
Manganese complex-catalysed α-alkylation of ketones with secondary alcohols enables the synthesis of β-branched carbonyl compounds†
Satyadeep
Waiba‡
,
Sayan K.
Jana‡
,
Ayan
Jati
,
Akash
Jana
and
Biplab
Maji
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur 741246, India. E-mail: bm@iiserkol.ac.in
First published on 19th May 2020
Abstract
Herein, β-branched carbonyl compounds were synthesised via the α-alkylation of ketones with secondary alcohols under “borrowing hydrogen” catalysis. A wide range of secondary alcohols, including various cyclic, acyclic, symmetrical, and unsymmetrical alcohols, have been successfully applied under the developed reaction conditions. A manganese(I) complex bearing a phosphine-free multifunctional ligand catalysed the reaction and produced water as the sole byproduct.
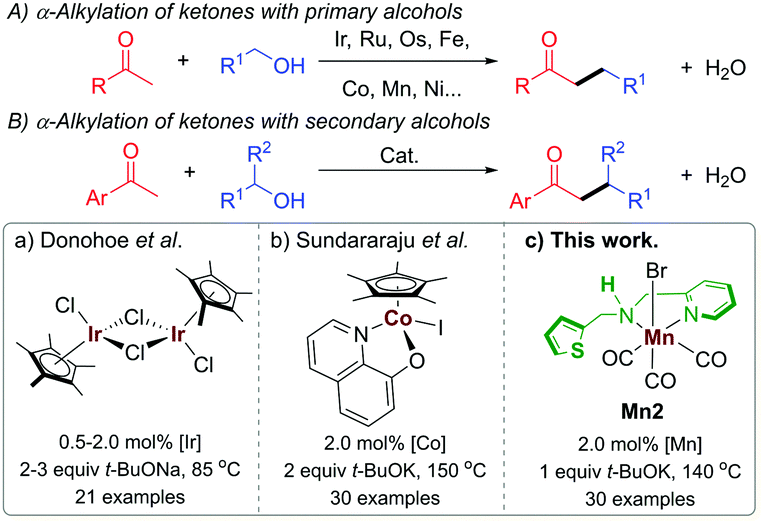
The development of catalytic methods for the construction of C–C bonds is one of the central goals in organic synthesis. The delicacy of “borrowing hydrogen” (BH) catalysis lies in the fact that it utilizes alcohol as an alternative carbon source for such processes under eco-friendly conditions avoiding substrate prefunctionalization, organohalides, cryogenic temperature, etc.1 In this process, alcohols get activated by dehydrogenation for a reaction with a nucleophile, which is then followed by hydrogenation of the resulting unsaturated intermediate by the “borrowed hydrogen”, thereby making the whole process atom economical, and benign.
In the recent past, BH catalysis has widely been applied for the α-alkylation of ketones with primary alcohols (Scheme 1A).1i,j,2 On the other hand, the use of secondary alcohols under BH catalysis to generate the β-branched molecular architecture is limited (Scheme 1B).3 Commonly, the β-branched ketones were made in the presence of lithium bases by using secondary alkyl halide as an electrophile and this unavoidably produced a large amount of waste.4 In a pioneering work, Donohoe and coworkers reported the α-alkylation of ketones with secondary alcohols leading to β-branched products using [Cp*IrCl2]2 (Cp* = (C5(CH3)5) as a catalyst (Scheme 1Ba).3a The steric factor of the used carbonyl substrate was exploited to limit the self-condensation of the carbonyl substrate used and the ketone, derived during the reaction from the secondary alcohol.3a,5 The replacement of precious metals by earth-abundant metals for this type of catalytic transformations is highly attractive in terms of toxicity, cost, and sustainability.6 In this regard, the only base metal-catalysed reaction of this type was reported by the Sundararaju group using [Cp*Co(N,O)I] ((N,O) = quinolin-8-olate) as a catalyst (Scheme 1Bb).3b However, the recent European Commission's list of critical raw materials (CRMs) suggested that except cobalt, all first-row transition metals exceed the economic importance threshold while their abundance is below the supply risk threshold.7 In this regard, lately, well-defined manganese(I) complexes have emerged as an attractive noble metal replacer in sustainable (de)hydrogenation reactions and other organic transformations.8 Manganese is the third-most abundant transition metal in the earth's crust (after iron and titanium) and it is less toxic than its higher analogs. Not long ago, selective α-alkylations of ketones was achieved by using Mn(I) catalysis by Beller,2h Rueping,2l Sortais,2m and our groups.2k Alkylations of esters and amides with primary alcohols were also disclosed under manganese catalysis.9 We have also reported the alkylations of nitriles using a well-defined phosphine-free manganese complex.10 However, to the best of our knowledge, manganese complex-catalysed alkylations of methyl ketones with secondary alcohols to form β-branched molecular structures have not been developed thus far.
