
Proteins as diverse, efficient, and evolvable scaffolds for artificial metalloenzymes
Woo Jae
Jeong†
,
Jaeseung
Yu†
and
Woon Ju
Song
 *
*
Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: woonjusong@snu.ac.kr
First published on 21st July 2020
Abstract
By combining synthetic catalysts and biochemical tools, numerous artificial metalloenzymes have been designed to exhibit high catalytic activity and selectivity in diverse chemical transformations. Out of the nearly infinite number of discovered or characterised proteins, however, only a handful of proteins have been employed as scaffolds for artificial metalloenzymes, implying that specific proteins are preferred owing to their native structural, functional, or biochemical properties. In the present review, we extract and group the biochemical and structural properties of proteins that are advantageous in the design of artificial metalloenzymes; protein stability, pre-existing metal centre, native binding affinity for small molecules, confined and empty space, well-defined secondary structure, and native cellular location. The desirable properties highlight proteins as the key players in the design of metal-dependent biocatalysts. We also propose rarely considered, yet promising, proteins that could be versatile and unique scaffolds for novel metalloenzymes.
 Woo Jae Jeong | Woo Jae Jeong earned a BS in Chemistry from Seoul National University in 2016 and is pursuing a PhD under the supervision of Professor Woon Ju Song. His current research topic is to design and evolve artificial metallo-hydrolases in novel protein scaffolds. |
 Jaeseung Yu | Jaeseung Yu received a BS in Chemistry from Kyungpook National University in 2016 and is pursuing a PhD in the Department of Chemistry at Seoul National University under the guidance of Professor Woon Ju Song. His current research topic lies in the controlled and efficient design and evolution of metallo-hydrolases. |
 Woon Ju Song | Woon Ju Song obtained her MS under the guidance of Professor Wonwoo Nam at Ewha Womans University and her PhD in the department of chemistry at MIT under the supervision of Professor Stephen J. Lippard. Then, she began her postgraduate studies in Professor Akif F. Tezcan group at the University of California, San Diego. Since she started her independent career at Seoul National University in 2016 as an assistant professor, her research group has been interested in the design and evolution of metalloproteins, protein self-assembly, and the discovery of novel proteins. |
1. Introduction
The discovery of numerous metalloenzymes has inspired us to synthesize inorganic compounds that resemble the structure or function of the active sites.1,2 The design of biomimetic inorganic complexes often starts from the identification and construction of the active sites of natural enzymes, allowing for simplification of essential inorganic environments and evaluation of reaction mechanisms in catalysis. Unlike proteins, which have multiple amino acid residues comprising the active sites, however, synthesis of multiple functional groups around the catalytic sites is often a great synthetic challenge. In this regard, reengineering proteins into artificial metalloenzymes might be viewed as an efficient route to understanding remarkably complex and tightly orchestrated actions of natural enzymes and to synthesise diverse metal-dependent biocatalysts.3–5Recently, remarkable progress has been made in the design of artificial metalloenzymes that harbour inorganic cofactors or metal ions in the protein matrix.5–7 To design catalytically active units with only 20 canonical amino acids, well-defined hydrogen bonds, electrostatic, and/or hydrophobic interactions should be properly incorporated to accommodate a kinetically and thermodynamically tuned series of reactions during catalysis, which is not trivial even with our current state-of-the-art in the understanding of protein sequence-structure–function relationships. In contrast, metal ions alone exhibit a wide range of nucleophilicity, electrophilicity, and/or Lewis acidity, giving rise to the basal requirements for catalytic reactivities upon complexation with appropriate ligands. In addition, metal ions possess multiple coordination numbers, providing vacant sites for substrate-binding. If metal elements are the key to resolving the great hurdle in the creation of catalytically active units, three questions arise in the design of artificial metalloenzymes: which chemical reactions to catalyse, how to introduce inorganic elements into proteins, and what protein scaffolds to start with? The first and second questions have been comprehensively covered in several reviews,8–11 whereas the last inquiry has not been explicitly discussed in detail.
As the ligand design is essential in biomimetic complexes and synthetic catalysts, the selection of proteins that function as metal coordination ligands and the locations for catalysis is critical in artificial metalloenzymes. In the present review, we highlight the specific protein scaffolds that have been effectively transformed into artificial metalloenzymes. Although some proteins are selected based on the method by which inorganic elements are introduced, several other factors should be carefully considered because the choice of a protein determines the structure and function of an artificial metalloenzyme. In addition, the protein scaffold provides unpredictable yet crucial properties that affect catalysis, such as protein expression yields, stability upon mutation, secondary coordination environments to inorganic centres, experimental conditions for activity assays, dynamic motion during catalysis, and evolvability. We have therefore assembled recent examples of artificial metalloenzymes based on the properties of their protein scaffolds. We then suggest additional properties of proteins for future studies to expand the scopes of artificial metalloenzymes.
2. Considerations in the selection of protein ligands
The properties of proteins adapted for novel metal-mediated catalytic activities are categorised into the following six factors: protein stability, pre-existing metal centre, native binding affinity, confined and empty space, well-defined secondary structure, and native cellular location (Fig. 1). These factors contribute to the successful transformation of naturally occurring proteins into nascent artificial metalloenzymes. Notably, successfully repurposed or designed protein scaffolds usually exhibit more than one of these factors, indicating that prior inspection of protein scaffolds is recommended for the efficient design of artificial metalloenzymes.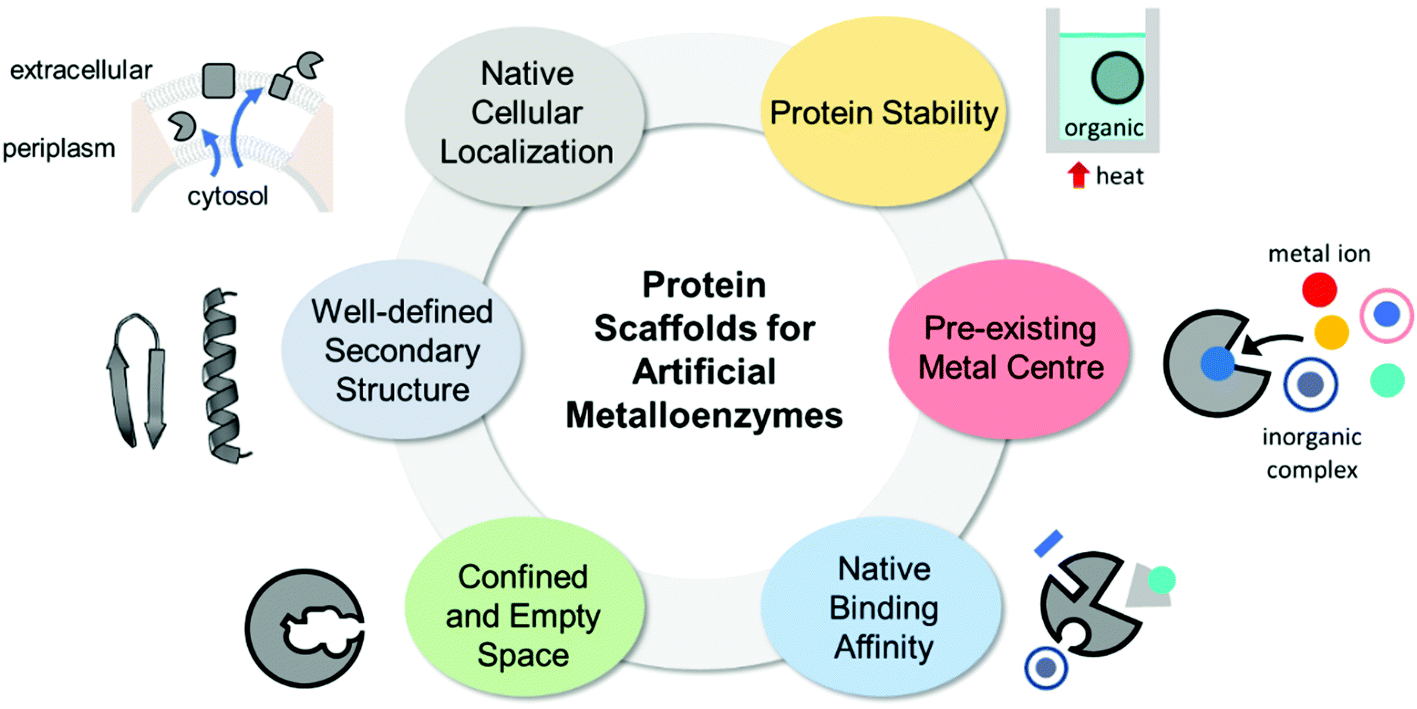 | ||
| Fig. 1 The desirable properties of proteins as the scaffolds for artificial metalloenzymes. | ||
2.1. Protein stability
Proteins are often considered as unstable, delicate, and unpredictable ligands relative to synthetic ligands, which is a valid concern for applications, where non-physiological temperature, pH, and organic solvents are often required.12 In addition, protein stability becomes even more essential in enzyme evolution because several rounds of mutation often lower protein stability by a trade-off for novel function.13,14 Fortunately, not every protein is unstable at nonbiological conditions (Table 1). Thermally stable and commercially available proteins, such as lysozymes15,16 and bovine serum albumin,17,18 have been adapted as feasible platforms on which to base catalytically active organometallic complexes such as a Grubbs catalyst (Fig. 2A) for ring-opening metathesis polymerisation19 or metal salts such as K2OsO2(OH)4, RuCl3, and Ti(OMe)4 for dihydroxylation and epoxidation activity.20 Similarly, lipase has been frequently used for the generation of artificial metalloenzymes.21,22 It is natively stable23,24 and commercially available so that covalent anchoring of a palladium pincer catalyst (Fig. 2B) resulted in catalytic activity for Heck cross-coupling.21 When immobilised on a sepharose support, lipase becomes durable even at high temperatures (70 °C) and in the presence of organic solvents (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF/H2O). Similarly, the integration of lipases with polymers yielded catalytic activity in the racemisation of (S)-1-phenylethylamine even at 55 °C.22 Another protein, mTFP1 from Clavularia sp., was also selected owing to its temperature and pH stability.25 The addition of a Cu-binding site to the protein resulted in Diels–Alderase and Friedel–Crafts alkylase activities.
:1 DMF/H2O). Similarly, the integration of lipases with polymers yielded catalytic activity in the racemisation of (S)-1-phenylethylamine even at 55 °C.22 Another protein, mTFP1 from Clavularia sp., was also selected owing to its temperature and pH stability.25 The addition of a Cu-binding site to the protein resulted in Diels–Alderase and Friedel–Crafts alkylase activities.
| Protein (described section) | Host | Thermal stability | pH stability | Solvent conditionb | ||
|---|---|---|---|---|---|---|
| Highest temperature (°C) | Methoda | pH range | Methoda | |||
| a CD, DSC, DSF, and FT-IR represent circular dichroism, differential scanning calorimetry, differential scanning fluorimetry, and Fourier-transform infrared spectroscopy respectively. b DCM, DMF, and DMSO indicate dichloromethane, N,N-dimethylformamide, and dimethyl sulfoxide, respectively. | ||||||
| Egg white lysozyme (2.1) | Gallus gallus | 7415 | CD | 3–1216 | FT-IR | |
| Bovine serum albumin (2.1) | Bos taurus | 6817 | DSC | >318 | CD | 100% DCM20 |
| Lipase, CAL-B (2.1, 2.3–2.4) | Candida antarctica | 54.823 | DSF | 5–924 | Activity | 100% DMF22 |
| mTFP* (2.1, 2.4.1) | Clavularia | 87–9025 | CD | 2.3–12.325 | CD | 60% DMSO25 |
| tHisF (2.1, 2.4.1) | Thermotoga maritima | 9526 | Far-UV CD | |||
| pVIII coat protein (2.1) | M13 bacteriophage | 7528 | CD | 98% DMSO28 | ||
| TM1459 (2.2) | Thermotoga maritima | 9229 | DSC | 2–1029 | CD | |
| Prolyl oligopeptidase (2.4.3) | Pyrococcus furiosus | 9530 | Activity | 4–931 | Activity | |
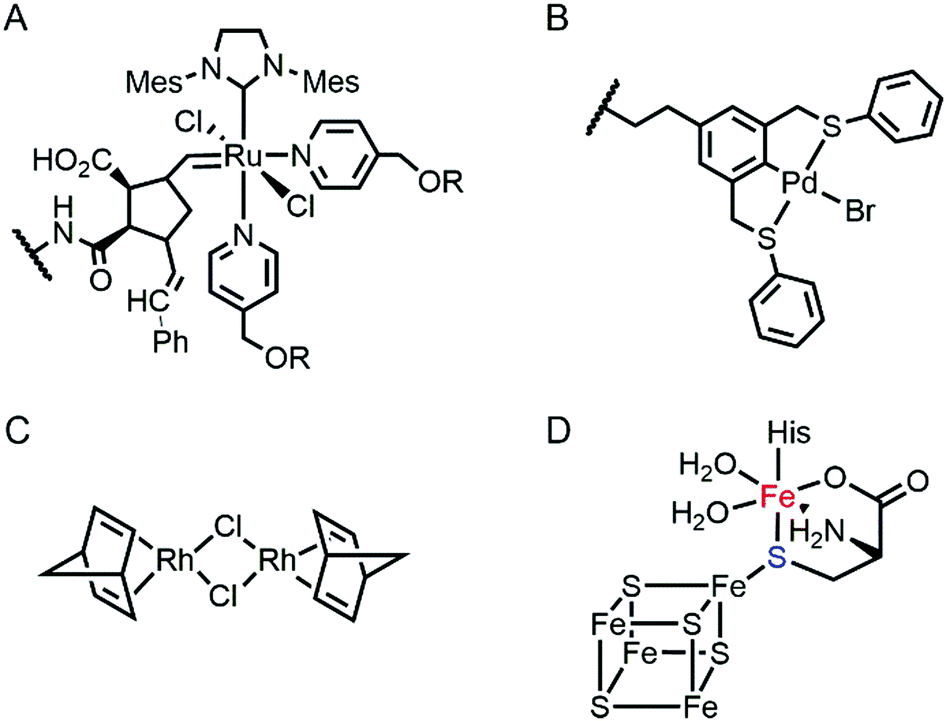 | ||
| Fig. 2 The structures of representative inorganic complexes described in Sections 2.1 and 2.2. (A) Grubbs catalyst for ring-opening metathesis polymerisation, where R stands for poly(ethylene glycol). (B) Pd pincer catalyst for Heck coupling. (C) Di-Rh dinorbonadiene complex for phenylacetylene polymerisation. (D) 5Fe–4S cluster in HydG in [FeFe]-hydrogenase. The exchangeable Fe and S atoms are coloured in red and blue, respectively. The wavy lines in (a) and (b) indicate the anchoring point on the proteins. | ||
The necessity for thermal stability has led to the discovery of novel proteins in thermophilic organisms or bacteriophages (Table 1). For example, tHisF protein26 from Thermotoga maritima was selected for asymmetric Diels–Alder reaction mediated by a mononuclear Cu-binding motif.27 In addition, an M13 bacteriophage pVIII coat protein was selected for Zn-dependent hydrolysis owing to its thermal stability, scalability, multi-valency, and ease of mutation.28 Therefore, these examples suggest that proteins with higher thermal stability are preferred to improve the feasibility of experiments and to expand the reaction to even harsh and non-physiological conditions.
For proteins for which stability have not been characterised, it would be useful to apply numerous in silico methods and simulation programmes.32 Applicable temperature ranges and alterations in melting temperature upon mutations can be predicted from protein sequence or structure, respectively.33–35
2.2. Pre-existing metal centres
One of the primary and long-standing methods to design novel metalloenzymes is to substitute inorganic elements, such as metal ions or metallocofactors, and/or to reshape the active site pockets. Then, natural metalloproteins that harbour pre-existing metal-active sites are the obvious starting materials for artificial metalloenzymes.This efficient approach is applicable when apo-proteins are somewhat stable even after pre-existing metal ion/cofactors are chelated prior to reconstitution with novel inorganic elements. At least moderate binding affinity for metal ions and cofactors other than the native ones is also necessary for the preparation of artificial metalloproteins. Such metal promiscuity might sound counter-intuitive to the general understanding of natural enzymes as being highly selective for the specific metal ion to perform dedicated biochemical functions. It is, however, more prevalent than expected,11,36–38 and protein promiscuity, in fact, can lead to the appearance and divergence of novel enzymes.39–41
To reconstitute metalloproteins, metal elements that favour similar inorganic environments to those of the native ones would be preferred, such as coordination number, geometry, and ligand properties (hard versus soft). The Metal-PDB database42 can be a valuable resource to search prevalent coordination geometry and ligands per each metal ion within proteinaceous environments. It is, however, worthy to note that proteins are flexible ligands, which may undergo multiple conformational changes and/or adapt alternative rotameric states depending on the neighbouring residues. Therefore, fine tailoring of proteins to construct novel metal-binding sites are still in great challenge.
Recent examples of replacing the native metal ions with novel ones have been carried out with structurally and spectroscopically well-characterised metalloenzymes (Fig. 3). A Cu in azurin (Fig. 3A)43 and Fe in rubredoxin (Fig. 3B)44 were replaced with Fe and Ni ion, yielding superoxide scavenging and hydrogenase activities, respectively. Notably, the Cu centre in azurin is a distorted trigonal bipyramidal geometry, which can be accommodated by Fe. Indeed, the Metal-PDB database indicates that Fe is the most prevalent metal element that replaces Cu-binding sites, followed by Zn ion. Similarly, the Ni-substituted rubredoxin was prepared presumably because such coordination geometry and the 4 cysteine ligands can be generated with Ni, as observed in [NiFe] hydrogenase.
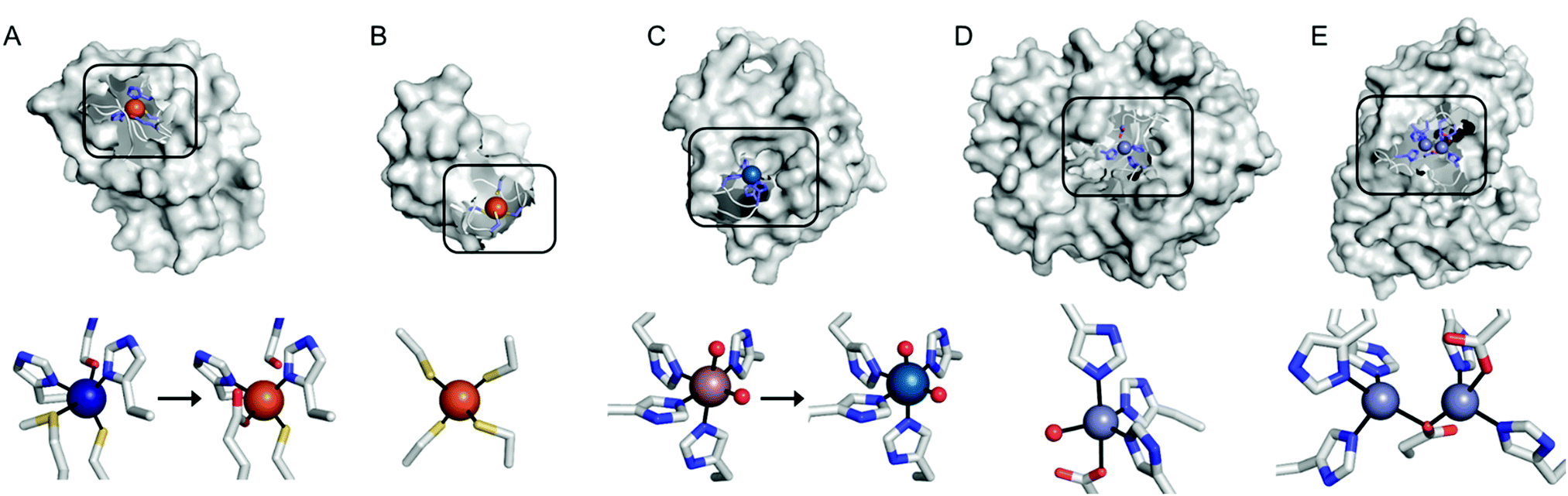 | ||
| Fig. 3 Transformation of pre-existing metal-binding sites into the active sites of artificial metalloenzymes. (A) Fe2+-Substituted azurin (4QLW) from Cu2+-azurin (4AZU). (B) The wild-type rubredoxin with a Fe3+-centre (6RXN). (C) TM1459 substituted with an Os3+ complex (5WSF) from the native form with Mn2+-bound form (1VJ2). (D) Zn2+-Dependent phosphatase PT3.1 (3T1G) derived from adenosine deaminase. (E) The wild-type glyoxalase II complexed with Zn2+ ions (PDB 1QH5). In (a) and (c), the native metal coordination sites before the metal-substitution are depicted bottom left. PDB codes are given in parentheses. Metal ions and metal-ligating amino acids are depicted with spheres and sticks, respectively. Cu, Fe, Mn, Os, and Zn atoms are coloured in navy, orange, purple, light blue, and light purple, respectively. Carbon, nitrogen, and oxygen atoms in metal-binding residues are coloured in grey, blue, and red, respectively. | ||
In addition, the structural resemblance between the native active sites and inorganic complexes can be adapted for repurposing metalloproteins. For example, a Mn-binding protein, TM1459 (Fig. 3C), was successfully reconstituted with an Os ion in its native metal centre.29 This work was initiated owing to the geometric resemblance of the active site to that of a previously characterised Os complex, Os-TPA (TPA = tris(2-pyridyl-methyl)amine). The chemo-mimetic enzyme exhibits peroxygenase activities with alkenes, resembling the catalytic activities of synthetic Os complexes. It is also worth noting that the enzyme activity assays were carried out at 70 °C, which was possible because of the native thermal stability of the protein (Table 1). This example suggests that protein scaffolds that satisfy more than one desirable property would increase the scope of biocatalysis.
Multinuclear metalloenzymes such as ferritin have also been adapted for artificial metalloenzymes. The native Fe ions were replaced with di-Rh norbornadiene complexes (Fig. 2C), resulting in biocatalysts that polymerize phenylacetylene.45 It is worthy to note that the successful transformation of ferritin have gained the benefit of using not only the pre-existing metal-coordination motifs but also well-defined cavities within the protein cage as a reaction vessel for substrate-binding and/or the formation and tuning of novel reactive intermediates.
In addition to the substitution of metal ions, native inorganic cofactors have been replaced to create artificial metalloenzymes (Fig. 4). Haem proteins are by far the most extensively and effectively repurposed metalloprotein scaffolds for biocatalysts.46–48 The native haem cofactors have been substituted with synthesised planar complexes such as Mn porphycene,49 Ru porphyrin,50 and Ir porphyrin complexes,51 catalysing C–H activation, N–H insertion of aniline, and enantio- and diastereoselective cyclopropanation of unactivated olefins, respectively (Fig. 4A). Notably, these transformations were carried out with a stable haem protein in the apo-state or with a protein from a thermophile, again, indicating that protein scaffolds that satisfy more than one of the listed criteria, such as protein stability, are beneficial to enzyme design.
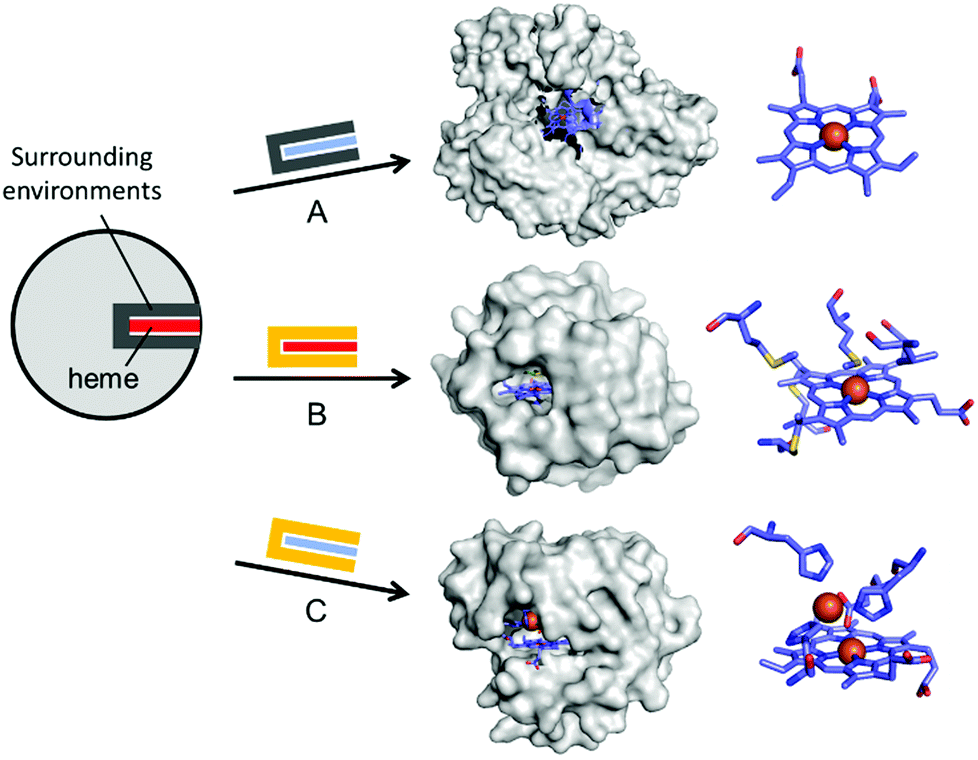 | ||
| Fig. 4 Representative examples in harnessing the native haem proteins for the design of artificial metalloenzymes. (A) Substitution of haem with synthetic planar complexes in thermophilic cytochrome P450 119 (1IO7). (B) Directed evolution of cytochrome c from Rhodothermus marinus (3CP5). (C) The alterations in both metal-active sites and the surrounding environments of myoglobin-based nitric oxide reductase, FeBMb (3M39). The native haem and synthetic analogues are depicted with red and blue rectangles, respectively. The native and altered active site pockets encapsulating the heme cofactors are depicted with grey and yellow claws, respectively. The reconstituted cofactors and the first metal-coordination sites are depicted with cyan sticks. PDB codes are given in parentheses. | ||
Recently, the substitution of more complicated and delicate metallocofactors has also been carried out by either tailoring the native multi-enzymatic biosynthetic machinery or chemically modifying the native cofactors. [FeFe]-hydrogenase, or HydA1, for example, innately harbours an H-cluster, composed of four CO, two CN− and a bridging dithiolate ligand between two Fe atoms.52 Because the metallocofactor is incorporated upon the action of a maturation enzyme, HydF, artificial hybrid hydrogenase HydA1 was prepared by pre-incubating a synthetic iron cluster analogues with HydF, followed by the addition of apo-HydA1. In addition, another enzyme associated with the bio-assembly of [FeFe]-hydrogenase, HydG, was created as an artificial metalloprotein having non-native Fe–S clusters.53 It natively contains a 4Fe–4S cluster with the fifth dangling Fe atom, resulting in a 5Fe–4S cluster (Fig. 2D). Upon chelation and metal exchange, the fifth metal ion was modified into Ni2+ and Co2+. Alternatively, the fifth metal-ligating S atom was exchanged into Se. The feasibility of these enzymatic and chemical alterations provide systematic tools to explore the inorganic properties of metalloenzymes and to develop tuneable biocatalysts.
Pre-existing metalloproteins can be harnessed by reshaping the active site pockets for modified substrates and/or reactivities, while the native inorganic elements are kept intact. For example, adenosine deaminase (Scheme 1A) was redesigned into organophosphate hydrolase (Scheme 1B) guided by computer-aided simulation and mutations (Fig. 3D).54 Alternatively, both metal elements and the surrounding protein environment can be optimized together, giving rise to even greater alterations in the catalytic reactivities. Glyoxalase II (Fig. 3E), for instance, natively hydrolyses the thiolester bond of S-D-lactoylglutathione (Scheme 1C).55 The active centre is composed of dinuclear metal-binding sites, where metal ion can be either Fe, Mn, or Zn ions, exhibiting some degrees of metal promiscuity. In addition, both glyoxalase II and β-lactamase have αβ/βα four-layer sandwich folds, implicating that they are evolutionarily related and novel β-lactamase can be created from glyoxalase II. With extensive loop engineering, insertion, deletion, and point mutations, glyoxalase II was transformed into an artificial β-lactamase (Scheme 1D), utilizing two Zn ions in the active site.
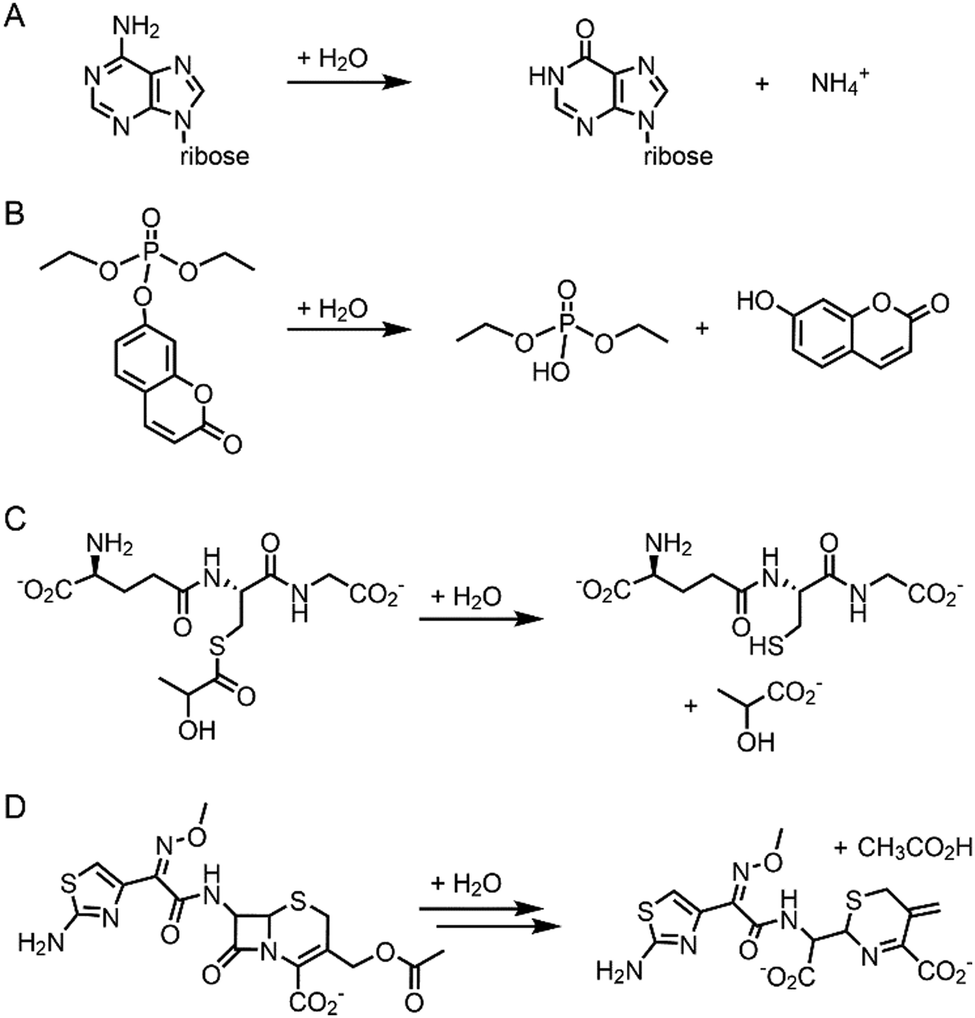 | ||
| Scheme 1 Chemical reactions catalysed by the native enzymes and the redesigned metalloenzymes. (A) Adenosine deaminase to (B) phosphatase (C) glyoxalase II to (D) β-lactamase. | ||
Haem proteins have also been iteratively mutated while the native cofactor is kept intact at the active sites. Arnold et al. have created a remarkable series of biocatalysts that exhibit catalytic activities in olefin cyclopropanation,56 carbon–silicon bond formation,57 chiral organoborane synthesis,58 and sp3 C–H functionalisation59via carbene transfer (Fig. 4B). In these examples, haem proteins have contributed greatly as scaffolds because the formation of a high-valent iron haem intermediate provides the molecular basis for abiological reactivities in the activation of various small molecules.
Rational modification of haem proteins in both metal centre and surrounding environments has also provided biocatalysts with novel activities. Lu et al. installed three histidine and one glutamate residues in myoglobin, or a [4Fe–4S] cluster in cytochrome c peroxidase, resulting in nitric oxide reductase60 and sulfite reductase,61 respectively (Fig. 4C). The authors also characterised the artificial nitric oxide reductase upon metal substitution to explore distinct inorganic reactivities of Cu versus Fe.62 This structure-based metalloprotein reconstruction allows the synthesis of novel biocatalysts as well as a systematic exploration of the nature and reactivity of metal ions in proteinaceous ligand sets.
2.3. Native binding affinity
Selective introduction of inorganic elements becomes the most critical step when a non-metalloprotein is employed as a starting material for the synthesis of artificial metalloenzymes. One successful approach is to harness biological host proteins with the native binding affinity to guest-like small molecules or ligands (Fig. 5). Ward et al. have employed streptavidin–biotin technology to incorporate inorganic catalysts at the desired position. The tight binding affinity of biotin to streptavidin was retained even upon structural modification of biotin and mutations in streptavidin, yielding a wide range of catalytic activities, such as transfer hydrogenation,63 Suzuki–Miyaura cross-coupling,64 and metathesis (Fig. 5A).65 The catalytic reactivities are derived from inorganic complexes such as Ir d6-piano stool complex (Fig. 6A), monophosphine Pd complex, and Hoveyda–Grubbs catalyst, respectively, which are selectively introduced by the covalent linkage to biotin, whereas the surrounding protein environment contributes to the determination of product distribution and turnover number. Notably, a monomeric version of streptavidin, originally a homotetramer, was created recently,66 diversifying the quaternary structure of the versatile protein scaffold for artificial metalloenzymes.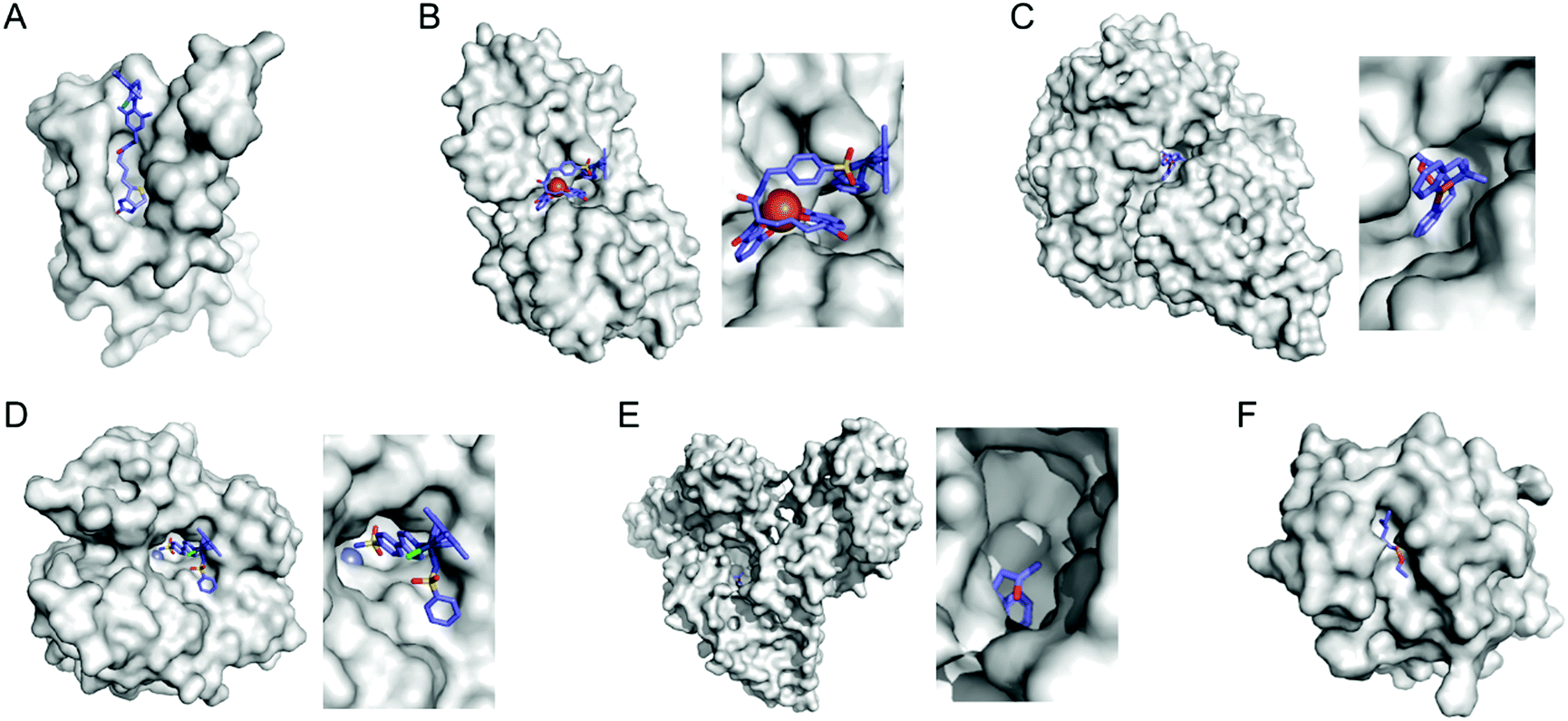 | ||
| Fig. 5 Representative proteins with the native binding properties. (A) Biotinylated Hoveyda–Grubbs catalyst bound in streptavidin (PDB 5IRA). (B) Periplasmic binding protein CeuE complexed with siderophore-Ir catalyst (5OD5). (C) An Fe complex bound inside nickel binding protein, NikA (4I9D). (D) α-Carbonic anhydrase complexed with an inhibitor-Ir catalyst (PDB 3ZP9). (E) A coumarin-derived molecule bound in human serum albumin (1H9Z). (F) The wild-type cutinase-inhibitor complex (1XZL). The active site pockets are enlarged for clarity. The small molecules that bind to the active site of the proteins are depicted with blue sticks. PDB codes are given in parentheses. | ||
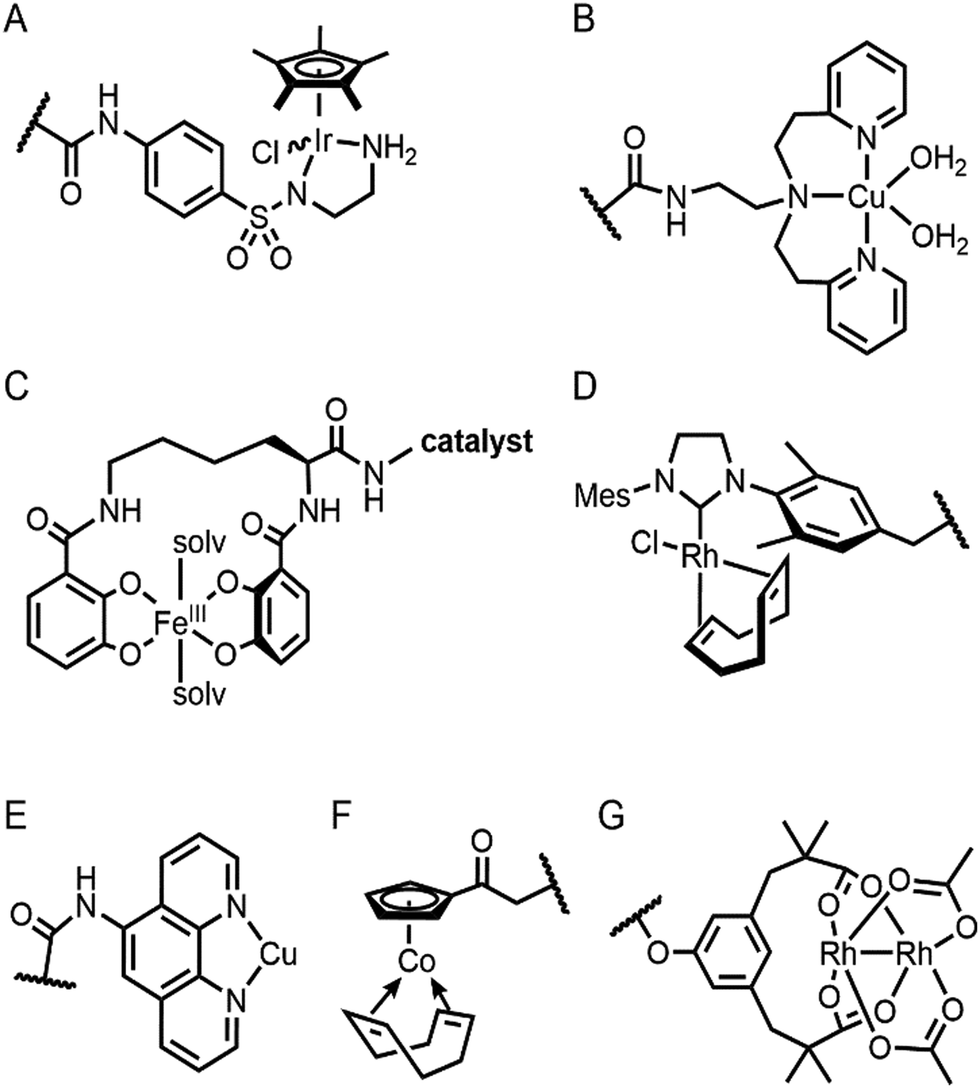 | ||
| Fig. 6 The structures of representative inorganic complexes described in Sections 2.3 and 2.4. (A) Ir d6-piano stool complex for transfer hydrogenation. (B) Cu(biot-et-dpea) complex, where biotin conjugated moiety is omitted for clarity. (C) Fe azotochelin linked to a catalyst of interest. The conjugated Ir catalyst is omitted for clarity. (D) Rh(NHC) phosphonate. (E) Cu(phen) complex. (F) Co half-sandwich complex. (G) Di-Rh complex. The wavy lines in (A), (B) and (D)–(G) indicate the anchoring point on the proteins. | ||
The streptavidin–biotin strategy enabled exploration of novel inorganic reactivity in proteinaceous environments. Borovik et al. recently introduced a mononuclear copper complex, Cu(biot-et-dpea), (Fig. 6B) into streptavidin.67 The chemical properties of the copper complex were previously investigated in non-aqueous solvents, and they are remarkably altered upon docking on the protein. In particular, hydrogen-bonding with adjacent residues significantly increase the stability of Cu-hydroperoxido species, indicating that proteins are evolvable platforms for metal-dependent biocatalysis.
Metal-trafficking proteins have also been exploited along these lines. These proteins exhibit high selectivity and binding affinity for dedicated metal ions or metal complexes, which can be readily utilized to introduce abiological inorganic elements into proteins. Siderophore is an innate chelator and transporter of Fe, exhibiting high selectivity for the ferric state over ferrous ion. As a result, Fe-coordination of azotochelin, a siderophore from Azotobacter vinelandii (Fig. 6C), was linked to a catalyst of interest, such as Ir imine reduction catalyst (Fig. 6A), resulting in selective association and dissociation upon oxidation and reduction of Fe, respectively.68 In addition, when an Ir catalyst was attached to siderophore, periplasmic binding proteins (PBPs) were transformed into artificial hydrogenase (Fig. 5B). Analogously, a nickel transporter NikA was also utilized as a scaffold to dock a Fe complex such as Fe(EDTA)(H2O)− for catalysis (Fig. 5C).69 The NikA–Fe hybrid protein functions as an artificial oxygenase for sulfide oxidation.70 Again, similarly to streptavidin, these examples indicate that catalytic activities are derived from metal complexes, when the secondary coordination environments, and consequently, the enzymatic activities, are tuned by protein matrix.
Proteins with inhibitors and strongly-binding ligands have also been utilized as biological hosts. Aryl-sulphonamide, for example, is an inhibitor of carbonic anhydrase (Fig. 5D).71 Human serum albumin is a carrier protein, exhibiting well-characterised interactions with coumarin-derivatives (Fig. 5E).72 After covalent linking of an IrCp* piano-stool complex or Hoveyda–Grubbs Ru catalyst to an inhibitor and a hydrophobic coumarin ligand, respectively, they became metalloenzymes with asymmetric transfer hydrogenation and ring-closing metathesis activities, respectively.
For serine or cysteine-dependent hydrolases, the catalytically active serine residues can function as nucleophiles to be covalently linked to abiological inorganic elements so that catalytic units can be steered to the native active site pockets. Various organometallic catalysts, such as Hoveyda–Grubbs catalyst, Ir d6-piano stool complex, Rh(NHC) phosphonate (Fig. 6D), and pincer-type Pt d8 complex have been attached to hydrolases such as α-chymotrypsin,73 papain,74 lipase,75 and cutinase (Fig. 5F),76 respectively. Similarly, a serine hydrolase with two catalytic triads was converted into an artificial enzyme for oxidation and Friedel–Crafts alkylation.77 One of its catalytic triads was conjugated with a Cu(bpy) complex, resulting in an artificial metalloenzyme with coupled catalytic activities.
In some cases, binding affinity to introduce external inorganic components to novel environments can be derived from the native protein–protein interaction. Douglas et al. have created artificial metalloenzymes by encapsulating [NiFe]-hydrogenase within the capsid of bacteriophage P22.78 The catalytic domain, [NiFe]-hydrogenase, was fused to a protein that exhibits protein–protein interaction with a coat protein. The self-assembled artificial metalloenzyme exhibits enhanced catalytic activity and stability against protease, thermal denaturation, and air exposure, indicating that a combination of two proteins can also give rise to artificial metalloenzymes.
2.4. Confined and empty spaces in proteins
Enzyme catalysis, in general, occurs in structurally well-defined active site pockets. In such confined spaces, multiple amino acids encapsulate catalytic units, producing orchestrated multiple steps associated with catalysis, such as substrate-binding, product-release, and protection from adventitious reactions. Substitution of inorganic elements with novel ones in pre-existing metalloproteins (described in Section 2.2), therefore, takes advantage of both the binding sites for catalytic units and the well-defined space. Some proteins natively possess these empty spaces, which can be adapted for artificial metalloenzyme catalysis.48 Once a protein with a potential cavity for catalysis is selected, the next step is to embed a catalytically active unit in the desired position of the void space. The examples below have employed either cysteine by chemical modification or unnatural amino acids by acyl-tRNA/t-RNA synthase pairs.79,80Adipocyte lipid binding protein (ALBP), for example, was selected because of its hydrophobic active sites to attach a Cu(phen) complex (Fig. 6E) for enantioselective ester hydrolysis (Fig. 7A).81 Sterol carrier protein type 2 (SCP-2L) also exhibits a cylindrically shaped hydrophobic cavity (Fig. 7B), providing a potential environment for biocatalysis. Bioconjugation of nitrogen-donor ligands to a cysteine inside of the cavity, followed by the addition of Cu or Fe ions, leads to the formation of biocatalysts for asymmetric Diels–Alder82 and oxidative lignin degradation,83 respectively. In addition, two thermally stable proteins described in Section 2.1 (Table 1), tHisF27 (Fig. 7C) and mTFP*25 (Fig. 7D) possess confined spaces within a TIM and a β-barrel structure, respectively. As a result, the concave regions could be beneficially transformed into the active site of artificial Cu enzymes by the installation of 2His/1carboxylate motif.25,27
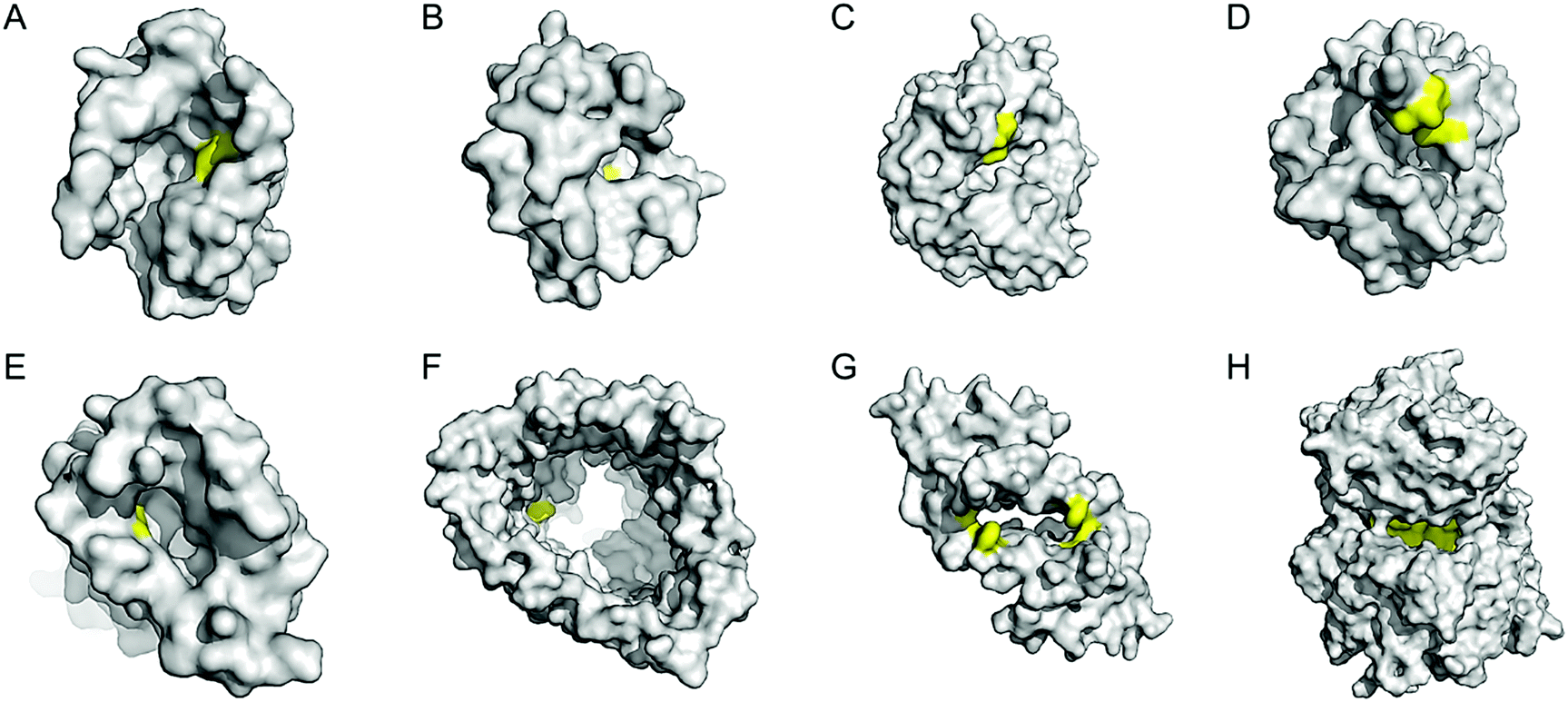 | ||
| Fig. 7 Representative proteins with the native confined and empty spaces. (A) Adipocyte lipid binding protein (1LIB). (B) Sterol carrier protein 2L (1IKT). (C) Glycerol phosphate synthase tHisF from Thermotoga maritima (1THF). (D) Monomeric cyan fluorescent protein mutant mTFP* from Clavularia sp. (6QSL). (E) Nitrobindin (3EMM). (F) Ferrichrome outer membrane transporter FhuA (1BY3). (G) Multidrug transcriptional regulator LmrR from Lactococcus lactis (3F8B). (H) Prolyl oligopeptidase from Pyrococcus furiosus (5T88). The cavities adapted for novel biocatalysis are coloured in yellow in surface representation. PDB codes are given in parentheses. | ||
Chaperonin has also been exploited as a protein nanoreactor owing to its shape: a hexadecameric protein with both open and closed conformations. By covalently linking a Cu-catalyst for atom-transfer radical polymerisation, the concurrent metalloenzyme yielded lower polydispersity indices than the catalyst alone.84 Analogously, various cage proteins have been adapted as versatile platforms for catalysis because of their compartmentalized internal space. For example, ferritin is used to encapsulate artificial metalloenzymes composed of streptavidin–biotin complexes.85 Significant improvements in the turnover of artificial transfer hydrogenases within ferritin were detected, indicating that proteins with a defined cavity provide a ternary coordination sphere for catalysis. Similarly, a capsid of bacteriophage P22 was also applied as a protein-reactor78 as described above.
Barrel-shaped proteins have also been used to generate artificial metalloenzymes because of their large void spaces. Nitrobindin (Fig. 7E), for example, is adapted for polymerisation, hydrogen evolution, and metathesis by anchoring an Rh-catalyst,86 a diiron complex,87 and a Hoveyda–Grubbs catalysts88 inside of the void space, respectively. The hybrid proteins produce either more selective products and/or higher efficiency owing to the unique environments. Similarly, another β-barrel protein, FhuA (Fig. 7F), was also transformed into artificial metalloenzymes by covalently conjugated to either a Hoveyda–Grubbs catalyst,89 Cu(NHC) complex,90 or cobalt half-sandwich complex (Fig. 6F).91 As a result, the inherently transmembrane protein that mediates Fe uptake now exhibits catalytic activities in metathesis, Diels–Alder, and cyclotrimerisation of phenylacetylene, respectively. Notably, FhuA is one of the few membrane proteins used for artificial metalloenzyme design, expanding the spectrum of protein scaffolds and reaction conditions for biocatalysis.
2.5. Well-defined secondary structures
Instead of using abiological catalysts, metal-binding sites can be directly created in a de novo approach. By appropriately placing metal-coordinating amino acids, such as glutamate, aspartate, histidine, and cysteine, on non-metalloprotein scaffolds, artificial metalloproteins can be generated. This bottom-up approach presumably resembles how natural metalloproteins might have emerged by utilizing natural metal elements and canonical metal-ligating amino acids. Although this approach can be applied for essentially any protein, a challenge arises owing to our incomplete understanding of protein sequence-structure–function relationship and protein–metal binding interactions. Therefore, de novo metal-binding sites have been synthesized with relatively small and well-characterised proteins, primarily in their secondary structures. Notably, self-assembled protein oligomers comprising of α-helixes and β-sheets have been employed because they form a crevice or pocket-shaped architecture, which is suitable for catalysis, as described in Section 2.4.Pecoraro et al. synthesized homo and hetero-α-helical trimers as artificial metalloenzymes (Fig. 8A).99 The peptides are templated by binding Hg2+, Cd2+, or Pb2+ where other metal ions such as Zn and Cu play catalytic roles, such as in CO2 hydration/ester hydrolysis100,101 and nitrite reduction,102,103 respectively. Alternatively, Fe2+ can be incorporated into the α-helical structure, resulting in a rubredoxin-like protein, facilitating electron transfer.104 Similarly, Tanaka et al. also have constructed an artificial dinuclear Cu-binding site by placing two histidine and two cysteine residues into a four-α-helical coiled-coil, yielding two copper atoms bridged with two cysteines (Fig. 8B).105 Spectroscopic characterisation suggests that the artificial metalloenzyme is a structural and functional mimic of cytochrome c oxidase.
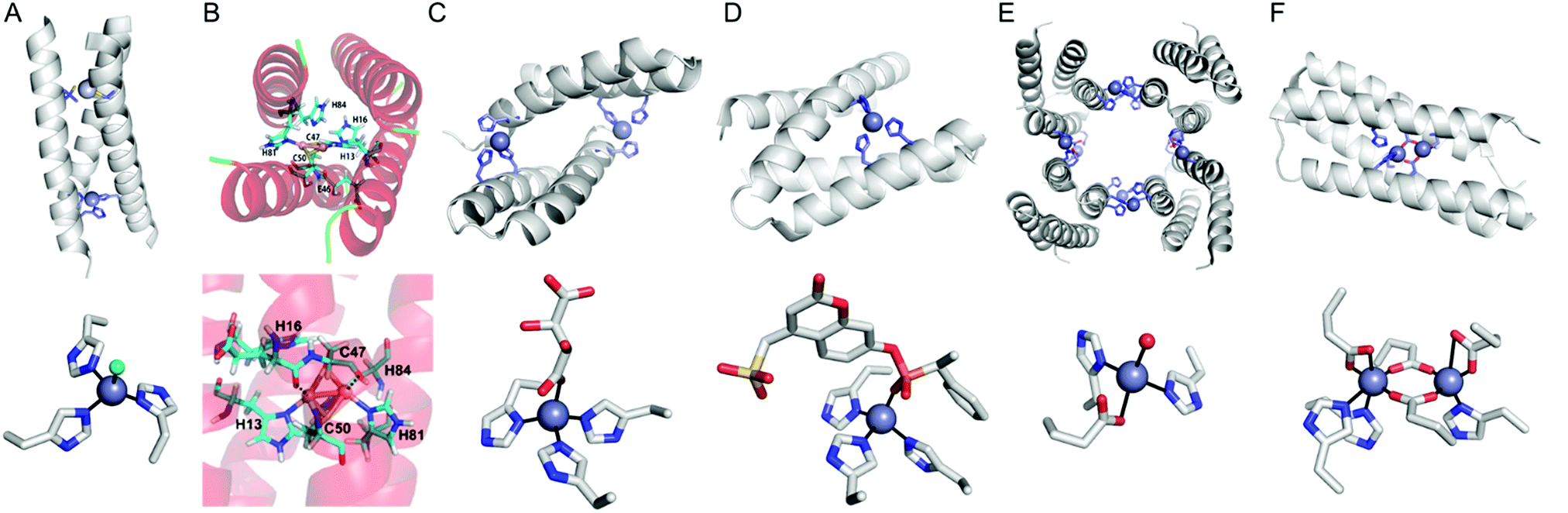 | ||
| Fig. 8 Representative structures of de novo metallo-α-helical peptides and proteins (A) Zn-dependent metallo-esterase (3PBJ). (B) A structural model of the dicopper-binding protein. Reprinted with permission from ref. 105. Copyright (2012) American Chemical Society. (C) Zn-Dependent hydrolase MID1-Zn (3V1C). (D) Zn-Dependent hydrolase MID1sc10 (5OD1). (E) Zn-Dependent β-lactamase (4U9E). (F) Diiron artificial metalloenzyme 3His-G2DFsc (2LFD). PDB codes are given in parentheses. Metal atoms and metal-ligating residues are shown with navy spheres and sticks, respectively. In (C) and (D), co-crystallized metal-binding molecules are also shown with sticks. The metal-bound chloride and water-derived molecule are coloured in green and red, respectively. | ||
In addition, α-helical peptides have been versatile platforms to anchor a dirhodium centre. Ball et al. created artificial metallopeptides with catalytic activity in enantioselective carbenoid insertion into Si–H bonds106 and asymmetric cyclopropanation.107 Kuhlman et al. have also designed metal-binding histidine residues on α-helical peptides, resulting in dimeric metalloproteins with crevice-like architectures (Fig. 8C).108 These peptides were further engineered and evolved into a globular metallo-esterase with high enantiospecificity and catalytic efficiency (Fig. 8D).109
Four α-helical bundle proteins have also been extensively employed for the design of artificial metalloenzymes. Dutton et al. have created maquettes that bind haem for O2 transport110 or exhibit oxidoreductase activity with haem, flavins, Fe–S clusters, Zn porphyrins, and Zn chlorines.111 The proteins were further engineered into artificial oxidases that react with a diverse array of substrates using hydrogen peroxide as an oxidant.112
Tezcan et al. employed a four α-helical bundle protein, cytochrome cb562, as a monomer to generate self-assembled tetramers as a 16 α-helical bundle protein.113,114 Rational and in silico redesign produced a tetrameric metalloprotein (Fig. 8E), creating protein–protein interfaces for the sites for metal-dependent hydrolysis even inside E. coli cells.115 The α-helical protein scaffold is so versatile that various proteins that bind a wide array of first-row transition metal ions can be effectively generated,116 and the diversion of evolutionary paths differed by local flexibility can be also explored.117
DeGrado and Lombardi et al. have designed a de novo Due Ferri protein (Fig. 8F),118,119 where the four α-helix bundles provide a dinuclear Fe-coordinating site for selective N-hydroxylation of arylamines118 and artificial phenol oxidase activity.119 Alternatively, the four α-helix bundle generated the sites for tetra-nuclear clusters120 and for highly electron-deficient non-natural zinc porphyrin, the latter of which was highly stable even at 100 °C.121
Other than soluble α-helical proteins, DeGrado et al. reported the de novo design of a functional four α-helix transmembrane protein.122 Despite great challenges in the design of transmembrane proteins,123,124 the group has designed two stable asymmetric states with a dynamic interface to transport selective metal ions, such as Zn2+ and Co2+ but not Ca2+, across membranes. Although the protein exhibits no catalytic activity, this work suggests that the scope of α-helical scaffolds can be extended to membrane proteins. In addition, the sequence, structure, and function of α-helical proteins can be further expanded as exemplified with a combinatory library of α-helical proteins and their emergence of peroxidase activity.125 Therefore, α-helical proteins are likely to contribute more versatile scaffolds for de novo artificial metalloenzymes in the future.
One of the challenges associated with β-sheet proteins is their propensity to aggregate and form fibril structures. Although such structural features can give rise to more heterogeneous and poorly defined states for catalysis, they can be catalytically active with exogenous substrates. For example, a seven-amino acid-long β-sheet peptide was converted into an artificial enzyme upon the addition of Zn and Cu, hydrolysing p-nitrophenyl acetate129 and paraoxon,130 respectively. The catalytic activity was measured in amyloid-like fibril structures, providing kinetic advantages in high local concentrations, as often shown in heterogeneous catalysts. Notably, analogous amyloid-like structures and hydrolase activity were observed with a Zn-coordinated phenylalanine amino acid, with no assistance of any peptide or protein.131 These data indicate that a heterogeneous biomaterial may generate catalytic environments, similar to the function of a protein matrix.
In addition, intermolecular space upon self-assembly of β-sheet proteins can be generated for catalysis, similar to supramolecular structures of α-helical proteins. A computationally redesigned β-propeller protein (Pizza6) demonstrated that a β-structural protein can produce 19-atom nanocrystals within the protein–protein interface.132 Although this work was carried out for biomineralisation rather than for catalysis, the successful positioning of metal centres in a concave pocket suggests that artificial metalloenzymes can be created in β-structural proteins.
2.6. Native cellular location of proteins
The accessibility of synthetic complexes and non-native substrates becomes essential for the in vivo activity of artificial metalloenzymes. Then, unnatural components should bypass the cell membrane to bind the target proteins. In addition, potential interference with other proteins, metabolites such as glutathione, and salts must also be overcome. To this end, reaction conditions can be implemented to resolve the issue.133,134 A native leader sequence has been also applied to several non-periplasmic proteins for translocation,65,115 including streptavidin and cytochrome b562, described in Sections 2.3 and 2.5.1, respectively. As a result, they were placed in the periplasm, where abiological catalysts, substrates, and metal ions at non-physiological concentrations can be more readily reached than in the cytosol.In addition, a protein of interest can be fused to a membrane protein such as a truncated lipoprotein and an outer membrane protein, resulting in the positioning of the artificial metalloenzyme on the cell surface (Fig. 9A).135 Alternatively, membrane protein by itself can be directly converted to artificial metalloenzymes. An A2A adenosine receptor is natively embedded in the cytoplasmic membranes of human embryonic kidney cells (Fig. 9B),136 displaying strong association with its antagonists. When a Cu catalyst covalently anchored to the antagonist was introduced, the A2A adenosine receptor became an artificial Diels–Alderase that is reactive with abiological substrates such as cyclopentadiene and 2-azachalcone.
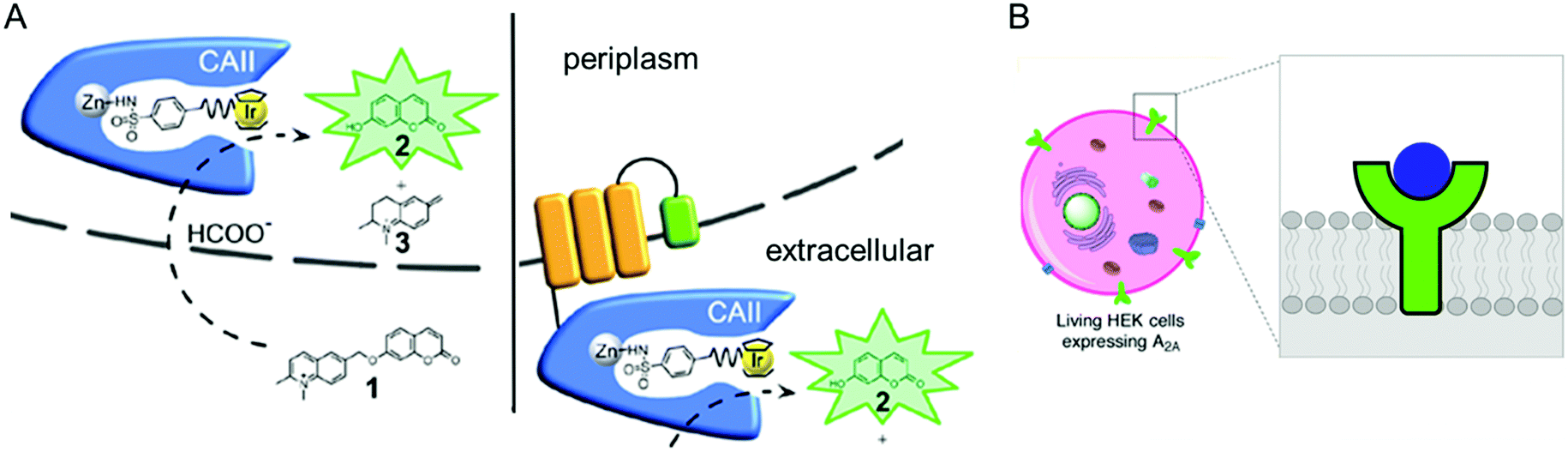 | ||
| Fig. 9 Representative examples of using native cellular localisation. (A) Signal peptide-induced periplasmic secretion (left) and E. coli cell-surface display (right) of human carbonic anhydrase II. Reprinted with permission from ref. 135. Copyright (2019) American Chemical Society. (B) HEK cell with a Cu complex bound A2A receptor. Reprinted with permission from ref. 136. Copyright (2018) American Chemical Society. | ||
3. Additional considerations in the selection of protein platforms
3.1. Native geometric parameters
An alternative promising approach for the design of artificial metalloenzymes would be to search for non-metalloproteins that possess a pre-arranged set of residues for metal coordination. Ward et al. have carried out data mining on facial triad motifs, which is one of the most extensively investigated mononuclear metal-binding sites,137,138 comprised of two histidine and one-carboxylate residues in facial position of octahedral geometry.139 From the motif search on the Protein Data Bank (PDB), they discovered 121 viable triads, implicating that they can be amendable for metal coordination and, possibly, novel metal-dependent catalysis.Lai and He et al. have also applied computational screening to identify proteins with a set of residues that potentially bind uranyl (UO22+) upon mutation.140 Because uranyl ion prefers pentagonal or hexagonal bipyramidal geometry upon coordination to five to six ligating oxygen atoms at the equatorial positions, protein structures were examined to find the sets of amino acids that can be mutated to two or three carboxylate in a plane to accommodate uranyl-binding. The protein was further engineered to a novel protein with femtomolar affinity for uranyl ions and greater than 104-fold selectivity over other metal ions.
In addition, protein symmetry can simplify the design of de novo metalloenzymes. When a metal-binding active site is created at the protein–protein interface, symmetry operation multiplies the metal-ligating residues, reducing the number of residues to design. Kerfeld et al., for example, engineered bacterial microcompartment shell protein (BMC) to create a 4Fe–4S cluster site that mediates multi-electron transfer.141 Site-directed mutagenesis of a residue at the site of three-fold symmetry leads to the formation of three equivalent cysteine residues for Fe-binding. The [4Fe–4S] clusters mimic the coordination environment of aconitase and radical S-adenosylmethionine (SAM) enzyme, whereas its redox properties are analogous to those of bacterial ferredoxins.
As protein structural databases expand and search algorithms become more accurate and finely detailed, geometric factors for metal coordination will become more important in the de novo design of metalloenzymes. Presumably, the orchestration with adjacent residues and/or dynamic motion during catalysis can be also simulated for artificial metalloenzymes, if the search target is beyond the first coordination sphere.
3.2. Native functions
If the native function of protein scaffolds, such as substrate binding affinity, electron/proton transfer pathway, and hydrogen-bonding network, can be adapted for the aimed novel catalytic activity of artificial metalloenzymes, the protein would be a privileged starting point. Although this sounds obvious, such properties related in enzyme catalysis have been rarely considered in the designing process. Presumably, this is because most protein platforms have been limited by the methods to introduce inorganic elements into the non-metalloproteins. De novo design of metal-binding sites, however, relieves the restrictions associated with pre-existing inorganic elements or the binding affinity to small molecules. One related example is the transformation of ferredoxin into artificial hydrogenase by anchoring [Ru(bpy)3]2+ and cobaloxime catalyst.142 The authors utilized the native electron transfer protein as a scaffold to mediate electron relay between the installed abiological elements. The protein scaffold presumably contributed to the biocatalyst by facilitating electron transfer in a thermodynamically and kinetically favourable manner.3.3. Protein dynamics and evolvability
Knowing how elegant and sophisticated natural enzymes are operative when achieving dedicated chemical transformation, steps other than the introduction of catalytically active inorganic elements may be necessary for the design of novel and efficient biocatalysts. One of the factors that are rarely considered is protein dynamics. A series of conformational changes upon substrate binding and consumption of co-substrates are well documented in numerous examples,143,144 implying that multi-conformations may be beneficial in the design of artificial metalloenzymes. Protein evolvability would be another factor to consider because it is a measure of adaptability for novel structure and functions.145 Although it is difficult to predict and quantitatively determine the degrees of evolvability, recent sequence analysis and prediction indicate that ancestor genes can be reconstructed and be efficiently diverged to exhibit novel catalytic activities,146,147 implying that evolvable protein scaffolds can be considered for the design of artificial metalloenzymes.3.4. Membrane proteins
Membrane proteins account for approximately a third of genomes and proteomes in many organisms ranging from Gram-negative bacteria to Homo sapiens.148,149 In addition to their natural abundance and diversity, numerous membrane proteins are responsible for essential chemical transformations and biochemical processes, including trafficking, signalling, and transport. As mentioned in Section 2.6, the cellular localisation of enzymes becomes important in the accessibility of abiological components for catalysis. Therefore, membrane proteins are attractive platforms for the generation of artificial metalloenzymes. Despite their natural abundance, diversity, and potential, very few membrane proteins have been utilized for enzyme design.It is presumably ascribed to the difficulties in the preparation and handling of membrane proteins. Heterologous expression of membrane proteins often leads to the formation of inclusion bodies, which requires extraction and purification of aggregated proteins using detergents. The additional steps are non-trivial and laborious, as reflected in the RCSB database, where only 1–3% datasets account for the structures of membrane proteins. Indeed, among the numerous examples discussed in the present review, only three are membrane proteins, a β-barrel protein, FhuA,89–91 a de novo designed α-helical coiled-coil,122 and an A2A adenosine receptor protein.136 Membrane proteins, however, would be prospective protein scaffolds because more atomic structural information has become available owing to tremendous advances and applications in the preparation of membrane proteins and determination of three-dimensional structures by cryo-electron microscopy. The availability of structural information, in particular, would link the sequence and molecular function of membrane proteins, allowing us to inspect more diverse scaffolds for artificial metalloenzymes. In addition, as we further explore the folding landscape and sequence-structure–function relationship of membrane proteins, it is expected that we will be able to utilize a diverse range of these proteins for the design of artificial metalloenzymes.
4. Conclusions
In the present review, we have highlighted recent studies on the design of artificial metalloenzymes. By extracting and categorising the essential and beneficial properties of proteins that are adapted as the scaffolds for metal-mediated biocatalysis, we compiled six underlying desirable factors that should be considered when choosing new ones. Because immense protein sequence and structural databases are being updated daily, including tailor-made proteins,150 the scope of targeting reactions, inorganic elements, and catalytic conditions is expected to increase dramatically in the near future. In addition, computer-aided metalloenzyme design, including protein–metal interaction, protein–substrate docking, energy calculation of metal-derived intermediates, would provide accurate and in-depth insights on how protein and metal cooperate in enzyme catalysis.151,152 The synergistic combination of chemo-synthetic and biochemical elements for the design of artificial metalloenzymes will expand the chemical repertoire of metal-mediated enzyme catalysis, aiming for high catalytic efficiency, selectivity, tunability, and stability. Simultaneously, it will provide a framework for the exploration of protein sequence–structure–function relationship, enzyme evolution, and inorganic reactivity in proteinaceous environments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Creative-pioneering researchers programme from Seoul National University (SNU) and National Research Foundation from Korea government (NRF-2019R1C1C1003863).References
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, 1994 Search PubMed.
- H. B. Gray, E. I. Stiefel, J. S. Valentine and I. Bertini, Biological Inorganic Chemistry: Structure and Reactivity, University Science Book, 2006 Search PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- V. Nanda and R. L. Koder, Nat. Chem., 2010, 2, 15–24 CrossRef CAS PubMed.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef CAS PubMed.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- J. C. Lewis, ACS Catal., 2013, 3, 2954–2975 CrossRef CAS.
- T. K. Hyster and T. R. Ward, Angew. Chem., Int. Ed., 2016, 55, 7344–7357 CrossRef CAS PubMed.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS PubMed.
- Y.-W. Lin, Coord. Chem. Rev., 2017, 336, 1–27 CrossRef CAS.
- G. D. Haki and S. K. Rakshit, Bioresour. Technol., 2003, 89, 17–34 CrossRef CAS PubMed.
- J. D. Bloom, S. T. Labthavikul, C. R. Otey and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5869–5874 CrossRef CAS PubMed.
- N. Tokuriki, F. Stricher, L. Serrano and D. S. Tawfik, PLoS Comput. Biol., 2008, 4, e1000002 CrossRef PubMed.
- P. Shih, D. R. Holland and J. F. Kirsch, Protein Sci., 1995, 4, 2050–2062 CrossRef CAS PubMed.
- S. Venkataramani, J. Truntzer and D. R. Coleman, J. Pharm. BioAllied Sci., 2013, 5, 148–153 CrossRef PubMed.
- V. A. Borzova, K. A. Markossian, N. A. Chebotareva, S. Y. Kleymenov, N. B. Poliansky, K. O. Muranov, V. A. Stein-Margolina, V. V. Shubin, D. I. Markov and B. I. Kurganov, PLoS One, 2016, 11, e0153495 CrossRef PubMed.
- R. Li, Z. Wu, Y. Wangb, L. Ding and Y. Wang, Biotechnol. Rep., 2016, 9, 46–52 CrossRef PubMed.
- S. A. Isarov and J. K. Pokorski, ACS Macro Lett., 2015, 4, 969–973 CrossRef CAS.
- M. Leurs, B. Dorn, S. Wilhelm, M. Manisegaran and J. C. Tiller, Chem. – Eur. J., 2018, 24, 10859–10867 CrossRef CAS PubMed.
- M. Filice, O. Romero, A. Aires, J. M. Guisan, A. Rumbero and J. M. Palomo, Adv. Synth. Catal., 2015, 357, 2687–2696 CrossRef CAS.
- X. Li, Y. Cao, K. Luo, Y. Sun, J. Xiong, L. Wang, Z. Liu, J. Li, J. Ma, J. Ge, H. Xiao and R. N. Zare, Nat. Catal., 2019, 2, 718–725 CrossRef CAS.
- Q. A. T. Le, J. C. Joo, Y. J. Yoo and Y. H. Kim, Biotechnol. Bioeng., 2012, 109, 867–876 CAS.
- J. Zdarta, M. Wysokowski, M. Norman, A. Kołodziejczak-Radzimska, D. Moszyński, H. Maciejewski, H. Ehrlich and T. Jesionowski, Int. J. Mol. Sci., 2016, 17, 1581 Search PubMed.
- J. Fischer, D. Renn, F. Quitterer, A. Radhakrishnan, M. Liu, A. Makki, S. Ghorpade, M. Rueping, S. T. Arold, M. Groll and J. Eppinger, ACS Catal., 2019, 9, 11371–11380 CrossRef CAS.
- L. Carstensen, G. Zoldák, F.-X. Schmid and R. Sterner, Biochemistry, 2012, 51, 3420–3432 CrossRef CAS PubMed.
- J. Podtetenieff, A. Taglieber, E. Bill, E. J. Reijerse and M. T. Reetz, Angew. Chem. Int. Ed., 2010, 49, 5151–5155 CrossRef CAS PubMed.
- J. P. Casey Jr, R. J. Barbero, N. Heldman and A. M. Belcher, J. Am. Chem. Soc., 2014, 136, 16508–16514 CrossRef PubMed.
- N. Fujieda, T. Nakano, Y. Taniguchi, H. Ichihashi, H. Sugimoto, Y. Morimoto, Y. Nishikawa, G. Kurisu and S. Itoh, J. Am. Chem. Soc., 2017, 139, 5149–5155 CrossRef CAS PubMed.
- V. J. Harwood, J. D. Denson, K. A. Robinson-Bidle and H. J. Schreier, J. Bacteriol., 1997, 179, 3613–3618 CrossRef CAS PubMed.
- M. N. Harris, J. D. Madura, L. J. Ming and V. J. Harwood, J. Biol. Chem., 2001, 276, 19310–19317 CrossRef CAS PubMed.
- R. Kazlauskas, Chem. Soc. Rev., 2018, 47, 9026–9045 CAS.
- C. Savojardo, P. Fariselli, P. L. Martelli and R. Casadio, Bioinformatics, 2016, 32, 2542–2544 CrossRef CAS PubMed.
- T. Ku, P. Lu, C. Chan, T. Wang, S. Lai, P. Lyu and N. Hsiao, Comput. Biol. Chem., 2009, 33, 445–450 CrossRef CAS PubMed.
- F. Pucci, R. Bourgeas and M. Rooman, Sci. Rep., 2016, 6, 23257 CrossRef CAS PubMed.
- C. E. Valdez, Q. A. Smith, M. R. Nechay and A. N. Alexandrova, Acc. Chem. Res., 2014, 47, 3110–3117 CrossRef CAS PubMed.
- H. Eom and W. J. Song, J. Biol. Inorg. Chem., 2019, 24, 517–531 CrossRef CAS PubMed.
- A. Pordea, Curr. Opin. Chem. Biol., 2015, 25, 124–132 CrossRef CAS PubMed.
- P. J. O'Brien and D. Herschlag, Chem. Biol., 1999, 6, R91–R105 CrossRef.
- I. Nobeli, A. D. Favia and J. M. Thornton, Nat. Biotechnol., 2009, 27, 157–167 CrossRef CAS PubMed.
- O. Khersonsky and D. S. Tawfik, Annu. Rev. Biochem., 2010, 79, 471–505 CrossRef CAS PubMed.
- V. Putignano, A. Rosato, L. Banci and C. Andreini, Nucleic Acids Res., 2018, 46, D459–d464 CrossRef CAS PubMed.
- J. Liu, K. K. Meier, S. Tian, J. L. Zhang, H. Guo, C. E. Schulz, H. Robinson, M. J. Nilges, E. Münck and Y. Lu, J. Am. Chem. Soc., 2014, 136, 12337–12344 CrossRef CAS.
- J. W. Slater and H. S. Shafaat, J. Phys. Chem. Lett., 2015, 6, 3731–3736 CAS.
- S. Abe, K. Hirata, T. Ueno, K. Morino, N. Shimizu, M. Yamamoto, M. Takata, E. Yashima and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 6958–6960 CrossRef CAS PubMed.
- L. Fruk, C.-H. Kuo, E. Torres and C. M. Niemeyer, Angew. Chem. Int. Ed., 2009, 48, 1550–1574 CrossRef CAS PubMed.
- T. Hayashi, Y. Hitomi, T. Takimura, A. Tomokuni, T. Mizutani, Y. Hisaeda and H. Ogoshi, Coord. Chem. Rev., 1999, 190-192, 961–974 CrossRef CAS.
- T. Ueno, S. Abe, N. Yokoi and Y. Watanabe, Coord. Chem. Rev., 2007, 251, 2717–2731 CrossRef CAS.
- K. Oohora, H. Meichin, Y. Kihira, H. Sugimoto, Y. Shiro and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 18460–18463 CAS.
- M. W. Wolf, D. A. Vargas and N. Lehnert, Inorg. Chem., 2017, 56, 5623–5635 CrossRef CAS PubMed.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- G. Rao, K. B. Alwan, N. J. Blackburn and R. D. Britt, Inorg. Chem., 2019, 58, 12601–12608 CrossRef CAS PubMed.
- S. D. Khare, Y. Kipnis, P. Greisen Jr., R. Takeuchi, Y. Ashani, M. Goldsmith, Y. Song, J. L. Gallaher, I. Silman, H. Leader, J. L. Sussman, B. L. Stoddard, D. S. Tawfik and D. Baker, Nat. Chem. Biol., 2012, 8, 294–300 CrossRef CAS PubMed.
- H.-S. Park, S.-H. Nam, J. K. Lee, C. N. Yoon, B. Mannervik, S. J. Benkovic and H.-S. Kim, Science, 2006, 311, 535–538 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- S. B. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed.
- S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132–136 CrossRef CAS PubMed.
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67–72 CrossRef CAS PubMed.
- N. Yeung, Y.-W. Lin, Y.-G. Gao, X. Zhao, B. S. Russell, L. Lei, K. D. Miner, H. Robinson and Y. Lu, Nature, 2009, 462, 1079–1082 CrossRef CAS PubMed.
- E. N. Mirts, I. D. Petrik, P. Hosseinzadeh, M. J. Nilges and Y. Lu, Science, 2018, 361, 1098–1101 CrossRef CAS PubMed.
- A. Bhagi-Damodaran, M. A. Michael, Q. Zhu, J. Reed, B. A. Sandoval, E. N. Mirts, S. Chakraborty, P. Moënne-Loccoz, Y. Zhang and Y. Lu, Nat. Chem., 2017, 9, 257–263 CrossRef CAS PubMed.
- V. Köhler, Y. M. Wilson, M. Dürrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knörr, D. Häussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93–99 CrossRef PubMed.
- A. Chatterjee, H. Mallin, J. Klehr, J. Vallapurackal, A. D. Finke, L. Vera, M. Marsh and T. R. Ward, Chem. Sci., 2016, 7, 673–677 RSC.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed.
- S. Wu, Y. Zhou, J. G. Rebelein, M. Kuhn, H. Mallin, J. Zhao, N. V. Igareta and T. R. Ward, J. Am. Chem. Soc., 2019, 141, 15869–15878 CrossRef CAS PubMed.
- S. I. Mann, T. Heinisch, T. R. Ward and A. S. Borovik, J. Am. Chem. Soc., 2017, 139, 17289–17292 CrossRef CAS PubMed.
- D. J. Raines, J. E. Clarke, E. V. Blagova, E. J. Dodson, K. S. Wilson and A.-K. Duhme-Klair, Nat. Catal., 2018, 1, 680–688 CrossRef CAS.
- M. V. Cherrier, L. Martin, C. Cavazza, L. Jacquamet, D. Lemaire, J. Gaillard and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2005, 127, 10075–10082 CrossRef CAS PubMed.
- C. Esmieu, M. V. Cherrier, P. Amara, E. Girgenti, C. Marchi-Delapierre, F. Oddon, M. Iannello, A. Jorge-Robin, C. Cavazza and S. Ménage, Angew. Chem. Int. Ed., 2013, 52, 3922–3925 CrossRef CAS PubMed.
- F. W. Monnard, E. S. Nogueira, T. Heinisch, T. Schirmer and T. R. Ward, Chem. Sci., 2013, 4, 3269–3274 RSC.
- S. Eda, I. Nasibullin, K. Vong, N. Kudo, M. Yoshida, A. Kurbangalieva and K. Tanaka, Nat. Catal., 2019, 2, 780–792 CrossRef CAS.
- T. Matsuo, C. Imai, T. Yoshida, T. Saito, T. Hayashi and S. Hirota, Chem. Commun., 2012, 48, 1662–1664 RSC.
- P. Haquette, M. Salmain, K. Svedlung, A. Martel, B. Rudolf, J. Zakrzewski, S. Cordier, T. Roisnel, C. Fosse and G. Jaouen, ChemBioChem, 2007, 8, 224–231 CrossRef CAS PubMed.
- M. Basauri-Molina, C. F. Riemersma, M. A. Würdemann, H. Kleijn and R. J. M. Klein Gebbink, Chem. Commun., 2015, 51, 6792–6795 RSC.
- C. A. Kruithof, M. A. Casado, G. Guillena, M. R. Egmond, A. van der Kerk-van Hoof, A. J. Heck, R. J. Klein Gebbink and G. van Koten, Chem. – Eur. J., 2005, 11, 6869–6877 CrossRef CAS PubMed.
- S. Alonso, G. Santiago, I. Cea-Rama, L. Fernandez-Lopez, C. Coscolín, J. Modregger, A. K. Ressmann, M. Martínez-Martínez, H. Marrero, R. Bargiela, M. Pita, J. L. Gonzalez-Alfonso, M. L. Briand, D. Rojo, C. Barbas, F. J. Plou, P. N. Golyshin, P. Shahgaldian, J. Sanz-Aparicio, V. Guallar and M. Ferrer, Nat. Catal., 2020, 3, 319–328 CrossRef CAS.
- P. C. Jordan, D. P. Patterson, K. N. Saboda, E. J. Edwards, H. M. Miettinen, G. Basu, M. C. Thielges and T. Douglas, Nat. Chem., 2016, 8, 179–185 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- C. Hu, S. I. Chan, E. B. Sawyer, Y. Yu and J. Wang, Chem. Soc. Rev., 2014, 43, 6498–6510 RSC.
- R. R. Davies and M. D. Distefano, J. Am. Chem. Soc., 1997, 119, 11643–11652 CrossRef CAS.
- P. J. Deuss, G. Popa, A. M. Z. Slawin, W. Laan and P. C. J. Kamer, ChemCatChem, 2013, 5, 1184–1191 CrossRef CAS.
- M. V. Doble, A. G. Jarvis, A. C. C. Ward, J. D. Colburn, J. P. Götze, M. Bühl and P. C. J. Kamer, ACS Sustainable Chem. Eng., 2018, 6, 15100–15107 CrossRef CAS.
- K. Renggli, M. G. Nussbaumer, R. Urbani, T. Pfohl and N. Bruns, Angew. Chem., Int. Ed., 2014, 53, 1443–1447 CrossRef CAS PubMed.
- M. Hestericová, T. Heinisch, M. Lenz and T. R. Ward, Dalton Trans., 2018, 47, 10837–10841 RSC.
- A. Onoda, K. Fukumoto, M. Arlt, M. Bocola, U. Schwaneberg and T. Hayashi, Chem. Commun., 2012, 48, 9756–9758 RSC.
- A. Onoda, Y. Kihara, K. Fukumoto, Y. Sano and T. Hayashi, ACS Catal., 2014, 4, 2645–2648 CrossRef CAS.
- D. F. Sauer, T. Himiyama, K. Tachikawa, K. Fukumoto, A. Onoda, E. Mizohata, T. Inoue, M. Bocola, U. Schwaneberg, T. Hayashi and J. Okuda, ACS Catal., 2015, 5, 7519–7522 CrossRef CAS.
- F. Philippart, M. Arlt, S. Gotzen, S.-J. Tenne, M. Bocola, H.-H. Chen, L. Zhu, U. Schwaneberg and J. Okuda, Chem. – Eur. J., 2013, 19, 13865–13871 CrossRef CAS PubMed.
- H. Osseili, D. F. Sauer, K. Beckerle, M. Arlt, T. Himiyama, T. Polen, A. Onoda, U. Schwaneberg, T. Hayashi and J. Okuda, Beilstein J. Org. Chem., 2016, 12, 1314–1321 CrossRef CAS PubMed.
- A. Thiel, D. F. Sauer, M. A. S. Mertens, T. Polen, H. H. Chen, U. Schwaneberg and J. Okuda, Org. Biomol. Chem., 2018, 16, 5452–5456 RSC.
- G. Roelfes, Acc. Chem. Res., 2019, 52, 545–556 CrossRef CAS PubMed.
- L. Villarino, K. E. Splan, E. Reddem, L. Alonso-Cotchico, C. Gutiérrez de Souza, A. Lledós, J.-D. Maréchal, A.-M. W. H. Thunnissen and G. Roelfes, Angew. Chem., Int. Ed., 2018, 57, 7785–7789 CrossRef PubMed.
- H. Yang, P. Srivastava, C. Zhang and J. C. Lewis, ChemBioChem, 2014, 15, 223–227 CrossRef CAS PubMed.
- P. Srivastava, H. Yang, K. Ellis-Guardiola and J. C. Lewis, Nat. Commun., 2015, 6, 7789 CrossRef CAS PubMed.
- H. Yang, A. M. Swartz, H. J. Park, P. Srivastava, K. Ellis-Guardiola, D. M. Upp, G. Lee, K. Belsare, Y. Gu, C. Zhang, R. E. Moellering and J. C. Lewis, Nat. Chem., 2018, 10, 318–324 CrossRef CAS PubMed.
- J. L. Beesley and D. N. Woolfson, Curr. Opin. Biotechnol, 2019, 58, 175–182 CrossRef CAS PubMed.
- O. Zozulia, M. A. Dolan and I. V. Korendovych, Chem. Soc. Rev., 2018, 47, 3621–3639 RSC.
- T. B. J. Pinter, K. J. Koebke and V. L. Pecoraro, Angew. Chem., Int. Ed., 2020, 59, 7678–7699 CrossRef CAS PubMed.
- M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118–123 CrossRef CAS PubMed.
- A. E. Tolbert, C. S. Ervin, L. Ruckthong, T. J. Paul, V. M. Jayasinghe-Arachchige, K. P. Neupane, J. A. Stuckey, R. Prabhakar and V. L. Pecoraro, Nat. Chem., 2020, 12, 405–411 CrossRef CAS PubMed.
- M. Tegoni, F. Yu, M. Bersellini, J. E. Penner-Hahn and V. L. Pecoraro, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21234–21239 CrossRef CAS PubMed.
- F. Yu, J. E. Penner-Hahn and V. L. Pecoraro, J. Am. Chem. Soc., 2013, 135, 18096–18107 CrossRef CAS PubMed.
- A. G. Tebo, T. B. J. Pinter, R. García-Serres, A. L. Speelman, C. Tard, O. Sénéque, G. Blondin, J.-M. Latour, J. Penner-Hahn, N. Lehnert and V. L. Pecoraro, Biochemistry, 2018, 57, 2308–2316 CrossRef CAS PubMed.
- D. Shiga, Y. Funahashi, H. Masuda, A. Kikuchi, M. Noda, S. Uchiyama, K. Fukui, K. Kanaori, K. Tajima, Y. Takano, H. Nakamura, M. Kamei and T. Tanaka, Biochemistry, 2012, 51, 7901–7907 CrossRef CAS PubMed.
- R. Sambasivan and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 9289–9291 CrossRef CAS PubMed.
- R. Sambasivan and Z. T. Ball, Angew. Chem. Int. Ed., 2012, 51, 8568–8572 CrossRef CAS PubMed.
- B. S. Der, M. Machius, M. J. Miley, J. L. Mills, T. Szyperski and B. Kuhlman, J. Am. Chem. Soc., 2012, 134, 375–385 CrossRef CAS PubMed.
- S. Studer, D. A. Hansen, Z. L. Pianowski, P. R. E. Mittl, A. Debon, S. L. Guffy, B. S. Der, B. Kuhlman and D. Hilvert, Science, 2018, 362, 1285–1288 CrossRef CAS PubMed.
- R. L. Koder, J. L. R. Anderson, L. A. Solomon, K. S. Reddy, C. C. Moser and P. L. Dutton, Nature, 2009, 458, 305–309 CrossRef CAS PubMed.
- T. A. Farid, G. Kodali, L. A. Solomon, B. R. Lichtenstein, M. M. Sheehan, B. A. Fry, C. Bialas, N. M. Ennist, J. A. Siedlecki, Z. Zhao, M. A. Stetz, K. G. Valentine, J. L. R. Anderson, A. J. Wand, B. M. Discher, C. C. Moser and P. L. Dutton, Nat. Chem. Biol., 2013, 9, 826–833 CrossRef CAS.
- D. W. Watkins, J. M. X. Jenkins, K. J. Grayson, N. Wood, J. W. Steventon, K. K. Le Vay, M. I. Goodwin, A. S. Mullen, H. J. Bailey, M. P. Crump, F. MacMillan, A. J. Mulholland, G. Cameron, R. B. Sessions, S. Mann and J. L. R. Anderson, Nat. Commun., 2017, 8, 358 CrossRef PubMed.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS PubMed.
- L. A. Churchfield and F. A. Tezcan, Acc. Chem. Res., 2019, 52, 345–355 CrossRef CAS PubMed.
- W. J. Song and F. A. Tezcan, Science, 2014, 346, 1525–1528 CrossRef CAS PubMed.
- J. Rittle, M. J. Field, M. T. Green and F. A. Tezcan, Nat. Chem., 2019, 11, 434–441 CrossRef CAS PubMed.
- W. J. Song, J. Yu and F. A. Tezcan, J. Am. Chem. Soc., 2017, 139, 16772–16779 CrossRef CAS PubMed.
- A. J. Reig, M. M. Pires, R. A. Snyder, Y. Wu, H. Jo, D. W. Kulp, S. E. Butch, J. R. Calhoun, T. Szyperski, E. I. Solomon and W. F. DeGrado, Nat. Chem., 2012, 4, 900–906 CrossRef CAS PubMed.
- M. Faiella, C. Andreozzi, R. T. M. de Rosales, V. Pavone, O. Maglio, F. Nastri, W. F. DeGrado and A. Lombardi, Nat. Chem. Biol., 2009, 5, 882–884 CrossRef CAS PubMed.
- S.-Q. Zhang, M. Chino, L. Liu, Y. Tang, X. Hu, W. F. DeGrado and A. Lombardi, J. Am. Chem. Soc., 2018, 140, 1294–1304 CrossRef CAS PubMed.
- N. F. Polizzi, Y. Wu, T. Lemmin, A. M. Maxwell, S.-Q. Zhang, J. Rawson, D. N. Beratan, M. J. Therien and W. F. DeGrado, Nat. Chem., 2017, 9, 1157–1164 CrossRef CAS PubMed.
- N. H. Joh, T. Wang, M. P. Bhate, R. Acharya, Y. Wu, M. Grabe, M. Hong, G. Grigoryan and W. F. DeGrado, Science, 2014, 346, 1520–1524 CrossRef CAS PubMed.
- J. M. Perez-Aguilar and J. G. Saven, Structure, 2012, 20, 5–14 CrossRef CAS PubMed.
- A. Niitsu, J. W. Heal, K. Fauland, A. R. Thomson and D. N. Woolfson, Philos. Trans. R. Soc., B, 2017, 372, 20160213 CrossRef PubMed.
- S. C. Patel and M. H. Hecht, Protein Eng., Des. Sel., 2012, 25, 445–452 CrossRef CAS PubMed.
- V. Nanda, M. M. Rosenblatt, A. Osyczka, H. Kono, Z. Getahun, P. L. Dutton, J. G. Saven and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 5804–5805 CrossRef CAS.
- R. Bau, D. C. Rees, D. M. Kurtz Jr, R. A. Scott, H. Huang, M. W. W. Adams and M. K. Eidsness, J. Biol. Inorg. Chem., 1998, 3, 484–493 CrossRef CAS.
- A. D'Souza, M. Mahajan and S. Bhattacharjya, Chem. Sci., 2016, 7, 2563–2571 RSC.
- C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303–309 CrossRef CAS PubMed.
- Z. Lengyel, C. M. Rufo, Y. S. Moroz, O. V. Makhlynets and I. V. Korendovych, ACS Catal., 2018, 8, 59–62 CrossRef CAS PubMed.
- P. Makam, S. S. R. K. C. Yamijala, K. Tao, L. J. W. Shimon, D. S. Eisenberg, M. R. Sawaya, B. M. Wong and E. Gazit, Nat. Catal., 2019, 2, 977–985 CrossRef CAS PubMed.
- A. R. Voet, H. Noguchi, C. Addy, K. Y. Zhang and J. R. Tame, Angew. Chem., Int. Ed., 2015, 54, 9857–9860 CrossRef CAS PubMed.
- Y. M. Wilson, M. Dürrenberger, E. S. Nogueira and T. R. Ward, J. Am. Chem. Soc., 2014, 136, 8928–8932 CrossRef CAS PubMed.
- Y. Okamoto, R. Kojima, F. Schwizer, E. Bartolami, T. Heinisch, S. Matile, M. Fussenegger and T. R. Ward, Nat. Commun., 2018, 9, 1943 CrossRef PubMed.
- J. G. Rebelein, Y. Cotelle, B. Garabedian and T. R. Ward, ACS Catal., 2019, 9, 4173–4178 CrossRef CAS PubMed.
- W. Ghattas, V. Dubosclard, A. Wick, A. Bendelac, R. Guillot, R. Ricoux and J.-P. Mahy, J. Am. Chem. Soc., 2018, 140, 8756–8762 CrossRef CAS PubMed.
- E. L. Hegg and L. Q. Jr, Eur. J. Biochem., 1997, 250, 625–629 CrossRef CAS.
- K. D. Koehntop, J. P. Emerson and L. Que, J. Biol. Inorg. Chem., 2005, 10, 87–93 CrossRef CAS PubMed.
- B. Amrein, M. Schmid, G. Collet, P. Cuniasse, F. Gilardoni, F. P. Seebeck and T. R. Ward, Metallomics, 2012, 4, 379–388 RSC.
- L. Zhou, M. Bosscher, C. Zhang, S. Ozçubukçu, L. Zhang, W. Zhang, C. J. Li, J. Liu, M. P. Jensen, L. Lai and C. He, Nat. Chem., 2014, 6, 236–241 CrossRef CAS PubMed.
- C. Aussignargues, M.-E. Pandelia, M. Sutter, J. S. Plegaria, J. Zarzycki, A. Turmo, J. Huang, D. C. Ducat, E. L. Hegg, B. R. Gibney and C. A. Kerfeld, J. Am. Chem. Soc., 2016, 138, 5262–5270 CrossRef CAS PubMed.
- S. R. Soltau, J. Niklas, P. D. Dahlberg, O. G. Poluektov, D. M. Tiede, K. L. Mulfort and L. M. Utschig, Chem. Commun., 2015, 51, 10628–10631 RSC.
- G. G. Hammes, Biochemistry, 2002, 41, 8221–8228 CrossRef CAS PubMed.
- S. Hammes-Schiffer and S. J. Benkovic, Annu. Rev. Biochem., 2006, 75, 519–541 CrossRef CAS PubMed.
- J. L. Payne and A. Wagner, Nat. Rev. Genet., 2019, 20, 24–38 CrossRef CAS PubMed.
- M. A. Siddiq, G. K. Hochberg and J. W. Thornton, Curr. Opin. Struct. Biol., 2017, 47, 113–122 CrossRef CAS PubMed.
- B. E. Clifton, J. A. Kaczmarski, P. D. Carr, M. L. Gerth, N. Tokuriki and C. J. Jackson, Nat. Chem. Biol., 2018, 14, 542–547 CrossRef CAS PubMed.
- E. Wallin and G. V. Heijne, Protein Sci., 1998, 7, 1029–1038 CrossRef CAS PubMed.
- L. Fagerberg, K. Jonasson, G. Von Heijne, M. Uhlén and L. Berglund, Proteomics, 2010, 10, 1141–1149 CrossRef CAS PubMed.
- P.-S. Huang, S. E. Boyken and D. Baker, Nature, 2016, 537, 320–327 CrossRef CAS.
- N. M. Gallup and A. N. Alexandrova, in Methods in Enzymology, ed. G. A. Voth, Academic Press, 2016, vol. 577, pp. 319–339 Search PubMed.
- L. Alonso-Cotchico, J. Rodríguez-Guerra, A. Lledós and J.-D. Maréchal, Acc. Chem. Res., 2020, 53, 896–905 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |