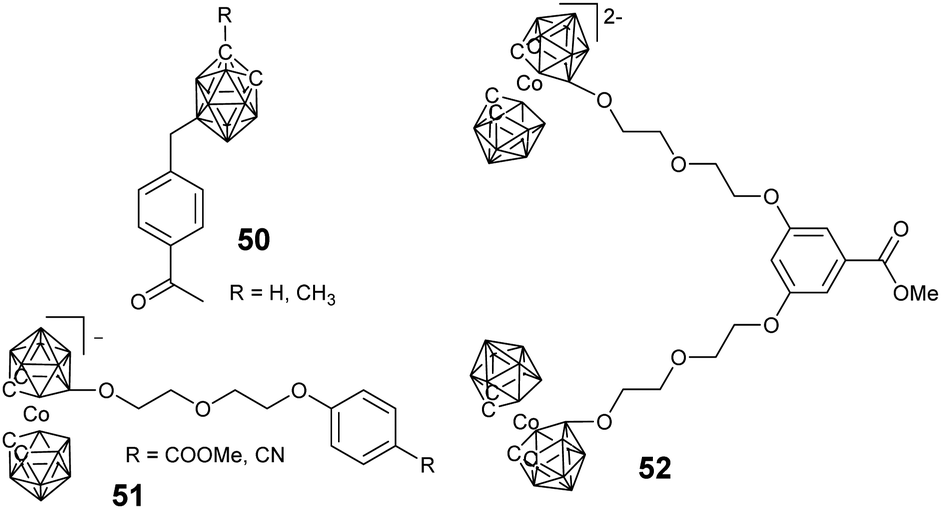 | ||
| Fig. 14 Different borane cluster-containing dendrons used in the convergent approach. | ||
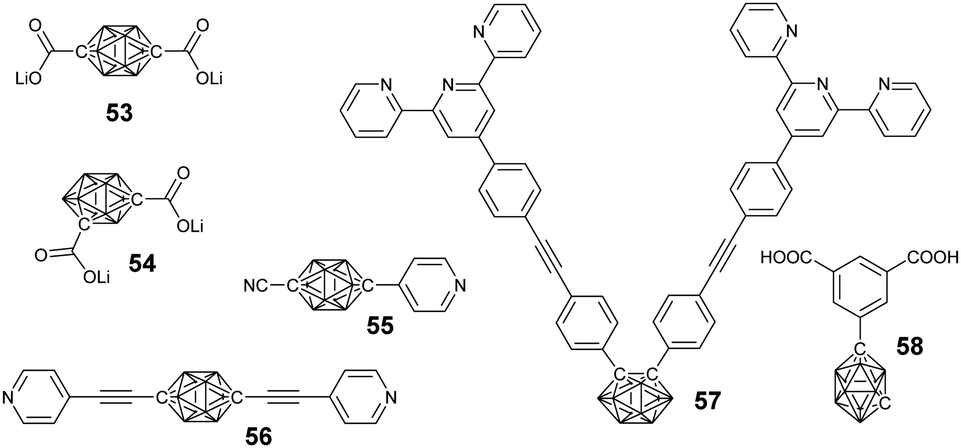 | ||
| Fig. 15 Representative coordination group-modified borane cluster-containing ligands used in the preparation of borane cluster-containing macrocycles. | ||
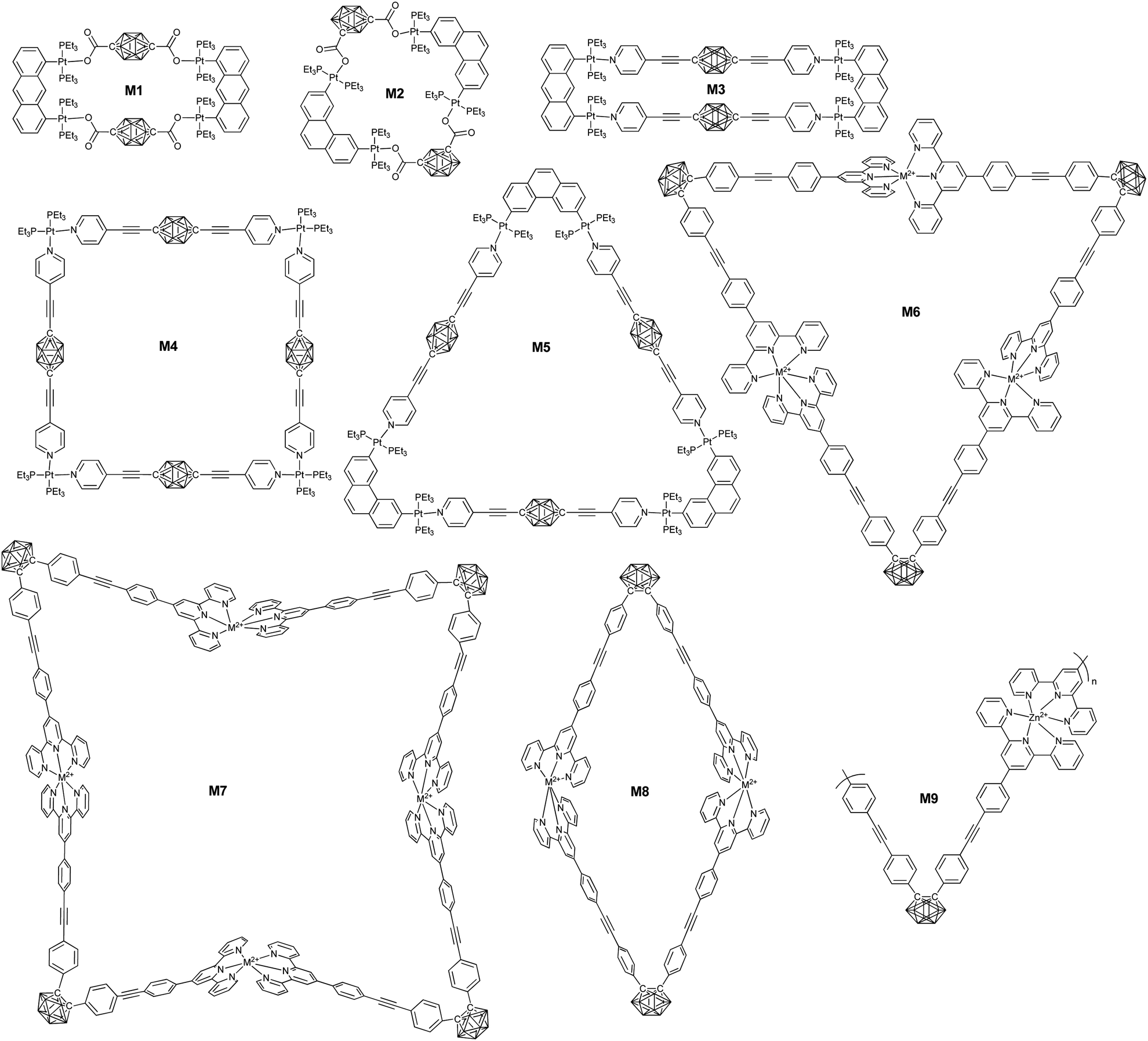 | ||
| Fig. 16 Representative borane cluster-containing the macrocycles prepared from coordination. | ||
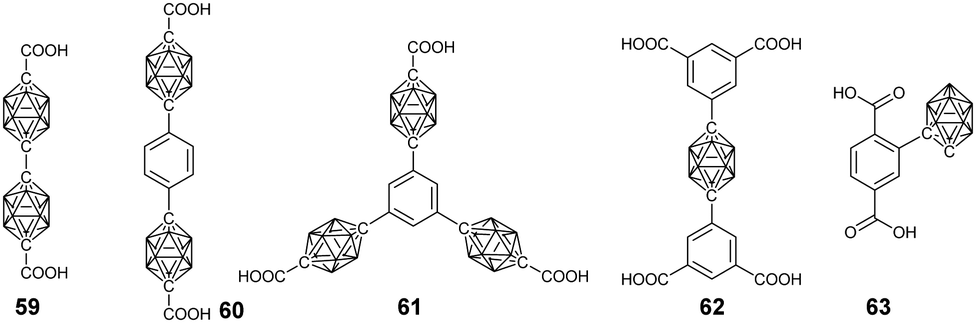 | ||
| Fig. 17 Representative borane cluster-containing ligands used in the preparation of the corresponding MOFs. | ||
 | ||
| Fig. 18 Synthetic route of borane cluster-containing POPs. | ||
3.1 Borane cluster-containing dendrimer-like structures
Considering their high boron content and precise structure, dendrimers have used widely to load borane clusters for the application of BNCT. In principle, both convergent and divergent methodologies have been developed.In the case of a divergent strategy, a core with functional groups and effective modification methods is necessary. Due to the facile and high modification efficiency, click chemistry involving copper-catalyzed azide alkyne coupling (CuAAC), thiol–ene, has been broadly used. Hosmane and co-workers carried out the CuAAC reaction between alkynl-modified borane cluster 47 and different generations of azide-terminated cores (D5).81 The highly efficient click reaction enabled the complete modification of the core and could afford dendrimers with a borane cluster up to 81, as indicated by the low polydispersity (Đ < 1.05). Alternately, CuAAC between the azide-modified borane cluster 43 and the alkynyl-terminated cores (D1–D3) can also afford the corresponding dendrimers.82 The reaction catalyzed by CuSO4/potassium ascorbate was almost quantitative with a yield higher than 75%. These borane-containing dendrimers can be accumulated in the human liver cancer cells (SK-Hep1), with the highest boron accumulation up to 2540 ng of boron/5 × 105 cells over a period of 20 h. To integrate the functionalized gold cluster and BNCT, PEGylated-gold nanoparticles (AuNPs, D4) with terminal azide groups were also used as the core.83 The successful CuAAC reaction can produce carboranes up to 282 per AuNPs. More importantly, the introduction of PEG greatly improved the water solubility of the resulting borane-containing dendrimer. Furthermore, the anti-HER2 antibody (61 IgG) was appended to these PEGylated borane cluster-containing AuNPs, which