 | ||
| Scheme 1 Alkylation of ketones. | ||
Multifunctional ligand design is an integral part of synthetic chemistry to augment the bond activation processes during the catalytic cycle.2d In this regard, most BH catalysts with base metals rely on metal–ligand bifunctional cooperation for alcohol (de)hydrogenation. We recently reported the synthesis of (1 + n)-membered cycloalkanes from methyl ketones and 1,n-diols by using a Mn(I)-catalyst Mn2 bearing a hybrid ligand.11 We have shown that in addition to protonation/deprotonation bifunctionality in the picolylamine fragment, a hemilabile thiophene sidearm that coordinates the manganese site with a relatively elongated Mn–S bond is instrumental for the success of the catalyst. In continuation of our recent efforts in developing phosphine-free manganese catalysis for the (de)hydrogenation reaction,2k,10–12 herein, we report the catalytic efficiency of Mn2 for the α-alkylation of ketones with secondary alcohols leading to the β-functionalised products with the elimination of water as the sole byproduct (Scheme 1Bc).
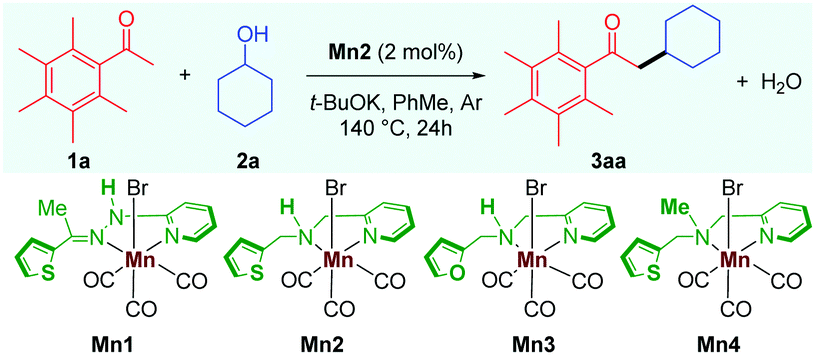
We started our investigation by carrying out the alkylation of 2,3,4,5,6-pentamethyl acetophenone 1a with cyclohexanol 2a as the model substrates (Table 1). Treatment of 1a and 2a in the catalytic system of Mn2 (2 mol%) and t-BuOK (1 equiv.) in toluene as solvent at 140 °C for 24 h gave the desired alkylated product 3aa in 85% isolated yield, and no other side products were observed (Table 1, entry 1). The previously reported catalyst Mn1, which displayed high catalytic activities for the α-alkylations of nitriles,10 was found to be ineffective for this reaction, giving the product 3aa in less than 10% yield (entry 2). The manganese catalyst Mn3 with a furan sidearm instead of thiophene performs poorly, delivering 3aa in 25% yield, thereby stressing the need of the soft donor in the ligand backbone (entry 3). No product was observed when Mn4 was used as a catalyst (entry 4), highlighting the importance of the NH proton for catalytic activity, vide infra. The manganese precursor MnBr(CO)5 did not catalyze the reaction (entry 5), which further validates the need for a proper ligand design for efficient catalysis. Further, the effects of other bases and solvents were tested, and, in all cases, they were found to be less effective (entries 6 and 7). Control reactions demonstrated the importance of the catalyst and the base in the reaction as the reaction could not be reproduced in the absence of either of them (entry 8).
|
|
||
|---|---|---|
| Entry | Deviation from above | Yield of 3aa |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), Mn-cat (2 mol%), and t-BuOK (0.2 mmol) under Ar at 140 °C in toluene (0.05 mL) for 24 h. Isolated yields. | ||
| 1 | None | 85 |
| 2 | Mn1 | <10 |
| 3 | Mn3 | 25 |
| 4 | Mn4 | <5 |
| 5 | MnBr(CO)5 | <5 |
| 6 | t-BuOH or 1,4-dioxane as solvent | <51 |
| 7 | t-BuOLi, t-BuONa or Cs2CO3 as base | <15 |
| 8 | No catalyst, no base | 5–10 |
With the optimised conditions in hand, we next proceeded to determine the scope of this manganese catalysed C–C bond formation reaction, and the results are summarised in Table 2. The cyclic secondary alcohols with different ring sizes were found to be viable coupling partners delivering the desired products 3aa–3ac in high yields. Interestingly, the 4-substituted cyclohexanols 2d–f were found to impart high cis-diastereoselectivity to the products 3ad–3af, which were obtained in 76–89% yields. An equatorial attack of the intermediate manganese hydride on the exocyclic enone species can explain the observed diastereoselectivity. Reactions with unsymmetrical cyclic alcohols such as 1-tetralol 2g, 1-indanol 2i, and their derivatives 2h,l also proceeded smoothly under the reaction conditions delivering the desired products 3ag–3al in 64–90% yields. The NMR analysis revealed cis-diastereoselectivity in the products 3ak and 3al.
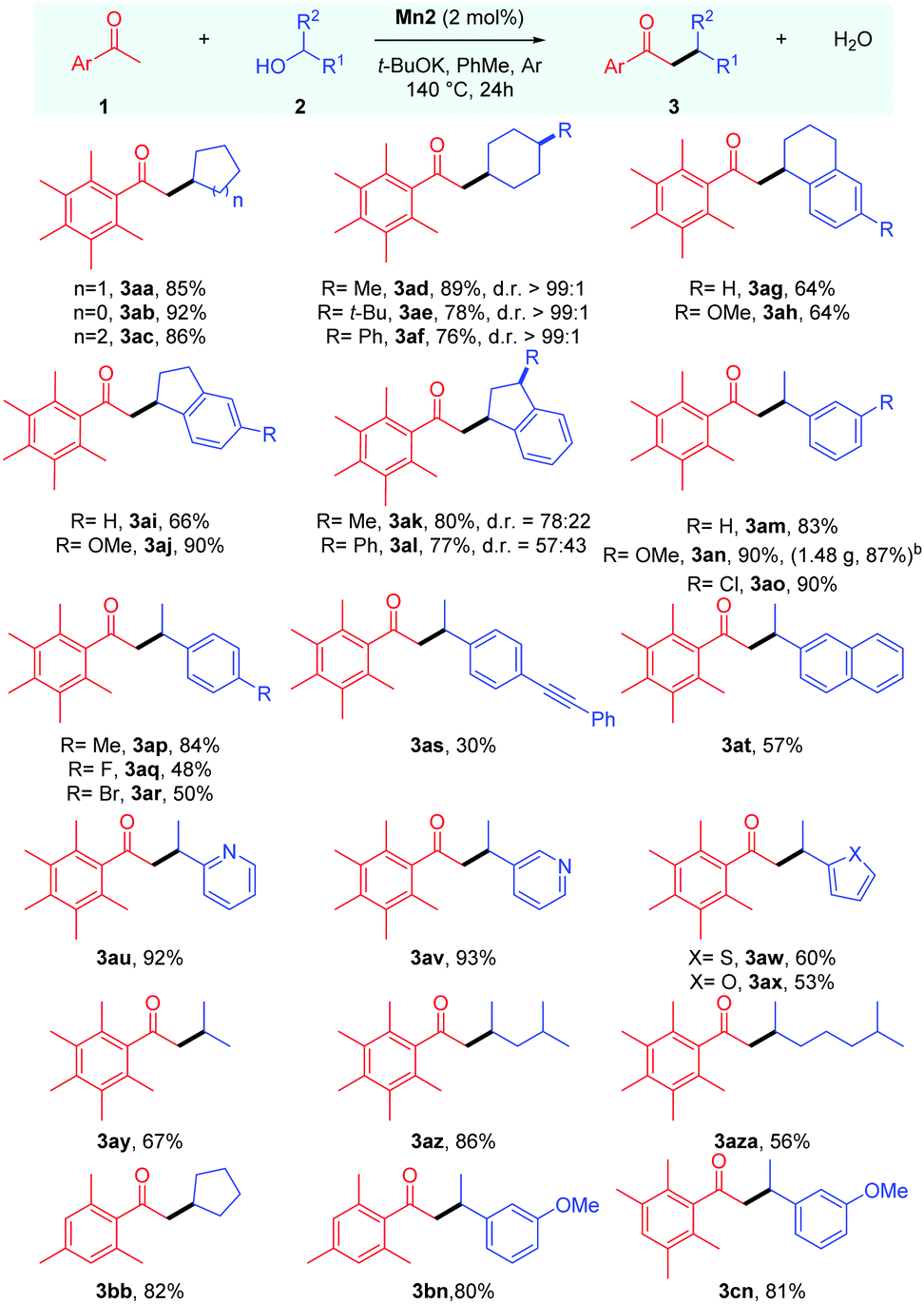
The scope of acyclic secondary alcohols was then examined. 1-Phenylethan-1-ol 2m and their derivatives 2n and 2p with electron-donating substituents underwent smooth α-alkylation reaction delivering the β-branched products 3am, 3an and 3ap in 93–90% yields. Gratifyingly, the reaction of 1a with 2n could be performed in gram scale, and the product 3an was isolated in high yield (1.48 g, 87%). This thus highlights the synthetic utility and practicality of the protocol. Aryl alkyl secondary alcohols with halogen substituents 2o, 2q and 2r also produced the desired products 3ao, 3aq and 3ar in good to moderate yields with the retention of the halo group, which leaves room for further functionalizations. The alcohol bearing a p-(phenylethynyl) group, 2s, selectively yielded the desired product in 30% yield where the triple bond remains intact. Similarly, α-methyl-2-naphthalenemethanol 2t could also be used as a coupling partner. We then shifted our focus towards aryl alkyl secondary alcohols bearing heteroaromatic rings. Secondary alcohols with 2-,3-pyridyl (2u,v), 2-thiophenyl (2w), and 2-furyl (2x) groups were found to be viable coupling partners delivering the desired products 3au–3ax in 60–93% yields. It was found that the reaction is equally tolerable to aliphatic secondary alcohols with different chain lengths delivering the desired alkylated products 3ay–3aza in 56–86% yields.
Then, we tried to examine the scope of the ketone coupling partner. As demonstrated earlier by Donohoe and coworkers,3a,5 the use of ortho disubstituted methyl ketones, which can sterically block the undesired aldol condensation and transfer hydrogenation reaction, was found to be necessary for the success of this process. Gratifyingly, 2,4,6-trimethyl acetophenone 1b was found to display similar reactivity and selectivity as 1a, giving the desired products 3bb and 3bn in 82% and 80% yields, respectively. Similarly, 2,3,5,6-tetramethyl acetophenone 1c delivered the desired alkylated product 3cn in 81% yield. However, 2-acetylnapthalene failed to give the desired product in a practically useful yield under these conditions.
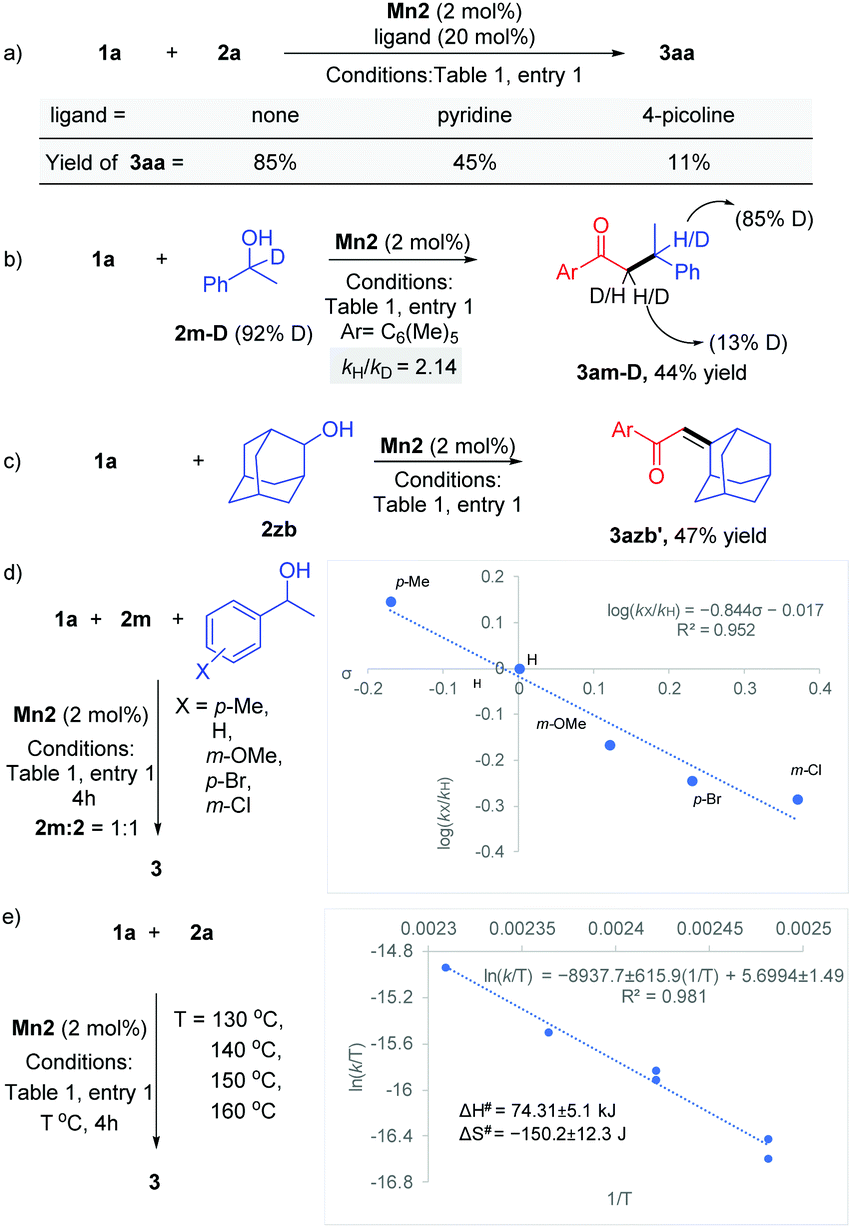
Various experimental studies to probe the probable reaction mechanism were then performed (Scheme 2). Initially, the importance of the M–L bifunctional activation of the ligand was examined. It is thought that the N–H bifunctionality in the ligand takes part via a base-mediated dehydrohalogenation to facilitate the alcohol dehydrogenation. Along this direction, the manganese complex Mn4 derived from the N–Me analog of the ligand failed to catalyze the reaction of 1a with 2a (Table 1, entry 4). Previously, we have delineated the role of the hemilabile thiophene arm for the success of Mn2 in (n + 1) annulation reaction with 1,n-diols (n = 4–6).11 To examine the importance of the hemilabile coordination of the soft S-donor atom for the alkylation reaction with secondary alcohols, we have performed the reaction of 1a with 2a using Mn3 as a catalyst. Supporting our hypothesis, the complex Mn3 with the furan side arm lacking the polarizable soft donor atom performed poorly, and only 25% 3aa was obtained (Table 1, entry 3). Further, when the Mn2 catalysed reaction was performed in the presence of exogenous Lewis bases (pyridine, 4-picoline), the yield of 3aa was substantially lowered, further supporting the necessity of the hemilabile coordination of the thiophene arm (Scheme 2a).13
 | ||
| Scheme 2 Mechanistic studies. | ||
Deuterium labeling experiments indicated the involvement of BH catalysis. The reaction of 1-phenylethan-1-d-1-ol 2m-D (92% D) with 1a delivered the product 3am-D in 44% yield (Scheme 2b). 1H NMR analysis of the product showed 85% D incorporation at the β-position of the carbonyl group and 13% D incorporation at the α-position. This points out a deuteride transfer at the β-position of the intermediate chalcone species from a manganese deuteride intermediate. The calculated primary kinetic isotope effect kH/kD = 2.14 from a parallel experiment suggests that the dehydrogenation step might be the rate-determining step. The intermediacy of the chalcone was further probed by the reaction between 1a and 2-adamantanol (2zb). The steric factor imparted due to the bulky adamantyl group slowed down the hydrogenation step and allowed the isolation of the unsaturated product 3azb′ in 47% yield (Scheme 2c). To study the electronic influence of the aryl alkyl secondary alcohol substituents on the reaction outcome, the Hammett correlation study was performed (Scheme 2d).14 We observed a linear correlation with a slope of −0.84, which suggests that in the transition state, electrons are flowing away, which is stabilised by the electron-donating substituents. Further, an Eyring analysis was carried out to predict the type of mechanism involved. We have found that ΔH# = +74.3 ± 5.1 kJ mol−1 and ΔS# = −150.2 ± 12.3 J mol−1 K−1, suggesting the involvement of an associative pathway (Scheme 2e).
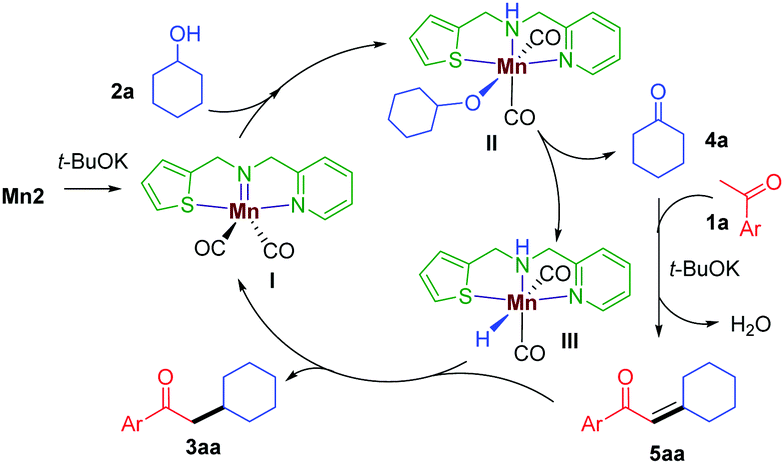
Based on the above experiments and previous reports,11 a plausible mechanism is proposed (Scheme 3). In the presence of t-BuOK, Mn2 generates the active imido intermediate I, which reacts with the secondary alcohol to give the intermediate II. The hemilabile thiophene arm facilitates the alcohol dehydrogenation to release the ketone 4a and the manganese hydride species III. The aldol condensation of 1a with 4a delivers the α,β-unsaturated intermediate 5aa, which is then subsequently reduced by III giving the desired saturated product 3aa and continuing the catalytic cycle.
 | ||
| Scheme 3 Proposed mechanism. | ||
In conclusion, we have demonstrated that a manganese(I) complex bearing a phosphine-free ligand catalysed the α-alkylation of ketones with secondary alcohols for the synthesis of diverse β-functionalised carbonyl compounds. The preliminary mechanistic studies indicated the involvement of “borrowing hydrogen” catalysis and delineated the role of the multifunctionality of the ligand backbone for the success of such catalysis. Water is produced as the sole byproduct of such a reaction. We hope that this report paves a way towards further development of molecular catalysts using earth-abundant transition metal catalysts for the synthesis of complex molecular architecture from simple building blocks.
The authors acknowledge IISER, Kolkata (ARF) and SERB (ECR/2016/001654) for the financial support. S. W., Ay. J., and Ak. J. thank CSIR, and S. K. J. thanks IISER Kolkata for the fellowships.
Conflicts of interest
There are no conflicts to declare.References
- (a) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS; (b) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS; (c) Y. Obora, Top. Curr. Chem., 2016, 374, 1–29 CrossRef CAS PubMed; (d) H. Fei, L. Zhuqing and Y. Zhengkun, Angew. Chem., Int. Ed., 2016, 55, 862–875 CrossRef; (e) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef; (f) A. Nandakumar, S. P. Midya, V. G. Landge and E. Balaraman, Angew. Chem., Int. Ed., 2015, 54, 11022–11034 CrossRef CAS; (g) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS; (h) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS; (i) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS; (j) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC.
- (a) Y. Iuchi, Y. Obora and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 2536–2537 CrossRef CAS; (b) M. L. Buil, M. A. Esteruelas, J. Herrero, S. Izquierdo, I. M. Pastor and M. Yus, ACS Catal., 2013, 3, 2072–2075 CrossRef CAS; (c) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS; (d) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS; (e) A. Quintard and J. Rodriguez, ChemSusChem, 2016, 9, 28–30 CrossRef CAS; (f) B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2019, 17, 1595–1607 RSC; (g) S. Elangovan, J.-B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef CAS; (h) M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 14967–14971 CrossRef; (i) G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Org. Lett., 2017, 19, 1080–1083 CrossRef CAS; (j) J. Das, K. Singh, M. Vellakkaran and D. Banerjee, Org. Lett., 2018, 20, 5587–5591 CrossRef CAS PubMed; (k) M. K. Barman, A. Jana and B. Maji, Adv. Synth. Catal., 2018, 360, 3233–3238 CrossRef CAS; (l) J. Sklyaruk, J. Borghs, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 775–779 CrossRef CAS; (m) A. Bruneau-Voisine, L. Pallova, S. Bastin, V. César and J.-B. Sortais, Chem. Commun., 2019, 55, 314–317 RSC; (n) K. Das, A. Mondal and D. Srimani, Chem. Commun., 2018, 54, 10582–10585 RSC.
- (a) W. M. Akhtar, C. B. Cheong, J. R. Frost, K. E. Christensen, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2017, 139, 2577–2580 CrossRef CAS; (b) P. Chakraborty, M. K. Gangwar, B. Emayavaramban, E. Manoury, R. Poli and B. Sundararaju, ChemSusChem, 2019, 12, 3463–3467 CrossRef CAS; (c) S. Thiyagarajan and C. Gunanathan, J. Am. Chem. Soc., 2019, 141, 3822–3827 CrossRef CAS.
- M. T. Reetz, Angew. Chem., Int. Ed. Engl., 1982, 21, 96–108 CrossRef.
- J. R. Frost, C. B. Cheong, W. M. Akhtar, D. F. J. Caputo, N. G. Stevenson and T. J. Donohoe, J. Am. Chem. Soc., 2015, 137, 15664–15667 CrossRef CAS.
- R. M. Bullock, Catalysis Without Precious Metals, Wiley-VCH, Weinheim, 2010 Search PubMed.
- British Geological Survey, Bureau de Recherches Géologiques et Minières, Deloitte Sustainability, Directorate-General for Internal Market, Industry, Entrepreneurship and SMEs (European Commission) and TNO, Study on the Review of the List of Critical Raw Materials, European Commission, 2017.
- (a) B. Maji and M. K. Barman, Synthesis, 2017, 3377–3393 CAS; (b) M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 4344–4362 CrossRef CAS; (c) N. Gorgas and K. Kirchner, Acc. Chem. Res., 2018, 51, 1558–1569 CrossRef CAS; (d) D. A. Valyaev, G. Lavigne and N. Lugan, Coord. Chem. Rev., 2016, 308, 191–235 CrossRef CAS; (e) A. Mukherjee and D. Milstein, ACS Catal., 2018, 11435–11469 CrossRef CAS; (f) S. Waiba and B. Maji, ChemCatChem, 2020, 12, 1891–1902 CrossRef CAS; (g) Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS; (h) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS; (i) X. Yang and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 923–928 CrossRef CAS; (j) Y.-F. Liang, R. Steinbock, A. Münch, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5384–5388 CrossRef CAS; (k) Y.-F. Liang, R. Steinbock, L. Yang and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10625–10629 CrossRef CAS; (l) S. Ali, J. Huo and C. Wang, Org. Lett., 2019, 21, 6961–6965 CrossRef CAS; (m) S. Cembellín, T. Dalton, T. Pinkert, F. Schäfers and F. Glorius, ACS Catal., 2020, 10, 197–202 CrossRef.
- (a) S. Chakraborty, P. Daw, Y. Ben David and D. Milstein, ACS Catal., 2018, 8, 10300–10305 CrossRef CAS; (b) Y. K. Jang, T. Krückel, M. Rueping and O. El-Sepelgy, Org. Lett., 2018, 20, 7779–7783 CrossRef CAS PubMed.
- A. Jana, C. B. Reddy and B. Maji, ACS Catal., 2018, 8, 9226–9231 CrossRef CAS.
- A. Jana, K. Das, A. Kundu, P. R. Thorve, D. Adhikari and B. Maji, ACS Catal., 2020, 10, 2615–2626 CrossRef CAS.
- M. K. Barman, S. Waiba and B. Maji, Angew. Chem., Int. Ed., 2018, 57, 9126–9130 CrossRef CAS.
- (a) P. M. Perez Garcia, T. Di Franco, A. Epenoy, R. Scopelliti and X. Hu, ACS Catal., 2016, 6, 258–261 CrossRef CAS; (b) A. Kumar, N. A. Espinosa-Jalapa, G. Leitus, Y. Diskin-Posner, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 14992–14996 CrossRef CAS; (c) S. Harris, Polyhedron, 1997, 16, 3219–3233 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc01460e |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |