
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A biomimetic hybrid material consisting of CaCO3 mesoporous microspheres and an alternating copolymer for reversed-phase HPLC†
Mai
Mochida
,
Yuta
Nagai
,
Hiroto
Kumagai
,
Hiroaki
Imai
 ,
Daniel
Citterio
and
Yuki
Hiruta
*
,
Daniel
Citterio
and
Yuki
Hiruta
*
Department of Applied Chemistry, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, 223-8522 Yokohama, Japan. E-mail: hiruta@applc.keio.ac.jp
First published on 10th July 2019
Abstract
We developed a biomineral-inspired hybrid material composed of CaCO3 and an organic polymer as a column packing material for HPLC. This material combines a hierarchical mesoporous structure and the functionality of the polymer. The surface of monodispersed mesoporous CaCO3 microspheres was modified with poly(maleic acid-alt-1-octadecene) (PMAcO) comprising hydrophobic alkyl chains and anionic carboxylate groups. PMAcO adsorbed onto the surface of CaCO3 through electrostatic interaction between Ca2+ sites and carboxylate groups, resulting in an octadecene coated microsphere interface. These microspheres were applied as a HPLC column and exhibited reversed-phase retention behavior in the separation of alkylbenzenes. This column showed high alkaline mobile phase resistance compared with the conventionally applied ODS column packing material. Quantitative analysis of the basic antidepressants clomipramine and imipramine spiked into whole blood was achieved with an alkaline mobile phase, demonstrating the potential of the biomineral-inspired material as a HPLC stationary phase for practical applications in routine analyses of basic drugs requiring alkaline mobile phases.
Introduction
Biominerals are inorganic–organic hybrid materials with hierarchically organized structures from nanoscopic to macroscopic scales.1 For example, eggshells, seashells and corals have mesostructures of CaCO3 nanocrystals, which are elaborately controlled by biopolymers.2,3 Focusing on eggshells as an example, their structure has pores that are necessary for gas exchange allowing respiration, and the presence of biopolymers prevents virus invasion. Furthermore, they provide relatively high mechanical strength to protect the inner shell.4 These characteristics of hierarchical structures combining CaCO3 nanocrystals and biopolymers enable the control of substance distribution and provide mechanical strength. Therefore, material scientists have been attracted by the elaborate architecture and formation processes of these biominerals, attempting to develop new fabrication technologies and to obtain novel functionalities through imitation of these features.5,6Paying attention to the hierarchical structure of biominerals to control the distribution of substances, a normal phase chromatography stationary phase based on biopolymer-removed crushed seashells has been developed.7 Although it was shown that the hierarchical structure of biominerals can be applied to a chromatographic separation material, the functionality of the biopolymer of biominerals like egg shells in terms of controlling substance distribution was not fully used for separation purposes.
In order to control the morphology of CaCO3, crystallization methods involving anionic small molecules8,9 or anionic polymers10–12 were studied by mimicking the formation process of biominerals. We have also reported that monodispersed mesoporous CaCO3 microspheres with 20 nm mesopores and controllable particle size can be obtained by a biomimetic process.13 In this method, an anionic polymer, poly(sodium 4-sulfonate) (PSS), controls crystal growth (i.e. assemblage of 20 nm CaCO3 nanocrystals) by electrostatic adsorption, resulting in the formation of a metastable phase vaterite crystal. In addition, we have modified these microspheres with thermo-responsive poly(N-isopropylacrylamide) (PNIPAAm) to transport chemical substances from an organic to an aqueous phase by temperature-switching.14 However, after modification with PNIPAAm, the inter-particular space was fully filled with the thermo-responsive polymer, resulting in the loss of the hierarchical pore structure that is important in controlling substance distribution.
Cooperative interaction between the hierarchical structure and organic molecules is important to obtain the functions of biominerals and the high mechanical strength as in the case of eggshells.15 To the best of our knowledge, a biomimetic material that synergistically combines the hierarchical structure and the function of organic molecules to enable substance distribution control has not been reported. Herein, we have developed biomineral-inspired amphiphilic polymer-modified monodisperse CaCO3 microspheres having a hierarchical pore structure and the chemical functionality of the polymer used for modification. In addition, due to the ordered assembled structures composed of polymers and abundant inorganic CaCO3 (Fig. 1), excellent physical properties are achieved. We thought that these properties of the biomineral-inspired hybrid material are suitable for controlling substance distribution similarly to egg shells, and we applied it as a column packing material for high performance liquid chromatography (HPLC).
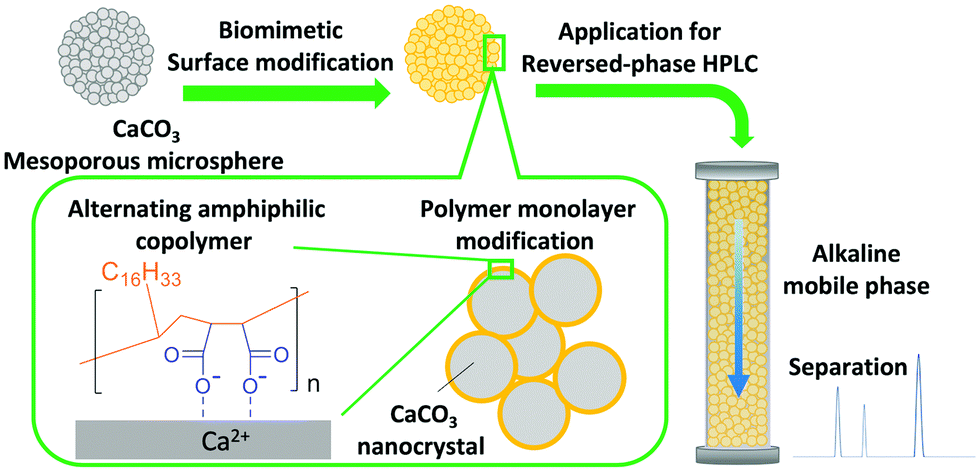 | ||
| Fig. 1 Concept image. | ||
Column chromatography was originally developed by the botanist Mikhail Tswett, using liquid-adsorption columns containing CaCO3 in the form of a powder of ground chalk (i.e. calcite crystals). This implies that synthetic CaCO3 mesoporous microspheres are applicable as a stationary phase packing material for HPLC.16 In current HPLC columns, octadecyl silica (ODS) is the most widely used stationary phase for reversed-phase chromatography. It consists of mesoporous inorganic silica gel modified with organic hydrophobic octadecyl moieties. Because biomineral-inspired materials also possess a hierarchical mesoporous structure, their application as a reversed-phase HPLC column packing material similar to ODS is possible by hydrophobic alkyl polymer modification of their surface.
As with CaCO3-based biominerals wherein hierarchical structures are controlled by electrostatic adsorption between CaCO3 crystals and biopolymers while stably maintaining the morphology,2 the imitation of their formation process allows the modification of synthetic CaCO3 mesoporous microspheres with an anionic polymer by electrostatic adsorption. By this method, mesoporous CaCO3 particles can be stably modified, due to polymer adsorption at multiple sites on the surface of CaCO3, while effectively retaining the mesopores.17
In addition, CaCO3 is stable against dissolution in alkaline solutions, since it is a basic salt. On the other hand, the use of ODS is limited due to poor hydrolytic stability against alkaline mobile phases. The fact that alkaline mobile phases have to be avoided when using low alkaline resistant silica gel column materials is a disadvantage leading to low separation performance for basic analytes.18 By replacing ODS with the polymer-modified CaCO3 particles, it is possible to realize an alkaline stable packing material overcoming the disadvantages of conventional stationary phases.
We have developed a biomineral-inspired inorganic–organic hybrid material by modifying the surface of monodisperse mesoporous CaCO3 microspheres through electrostatic adsorption of a regularly alternating copolymer having both anionic groups and functional groups, while maintaining the mesoporous structure. Whereas the anionic groups serve the purpose of adsorbing the polymer to the crystal surface, the alternating functional groups impart their functionality. Thus, efficient modification and functionalization of the surface of the monodisperse mesoporous CaCO3 microspheres were simultaneously achieved. Especially, maleic anhydride based alternating copolymers can obtain various functionality by radical copolymerization of maleic anhydride with styrene, α-olefins, or vinyl ethers.19 In this work, poly(maleic acid-alt-1-octadecene) (PMAcO), consisting of regularly alternating hydrophobic alkyl chains and two carboxylate units, was used (Fig. 1). The presence of the octadecyl groups on the surface of the monodisperse mesoporous CaCO3 microspheres imparts a strong hydrophobic character, similar to the octadecyl groups of ODS, and enables the application as a versatile packing material in reversed-phase HPLC.
Results and discussion
Preparation and characterization of amphiphilic polymer modified monodisperse CaCO3 microspheres
Bare monodisperse mesoporous CaCO3 microspheres were prepared by a simple mixing method of two solutions at room temperature according to the literature.13 CaCO3 crystal growth during microsphere synthesis was controlled with PSS, followed by the decomposition of PSS using aqueous NaClO solution and calcination. PMAcO was obtained by the hydrolysis of repeating maleic anhydride units in poly(maleic anhydride-alt-1-octadecene) (PMAO), as shown in Scheme 1.20 Hydrolysis was confirmed by FT-IR spectra (Fig. S1, ESI†), in which the peak at 1780 cm−1 derived from maleic anhydride decreased, while the peak at 1729 cm−1 derived from maleic acid was enhanced. The surface of the bare CaCO3 microspheres was modified with PMAcO by simply dispersing the microspheres in an organic solution of PMAcO. PMAcO adsorbed onto the surface of CaCO3 through electrostatic interaction between cationic calcium sites and anionic carboxylate groups, resulting in an octadecyl-coated microsphere interface referred to as “CaCO3–PMAcO”.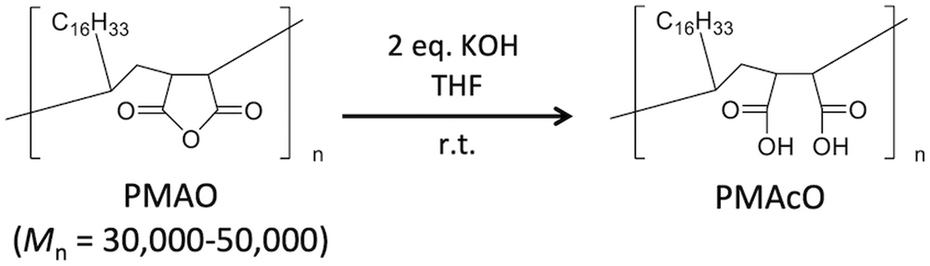 | ||
| Scheme 1 Hydrolysis reaction of PMAO to form PMAcO. | ||
Particle morphologies of bare CaCO3 and CaCO3–PMAcO particles were observed by scanning electron microscopy (SEM), as shown in Fig. 2. Both before and after modification with PMAcO, monodisperse spherical particles with identical average particle diameters of 3.5 ± 0.4 μm and 3.5 ± 0.6 μm, respectively, were observed (n = 400, measured from Fig. 2D and H). The particle size distributions are shown in Fig. S2 (ESI†). CaCO3 microspheres obtained by the applied synthesis method have been reported to consist of aggregates of 20 nm-sized CaCO3 crystals,21 which was also observed for the modified CaCO3–PMAcO particles (Fig. S3, ESI†), confirming that the morphology of the mesoporous microspheres was retained after modification with PMAcO. According to the XRD patterns (Fig. 3A), the bare CaCO3 particles were assigned to be pure vaterite, with the crystal structure unchanged by modification with PMAcO. FT-IR spectra (Fig. 3B) showed the appearance of peaks at 2924 and 2858 cm−1 derived from –CH2– moieties after modification with PMAcO. In addition, the mass reduction in the thermogravimetric (TG) curves in the 240–540 °C range (Fig. 3C) indicated the presence of 5 wt% PMAcO on the surface of CaCO3 microspheres, which was comparable to oleic acid for CaCO3 surface modification prepared in analogy to the CaCO3–PMAcO particles (Fig. 3D). These results demonstrate that PMAcO was successfully modified onto the surface of CaCO3 microspheres while maintaining their properties. The pore size distributions of CaCO3 microspheres before and after PMAcO modification were obtained by nitrogen adsorption–desorption isotherms (Fig. 3E). While the pore size distributions hardly changed upon modification with the polymer, the pore diameter decreased from 22 to 16 nm (Fig. 3F). The thickness of an octadecanoic acid monolayer on CaCO3 has been reported to be ≈2.6 nm.22 Therefore, these results infer that a uniform monolayer of repeating octadecene units was formed on the surface of the pores by PMAcO modification. Since ODS particles used in HPLC column packings have a uniform particle size of 2–5 μm with 6–30 nm pores, the current CaCO3–PMAcO microspheres with comparable uniform particle size and pore size fulfil the basic requirements to be applied as a HPLC column packing material. For successful application as a packing material for long-term reproducible HPLC analysis, durability against the mobile phase solvent is required. Since the PMAcO polymer has a plurality of adsorption sites, it is expected to be stably adsorbed onto the surface of CaCO3. The durability of the PMAcO modification was experimentally evaluated by comparing the amount of organic components by means of the TG curves in the 240–540 °C regime recorded before and after washing with a water/methanol mixture (50/50, v/v) representing a common mobile phase. The results shown in Fig. 3D indicate that the amount of PMAcO did not change during the washing process. On the other hand, in a comparative experiment using oleic acid for CaCO3 surface modification (prepared in analogy to the CaCO3–PMAcO particles), a drastic decrease of the organic compound from 5 to 1 wt% upon washing was observed, corresponding to an 80% reduction. This significant difference is attributed to the fact that oleic acid only has a single carboxylate residue to interact with the CaCO3 surface, in contrast to PMAcO with multiple binding sites, leading to the higher durability of the latter. Therefore, the biomineral inspired surface modification with the polymer by interaction between polymeric functional groups and calcium sites seems to contribute to the long-term durability of CaCO3–PMAcO and, hence, its applicability as a packing material for HPLC.
 | ||
| Fig. 2 SEM images of (A–D): bare CaCO3 particles and (E–H): polymer-modified CaCO3–PMAcO particles. | ||
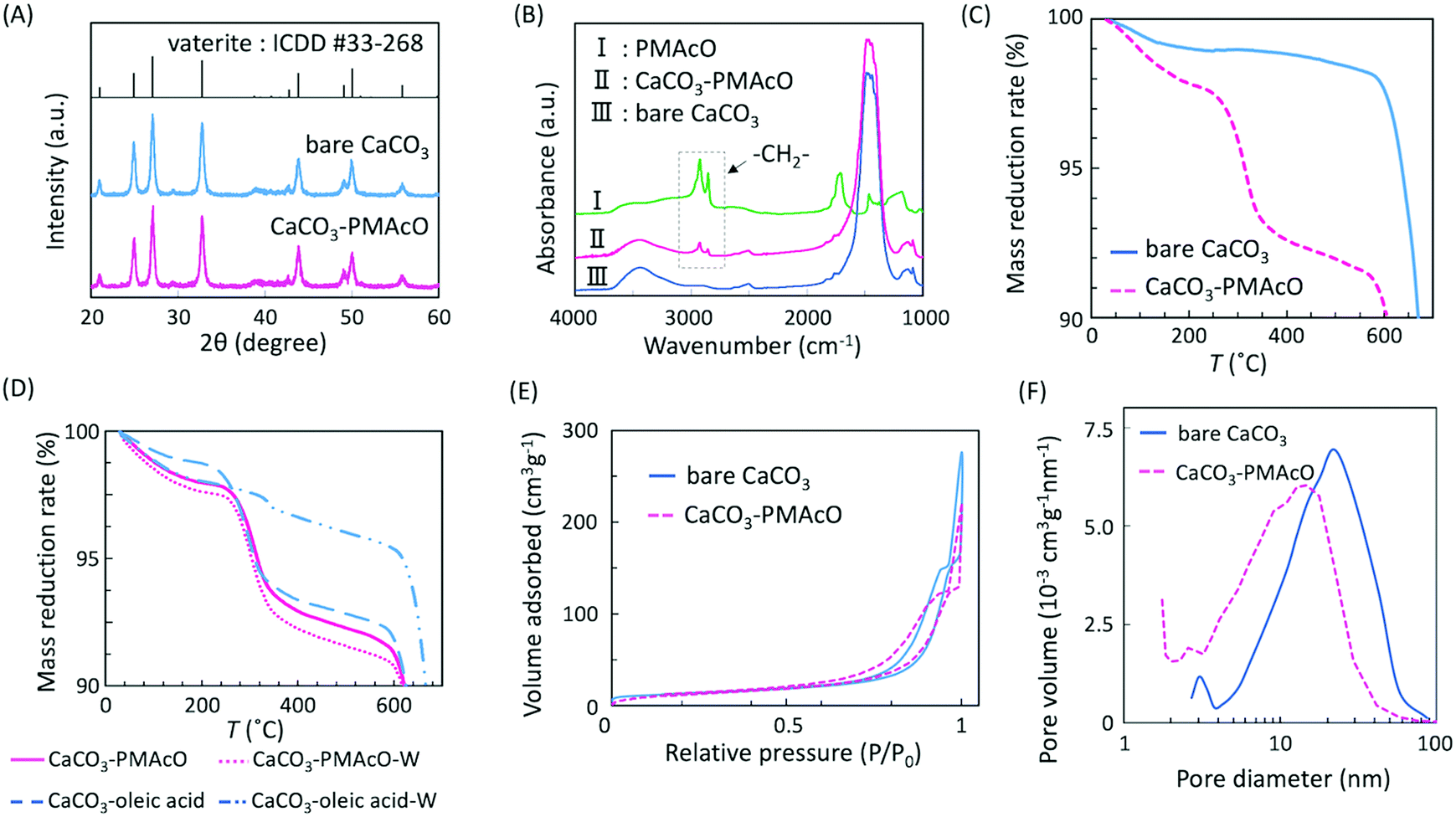 | ||
| Fig. 3 Physical properties of bare CaCO3 and CaCO3–PMAcO microspheres: (A) XRD spectra, (B) FT-IR spectra, (C) TG curves, (D) TG curves of CaCO3–PMAcO and CaCO3–oleic acid before and after washing; “-W” indicates samples after washing, (E) N2 adsorption–desorption isotherms, and (F) pore-size distributions. | ||
Evaluation of CaCO3–PMAcO microspheres as a column packing material for HPLC
CaCO3–PMAcO microspheres were packed into a column (100 × 2.1 mm I.D.) and their performance as a stationary phase for HPLC was evaluated. Fig. S4A (ESI†) shows the relationship between the mobile phase mixing ratio and the back pressure for methanol/water mixtures. At a flow rate of 0.4 mL min−1, the back pressure of the column was below 15 MPa (Fig. S4B, ESI†). Furthermore, the back pressure was found to be stable and, therefore, suitable for use with a conventional HPLC system. The separation mode of this column was evaluated through the elution behavior of naphthalene and alkylbenzenes (C0–C10) depending on the mixing ratio of methanol/water as the mobile phase. The retention factors of naphthalene and alkylbenzenes increased with decreasing proportions of methanol (Fig. S5, ESI,† and Fig. 4A) and longer alkyl chains resulted in higher hydrophobicity (Fig. 4A). These results indicate that CaCO3–PMAcO behaved as a reversed-phase mode stationary phase for the separation of naphthalene and alkylbenzenes, while pure CaCO3 generally works as normal-phase mode stationary phase.7 The octadecene groups of the CaCO3–PMAcO microspheres at the stationary phase to mobile phase interface performed a role identical to the octadecyl groups of ODS, resulting in reversed-phase characteristics. During the separation of a mixture of 11 alkylbenzenes, all peaks could be clearly observed with a baseline resolution (peak resolution Rs = 1.8–4.2 > 1.5) in the chromatogram (Fig. 4B).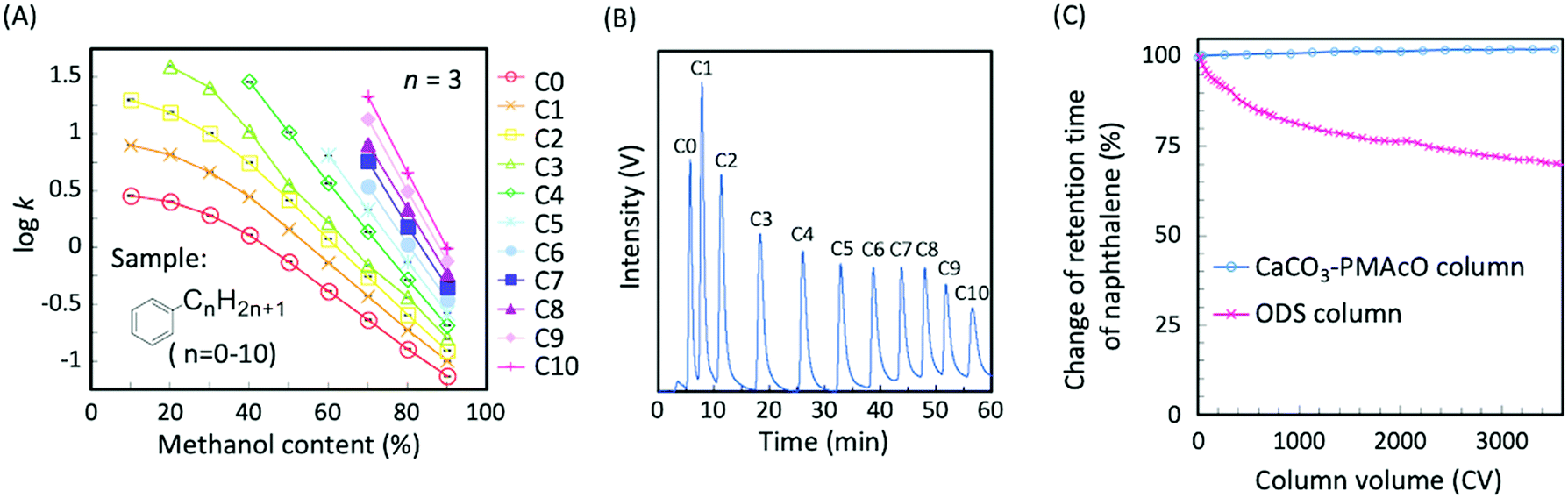 | ||
| Fig. 4 (A) Retention factors of C0–10 alkylbenzenes obtained with a CaCO3–PMAcO packed column. Each sample was eluted at each mixing ratio of the methanol/water mobile phase ranging from 10/90 (v/v) to 90/10 (v/v); error bars represent mean values ±1σ (n = 3); flow rate of mobile phase 0.3 mL min−1. (B) Chromatogram showing the separation of a mixture of C0–10 alkylbenzenes by the CaCO3–PMAcO column. Gradient elution was applied at a flow rate of 0.1 mL min−1 starting with methanol/water (50/50, v/v) for 4 min, followed by the gradient program up to 80/20 (v/v) over 36 min and finally elution with 80/20 (v/v) for 20 min. (C) Relative change of the retention time of naphthalene upon continuous running of alkaline mobile phase (50/50, v/v methanol/pH 10.8 Na2B4O7 buffer) in the case of CaCO3–PMAcO and ODS columns; flow rate of mobile phase 0.3 mL min−1. | ||
The durability of the CaCO3–PMAcO column against an alkaline mobile phase (mixture of pH 10.8 aqueous Na2B4O7 buffer and methanol) was evaluated by monitoring the elution behavior of naphthalene. The deterioration of the column was estimated by the relative change of the retention time during the continuous flow of the mobile phase. A comparative study was carried out using an ODS column prepared from a commercially available 3 μm ODS silica packing material through the same procedure as that for the CaCO3–PMAcO column. As shown in Fig. 4C and Fig. S6 (ESI†), the retention time and the number of theoretical plates of naphthalene were retained over 3500 column volumes (CVs) of alkaline mobile phase purging in the case of the CaCO3–PMAcO column (RSD of retention time: 0.7%), while it continuously decreased when using the ODS packed column (over 35% decrease after 3500 CV purges). It is assumed that the amount of octadecyl groups on the silica surface gradually decreased due to the inherent solubility of silica in alkaline media, resulting in a reduction of hydrophobic interaction with naphthalene. In contrast, the performance of the CaCO3–PMAcO packing in the alkaline mobile phase was preserved, since the solubility of CaCO3 decreases with increasing pH. These results indicated that CaCO3–PMAcO packed columns show excellent durability at high pH and could be used for routine analyses requiring alkaline mobile phases.
Quantitative analysis of basic antidepressants spiked into whole blood using a CaCO3–PMAcO packed column
As a proof-of-concept demonstration, we conducted the analysis of basic compounds requiring an alkaline mobile phase for successful separation. When a neutral mobile phase is used, basic compounds become positively charged, resulting in unwanted electrostatic and hydrogen bond interactions with the stationary phase in addition to hydrophobic interactions. On the other hand, these are eliminated by deprotonation of basic compounds in alkaline mobile phases, allowing the separation of basic compounds as free bases, which is attractive for routine analysis. Silica-based columns are reluctantly used because of their inherent instability to alkaline mobile phases.23 Herein, we demonstrate the analysis of the basic antidepressants clomipramine and imipramine (pKa of 9.3 and 9.5, respectively,24 shown in Fig. 5A), known as tricyclics. Since addiction to psychotropic drugs is a serious problem, the routine analysis of these substances in biological fluids is of significance.25 First, the effect of the mobile phase pH on the elution behavior of clomipramine using a CaCO3–PMAcO packed column was investigated. Fig. 5B shows the chromatograms of clomipramine on a CaCO3–PMAcO stationary phase eluted with a neutral or alkaline mobile phase. A sharper peak was obtained when using the alkaline mobile phase compared to the neutral mobile phase (1.4-fold number of theoretical plates; 0.7-fold peak asymmetry factor). Protonation of clomipramine in the neutral mobile phase resulted in the interaction with anionic carboxylates or carbonates, causing tailing and extension of the retention time. The suppression of clomipramine protonation eliminated these unwanted interactions, leading to a sharp chromatographic peak with simple retention behavior only governed by hydrophobic interactions between clomipramine and octadecene groups. In addition, clomipramine and imipramine were successfully separated by the CaCO3–PMAcO column in combination with the alkaline mobile phase (Fig. 5C). Linear calibration curves for clomipramine and imipramine were obtained based on the peak areas or heights from chromatograms of mixed samples of the two basic antidepressants (Fig. S7A and B, ESI†). Finally, porcine whole blood spiked with clomipramine and imipramine was analyzed using the CaCO3–PMAcO packed column and the alkaline mobile phase. The sample was prepared according to a general whole blood pretreatment method26 before HPLC analysis. Fig. 5D shows the corresponding chromatogram. Blood constituents (mainly serum albumin) and the two basic antidepressants were successfully separated. Furthermore, good recovery values (95.6–97.6%) and small relative standard deviations (RSD < 0.7%) calculated from the calibration curves (Fig. S7A and B, ESI†) were achieved (Table 1). On the other hand, the two basic antidepressants were not successfully separated with the ODS packed column in a neutral mobile phase (Fig. S8, ESI†). These results indicated that HPLC analysis with CaCO3–PMAcO packed columns in an alkaline mobile phase is practically applicable to the routine analysis of basic antidepressants.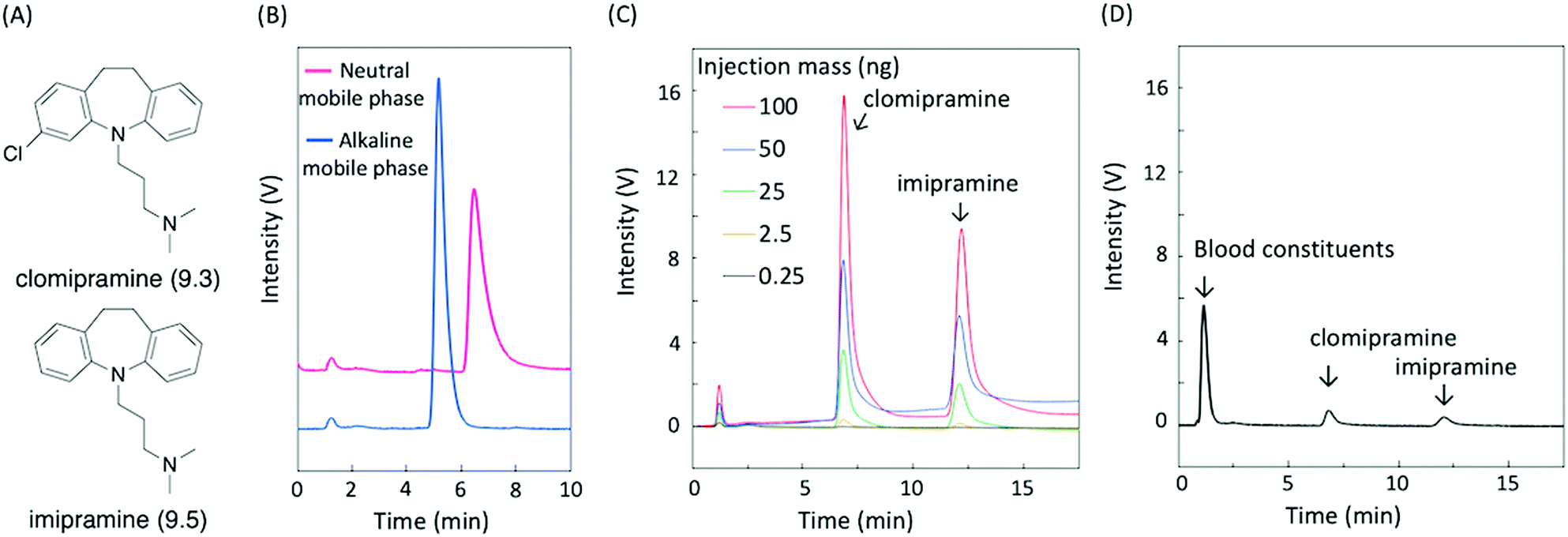 | ||
| Fig. 5 (A) Chemical structures and pKa of the basic antidepressants clomipramine and imipramine; chromatograms of basic antidepressants for the CaCO3–PMAcO column flowed at 0.3 mL min−1: (B) clomipramine (0.1 mg mL−1) with neutral and alkaline mobile phases. Methanol/water (63/37, v/v) was used as a neutral mobile phase and methanol/pH 10.8 Na2B4O7 buffer (63/37, v/v) was used as an alkaline mobile phase. (C) Mixture of two basic antidepressants (clomipramine and imipramine) with a methanol/pH 10.8 Na2B4O7 buffer (55/45, v/v). (D) Basic antidepressants (clomipramine and imipramine) spiked into whole blood of porcine with a methanol/pH 10.8 Na2B4O7 buffer (55/45, v/v). | ||
| Basic drug | Level | From peak area | From peak height | ||
|---|---|---|---|---|---|
| Recovery (%, n = 3) | RSD (%, n = 3) | Recovery (%, n = 3) | RSD (%, n = 3) | ||
| (ng μL−1) | |||||
| Imipramine | 5.0 | 96.4 | 0.2 | 95.6 | 0.3 |
| Clomipramine | 5.0 | 97.6 | 0.7 | 96.2 | 0.7 |
Conclusions
This work is to the best of our knowledge the first example of a biomimetic material in which the hierarchical structure and the polymer function are fused to enable effective control of substance distribution. By mimicking the formation process of biominerals, mesoporous CaCO3 microspheres were fabricated and surface-modified with an alternating amphiphilic copolymer consisting of hydrophobic alkyl chains and anionic carboxylate groups. Because of the monodisperse and mesoporous structure modified with hydrophobic octadecene groups on its surface, this biomineral-inspired hybrid material was found to be suitable for application in HPLC. When applied as a HPLC column packing material, it showed reversed-phase retention behavior and high resistance against alkaline mobile phases. In a proof-of-concept application, whole blood spiked with basic antidepressants clomipramine and imipramine was successfully analyzed with an alkaline mobile phase, in which conventional ODS packings have significant stability limitations. Thus, the present work clearly demonstrates the potential of biomineral-inspired PMAcO-modified mesoporous CaCO3 microspheres as a HPLC column packing material. By decreasing the particle size of the mesoporous CaCO3 microspheres, the pore size distribution can be narrowed. Furthermore, since decreasing the particle diameter leads to an expansion of the specific surface area and an improvement of packing density, it is expected that a higher separation ability can be obtained by decreasing the particle diameter. Furthermore, it is expected that various retention modes can be imparted to this packing material with a variety of maleic acid-based alternating polymers. Therefore, we expect that this work opens a new route for biomimetic materials in separation applications.Experimental section
Materials and instruments
Calcium chloride (CaCl2), sodium carbonate (Na2CO3), 5 wt% aqueous sodium hypochlorite (NaClO) solution, tetrahydrofran (THF) and HPLC-grade methanol were purchased from Kanto Chemical (Tokyo, Japan). Toluene, acetone, potassium hydroxide (KOH), trifluoroacetic acid (TFA), oleic acid, sodium tetraborate decahydrate (Na2B4O7·10H2O), sodium hydroxide (NaOH), naphthalene and clomipramine hydrochloride were purchased from Wako Pure Chemical (Osaka, Japan). Poly(sodium 4-sulfonate) (PSS, Mw: ∼70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and poly(maleic anhydride-alt-1-octadecene) (PMAO, Mn: 30000–50000) were purchased from Sigma-Aldrich (St. Louis, MO). Benzene, methylbenzene, ethylbenzene, amylbenzene, butylbenzene, hexylbenzene, heptylbenzene, n-octylbenzene, nonylbenzene, decylbenzene and imipramine hydrochloride were purchased from Tokyo Chemical Industry (Tokyo, Japan). A 3 μm fully porous ODS silica gel (CHEMCOSORB 3-ODS-L) was purchased from Chemco Scientific (Osaka, Japan). Porcine whole blood with 0.3 wt% citric acid was purchased from Tokyo Shibaura Zouki CO (Tokyo, Japan). Ultrapure water (18.2 MΩ cm) was obtained from a PURELAB flex water purification system (ELGA, Veolia Water, Marlow, U.K.).
000) and poly(maleic anhydride-alt-1-octadecene) (PMAO, Mn: 30000–50000) were purchased from Sigma-Aldrich (St. Louis, MO). Benzene, methylbenzene, ethylbenzene, amylbenzene, butylbenzene, hexylbenzene, heptylbenzene, n-octylbenzene, nonylbenzene, decylbenzene and imipramine hydrochloride were purchased from Tokyo Chemical Industry (Tokyo, Japan). A 3 μm fully porous ODS silica gel (CHEMCOSORB 3-ODS-L) was purchased from Chemco Scientific (Osaka, Japan). Porcine whole blood with 0.3 wt% citric acid was purchased from Tokyo Shibaura Zouki CO (Tokyo, Japan). Ultrapure water (18.2 MΩ cm) was obtained from a PURELAB flex water purification system (ELGA, Veolia Water, Marlow, U.K.).
Fabrication of CaCO3 microspheres
128 mL of 1 M CaCl2 solution was added to 4.0 mL of 16 mM Na2CO3 solution containing 1.0 g L−1 PSS. After mixing, the solution was stirred at 1500 rpm for 90 s with a discoid shape magnetic stirrer tip (diameter: 30.5 mm, and height: 12 mm) and then left for 24 h. After filtration, the organic components included in the products were extracted by immersion in 5 wt% NaClO aqueous solution for 48 h. The resultant microspheres were washed extensively with purified water and annealed at 400 °C, leading to bare CaCO3 microspheres.Preparation of PMAcO by hydrolysis of PMAO
315 mg of solid KOH was added into 10 mL of a solution of 1.0 g of PMAO in THF, followed by vigorous stirring for 2 hours at room temperature. After evaporating the solvent, the residue was dissolved in a small amount of acetone, and an excess amount of TFA was added. Aggregates were re-dissolved in 5 mL of acetone and slowly poured into 100 mL of water to precipitate the polymer. This purified polymer was collected by suction filtration with water washing and dried in a vacuum.Modification of PMAcO onto the surface of CaCO3 microspheres
100 mg of PMAcO was dissolved in 20 mL of THF and 30 mL of toluene. After addition of 1.0 g of bare CaCO3 microspheres, stirring was vigorously continued for 2 hours at 60 °C. The collected “CaCO3–PMAcO” microspheres were washed with a small amount of THF and dried at 60 °C. Oleic acid-modified CaCO3 microspheres were obtained by the same procedure. 100 mg of oleic acid was dissolved in 50 mL of toluene. After addition of 1.0 g of bare CaCO3 microspheres, stirring was vigorously continued for 2 hours at 60 °C. The collected “CaCO3–oleic acid” microspheres were washed with a small amount of THF and dried at 60 °C.Characterization of PMAcO, CaCO3 particles and modified CaCO3 particles
The morphologies of bare CaCO3 and CaCO3–PMAcO were observed using scanning electron microscopy (SEM, S-4700, Hitachi, Tokyo, Japan). Before observation, samples were coated with osmium using an osmium coater (HPC-1S, Vacuum Device, Ibaraki, Japan) for 15 s at 10 mA. For the observation of fractured CaCO3–PMAcO, the sample was prepared by a thick coating with osmium using an osmium coater for 60 s at 10 mA, followed by crushing with a microspatula. The size of microspheres was measured using the image processing software ImageJ (National Institutes of Health). The Brunauer–Emmett–Teller (BET) surface area, pore volume and pore size distributions were determined by N2 sorption (3Flex, Micromeritics, SHIMADZU, Kyoto, Japan). Before measurements, samples were degassed for 6 h at 160 °C. The crystal structures of the samples were confirmed by X-ray diffraction (XRD, MiniFlex II diffractometer, Rigaku, Tokyo, Japan) using CuKα radiation. Organic components of samples were also characterized using high throughput Fourier transform infrared spectroscopy (FT-IR, model-Alpha, Bruker, Germany). The contents of organic components of microspheres were measured by thermogravimetric analysis (TG, TG/DTA7200, SII, Chiba, Japan). Durability of PMAcO modification was evaluated by comparing the amounts of organic components in the 240–540 °C regime of the TG curves before and after washing with solvent. CaCO3–PMAcO was washed 10 times with 10 mL (total 100 mL) of a mixed solvent of water and methanol (50/50, v/v) with suction filtration and dried in a vacuum. CaCO3–oleic acid was evaluated according to the same procedure.Column packing
A slurry of CaCO3–PMAcO was packed into stainless steel columns (100 mm × 2.1 mm I.D., Chemco Scientific). The slurry of CaCO3–PMAcO beads (0.7 g) in methanol (13 mL) was poured into a slurry reservoir (Chemco Scientific) connected to the stainless steel column. Methanol was flowed through the slurry reservoir using a HPLC pump (LC-6AD, SHIMADZU) for 30 min, followed by flowing methanol/water (50/50, v/v) for 60 min with a constant pressure of 35 MPa. CHEMCOSORB 3-ODS-L was packed by the same procedure.Chromatographic analyses
Chromatographic analyses were carried out using a Prominence-I LC2030C (SHIMADZU) system equipped with a UV-Vis detector. All measurements were conducted at 25 °C with 1 μL injection volume. Elution behavior of all samples was monitored at 254 nm. All samples were dissolved in methanol and filtered through a 0.2 μm membrane filter. The pH of the 50 mM Na2B4O7 buffer was adjusted to 10.8 with 100 mM NaOH aq.Chromatographic parameters were calculated using the following equations:
Retention factor: k = (tR − t0)/t0, where t0 and tR are the retention times of solvent peak and the target analyte, respectively.
Peak resolution: RS = 2(tR2 − tR1)/(W1 + W2), where tR is the retention time (tR2 > tR1), W is the peak width of the analyte, and Rs > 1.5 indicates complete separation.
Theoretical plate: N = 16(tR/W)2
Peak asymmetry factor: As = b/a, where a is the time from the leading edge of the peak to the peak midpoint, and b is the time from the peak midpoint to the tailing edge.
Preparation of whole blood samples
Porcine whole blood samples were pretreated using a protein precipitation method.26 To a 2 mL centrifuge tube, 200 μL of whole blood, 100 μL of basic drug mixture (50 μg mL−1 imipramine, and 50 μg mL−1 clomipramine in methanol) or 100 μL of methanol as a blank sample, and 700 μL of acetonitrile were added. These solutions were mixed using a vortex mixer for 10 minutes and centrifuged at 10000 rpm for 10 minutes. The supernatant was filtered through a 0.2 μm membrane filter.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Mann, Nature, 1993, 365, 499–505 CrossRef CAS.
- Y. Oaki, A. Kotachi, T. Miura and H. Imai, Adv. Funct. Mater., 2006, 16, 1633–1639 CrossRef CAS.
- H. B. Yao, J. Ge, L. B. Mao, Y. X. Yan and S. H. Yu, Adv. Mater., 2014, 26, 163–188 CrossRef CAS PubMed.
- O. D. Wangensteen, D. Wilson and H. Rahn, Respir. Physiol., 1970, 11, 16–30 CrossRef CAS PubMed.
- S. Mann, B. R. Heywood, S. Rajam and J. D. Birchall, Nature, 1988, 334, 692–695 CrossRef CAS.
- S. Matsumura, S. Kajiyama, T. Nishimura and T. Kato, Small, 2015, 11, 5127–5133 CrossRef CAS PubMed.
- K. Sato, Y. Oaki, D. Takahashi, K. Toshima and H. Imai, Chem. – Eur. J., 2015, 21, 5034–5040 CrossRef CAS PubMed.
- S. Kim and C. B. Park, Langmuir, 2010, 26, 14730–14736 CrossRef CAS PubMed.
- Y. Lai, L. Chen, W. Bao, Y. Ren, Y. Gao, Y. Yin and Y. Zhao, Cryst. Growth Des., 2015, 15, 1194–1200 CrossRef CAS.
- A. W. Xu, M. Antonietti, S. H. Yu and H. Cölfen, Adv. Mater., 2008, 20, 1333–1338 CrossRef CAS.
- M. Abebe, N. Hedin and Z. Bacsik, Cryst. Growth Des., 2015, 15, 3609–3616 CrossRef CAS.
- G. D. Profio, S. M. Salehi, R. Caliandro, P. Guccione, G. Nico, E. Curcio and E. Fontananova, Adv. Mater., 2016, 28, 610–616 CrossRef PubMed.
- H. Imai, N. Tochimoto, Y. Nishino, Y. Takezawa and Y. Oaki, Cryst. Growth Des., 2012, 12, 876–882 CrossRef CAS.
- A. Inoue, H. Tamagawa, Y. Oaki, S. Aoshima and H. Imai, J. Mater. Chem. B, 2015, 3, 3604–3608 RSC.
- T. Kato, Adv. Mater., 2000, 12, 1543–1546 CrossRef CAS.
- M. Tswett, Ber. Dtsch. Bot. Ges., 1906, 316–323 CAS.
- J. Zhang, J. Guo, T. Li and X. Li, Int. J. Green Nanotechnol. Phys. Chem., 2010, 1, P65–P71 CrossRef.
- H. A. Claessens and M. A. van Straten, J. Chromatogr. A, 2004, 1060, 23–41 CrossRef CAS PubMed.
- Z. M. O. Rzaev, Prog. Polym. Sci., 2000, 25, 163–217 CrossRef CAS.
- W. Lee, A. R. Esker and H. Yu, Colloids Surf., A, 1995, 102, 191–201 CrossRef CAS.
- H. Tamagawa, H. Kageyama, Y. Oaki, Y. Hoshino, Y. Miura and H. Imai, Chem. Lett., 2015, 44, 1425–1427 CrossRef CAS.
- P. Fenter and N. C. Sturchio, Geochim. Cosmochim. Acta, 1999, 63, 3145–3152 CrossRef CAS.
- J. J. Kirkland, M. A. van Straten and H. A. Claessens, J. Chromatogr. A, 1998, 797, 111–120 CrossRef CAS.
- C. Alves, C. Fernandes, A. Jose dos Santos Neto, J. C. Rodrigues, M. E. Costa Queiroz and F. M. Lancas, J. Chromatogr. Sci., 2006, 44, 340–346 CAS.
- K. Kudo, T. Ishida, W. Hikiji, Y. Usumoto, T. Umehara, K. Nagamatsu, A. Tsuji and N. Ikeda, Forensic Toxicol., 2010, 28, 25–32 CrossRef CAS.
- A. E. Steuer, M. Poetzsch, M. Koenig, E. Tingelhoff, S. N. Staeheli, A. T. Roemmelt and T. Kraemer, J. Chromatogr. A, 2015, 1381, 87–100 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional material characterizations, chromatographic results. See DOI: 10.1039/c9tb01014a |
| This journal is © The Royal Society of Chemistry 2019 |