
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene oxide prevents mycobacteria entry into macrophages through extracellular entrapment
Flavio
De Maio
a,
Valentina
Palmieri
 *b,
Alessandro
Salustri
a,
Giordano
Perini
b,
Maurizio
Sanguinetti
a,
Marco
De Spirito
b,
Giovanni
Delogu
a and
Massimiliano
Papi
b
*b,
Alessandro
Salustri
a,
Giordano
Perini
b,
Maurizio
Sanguinetti
a,
Marco
De Spirito
b,
Giovanni
Delogu
a and
Massimiliano
Papi
b
aInstitute of Microbiology, Università Cattolica del Sacro Cuore, Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma, Italy
bInstitute of Physics, Università Cattolica del Sacro Cuore, Fondazione Policlinico Universitario A. Gemelli IRCCS, Largo A. Gemelli, 8, 00168, Roma, Italy. E-mail: valentina.palmieri@unicatt.it
First published on 16th January 2019
Abstract
Tuberculosis (TB) remains a global threat and there is an urgent need for improved drugs and treatments, particularly against the drug-resistant strains of Mycobacterium tuberculosis (Mtb). Graphene oxide (GO) is an innovative bi-dimensional nanomaterial that when administered in vivo accumulates in the lungs. Further, GO is readily degraded by peroxidases and has a high drug loading capacity and antibacterial properties. In this study, we first evaluated the GO anti-mycobacterial properties using Mycobacterium smegmatis (Ms) as a model. We observed that GO, when administered with the bacteria, was able to trap Ms in a dose-dependent manner, reducing entry of bacilli into macrophages. However, GO did not show any anti-mycobacterial activity when used to treat infected cells or when macrophages were pre-treated before infection. Similar results were obtained when the virulent Mtb strain was used, showing that GO was able to trap Mtb and prevent entry into microphages. These results indicate that GO can be a promising tool to design improved therapies against TB.
Introduction
Tuberculosis (TB) causes approximately 1.8 million deaths each year and around 10.4 million people fell ill with TB in 2016.1 Interestingly, around a quarter of the global population is estimated to be infected by Mycobacterium tuberculosis (Mtb), the etiologic agent of human TB.1,2When Mtb reaches the alveolus it is phagocytized by alveolar macrophages that can block the infection.3 If macrophages fail, active Mtb replication ensues with infection of nearby cells and the bacteria spread to other tissues.3,4 After these early events, the emergence of the host adaptive immune response usually contains Mtb replication, preventing the development of active disease and leading to a latent state of infection where Mtb persists in a dynamic equilibrium with the host without inducing symptoms or signs of disease.5–7 For reasons that still remain unknown, TB reactivation may occur years or decades following primary infection.3,8,9 A rapid diagnosis and the prompt start of the drug regimen can greatly contribute to reducing TB incidence and associated deaths, as clearly stated by the target goals of the End TB strategy.1
Anti-TB treatments are complex and long-lasting; guidelines recommend the establishment of a 6 month regimen of four first-line drugs (isoniazid, rifampicin, ethambutol and pyrazinamide) or four first-line drugs for 2–3 months followed by isoniazid and rifampicin for 2 months.1 These drug regimens can be associated with toxic events that may contribute to non-compliance and poor treatment outcomes.10 Treatment of multidrug resistant (MDR-TB) or extensively drug resistant (XDR-TB) strains requires the use of second-line drugs which are more toxic, expensive and less effective, as shown by the success rate of 54% for MDR-TB compared to 85% for drug-susceptible TB.1,11
New approaches based on nanomaterials have been proposed in TB treatment to increase drug efficacy and/or to maximize organ localization.12 The innovative bi-dimensional nanomaterial graphene oxide (GO) made of a carbon honeycomb and oxygen functional groups might represent an important support to treat mycobacterial infections.13,14 First, it has been demonstrated that GO accumulates in the lungs of mice when intratracheally15 or intravenously administered.16 GO inhalation did not show any toxic effects on rat health or any significant changes in bronchoalveolar proteins, cellular composition and inflammatory markers.17,18 Second, myeloperoxidases, well known anti-bacterial enzymes, can perform clearance of carbon nanomaterials in the lungs.19 GO is indeed degraded by peroxidases which create holes in the basal plane of this nanomaterial.20 Finally, GO does not penetrate large microorganisms with thick cell walls (like fungi) but displays a wrapping/trapping effect: GO sheets build a blanket/web surrounding and isolating microorganisms from the external environment, hindering proliferation and nutrient consumption.21 This phenomenon could effectively limit Mtb replication and its spreading.
In this study, we characterize the GO anti-mycobacterial activity against the non-pathogenic species Mycobacterium smegmatis (Ms) and the human pathogen Mtb.
Results
Graphene oxide characterization
GO characterization was performed with several techniques and the data are summarized in Fig. 1a. As shown in Fig. 1b, the morphology of GO samples was visualized by AFM. The size distribution analysis showed that most GO sheets (>95%) had sizes comprised between 450 nm and 870 nm and an average thickness of 0.78 ± 0.03 nm, indicating single-layered GO sheets in solution. The size of GO samples was confirmed by dynamic light scattering (average hydrodynamic radius of 511 ± 37 nm). The UV-vis spectrum in Fig. 1c shows an absorption peak at 230 nm, and a shoulder at ∼300 nm as previously reported.22–24 According to the literature, the λmax is determined by π → π* transitions while the shoulder peak is attributed to n → π* transitions of the carbonyl groups.25,26 GO showed a zeta potential of −36.9 ± 9.6 mV and a C/O ratio of 1.9 ± 0.1, according to previous reports.27 All these data confirmed that the GO used was monodisperse and highly oxidized.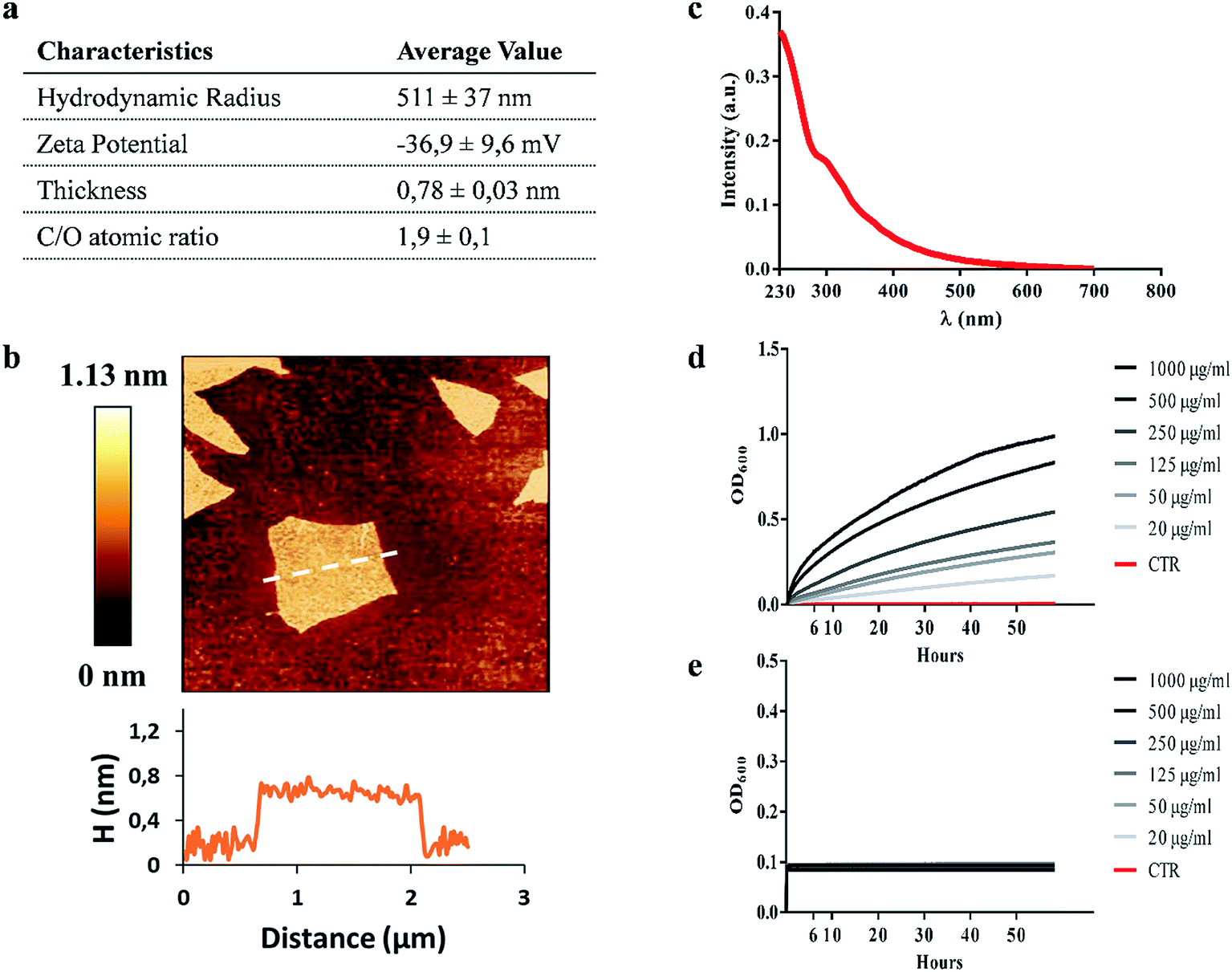 | ||
| Fig. 1 Graphene oxide characterization. Table summarizing the characterization of graphene oxide (GO) size, surface charge and oxidation degree of GO nanomaterial (a). A representative image of GO flakes and height profile obtained by Atomic Force Microscopy (AFM) (b). UV-vIS spectrum of GO characterized by two absorption peak maxima at ∼230 nm and ∼300 nm (c). GO stability at different concentrations (1000 μg ml−1 to 20 μg ml−1) in 7H9 mycobacterial medium (d) and ddH2O (e) obtained using Optical Density (OD) measurements. | ||
GO reduces Ms colony forming units (CFUs), in the absence of a direct mycobactericidal effect
We assessed the anti-mycobacterial activity of GO by first measuring bacterial viability following incubation of the non-pathogenic strain Ms, with serial dilutions of GO in a specific mycobacterial liquid medium (7H9). Firstly, we assayed the GO stability after incubation in 7H9 or in ddH2O. As shown in Fig. 1d, GO aggregates in a dose-dependent manner in 7H9. Conversely GO is stable in ddH2O (Fig. 1e). This aggregation masks GO edges and can be exploited to facilitate trapping of bacteria. Indeed, in previous studies it was demonstrated that salts and nutrients in media used for bacterial and cell culture cause GO aggregation and that bivalent cations favour the tight attachment of bacteria to the surface.13,27The results in Fig. 2a indicate that incubation of Ms with GO for 6 h significantly reduced bacterial CFUs in the medium in a dose-dependent manner. Particularly, concentrations of 1000 μg ml−1 and 500 μg ml−1 significantly reduced bacterial replication (p < 0.0001).
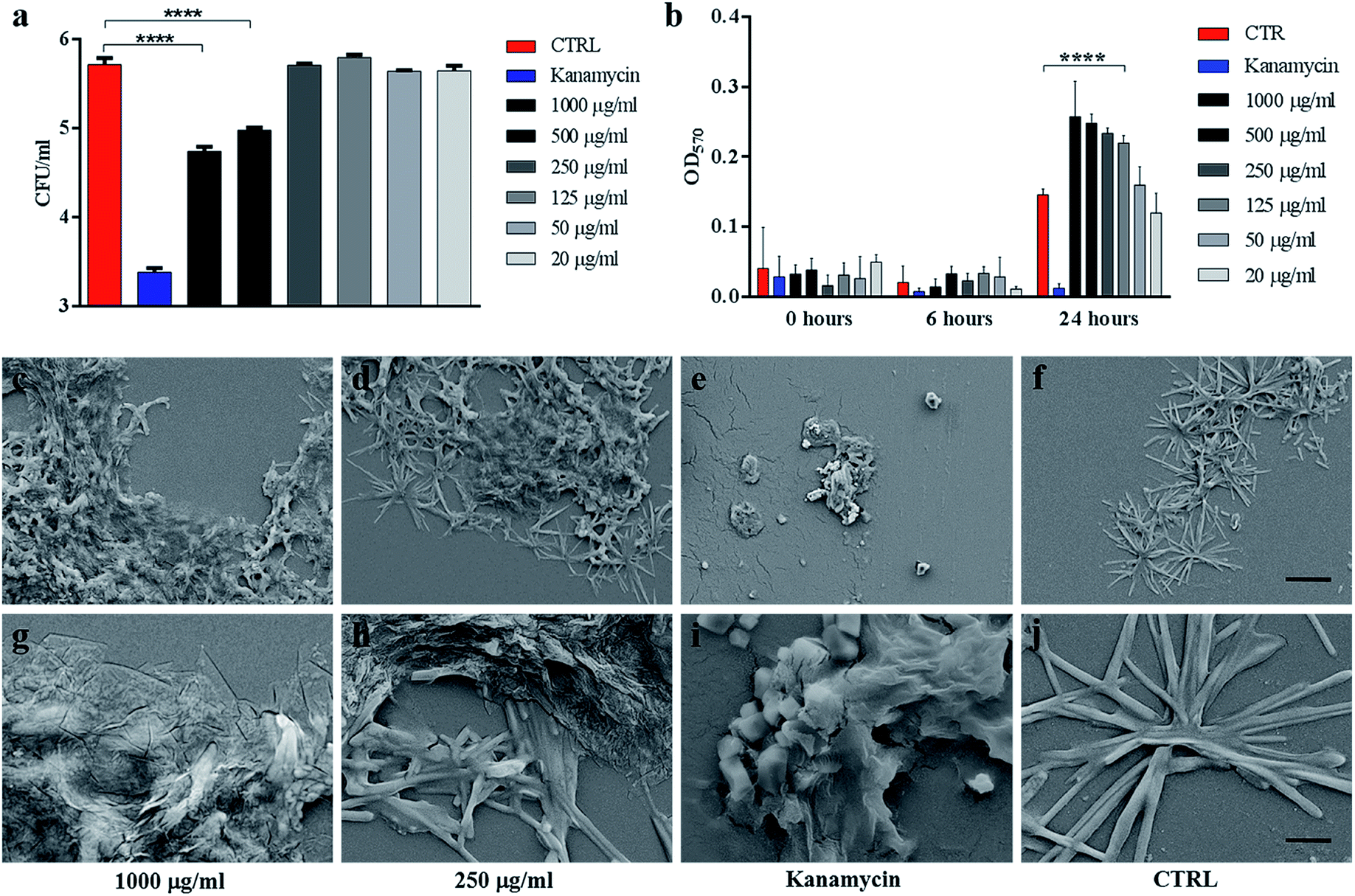 | ||
| Fig. 2 . GO reduces Ms colony forming units without affecting its viability. Ms was incubated with serial dilutions of GO (1000 μg ml−1 to 20 μg ml−1), kanamycin or with the medium alone for six hours. Colony forming units (CFUs) and bacterial viability, measured by the Alamar assay, were quantified (a–b). Aliquots of Ms incubated with GO (1000 μg ml−1 and 250 μg ml−1), kanamycin or with the medium alone were analysed by SEM to evaluate direct bacterial damage (c–l). One-way ANOVA was used to analyse the Alamar assay results and CFUs. CFUs are reported in log10 scale. | ||
The impact of GO on mycobacterial metabolism was assessed by the Alamar assay. As shown in Fig. 2b, the reduction of resazurin, which indicates the presence of metabolically active mycobacteria, was measured in all the samples regardless of GO concentration with the only exception of bacteria treated with the antibiotic kanamycin. Additionally, GO appeared to stimulate mycobacterial metabolism at 24 h post treatment in line with the GO probiotic effect observed on other bacteria.27,28
Ms treated with GO was harvested and analysed using scanning electron microscopy (SEM) to assess cell integrity and bacterial and GO interaction in the medium. As shown in Fig. 2(c, d, g and h), mycobacteria aggregates were detected at increasing concentrations of GO with no significant damage to the mycobacterial cells. Mycobacteria treated with kanamycin were visibly lysed or damaged and showed altered morphology as shown in Fig. 2(e and i).
To confirm the trapping effect, GO–Ms suspensions were sonicated at a power that promotes rupture of bacterial aggregates (Fig. 3a) and GO sheets,29 allowing the dispersal of bacterial cells. As shown in Fig. 3b, the increase of CFUs following sonication indicates that GO does not exert direct mycobactericidal activity and confirms that mycobacteria were alive and entrapped by GO sheets.
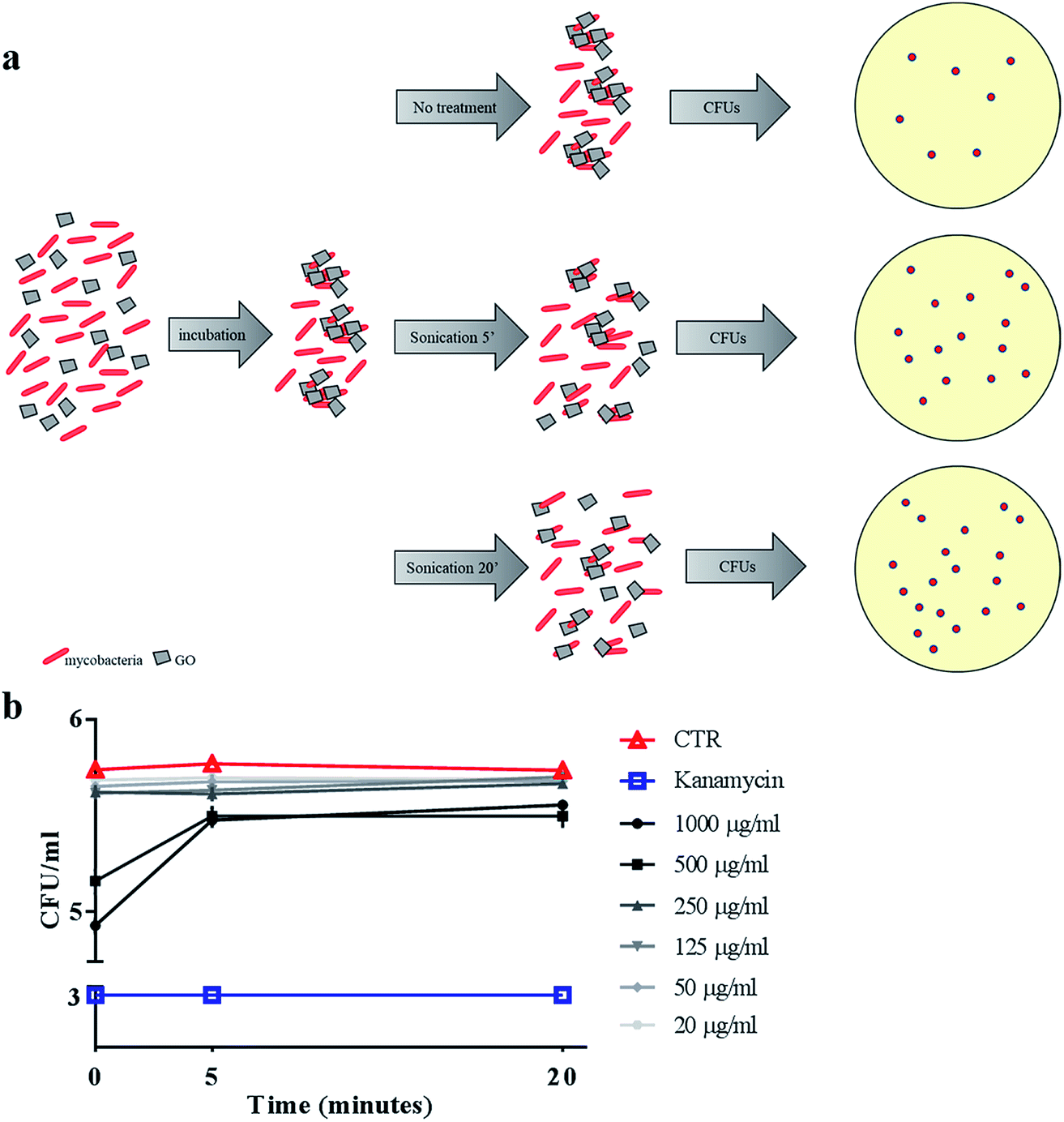 | ||
| Fig. 3 Sonication disrupts GO nets releasing live Ms. Ms was incubated with serial dilutions of GO (1000 μg ml−1 to 20 μg ml−1), kanamycin or with the medium alone. After six hours, suspensions were sonicated for five or twenty minutes (a) and colony forming units (CFUs) were quantified (b). CFUs are reported in log10 scale. | ||
Taken together, these results indicate that GO treatment promotes trapping of mycobacteria without affecting metabolism or cell integrity, suggesting that the reduction in CFUs observed is not the result of direct GO antimicrobial activity but is caused by bacterial trapping in GO webs formed in 7H9 medium.
GO reduces Ms entry into macrophages
A main feature of mycobacteria is their ability to enter into and survive in macrophages. To test the impact of GO on the mycobacterial entry into phagocytic cells, murine macrophages (J774) were infected with Ms at a MOI of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. We assessed GO effects under 3 different experimental conditions, i.e. by infecting cells (i) simultaneously with GO and bacteria, (ii) first with bacteria and subsequently with GO and (iii) first with GO and subsequently with bacteria (see Materials and methods for further details). Under all the conditions, intracellular CFUs were quantified and GO aggregate–bacterial interaction was assessed by confocal microscopy using a fluorescent Ms strain expressing cytosolic GFP.30
:1. We assessed GO effects under 3 different experimental conditions, i.e. by infecting cells (i) simultaneously with GO and bacteria, (ii) first with bacteria and subsequently with GO and (iii) first with GO and subsequently with bacteria (see Materials and methods for further details). Under all the conditions, intracellular CFUs were quantified and GO aggregate–bacterial interaction was assessed by confocal microscopy using a fluorescent Ms strain expressing cytosolic GFP.30
First, to better highlight the ability of GO to block extracellular mycobacterial entry, GO at three different concentrations (1000 μg ml−1, 100 μg ml−1 and 10 μg ml−1) was added in the infecting solution, i.e. together with Ms (Fig. 4a). In confocal microscopy experiments, we observed a reduction of intracellular bacteria in macrophages and the formation of bacterial clusters in the extracellular environment (Fig. 4b). Quantification of the number and size of clusters by image analysis showed a reduction in the number of larger clusters at higher GO concentrations (Fig. 4c). CFU analysis showed that the presence of GO in the infecting solution caused a reduction in Ms entry in a dose-dependent manner (Fig. 4d) which was confirmed by confocal analysis. Additionally, no direct GO cytotoxic effect on J774 macrophages was observed as assessed by measuring cell viability (Fig. 4e).
 | ||
| Fig. 4 GO prevents Ms entry into macrophages. To elucidate the Ms-blocking properties of graphene oxide (GO), murine macrophages (J774) were infected with Ms expressing GFP (MsGFP) at an MOI of 10:1 and GO (1000 μg ml−1, 100 μg ml−1 and 10 μg ml−1) or gentamycin. Four hours post infection, the infection solution was removed, and the cells were washed two times (a). Infected cells were fixed by using 4% PFA, permeabilized and marked with red phalloidin in order to perform confocal microscopy analysis (b). Finally, colony forming units (CFUs) were enumerated (d). To exclude that GO-toxicity affected CFUs, a viability assay was performed on GO-treated macrophages (e). One-way ANOVA was used to analyse CFUs, cluster results and the viability assay. CFUs are reported in log10 scale. | ||
To determine whether GO was able to restrict mycobacteria intracellularly, infected J774 murine macrophages were washed and then treated with different concentrations of GO (Fig. 5a). As expected, when GO was added on the macrophages already infected with Ms, no reduction in CFUs or decrease in fluorescent aggregates was measured (Fig. 5b and c), indicating that GO and bacteria interaction is necessary to obtain the antimicrobial effect.
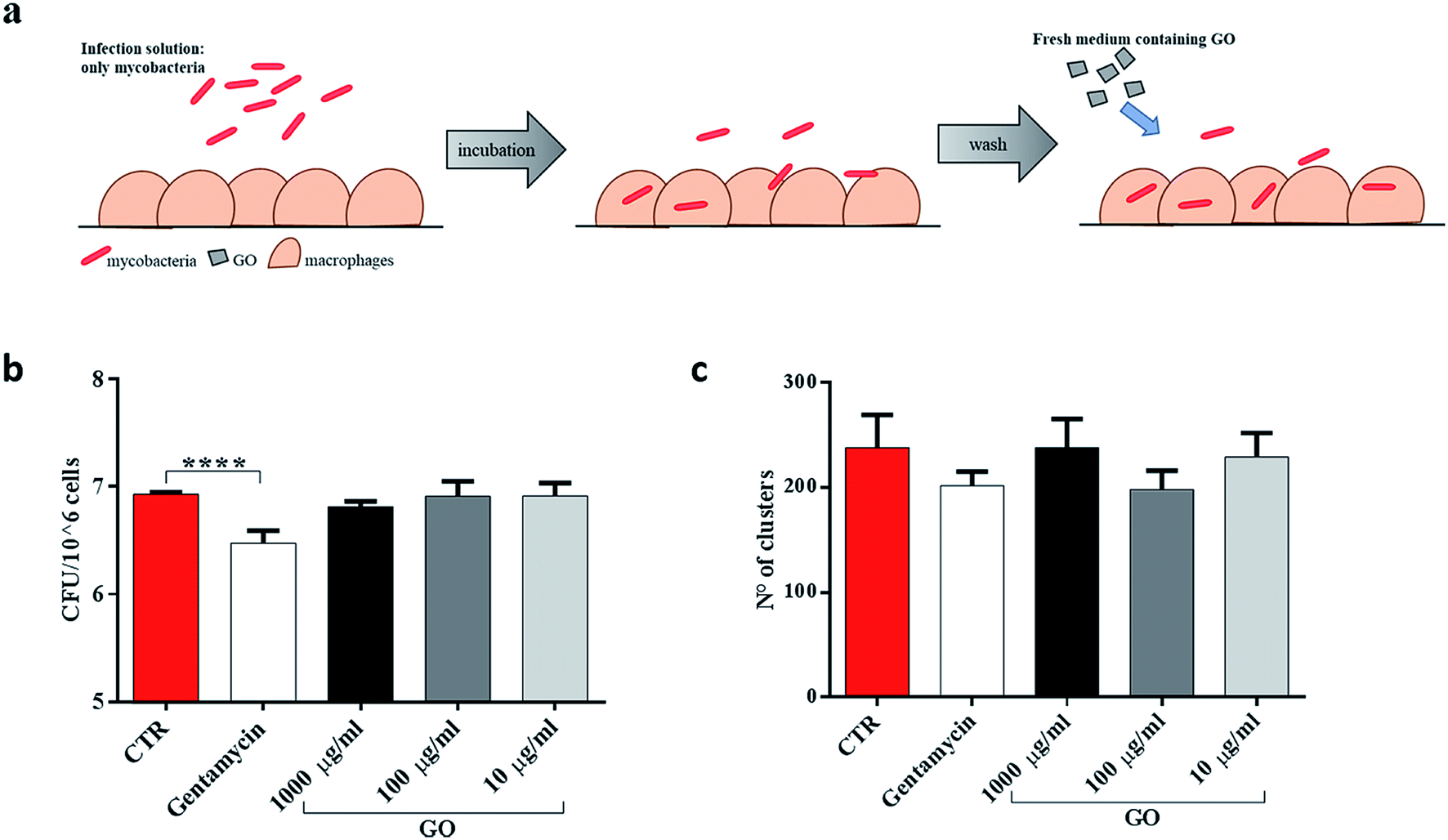 | ||
| Fig. 5 GO prevents entry only when mycobacteria are in the extracellular space. To distinguish the role of GO in preventing mycobacterial entry from cytosolic activity, murine macrophages (J774) were infected with Ms expressing GFP (MsGFP) (MOI 10:1). Four hours later, the infection solution was removed, the cells were washed two times and new medium containing GO (1000 μg ml−1, 100 μg ml−1 and 10 μg ml−1) or gentamycin and fresh medium alone was added (a). CFU analysis was carried out two hours later, as well as confocal microscopy analysis (b and c). One-way ANOVA was used to analyse CFUs or cluster results. CFUs are reported in log10 scale. | ||
Finally, since recent studies have demonstrated that large GO sheets are adsorbed on the macrophage plasma membrane,31 we hypothesized that adsorbed GO creates a barrier which potentially reduces mycobacterial entry. To assess this hypothesis, macrophages were first treated with a medium containing GO or gentamycin and then infected with Ms (Fig. 6a). No visible reduction of CFUs was observed when GO pre-treated macrophages were infected with the Ms strain (Fig. 6b and c).
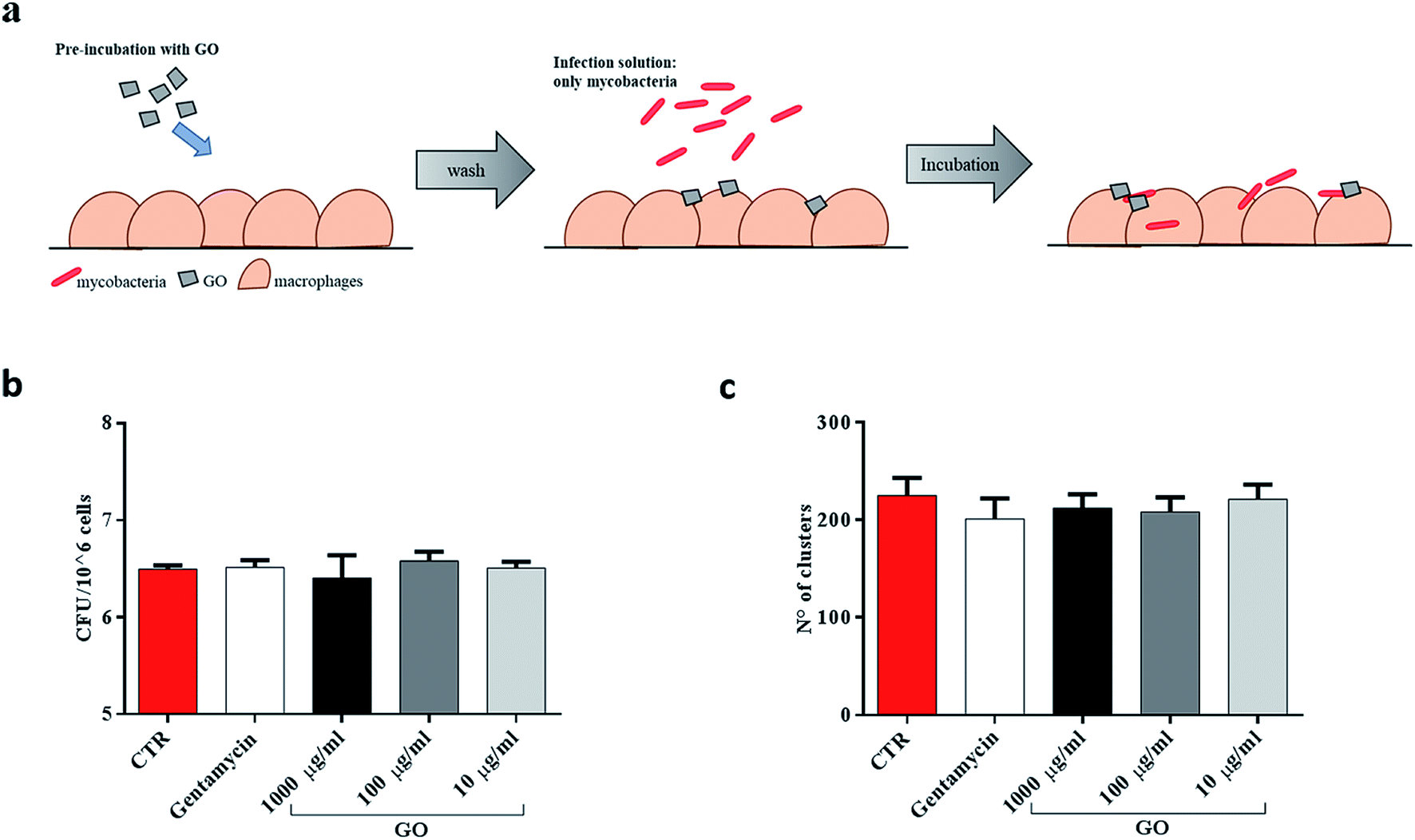 | ||
| Fig. 6 GO does not create a barrier to hinder mycobacteria entry into macrophages. To disprove the fact that GO creates a barrier on the cell surface, murine macrophages (J774) were pre-treated with medium containing GO (1000 μg ml−1, 100 μg ml−1 and 10 μg ml−1) or gentamycin and medium alone (a). Two hours later, J774 were infected with Ms expressing GFP (MsGFP) (MOI 10:1). Four hours post infection, CFUs and confocal images were analysed (b and c). One-way ANOVA was used to analyze CFUs or clusters results. CFUs are reported in log10 scale. | ||
Taken together, these results suggest that GO can inhibit Ms entry into macrophages by trapping mycobacteria while they are in the extracellular space, and it does not show any anti-mycobacterial activity inside the cells or any formation of a physical barrier to limit the infection.
GO decreases Mtb CFUs in an antimicrobial assay and reduces its entry into macrophages
Ms is a model organism for mycobacteria, though the pathogenic and virulence properties of the human pathogen Mtb are much higher. To confirm the observed trapping effects of GO, the same antimicrobial assay was carried out by using virulent Mtb. While GO treatment reduced Ms CFUs at up to 500 μg ml−1, its efficacy on Mtb was evident only at 1000 μg ml−1 (Fig. 7a). The trapping phenomenon and the consequent tight association between bacterial cells can affect the linear relationship between viable bacteria in the culture and CFUs. To weigh the impact of trapping on the observed CFU counting, we incubated Mtb with GO as illustrated in Fig. 3a, but before plating for CFU counting the suspension was sonicated (5 or 20 minutes) to disrupt bacterial aggregates with the GO mesh. The results indicate again that the CFUs counted from the mycobacterial–GO suspension increase with the sonication time (Fig. 7b). Hence, GO does not show a bactericidal effect against Mtb.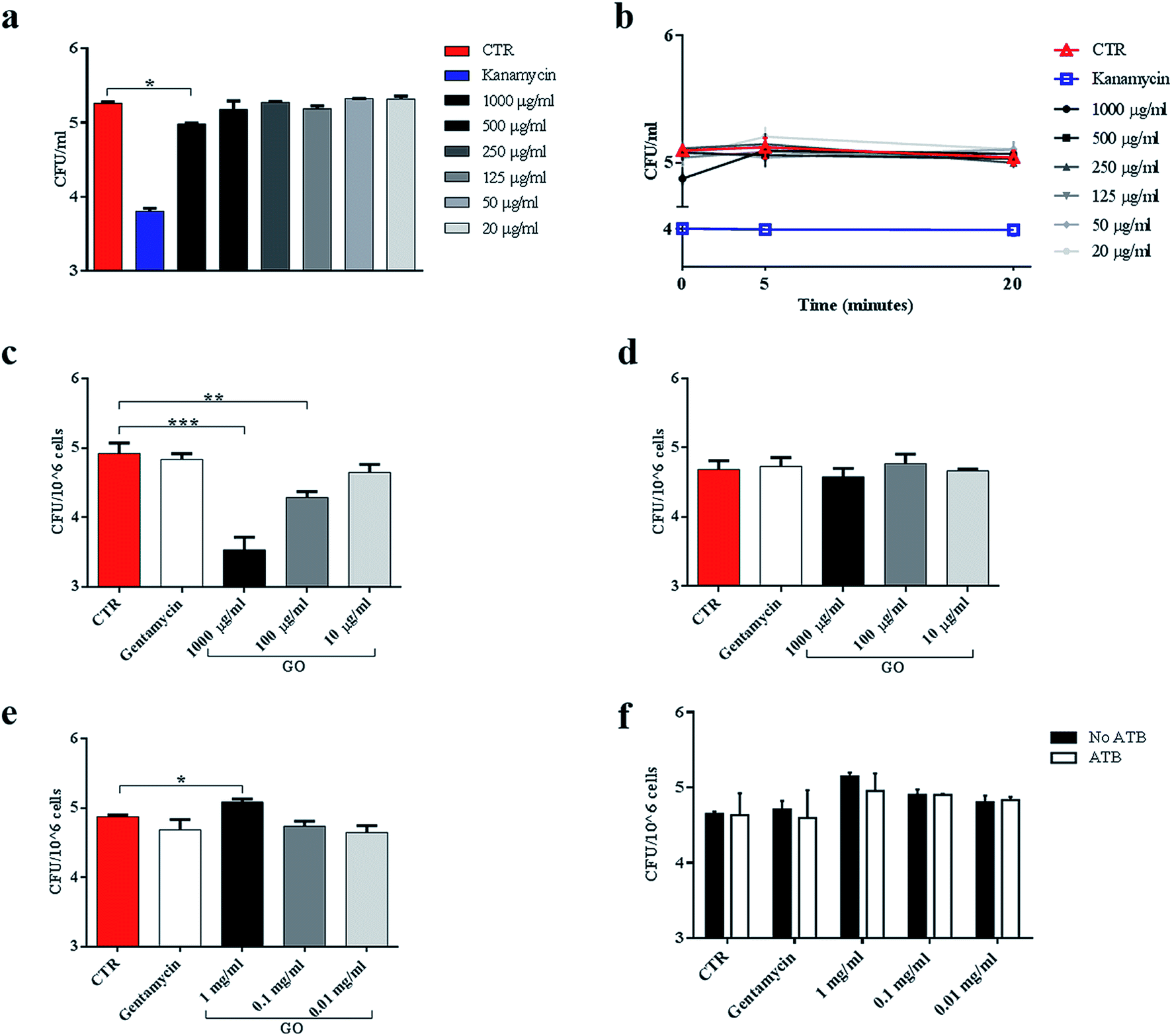 | ||
| Fig. 7 GO causes trapping of Mycobacterium tuberculosis and significantly reduces entry into macrophages. To evaluate the trapping effect on the virulent Mtb, the reference Mtb H37Rv expressing cytosolic GFP (MtbGFP) was incubated with serial concentrations of GO (1000 μg ml−1 to 20 μg ml−1), kanamycin or medium alone, and colony forming units (CFUs) were enumerated six hours later directly from the suspension (a) or after sonication (b). Murine macrophages (J774) were infected with MtbGFP (MOI 1:1, 1 hour) in a medium containing GO (1000 μg ml−1, 100 μg ml−1 and 10 μg ml−1) or gentamycin, and finally incubated for 5 h (c). In the second experimental setting, J774 were infected with a suspension of Mtb (MOI 1:1) and after 1 h were washed and GO was added after 5 h (d). Finally, J774 were pre-treated with GO and after 4 h were infected with Mtb (MOI 1:1) (e). Finally, pre-treated infected cells were also incubated with gentamycin and CFU analysis was carried out two hours later (f). One-way ANOVA was used to analyse CFUs or clusters results. CFUs are reported in log10 scale. | ||
To assess the impact of GO on Mtb entry into macrophages, J774 were infected with a solution containing Mtb and GO as previously described. As shown in Fig. 7c, a dose-dependent inhibition of Mtb entry into macrophages was observed, which was consistent with a reduction in fluorescent bacteria in/on macrophages (data not shown). Again, when GO was added to the supernatants of cells previously infected with Mtb, no reduction in intracellular CFUs (Fig. 7d) or fluorescent bacteria on cells was observed (data not shown) as for Ms. These results confirm the ability of GO to reduce mycobacterial cell internalization by trapping Mtb in the extracellular space.
Finally, macrophages were pre-treated with GO and then infected. Interestingly, at high GO concentration (1000 μg ml−1), a slight increase in CFUs was observed (Fig. 7e). To assess whether this increase was caused by a promotion of adhesion of Mtb to cell membranes mediated by GO, we repeated the experiment and added an antibiotic 1 h post infection to eliminate possible Mtb adhering to macrophages. No differences were observed between antibiotic treated or untreated specimens and comparable CFUs were measured (Fig. 7f).
These results indicate that GO at high dose could promote the entry of Mtb into macrophages, while this effect was not observed when macrophages were infected with the non-virulent Ms species.