can avoid non-specific accumulation in RES-rich organs and therefore improve the BNCT effect.84 In addition to the nanoparticles, different functional cores were also introduced, such as branched PEG,85o-carborane cluster,86 POSS,87 and luminescent tetraphenylethylene (TPE).88 For example, the six-armed hydroxyl group-terminated carborane core D6 was synthesized through subsequent nucleophilic substitution with allyl bromide and hydroboration/oxidation.86 Then, nucleophilic oxonium ring opening reaction was initiated from the periphery hydroxyl groups of such a core to afford the dendrimer. Heck coupling between the eight-armed bromide-terminated POSS (D7) and TPE (D8) cores and styrene-functionalized carborane cluster 46 can afford the corresponding dendrimers with reasonable yields. As a result of the space separation of the carborane clusters, the fluorescence emission in solution was significantly enhanced with respect to the non-functionalised POSS vinyl stilbene derivatives.87 More importantly, the enhanced conjugation and restriction further reduced the internal conversion, resulting in bright fluorescence emission with high quantum yield.88
Due to their excellent characteristics such as potential high water solubility and biocompatibility, Fréchet-type aliphatic polyester dendrimer (D9–D14) were used to load the carborane for BNCT. The bifunctional carborane precursor 49 with a carboxylic acid and a protected alcohol group was designed and easily synthesized from p-carborane.89 On the one hand, such a precursor can be attached to the hydroxyl group terminated Fréchet-type dendrimer via facile EDC coupling; on the other hand, the deprotection of the benzyl ester groups can also release new reactive hydroxyls, which allowed the further formation of a new Fréchet-type dendrimer on the periphery. The advantage of such a methodology is that it not only increases the boron content but also improves the water solubility of the resulting BCCPs to 5 mg mL−1. The tentative irradiation of these materials with thermal neutrons resulted in the emission of gamma radiation, indicating the boron neuron capture activity of these dendrimers. Interestingly, the water solubility of these dendrimers exhibited a lower critical solution temperature, depending on the generation of dendrimers as well as the number of heating/cooling cycles.90 The solubility of these dendrimers increased upon increasing the heating/cooling cycle, which is attributed to the gradual break-up of the large insoluble aggregates.
For the strategy of convergence, facile silicon tetrachloride-mediated cyclotrimerization reaction was developed. Typically, benzylated carborane cluster-containing the dendron-like precursor 50 was synthesized via electrophilic alkylation with benzyl halide, which can undergo facile trimerization with the aid of silicon tetrachloride and ethanol.91 The resultant borane cluster-containing dendrimers can be further functionalized through reaction with various silanes and aliphatic halides. Similarly, the negatively-charged cobaltabis(dicarbollide) can be introduced at the periphery of the dendrimers with either a phenylene or a triazine core via the trimerization of keto and cyano derivatives (51), respectively.92
3.2 Borane cluster-containing macrocycles
Taking advantage of the precise coordination chemistry of precious metal such as Pt and Ag, different hybrid macrocycles were developed from ligand-modified borane clusters. Housecroft summarized a variety of carborane cluster-containing coordination polymers.93 Herein, we will emphasize the borane-containing macrocycles and framework structures built based on coordination.In principle, the substitution position on carborane and the geometry of the metal precursors can be used to control the structure of the resultant macrocycles. For example, the coordination between the dilithium salt of p-carborane acid (53) and Pt(II)-based 90° building block gave a neutral rectangular (M1) macrocycle in quantitative yield. On the other hand, the reaction of o-carborane acid (54) and Pt(II)-based 60° building block gave a rhomboidal (M2) assembly.94
Similarly, more complicated structures such as rectangular (M3), triangle (M4), and square (M5) can be prepared based on the merit of the linear coordination geometry of pyridine-Pt(PEt3)2.95 Besides the geometry of both the ligand and the metal precursor, the structure of the resulting borane-containing macrocycles can also be tuned by the coordination between the metal and ligand through either kinetic or thermodynamic pathways.96 For example, the coordination of o-carborane-based bisterpyridyl monomer 57 with different kinds of metal ions may generate cyclic dimer (M8), trimer (M6), and tetramer (M7). In case of non-labile FeCl2, the main product was triangular due to the typical 60°-based geometry. By using more labile transition metal ions, such as Zn2+, the coordinated product was a dimer, as driven by entropy.
A large supramolecular polyhedron can also be expected through the combination of carborane cluster-based bisisophthalic acid and biscopper(II) ‘paddle-wheel’ nodes. For example, the coordination between the bisacid ligand 58 and Cu2(OAc)4·2H2O gave a cuboctahedron with 240 boron atoms.97
Ideally, the coordination of ditopic ligands and metal ions preferentially generates a one-dimensional supramolecular polymer. Chujo's group reported the very first supramolecular polymers (M9) based on infinite coordination between monomer 57 and Zn(OAc)2.98 As expected, coordination with Zn2+ was able to change the emission from green-yellow to light blue due to the switching of the emission mode from AIE to intra-ligand charge transfer. Taking advantage of the coordination property from different ligands, Duttwyler's group reported a coordination polymer based on an asymmetrical modified carborane-containing ligand (55).99 In the resulting zig-zag supramolecular polymer, tetrahedral coordination of the Cu center was observed, which coordinated with both the –CN and pyridyl groups.
3.3 Borane cluster-containing frameworks
As an important class of supramolecular polymers, the advantage of introducing a carborane cluster into the metal–organic frameworks (MOFs) can be easily envisaged due to its multiple functions. Mirkin's group conceptualized and reported the first carborane-based MOFs by reacting diacid derivatives of p-carborane 59 and Zn(NO3)2·6H2O in diethylformamide (DEF).100 In comparison with the benzene analogue, the introduction of a three-dimensional carborane can increase the rigidity of the resultant MOFs and decrease the π–π interaction, thus reducing material collapse as well as increasing the stability. More importantly, this method was able to generate smaller pores, which might be potential H2 storage materials. Thanks to the increased stability, thermal treatment to remove the free and coordinated solvent molecules formed a constricted pore structure with a higher surface area. Such a material was found to be a promising candidate for the separation and purification of CO2 from CO2/CH4 mixtures, which may be attributed to the quadrupolar moment in CO2.101 As expected, extending the length of the ligand provided a possible way to enhance the porosity. The solvothermal treatment of ligands 60 and 61 with Zn(NO3)2·6H2O produced Zn(II)-based MOFs with 2D- and 3D-open framework structures, which displayed a BET surface area of up to 1180 m2 g−1 after thermal activation.102For practical application as a gas storage material, both the pore volume and internal surface area should be well controlled. Thus, the carborane cluster-based triacid (62)103 and tetraacid were developed and used to prepare the corresponding MOFs. For example, the coordination between the tetraacid ligand X and Cu(NO3)2·2.5H2O produced an MOF with a pore volume of 1.02 cm3 g−1 and a gravimetric BET surface area of ca. 2600 m2 g−1, which displayed high CH4 and H2 storage capacity.104
To explore the photophysical property of the carborane in MOFs, ligand 63 was designed. However, the direct synthesis of a UiO-66-like structure using ZrCl4 and 63 failed to give the target product. Even the attempt of using mixed ligands only gave non-functionalized UiO-66. Alternately, post-synthetic ligand exchange strategy was explored, which was able to insert the carborane-functionalized ligand into UiO-66 with a percentage of 14%.105 More importantly, the incorporation of luminescent o-carborane endowed this MOF material with AIE features.
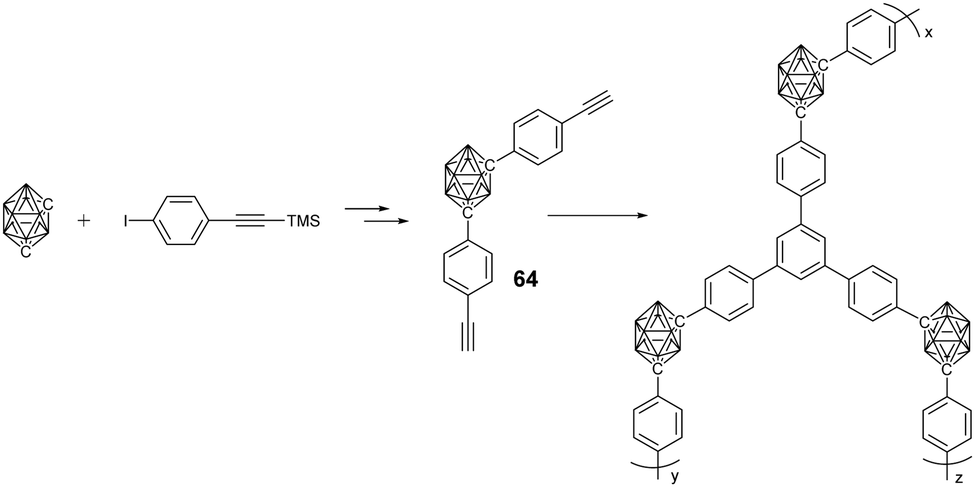
In addition to MOFs, carborane-containing porous organic polymers (POPs) with improved chemical stability were also developed. Generally, m-carborane-containing monomer 64 with either two ethynyl or chloromethyl groups was used to prepare the corresponding POPs via ethynyl trimerization and Friedel–Crafts alkylation, respectively.106 As expected, these POPs were much more stable than the above MOFs, as indicated by the almost no weight loss up to 500 °C. Furthermore, these POPs demonstrated higher hydrogen adsorption capacity than the carborane-containing MOFs, which is attributed to the electron-deficient characteristic of the carborane cluster as well as the narrow pore size distribution of the POPs.
4. Applications of borane cluster-containing polymers
The introduction of borane clusters into polymeric frameworks not only generates novel types of hybrid polymers but also produces materials with different functionalities, mainly including precursors for functional ceramics, biomedical application, and nuclear shielding materials. Since this feature article mainly focuses on the synthesis and architecture of BCCPs, the applications of BCCPs will be introduced briefly.4.1 Functional ceramics from borane cluster-containing polymers
BCCPs have been also widely used as precursors for functional ceramic materials, which are used as ablation-resistant coatings and oxidation-resistant materials. In principle, the pyrolysis of BCCPs in an inert atmosphere could generate ceramic materials composed of B4C and C.107 The advantage of such a method is that the ceramic phases formed during pyrolysis are homogeneously dispersed in the carbon matrix. For example, the pyrolysis of carborane-containing poly(silyleneethynylenephenyleneethynylene) in argon could generate ceramics with a composition of SiC/B4C/C.108 This ceramic showed excellent thermo-oxidative stability in air due to the formation of the borosilicate layer. By using the strategy of precursor infiltration-pyrolysis (PIP), Li and co-workers achieved surface coating of carbon fibers with different kinds of BCCPs.109,110 The PIP procedure allowed tunable thickness of the resulting ceramic coating as well as oxidation resistance performance.Besides the linear BCCPs, borane cluster-containing block copolymers were also developed in order to prepare nanostructured ceramic materials. Malenfant and co-workers found that polynorbornene-b-polynorbornenedecaborane can form various morphologies after the evaporation of different solvents, which can be further used to prepared ceramic materials with corresponding nanostructures.111 For example, the use of THF as a solvent generated a hexagonally-packed cylindrical morphology, which could therefore form meso-porous boron nitride with a similar nanostructure after pyrolysis in ammonia atmosphere, while in the case of chloroform, the lamellar structure formed after solvent evaporation could be transferred into layered boron carbonitride/carbon ceramic composites after pyrolysis in nitrogen atmosphere.
4.2 Biomedical application of borane cluster-containing polymers
Due to the high neutron capture cross section of the 10B isotope, borane clusters are potential candidates for non-invasive BNCT, for example, disodium mercaptoundecahydro-closo-dodecaborate (BSH) has already been used in clinical trials.112 BCCP provides opportunities to improve the biocompatibility and selectivity as well as the loading efficiency of BNCT agents, which are challenges in BNCT. In order to overcome these issues, the covalent attachment of borane clusters to peptides was used. For example, the carborane cluster was used to form conjugates with the gastrin-releasing peptide receptor (GSPR), which is a well-known target in cancer diagnosis. These BCCPs displayed high receptor activity, induced receptor internalization, and exhibited no intrinsic cytotoxicity.113 To increase the cellular uptake and to control the intracellular location of the BNCT agent, dodecaborates were conjugated to the organelle-targeted peptide. The in vitro BNCT assay demonstrated that such a BCCP exhibited anticancer effects with ATP reduction and apoptosis.114As demonstrated by Huang's group, BNCT can also be used in combination with chemotherapy.115 They prepared the corresponding BCCP through the reaction between decaborane and side alkynyl groups from the polymer precursor. This amphiphilic BCCP can self-assemble into micelles and load doxorubicin (DOX) through the characteristic B–H⋯H–O(N) interactions. Such a system could not only suppress the leakage of the boron compounds into the bloodstream but also protect DOX from initial burst release under the physiological conditions.
4.3 Nuclear shielding application of borane cluster-containing polymers
The efficient absorption of thermal neutrons by 10B also makes BCCPs potential neutron shielding materials. For example, Carturan and co-workers found that carborane-containing polysiloxane-based scintillators exhibited good radiation resistance property.116 To achieve better processability of BCCPs-based shielding materials, Yang and co-workers prepared o-carborane cluster-based polyesters.117 These BCCPs can be processed into fibers through melt spinning and exhibited a high neutron absorption cross-section of 17.04 cm−1, which is ca. 50 times higher than that of neat polyesters. Unlike the shielding materials prepared from physical blending, the covalent linkage of the borane clusters to the polymeric frameworks can avoid a decrease in the absorption efficiency due to the leakage of boron particles.118 Nadkarni and co-workers found that the copolymer of allyl propargyl carbonate carborane and allyl diglycol carbonate can be used as an efficient polymeric detector in neutron dosimetry.119 The advantage of this method is that the thickness of the detector film can be easily controlled, therefore varying the concentration of natural boron.5. Conclusion and outlook
Borane clusters, as new building blocks with unique structure and multiple functions, have been widely used in the preparation of polymers with novel architectures and properties. In this feature article, we have reviewed the preparation methodology of BCCPs with different topological structures and the most recent progress of the four main BCCPs, including linear, dendritic, macrocyclic, and frameworked polymers has been summarized in detail.Although different monomers and polymers have been reported in the area of BCCPs, one can find that there are still some challenges in the synthetic chemistry of BCCPs. Firstly, the facile and mild synthesis of borane clusters and easy scale-up is necessary, which is also very important for practical applications such as aerospace coatings. Meanwhile, the development of new stable borane clusters and related derivatives is also urgent, which is a source for new functions; secondly, more non-radical polymerization methods should be developed to overcome the drawbacks of potential radical quenching activity, especially for the negatively-charged borane clusters. The preparation of boron cluster-containing block copolymers and the control of the block sequence should be more rational. For example, in view of the BNCT application, how to integrate with other therapy more effectively, and how to achieve better biocompatibility and targeting through effective block copolymerization still need to be understood. Also, how to balance the effect of different components and achieve synergetic effect is still a critical challenge faced during the construction of task-specific BCCPs.
In light of their broad applications in luminescent materials, chemical sensors, BNCT agents, and precursors for high performance ceramic materials, BCCPs are a new interest of polymer chemists and material scientists. As mentioned above, most of these properties are related to the supramolecularly self-assembled structure of the BCCPs, especially for the preparation of ordered nanostructures. Therefore, the relationship between the architecture of BCCPs and their self-assembly behaviors should be studied in detail. Unlike other giant molecules, the rigid nature of the borane cluster may bring about a different influence during the self-assembly of BCCPs. More importantly, as we have mentioned, both the synthesis and the self-assembly of the negatively-charged BCCPs are undeveloped. One can expect that these negatively-charged BCCPs could be a new class of polyelectrolytes and potential BNCT agents with better water solubility.
As a promising material system, more functions of BCCPs should be explored in close combination with the characteristics of the borane cluster itself and the nature of polymeric frameworks, such as the application as hypergolic materials in propellant systems.120
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the funding support from NSFC (21975205, 21504068), Shaanxi National Science Foundation (2020JM-138, 2019JQ-416), Fundamental Research Funds for the Central Universities (3102017jc01001, 310201911cx031), Fundamental Research Program of Taicang (TC2018JC01), and Open Project of State Key Laboratory of Supramolecular Structure and Materials (sklssm202029). Dedicated to Prof. Dr Lixin Wu on the occasion of his 60th birthday.Notes and references
- R. B. Grubbs and R. H. Grubbs, Macromolecules, 2017, 50, 6979–6997 CrossRef CAS.
- Y. Yan, J. Zhang, L. Ren and C. Tang, Chem. Soc. Rev., 2016, 45, 5232–5263 RSC.
- C.-L. Ho, Z.-Q. Yu and W.-Y. Wong, Chem. Soc. Rev., 2016, 45, 5264–5295 RSC.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS.
- P. Gurnani and S. Perrier, Prog. Polym. Sci., 2020, 102, 101209 CrossRef CAS.
- D. Konkolewicz, P. Krys and K. Matyjaszewski, Acc. Chem. Res., 2014, 47, 3028–3036 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- V. D. Punetha, S. Rana, H. J. Yoo, A. Chaurasia, J. T. McLeskey, Jr., M. S. Ramasamy, N. G. Sahoo and J. W. Cho, Prog. Polym. Sci., 2017, 67, 1–47 CrossRef CAS.
- B. C. Thompson and J. M. J. Frechet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS.
- W. Zhang, G. Camino and R. Yang, Prog. Polym. Sci., 2017, 67, 77–125 CrossRef CAS.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- J. Yan, X. W. Zheng, J. H. Yao, P. Xu, Z. L. Miao, J. L. Li, Z. D. Lv, Q. Y. Zhang and Y. Yan, J. Organomet. Chem., 2019, 884, 1–16 CrossRef CAS.
- Boron Science: New Technologies and Applications, ed. N. S. Hosmane, 2012 Search PubMed.
- Z. J. Lesnikowski, Expert Opin. Drug Discovery, 2016, 11, 569–578 CrossRef CAS.
- F. Cheng and F. Jaekle, Polym. Chem., 2011, 2, 2122–2132 RSC.
- R. F. Barth, J. A. Coderre, M. G. H. Vicente and T. E. Blue, Clin. Cancer Res., 2005, 11, 3987–4002 CrossRef CAS.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef.
- N. Matsumi and Y. Chujo, Polym. J., 2008, 40, 77–89 CrossRef CAS.
- A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903–919 CAS.
- V. I. Bregadze, Russ. Chem. Bull., 2014, 63, 1021–1026 CrossRef CAS.
- N. S. Hosmane, Y. Zhu, J. A. Maguire, W. Kaim and M. Takagaki, J. Organomet. Chem., 2009, 694, 1690–1697 CrossRef CAS.
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS.
- B. R. S. Hansen, M. Paskevicius, H.-W. Li, E. Akiba and T. R. Jensen, Coord. Chem. Rev., 2016, 323, 60–70 CrossRef CAS.
- R. Nunez, I. Romero, F. Teixidor and C. Vinas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC.
- D. M. Badgujar, M. B. Talawar, V. E. Zarko and P. P. Mahulikar, Combust., Explos. Shock Waves, 2017, 53, 371–387 CrossRef.
- Carborane Polymers and Dendrimers, in Carboranes, ed. R. N. Grimes, Elsevier, 2011 Search PubMed.
- N. Li, F. Zeng, D. Qu, J. Zhang, L. Shao and Y. Bai, J. Appl. Polym. Sci., 2016, 133 Search PubMed.
- Y. P. Yampolskii, S. A. Legkov, B. F. Shklyaruk, A. L. Rusanov and V. I. Bregadze, Pet. Chem., 2017, 57, 153–158 CrossRef CAS.
- Y. Luo, Y. Lu, N. Li, Y. Li, X. Zhang and S. Qi, J. Appl. Polym. Sci., 2015, 132, 42227–42238 Search PubMed.
- T. Xing, Y. Huang, K. Zhang and J. Wu, RSC Adv., 2014, 4, 53628–53633 RSC.
- T. Xing and K. Zhang, Polym. Int., 2015, 64, 1715–1721 CrossRef CAS.
- Y. Wu, G. Chen, C. Feng and J. Yang, Macromol. Rapid Commun., 2018, 39, 1800484 CrossRef.
- F. Liu, G. Fang, H. Yang, S. Yang, X. Zhang and Z. Zhang, Polymers, 2019, 11, 1930 CrossRef CAS.
- J. Yue, Y. Li, H. Li, Y. Zhao, C. Zhao and X. Wang, RSC Adv., 2015, 5, 98010–98019 RSC.
- J. Yue, Y. Li, Y. Zhao, D. Xiang and Y. Dai, Polym. Degrad. Stab., 2016, 129, 286–295 CrossRef CAS.
- S. Cheng, J. Han, X. Wang, K. Yuan, X. Jian and J. Wang, Polymer, 2017, 115, 96–105 CrossRef CAS.
- X. Men, Y. Cheng, G. Chen, J. Bao and J. Yang, High Perform. Polym., 2015, 27, 497–509 CrossRef CAS.
- J. Zhao, N. Qing, S. Jiang and S. Wu, High Perform. Polym., 2018, 30, 1094–1100 CrossRef CAS.
- J. Zhao, S. Jiang, Y. Chen, X. Chen, Q. Yang and C. Yin, Polyhedron, 2018, 142, 105–109 CrossRef CAS.
- M. Patel and A. C. Swain, Polym. Degrad. Stab., 2004, 83, 539–545 CrossRef CAS.
- J. Xie, H.-J. Sun, X.-Z. Zhang, Z.-M. Xie and Z.-J. Zhang, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 277–291 CrossRef CAS.
- B. A. Izmaylov, V. A. Vasnev and G. D. Markova, Dokl. Chem., 2019, 488, 249–251 CrossRef CAS.
- X. Huang, Q. Zhang, G. Deng, Z. Meng, X. Jia and K. Xi, RSC Adv., 2016, 6, 24690–24697 RSC.
- C. Masalles, S. Borrós, C. Viñas and F. Teixidor, Adv. Mater., 2000, 12, 1199–1202 CrossRef CAS.
- C. Masalles, J. Llop, C. Viñas and F. Teixidor, Adv. Mater., 2002, 14, 826–829 CrossRef CAS.
- B. Fabre, S. Chayer and M. G. H. Vicente, Electrochem. Commun., 2003, 5, 431–434 CrossRef CAS.
- B. Fabre, J. C. Clark and M. G. H. Vicente, Macromolecules, 2006, 39, 112–119 CrossRef CAS.
- E. Hao, B. Fabre, F. R. Fronczek and M. G. H. Vicente, Chem. Mater., 2007, 19, 6195–6205 CrossRef CAS.
- E. Hao, B. Fabre, F. R. Fronczek and M. G. H. Vicente, Chem. Commun., 2007, 4387–4389 RSC.
- F. Barrière, B. Fabre, E. Hao, Z. M. LeJeune, E. Hwang, J. C. Garno, E. E. Nesterov and M. G. H. Vicente, Macromolecules, 2009, 42, 2981–2987 CrossRef.
- E. G. Cansu-Ergun and A. Cihaner, J. Electroanal. Chem., 2013, 707, 78–84 CrossRef CAS.
- Y. C. Simon, J. J. Peterson, C. Mangold, K. R. Carter and E. B. Coughlin, Macromolecules, 2009, 42, 512–516 CrossRef CAS.
- J. J. Peterson, M. Werre, Y. C. Simon, E. B. Coughlin and K. R. Carter, Macromolecules, 2009, 42, 8594–8598 CrossRef CAS.
- J. J. Peterson, Y. C. Simon, E. B. Coughlin and K. R. Carter, Chem. Commun., 2009, 4950–4952 RSC.
- K. L. Martin, J. N. Smith, E. R. Young and K. R. Carter, Macromolecules, 2019, 52, 7951–7960 CrossRef CAS.
- J. J. Peterson, A. R. Davis, M. Werre, E. B. Coughlin and K. R. Carter, ACS Appl. Mater. Interfaces, 2011, 3, 1796–1799 CrossRef CAS.
- K. Kokado and Y. Chujo, Macromolecules, 2009, 42, 1418–1420 CrossRef CAS.
- K. Kokado, Y. Tokoro and Y. Chujo, Macromolecules, 2009, 42, 2925–2930 CrossRef CAS.
- K. Kokado, Y. Tokoro and Y. Chujo, Macromolecules, 2009, 42, 9238–9242 CrossRef CAS.
- J. Marshall, Z. Fei, C. P. Yau, N. Yaacobi-Gross, S. Rossbauer, T. D. Anthopoulos, S. E. Watkins, P. Beavis and M. Heeney, J. Mater. Chem. C, 2014, 2, 232–239 RSC.
- J. Marshall, J. Hooton, Y. Han, A. Creamer, R. S. Ashraf, Y. Porte, T. D. Anthopoulos, P. N. Stavrinou, M. A. McLachlan, H. Bronstein, P. Beavis and M. Heeney, Polym. Chem., 2014, 5, 6190–6199 RSC.
- Q. Zhou, Z. Mao, L. Ni and J. Chen, J. Appl. Polym. Sci., 2007, 104, 2498–2503 CrossRef CAS.
- Y. Pan, S. Cheng, H. Dong, N. Li, F. Bao, J. Wang and X. Jian, Corros. Sci., 2019, 154, 1–10 CrossRef CAS.
- T. B. Yisgedu, X. Chen, S. Schricker, J. Parquette, E. A. Meyers and S. G. Shore, Chem. – Eur. J., 2009, 15, 2190–2199 CrossRef CAS.
- M. C. Parrott, E. B. Marchington, J. F. Valliant and A. Adronov, Macromol. Symp., 2003, 196, 201–211 CrossRef CAS.
- S. E. A. Gratton, M. C. Parrott and A. Adronov, J. Inorg. Organomet. Polym. Mater., 2005, 15, 469–475 CrossRef CAS.
- Z. Ruan, P. Yuan, L. Liu, T. Xing and L. Yan, Int. J. Polym. Mater. Polym. Biomater., 2018, 67, 720–726 CrossRef CAS.
- D. Qu, Y. Bai and N. Li, J. Inorg. Organomet. Polym. Mater., 2019, 29, 1496–1502 CrossRef CAS.
- Z. Ruan, L. Liu, L. Fu, T. Xing and L. Yan, Polym. Chem., 2016, 7, 4411–4418 RSC.
- Z. Ruan, P. Yuan, T. T. Jing, T. Xing and L. F. Yan, Macromol. Res., 2018, 26, 270–277 CrossRef CAS.
- R. Fernandez-Alvarez, E. Hlavatovičová, K. Rodzeń, A. Strachota, S. Kereïche, P. Matějíček, J. Cabrera-González, R. Núñez and M. Uchman, Polym. Chem., 2019, 10, 2774–2780 RSC.
- M. S. Messina, C. T. Graefe, P. Chong, O. M. Ebrahim, R. S. Pathuri, N. A. Bernier, H. A. Mills, A. L. Rheingold, R. R. Frontiera, H. D. Maynard and A. M. Spokoyny, Polym. Chem., 2019, 10, 1660–1667 RSC.
- S. R. Benhabbour, M. C. Parrott, S. E. A. Gratton and A. Adronov, Macromolecules, 2007, 40, 5678–5688 CrossRef CAS.
- Y. C. Simon, C. Ohm, M. J. Zimny and E. B. Coughlin, Macromolecules, 2007, 40, 5628–5630 CrossRef CAS.
- Y. C. Simon and E. B. Coughlin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2557–2563 CrossRef CAS.
- G. Kahraman, D.-Y. Wang, J. von Irmer, M. Gallei, E. Hey-Hawkins and T. Eren, Polymers, 2019, 11, 613 CrossRef.
- M. H. Park, K. M. Lee, T. Kim, Y. Do and M. H. Lee, Chem. – Asian J., 2011, 6, 1362–1366 CrossRef CAS.
- R. Djeda, J. Ruiz, D. Astruc, R. Satapathy, B. P. Dash and N. S. Hosmane, Inorg. Chem., 2010, 49, 10702–10709 CrossRef CAS.
- B. P. Dash, R. Satapathy, B. P. Bode, C. T. Reidl, J. W. Sawicki, A. J. Mason, J. A. Maguire and N. S. Hosmane, Organometallics, 2012, 31, 2931–2935 CrossRef CAS.
- N. Li, P. Zhao, L. Salmon, J. Ruiz, M. Zabawa, N. S. Hosmane and D. Astruc, Inorg. Chem., 2013, 52, 11146–11155 CrossRef CAS.
- C.-Y. Wu, J.-J. Lin, W.-Y. Chang, C.-Y. Hsieh, C.-C. Wu, H.-S. Chen, H.-J. Hsu, A.-S. Yang, M.-H. Hsu and W.-Y. Kuo, Colloids Surf., B, 2019, 183, 110387 CrossRef CAS.
- P. Matejicek, M. Uchman, M. Lepsik, M. Srnec, J. Zednik, P. Kozlik and K. Kalikova, ChemPlusChem, 2013, 78, 528–535 CrossRef CAS.
- F. Teixidor, A. Pepiol and C. Vinas, Chem. – Eur. J., 2015, 21, 10650–10653 CrossRef CAS.
- J. Cabrera-Gonzalez, A. Ferrer-Ugalde, S. Bhattacharyya, M. Chaari, F. Teixidor, J. Gierschner and R. Nunez, J. Mater. Chem. C, 2017, 5, 10211–10219 RSC.
- J. Cabrera-Gonzalez, S. Bhattacharyya, B. Milian-Medina, F. Teixidor, N. Farfan, R. Arcos-Ramos, V. Vargas-Reyes, J. Gierschner and R. Nunez, Eur. J. Inorg. Chem., 2017, 4575–4580 CrossRef CAS.
- M. C. Parrott, E. B. Marchington, J. F. Valliant and A. Adronov, J. Am. Chem. Soc., 2005, 127, 12081–12089 CrossRef CAS.
- M. C. Parrott, J. F. Valliant and A. Adronov, Langmuir, 2006, 22, 5251–5255 CrossRef CAS.
- B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, Org. Lett., 2008, 10, 2247–2250 CrossRef CAS.
- B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, Organometallics, 2010, 29, 5230–5235 CrossRef CAS.
- C. E. Housecroft, J. Organomet. Chem., 2015, 798, 218–228 CrossRef CAS.
- N. Das, P. J. Stang, A. M. Arif and C. F. Campana, J. Org. Chem., 2005, 70, 10440–10446 CrossRef CAS.
- H. Jude, H. Disteldorf, S. Fischer, T. Wedge, A. M. Hawkridge, A. M. Arif, M. F. Hawthorne, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 12131–12139 CrossRef CAS.
- J. M. Ludlow, III, M. Tominaga, Y. Chujo, A. Schultz, X. Lu, T. Xie, K. Guo, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, Dalton Trans., 2014, 43, 9604–9611 RSC.
- D. J. Clingerman, R. D. Kennedy, J. E. Mondloch, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, Chem. Commun., 2013, 49, 11485–11487 RSC.
- K. Kokado and Y. Chujo, Dalton Trans., 2011, 40, 1919–1923 RSC.
- K. Zhang, Y. Shen, J. Liu, B. Spingler and S. Duttwyler, Chem. Commun., 2018, 54, 1698–1701 RSC.
- O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680–12681 CrossRef CAS.
- Y.-S. Bae, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2008, 4135–4137 RSC.
- A. M. Spokoyny, O. K. Farha, K. L. Mulfort, J. T. Hupp and C. A. Mirkin, Inorg. Chim. Acta, 2010, 364, 266–271 CrossRef CAS.
- D. J. Clingerman, W. Morris, J. E. Mondloch, R. D. Kennedy, A. A. Sarjeant, C. Stern, J. T. Hupp, O. K. Farha and C. A. Mirkin, Chem. Commun., 2015, 51, 6521–6523 RSC.
- R. D. Kennedy, V. Krungleviciute, D. J. Clingerman, J. E. Mondloch, Y. Peng, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, J. T. Hupp, T. Yildirim, O. K. Farha and C. A. Mirkin, Chem. Mater., 2013, 25, 3539–3543 CrossRef CAS.
- S. Choi, H.-E. Lee, C. H. Ryu, J. Lee, J. Lee, M. Yoon, Y. Kim, M. H. Park, K. M. Lee and M. Kim, Chem. Commun., 2019, 55, 11844–11847 RSC.
- S. Yuan, D. White, A. Mason and D.-J. Liu, Int. J. Energy Res., 2013, 37, 732–740 CrossRef CAS.
- C. Wang, F. Huang, Y. Jiang, J. Li, Y. Zhou and L. Du, Ceram. Int., 2012, 38, 3081–3088 CrossRef CAS.
- C. Wang, F. Huang, Y. Jiang, Y. Zhou and L. Du, J. Am. Ceram. Soc., 2012, 95, 71–74 CrossRef CAS.
- Y. Zong, D. Tong and Z. Li, High Perform. Polym., 2019, 31, 1085–1100 CrossRef CAS.
- Y. Sun, D. Tong and Z. Li, Fibers Polym., 2019, 20, 1564–1576 CrossRef CAS.
- P. R. L. Malenfant, J. Wan, S. T. Taylor and M. Manoharan, Nat. Nanotechnol., 2007, 2, 43–46 CrossRef CAS.
- R. F. Barth, J. A. Coderre, M. G. H. Vicente and T. E. Blue, Clin. Cancer Res., 2005, 11, 3987 CrossRef CAS.
- P. Hoppenz, S. Els-Heindl, M. Kellert, R. Kuhnert, S. Saretz, H.-G. Lerchen, J. Köbberling, B. Riedl, E. Hey-Hawkins and A. G. Beck-Sickinger, J. Org. Chem., 2020, 85, 1446–1457 CrossRef CAS.
- I. Nakase, M. Katayama, Y. Hattori, M. Ishimura, S. Inaura, D. Fujiwara, T. Takatani-Nakase, I. Fujii, S. Futaki and M. Kirihata, Chem. Commun., 2019, 55, 13955–13958 RSC.
- H. Xiong, D. Zhou, Y. Qi, Z. Zhang, Z. Xie, X. Chen, X. Jing, F. Meng and Y. Huang, Biomacromolecules, 2015, 16, 3980–3988 CrossRef CAS.
- S. Carturan, A. Quaranta, T. Marchi, F. Gramegna, M. Degerlier, M. Cinausero, V. L. Kravchuk and M. Poggi, Radiat. Prot. Dosim., 2011, 143, 471 CrossRef CAS.
- Y. Wu, J. Hu, C. Feng, G. Chen and J. Yang, Mater. Des., 2019, 172, 107772 CrossRef CAS.
- X. Li, J. Wu, C. Tang, Z. He, P. Yuan, Y. Sun, W.-M. Lau, K. Zhang, J. Mei and Y. Huang, Composites, Part B, 2019, 159, 355–361 CrossRef CAS.
- V. M. Pawar, M. Beck, A. D. Shetgaonkar, R. Pal, A. K. Bakshi and V. S. Nadkarni, Nucl. Instrum. Methods Phys. Res., Sect. B, 2020, 462, 169–176 CrossRef CAS.
- Q.-Y. Wang, J. Wang, S. Wang, Z.-Y. Wang, M. Cao, C.-L. He, J.-Q. Yang, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2020, 142, 12010–12014 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |