
Control of local flexibility towards p-xylene sieving in Hofmann-type porous coordination polymers†
Mohana
Shivanna
 a,
Ken-ichi
Otake
a,
Jia-Jia
Zheng
ab,
Shigeyoshi
Sakaki
b and
Susumu
Kitagawa
*a
a,
Ken-ichi
Otake
a,
Jia-Jia
Zheng
ab,
Shigeyoshi
Sakaki
b and
Susumu
Kitagawa
*a
aInstitute for Integrated Cell-Material Sciences (iCeMS), Institute for Advanced Study, Kyoto University (KUIAS), Yoshida Ushinomiyacho, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: kitagawa@icems.kyoto-u.ac.jp
bElement Strategy Initiative for Catalyst and Batteries, Kyoto University, Goryo-Ohara 1-30, Nishikyo-ku, Kyoto 615-8245, Japan
First published on 23rd July 2020
Abstract
Adsorption-based xylene isomer separation is more energy efficient than conventional processes. Herein, three isostructural Hofmann-type porous coordination polymers (PCPs), {M(Pz)[Ni(CN)4]n} (M = Fe, FePzNi, Co, CoPzNi, and Ni, NiPzNi; Pz = pyrazine) were synthesized and shown to exhibit coordination-dependent lability for the selectivity toward p-xylene over m- and o-xylene.
The separation and purification of important industrial commodities (e.g., gases or solvent vapors) account for 15% of global energy consumption.1 Among these, xylene isomers (para-, meta- and ortho-xylene abbreviated as px, mx, and ox, respectively) are currently separated from crude oil via distillation. This technology involves energy-intensive and time-consuming processes because the similar physicochemical properties of the isomers (boiling point, molecular size, and shape; Table S1, ESI†) make the separation process difficult. However, their separation is crucial for the production of chemical intermediates used to produce many valuable products such as polymer fibers, films, plasticizers, resins, and pigments.2 Therefore, other approaches have been employed, such as adsorption, crystallization, complexation, and isomerization.3 In adsorption, physisorbents can effectively resolve the issue of the selective adsorption of one isomer over others because of their pore size and/or pore functionality, which reduces the energy associated with sorbent recycling. In this context, traditional classes of porous solids such as zeolites or silica have been investigated, but are limited to their application as molecular sieves because of the high uptake of all isomers with small difference. For example, FAU zeolite has a selectivity coefficient of only 3–54 and MFI zeolite has a selectivity of 104 for px/ox.5 A new subclass of porous materials known as porous coordination polymers (PCPs),6,7 or metal organic frameworks (MOFs)8 constructed from metal nodes and organic linkers has gained considerable research interest because of the high surface area and extraordinary diverse compositions, making them potential candidates for xylene separation and other applications.9,10 The selective uptake of one xylene isomer has been found to be significantly improved compared to that of traditional porous materials.11,12 Hofmann clathrates13 and Warner complexes14 are also considered as promising candidates for the separation of xylene isomers.15 Various strategies have been employed to evaluate their performance, such as their interaction with open metals,16 pore-size-dependent selectivity in rigid materials,11 breathing induced separation,17 gate open and close phenomena in switching layered materials,18 and others.19,20 For example, to affirm their flexibility, temperature-dependent tuning of the window aperture of ZIF-8 was shown to demonstrate high selectively toward px over mx and ox.21 Such stimulus-induced transformation of the host network has been exemplified toward guest insertion and removal.22,23 Our idea was to utilize pillar rotation to control the diffusion pathway in a pillared layer structure under ambient conditions. Therefore, we chose Hofmann-type three-dimensional PCPs to tune the local flexibility using different metal combinations, that is, tuning the pore window opening toward the recognition of specific xylene isomers. Herein, three isostructural Hofmann-type PCPs with the formula M(Pz)[Ni(CN)n] (M = Fe, FePzNi, Co, CoPzNi, and Ni, NiPzNi; Pz = pyrazine) were synthesized; the Brunauer–Emmett–Teller (BET) surface areas of these PCPs were in the order FePzNi < CoPzNi < NiPzNi. Interestingly, the observed high px adsorption affinity followed the reverse trend (FePzNi > CoPzNi > NiPzNi) and excluded the mx and ox isomers from the pores (Fig. 1).
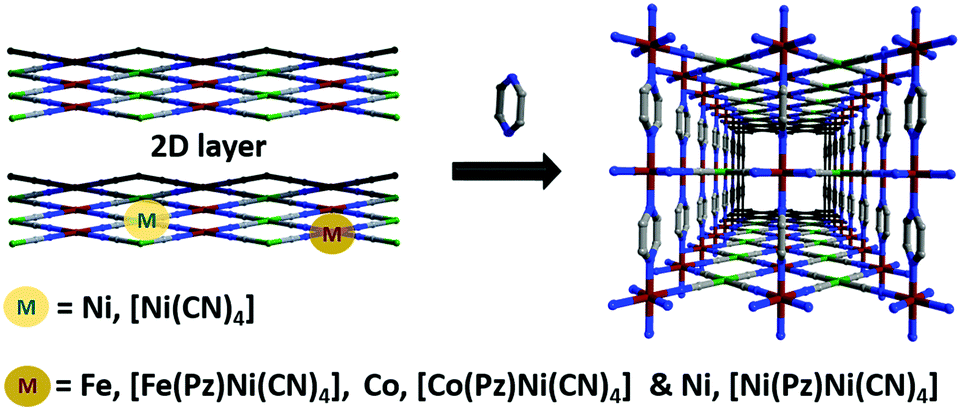 | ||
| Fig. 1 Schematic representation of the Hofmann-type 2D network pillared by pyrazine used to form a 3D porous coordination polymer. The three isostructural networks are synthesized upon varying the octahedral metal (Fe, Co, Ni) and maintaining the same square planar metal center. | ||
Three isostructural PCPs were synthesized using a slight modification of a previously reported procedure24 (a more detailed synthetic procedure is available in the ESI†). The purities of the as-synthesized phases consisting of FePzNi, CoPzNi, and NiPzNi were confirmed by comparing their powder X-ray diffraction (PXRD) patterns to those calculated from their previously reported crystal structures (Fig. S1–S3, ESI†).24–26 Thermogravimetric analysis proved that FePzNi, CoPzNi and NiPzNi were stable up to ∼250, ∼280 and ∼350 °C (Fig. S4–S6, ESI†). All three PCPs were activated at 120 °C for 12 h under vacuum prior to the sorption measurements. To confirm their porosity, gas sorption isotherm measurements using N2 at 77 K and CO2 at 195 K were performed. The three isostructural PCPs exhibit typical type-I profiles toward both N2 and CO2. The observed CO2 and N2 uptakes were found to be 120 cm3 g−1 for FePzNi with a BET surface area of ∼330 m2 g−1 (Fig. 2a). In the case of CoPzNi, the CO2 uptake was slightly lower (140 cm3 g−1) than that observed for N2 (150 cm3 g−1, BET ∼430 m2 g−1), but higher than FePzNi (Fig. 2b). NiPzNi exhibited a slightly higher uptake for CO2 (130 cm3 g−1) and N2 (160 cm3 g−1), but its BET surface area was found to be ∼500 m2 g−1 (Fig. 2c).
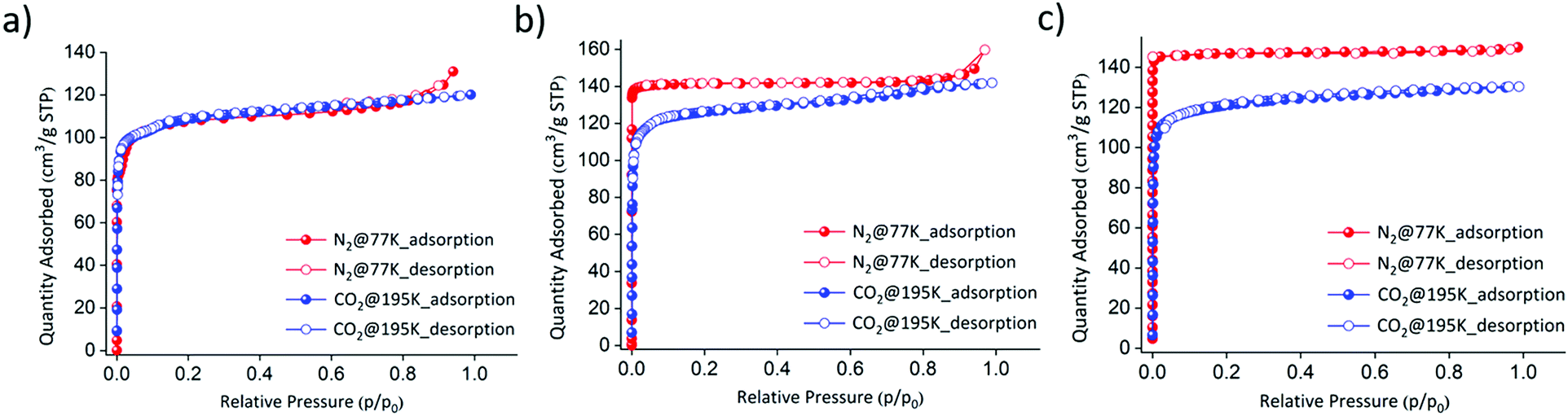 | ||
| Fig. 2 Gas sorption (blue, CO2 at 195 K; red, N2 at 77 K) on three isostructural PCPs: (a) FePzNi (calculated BET surface area using N2 was found to be ∼330 m2 g−1), (b) CoPzNi (BET surface area using N2 ∼430 m2 g−1), and (c) NiPzNi (BET surface area using N2 ∼500 m2 g−1). | ||
The analysis of the pore sizes of the crystal structures prompted us to study their xylene vapor sorption isotherms. Interestingly, under vapor pressures of the three xylene isomers, FePzNi exhibits selectivity toward the px isomer over the other two, namely mx and ox. The vapor sorption profile obtained for the px isomer was a typical type-I isotherm with an uptake of 40 cm3 g−1 at P/P0 = 1.0. However, mx (10 cm3 g−1) and ox (5 cm3 g−1) showed negligible uptake, which was limited to surface adsorption only (Fig. 3a). This indicates that FePzNi shows high selectivity toward px (6.7 Å), which has a slightly smaller kinetic diameter when compared to mx (7.1 Å) and ox (7.4 Å).4 In contrast, CoPzNi exhibits S-shaped or gate opening27 type adsorption behavior toward the px isomer; a slight uptake from 0 to 5 cm3 g−1 was observed upon increasing the pressure up to P/P0 = 0.18 (the gate-open pressure), followed by a sudden increase in the uptake to 30 cm3 g−1 prior to P/P0 = 0.22, which saturated to 40 cm3 g−1 at P/P0 = 1.0. The desorption process follows the adsorption profile and finally reverts to the activated phase, as verified by PXRD (Fig. S2, ESI†). CoPzNi exhibited a low affinity toward the other two isomers with uptakes of 9 cm3 g−1 (mx) and 4 cm3 g−1 (ox) (Fig. 3b). NiPzNi exhibits a small non-selective uptake of the three isomers of 7 (px), 5 (mx), and 3 cm3 g−1 (ox) (Fig. 3c). When the temperature was increased to 308 K during the vapor sorption of px, the gate-open pressure observed for CoPzNi showed a slight increase to P/P0 = 0.2 (Fig. S9, ESI†). FePzNi and NiPzNi exhibit no difference in their sorption performance when compared to those observed at 298 K. The recyclability tests using FePzNi and CoPzNi indicate that their px sorption profiles were reproducible with the same uptake maintained over 5 cycles (Fig. S10 and S11, ESI†). Importantly, the adsorption affinity toward px follows the reverse trend (FePzNi > CoPzNi > NiPzNi) compared to the trend for the BET surface area (FePzNi < CoPzNi < NiPzNi). To determine the binary mixture separation performance, we conducted vapor phase experiments using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of xylene isomers. The selectivities were estimated by the integration area ratio of the adsorbed components by the 1H NMR spectra (Fig. S15–S18; see ESI† for the detail). Interestingly, FePzNi exhibited an exceptional px selectivity when exposed to px/mx mixture. While upon px/ox mixture, FePzNi showed the selectivity of 2. Whereas, CoPzNi showed px selectivities of 1.2 and 1.15 over px/mx and px/ox mixture, respectively. Except for FePzNi towards px/mx, these calculated selectivities are slightly higher or comparable to previously reported materials28 (Table S2, ESI†).
:1 mixture of xylene isomers. The selectivities were estimated by the integration area ratio of the adsorbed components by the 1H NMR spectra (Fig. S15–S18; see ESI† for the detail). Interestingly, FePzNi exhibited an exceptional px selectivity when exposed to px/mx mixture. While upon px/ox mixture, FePzNi showed the selectivity of 2. Whereas, CoPzNi showed px selectivities of 1.2 and 1.15 over px/mx and px/ox mixture, respectively. Except for FePzNi towards px/mx, these calculated selectivities are slightly higher or comparable to previously reported materials28 (Table S2, ESI†).
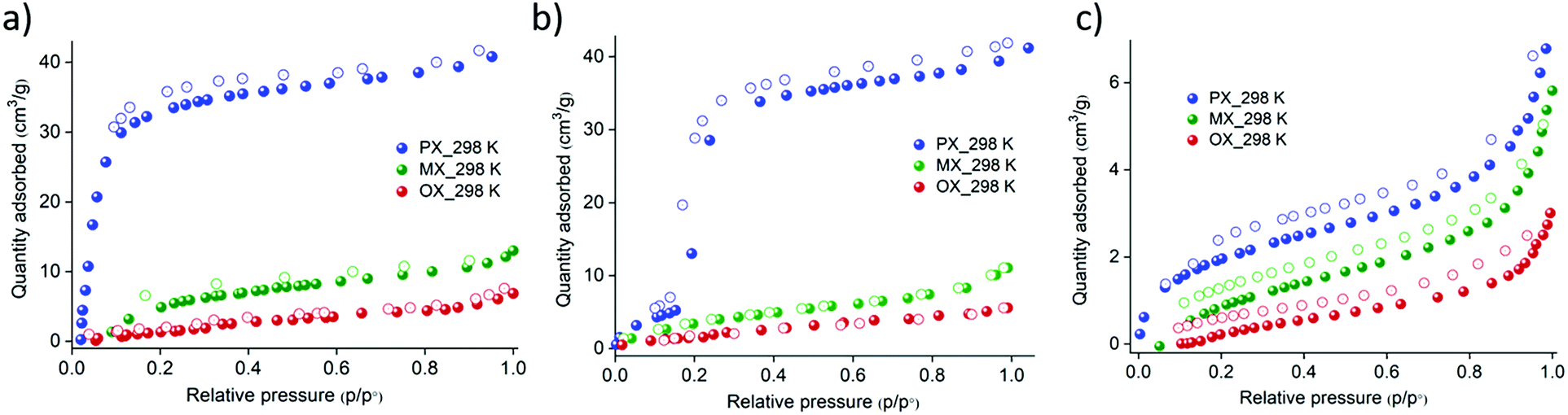 | ||
| Fig. 3 Pure component xylene isomer (px-blue, mx-green, and ox-red) sorption on the three isostructural PCPs measured at 298 K (a) FePzNi exhibits a typical type-I isotherm for px with an uptake of 40 cm3 g−1. (b) CoPzNi exhibits a stepped profile during the sorption of px with the same uptake as FePzNi. (c) No gate opening was observed for px when using NiPzNi. All three PCPs shown negligible uptake of mx and ox. | ||
To gain insight into mechanism of px selective adsorption and the structural transformation, we collected PXRD data by soaking a few milligrams of each PCP in three different vials containing the pure liquid (px isomer) for 24 h. The PXRD patterns indicate negligible changes in FePzNi and NiPzNi (Fig. S1 and S3, ESI†). CoNiPz exhibits some shifts in the peaks corresponding to (001) and (002), which are directed toward the pyrazine and 2D metal layers (Fig. S2 and S19, ESI†). This implies that upon px adsorption pyrazine undergoes rotation and that the subtle transformation of the 2D layer leads to a stepped profile. We further analyzed the crystal structures in order to gain an insight into the lability of the coordination bonds. In the as-synthesized form of FePzNi, the pyrazine molecules align parallel to the channel along the b-axis, while upon removal of the guest molecules (water), the pyrazine molecules are distorted to close the channel window (Fig. 4a). When the pyrazine molecules align parallel to the channel direction, the centroid distance between the two aromatic moieties (C–C⋯π with DC⋯C = 7.257(5) Å) is suitable for the px isomer when compared to mx and ox. In the perpendicular direction, the π⋯π distance was reduced to 5.1(3) Å. The Fe–Fe distances along the diagonal and between the 2D layers were found to be DFe1⋯Fe3 = 10.262(23) Å and DFe1⋯Fe4 = 7.256(3) Å, respectively (Fig. S20, ESI† and Table 1). The distances between the octahedral center of Fe and the N atom of the cyanide was DFe⋯N = 2.117(44) Å and N atom of the pyrazine was DFe⋯N = 2.212(116) Å. The square planar metal center (NiCN4) was aligned with DNi⋯C = 1.864(44) Å (Fig. S20, ESI† and Table 1). Most of these bond distances are slightly shorter in CoPzNi and much shorter in NiPzNi structures (Fig. S21 and S22, ESI† and Table 1). Therefore, as shown in Fig. 4a, pyrazine rotation is a key and controlling factor to open the channel gate toward px selectivity. The 2D interlayer distance (7.256(3) Å) can allow facile ring rotation in FePzNi, but the slightly reduced distance (7.107(6) Å), which pushes to higher pressures in CoPzNi and much smaller distance (7.024(4) Å) makes it difficult for the NiPzNi structure.
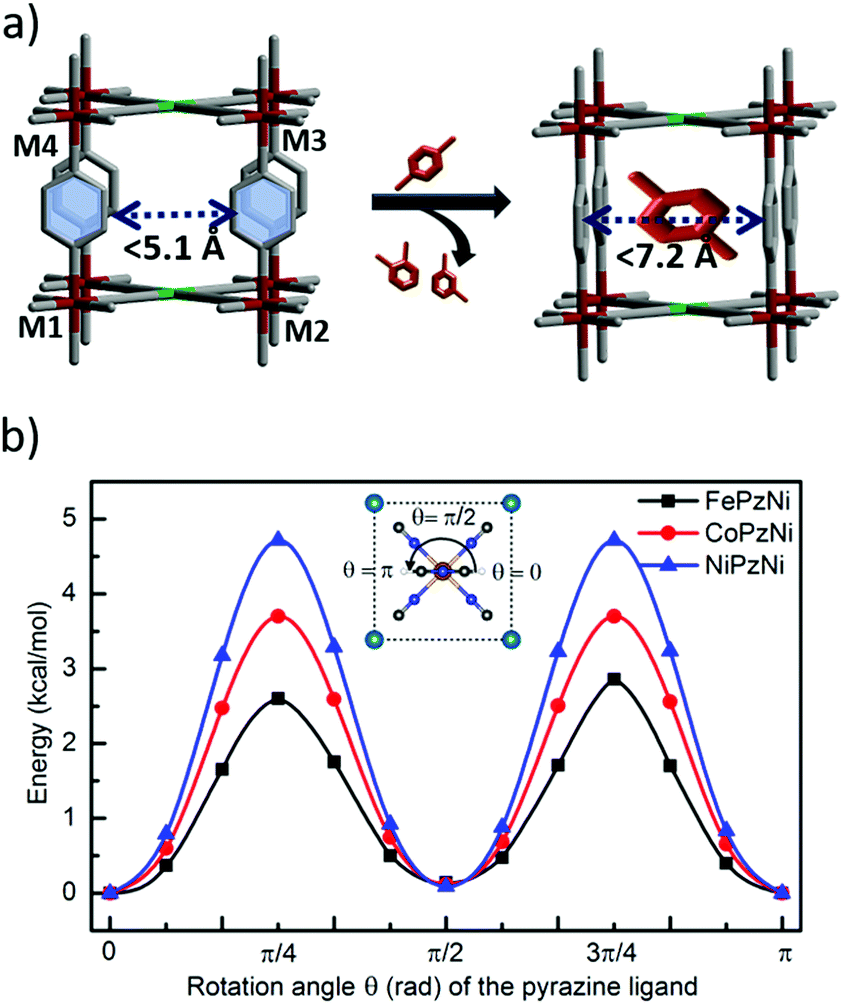 | ||
| Fig. 4 (a) A schematic representation of the pillar rotation toward the px isomer. (b) The potential energy surface for the rotation of the pyrazine pillars in FePzNi, CoPzNi, and NiPzNi. | ||
| D M⋯M | D M⋯N(CN) | D M⋯N(PZ) | D Ni⋯C(CN4) | |
|---|---|---|---|---|
| FePzNi | D Fe1⋯Fe3 = 10.262(23), DFe1⋯Fe4 = 7.256(3) Å | D Fe⋯N = 2.117(44) Å | D Fe⋯N = 2.212(116) Å | 1.864(44) Å |
| CoPzNi | D Co1⋯Co3 = 10.131(6), DCo1⋯Co4 = 7.107(6) Å | D Co⋯N = 2.089(36) Å | D Co⋯N = 2.167(64) Å | 1.858(43) Å |
| NiPzNi | D Ni1⋯Ni3 = 10.027(4), DNi1⋯Ni4 = 7.024(4) Å | D Ni⋯N = 2.160(215) Å | D Ni⋯N = 2.037(7) Å | 1.825(209) Å |
The rotational barrier of the Pz pillar (Fig. 4b) and deformation energy increases in the order FePzNi (2.6 kcal mol−1) < CoPzNi (3.7 kcal mol−1) < NiPzNi (4.7 kcal mol−1) and FePzNi (2.5 kcal mol−1) < CoPzNi (2.7 kcal mol−1) < NiPzNi (3.2 kcal mol−1) (Table S3, ESI†) which is consistent with these geometrical features and indicates that this steric effect plays an important role in the rotation of the pyrazine molecules in these PCPs, as discussed in our previous work.23 In addition, the large space in FePzNi makes it the best for px adsorption among the three Hofmann-type PCPs studied, where the binding energy (see ESI† for the definition of the binding energy and computational details) of px decreases in the order of FePzNi (−18.3 kcal mol−1) > CoPzNi (−16.1 kcal mol−1) > NiPzNi (−13.8 kcal mol−1). While interaction energy between px and PCPs follows same trend, FePzNi (−20.8 kcal mol−1) > CoPzNi (−18.8 kcal mol−1) > NiPzNi (−17.0 kcal mol−1) (Table S3, ESI†), indicating the largest affinity of FePzNi toward px. These results suggest that an appropriate distance between the pyrazine rings in FePzNi was beneficial for px adsorption both kinetically and thermodynamically.
In conclusion, three isostructural Hofmann-type PCPs exhibit control over pillar rotation to recognize specific xylene isomers. When FePzNi is exposed to xylene vapor, the pyrazine rotation showed a high affinity toward the px isomer with a typical type-I isotherm. In the case of the CoPzNi structure upon px sorption, pyrazine rotation required a slightly higher energy and led to a stepped or gate opening isotherm with no considerable hysteresis. The high uptake and selectivity due to the pore size is a perfect fit for the px isomer after pillar rotation. However, NiPzNi behaves as a highly rigid material and does not induce any structural transformation of the pillar under a partial pressure of any of the three xylene isomers. The results indicate that coordination is a key factor in controlling these subtle transformations, which can allow specific guest molecules into the pores. Therefore, this work demonstrates that the construction of a framework with desirable metal coordination plays an important role in the separation of xylene isomers. Moreover, their ease of synthesis from common and inexpensive starting chemicals may lead to the design of potential candidates that can be applied for industrial-related molecular recognition. Separation studies of C2 gas mixtures with NiPzNi are in progress.
Authors thank the financial support of KAKENHI, Grant-in-Aid for Scientific Research (S) (JP18H05262), and Early-Career Scientists (JP19K15584) from the Japan Society of the Promotion of Science (JSPS). Synchrotron XRD measurements were supported by the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2019A1600, 2019B1554, 2020A1496). We thank Dr Kawaguchi of JASRI and Prof. Kubota of Osaka Prefecture University for their experimental help at SPring-8. NMR facility was used at the iCeMS Analysis Center, KUIAS.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- N. Sun, S.-Q. Wang, R. Zou, W.-G. Cui, A. Zhang, T. Zhang, Q. Li, Z.-Z. Zhuang, Y.-H. Zhang, J. Xu, M. J. Zaworotko and X.-H. Bu, Chem. Sci., 2019, 10, 8850–8854 RSC.
- M. du Plessis, V. I. Nikolayenko and L. J. Barbour, J. Am. Chem. Soc., 2020, 142, 4529–4533 CrossRef CAS PubMed.
- Y. Yang, P. Bai and X. Guo, Ind. Eng. Chem. Res., 2017, 56, 14725–14753 CrossRef CAS.
- G.-Q. Guo, H. Chen and Y.-C. Long, Microporous Mesoporous Mater., 2000, 39, 149–161 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem. Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination polymers: design, analysis and application introduction, RSC Publishing, Cambridge, UK, 2009 Search PubMed.
- M. Schroder, Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis, in Topics in Current Chemistry, ed. M. Schroder, 2010, vol. 293, pp. 1–262 Search PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- B. Saccoccia, A. M. Bohnsack, N. W. Waggoner, K. H. Cho, J. S. Lee, D.-Y. Hong, V. M. Lynch, J.-S. Chang and S. M. Humphrey, Angew. Chem. Int. Ed., 2015, 54, 5394–5398 CrossRef CAS PubMed.
- J. E. Warren, C. G. Perkins, K. E. Jelfs, P. Boldrin, P. A. Chater, G. J. Miller, T. D. Manning, M. E. Briggs, K. C. Stylianou, J. B. Claridge and M. J. Rosseinsky, Angew. Chem. Int. Ed., 2014, 53, 4592–4596 CrossRef CAS PubMed.
- H. M. Powell, J. Am. Chem. Soc., 1948, 61–73 RSC.
- W. D. Schaeffer, W. S. Dorsey, D. A. Skinner and C. G. Christian, J. Am. Chem. Soc., 1957, 79, 5870–5876 CrossRef CAS.
- M. Lusi and L. J. Barbour, Angew. Chem. Int. Ed., 2012, 51, 3928–3931 CrossRef CAS PubMed.
- M. I. Gonzalez, M. T. Kapelewski, E. D. Bloch, P. J. Milner, D. A. Reed, M. R. Hudson, J. A. Mason, G. Barin, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2018, 140, 3412–3422 CrossRef CAS PubMed.
- V. Finsy, C. E. A. Kirschhock, G. Vedts, M. Maes, L. Alaerts, D. E. De Vos, G. V. Baron and J. F. M. Denayer, Chem. – Eur. J., 2009, 15, 7724–7731 CrossRef CAS PubMed.
- S.-Q. Wang, S. Mukherjee, E. Patyk-Kaźmierczak, S. Darwish, A. Bajpai, Q.-Y. Yang and M. J. Zaworotko, Angew. Chem. Int. Ed., 2019, 58, 6630–6634 CrossRef CAS PubMed.
- F. Vermoortele, M. Maes, P. Z. Moghadam, M. J. Lennox, F. Ragon, M. Boulhout, S. Biswas, K. G. M. Laurier, I. Beurroies, R. Denoyel, M. Roeffaers, N. Stock, T. Düren, C. Serre and D. E. De Vos, J. Am. Chem. Soc., 2011, 133, 18526–18529 CrossRef CAS PubMed.
- V. Finsy, H. Verelst, L. Alaerts, D. De Vos, P. A. Jacobs, G. V. Baron and J. F. M. Denayer, J. Am. Chem. Soc., 2008, 130, 7110–7118 CrossRef CAS PubMed.
- D. M. Polyukhov, A. S. Poryvaev, S. A. Gromilov and M. V. Fedin, Nano Lett., 2019, 19, 6506–6510 CrossRef CAS PubMed.
- M. Ohba, K. Yoneda, G. Agustí, M. C. Muñoz, A. B. Gaspar, J. A. Real, M. Yamasaki, H. Ando, Y. Nakao, S. Sakaki and S. Kitagawa, Angew. Chem. Int. Ed., 2009, 48, 4767–4771 CrossRef CAS PubMed.
- H. Ando, Y. Nakao, H. Sato, M. Ohba, S. Kitagawa and S. Sakaki, Chem. Phys. Lett., 2011, 511, 399–404 CrossRef CAS.
- P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef CAS PubMed.
- J. Rodríguez-Hernández, A. A. Lemus-Santana, J. Ortiz-López, S. Jiménez-Sandoval and E. Reguera, J. Solid State Chem., 2010, 183, 105–113 CrossRef.
- J. Pei, K. Shao, J.-X. Wang, H.-M. Wen, Y. Yang, Y. Cui, R. Krishna, B. Li and G. Qian, Adv. Mater., 2020, 1908275 CrossRef CAS PubMed.
- Q. Y. Yang, P. Lama, S. Sen, M. Lusi, K. J. Chen, W. Y. Gao, M. Shivanna, T. Pham, N. Hosono, S. Kusaka, J. J. T. Perry, S. Ma, B. Space, L. J. Barbour, S. Kitagawa and M. J. Zaworotko, Angew. Chem. Int. Ed., 2018, 57, 5684–5689 CrossRef CAS PubMed.
- L. Alaerts, C. E. A. Kirschhock, M. Maes, M. A. van der Veen, V. Finsy, A. Depla, J. A. Martens, G. V. Baron, P. A. Jacobs, J. F. M. Denayer and D. E. De Vos, Angew. Chem. Int. Ed., 2007, 46, 4293–4297 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD, TGA, sorption, and NMR data, and additional experimental and computational details. See DOI: 10.1039/d0cc03854g |
| This journal is © The Royal Society of Chemistry 2020 |