
MoS2-covered SnS nanosheets as anode material for lithium-ion batteries with high capacity and long cycle life†
Qichang
Pan
a,
Fenghua
Zheng
a,
Yanan
Wu
a,
Xing
Ou
a,
Chenghao
Yang
 *a,
Xunhui
Xiong
a and
Meilin
Liu
ab
*a,
Xunhui
Xiong
a and
Meilin
Liu
ab
aGuangzhou Key Laboratory for Surface Chemistry of Energy Materials, New Energy Research Institute, School of Environment and Energy, South China University of Technology, Guangzhou 510006, PR China. E-mail: esyangc@scut.edu.cn
bSchool of Materials Science & Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332-0245, USA
First published on 30th November 2017
Abstract
A designed hierarchical nanostructure consisting of SnS nanosheets and ultrathin MoS2 nanosheets was achieved by a facile hydrothermal process with the assistance of fluoride and glucose. In this unique architecture, on the one hand, SnS and MoS2 nanosheets can greatly reduce the Li-ion and electron diffusion distance in the electrode. On the other hand, MoS2 nanosheets and amorphous carbon can not only prevent the direct exposure of SnS to the electrolyte but also maintain the structural stability of the electrode. In addition, the MoS2 nanosheets can offer more active sites for hosting lithium ions, resulting in higher capacity. When evaluated as anode material for lithium-ion batteries (LIBs), this SnS/MoS2–C composite exhibited stable cycling performance (989.7 mA h g−1 at 0.2 A g−1 after 60 cycles), superior rate capability (675 mA h g−1 even at 5.0 A g−1) and a long cycle life (718 mA h g−1 at 2.0 A g−1 after 700 cycles). Therefore, this SnS/MoS2–C composite is a promising candidate as anode material for next-generation high-performance LIBs.
Introduction
Currently, rechargeable secondary batteries are one of the most important energy storage technologies.1–4 Especially, lithium-ion batteries (LIBs) are currently the mainly power sources for portable electronic devices. However, developing anode materials with high energy/power density is one of the hot topics in LIB research owing to the dominant commercially used graphite anode having low theoretical capacity (372 mA h g−1) and poor rate capability.5,6 Therefore, to satisfy the demand for increasing energy/power density for LIBs, various anode materials such as transition metal oxides, metal sulfides, and metal/nonmetal have been investigated widely as promising candidate anodes for LIBs owing to their high capacity.7–10 Among all of them, SnS is considered the most likely to be an alternative to graphite as anode for LIBs owing to its high capacity, abundance, and low cost. However, the large volumetric expansion of SnS, which is mainly caused by the Li–Sn alloying process during the charge and discharge process, results in pulverization and rapid, severe capacity fade, and has seriously hampered the practical application in LIBs.11,12Researchers have focused on finding an effective way to improve the performance of the SnS-based anodes, by designing various SnS nanostructures, such as nanorods, nanosheets, nanobelts, nanoflowers and 3D-hierarchical nanostructures, which have all been extensively studied.13–17 In the end, significantly improved electrochemical performance of SnS-based anodes has been achieved because altering the nanostructure can efficiently address the internal strain induced by volume change during the lithium ion insertion/extraction process. In addition, it also can significantly decrease the diffusion length for Li-ions, which results in high rate performance. Another effective strategy is to construct hybrid anodes consisting of SnS and carbon. Therefore, some SnS/C and SnS/graphene composites have been developed, showing enhanced electrochemical performance compared with the bare SnS anode,11,12,18 as these carbon matrices can not only improve the structural stability but also enhance the electrical conductivity of the overall electrode.
Recently, MoS2 have been extensively investigated owing to the fact that it can deliver high capacity as anode material for LIBs.19 Nevertheless, the quickly fading capacity and poor rate performance seriously limit its practical application in LIBs.20 To solve these issues, various efforts have been devoted to ameliorate the performance of MoS2 by designing a variety of nanostructures and combination with carbonaceous materials or other active materials.21–24 Moreover, MoS2 can cushion the mechanical stress caused by the volume expansion of other active materials during the lithium ion insertion/extraction process thanks to its having high mechanical strength.25,26
In this study, we demonstrate a facile approach to fabricate a hierarchical nanostructure of SnS/MoS2–C composite. Firstly, the SnS2 nanosheets were synthesized via a facile fluoride-mediated hydrothermal process. After that, the ultrathin MoS2 nanosheets were directly grown on the surface of the SnS2via a hydrothermal process with the assistance of glucose, and then annealed in an Ar atmosphere to obtain the SnS/MoS2–C composite. When evaluated as anode for LIBs, this structure endows the SnS/MoS2–C composite with the following advantages: (1) the SnS nanosheets can greatly reduce the diffusion distance for Li-ions and electrons during the charge/discharge process, which can achieve ultrafast charging/discharging; (2) the ultrathin MoS2 nanosheets result in high capacity owing to the large number of available sites for Li+ storage and high surface contact area with electrolyte; (3) MoS2 and C can maintain the structural stability of the composite, avoiding the volume expansion of SnS nanosheets during the charge/discharge process because MoS2 and C endow good structural stability via the C–O–Mo bonds.
Results and discussion
The fabrication process of the SnS/MoS2–C composite is shown in detail in Fig. 1A. First, SnS2 nanosheets were prepared by a simple hydrothermal process with assistance of NH4F. In this process, the added NH4F contributed to dissolution of the tin source, as F− can reduce the chemical reactivity of the precursor because it has higher electronegativity. Therefore, F− could effectively reduce the hydrolysis and condensation rates of Sn2+ by complexation.27–29 Also, the added fluoride anion helps the subsequent mass transfer from the interior to the surface. During this hydrothermal process, SnS2 nanosheets were obtained. Second, uniform ultrathin MoS2 nanosheets covering the surface of the SnS2 nanosheets were synthesized by a hydrothermal process with assistance of glucose. After this process, the obtained SnS2/MoS2–C composite was annealed in an Ar atmosphere; the SnS2 was transformed into SnS, increasing the crystalline quality of the MoS2, so that a SnS/MoS2–C composite was obtained.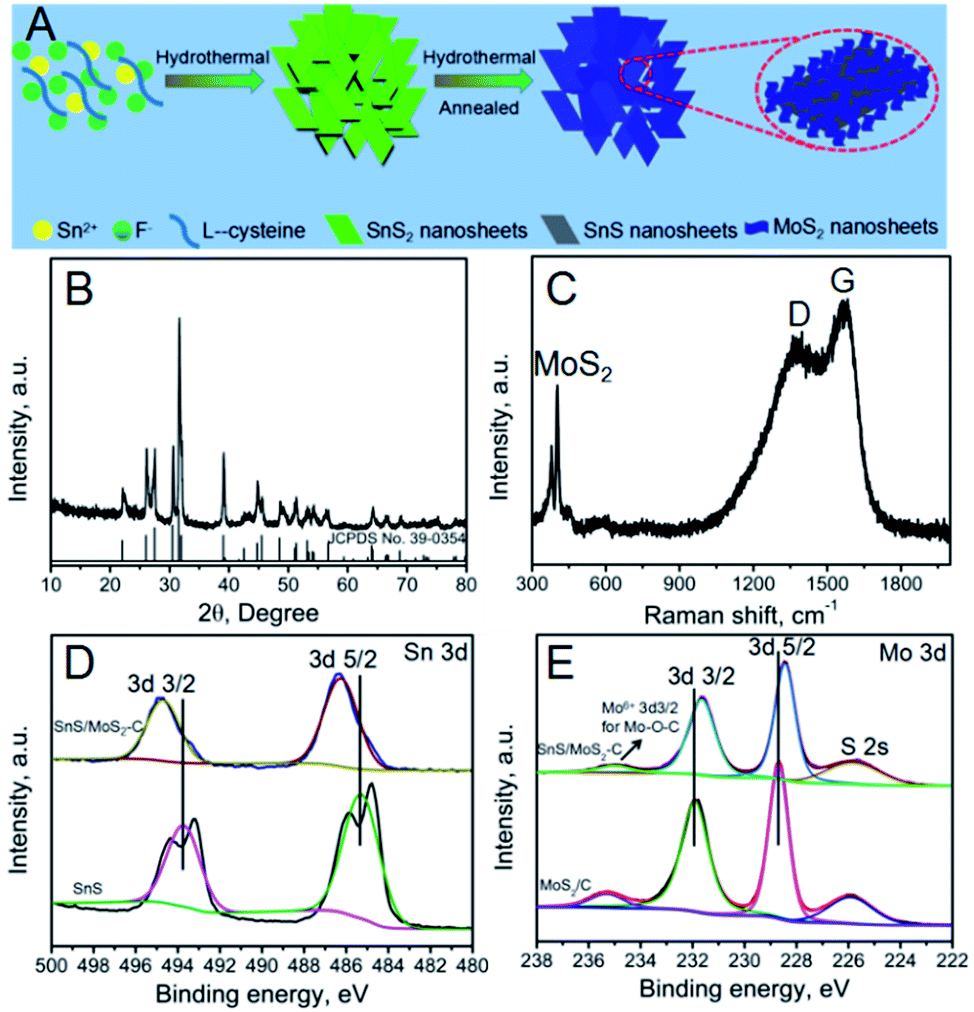 | ||
| Fig. 1 (A) Schematic illustration of the preparation of SnS/MoS2–C composite; (B) XRD pattern of SnS/MoS2–C composite; (C) Raman spectrum of SnS/MoS2–C composite; (D) Sn 3d spectra of SnS and SnS/MoS2–C composite; (E) Mo 3d spectra of MoS2/C and SnS/MoS2–C composite. | ||
The X-ray diffraction (XRD) pattern of the SnS2 is shown in Fig. S1;† apart from a small amount of SnS impurity existing in the as-prepared SnS2, all of the peaks accord well with hexagonal SnS2 (JCPDS card no. 79-5206).12,30 Fig. S2† shows the XRD pattern of SnS. After annealing at 600 °C under Ar atmosphere, the XRD pattern of the SnS is consistent with that of orthorhombic SnS, indicating that the hexagonal SnS2 was transformed into orthorhombic SnS during the annealing process.12,15 The XRD pattern of MoS2/C is shown in Fig. S3†; all reflections due to MoS2 are in good agreement with a hexagonal structure (JCPDS 37-1492). The XRD pattern of the SnS/MoS2–C composite, as shown in Fig. 1B, is similar to the XRD pattern of SnS; all of the diffraction peaks can be indexed to orthorhombic SnS. And, the (002) plane of MoS2 was not detected in the XRD pattern of SnS/MoS2–C, suggesting that the MoS2 nanosheets lie in the (002) direction and that the composite prepared with the assistance of the glucose only contains few layers, which are too thin to be detected by XRD.31,32
To further explore the properties of the SnS/MoS2–C composite, Raman spectroscopy was carried out; the result is shown in Fig. 1C. Two peaks located at around 368 and 389 cm−1 clearly verify the presence of MoS2 in the SnS/MoS2–C; these peaks are attributed to the representative modes of E2g and A1g of MoS2, respectively.33 Moreover, the characteristic D- and G-bands of carbon at around 1350 and 1580 cm−1 can be found in the spectrum of the SnS/MoS2–C composite.34 Therefore, the results indicate that SnS/MoS2–C composite was successfully synthesized.
In order to confirm the content of MoS2 and carbon in SnS/MoS2–C, thermogravimetric analysis (TGA) and inductively coupled plasma optical emission spectroscopy (ICP-OES) were carried out for SnS/MoS2–C. The TGA curves of the SnS/MoS2–C display a weight loss in the range of 300–600 °C, which corresponds to the oxidation of MoS2 to MoO3 or C to CO2 (Fig. S4†). Therefore, based on the ICP-OES (Table S1†) and TGA results for SnS/MoS2–C, the contents of MoS2 and carbon in SnS/MoS2–C are about 12.7% and 18.9%, respectively.
To clarify the chemical bonding states of the elements in the samples, X-ray photoelectron spectroscopy (XPS) measurements were investigated. Fig. S5† shows the survey spectra of SnS, MoS2/C and SnS/MoS2–C composite, respectively. The SnS/MoS2–C composite is composed of the elements Sn, S, Mo, O and C according to the survey spectra of the SnS/MoS2–C composite, and the MoS2/C composite is composed of the elements Mo, S, O and C. However, the element C is detected in SnS, which may be attributed to the conductive plastic. In Fig. 1D, two peaks at around 495 and 487 eV for Sn 3d can be observed in the SnS/MoS2–C sample, which exhibits a positive shift in the binding energy compared with the SnS sample. In the high-resolution Mo 3d spectrum of SnS/MoS2–C (Fig. 1E), two peaks located at around 231.6 and 228.4 eV are observed, which can be ascribed to the Mo 3d3/2 and Mo 3d5/2 binding energies, respectively, and correspond to the characteristic peaks of Mo4+ in MoS2.35 However, compared with the MoS2/C sample, the two characteristic peaks of Sn 3d in the SnS/MoS2–C sample are shifted towards the low binding energy region. Therefore, these results suggest an increased density of electron clouds around the ultrathin MoS2 nanosheets. The XPS results clearly indicate that there is strong electron interaction between the SnS nanosheets and MoS2 nanosheets, which is expected to endow the SnS/MoS2–C composite with a stable structure for long-term cycling in LIB applications.36 Furthermore, the peak at around 234.9 eV is related to the Mo 3d3/2 of Mo6+ (typical of the Mo–O bond).37 Another small peak at around 225.8 eV corresponds to the S 2s component. The N2 sorption isotherms and the pore size distributions of SnS2, SnS and SnS/MoS2–C composite are shown in Fig. S6.† The size of the pores for the three samples is mainly centered below 30 nm with a relatively narrow distribution based on desorption data (Fig. S6B, D and F†).27 The specific surface area of the SnS/MoS2–C composite is larger than that of SnS2 and SnS, which can be attributed to MoS2 nanosheets growing vertically on the SnS nanosheets; these ultrathin MoS2 nanosheets may contribute to the increase in specific surface area.
The morphology of all samples was first investigated by scanning electron microscopy (SEM). Fig. 2A and B show the SEM images of the SnS2 with added NH4F; it can be clearly seen that the SnS2 is composed of nanosheets and the thickness of the nanosheets is around 50 nm. In contrast, only bulk SnS2 was obtained without added NH4F (Fig. S7†). Moreover, after being annealed in an Ar atmosphere at 600 °C, the sample still exhibits the same nanosheet structure when the SnS2 is transformed into SnS, but the nanosheets are thinner than in the SnS2 (Fig. 2C and D). In addition, the SEM images of SnS/MoS2–C reveal the same structure with nanosheets. However, in the SnS/MoS2–C composite, ultrathin MoS2 nanosheets are clearly visible, fully covering the surface of the SnS nanosheets (Fig. 3E and F). On the other hand, the thickness of nanosheets in the SnS/MoS2–C composite is greater (at about 100 nm) than in the bare SnS2 and SnS, which may be due to the introduction of the carbon and MoS2 nanosheets. In addition, the SEM images of the SnS2/MoS2–C composite are similar to those of the SnS/MoS2–C composite (Fig. S8†). Moreover, the morphology of the MoS2/C composite is shown in Fig. S9,† which exhibits large micrometer-sized sphere-like architecture with nanosheets owing to the glucose playing a role in the annealing of the sphere-like MoS2/C composite during the hydrothermal process.36
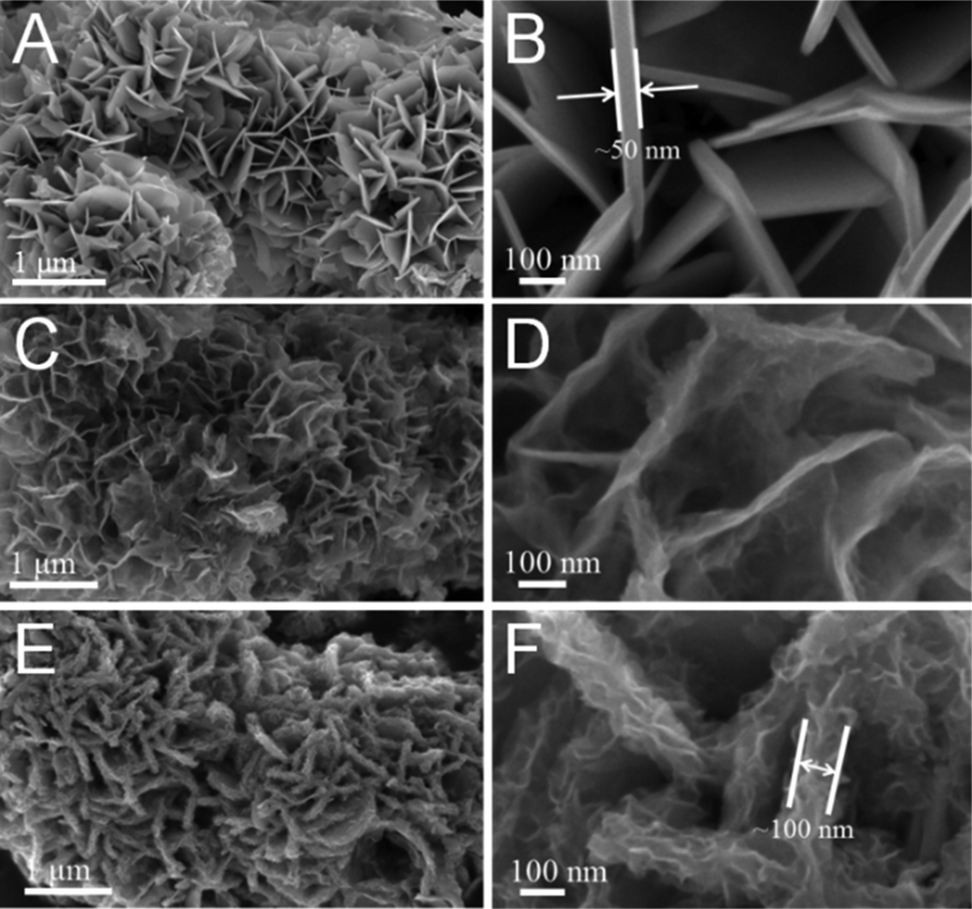 | ||
| Fig. 2 SEM images of (A, B) SnS2, (C, D) SnS and (E, F) SnS/MoS2–C composite. | ||
 | ||
| Fig. 3 (A–D) Transmission electron microscopy (TEM) and (E and F) high-resolution TEM (HRTEM) images of SnS/MoS2–C composite. (G) TEM elemental mapping of SnS/MoS2–C composite. | ||
The microstructures of the SnS/MoS2–C composite were further investigated by transmission electron microscopy (TEM) and high-resolution TEM (HRTEM). As shown in Fig. 3A and B, the SnS nanosheets are fully covered by MoS2 and carbon, which is similar to the observation in the SEM images (Fig. 2E and F). On the other hand, the MoS2 nanosheets stand vertically on the surface of the SnS nanosheets according to Fig. 3C and D. Moreover, the HRTEM images show lattice fringes of 0.63 nm and 0.28 nm (Fig. 3E and F), which correspond to the (002) plane for MoS2 and (400) plane for SnS, respectively.6,11 Further, energy-dispersive X-ray spectroscopy (EDS) mapping (Fig. 3G) shows the uniform distribution of the elements C, S, Sn, and Mo in the SnS/MoS2–C composite.
The electrochemical properties of the as-prepared SnS, MoS2/C and SnS/MoS2–C were evaluated by cyclic voltammetry (CV) firstly. Fig. S10A† shows the CV curves of the SnS; during the initial anodic scan, a series of peaks between 0.4 V and 0.8 V can be observed, which can be attributed to the delithiation of Li+ from the LixSn alloy.12,13 And a broad peak around 1.9 V may be ascribed to the reversible conversion of Sn and Li2S into SnS. In the initial cathodic scan, the peak at around 1.2 V is assigned to the formation of the solid-electrolyte interphase (SEI) layer and the decomposition of SnS into Sn and Li2S. The peaks at around 0.1 V and 0.25 V are referenced to the alloying process of Li into Sn, forming LixSn. Fig. S11A† exhibits the CV curves of MoS2/C; two anodic peaks at around 1.45 and 2.22 V are obtained, which correspond to a partial oxidation of Mo to form MoS2, and Li+ extraction by the oxidation of Li2S, respectively.38 Moreover, a peak is present at around 0.6 V during the first cathodic scan, which corresponds to the decomposition of MoS2 into Mo nanoparticles embedded in a Li2S matrix by a series of reactions. The CV curves of SnS/MoS2–C composite are shown in Fig. 4A; a series of peaks between 0.4 V and 0.8 V can be observed, which can be ascribed to the delithiation of Li+ from the LixSn alloy. Two peaks at around 1.9 V and 2.25 V are due to the reversible conversion of Sn and Li2S into SnS and Li+ extraction by the oxidation of Li2S. Turning to the initial cathodic scan, the peak at around 1.2 V is assigned to the formation of the SEI layer and the decomposition of SnS into Sn and Li2S, and a series of peaks at around 0.1 V and 0.25 V are referenced to the alloying process of Li into Sn, forming LixSn.23,24 The charge and discharge curves of the as-prepared samples over a voltage range of 0.01–3.0 V were also investigated. As shown in Fig. S10B† and 4B, in the discharge curve, two poorly defined plateaus at around 1.3 V and 0.3 V can be identified, which are attributed to formation of the SEI layer as well as the decomposition of SnS into Sn and formation of Li–Sn alloy, respectively. In the charge process, a plateau at around 0.3 V–1.0 V is due to de-alloying of the Li–Sn alloy. And for MoS2/C and SnS/MoS2–C (Fig. S11B† and 4B), a plateau around 2.2 V also can be identified in the charge process, which can be ascribed to Li+ extraction by the oxidation of Li2S. Therefore, all of the charge and discharge curves are in good agreement with the CV results.
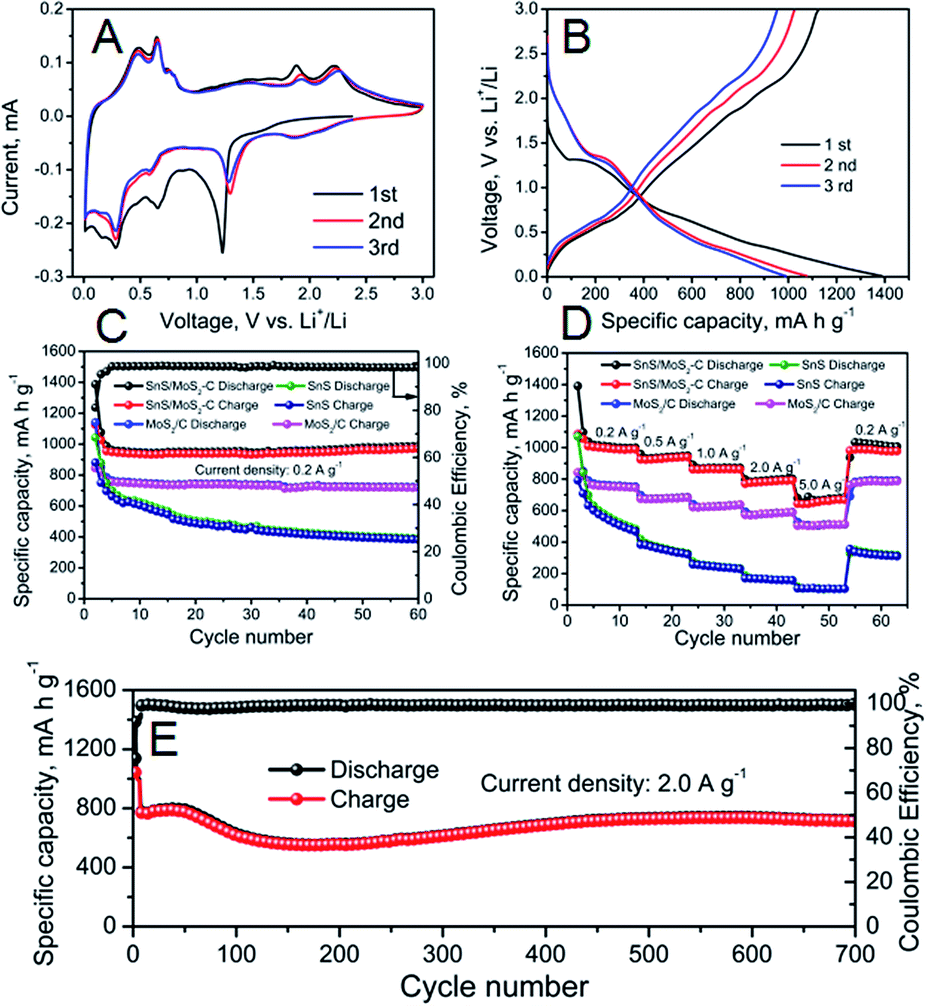 | ||
| Fig. 4 (A) CV curves at a scanning rate of 0.1 mV s−1 of the first three cycles for SnS/MoS2–C composite. (B) Charge–discharge curves of the initial three cycles for SnS/MoS2–C composite. (C) Cycling performance of SnS, MoS2/C and SnS/MoS2–C composite at 0.2 A g−1. (D) Rate capability of SnS, MoS2/C and SnS/MoS2–C at various current densities. (E) Long-term cycling performance of SnS/MoS2–C electrode at 2.0 A g−1. | ||
The cycling performance of the SnS, MoS2/C and SnS/MoS2–C electrode were evaluated, and the results are shown in Fig. 4C. SnS/MoS2–C exhibited excellent cycling performance, and a high discharge capacity of 989.7 mA h g−1 could be obtained after 60 cycles at 0.2 A g−1. In contrast, SnS and MoS2/C delivered a lower discharge capacity of 391.8 and 722 mA h g−1, respectively. Furthermore, SnS/MoS2–C exhibited a high initial coulombic efficiency of 81%, but the SnS and MoS2/C had lower initial coulombic efficiency of 73% and 74%, respectively. These results indicate that the SnS/MoS2–C electrode exhibited greatly enhanced lithium storage and cycling stability compared with the SnS and MoS2/C electrodes. Moreover, the SnS/MoS2–C electrode also exhibited much improved rate capability when used as anode in LIBs (Fig. 4D). The SnS/MoS2–C electrode can deliver a high capacity of 1002, 940, 870 and 790 mA h g−1 when the current density increases from 0.2 to 2.0 A g−1. Even at the high current density of 5.0 A g−1, the SnS/MoS2–C electrode can deliver a high capacity of 675 mA h g−1, which is 67.37% of the current density at 0.2 A g−1, exhibiting an excellent rate capability. After undergoing 50 cycles at various current density values even at a high rate of 5.0 A g−1, a high discharge capacity of 1020 mA h g−1 also can be obtained when the current density is returned to 0.2 A g−1. On the other hand, the other samples show lower rate capability than the SnS/MoS2–C composite; SnS and MoS2/C can deliver a discharge capacity of around 500 and 757 mA h g−1, respectively, at 0.2 A g−1. However, when tested at 5.0 A g−1, they deliver a discharge capacity of 100 and 505 mA h g−1, respectively. More importantly, the SnS/MoS2–C exhibited excellent long cycling stability even at high current density; the result is shown in Fig. 4E. A high discharge capacity of 718 mA h g−1 can be retained even after 700 cycles at 2.0 A g−1, and the coulombic efficiency of the SnS/MoS2–C is also excellent, remaining at nearly 100% in the subsequently cycling process. However, note that the capacity exhibits a rise and fall curve during the cycling process, which can be attributed to more and more available sites in both SnS and MoS2 and possibly interfacial lithium storage as well as electrochemical activation of the hybrid during the cycling process according to previous literature.24,39,40 The results indicate that this Sn/MoS2–C composite displays high reversible capacity, excellent rate capability and long cycling life.
The Nyquist plots of the as-prepared samples were investigated. Each plot consists of a semicircle at high frequency and a long line at low frequency.41 As displayed in Fig. S12,† the diameter of the semicircle at high frequency is reduced in the plot for the SnS/MoS2–C compared with that for the bare SnS, indicating the decreased charge-transfer resistance at the electrode/electrolyte interface due to robust binding and a complete coating of high electrical conductivity carbon. However, the diameter of the semicircle of the MoS2/C is increased compared with SnS and SnS/MoS2–C, which is due to the low conductivity of MoS2.42 Therefore, the enhanced charge-transfer in the SnS/MoS2–C could improve the electrochemical performance.
The excellent rate performance and superior long-life cycling stability of the SnS/MoS2–C composite can be attributed to the synergistic effect between a large number of defects in the few-layer MoS2 nanosheets and abundant interfaces in the composites. First, the grown MoS2 nanosheets cover the surface of the SnS nanosheets via face-to-face contact with assistance of carbon, leading to Li+ conducting channels fully connected from MoS2 to SnS and favorable electron transfer paths in MoS2 and SnS (Fig. 5A).32 Second, the MoS2 shell with an adequate degree of defects offers large amounts of additional active sites for Li+ storage, shortening the diffusion pathway and promoting the transport kinetics of Li-ions (Fig. 5B).43,44 Furthermore, the C–O–Mo bonds formed endow the SnS/MoS2–C composite with a highly stable structure for long-term cycling in LIB applications. The C–O–Mo bond also provides a good electron transfer path between MoS2 and amorphous carbon.22 Third, MoS2 and C can not only maintain the structural stability of the composite, avoiding the volume expansion of SnS during the charge/discharge process, but also provide an effective electron transfer path between MoS2, carbon and SnS because of good adhesion of MoS2 with carbon via the C–O–Mo bonds.22 Finally, in the first discharge process, the defect-rich MoS2 in the SnS/MoS2–C composite shows abundant additional edge sites, which can shorten the Li-ion diffusion path length and provide more channels for Li-ion diffusion, facilitating the transport of Li-ions into the interfaces and deep locations of the electrode.45–48 However, the MoS2 and SnS nanosheets break down into small nanoparticles after discharge, and after the first cycle, MoS2 and SnS nanosheets have disappeared, while Mo, S and Li4.4Sn have been formed, and the reversible reactions are S to Li2S and Sn to Li4.4Sn.49–54 Therefore, it can be supposed that smaller amorphous Mo, S nanoclusters and Li4.4Sn nanoparticles would be formed and anchored onto the amorphous carbon, which could produce larger contact area between active material and electrolyte and more channels for diffusion of Li-ions, thus facilitating the lithiation kinetics. On the other hand, the amorphous carbon can suppress the aggregation of nanoparticles as well as enhance structural stability during the follow cycling process.24,55–58 Therefore, all of these factors result in the SnS/MoS2–C anode exhibiting excellent electrochemical performance, and it should be noted that this SnS/MoS2–C composite exhibits a superior rate performance and high reversible capacity compared with other SnS-based composites (Fig. 5C and Table S2†) and MoS2-based composites (Fig. 5D).
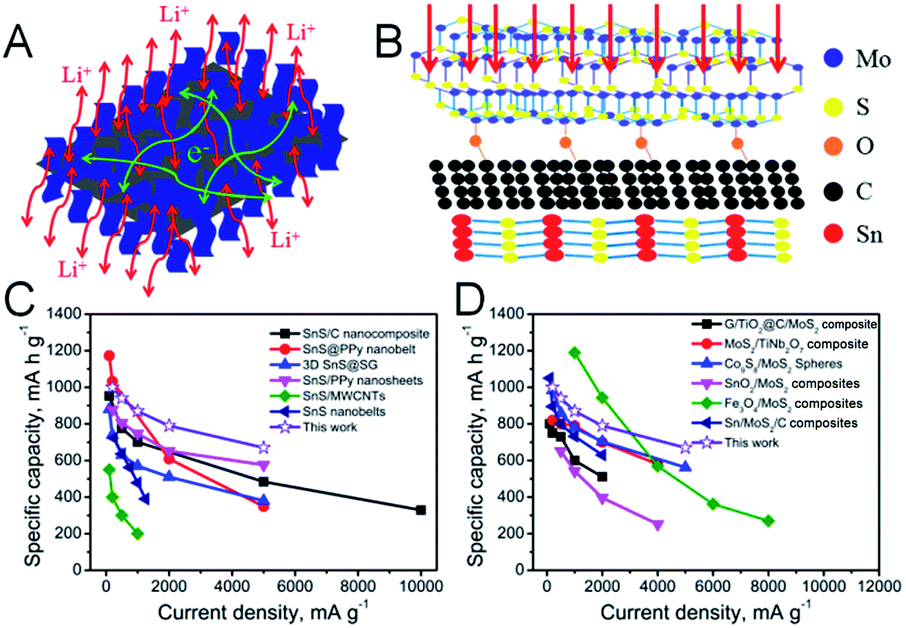 | ||
| Fig. 5 (A) The pathways of Li+ ions and electrons are displayed for the Sn/MoS2–C composite. (B) Atomistic and mesoscopic views of the SnS/MoS2–C composite. (C) Comparison of the average capacity values at various current densities from previously reported results related to SnS-based composites and our work. (D) Comparison of the average capacity values at various current densities from previously reported results related to MoS2-based composites and our work. | ||
Conclusions
In summary, a novel SnS/MoS2–C composite composed of SnS nanosheets and MoS2 was designed and synthesized by a facile hydrothermal process. Nanosheet-assembled SnS2 was fabricated by a fluoride-mediated hydrothermal process, and MoS2 was grown on the SnS nanosheets with the assistance of glucose. The designed architecture prevents the agglomeration of MoS2 nanosheets and maintains the structural stability during the cycling process. When evaluated as anode material for LIBs, SnS/MoS2–C exhibited a high reversible capacity (989.7 mA h g−1 at 0.2 A g−1 after 60 cycles), a superior rate capability (675 mA h g−1 even at 5.0 A g−1) and long cycle life (718 mA h g−1 at 2.0 A g−1 after 700 cycles). The obtained excellent performance can be ascribed to a strong synergistic effect between SnS nanosheets and ultrathin MoS2 nanosheets. As a result, the SnS/MoS2–C composite can be considered a promising anode material for next-generation high-performance LIBs.Experimental
Synthesis of SnS2 nanosheets
In a typical synthesis, 3.6 g SnCl2·2H2O was added to deionized water, and then an appropriate amount of NH4F was added under stirring to obtain a clear transparent solution. After that, 3.6 g L-cysteine was added to the solution and stirred for another 30 min. Then the solution was transferred into a 100 mL Teflon-lined stainless steel autoclave. Finally, the autoclave was sealed and maintained at 180 °C for 24 h. The as-prepared yellow precipitate was washed with water when the autoclave had cooled down to room temperature, and finally dried at 80 °C.Synthesis of SnS, and MoS2/C and SnS/MoS2–C composites
In order to synthesize SnS/MoS2–C, firstly, 0.5 g of SnS2 nanosheets was added to 80 mL deionized water and ultrasonicated for 30 min; then 0.15 g Na2MoO4·2H2O, 0.3 g L-cysteine and 0.5 g glucose were added and stirring continued for another 30 min. Finally, the solution was transferred into a 100 mL Teflon-lined stainless steel autoclave and maintained at 180 °C for 24 h. The final products were collected by filtration, washed with deionized water and then dried at 80 °C overnight. Then, the precipitate was further treated at 600 °C in an Ar atmosphere for 2 h with a heating rate of 5 °C min−1 to obtain SnS/MoS2–C. For comparison, synthesis of MoS2/C was carried out using 1.0 g glucose as carbon source, 0.3 g Na2MoO4·2H2O as molybdenum source and 0.6 g L-cysteine as sulfur source, respectively. To prepare SnS, SnS2 was directly annealed under the same conditions.Material characterization
The composition and phase purity of these composites were characterized by X-ray diffraction (XRD). The morphology of these composites was characterized using FESEM and TEM (HRTEM). X-ray photoelectron spectroscopy (XPS) experiments were recorded by an AXIS ULTRA DLD instrument with aluminum Kα X-ray radiation. Brunauer–Emmett–Teller.(BET) surface areas and porosities of the products were determined by nitrogen adsorption and desorption using a Micromeritics ASAP 2020 analyzer. Thermogravimetric analysis (TGA) was performed with a Mettler-Toledo TGA/DSC1 with air flowing over the samples heated at 5 °C min−1. The exact Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn molar ratio in SnS/MoS2–C was investigated using an ICP optical emission spectrometer (ICP-OES) (Agilent 730). Raman measurement of the composite was performed using a laser Raman spectrometer (Jobin Yvon, T6400).
:Sn molar ratio in SnS/MoS2–C was investigated using an ICP optical emission spectrometer (ICP-OES) (Agilent 730). Raman measurement of the composite was performed using a laser Raman spectrometer (Jobin Yvon, T6400).
Electrochemical measurements
The working electrode consisted of active materials (SnS, MoS2/C and SnS/MoS2–C) mixed with carboxymethyl cellulose (CMC) and acetylene black with a weight ratio of 70:15:15 and sufficient deionized water to form a slurry. The electrolyte was composed of LiPF6 (1 M), ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1, v/v). Electrochemical measurements were made using a CR2025-type coin cell with Li metal as counter and reference electrode. The cycling performance and rate performance of the as-prepared samples were tested on a LAND-BT2013A measurement system with a voltage range from 0.01 to 3.0 V at 25 °C. CV measurements were conducted on a CHI 660E workstation at 0.1 mV s−1 with a voltage range of 0.01–3.0 V. Electrochemical impedance spectroscopy (EIS) analysis was done using a CHI 660E workstation in the frequency range from 100 kHz to 0.01 Hz.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Natural Science Foundation of China (51402109), Project of Public Interest Research and Capacity Building of Guangdong Province (2014A010106007), Pearl River S & T NovaProgram of Guangzhou (201506010030), Guangdong Innovative and Entrepreneurial Research Team Program (2014ZT05N200), Guangdong Natural Science Funds for Distinguished Young Scholar (2016A030306010) and Fundamental Research Funds for Central Universities, China (2017ZX010).Notes and references
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444 CrossRef CAS PubMed.
- F. H. Zheng, C. H. Yang, X. H. Xiong, J. W. Xiong, R. Z. Hu, Y. Chen and M. L. Liu, Angew. Chem., Int. Ed., 2015, 54, 13058 CrossRef CAS PubMed.
- C. H. Yang, X. Ou, X. H. Xiong, F. H. Zheng, R. Z. Hu, Y. Chen, M. L. Liu and K. V. Huang, Energy Environ. Sci., 2017, 10, 107 CAS.
- X. H. Xiong, G. H. Wang, Y. W. Lin, Y. Wang, X. Ou, F. H. Zheng, C. H. Yang, J. H. Wang and M. L. Liu, ACS Nano, 2016, 10, 10953 CrossRef CAS PubMed.
- Z. L. Yu, S. Xin, Y. You, L. Yu, Y. Lin, D. W. Xu, C. Qiao, Z. H. Huang, N. Yang, S. H. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 14915 CrossRef CAS PubMed.
- H. Q. Wang, Q. C. Pan, Q. Wu, X. H. Zhang, Y. G. Huang, A. Lushington, Q. Y. Li and X. L. Sun, J. Mater. Chem. A, 2017, 5, 4576 CAS.
- Y. F. Dong, M. L. Yu, Z. Y. Wang, Y. Liu, X. Z. Wang, Z. B. Zhao and J. S. Qiu, Adv. Funct. Mater., 2016, 26, 7590 CrossRef CAS.
- L. Yu, J. F. Yang and X. W. Lou, Angew. Chem., Int. Ed., 2016, 55, 13422 CrossRef CAS PubMed.
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255 CrossRef CAS PubMed.
- L. D. Lin, X. N. Xu, C. X. Chu, M. K. Majeed and J. Yang, Angew. Chem., Int. Ed., 2016, 55, 14063 CrossRef CAS PubMed.
- C. B. Zhu, P. Kopold, W. H. Li, P. A. van Aken, J. Maier and Y. Yu, Adv. Sci., 2015, 2, 1500200 CrossRef PubMed.
- D. H. Youn, S. K. Stauffer, P. H. Xiao, H. M. Park, Y. J. Nam, A. Dolocan, G. Henkelman, A. Heller and C. B. Mullins, ACS Nano, 2016, 10, 10778 CrossRef CAS PubMed.
- J. J. Cai, Z. S. Li and P. K. Shen, ACS Appl. Mater. Interfaces, 2012, 4, 4093 CAS.
- S. K. Li, J. X. Zheng, S. Y. Zuo, Z. G. Wu, P. X. Yan and F. Pan, RSC Adv., 2015, 5, 46941 RSC.
- J. Liu, Y. R. Wen, P. A. van Aken, J. Maier and Y. Yu, J. Mater. Chem. A, 2015, 3, 5259 CAS.
- D. D. Vaughn, O. D. Hentz, S. R. Chen, D. H. Wang and R. E. Schaak, Chem. Commun., 2012, 48, 5608 RSC.
- S. K. Li, J. X. Zheng, Z. X. Hu, S. Y. Zuo, Z. G. Wu, P. X. Yan and F. Pan, RSC Adv., 2015, 5, 72857 RSC.
- B. Zhao, Z. X. Wang, F. Chen, Y. Q. Yang, Y. Gao, L. Chen, Z. Jiao, L. L. Cheng and Y. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 1407 CAS.
- Y. Fang, Y. Y. Lv, F. Gong, A. A. Elzatahry, G. F. Zheng and D. Y. Zhao, Adv. Mater., 2016, 28, 9385 CrossRef CAS PubMed.
- T. T. Shan, S. Xin, Y. You, H. P. Cong, S. H. Yu and A. Manthiram, Angew. Chem., Int. Ed., 2016, 5, 12783 CrossRef PubMed.
- X. X. Zuo, K. Chang, J. Zhao, Z. Z. Xie, H. W. Tang, B. Li and Z. R. Chang, J. Mater. Chem. A, 2016, 4, 51 CAS.
- S. P. Zhang, B. V. R. Chowdari, Z. Y. Wen, J. Jin and J. H. Yang, ACS Nano, 2015, 9, 12464 CrossRef CAS PubMed.
- Q. Y. Li, Q. C. Pan, G. H. Yang, X. L. Lin, Z. X. Yan, H. Q. Wang and Y. G. Huang, J. Mater. Chem. A, 2015, 3, 20375 CAS.
- Y. G. Huang, Q. C. Pan, H. Q. Wang, C. Ji, X. M. Wu, Z. Q. He and Q. Y. Li, J. Mater. Chem. A, 2016, 4, 7185 CAS.
- Q. C. Pan, Y. G. Huang, H. Q. Wang, G. H. Yang, L. C. Wang, J. Chen, Y. H. Zan and Q. Y. Li, Electrochim. Acta, 2016, 197, 50 CrossRef CAS.
- Q. C. Pan, F. H. Zheng, X. Ou, C. H. Yang, X. H. Xiong and M. L. Liu, Chem. Eng. J., 2017, 316, 393 CrossRef CAS.
- X. Y. Zhao, M. H. Cao and C. W. Hu, RSC Adv., 2012, 2, 11737 RSC.
- J. G. Yu, H. T. Guo, S. A. Davis and S. Mann, Adv. Funct. Mater., 2006, 16, 2035 CrossRef CAS.
- S. W. Liu, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914 CrossRef CAS PubMed.
- W. P. Sun, X. H. Rui, D. Yang, Z. Q. Sun, B. Li, W. Y. Zhang, Y. Zong, S. Madhavi, S. X. Dou and Q. Y. Yan, ACS Nano, 2015, 9, 11371 CrossRef CAS PubMed.
- X. D. Li, W. Li, M. C. Li, P. Cui, D. H. Chen, T. Gengenbach, L. H. Chu, H. Y. Liu and G. S. Song, J. Mater. Chem. A, 2015, 3, 2762 CAS.
- D. P. Cong, J. H. Choi, J. Yun, A. S. Bandarenka, J. Kim, P. V. Braun, S. Young Jeong and C. R. Cho, ACS Nano, 2017, 11, 1026 CrossRef PubMed.
- J. Wang, J. L. Liu, D. L. Chao, J. X. Yan, J. Y. Lin and Z. X. Shen, Adv. Mater., 2014, 26, 7162 CrossRef CAS PubMed.
- J. X. Zhu, K. Sakaushi, G. Clavel, M. Shalom, M. Antonietti and T. P. Fellinger, J. Am. Chem. Soc., 2015, 137, 5480 CrossRef CAS PubMed.
- Q. C. Pan, F. H. Zheng, X. Ou, C. H. Yang, X. H. Xiong, Z. H. Tang, L. Z. Zhao and M. L. Liu, ACS Sustainable Chem. Eng., 2017, 5, 4739 CrossRef CAS.
- F. H. Zheng, Q. C. Pan, C. H. Yang, X. H. Xiong, X. Ou, R. Z. Hu, Y. Chen and M. L. Liu, Chem.–Eur. J., 2017, 23, 5051 CrossRef CAS PubMed.
- Y. Q. Teng, H. L. Zhao, Z. J. Zhang, Z. L. Li, Q. Xia, Y. Zhang, L. N. Zhao, X. F. Du, Z. H. Du, P. P. Lv and K. Swierczek, ACS Nano, 2016, 10, 8526 CrossRef CAS PubMed.
- B. Chen, E. Z. Liu, T. T. Cao, F. He, C. S. Shi, C. N. He, L. Y. Ma, Q. Y. Li, J. J. Li and N. Q. Zhao, Nano Energy, 2017, 33, 247 CrossRef CAS.
- Y. Chen, B. H. Song, X. S. Tang, L. Lu and J. M. Xue, Small, 2014, 10, 1536 CrossRef CAS PubMed.
- R. H. Wang, C. H. Xu, J. Sun, Y. Q. Liu, L. Gao, H. L. Yao and C. C. Lin, Nano Energy, 2014, 8, 183 CrossRef CAS.
- D. A. Agyeman, K. Song, G. H. Lee, M. H. Park and Y. M. Kang, Adv. Energy Mater., 2016, 6, 1600904 CrossRef.
- H. H. Zhao, H. Zeng, Y. Wu, S. G. Zhang, B. Li and Y. H. Huang, J. Mater. Chem. A, 2015, 3, 10466 CAS.
- Y. Liu, X. Z. Wang, X. D. Song, Y. F. Dong, L. Yang, L. X. Wang, D. Z. Jia, Z. B. Zhao and J. S. Qiu, Carbon, 2016, 109, 461 CrossRef CAS.
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. (David) Lou, Energy Environ. Sci., 2014, 7, 3302 CAS.
- B. Chen, E. Z. Liu, F. He, C. S. Shi, C. N. He, J. J. Li and N. Q. Zhao, Nano Energy, 2016, 26, 541 CrossRef CAS.
- J. Y. Li, Y. Hou, X. F. Gao, D. S. Guan, Y. Y. Xie, J. H. Chen and C. Yuan, Nano Energy, 2015, 16, 10 CrossRef CAS.
- S. B. Wang, B. Y. Guan, L. Yu and X. W. (David) Lou, Adv. Mater., 2017, 1702724 CrossRef PubMed.
- Y. M. Chen, X. Y. Yu, Z. Li, U. Paik and X. W. (David) Lou, Sci. Adv., 2016, 2, e1600021 Search PubMed.
- Z. Y. Zeng, X. W. Zhang, K. Bustillo, K. Y. Niu, C. Gammer, J. Xu and H. M. Zheng, Nano Lett., 2015, 15, 5214 CrossRef CAS PubMed.
- L. L. Luo, B. L. Zhao, B. Xiang and C. M. Wang, ACS Nano, 2016, 10, 1249 CrossRef CAS PubMed.
- X. H. Xiong, C. H. Yang, G. H. Wang, Y. W. Lin, X. Ou, J. H. Wang, B. T. Zhao, M. L. Liu, Z. Lin and K. V. Huang, Energy Environ. Sci., 2017, 10, 1757 CAS.
- Y. W. Wang, L. Yu and X. W. (David) Lou, Angew. Chem., Int. Ed., 2016, 55, 7423 CrossRef CAS PubMed.
- X. Y. Yu, H. Hu, Y. W. Wang, H. Y. Chen and X. W. (David) Lou, Angew. Chem., Int. Ed., 2015, 54, 7395 CrossRef CAS PubMed.
- X. Xu, Z. Y. Fan, X. Y. Yu, S. J. Ding, D. M. Yu and X. W. (David) Lou, Adv. Energy Mater., 2014, 4, 1400902 CrossRef.
- S. Q. Zhao, Z. W. Wang, Y. J. He, B. B. Jiang, Y. W. Harn, X. Q. Liu, F. Q. Yu, F. Feng, Q. Shen and Z. Q. Lin, ACS Energy Lett., 2017, 2, 111 CrossRef CAS.
- B. B. Jiang, C. P. Han, B. Li, Y. J. He and Z. Q. Lin, ACS Nano, 2016, 10, 2728 CrossRef CAS PubMed.
- B. B. Jiang, Y. J. He, B. Li, S. Q. Zhao, S. Wang, Y. B. He and Z. Q. Lin, Angew. Chem., Int. Ed., 2017, 56, 1869 CrossRef CAS PubMed.
- C. Wu, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1604015 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta08346g |
| This journal is © The Royal Society of Chemistry 2018 |