Discussion
Among the new therapeutic approaches, nanomaterials have been proposed as new anti-bacterial drugs or as carriers for existing antibiotics to increase efficacy and targeted delivery.12 In this context, GO could be seen as a potential anti-mycobacterial agent due to its bactericidal and bacteriostatic properties observed on several bacterial species.32,33 In this study, we investigated for the first time the anti-mycobacterial activity of GO flakes with large lateral sizes (∼400–800 nm) in axenic cultures and in the in vitro macrophage infection model. The GO flake size range was chosen to comply with that in previous studies which showed that large GO flakes are poorly phagocytized and do not affect the viability of macrophages and their functions (see Fig. 4e).29,34We tested GO concentrations starting from 10 μg ml−1 up to 1000 μg ml−1, which are concentrations that can be of significance for TB since GO is known to preferentially accumulate in the lungs when administered intratracheally15 or intravenously.16 Generally, the GO antibacterial effect is thought to be mediated by a mechanical cutting or perturbation of cell membranes followed by oxidative stress induction.21 Conversely, we demonstrate the GO trapping effect both on Ms and on the human pathogen Mtb. The trapping effect occurs when GO is well dispersed in solution, but its instability and aggregation can favor the formation of a web. This phenomenon depends on the inability of GO to penetrate microorganisms, especially large microorganisms with thick cell walls like fungi, determining the building of a blocking web.21
We first tested the interaction of GO with Ms, an environmental non-pathogenic fast-growing mycobacterium. We observed a significant reduction of CFUs after incubation with GO in a dose-dependent manner. SEM analysis indicated that this reduction was mediated by a trapping mechanism and not by direct bacterial cell damage, as observed when mycobacteria were incubated with kanamycin. Indeed, measurements of cell metabolism and sonication of the GO/Ms suspension proved that the interaction with GO flakes did not affect mycobacterial growth or cell integrity, in line with previous observations obtained in larger microorganisms as fungi.13 We confirmed the trapping properties of GO with Mtb even though this effect was significant only at the highest GO concentration. Mtb and mycobacteria in general possess a unique cell wall, with a chemical composition and features that set them apart from most of the other bacteria.35 Furthermore, Mtb is a rod-shaped bacillus that can reach several microns in length, and with bacilli organized in clumps (tight bacterial aggregates) which de facto further extend bacterial length, the ratio between bacterial size and GO flake lateral dimensions becomes unbalanced. Hence, the observed anti-mycobacterial activity could be affected by the GO “blade” effect which is largely dependent on species cell wall, cell size and shape.13,36 Under our experimental conditions, the GO instability and aggregation in the medium used for bacterial/cell culturing, favor the trapping effect due to the presence of cations and proteins adsorbed on the GO surface.37 Aggregation masks the GO blades and cations act as “bridges” between the negatively charged GO surface and the mycobacterial surface, which is also negatively charged.38
In a series of experiments, we also showed that treatment with GO impairs mycobacterial entry into macrophages. Interestingly, GO was able to reduce the intracellular mycobacterial load as demonstrated by CFU analysis and confocal microscopy by trapping extracellular mycobacteria. Confining bacteria in the extracellular milieu is a well characterized host defence strategy involving neutrophils and macrophages that generate neutrophilic and macrophagic extracellular traps (NETs and METs, respectively).39–43 This local host immune response is associated with the secretion of chemokines and cytokines that promote a strong inflammatory response.44 Interestingly, Mtb can promote NET and MET formation in an ESX-1 dependent mechanism but it can evade the associated antimicrobial activity, thereby exploiting this process to promote inflammation and tissue damage while avoiding phagocytosis by activated macrophages.43,45 The results of our study, indicating that GO can exert the trapping effect in the absence of significant inflammation,34 may open new avenues to exploit the use of GO in TB treatment. For instance, hampering Mtb entry into macrophages may prevent the activation of the intracellular pathways that promote inflammation.44 Second, trapping Mtb in the extracellular space may increase the activity of the currently used anti-TB drugs, which can have easier access to the bacillus.
One possible approach may involve the combined use of GO with anti-mycobacterial drugs to enhance antimicrobial activity. Future studies could test this treatment in the murine model of pulmonary TB46 or in the ex vivo model of human granuloma-like structures (GLSs) that is based on the infection of peripheral blood leukocytes.47,48 In our opinion, testing the activity of GO with anti-TB drugs in this model would be extremely useful, since it involves different cell types and can be used to assess the impact of GO on the type of cellular organizations of the GLSs.
Conclusions
A variety of nanomaterials with dimensions of tens to hundreds of nanometres are currently emerging as promising therapeutic agents. We demonstrated the ability of GO to reduce Ms or Mtb entry into macrophages by trapping extracellular mycobacteria in a dose dependent manner. This ability could be used to design therapies for the acute phases of Mtb infection when mycobacteria actively replicate and infect new cells. GO therapy would also take advantage of its natural accumulation in the lungs31,34 reducing the need for high dose antibiotics and consequently the side effects on patients.Materials and methods
Bacterial strain manipulation
All minimal inhibitory concentration (MIC) assays and infection studies were performed using the rapid growth Mycobacterium smegmatis strain mc2 155 (Ms) and the virulent reference strain Mtb H37Rv. Both strains expressed the cytoplasmic green fluorescent protein (GFP) under the control of hps60 promoter (MsGFP and MtbGFP).30,49 All manipulations of the virulent MtbGFP were performed inside a biosafety level 3 (BSL-3) laboratory. Both MsGFP and MtbGFP were grown to their logarithmic phase peak (OD600: 0.5–0.8) in 7H9 liquid medium (Difco) supplemented with 10% Albumin Dextrose Catalase (ADC) (Sigma-Aldrich) and 0.05% Tween 80 (Sigma-Aldrich) at 37 °C and 110 rpm. The bacterial suspension was harvested and frozen by adding 20% sterile pure glycerol (Carlo Erba reagents) and keeping it at −80 °C. The total number of bacteria in the frozen suspension was established by plating.In vitro antimicrobial assay
The MIC assay was carried out by incubating MsGFP with scalar dilution of GO in 7H9 medium supplemented with 10% ADC and 0.05% Tween 80, as previously described. A suspension of 10 ml of 105 bacteria per ml was incubated with 1000 μg ml−1 to 20 μg ml−1 GO at 37 °C with continuous agitation. Suspensions of bacteria and bacteria plus kanamycin, at a final concentration of 0.5 mg ml−1, were used as positive and negative growth controls, respectively. Six hours post incubation colony forming units (CFUs) were determined by plating serial dilutions of the suspensions on 7H11 solid medium (Difco) supplemented with 10% Oleic Albumin Dextrose Catalase (OADC) (Sigma-Aldrich).38 To measure the GO trapping effect, suspensions of mycobacteria/GO were sonicated in a water bath (Becton Dickinson, output puissance 100 W to 42 kHz ±6%) for different time points (5′ and 20′) and the CFU was determined as above.Cell culture
Murine macrophages (J774 A.1 cell line) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Euroclone) supplemented with 10% inactivated fetal bovine serum (FBS) (Euroclone), 1% L-glutamine (Euroclone) and 1% streptomycin–penicillin (Euroclone) at 37 °C and 5% CO2. Adherent cells were washed with sterile warm phosphate buffered saline (PBS) (Euroclone) and removed for experiments by using 1× trypsin in PBS (Euroclone). Cells were counted and suspended in DMEM supplemented with 2% FCS and 1% L-glutamine. Finally, cells were seeded in sterile 48 well plates (Euroclone) at concentration of 1.2 × 106 cells per ml and incubated overnight until infection.Mycobacterial infection
J774 macrophages were infected with MsGFP using a multiplicity of infection (MOI) of 10 (10 bacteria to 1 cell) in three different ways schematically represented (Fig. 4a, 5a and 6a). In the first experiment a suspension of bacteria, bacteria and GO (1000 μg ml−1, 100 μg ml−1 and 10 μg ml−1) and bacteria plus gentamicin, at a final concentration of 1000 μg ml−1, was used to infect macrophages. Four hours post infection, cells were washed three times with sterile warm PBS and fresh medium was added. Six hours post infection, cells were washed with PBS, harvested with 0.01% Triton X-100, and colony forming units (CFUs) were quantified. In the second experimental setting, macrophages were infected with MsGFP alone. Four hours post infection, cells were washed three times with sterile warm PBS and fresh medium containing GO (1000 μg ml−1, 100 μg ml−1 and 10 μg ml−1) or gentamycin (1 mg ml−1) was added to each well. Finally, the plate was incubated for two hours until the cells were harvested and CFUs were obtained as described. Conversely, Mtb infection was carried out using an MOI of 1 (1:1 bacterium to cell ratio) and the initial infection was for 1 hour instead of 4 hours, in line with our previous research.50,51 In the third experimental setting, macrophages were pre-treated with medium alone and medium containing GO (1000 μg ml−1, 100 μg ml−1 and 10 μg ml−1) or gentamycin (10 μg ml−1). Four hours later, the macrophages were infected with MsGFP (MOI 10) or MtbGFP (MOI 1) and CFUs were obtained as previously described. A series of Mtb infected cells were also incubated with gentamycin two hours post infection, until CFUs were obtained. During infection, the cells were incubated under standard atmospheric conditions in all experimental settings.
Mycobacteria viability assay
To assess the viability of MsGFP during the MIC assay, aliquots of the suspension containing only bacteria, bacteria and GO, or bacteria plus kanamycin were collected and plated in a 96 well plate. Alamar Blue reagent (Biosource) was added in each well according to the manufacturer's instructions. Alamar reduction was assessed at 470 nm at 0, 6 and 24 hours after infection. Values of the control wells containing GO, kanamycin and medium alone were acquired to normalize the assay.Cell viability
The cytotoxic effects of GO and antibiotics were studied using a commercial CellTiter-Blue® Cell Viability Assay (Promega, WI, U.S.A.). Cellular viability was evaluated 24 hours after addition of GO or antibiotic solutions. The fluorescence (560Ex/590Em) of the solution was determined using an automatic microplate reader, Cytation 3 Cell Imaging Multi-Mode Reader (Biotek Instruments). The readings were obtained after an incubation of 2.5 h at 37 °C with the reagent. Each experiment was performed in triplicate.Dynamic light scattering and zeta potential
GO solutions (Graphenea) were prepared in ddH2O at concentrations between 0.01 mg ml−1 and 1 mg ml−1. Solutions were characterized by dynamic light scattering with a Zetasizer Nano ZS (Malvern, Herrenberg, Germany) equipped with a 633 nm He–Ne laser and operating at an angle of 173°. Solvent-resistant micro cuvettes (ZEN0040, Malvern, Herrenberg, Germany) were used for experiments with a sample volume of 40 μl. The measurements were performed at a fixed position with an automatic attenuator and at a controlled temperature as reported previously.52 For each sample, five measurements were averaged. The ζ-potential was calculated from the electrophoretic mobility by means of the Henry correction to Smoluchowski's equation.53Atomic force microscopy (AFM)
The samples were prepared as described elsewhere.54 Briefly, 20 μl of the samples were deposited on sterile, freshly cleaved mica disks and air-dried. After sample preparation, measurements were carried out with a NanoWizard II atomic force microscope (JPK Instruments AG, Berlin, Germany). Images were acquired using a standard AFM probe featuring pyramidal silicon tips, beam silicon cantilevers and rectangular silicon chips (CSC37 Mikro-Masch, Tallinn, Estonia). The tip was characterized by an end radius of about 10 nm and a half conical angle of 20°. The height of the tip was 15 μm. Cantilevers with a nominal spring constant of about k = 0.4 N m−1 were accurately calibrated as previously reported.27Confocal microscopy
To perform microscope analysis, J774 macrophages were plated on sterile chamber slides (ibidi) at a concentration of 1.2 × 106 cell per ml and then incubated overnight under standard atmospheric conditions. The chamber slides were infected following the previously indicated experimental settings. After 6 hours post infection the chamber slides were washed with PBS and then treated with 4% paraformaldehyde (PFA) overnight. Finally, the chamber slides were washed with PBS and analysed by confocal microscopy.38Images were collected by using an inverted confocal microscope (DMIRE2, Leica Microsystems, Wetzlar, Germany) equipped with a 63× oil immersion objective. For GFP excitation a He/Ne laser at 476 nm was used. Internal photon multiplier tubes collected 8 bit unsigned images at a 400 Hz scan speed in an emission range between 510 nm and 600 nm. Imaging was performed at room temperature. Image processing was performed with ImageJ software; image background values (defined as intensities below 7% of the maximum intensity) were set to zero and coloured in black.
Images of the infected cells, previously permeabilized and marked with phalloidin-red (Life Technologies), were collected using an inverted microscope (Nikon A1 MP+, Nikon, Japan) with an ADL 40× objective lens. Cells were imaged with a laser wavelength of excitation of 561 nm (emission GaAsp Detector 595/50 nm). For bacteria expressing GFP, a laser wavelength of excitation of 488 nm was used (emission GaAsp Detector 525/50 nm). Images were then analysed with ImageJ software.
Scanning electron microscopy (SEM)
For scanning electron microscopy, samples were fixed in glutaraldehyde, dehydrated in ethanol and sputter coated as reported previously.55 Cells and bacteria were imaged with a SEM Supra 25 (Zeiss, Germany).Data analysis
All experiments were replicated at least three times. Microsoft Excel (Office 2016) and Graphpad Prism software version 6 (GraphPad software) were used to collect and to analyse the data. All data were analysed by a one-way ANOVA comparison test, followed by the appropriate correction, and expressed as mean plus SD as specified in the caption under each figure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Experiments were performed at the LABCEMI Microscopy Facility (UCSC, Rome, Italy). We are extremely thankful to Mario Amici for the technical support during experiments.References
- World Health Organization, 2017, Global tubercolosis report 2017 Search PubMed.
- R. M. Houben and P. J. Dodd, PLoS Med., 2016, 13, e1002152 CrossRef PubMed.
- G. Delogu, M. Sali and G. Fadda, Mediterr J Hematol Infect Dis., 2013, 5, e2013070 CrossRef PubMed.
- O. Neyrolles, R. Hernández-Pando, F. Pietri-Rouxel, P. Fornès, L. Tailleux, J. A. B. Payán, E. Pivert, Y. Bordat, D. Aguilar, M. C. Prévost, C. Petit and B. Gicquel, PLoS One, 2006, 1, e43 CrossRef PubMed.
- D. G. Russell, P. J. Cardona, M. J. Kim, S. Allain and F. Altare, Nat. Immunol., 2009, 10, 943 CrossRef CAS PubMed.
- M. Gengenbacher and S. H. Kaufmann, FEMS Microbiol. Rev., 2012, 36, 514–532 CrossRef CAS PubMed.
- V. Peddireddy, S. N. Doddam and N. Ahmed, Front. Immunol., 2017, 8, 84 Search PubMed.
- D. G. Russell, C. E. Barry and J. L. Flynn, Science, 2010, 328, 852–856 CrossRef CAS PubMed.
- D. G. Russell, Immunol. Rev., 2011, 240, 252–268 CrossRef CAS PubMed.
- J. Heyckendorf, S. Andres, C. U. Köser, I. D. Olaru, T. Schön, E. Sturegård, P. Beckert, V. Schleusener, T. A. Kohl, D. Hillemann, D. Moradigaravand, J. Parkhill, S. J. Peacock, S. Niemann, C. Lange and M. Merker, Antimicrob. Agents Chemother., 2018, 62, e01550 Search PubMed.
- M. G. Gomes, M. L. Barreto, P. Glaziou, G. F. Medley, L. C. Rodrigues, J. Wallinga and S. B. Squire, BMC Infect. Dis., 2016, 16, 132 CrossRef PubMed.
- K. Xu, Z. C. Liang, X. Ding, H. Hu, S. Liu, M. Nurmik, S. Bi, F. Hu, Z. Ji, J. Ren, S. Yang, Y. Y. Yang and L. Li, Adv. Healthcare Mater., 2018, 7, 1700509 CrossRef PubMed.
- V. Palmieri, M. C. Lauriola, G. Ciasca, C. Conti, M. De Spirito and M. Papi, Nanotechnology, 2017, 28, 152001 CrossRef PubMed.
- M. Papi, V. Palmieri, F. Bugli, M. De Spirito, M. Sanguinetti, C. Ciancico, M. C. Braidotti, S. Gentilini, L. Angelani and C. Conti, Sci. Rep., 2016, 6, 12 CrossRef PubMed.
- E. J. Park, G. H. Lee, B. S. Han, B. S. Lee, S. Lee, M. H. Cho, J. H. Kim and D. W. Kim, Arch. Toxicol., 2015, 89, 1557–1568 CrossRef CAS PubMed.
- X. Zhang, J. Yin, C. Peng, W. Hu, Z. Zhu, W. Li, C. Fan and Q. Huang, Carbon, 2011, 49, 986–995 CrossRef CAS.
- S. G. Han, J. K. Kim, J. H. Shin, J. H. Hwang, J. S. Lee, T. G. Kim, J. H. Lee, G. H. Lee, K. S. Kim, H. S. Lee, N. W. Song, K. Ahn and I. J. Yu, BioMed Res. Int., 2015, 2015, 376756 Search PubMed.
- Y. H. Kim, M. S. Jo, J. K. Kim, J. H. Shin, J. E. Baek, H. S. Park, H. J. An, J. S. Lee, B. W. Kim, H. P. Kim, K. H. Ahn, K. Jeon, S. M. Oh, J. H. Lee, T. Workman, E. M. Fustman and I. J. Yu, Nanotoxicology, 2018, 12, 224–238 CrossRef CAS PubMed.
- A. A. Shvedova, A. A. Kapralov, W. H. Feng, E. R. Kisin, A. R. Murray, R. R. Mercer, C. M. S. Croix, M. A. Lang, S. C. Watkins, N. V. Konduru, B. L. Allen, J. Conroy, G. P. Kotchey, B. M. Mohamed, A. D. Meade, Y. Volkov, A. Star, B. Fadeel and V. E. Kagan, PLoS One, 2012, 7, e30923 CrossRef CAS PubMed.
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, ACS Nano, 2011, 5, 2098–2108 CrossRef CAS PubMed.
- V. Palmieri, M. Papi, C. Conti, G. Ciasca, G. Maulucci and M. De Spirito, Expert Rev. Med. Devices, 2016, 13, 1013–1019 CrossRef CAS PubMed.
- S. Gurunathan, J. W. Han, A. A. Dayem, V. Eppakayala and J. H. Kim, Int. J. Nanomed., 2012, 7, 5901–5914 CrossRef CAS PubMed.
- P. Khanra, T. Kuila, N. H. Kim, S. H. Bae, D. Yu and J. H. Lee, Chem. Eng. J., 2012, 183, 526–533 CrossRef CAS.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. Sanchez Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- Z. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS PubMed.
- V. Palmieri, F. Bugli, M. C. Lauriola, M. Cacaci, R. Torelli, G. Ciasca, C. Conti, M. Sanguinetti, M. Papi and M. De Spirito, ACS Biomater. Sci. Eng., 2017, 3, 619–627 CrossRef CAS.
- O. N. Ruiz, K. A. S. Fernando, B. Wang, N. A. Brown, P. G. Luo, N. D. McNamara, M. Vangsness, Y. P. Sun and C. E. Bunker, ACS Nano, 2011, 5, 8100–8107 CrossRef CAS PubMed.
- J. Russier, E. Treossi, A. Scarsi, F. Perrozzi, H. Dumortier, L. Ottaviano, M. Meneghetti, V. Palermo and A. Bianco, Nanoscale, 2013, 5, 11234–11247 RSC.
- F. De Maio, G. Maulucci, M. Minerva, S. Anoosheh, I. Palucci, R. Iantomasi, V. Palmieri, S. Camassa, M. Sali, M. Sanguinetti, W. Bitter, R. Manganelli, M. De Spirito and G. Delogu, PLoS One, 2014, 9, e112482 CrossRef PubMed.
- S. Vranic, A. F. Rodrigues, M. Buggio, L. Newman, M. R. H. White, D. G. Spiller, C. Bussy and K. Kostarelos, ACS Nano, 2017, 12, 1373–1389 CrossRef PubMed.
- P. C. Henriques, I. Borges, A. M. Pinto, F. D. Magalhães and I. C. Gonçalves, Carbon, 2018, 132, 709–732 CrossRef CAS.
- D. P. Singh, C. E. Herrera, B. Singh, S. Singh, R. K. Singh and R. Kumar, Mater. Sci. Eng. C, 2018, 86, 173–197 CrossRef CAS PubMed.
- A. Schinwald, F. A. Murphy, A. Jones, W. MacNee and K. Donaldson, ACS Nano, 2012, 6, 736–746 CrossRef CAS PubMed.
- L. J. Alderwick, J. Harrison, G. S. Lloyd and H. L. Birch, Cold Spring Harbor Perspect. Med., 2015, 5, a021113 CrossRef PubMed.
- V. D. Pham, V. Repain, C. Chacon, A. Bellec, Y. Girard, S. Rousset, E. Abad, Y. J. Dappe, A. Smogunov and J. Lagoute, ACS Nano, 2017, 11, 10742–10749 CrossRef CAS PubMed.
- V. Palmieri, G. Perini, M. De Spirito and M. Papi, Nanoscale Horiz., 2019 10.1039/C8NH00318A.
- F. De Maio, B. Battah, V. Palmieri, L. Petrone, F. Corrente, A. Salustri, I. Palucci, S. Bellesi, M. Papi, S. Rubino, M. Sali, D. Goletti, M. Sanguinetti, R. Manganelli, M. De Spirito and G. De Logu, Cell. Microbiol., 2018, 20, e12952 CrossRef PubMed.
- T. Dallenga and U. E. Schaible, Pathog. Dis., 2016, 74, ftw012 CrossRef PubMed.
- R. Appelberg, Trends Microbiol., 2007, 15, 87–92 CrossRef CAS PubMed.
- D. M. Lowe, P. S. Redford, R. J. Wilkinson, A. O'Garra and A. R. Martineau, Trends Immunol., 2012, 33, 14–25 CrossRef CAS PubMed.
- C. T. Yang, C. J. Cambier, J. M. Davis, C. J. Hall, P. S. Crosier and L. Ramakrishnan, Cell Host Microbe, 2012, 12, 301–312 CrossRef CAS PubMed.
- K. W. Wong and W. R. Jacobs Jr, J. Infect. Dis., 2013, 208, 109–119 CrossRef CAS PubMed.
- S. H. Kaufmann and A. Dorhoi, Curr. Opin. Immunol., 2013, 25, 441–449 CrossRef CAS PubMed.
- T. Dallenga, U. Repnik, B. Corleis, J. Eich, R. Reimer, G. W. Griffiths and U. E. Schaible, Cell Host Microbe, 2017, 22, 519–530 CrossRef CAS PubMed.
- G. Delogu, A. Li, C. Repique, F. Collins and S. L. Morris, Infect. Immun., 2002, 70, 292–302 CrossRef CAS PubMed.
- L. E. Fitzgerald, N. Abendaño, R. A. Juste and M. Alonso-Hearn, BioMed Res. Int., 2014, 2014, 623856 Search PubMed.
- E. Guirado and L. Schlesinger, Front. Immunol., 2013, 4, 98 Search PubMed.
- G. Delogu, C. Pusceddu, A. Bua, G. Fadda, M. J. Brennan and S. Zanetti, Mol. Microbiol., 2004, 52, 725–733 CrossRef CAS PubMed.
- A. Zumbo, I. Palucci, A. Cascioferro, M. Sali, M. Ventura, P. D’alfonso, R. Iantomasi, G. Di Sante, F. Ria, M. Sanguinetti, G. Fadda, R. Manganelli and G. De Logu, Pathog. Dis., 2013, 69, 232–239 CrossRef CAS PubMed.
- M. Minerva, F. De Maio, S. Camassa, B. Battah, P. Ivana, R. Manganelli, M. Sanguinetti, M. Sali and G. Delogu, Pathog. Dis., 2017, 75, 4082733 Search PubMed.
- V. Palmieri, D. Lucchetti, I. Gatto, A. Maiorana, M. Marcantoni, G. Maulucci, M. Papi, R. Pola, M. De Spirito and A. Sgambato, J. Nanopart. Res., 2014, 16, 2583 CrossRef.
- M. Papi, M. C. Lauriola, V. Palmieri, G. Ciasca, G. Maulucci and M. De Spirito, RSC Adv., 2015, 5, 81638–81641 RSC.
- F. Bugli, B. Posteraro, M. Papi, R. Torelli, A. Maiorana, F. P. Sterbini, P. Posteraro, M. Sanguinetti and M. De Spirito, Antimicrob. Agents Chemother., 2013, 57, 1275–1282 CrossRef CAS PubMed.
- V. Palmieri, M. Barba, L. Di Pietro, S. Gentilini, M. C. Braidotti, C. Ciancico, F. Bugli, G. Ciasca, R. Larciprete, W. Lattanzi, M. Sanguinetti, M. De Spirito, C. Conti and M. Papi, 2D Mater., 2017, 5, 15027 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |