
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanisms of catalytic reduction of CO2 with heme and nonheme metal complexes
Shunichi
Fukuzumi
 *ab,
Yong-Min
Lee
*ac,
Hyun S.
Ahn
*d and
Wonwoo
Nam
*ae
*ab,
Yong-Min
Lee
*ac,
Hyun S.
Ahn
*d and
Wonwoo
Nam
*ae
aDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 03760, Korea. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp; yomlee@ewha.ac.kr; wwnam@ewha.ac.kr
bGraduate School of Science and Engineering, Meijo University, Nagoya, Aichi 468-8502, Japan
cResearch Institute for Basic Sciences, Ewha Womans University, Seoul 03760, Korea
dDepartment of Chemistry, Yonsei University, Seoul 03722, Korea. E-mail: ahnhs@yonsei.ac.kr
eSchool of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, P. R. China
First published on 2nd July 2018
Abstract
The catalytic conversion of CO2 into valuable chemicals and fuels has attracted increasing attention, providing a promising route for mitigating the greenhouse effect of CO2 and also meeting the global energy demand. Among many homogeneous and heterogeneous catalysts for CO2 reduction, this mini-review is focused on heme and nonheme metal complexes that act as effective catalysts for the electrocatalytic and photocatalytic reduction of CO2. Because metalloporphyrinoids show strong absorption in the visible region, which is sensitive to the oxidation states of the metals and ligands, they are suited for the detection of reactive intermediates in the catalytic CO2 reduction cycle by electronic absorption spectroscopy. The first part of this review deals with the catalytic mechanism for the one-electron reduction of CO2 to oxalic acid with heme and nonheme metal complexes, with an emphasis on how the formation of highly energetic CO2˙ is avoided. Then, the catalytic mechanism of two-electron reduction of CO2 to produce CO and H2O is compared with that to produce HCOOH. The effect of metals and ligands of the heme and nonheme complexes on the CO or HCOOH product selectivity is also discussed. The catalytic mechanisms of multi-electron reduction of CO2 to methanol (six-electron reduced product) and methane (eight-electron reduced product) are also discussed for both electrocatalytic and photocatalytic systems.
 Shunichi Fukuzumi | Shunichi Fukuzumi earned a bachelor's degree and a Ph.D. degree in applied chemistry from the Tokyo Institute of Technology in 1973 and 1978, respectively. After working as a postdoctoral fellow from 1978 to 1981 at Indiana University in USA, he became an Assistant Professor in 1981 at Osaka University where he was promoted to a Full Professor in 1994. His research has been focused on electron transfer chemistry, particularly artificial photosynthesis. He is currently a Distinguished Professor at Ewha Womans University, a Designated Professor at Meijo University, and a Professor Emeritus at Osaka University. |
 Yong-Min Lee | Yong-Min Lee received his M.S. and Ph.D. degrees in Inorganic Chemistry from Pusan National University, Republic of Korea, under the supervision of Professor Sung-Nak Choi in 1999. After he worked at the Magnetic Resonance Centre (CERM) at the University of Florence, Italy, as a Postdoctoral fellow and Researcher under the direction of Professors Ivano Bertini and Claudio Luchinat from 1999 to 2005, he joined the Centre for Biomimetic Systems at Ewha Womans University, as a Research Professor in 2006. He is currently a Special Appointment Professor at Ewha Womans University since 2009. |
 Hyun S. Ahn | Hyun S. Ahn earned his undergraduate degrees in chemistry and chemical engineering from the University of Washington in Seattle (2008). He then pursued his Ph.D. studies at the University of California at Berkeley, in the field of inorganic chemistry under the supervision of Prof. T. Don Tilley. Upon conferral of his Ph.D. in 2013, Hyun continued his research as a postdoctoral scholar at the University of Texas at Austin with Prof. Allen J. Bard. He is currently an assistant professor of chemistry at Yonsei University. Hyun's research interest spans across the fields of catalysis, photoelectrochemistry, bio-electrochemistry, and energy conversion technologies. |
 Wonwoo Nam | Wonwoo Nam earned his B.S. (Honours) degree in Chemistry from California State University, Los Angeles, and his Ph.D. degree in Inorganic Chemistry from the University of California, Los Angeles (UCLA) under the supervision of Professor Joan S. Valentine in 1990. After working as a postdoctoral fellow at UCLA for one year, he became an Assistant Professor at Hong Ik University in 1991. In 1994, he moved to Ewha Womans University, where he is currently a Distinguished Professor. His current research has been focused on the dioxygen activation, water oxidation, and important roles of metal ions in bioinorganic chemistry. |
1 Introduction
Solar-driven reduction of CO2 has merited increasing attention due to the hike in the world-wide consumption of fossil fuels and the consequential escalation in the atmospheric CO2 level.1–9 There have so far been extensive studies on solar-driven reduction of CO2 (ref. 7–21) as well as electrocatalytic reduction22–36 and catalytic hydrogenation of CO2 in aprotic solvents and also in water.37–51 Carbon dioxide can be reduced by one electron and one proton to produce a half equivalent of oxalic acid (H2C2O4) with a standard reduction potential of −0.50 V vs. SHE [eqn (1)].26,52 The two-electron reduction of CO2 with two protons affords formic acid (HCOOH) [eqn (2)] or CO and H2O [eqn (3)] with the standard reduction potentials of −0.25 and −0.11 V, respectively.52 Carbon dioxide can be further reduced by four, six, and eight electrons with four, six, and eight protons to produce formaldehyde [HCHO: eqn (4)], methanol [CH3OH: eqn (5)] or ethylene [C2H4: eqn (6)], and methane [CH4: eqn (7)] with the standard reduction potentials of −0.07, +0.02 or +0.06 V, and +0.17 vs. SHE, respectively.52 Therefore, the standard reduction potential is anodically shifted with an increased number of electrons and protons for CO2 reduction, indicating that the involvement of a higher number of electrons and protons favours the CO2 reduction thermodynamically. However, because the kinetic barrier generally increases with an additional number of electrons and protons involved in the reaction, appropriate catalysts are required to facilitate turnovers.| CO2 + e− + H+ → (1/2)H2C2O4, E0 = −0.50 V vs. SHE | (1) |
| CO2 + 2e− + 2H+ → HCOOH, E0 = −0.25 V vs. SHE | (2) |
| CO2 + 2e− + 2H+ → CO + H2O, E0 = −0.11 V vs. SHE | (3) |
| CO2 + 4e− + 4H+ → HCHO + H2O, E0 = −0.07 V vs. SHE | (4) |
| CO2 + 6e− + 6H+ → CH3OH + H2O, E0 = +0.02 V vs. SHE | (5) |
| CO2 + 6e− + 6H+ → (1/2)C2H4 + 4H2O, E0 = +0.06 V vs. SHE | (6) |
| CO2 + 8e− + 8H+ → CH4 + 2H2O, E0 = +0.17 V vs. SHE | (7) |
Among many catalysts for CO2 reduction, metalloporphyrinoid complexes are suitable for mechanistic studies, because metalloporphyrinoids such as heme have intense absorption bands, which are sensitive to the oxidation states of metals and porphyrinoid ligands.53–58 Not only heme but also nonheme metal complexes have merited significant interest as bioinspired catalysts, which are studied in many redox reactions.26,58–73 This review is intended to focus on the mechanisms of both electrocatalytic and photocatalytic reduction of CO2 with heme and nonheme metal complexes. The catalytic mechanisms are discussed not only for the one-electron/one-proton and two-electron/two-proton reductions of CO2 [eqn (1) and (3)] but also for multi-electron/multi-proton pathways [eqn (4)–(7)]. The electrocatalytic and photocatalytic efficiencies are discussed based on the overpotentials and quantum yields, respectively.
2 Catalytic one-electron reduction of CO2 to oxalic acid
The standard one electron reduction potential of CO2 to CO2˙ is as negative as −2.21 V vs. SCE (=−1.97 V vs. SHE) in N,N′-dimethylformamide (DMF)74 and −1.90 V vs. SCE in water.75 The dimerization of CO2˙ affords oxalate dianions (C2O42−).76 Therefore, catalysts are required to avoid the formation of highly energetic CO2˙ and secure a low energy pathway en route to oxalic acid.AgII and PdII complexes of both 2,3,7,8,12,13,17,18-octaethylporphyrin (OEP) and 5,10,15,20-tetraphenylporphyrin (TPP) were reported to act as catalysts for one-electron reduction of CO2 to produce oxalic acid in CH2Cl2 containing 0.10 M tetrabutylammonium fluoride (TBAF) at an applied potential of −1.65 and −1.80 V vs. SCE, respectively.77 In comparison with AgII(TPP) and PdII(TPP), neither CuII(TPP) nor NiII(TPP) showed catalytic activity for the electrochemical CO2 reduction.77
In contrast to CuII(TPP), a dinuclear nonheme copper(II) complex ([4]4+) can catalyse the one-electron reduction of CO2 in acetonitrile (MeCN) in the presence of LiClO4 to produce lithium oxalate at an applied potential of −0.03 V vs. NHE.78 The electrochemical reduction of [4]4+ at a cathodic peak potential (Epc) of +0.06 V vs. NHE (=+0.06 V vs. SHE) produced a dinuclear copper(I) complex ([1]2+) that is oxidized in air selectively by CO2 (rather than O2) to yield a tetranuclear copper(II) complex containing two bridging CO2-derived oxalate groups ([2]4+) as shown in Scheme 1.78 The treatment of the copper(II) oxalate complex in MeCN with a soluble lithium salt results in the quantitative precipitation of lithium oxalate.78 DFT calculations suggest the catalytic mechanism (Scheme 2) in which one CO2 molecule is first reduced cooperatively by two Cu(I) metals to give a fully delocalized mixed-valence CuI/CuII(CO2˙) radical anion intermediate, followed by further partial reduction of the metal-ligated CO2 molecule and (metal-mediated) nucleophilic-like attack on the carbon atom of an incoming second CO2 molecule to afford the dinuclear Cu(II)-oxalate product ([2]4+).79
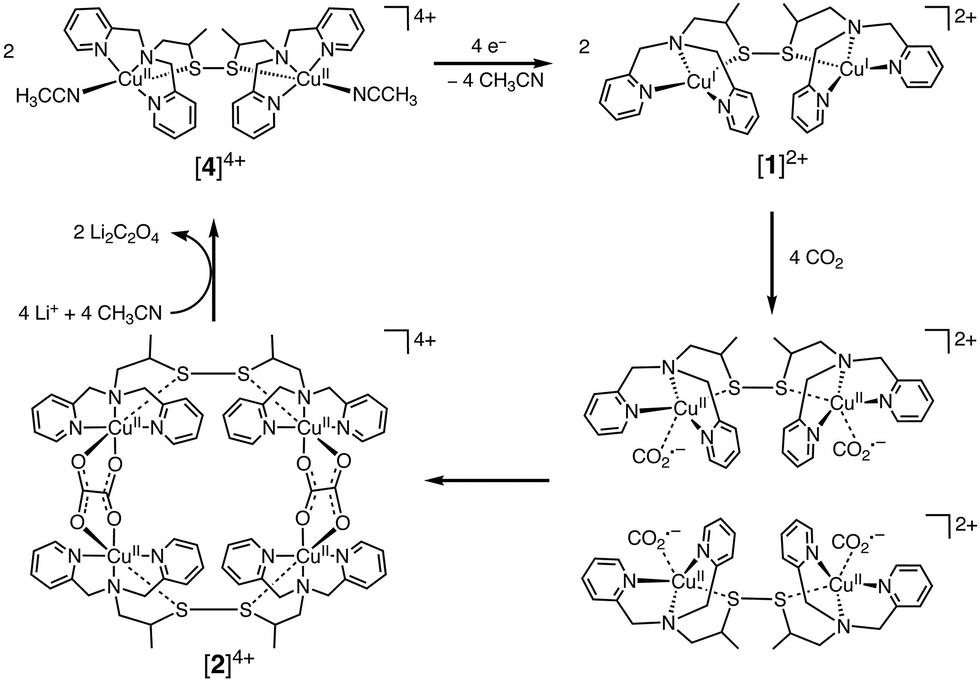 | ||
| Scheme 1 Catalytic one-electron reduction of CO2 to oxalate with a dinuclear copper(I) complex ([1]2+). Reprinted with permission from ref. 78. Copyright 2010, The American Association for the Advancement of Science. | ||
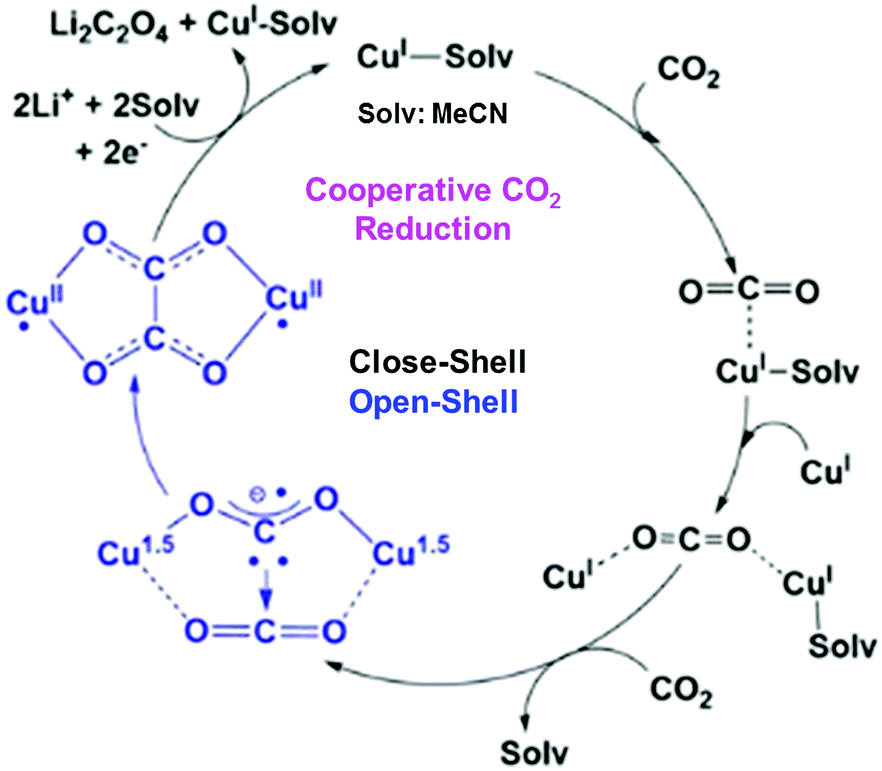 | ||
| Scheme 2 Proposed catalytic cycle for one-electron reduction of CO2 to oxalate with a copper(I) complex. Reprinted with permission from ref. 79. Copyright 2017, American Chemical Society. | ||
A binuclear metallacyclic copper complex was reported to be capable of selectively capturing CO2 from air and reduce CO2 to oxalate, in the form of an oxalate-bridged complex. The oxalate-bridged complex releases oxalic acid when it is treated with dilute mineral acid to regenerate the original copper complex.80
It has also been reported that the one-electron reduction of chalcogen-bridged tricopper cyclophanates (Cu3EL: L3− = tris(β-diketiminate; E = S, Se) (the one-electron reduction potential: Ered = −0.89 and −1.04 V vs. SCE, respectively) by CoCp2* (one-electron oxidation potential: Eox = −1.94 V vs. Fc/Fc+), FeCp(C6Me6) (Eox = −2.07 V vs. Fc/Fc+), or KC8 (Eox < −3.7 V vs. Fc/Fc+) in the presence of KPF6 in DMF afforded [Cu3EL]−, which reacts with CO2 to yield exclusively C2O42− (95% yield, TON = 24) and regenerate Cu3EL.81 Catalysis is observed employing KC8 and FeCp(C6Me6) as reductants, but only in the presence of KPF6.81
3 Electrocatalytic reduction of CO2 to CO with metalloporphyrins
Iron tetraphenylporphyrin [Fe(TPP)] (Fig. 1) was reported to catalyse the electrochemical reduction of CO2 at the FeI/Fe0 redox potential (−1.64 V vs. SCE) in DMF.82 However, the catalytic efficiency was very low and the catalytic activity of Fe(TPP) was rapidly lost during preparative-scale electrolysis.82 The addition of Mg2+ ions to the solution resulted in improved catalytic efficiency, resulting in a CO faradaic yield of ca. 60–70% and the rest in formate.83 The presence of Lewis acids, such as Li+, Na+, Ba2+, and Al3+ ions, also improved the catalytic efficiency. The addition of CF3CH2OH (1.47 M) resulted in a large increase of the FeI/Fe0 current to reach an ip/ip0 value of 131.82 The ip/ip0 value is given by eqn (8),| ip/ip0 = 2.24(kapp[CO2]RT/Fv)1/2 | (8) |
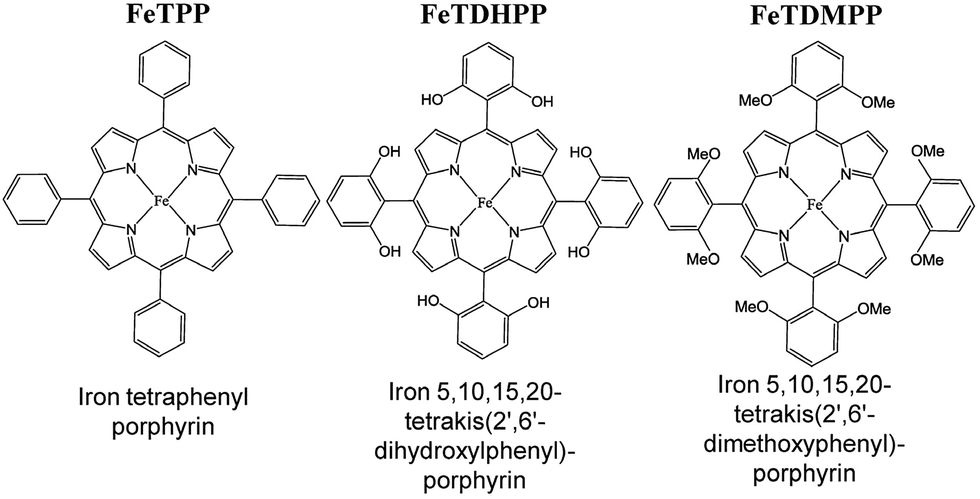 | ||
| Fig. 1 Iron tetraphenylporphyrin derivatives used for electrocatalytic reduction of CO2 to CO. Reprinted with permission from ref. 86. Copyright 2012, The American Association for the Advancement of Science. | ||
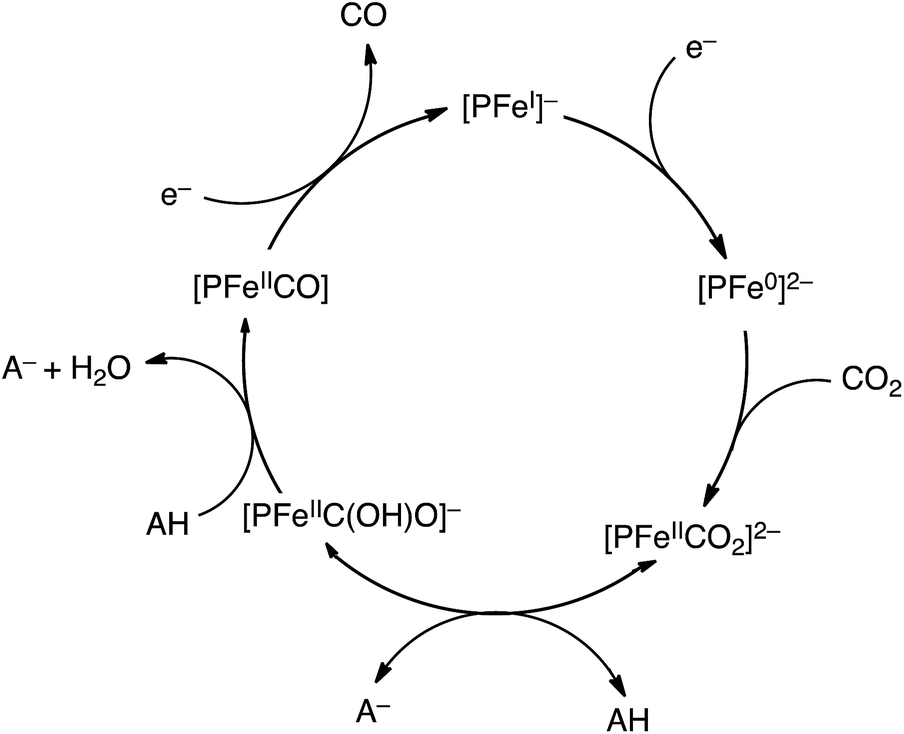 | ||
| Scheme 3 Catalytic cycle for two-electron reduction of CO2 to CO with an iron porphyrin ([PFeI]−).84 | ||
The electrocatalytic efficiency of CO2 reduction to CO was improved by the introduction of phenolic groups in all ortho and ortho′ positions of the phenyl groups of iron tetraphenylporphyrin.86 The electrocatalytic reduction of CO2 with iron 5,10,15,20-tetrakis(2′,6′-dihydroxyphenyl)porphyrin (FeTDHPP in Fig. 1) in the presence of 2 M H2O in CO2-saturated DMF ([CO2] = 0.23 M) afforded a CO faradaic yield of 94% with a turnover number (TON) of 5.0 × 107 for 4 hours of electrolysis at −1.16 V vs. SHE with no observed degradation.86 The average current density was 0.31 mA cm−2 that corresponds to a turnover frequency (TOF) of 3.2 × 103 s−1 at an overpotential of 0.466 V, which is the most efficient among other CO2 reduction catalysts.86–90 When the hydroxy groups in FeTDHPP were replaced by methoxy groups (FeTDMPP in Fig. 1), the catalytic activity of FeTDMPP was decreased by a factor of around 1 billion as compared to that of FeTDHPP with hydroxyl groups.86 Therefore, the enhanced catalytic activity of FeTDHPP was attributed to the high local concentration of protons associated with the phenolic hydroxy substituents.86
The introduction of four positively charged trimethylanilinium groups on the phenyl groups of iron tetraphenylporphyrin resulted in further improvement of the electrocatalytic activity of CO2 reduction by means of coulombic stabilization of the initial Fe0–CO2 adduct.88 When four positively charged trimethylanilinium groups were introduced at the ortho positions of the TPP groups (Fe-o-TMA in Fig. 2), the maximum TOF was as high as 106 s−1 at a low overpotential of 0.220 V.88 The catalyst standard potential (E0cat) of Fe-o-TMA was determined to be −0.944 V vs. SHE, which is the most positive ever reported for an iron porphyrin CO2-reduction catalyst.88 The para-substituted analogue (Fe-p-TMA in Fig. 2) exhibited an E0cat value of −1.263 V vs. SHE, which is much more negative than that of Fe-o-TMA, due to smaller coulombic stabilization of the initial Fe0–CO2 adduct.88 The importance of the coulombic stabilization for the electrocatalytic activity of CO2 reduction was further demonstrated by the introduction of four negatively charged sulfonate groups on the phenyl groups of iron tetraphenylporphyrin (Fe-p-PSULF in Fig. 2), which resulted in a more negative E0cat value (−1.428 V vs. SHE).88
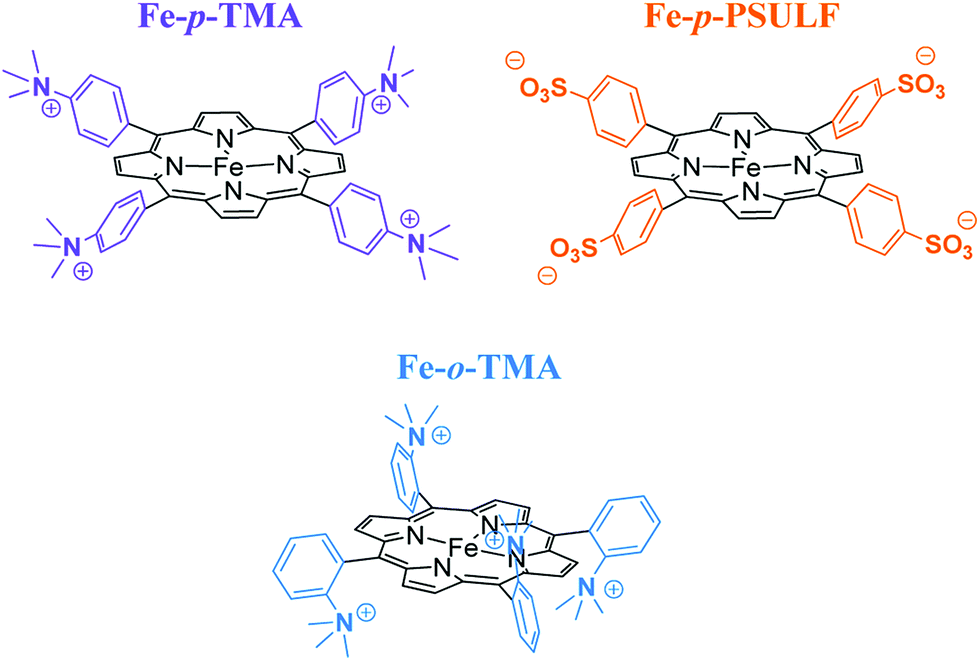 | ||
| Fig. 2 Iron tetraphenylporphyrin derivatives with positive and negative charges used for electrocatalytic reduction of CO2 to CO. Reprinted with permission from ref. 88. Copyright 2016, American Chemical Society. | ||
In contrast to the electrocatalytic reduction of CO2 in nonaqueous aprotic solvents such as DMF (vide supra), the electrocatalytic reduction of CO2 in water always competes with the reduction of aqueous protons to hydrogen (H2). The TOF of the electrocatalytic CO2 reduction in an aqueous solution was reported to be much smaller compared to that in DMF solution because of the lower solubility of CO2 in water.88 A successful example of selective electrocatalytic reduction of CO2 to CO over proton reduction was reported by employing Fe-p-TMA (Fig. 2) as a catalyst. This preparative-scale electrolysis was performed in water at pH 6.7 (adjusted by KOH addition) with an applied potential of −0.97 V vs. NHE for 4 h.88 The average faradaic yields were CO (90%), H2 (7%), acetate (1.4%), formate (0.7%) and oxalate (0.5%).88 When pH was adjusted to 3.7 by the addition of 0.1 M formic acid buffer, the electrolysis with Fe-p-TMA resulted in the exclusive proton reduction to produce H2 over the CO2 reduction.88
The selective electrocatalytic reduction of CO2 to CO (over proton reduction) under acidic conditions was made possible with a cobalt(II) chlorin complex, CoII(Ch), adsorbed on multi-walled carbon nanotubes (MWCNTs) as a catalyst (Fig. 3).91 The electrolysis of a CO2-saturated aqueous solution (pH = 4.6 with Na2SO4) employing a glassy carbon working electrode modified with CoII(Ch)@MWCNT (0.01 μmol) afforded mainly CO with a maximum TON of 1500 and TOF of 100 h−1 and little H2 at −1.1 V vs. NHE (=−1.34 V vs. SCE).91 The faradaic yield of CO for the initial 2 h was determined to be as high as 89%, whereas that for H2 production was only 11% at pH 4.6.91 The high selectivity for CO was maintained at pH 3.6, although H2 became the main product at pH 2.0.91
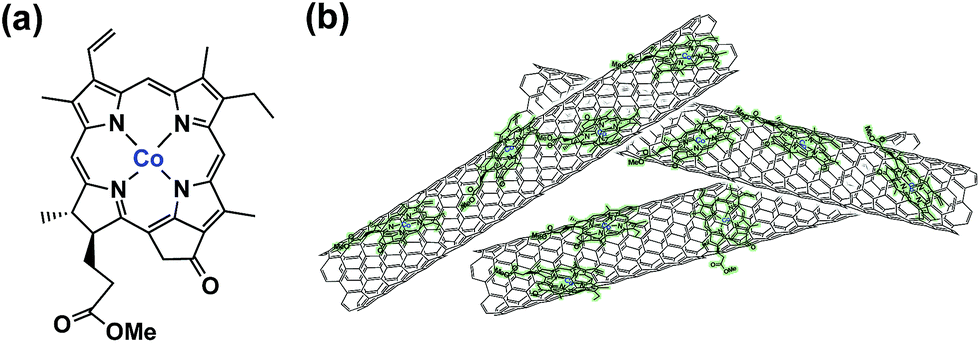 | ||
| Fig. 3 (a) Structure of CoII(Ch) and (b) schematic image of CoII(Ch) on MWCNTs. Reprinted with permission from ref. 91. Copyright 2015, Royal Society of Chemistry. | ||
When MWCNTs were replaced by reduced graphene oxide (rGO), which is a planar π-system, as a support material of CoII(Ch), the CO yield became significantly smaller (TON = 350 for CO and 250 for H2 at 20 h) (Fig. 4b) compared to that with MWCNTs (Fig. 4a).91 Thus, the three dimensional assembly of MWCNTs with CoII(Ch) (Fig. 3b) on the electrode surface is essential for the selective electrocatalytic reduction of CO2 to CO.91 The π–π interaction between MWCNTs and CoII(Ch) is presumed to provide a suitable hydrophobic environment for more selective binding of CO2 over protons compared to the system with two-dimensional rGO as a support material.91
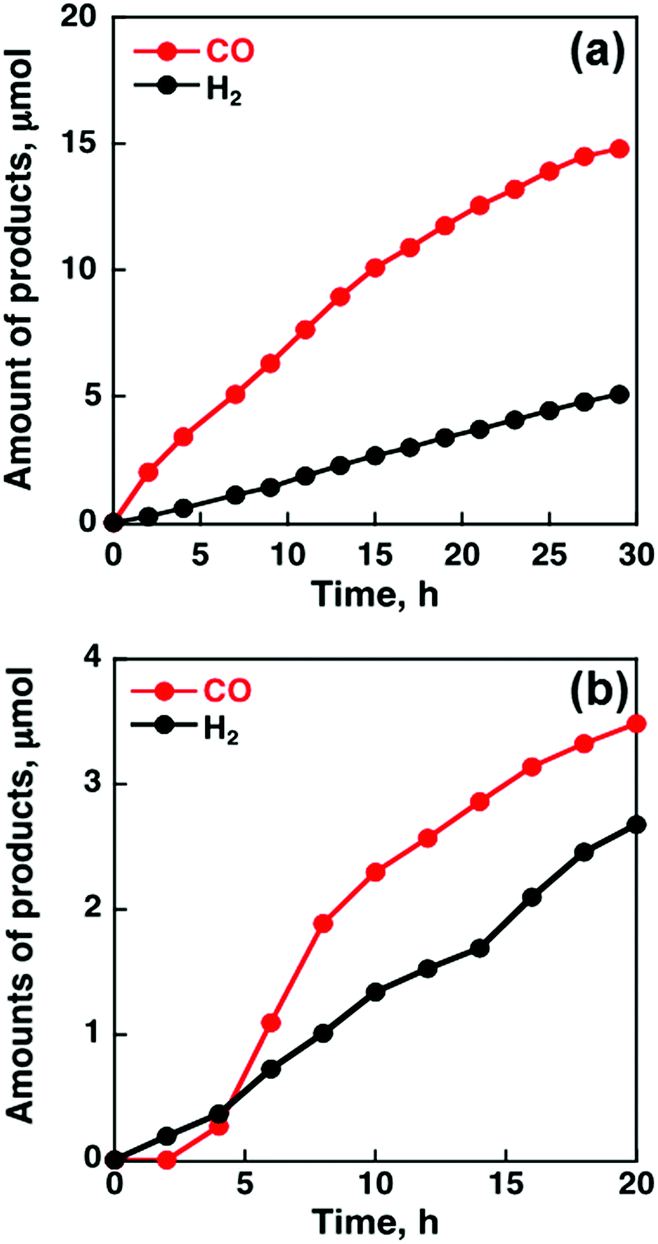 | ||
| Fig. 4 Time profiles of the formation of CO and H2 in the electrocatalytic reduction of CO2 on a glassy carbon electrode modified with CoII(Ch) (0.01 μmol) adsorbed on (a) MWCNTs (13 μg) and (b) rGO (13 μg) in a CO2-saturated aqueous solution (pH 4.6) containing Na2SO4 (5.0 mM) at an applied potential of −1.34 V vs. SCE. Reprinted with permission from ref. 91. Copyright 2015, Royal Society of Chemistry. | ||
FeTDHPP (Fig. 1), a known CO2 reduction catalyst in DMF, also exhibits electrocatalysis in a CO2-saturated aqueous solution (pH 7.3, NaHCO3 0.5 M) at an applied potential of −1.03 V vs. NHE (η = 480 mV), selectively producing CO over H2.92 A surface immobilisation tactic was implemented for the FeTDHPP catalyst on carbon surfaces by removing one phenyl group and appending a pyrene unit through a short linker.92 This immobilisation boosted both selectivity (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 CO:H2 ratio) and overall conversion (97% total faradaic yield).92
:4 CO:H2 ratio) and overall conversion (97% total faradaic yield).92
The cobalt phthalocyanine (CoPc) complex when adsorbed on MWCNTs was demonstrated to be an electrocatalyst for CO2 reduction to CO at −0.63 V vs. RHE, with a remarkable TON of 9.7 × 104 and a faradaic yield exceeding 90%.93 Compared to CoPc/MWCNT (2.5%), CoPc/rGO (2.2%) and CoPc/CB (3.3%) (CB: carbon black) exhibited less than one-third of the current density at −0.59 V vs. RHE with a 10% lower faradaic yield of CO and inferior stability.93 The higher graphitic nature of CNTs compared to those of either rGO or CB allows stronger π–π interactions with CoPc and higher electron conduction, resulting in superior electrocatalysis.93 A Pc/CNT hybrid without Co afforded a much lower faradaic yield for CO (only 19%), indicating that the Co centres in the CoPc/CNT act as the primary catalyst active sites.85 Low but non-zero conversion of CO2 to CO on Pc/CNT is attributed to the catalytic activity of Pc itself.93
An iron tetraphenylporphyrin bearing six pendant OH groups in ortho and ortho′ positions on three of the phenyl rings and one carboxylic acid group in the para position of the fourth phenyl was covalently attached to MWCNTs (CATCO2H, Scheme 4).94 The covalent grafting of an Fe porphyrin on MWCNTs also led to efficient electrocatalytic reduction of CO2 to CO selectively in water (pH 7.3) at an overpotential of 0.5 V.94
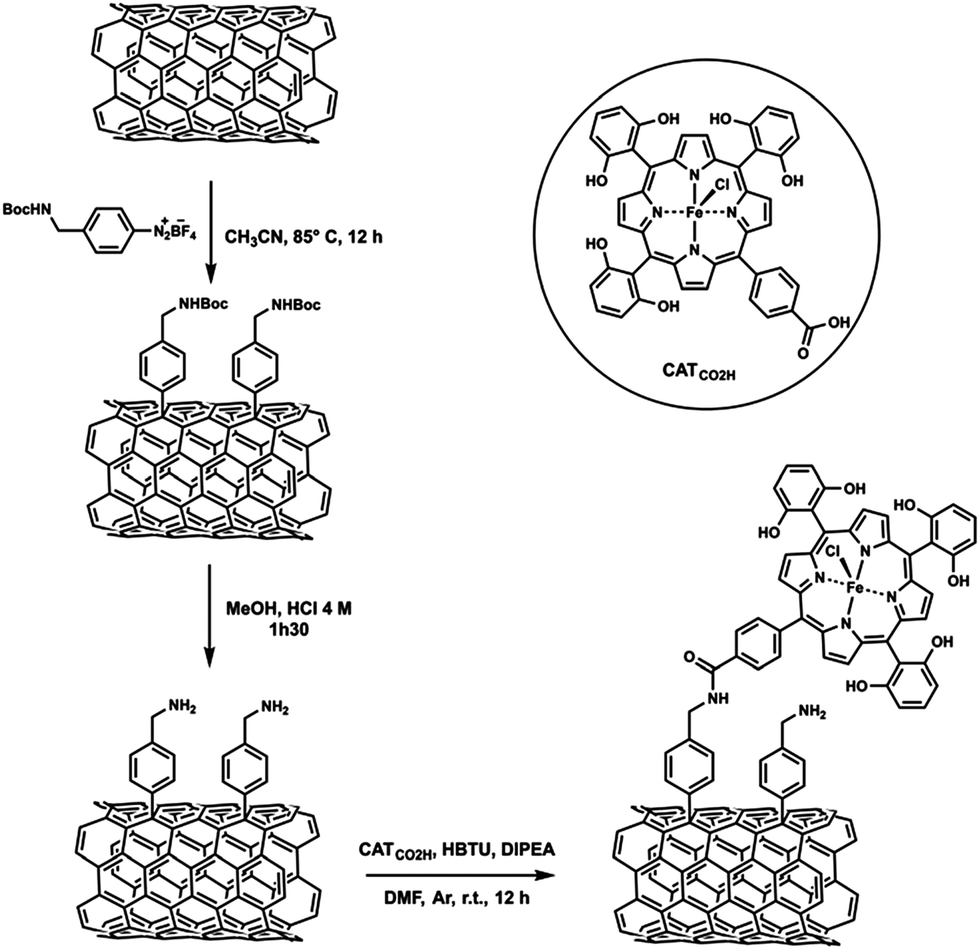 | ||
| Scheme 4 An iron tetraphenylporphyrin bearing six pendant OH groups in ortho and ortho′ positions on three of the phenyl rings and one carboxylic acid group in the para position of the fourth phenyl covalently attached to MWCNTs. Reprinted with permission from ref. 94. Copyright 2016, Royal Society of Chemistry. | ||
Remarkably, the redox silent zinc(II) complex of 5,10,15,20-tetramesitylporphyrin (ZnTPP) was also reported as an effective electrocatalyst that afforded faradaic efficiencies as high as 95% for CO2 reduction to CO at −1.7 V vs. SHE in DMF/H2O (9:1 v/v).95 The UV-vis spectrum of the reduced ZnTPP species exhibits absorption bands at 710, 820, and 920 nm (Fig. 5, red line), which are the characteristic features of a transiently generated Zn complex with reduced ligands (i.e., TPP˙).95 ZnTPP˙ can react with CO2 to regenerate ZnTPP and unidentified species.95 The exact mechanism of the CO2 reduction by ZnTPP˙ is yet to be clarified.
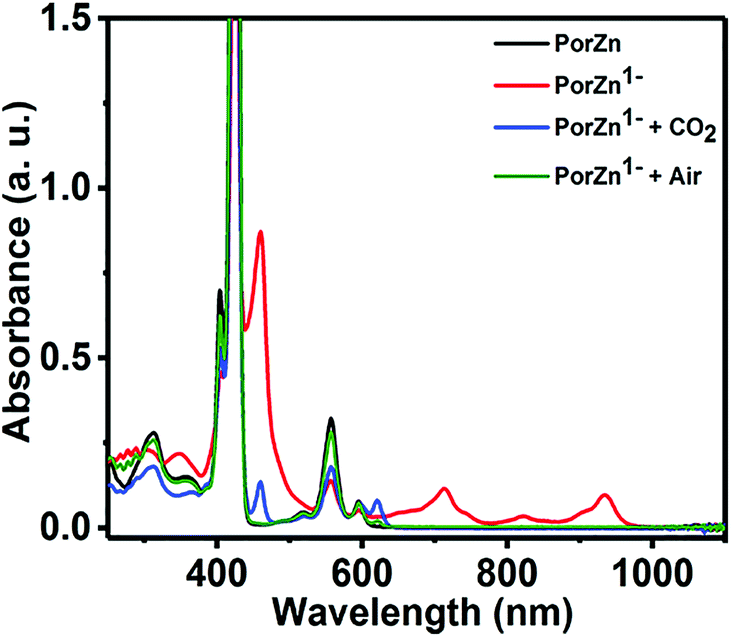 | ||
| Fig. 5 Absorption spectra of ZnTPP (black line), its reduced species (red line) produced by the reduction of ZnTPP by sodium naphthalenide, and the reduced species after exposure to CO2 (blue line) or air (green line) in THF. Reprinted with permission from ref. 95. Copyright 2017, American Chemical Society. | ||
4 Electrocatalytic reduction of CO2 to CO with nonheme metal complexes
A nonheme cobalt complex with a macrocyclic aminopyridine (I in Scheme 5) can also catalyse the electrochemical reduction of CO2 to CO with a faradaic efficiency of 98 ± 2% and 1.22(1) million TON at an applied potential of −2.8 V in a DMF solution containing [nBu4N][PF6] (0.1 M) and trifluoroethanol (1.2 M) under a CO2 atmosphere.96 When the pendant N–H group was replaced by a N–Me group, TONs became 300 times lower than that of I, indicating that the presence of the pendant NH moiety of the secondary amine is crucial for catalysis.96,97 Moreover, the presence of NH groups leads to a positive shift in the reduction potential of the CoI/0 couple, resulting in a decrease in the overpotential for CO2 reduction. Complex I is reduced by one electron to generate the CoI complex, which is further reduced to the Co0 form. CO2 binds to the Co0 complex to produce the CO2 adduct ([Co(CO2)]0, III in Scheme 5), which is stabilised by intramolecular H-bonds of the pendant secondary amines.96 The successive protonation of the CO2 complex affords CO and H2O to regeneratecomplex I.96,97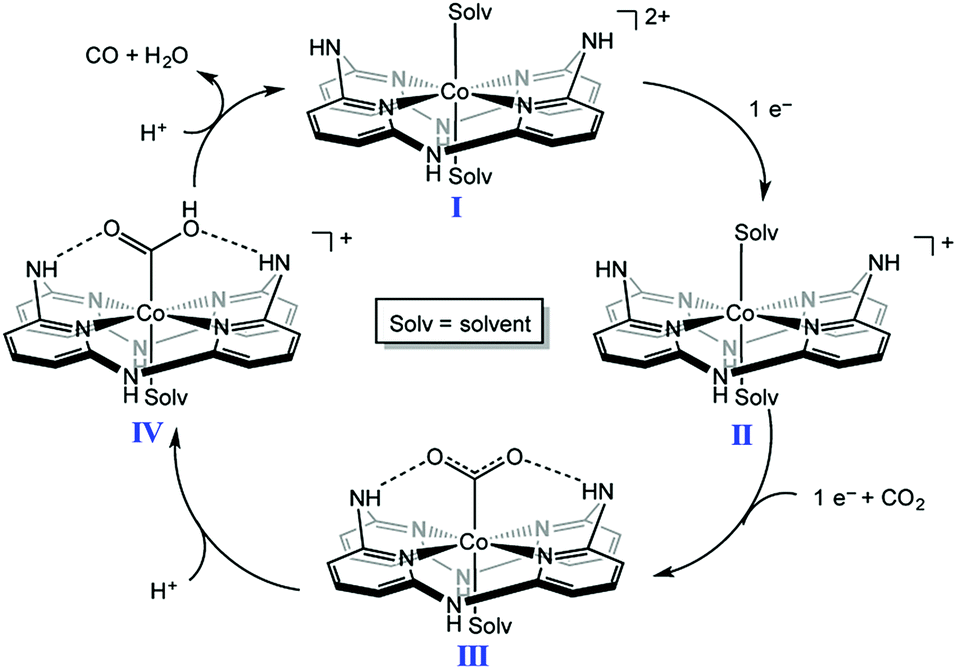 | ||
| Scheme 5 Catalytic cycle of two-electron reduction of CO2 to CO with a nonheme cobalt complex with a macrocyclic aminopyridine (I). Reprinted with permission from ref. 96. Copyright 2016, American Chemical Society. | ||
The [CoII(qpy)(H2O)2]2+ (qpy = 2,2′:6′,2′′:6′′,2′′′-quaterpyridine) complex also efficiently catalyses the electrochemical CO2-to-CO conversion in an MeCN solution in the presence of weak Brønsted acids.98 Controlled potential electrolysis (CPE) was performed at −1.1 V vs. SCE in the presence of 3 M PhOH, corresponding to 140 mV overpotential. CO was produced with 96% catalytic selectivity (a small amount of H2 (4%) was obtained as the only byproduct) and 94% faradaic efficiency.98 The [CoII(qpy)(H2O)2]2+ complex is an excellent catalyst, at a very low overpotential, better than active Mn complexes (vide infra),99–103 being only surpassed by the Fe tetraphenylporphyrin bearing four trimethylammonium groups in the ortho position of each phenyl (complex a in Fig. 6).88
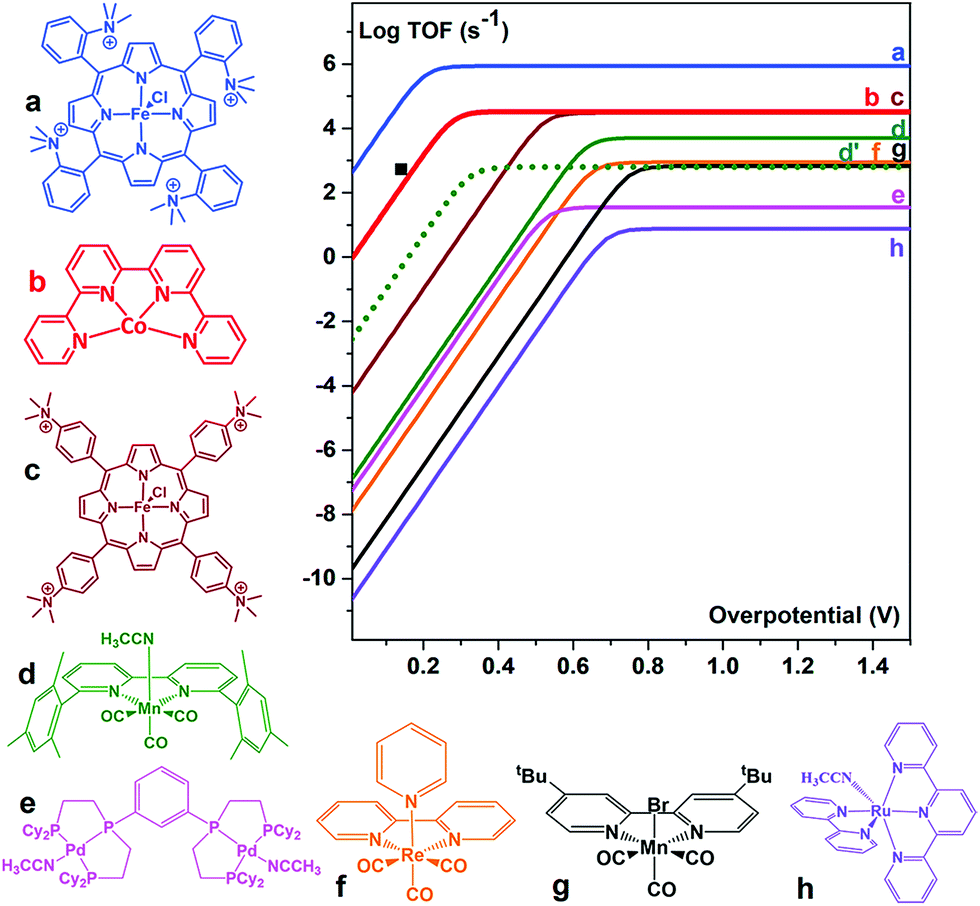 | ||
| Fig. 6 Comparison of the metal catalysts (a,88 b,98 c,88 d,99 e,100 f,101 g,102 and h103) for the CO2-to-CO electrochemical conversion in DMF or MeCN by means of their catalytic Tafel plots (log TOF as a function of the overpotential (η = E0(CO2/CO) − E). d′ dotted lines: data for complex d in the presence of 0.1 M Mg2+.99 ■: TOF value for b. Reprinted with permission from ref. 98. Copyright 2018, American Chemical Society. | ||
Rhenium bipyridine complexes (Re(bpy)(CO)3Cl and its derivatives)104–108 and manganese complexes (Mn(bpy)(CO)3Br and its derivatives)109–114 are known to act as catalysts for electrocatalytic reduction of CO2 to CO. The catalytic mechanisms have been investigated extensively using various spectroscopic methods including UV/Vis absorption and pulsed-EPR techniques (2P-ESEEM and HYSCORE) combined with DFT calculations.113 A key intermediate in the catalytic cycle of CO2 reduction is demonstrated to be a low spin MnII–hydroxycarbonyl complex after the oxidative addition of CO2 and H+ to a Mn0 carbonyl dimer (Scheme 6).113
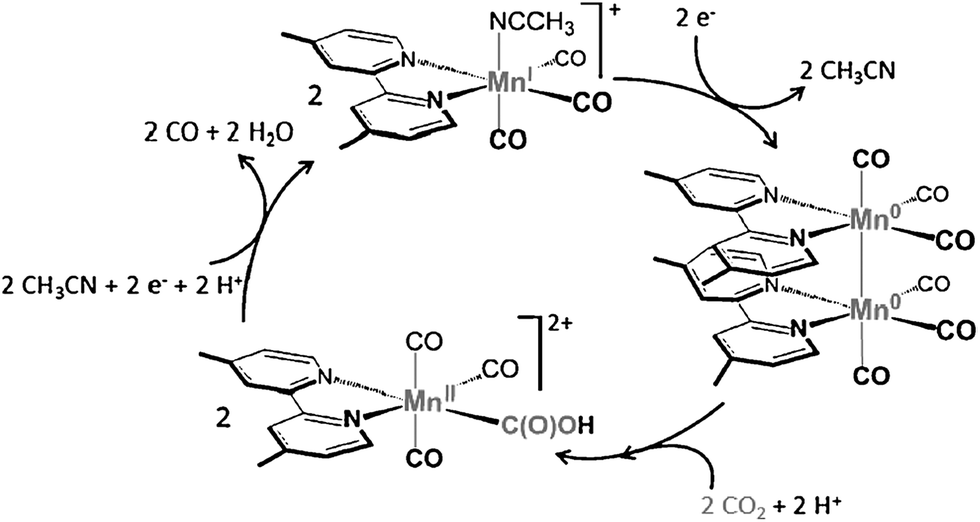 | ||
| Scheme 6 The catalytic cycle for two-electron reduction of CO2 to CO with [Mn(bpy)(CO)3]+. Reprinted with permission from ref. 113. Copyright 2014, WILEY-VCH Verlag GmbH. | ||
The catalytic activity of a manganese complex fac-[MnBr(4,4′-bis(phosphonic acid)-2,2′-bipyridine)(CO)3] (MnP) for CO production was improved when immobilized on a mesoporous TiO2 electrode (TiO2/MnP).114 Controlled potential electrolysis of TiO2/MnP at Eappl = −1.7 V vs. Fc+/Fc (η = 0.42 V) under CO2 in MeCN/H2O (19/1) for 2 h passed an average charge of 1.10 ± 0.25 Coulombs with a faradaic efficiency of 67 ± 5% and a TON of 112 ± 17 for CO. The faradaic yield of H2 for this system was 12.4 ± 1.4% and formate was not detected by ion chromatography.114 The high activity and low overpotential of TiO2/MnP are suggested to result from temporary desorption of the catalyst, followed by dimerization and re-anchoring within mesoporous TiO2, because phosphonic acid modified molecules, such as MnP, display some lability when bound to TiO2.115 The high local concentration of MnP may also place the metal centres in an environment where they are predisposed to dimerization upon reduction.114
The lowest overpotential for the electrocatalytic reduction of CO2 to CO was achieved by using a manganese complex, [Mn{4,4′-di(1H-pyrrolyl-3-propyl carbonate)-2,2′-bipyridine}(CO)3-MeCN]+(PF6)− loaded onto conductive MWCNTs ([Mn-MeCN]/MWCNT), which exhibited catalysis at −0.21 V vs. SHE (overpotential of about 100 mV for CO production).116 The MWCNTs together with surface adsorbed K+ ions provided an environment to stabilize CO2 adjacent to the Mn complex and significantly lowered the overpotential for CO2 reduction in an aqueous solution.116
A case of selectivity tuning was observed as a function of ligand type when Ni(II) complexes in Fig. 7 were employed as catalysts for CO2 reduction.117 Controlled potential electrolyses were carried out with each member of the catalyst series (Fig. 7) in CO2-saturated MeCN solutions containing 2% H2O.117 The 15-membered macrocyclic complex (3-Ni) gave high selectivity for CO2 reduction to CO with a faradaic efficiency of 87%, the remaining 11% faradaic yield resulted in H2 generation at an applied potential of 2.44 V vs. Fc+/Fc.116 On the other hand, 1-Ni supported by a non-macrocyclic ligand gave a faradaic efficiency of 93% for H2 and only 5% for CO, whereas the less rigid 16-membered macrocyclic complex (2-Ni) exhibited moderate selectivity for both CO (56%) and H2 (43%) evolution in the presence of CO2 and H2O.117 Thus, increased rigidity of the redox-active macrocycle leads to enhanced selectivity for CO2 reduction to CO over the competing hydrogen evolution reaction.117
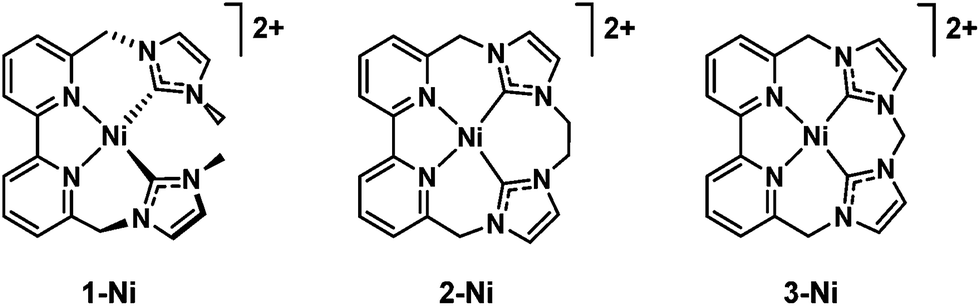 | ||
| Fig. 7 Nickel(II) catalysts supported by non-macrocyclic and macrocyclic bipyridyl-NHC ligands. Reprinted with permission from ref. 117. Copyright 2018, Royal Society of Chemistry. | ||
The first electron reduction of 1-Ni is calculated to be that of the metal center and the second electron reduction is calculated to be ligand-based to produce the [NiI(L˙)] species. The [NiI(L˙)] complex reacts with H+ to form a metal hydride, from which H2 is produced through a reaction with another H+.117 In contrast, the first and second reductions of 3-Ni are calculated to be both ligand-centered, and the resulting [NiII(L2˙)] complex is not capable of metal hydride generation, and therefore leads to no H2 production. The two-electron reduced species of 2-Ni is best described by resonance forms [NiII(L2˙)] ↔ [NiI(L˙)], which are capable of producing both CO and H2.117
A nickel complex supported by a pincer-type carbene–pyridine–carbene ligand also exhibited high selectivity for the electrocatalytic reduction of CO2 to CO over proton reduction.118 A series of NiII macrocycle complexes structurally similar to [Ni(cyclam)]2+ (cyclam = 1,4,8,11-tetraazacyclotetradecane) also act as selective electrocatalysts for CO production over H2 at a mercury pool working electrode in an aqueous solution.119–121 At pH 5, with an applied potential of −0.96 V vs. NHE (overpotential of −0.55 V), the Ni complexes are efficient, having high faradaic efficiencies for the selective reduction of CO2 to CO.118 Hg provides favorable noncovalent dispersive interactions with the cyclam ligand to destabilise the poisoned CO-bound form of the catalyst, leading to enhanced catalytic reactivity.122 The binding of CO2 to Ni(I) complexes has been extensively studied by pulse radiolysis measurements to demonstrate that the formation of the Ni(I)–CO2 adduct is in equilibrium between the Ni(I) complex and free CO2.123,124
5 Electrocatalytic reduction of CO2 to HCOOH with heme and nonheme metal complexes
The two-electron/two-proton reduction of CO2 also affords formate (HCOO−) as well as CO.125 The reduced metal complex (M−) may react directly with CO2 to produce the M–CO2− adduct that is further reduced with H+ to evolve CO and H2O (Scheme 7) as in the case of the iron porphyrin in Scheme 3.125 Alternatively M− can react with H+ to generate the hydride complex (MH) capable of direct reaction with a proton to produce H2, or alternatively couple with CO2 to produce HCOO− (Scheme 7).125 Whether the catalytic two-electron/two-proton reduction of CO2 affords CO or HCOO− depends on the interplay of metals and ligands of the implemented catalyst.126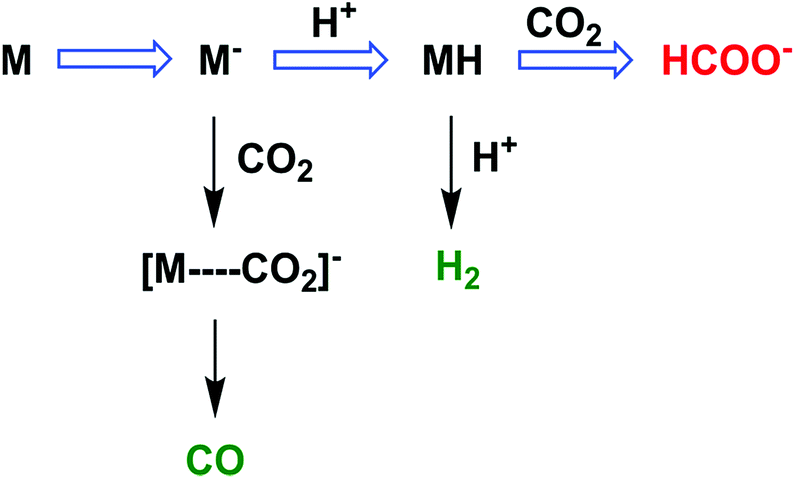 | ||
| Scheme 7 The reaction pathway of two-electron reduction of CO2 with a metal complex (M) to produce HCOO− or CO. Reprinted with permission from ref. 125. Copyright 2017, American Chemical Society. | ||
A series of metalloprotoporphyrins (MPPs in Fig. 8) were employed as catalysts for the electrochemical reduction of CO2 in an aqueous solution. Formic acid was not obtained when CrPP, MnPP, CoPP and FePP were used as catalysts. In the case of CoPP, CO was produced selectively at pH 3 with high faradaic efficiency.126 Exercising the reaction with InPP, CrPP, SnPP and GaPP as catalysts, no gaseous products other than H2 were observed. When SnPP, InPP and RhPP were employed as catalysts, however, significant amounts of formic acid were produced depending on the pH as shown in Fig. 9, where the faradaic efficiency of HCOO− with InPP at pH 9.6 was optimal, reaching a value close to 70%.126 At very low pHs, hydrogen evolution dominated, resulting in little or no HCOOH production.126 At very alkaline pHs, the H2 production also seemed to be dominant, leading to poor selectivity towards HCOO−.126 The catalytic mechanisms for HCOO− production with InPP, SnPP and RhPP have not yet been clarified.
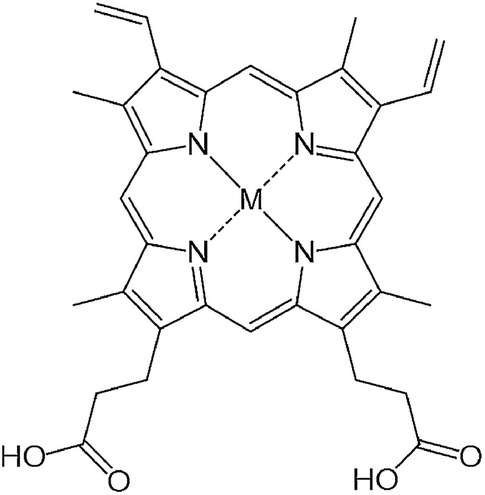 | ||
| Fig. 8 Chemical structure of metalloprotoporphyrins (MPP). Reprinted with permission from ref. 126. Copyright 2017, Elsevier. | ||
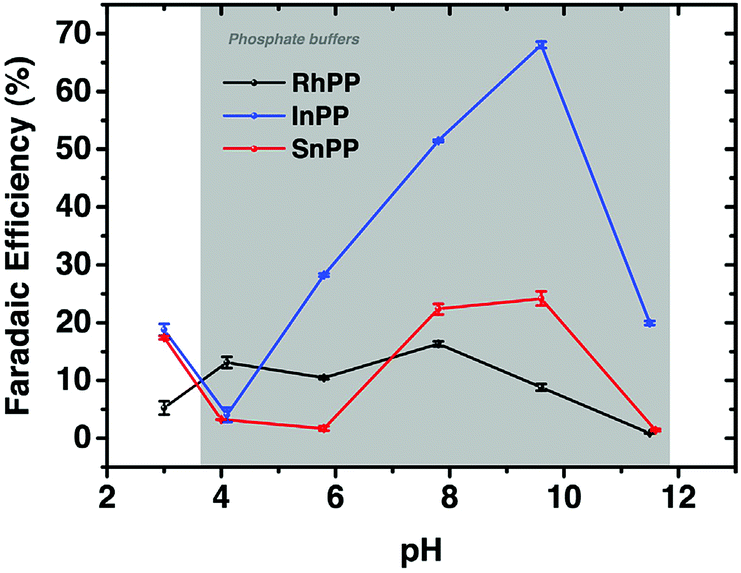 | ||
| Fig. 9 Faradaic efficiencies for the electrocatalytic reduction of CO2 to HCOOH or HCOO− with RhPP, InPP and SnPP, determined at t = 10 min, at E = −1.5 V vs. RHE as function of pH. Reprinted with permission from ref. 126. Copyright 2017, Elsevier. | ||
In contrast to iron porphyrins that catalyse the two-electron reduction of CO2 to CO (Scheme 1), a nonheme iron(III) chloride complex bearing a 6,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-2,2′-bipyridine (tbudhbpy) ligand, Fe(tbudhbpy)Cl, catalyses the electrochemical two-electron reduction of CO2 to formate in the presence of phenol (0.50 M) as a proton source in DMF with a faradaic efficiency of 68 ± 4% together with H2 as a minor product (30 ± 10% faradaic efficiency) and minimal CO (1.1 ± 0.3% faradaic efficiency) at −2.5 V vs. Fc/Fc+.127 The first one-electron reduction of Fe(tbudhbpy)Cl (FeIII/II; E0 = −0.89 V vs. Fc/Fc+) exhibited a Nernstian PhOH-dependent electrochemical response, which suggests that the reduction is coupled with protonation of a bound phenolate moiety of the ligand framework by a PhOH proton donor as shown in Scheme 8.127 At the second reduction potential (E0 = −2.09 V vs. Fc/Fc+), a second protonation event occurs at the metal centre to produce the iron(III)–hydride complex. At the third reduction potential (E0 = −2.65 V vs. Fc/Fc+), the catalytic activity was observed in the presence of PhOH as a sacrificial proton donor when the Fe(II)–hydride complex reacts with CO2 to produce the formate complex from which formate is released to regenerate the Fe(II) complex (Scheme 8).127 A kinetic isotope effect (KIE) of 4.8 ± 0.9 was observed when PhOD is used instead of PhOH, indicating that the insertion of CO2 into the Fe(II)–hydride complex is the rate-determining step.127 Fe(II)–hydride complexes were reported to hydrogenate CO2 to produce formate.128–130 An iron cluster hydride [HFe4N(CO)12]− is also reported as an effective catalyst for the electrochemical reduction of CO2 to formate under mild conditions of pH 7 buffered aqueous solution and an applied potential of −1.2 V vs. SCE.131,132
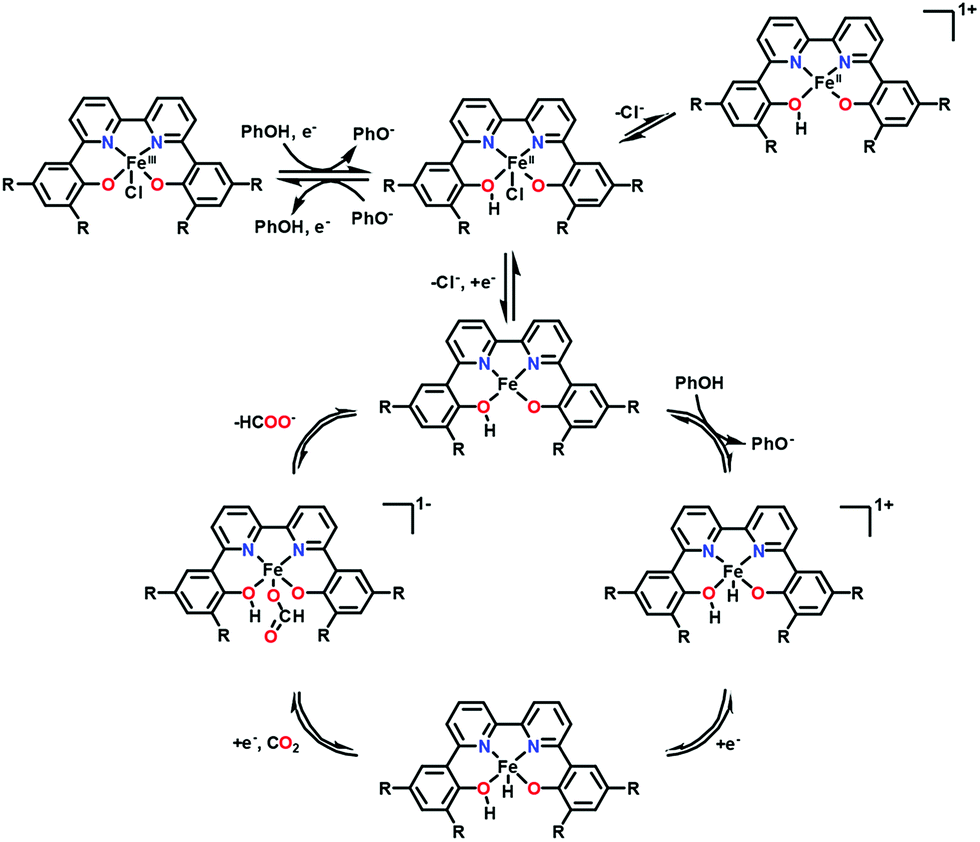 | ||
| Scheme 8 Catalytic cycle for two-electron reduction of CO2 to CO with a nonheme iron(III) chloride complex bearing a 6,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-2,2′-bipyridine ligand (Fe(tbudhbpy)Cl) via protonation by phenol. Reprinted with permission from ref. 127. Copyright 2018, American Chemical Society. | ||
Cobalt complexes ([CpCo(PR2NR′2)I]+) containing diphosphine ligands (PR2NR′2) with two pendant amine residues catalyse the production of formic acid in DMF/water mixtures with a faradaic efficiency of 90 ± 10% at 500–700 mV overpotentials.133 The [CpCo(PR2NR′2)I]+ complex with R = cyclohexyl and R′ = benzyl exhibited the best catalytic activity with a TON for HCOOH of 23 and 15 in the presence of 1.1 and 0.56 M water, respectively, after electrolyses at −2.25 V vs. Fc/Fc+ for 1 h.133 The electrocatalytic mechanism for CO2 reduction to formate is proposed as shown in Scheme 9, where a CoII–hydride intermediate hydrogenates CO2 to produce formate. After two successive electron transfers, a CoI species (complex III in Scheme 9) is generated. This species is then protonated and further reduced to form a cobalt(II)–hydride (CoII–H) complex (complex V in Scheme 9).133 DFT calculations indicated that the protonation of the pendant amine in either the CoII or CoI states allows two other pathways via intermediates III′ and IV, respectively, through intramolecular proton transfer to the cobalt centre with the reduction of the metal center.133 The CoII–H species V then reacts with CO2, generating complex VI. Internal hydride transfer from cobalt to CO2 then yields complex VII.133 The release of formic acid from complex VII regenerates the CoII species (complex II in Scheme 9) and completes the catalytic cycle.133
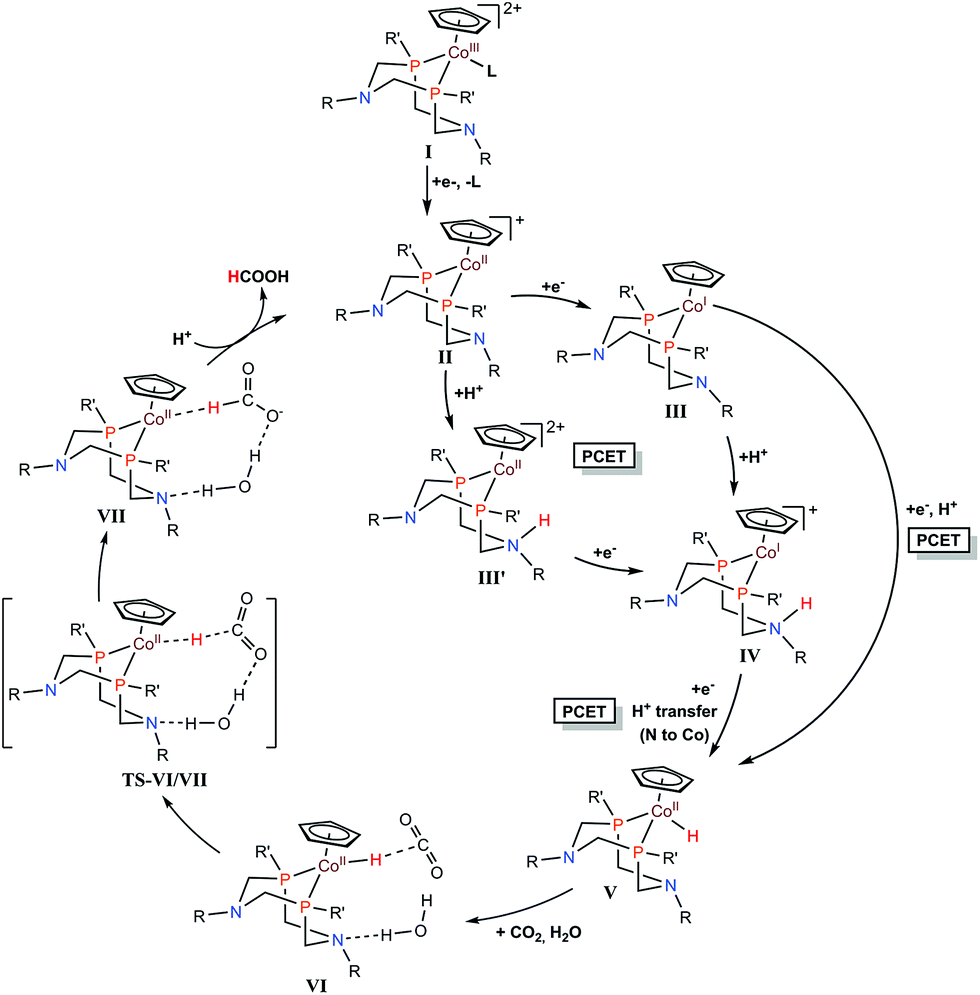 | ||
| Scheme 9 Catalytic cycle for two-electron reduction of CO2 to CO with [CpCo(PR2NR′2)I]+ complexes containing diphosphine ligands (PR2NR′2) with two pendant amine residues. Reprinted with permission from ref. 133. Copyright 2017, American Chemical Society. | ||
The product selectivity of electrocatalytic reduction of CO2 to CO vs. HCOOH with the assembly of a [MnBr(2,2′-bipyridine)(CO)3] complex anchored to a carbon nanotube electrode via a pyrene unit was examined, and reported to be tuned by variation of metal complex density on the nanotube surface, where CO was observed as the main product at high catalyst loadings, and formate was the dominant CO2 reduction product at low catalyst loadings.134 The formation of a dimeric Mn0 species at higher surface loading preferentially leads to CO formation, whereas at lower surface loading the electrochemical generation of a monomeric Mn–hydride is suggested to enhance the production of formate (Scheme 10).134 The Mn–CNT hybrid catalyst exhibited much higher activity for electrocatalytic reduction of aqueous CO2 with TONs of up to 1790 ± 290 for CO (η = 550 mV) and up to 3920 ± 230 for formate (η = 590 mV),134 compared to electrolyses in the absence of carbon nanotubes.135–137
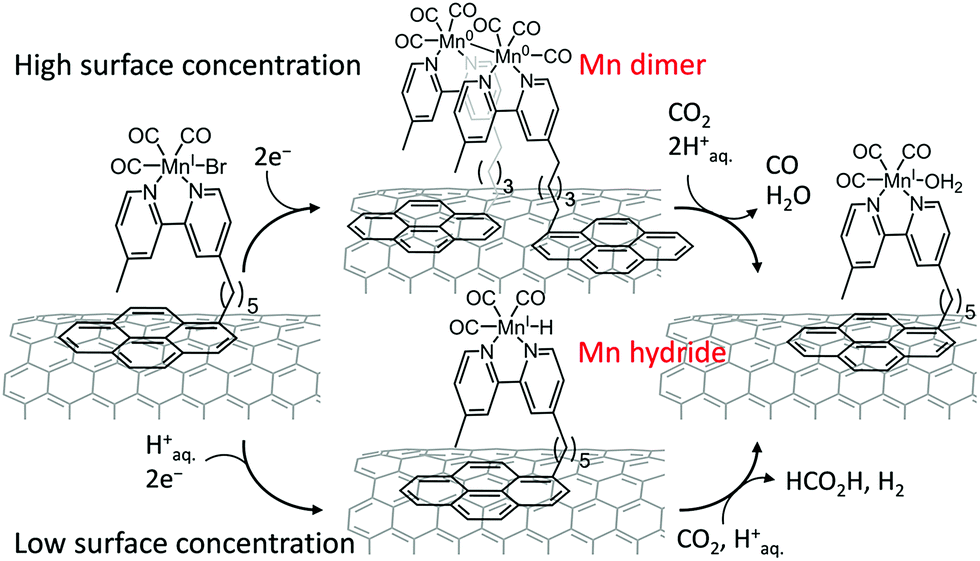 | ||
| Scheme 10 Two-electron reduction of CO2 to CO vs. HCOOH with the [MnBr(2,2′-bipyridine)(CO)3] complex anchored to a carbon nanotube electrode via a pyrene unit depending on the surface concentration. Reprinted with permission from ref. 134. Copyright 2017, American Chemical Society. | ||
When a Ni(bis-dithiolene) complex with a quinoxaline-pyran-fused dithiolene ligand (qpdt2−) (see the X-ray crystal structure in Fig. 10) was employed as a CO2 reduction catalyst using a Hg/Au amalgam electrode, formate as the major product together with minor amounts of CO and H2 was observed at reasonable overpotentials with good faradaic yield and notable stability.138 The onset potential of the catalytic wave with [Ni(qpdt)2]− was observed at approximately −1.7 V vs. Ag/AgCl (=−1.5 V vs. SHE), with a halfwave potential at −1.80 V and a peak at −2 V.138 The overpotential was determined to be ∼340 mV by comparing the experimental onset potential to the standard potential of the CO2/HCOOH couple in MeCN in the presence of trifluoroethanol as a proton source.138 During the electrolysis, a complete reduction of the ligand occurred, followed by the subsequent pyran ring opening to produce the real catalyst ([NiII(L)2]2−; L is a pyran ring-opened ligand produced from qpdt2− by electrolysis) as shown in Scheme 11.138 A Ni–hydride intermediate [NiIII(H)(L)2]2− was proposed to be a catalytically relevant species for CO2 reduction to formate, which was produced by one-electron reduction of [NiII(L)2]2−, followed by the reaction of [NiI(L)2]3− with H+.138 The [NiIII(H)(L)2]2− species reacts with CO2, followed by a release of HCOO− and regeneration of [NiII(L)2]2− upon the one-electron reduction at −0.51 V vs. Ag/AgCl.138
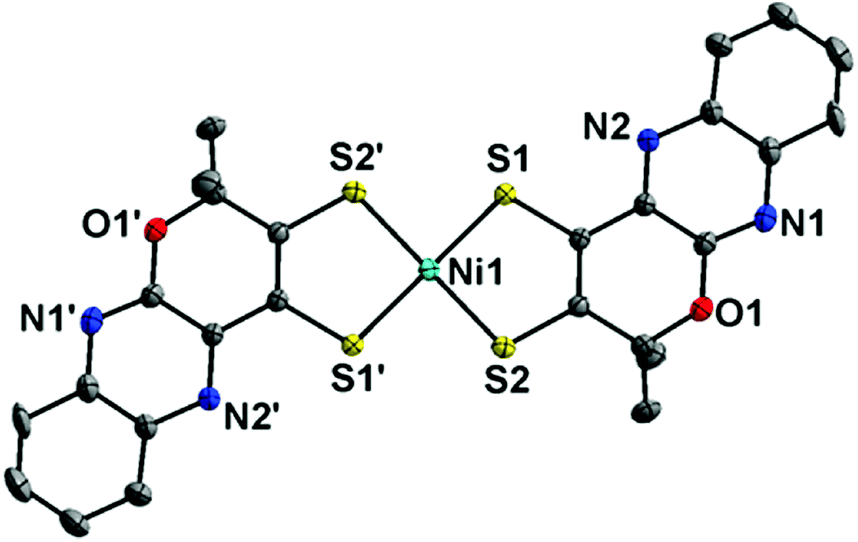 | ||
| Fig. 10 X-ray crystal structure of a NiIII(bis-dithiolene) complex with a quinoxaline-pyran-fused dithiolene dianion ligand (qpdt2−) ([NiIII(qpdt)2]−) at 50% probability. Hydrogen atoms are omitted for clarity. Reprinted with permission from ref. 138. Copyright 2018, American Chemical Society. | ||
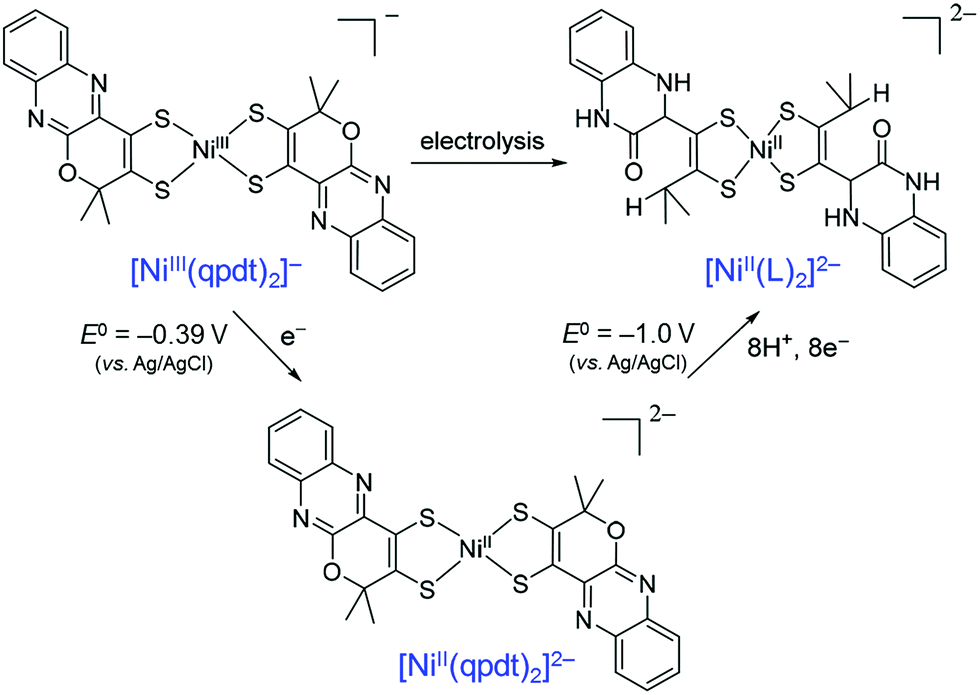 | ||
| Scheme 11 Electrochemical reduction of [Ni(qpdt)2]−, followed by the subsequent pyran ring opening to produce the real catalyst ([NiII(L)2]2−; L is a pyran ring-opened ligand produced from qpdt2− by electrolysis) during electrolysis. Reprinted with permission from ref. 138. Copyright 2018, American Chemical Society. | ||
A small amount of CO observed during the electrolysis was proposed to be produced via the η1-CO2 species based on the DFT calculations.138 Although many η2-CO2 nickel structures have been known to date,139–143 a rare η1-κC binding mode of CO2 to Ni was also reported by utilizing an anionic tridentate PNP ligand (PNP− = N[2-PiPr2-4-Me-C6H3]2−).144,145 The protonation of the η1-CO2 species affords the carboxylate intermediate ([Ni(C(O)OH)(L)2]2−), which undergoes a heterolytic C–O bond cleavage to generate [Ni(CO)(L)2]− upon protonation, followed by the release of CO.138,146
6 Further electrocatalytic reduction of CO to fuels with metalloporphyrins
The electrochemical reduction of CO2 to CO and its further reduction to methane were made possible by using a simple Co protoporphyrin molecular catalyst immobilized onto a pyrolytic graphite (PG) electrode (CoPP-PG).147 In an aqueous solution with a pH value of 1, the faradaic efficiency for methane production is larger than that for CO, but the dominant product is H2.147 At pH = 3, CO became a major product, especially at less cathodic potentials, where the faradaic efficiency for CO was 40%.147 The efficiency towards CO can be further increased by performing the experiment at higher CO2 pressure (10 atm), which leads to a faradaic efficiency of 60% at a potential of −0.6 V (Fig. 11).147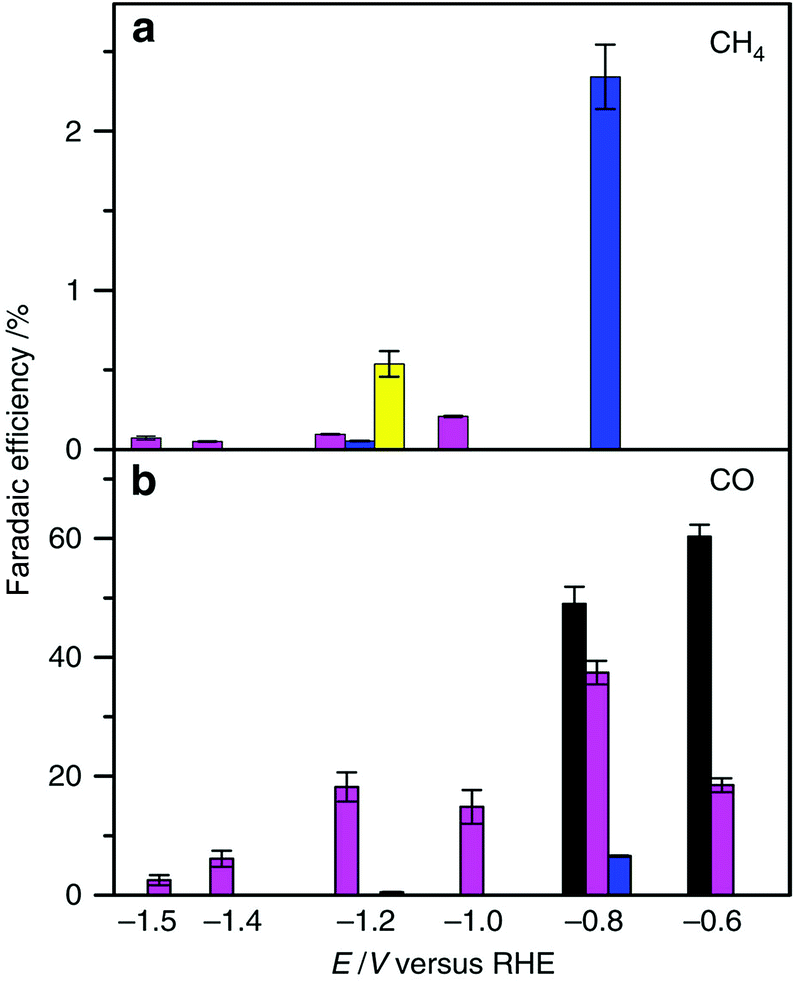 | ||
| Fig. 11 Faradaic efficiency (FE) for the electrocatalytic CO2 reduction to (a) CH4 and (b) CO in 0.1 M perchlorate solution saturated with CO2 conducted at each applied potential for 1 h and 90 min at PCO2 = 1 atm and 10 atm, respectively [yellow bars: pH = 1, PCO2 = 1 atm; blue bars: pH = 1, PCO2 = 10 atm; magenta bars: pH = 3, PCO2 = 1 atm and black bars pH = 3, PCO2 = 10 atm]. Reprinted with permission from ref. 147. Copyright 2015, Nature Publishing Group. | ||
The mechanism of catalytic CO2 reduction to CO by CoPP may be similar to that of iron porphyrins in Scheme 1. The reduction of CoIIPP to [CoIPP]− is coupled with CO2 binding to afford the CO2 adduct that reacts with water to produce the CoIIIPP–C(O)OH adduct, which is further reduced to produce the CoIIPP–CO adduct as shown in Scheme 12.147 The two-electron/two-proton reduction of the CoIIPP–CO adduct affords the CoIIPP–HCHO adduct that is further reduced by four electrons with four protons to finally yield methane and water, accompanied by the regeneration of CoIIPP (Scheme 12).147 The formation of CO is more favoured than H2 at higher pHs.147 However, further reduction of CO is more favoured at a low pH.147 Thus, the faradaic yield of methane was higher than that of CO at pH 1, while H2 remained the major product.147
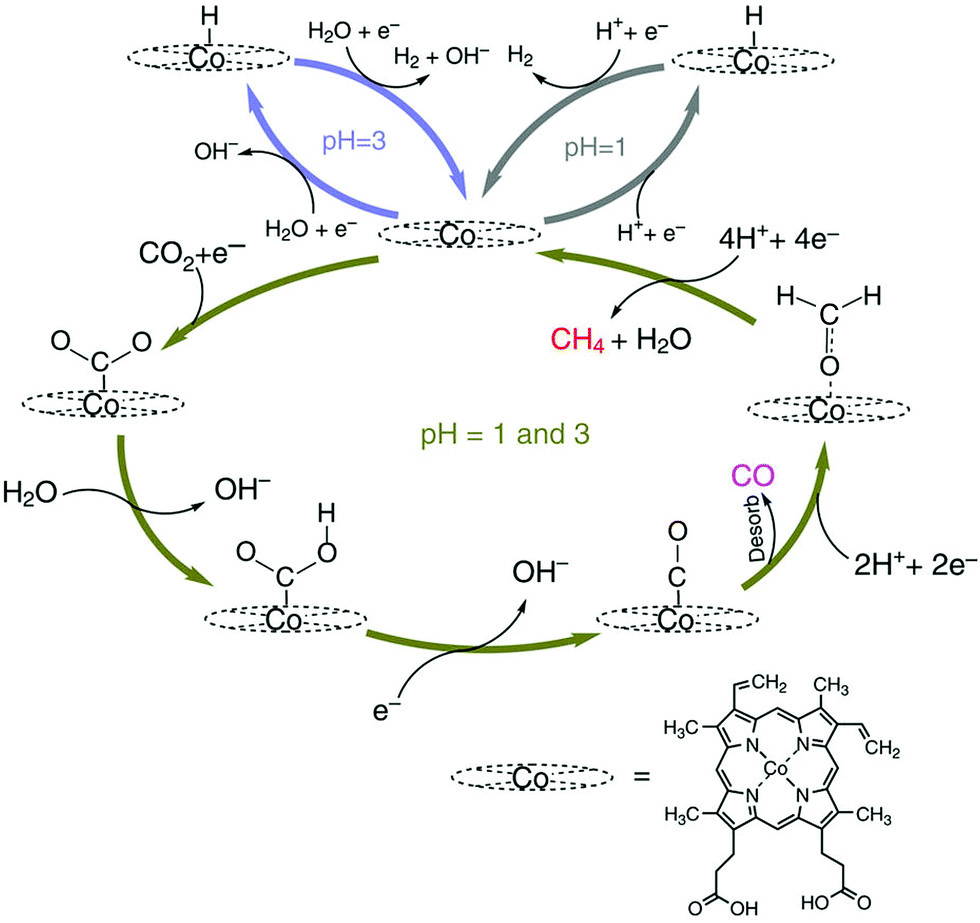 | ||
| Scheme 12 Catalytic cycle for two electron reduction of CO2 to CO and further reduction to CH4 with a Co protoporphyrin molecular catalyst (CoPP) immobilized onto a pyrolytic graphite (PG) electrode (CoPP-PG). Reprinted with permission from ref. 147. Copyright 2015, Nature Publishing Group. | ||
Crystallized copper phthalocyanine supported on carbon black (CuPc/CB) was reported to exhibit high selectivity for C2H4 with a maximum faradaic efficiency of 25% under atmospheric pressure at −1.6 V vs. Ag/AgCl (=−1.4 V vs. SHE), while CH4 and CO were also generated as minor products (Fig. 12).148 In contrast to the crystalline form of CuPc/CB, the noncrystalline CuPc/CB catalyst showed a much lower selectivity and reactivity for C2H4 production.148 When noncrystalline CuPc was treated with Milli-Q water and chloroform to completely restore its crystallinity, the restored crystalline CuPc/CB catalyst afforded faradaic efficiency and partial current density values as high as those of the original crystalline CuPc/CB catalyst.148 These results indicate that catalyst crystallinity is crucial for the selective conversion of CO2 to C2H4, because the C–C bond forming step to produce C2H4 from CO is likely mediated by two metal centers.148
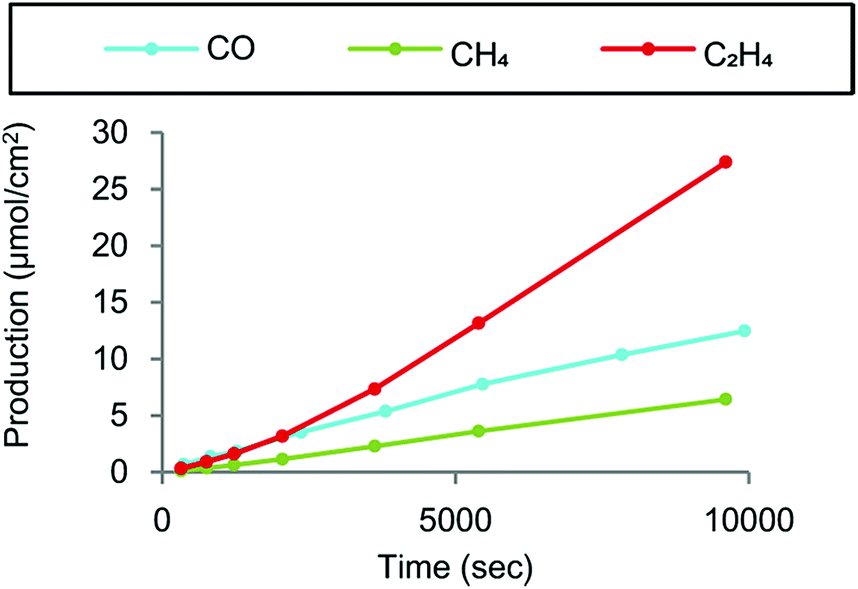 | ||
| Fig. 12 Products derived from CO2 electrochemical reduction by crystalline CuPc/C at −1.6 V vs. Ag/AgCl in a two-compartment H-cell. Reprinted with permission from ref. 148. Copyright 2017, American Chemical Society. | ||
Heterobimetallic cavities formed by the face-to-face coordination of thiol-terminated iron porphyrins on copper electrodes via self-assembly of supramolecular cages (M = Fe in Fig. 13) have made it possible to convert CO to C2 products via C–C bond formation in aqueous electrochemical CO reduction with high faradaic efficiency (83% total with 57% to ethanol) and current density (1.34 mA cm−2) at a potential of −0.40 V vs. RHE.149 The cage-functionalized electrodes afforded an order of magnitude improvement in both selectivity and activity for electrocatalytic CO reduction compared to their parent copper surfaces.149 Control analogues that lack thiol binding groups as well as positional isomers favouring edge-on binding or direct van der Waals stacking exhibited reduced surface access and negligible CO over water reduction selectivity, suggesting the critical role of the three-dimensional pockets in catalysis, where the Fe centre can aid in the cooperative reduction of potential acetaldehyde intermediates.149
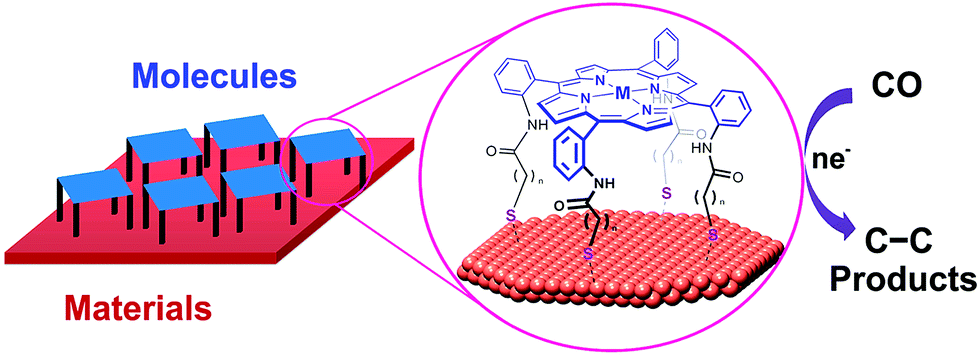 | ||
| Fig. 13 Schematic illustration of supramolecular assembly of cages between molecular components and supramolecular assembly of cages between molecular components and a copper electrode. Reprinted with permission from ref. 149. Copyright 2017, American Chemical Society. | ||
7 Photocatalytic reduction of CO2 with heme and nonheme metal complexes
The electrocatalytic reduction of CO2 can be replaced by photocatalytic reduction using photocatalysts instead of electrocatalysts. For example, the selective electrocatalytic reduction of CO2 to CO with a glassy carbon electrode modified with a cobalt(II) chlorin complex (CoII(Ch)) adsorbed on MWCNTs (CoII(Ch)/MWCNTs; see Fig. 3 for the schematic image) is replaced by the photocatalytic reduction of CO2 with trimethylamine (TEA) using CoII(Ch)/MWCNTs as a CO2 reduction catalyst and [RuII(Me2phen)3]2+ (Me2phen = 4,7-dimethyl-1,10-phenanthroline) as a sensitizer.150 The photocatalytic mechanism of the CO2 reduction is shown in Scheme 13, where electron transfer from TEA to the excited state of [RuII(Me2phen)3]2+ ([RuII(Me2phen)3]2+*: * denotes the excited state) occurs to produce TEA radical cations and [RuI(Me2phen)3]+ that reduces CoII(Ch) to [CoI(Ch)]−.150 [CoI(Ch)]− reacts with CO2 to produce the [(Ch)CoIII(CO2)]− complex that is protonated to produce CoII(Ch)COOH, from which water is released by the protonation to produce the [CoIII(Ch)CO]+ complex.150 After the CO release from the Co(III)CO complex, the Co(III) complex is reduced by TEA to regenerate CoII(Ch). Hydrogen evolution simultaneously occurs as a side reaction, much like in the electrocatalytic case, because [CoI(Ch)]− can also react with H+ to produce the hydride complex ([CoIII(Ch)(H)]), which then can couple to a H+ to produce H2.151 The TON of the photocatalytic reduction was determined to be 710 with the ratio of CO to H2 of 2.4:1 in 20 h when 5.0 μM of CoII(Ch) and 1.0 mg of MWCNTs were used.150 The π–π interaction between MWCNTs and CoII(Ch) may provide a suitable hydrophobic environment for the selective binding of CO2 over protons, because the binding of CO2 to the Co(I) complex is required for the formation of CO (Scheme 13).150
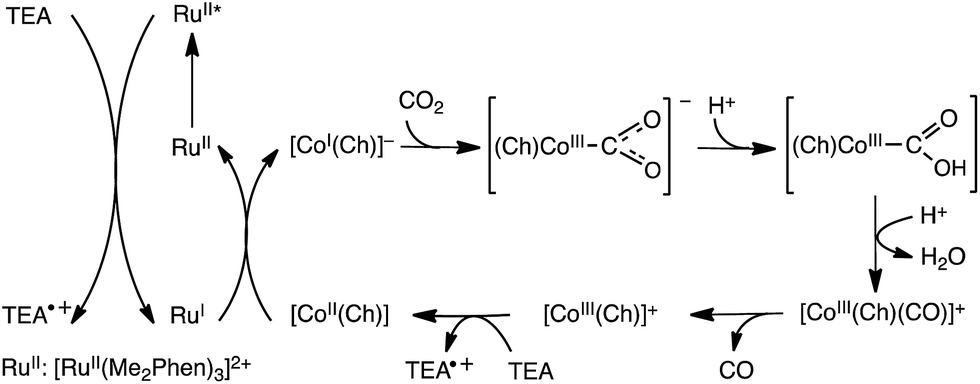 | ||
| Scheme 13 Photocatalytic cycle for two-electron reduction of CO2 to CO with a cobalt(II) chlorin complex (CoII(Ch)) adsorbed on multi-walled carbon nanotubes (CoII(Ch)/MWCNTs). Reprinted with permission from ref. 150. Copyright 2016, Royal Society of Chemistry. | ||
An iron tetraphenylporphyrin complex functionalized with trimethylanilinium groups (Fe-p-TMA in Fig. 2), which is an efficient and selective molecular electrocatalyst for converting CO2 to CO,88 can also act as an efficient catalyst in photocatalytic reduction of CO2 to methane using Ir(ppy) (ppy = phenylpyridine) as a dye under visible light irradiation at ambient temperature and pressure.152,153 The catalytic system, which was evaluated in an acetonitrile solution containing a photosensitizer (Ir(ppy)), sacrificial electron donor (TEA) and proton source (trifluoroethanol), operates stably under irradiation over several days to produce CO, CH4, and H2 with turnover numbers (and selectivities) of 367 (78%), 79 (17%) and 26 (5%), respectively in 107 h.152 These values correspond to a methane production rate of 763 μmol per hour per gram of catalyst (μmol h−1 g−1), which is larger than those obtained using other catalysts154–159 that generate methane from CO2. Trifluoroethanol facilitates the C–O bond cleavage step as observed in the electrocatalytic reduction of CO2 (Scheme 3).84 No other gaseous product, methanol, formaldehyde or formate was produced.152 Isotopic labelling experiments conducted under a 12CO2 or a 13CO2 atmosphere confirmed that methane was produced from CO2. A two-step procedure, that first reduces CO2 to CO and then reduces CO to further reduced species of CO, generates methane with a selectivity of up to 82% and a quantum yield (light-to-product efficiency) of 0.18%.152
The photocatalytic mechanism for CO2 reduction to methane is proposed as shown in Scheme 14, where the starting FeIII porphyrin is reduced with three electrons by Ir(ppy)* to the catalytically active Fe0 species.152 The Fe0 species reduces CO2 to CO through a two-step protonation of the Fe0 species by CF3CH2OH with dehydration (right-hand iron cycle) as in the case of the electrocatalytic reduction of CO2 to CO in Scheme 3, where AH = trifluoroethanol.84 The CO produced binds to FeII and is further reduced by a total of six electrons by electron transfer from Ir(ppy)* and six protons to generate CH4 (and H2O), through a postulated FeI-formyl (FeICHO) intermediate (left-hand iron cycle).152
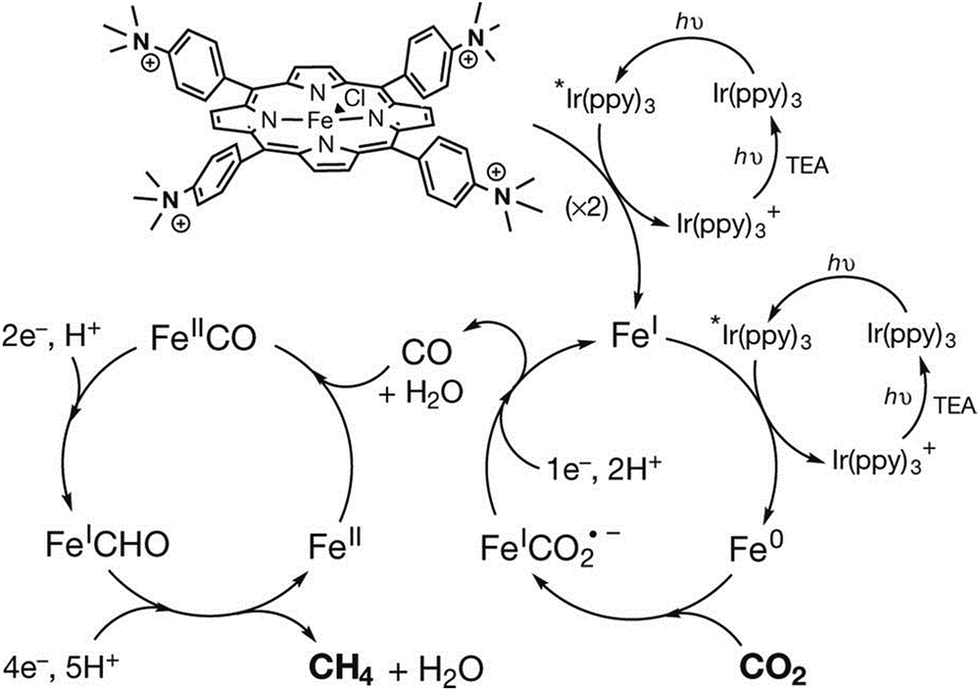 | ||
| Scheme 14 Photocatalytic cycles for two-electron reduction of CO2 to CO and further reduction to CH4 with Fe-p-TMA and Ir(ppy)3. Reprinted with permission from ref. 152. Copyright 2017, Macmillan Publishers Limited, part of Springer Nature. | ||
Not all photocatalytic systems employ separately a catalyst and a photosensitizer. Fe-p-TMA can also catalyse the photocatalytic reduction of CO2 to CO with 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]-imidazole (BIH) selectively under visible light irradiation without the assistance of an additional photosensitizer.160 BIH was reported to enhance the photocatalytic formation of CO from CO2,161,162 due to its high reductive ability (Eox = +0.33 V vs. SCE = +0.57 vs. SHE),163 its fast deprotonation to BI˙ once oxidized and its overall two-electron donation capacity (BI˙ could be in turn easily oxidized to BI+).
BIH was also used as a sacrificial reductant together with triethanolamine (TEOA) for visible-light driven CO2 reduction in MeCN using [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) as a photosensitizer and a CuII complex bearing 2,2′:6′,2′′:6′′,2′′′-quaterpyridine (qpy) ([Cu(qpy)]2+) as a selective reduction catalyst.164 The photocatalytic reaction is greatly enhanced by the presence of H2O (1–4% v/v), and a TON of >12400 for CO production has been achieved with 97% selectivity, which is among the highest of molecular 3d-metal CO2 reduction catalysts.164 It was confirmed that the photocatalyst remains homogeneous based on results from Hg poisoning and dynamic light scattering experiments.164
BIH alone acts as an electron donor in the photocatalytic reduction of CO2 to CO with a Ni(II) complex bearing an S2N2-type tetradentate ligand, [NiII(bpet)]2+ (bpet = bis(2-pyridylmethyl)-1,2-ethanedithiol), as a CO2 reduction catalyst and [Ru(bpy)3]2+ as a photosensitizer in a DMA/H2O solution mixture (9:1 v/v) under a CO2 atmosphere at 298 K.165 In this photocatalytic system, the [NiII(bpet)]2+ complex showed a high TON exceeding 700 with a very high CO selectivity of >99% and a quantum yield of 1.42%.165
Composite electrodes formed by a combination of an organic-semiconductor with a metal-porphyrinoid catalyst were also evaluated as photocatalysts for CO2 reduction. Graphite carbon nitride (g-C3N4), which has frequently been used as an organic semiconductor photocatalyst,166–169 was modified with a carboxyl group of tetra(4-carboxyphenyl)porphyrin iron(III) chloride (FeTCPP) to prepare a g-C3N4 nanosheets/FeTCPP (g-C3N4/FeTCPP) heterogeneous catalyst. The material was then implemented for the photoreduction of CO2 to CO with TEOA under visible light illumination in MeCN:H2O:TEOA (3:1:1) (Scheme 15).170 Similar to the case shown in Scheme 13, FeIIITCPP was reduced by the photo-induced electrons produced in g-C3N4 nanosheets up to Fe0TCPP that reduced CO2 to CO coupled with proton transfer, whereas the holes left in g-C3N4 nanosheets were reduced by TEOA.170 A maximum rate of 6.52 mmol g−1 in 6 h and a selectivity up to 98% for CO production have been achieved using the g-C3N4/FeTCPP heterogeneous catalyst.170
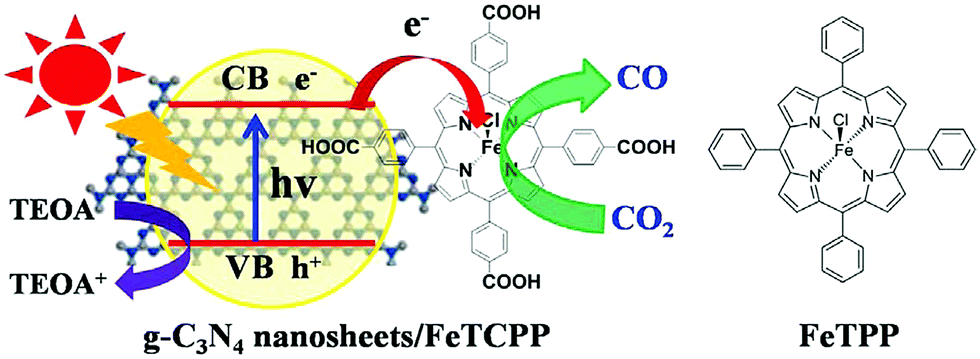 | ||
| Scheme 15 Photocatalytic reduction of CO2 to CO with TEOA using g-C3N4 nanosheets/FeTCPP (g-C3N4/FeTCPP). Reprinted with permission from ref. 170. Copyright 2018, Elsevier. | ||
To further improve the catalytic performance of composite electrodes, rutile TiO2 nanoparticles were employed as modifiers to enhance interfacial charge transfer between semiconducting carbon nitride nanosheets (NS–C3N4) and a catalyst (supramolecular Ru(II)–Re(I) binuclear complex (RuRe)).171 The RuRe/TiO2/NS-C3N4 hybrid photocatalyzed CO2 reduction to CO with high selectivity under visible light (λ > 400 nm) irradiation, exhibiting higher catalytic activity compared to an analogue without TiO2 by a factor of 4, in terms of both the CO formation rate and the TON.171 The enhanced photocatalytic activity was attributed mainly to the prolonged lifetime of free and/or shallowly trapped electrons generated in TiO2/NSC3N4 under visible-light irradiation, as revealed by transient absorption spectroscopy.171
A case of metal porphyrinoid catalysis was reported in conjunction with an inorganic semiconductor photoanode, completing a full photoelectrochemical cell. The photoelectrochemical reduction of CO2 was performed in a two-compartment cell composed of an FeO(OH)/BiVO4/FTO photoanode and a CoII(Ch)/MWCNT cathode, which are connected with conducting wire as an external circuit and separated by a Nafion membrane (Fig. 14).172 A photo-driven oxidation reaction (of water) occurs at the photoanode, and the generated electrons are transported through the external circuit, being supplied as reducing equivalents to the CoII(Ch)/MWCNT catalyst at the cathode. The photocatalytic controlled potential electrolysis of a CO2-saturated aqueous solution (pH 4.6) using a photoelectrochemical cell in Fig. 14 at an applied bias voltage of −1.3 V at the cathode versus the photoanode resulted in the formation of CO and H2 as shown in Fig. 15a, where the CO yield is significantly higher than the H2 yield.172 The maximum current efficiency for CO production for the initial 2 h was 83% at pH 4.6.172 The amount of O2 produced in the photoelectrochemical oxidation of water is one-half of the amounts of the sum of CO and H2 produced in the electrochemical reduction of CO2 and H2O on the cathode (Fig. 15b).172
 | ||
| Fig. 14 Schematic illustration of a photoelectrochemical cell composed of an FeO(OH)/BiVO4/FTO photoanode for the photocatalytic oxidation of water to O2 and a CoII(Ch)/MWCNT cathode for the catalytic reduction of CO2 to CO. Cathode and anode compartments are separated by a Nafion membrane. Reprinted with permission from ref. 172. Copyright 2017, American Chemical Society. | ||
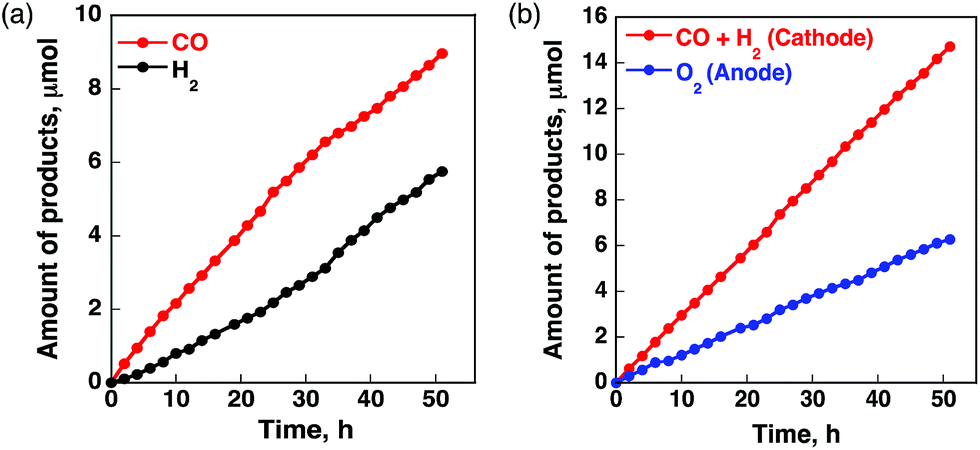 | ||
| Fig. 15 Time profiles of (a) the formation of CO (red circles) and H2 (black circles) and (b) the formation of O2 (blue circles) and CO plus H2 (red circles) in the photo-assisted CPE with the CoII(Ch)-modified cathode at an applied bias voltage of −1.3 V at the cathode versus the FeO(OH)/BiVO4/FTO photoanode in a CO2-saturated aqueous solution containing Na2SO4 (5.0 mM) at pH 4.6 under simulated 1 sun (AM 1.5G) illumination at 298 K. Reprinted with permission from ref. 172. Copyright 2017, American Chemical Society. | ||
A direct covalent linkage between a photosensitizer and a catalyst is a popular strategy due to the ease of electron transfer induced by proximity, and also one that is well suited for metal porphyrinoid catalysts owing to their synthetic versatility and flexibility. Such a case is shown in Scheme 16, where a Ru-based dye and a Re-based catalyst pair is synthesised and immobilised on a semiconductor surface. The photoelectrochemical reduction of CO2 to CO was performed by using a RuRe/CuGaO2 electrode under irradiation (λex > 460 nm), which can be selectively absorbed by the Ru photosensitizer unit of RuRe, in an aqueous solution containing NaHCO3 (50 mM) saturated with CO2 as shown in Scheme 16.173 The difference in the observed current between the irradiation and dark conditions indicated that the cathodic photoresponse of the RuRe/CuGaO2 electrode started at +0.30 V vs. Ag/AgCl (=+0.50 V vs. SHE), which was approximately 0.4 V more positive (due to the photovoltage supplied by the underlying semiconductor) than that of RuRe/NiO (ca. −0.1 V vs. Ag/AgCl = +0.1 vs. SHE).173 The TON for the formation of CO was 125 based on the RuRe loading, and the total faradaic efficiency for the production of CO plus H2 was 81%.173 The wavelength dependence of the incident-photon-to-current efficiencies of the RuRe/CuGaO2 electrode agreed well with the absorption spectrum of the electrode, whereas the CuGaO2 electrode alone (without the dye and catalyst) exhibited almost no photoresponse under irradiation (λex > 460 nm).173 The photocurrent was generated by the injection of the electrons from the CuGaO2 electrode into the excited Ru photosensitizer unit of RuRe, as shown in Scheme 16, because the flat band potential of the CuGaO2 electrode in the reaction solution (+0.47 V vs. Ag/AgCl = +0.67 V vs. SHE) is less positive than the one-electron reduction potential of the excited state of RuRe (Ered = +0.49 V vs. Ag/AgCl = +0.69 V vs. SHE).173
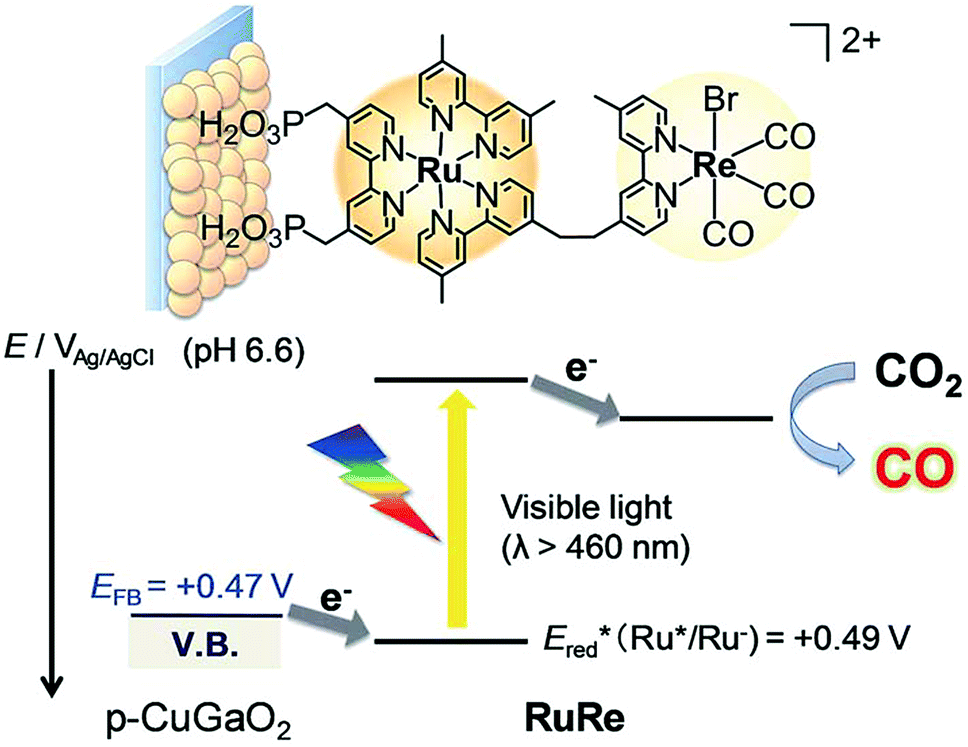 | ||
| Scheme 16 Schematic illustration for the photocatalytic reduction of CO2 to CO with the RuRe/CuGaO2 electrode. Reprinted with permission from ref. 173. Copyright 2017, Royal Society of Chemistry. | ||
8 Conclusions
The one-electron reduction of CO2 to oxalic acid is catalysed not only by metalloporphyrins such as AgII(TPP) an PdII(TPP) but also by nonheme dicopper and tricopper complexes. Iron porphyrins act as efficient catalysts for both the electrocatalytic and photocatalytic reduction of CO2 to CO in competition with the proton reduction to H2. When four positively charged trimethylanilinium groups were introduced at the ortho positions of the TPP phenyls of an iron tetraphenylporphyrin (Fe-o-TMA in Fig. 2), the lowest overpotential of 0.220 V for CO2 reduction to CO was achieved with a maximum TOF of 106 s−1. A variety of metal complexes such as cobalt and nickel complexes also act as effective catalysts for two-electron reduction of CO2 to CO. When indium protoporphyrin was employed as an electrocatalyst, formate (HCOO−) was the main product instead of CO and the faradaic efficiency of HCOO− at pH 9.6 was close to 70%. Whether CO or HCOOH is produced depends on the metals and ligands of metalloporphyrins.Further reduction of CO to methane has been made possible by using a Co protoporphyrin molecular catalyst immobilized onto a pyrolytic graphite (PG) electrode (CoPP-PG) in a purely aqueous electrolyte solution. The reduction of CO to C2H4 was also made possible by using crystallized copper phthalocyanine supported on carbon black (CuPc/CB) with a maximum faradaic efficiency of 25% under atmospheric pressure at −1.4 V vs. SHE, while CH4 and CO were produced as minor products. The photocatalytic reduction of CO2 to CO occurs in competition with the proton reduction to H2 using triethylamine as a reductant, CoII(Ch)/MWCNTs as a CO2 reduction catalyst, and [RuII(Me2phen)3]2+ as a photosensitizer. When Fe-p-TMA with four positively charged trimethylanilinium groups introduced at the para positions of the TPP phenyls was employed as a catalyst, the photocatalytic reduction of CO using triethylamine as a reductant and Ir(ppy) afforded methane with a selectivity of up to 82% and a quantum yield of 0.18% under visible light irradiation at ambient temperature and pressure. Water can be used as an electron and proton source for the photocatalytic reduction of CO2 to CO using a two-compartment cell composed of an FeO(OH)/BiVO4/FTO photoanode and a CoII(Ch)/MWCNT cathode. There are many reports on the photocatalytic reduction of CO2 with water as an electron and proton source to produce CO, HCOOH and CH4 using heterogenous catalysts.174–184 It is highly desired to develop photocatalytic systems of CO2 reduction to fuels such as ethylene, methanol and methane using water as an electron and proton source using homogeneous molecular catalysts as well.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the contributions of their collaborators and coworkers cited in the listed references, and support through a SENTAN project (to S. F.) from the Japan Science and Technology Agency (JST), JSPS KAKENHI (No. 16H02268 to S. F.), the NRF of Korea through CRI (NRF-2012R1A3A2048842 to W. N.), GRL (NRF-2010-00353 to W. N.), and the Basic Science Research Program (2017R1D1A1B03029982 to Y. M. L., 2017R1D1A1B03032615 to S. F., and 2017R1C1B2011074 to H. S. A.).Notes and references
- N. S. Lewis, Energy Environ. Sci., 2016, 9, 2172–2176 RSC.
- M. Robert, ACS Energy Lett., 2016, 1, 281–282 CrossRef.
- Y. Zeng, J. Chen, T. Yu, G. Yang and Y. Li, ACS Energy Lett., 2017, 2, 357–363 CrossRef.
- J. Su and L. Vayssieres, ACS Energy Lett., 2016, 1, 121–135 CrossRef.
- J. M. Thomas and K. D. M. Harris, Energy Environ. Sci., 2016, 9, 687–708 RSC.
- N. S. Lewis, Nat. Nanotechnol., 2016, 11, 1010–1019 CrossRef PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 RSC.
- S. Fukuzumi, Joule, 2017, 1, 689–738 CrossRef.
- S. Perathoner and G. Centi, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.03.005.
- K. K. Sakimoto, N. Kornienko and P. Yang, Acc. Chem. Res., 2017, 50, 476–481 CrossRef PubMed.
- P. V. Kamat, Acc. Chem. Res., 2017, 50, 527–531 CrossRef PubMed.
- B. Wang, W. Chen, Y. Song, G. Li, W. Wei, J. Fanga and Y. Sun, Catal. Today, 2018, 311, 23–39 CrossRef.
- K. Maeda, Prog. Solid State Chem., 2018 DOI:10.1016/j.progsolidstchem.2017.11.003.
- G. Zhao, X. Huang, X. Wang and X. Wang, J. Mater. Chem., 2017, 5, 21625–21649 RSC.
- Y. Zheng, W. Zhang, Y. Li, J. Chen, B. Yu, J. Wang, L. Zhang and J. Zhang, Nano Energy, 2017, 40, 512–539 CrossRef.
- Y. Sohna, W. Huang and F. Taghipour, Appl. Surf. Sci., 2017, 396, 1696–1711 CrossRef.
- S. R. Lingampalli, M. M. Ayyub and C. N. R. Rao, ACS Omega, 2017, 2, 2740–2748 CrossRef.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef PubMed.
- W. Kim, E. Edri and H. Frei, Acc. Chem. Res., 2016, 49, 1634–1645 CrossRef PubMed.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef.
- J. Bonin, A. Maurin and M. Robert, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef.
- Y. Yang, S. Ajmal, X. Zheng and L. Zhang, Sustainable Energy Fuels, 2018, 2, 510–537 RSC.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, Green Chem., 2017, 19, 2406–2415 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef PubMed.
- R. Daiyan, X. Lu, T. H. Ng and R. Amal, ChemSusChem, 2017, 10, 4342–4358 CrossRef PubMed.
- B. Khezri, A. C. Fisher and M. Pumera, J. Mater. Chem. A, 2017, 5, 8230–8246 RSC.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- S. Ponnurangam, I. V. Chernyshova and P. Somasundaran, Adv. Colloid Interface Sci., 2017, 244, 184–198 CrossRef PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef PubMed.
- A. J. Martín, G. O. Larrazábal and J. Pérez-Ramírez, Green Chem., 2015, 17, 5114–5130 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2015, 48, 2996–3006 CrossRef PubMed.
- K. Mase, S. Aoi, K. Ohkubo and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2016, 20, 935–949 CrossRef.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef PubMed.
- N. Onishi, G. Laurenczy, M. Beller and Y. Himeda, Coord. Chem. Rev., 2018 DOI:10.1016/j.ccr.2017.11.021.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef PubMed.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef PubMed.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef PubMed.
- W. H. Bernskoetter and N. Hazari, Acc. Chem. Res., 2017, 50, 1049–1058 CrossRef PubMed.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef PubMed.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef PubMed.
- S. Fukuzumi and T. Suenobu, Dalton Trans., 2013, 42, 18–28 RSC.
- E. Fujita, J. T. Muckerman and Y. Himeda, Biochim. Biophys. Acta, 2013, 1827, 1031–1038 CrossRef PubMed.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7367 RSC.
- S. Fukuzumi, Eur. J. Inorg. Chem., 2008, 1351–1362 CrossRef.
- A. J. Bard, R. Parsons and J. Jordan, Standard potentials in aqueous solutions, CRC press, 1985 Search PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef PubMed.
- P. J. Mak and I. G. Denisov, Biochim. Biophys. Acta, 2018, 1866, 178–204 CrossRef PubMed.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef PubMed.
- S. Fukuzumi and W. Nam, J. Porphyrins Phthalocyanines, 2016, 20, 35–44 CrossRef.
- R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef PubMed.
- S. Fukuzumi, T. Honda and T. Kojima, Coord. Chem. Rev., 2012, 256, 2488–2502 CrossRef.
- W. Nam, Acc. Chem. Res., 2015, 48, 2415–2423 CrossRef PubMed.
- X. Engelmann, I. Monte-Perez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef PubMed.
- G. Olivo, O. Lanzalunga and S. Di Stefano, Adv. Synth. Catal., 2016, 358, 843–863 CrossRef.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef PubMed.
- O. Cusso, X. Ribas and M. Costas, Chem. Commun., 2015, 51, 14285–14298 RSC.
- Z. Chen and G. Yin, Chem. Soc. Rev., 2015, 44, 1083–1100 RSC.
- M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef PubMed.
- P. P. Chandrachud and D. M. Jenkins, Tetrahedron Lett., 2015, 56, 2369–2376 CrossRef.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef PubMed.
- S. Fukuzumi, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2018, 355, 54–73 CrossRef.
- S. Fukuzumi, Y.-M. Lee and W. Nam, ChemCatChem, 2018, 10, 9–28 CrossRef.
- S. J. C. Robinson and D. M. Heinekey, Chem. Commun., 2017, 53, 669–676 RSC.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865–878 RSC.
- Z. Han and R. Eisenberg, Acc. Chem. Res., 2014, 47, 2537–2544 CrossRef PubMed.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- E. Lamy, L. Nadjo and J. M. Savéant, J. Electroanal. Chem., 1977, 78, 403–407 CrossRef.
- H. A. Schwarz and R. W. Dodson, J. Phys. Chem., 1989, 93, 409–414 CrossRef.
- A. Gennaro, A. A. Isse, M.-G. Severin, E. Vianello, I. Bhugun and J.-M. Savéant, J. Chem. Soc., Faraday Trans., 1996, 92, 3963–3968 RSC.
- J. Y. Becker, B. Vainas, R. Eger and L. Kaufman, J. Chem. Soc., Chem. Commun., 1985, 1471–1472 RSC.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313–315 CrossRef PubMed.
- J. Lan, T. Liao, T. Zhang and L. W. Chung, Inorg. Chem., 2017, 56, 6809–6819 CrossRef PubMed.
- U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Nat. Commun., 2014, 5, 5883 CrossRef PubMed.
- B. J. Cook, G. N. Di Francesco, K. A. Abboud and L. J. Murray, J. Am. Chem. Soc., 2018, 140, 5696–5700 CrossRef PubMed.
- M. Hammouche, D. Lexa, M. Momenteau and J.-M. Savéant, J. Electroanal. Chem., 1988, 249, 347–351 CrossRef.
- M. Hammouche, D. Lexa, M. Momenteau and J.-M. Savéant, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef.
- I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1994, 116, 5015–5016 CrossRef.
- B. Mondal, A. Rana, P. Sen and A. Dey, J. Am. Chem. Soc., 2015, 137, 11214–11217 CrossRef PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef PubMed.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2014, 136, 11821–11829 CrossRef PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef.
- C. Costentin, M. Robert, J.-M. Savéant and A. Tatin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882–6886 CrossRef PubMed.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Chem. Commun., 2015, 51, 10226–10228 RSC.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef PubMed.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- A. Maurin and M. Robert, Chem. Commun., 2016, 52, 12084–12087 RSC.
- Y. Wu, J. Jiang, Z. Weng, M. Wang, D. L. J. Broere, Y. Zhong, G. W. Brudvig, Z. Feng and H. Wang, ACS Cent. Sci., 2017, 3, 847–852 CrossRef PubMed.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, J. Am. Chem. Soc., 2016, 138, 5765–5768 CrossRef PubMed.
- A. Chapovetsky, M. Welborn, J. M. Luna, R. Haiges, T. F. Miller III and S. C. Marinescu, ACS Cent. Sci., 2018, 4, 397–404 CrossRef PubMed.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, ACS Catal., 2018, 8, 3411–3417 CrossRef.
- M. D. Sampson and C. P. Kubiak, J. Am. Chem. Soc., 2016, 138, 1386–1393 CrossRef PubMed.
- J. W. Raebiger, J. W. Turner, B. C. Noll, C. J. Curtis, A. Miedaner, B. Cox and D. L. DuBois, Organometallics, 2006, 25, 3345–3351 CrossRef.
- K.-Y. Wong, W.-H. Chung and C.-P. Lau, J. Electroanal. Chem., 1998, 453, 161–169 CrossRef.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef PubMed.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607–12609 RSC.
- M. D. Sampson, J. D. Froehlich, J. M. Smieja, E. E. Benson, I. D. Sharp and C. P. Kubiak, Energy Environ. Sci., 2013, 6, 3748–3755 RSC.
- J. Agarwal, E. Fujita, H. F. Schaefer III and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef PubMed.
- J. A. Keith, K. A. Grice, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2013, 135, 15823–15829 CrossRef PubMed.
- T. W. Schneider, M. Z. Ertem, J. T. Muckerman and A. M. Angeles-Boza, ACS Catal., 2016, 6, 5473–5481 CrossRef.
- M. L. Clark, P. L. Cheung, M. Lessio, E. A. Carter and C. P. Kubiak, ACS Catal., 2018, 8, 2021–2029 CrossRef.
- Y. C. Lam, R. J. Nielsen, H. B. Gray and W. A. Goddard III, ACS Catal., 2015, 5, 2521–2528 CrossRef.
- C. Riplinger and E. A. Carter, ACS Catal., 2015, 5, 900–908 CrossRef.
- K. T. Ngo, M. McKinnon, B. Mahanti, R. Narayanan, D. C. Grills, M. Z. Ertem and J. Rochford, J. Am. Chem. Soc., 2017, 139, 2604–2618 CrossRef PubMed.
- J. E. Vandezande and H. F. Schaefer III, Organometallics, 2018, 37, 337–342 CrossRef.
- M. Bourrez, M. Orio, F. Molton, H. Vezin, C. Duboc, A. Deronzier and S. Chardon-Noblat, Angew. Chem., Int. Ed., 2014, 53, 240–243 CrossRef PubMed.
- T. E. Rosser, C. D. Windle and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 7388–7392 CrossRef PubMed.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef PubMed.
- S. Sato, K. Saita, K. Sekizawa, S. Maeda and T. Morikawa, ACS Catal., 2018, 8, 4452–4458 CrossRef.
- X. Su, K. M. McCardle, J. A. Panetier and J. W. Jurss, Chem. Commun., 2018, 54, 3351–3354 RSC.
- M. Sheng, N. Jiang, S. Gustafson, B. You, D. H. Ess and Y. Sun, Dalton Trans., 2015, 44, 16247–16250 RSC.
- J. Schneider, H. Jia, K. Kobiro, D. E. Cabelli, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2012, 5, 9502–9510 RSC.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef PubMed.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- Y. Wu, B. Rudshteyn, A. Zhanaidarova, J. D. Froehlich, W. Ding, C. P. Kubiak and V. S. Batista, ACS Catal., 2017, 7, 5282–5288 CrossRef.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- D. C. Grills, D. E. Polyansky and E. Fujita, ChemSusChem, 2017, 10, 4359–4373 CrossRef PubMed.
- N. D. Loewen, T. V. Neelakantan and L. A. Berben, Acc. Chem. Res., 2017, 50, 2362–2370 CrossRef PubMed.
- Y. Y. Birdja, J. Shen and M. T. M. Koper, Catal. Today, 2017, 288, 37–47 CrossRef.
- A. W. Nichols, S. Chatterjee, M. Sabat and C. W. Machan, Inorg. Chem., 2018, 57, 2111–2121 CrossRef PubMed.
- F. Bertini, N. Gorgas, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, ACS Catal., 2016, 6, 2889–2893 CrossRef.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef PubMed.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, R. Baumann, W. A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef PubMed.
- A. Taheri, E. J. Thompson, J. C. Fettinger and L. A. Berben, ACS Catal., 2015, 5, 7140–7151 CrossRef.
- A. Taheri, N. D. Loewen, D. B. Cluff and L. A. Berben, Organometallics, 2018, 37, 1087–1091 CrossRef.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef PubMed.
- B. Reuillard, K. H. Ly, T. E. Rosser, M. F. Kuehnel, I. Zebger and E. Reisner, J. Am. Chem. Soc., 2017, 139, 14425–14435 CrossRef PubMed.
- A. Sinopoli, N. T. La Porte, J. F. Martinez, M. R. Wasielewski and M. Sohail, Coord. Chem. Rev., 2018, 365, 60–74 CrossRef.
- J. J. Walsh, C. L. Smith, G. Neri, G. F. S. Whitehead, C. M. Robertson and A. J. Cowan, Faraday Discuss., 2015, 183, 147–160 RSC.
- M. Stanbury, J.-D. Compain and S. Chardon-Noblat, Coord. Chem. Rev., 2018, 36, 120–137 CrossRef.
- T. Fogeron, T. K. Todorova, J.-P. Porcher, M. Gomez-Mingot, L.-M. Chamoreau, C. Mellot-Draznieks, Y. Li and M. Fontecave, ACS Catal., 2018, 8, 2030–2038 CrossRef.
- Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458–11461 RSC.
- C. Yoo, Y.-E. Kim and Y. Lee, Acc. Chem. Res., 2018, 51, 1144–1152 CrossRef PubMed.
- J. S. Anderson, V. M. IIuc and G. L. Hillhouse, Inorg. Chem., 2010, 49, 10203–10207 CrossRef PubMed.
- R. Beck, M. Shoshani, J. Krasinkiewicz, J. A. Hatnean and S. A. Johnson, Dalton Trans., 2013, 42, 1461–1475 RSC.
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636–637 RSC.
- C. Yoo and Y. Lee, Chem. Sci., 2017, 8, 600–605 RSC.
- C. Yoo, J. Kim and Y. Lee, Organometallics, 2013, 32, 7195–7203 CrossRef.
- D. Sahoo, C. Yoo and Y. Lee, J. Am. Chem. Soc., 2018, 140, 2179–2185 CrossRef PubMed.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- S. Kusama, T. Saito, H. Hashiba, A. Sakai and S. Yotsuhashi, ACS Catal., 2017, 7, 8382–8385 CrossRef.
- M. Gong, Z. Cao, W. Liu, E. M. Nichols, P. T. Smith, J. S. Derrick, Y.-S. Liu, J. Liu, X. Wen and C. J. Chang, ACS Cent. Sci., 2017, 3, 1032–1040 CrossRef PubMed.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Catal. Sci. Technol., 2016, 6, 4077–4080 RSC.
- S. Aoi, K. Mase, K. Ohkubo and S. Fukuzumi, Chem. Commun., 2015, 51, 15145–15148 RSC.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef PubMed.
- C. Steinlechner and H. Junge, Angew. Chem., Int. Ed., 2018, 57, 44–45 CrossRef PubMed.
- T. Wu, L. Zou, D. Han, F. Li, Q. Zhang and L. Niu, Green Chem., 2014, 16, 2142–2146 RSC.
- B. AlOtaibi, S. Fan, D. Wang, J. Ye and Z. Mi, ACS Catal., 2015, 5, 5342–5348 CrossRef.
- X. Liu, S. Inagaki and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 14924–14950 CrossRef PubMed.
- Y. Wang, X. Bai, H. Qin, F. Wang, Y. Li, X. Li, S. Kang, Y. Zuo and L. Cui, ACS Appl. Mater. Interfaces, 2016, 8, 17212–17219 CrossRef PubMed.
- L. Yu, G. Li, X. Zhang, X. Ba, G. Shi, Y. Li, P. K. Wong, J. C. Yu and Y. Yu, ACS Catal., 2016, 6, 6444–6454 CrossRef.
- H. Wang, Y. Chen, X. Hou, C. Ma and T. Tan, Green Chem., 2016, 18, 3250–3256 RSC.
- H. Rao, J. Bonin and M. Robert, Chem. Commun., 2017, 53, 2830–2833 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef.
- Y. Kuramochi and O. Ishitani, Inorg. Chem., 2016, 55, 5702–5709 CrossRef PubMed.
- E. Hasegawa, S. Takizawa, T. Seida, A. Yamaguchi, N. Yamaguchi, N. Chiba, T. Takahashi, H. Ikeda and K. Akiyama, Tetrahedron, 2006, 62, 6581–6588 CrossRef.
- Z. Guo, F. Yu, Y. Yang, C.-F. Leung, S.-M. Ng, C.-C. Ko, C. Cometto, T.-C. Lau and M. Robert, ChemSusChem, 2017, 10, 4009–4013 CrossRef PubMed.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2017, 139, 6538–6541 CrossRef PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef PubMed.
- L. Wang, R.-J. Xie, T. Suehiro, T. Takeda and N. Hirosaki, Chem. Rev., 2018, 118, 1951–2009 CrossRef PubMed.
- M. Mousavi, A. Habibi-Yangjeh and S. R. Pouran, J. Mater. Sci.: Mater. Electron., 2018, 29, 1719–1747 CrossRef.
- H. J. Yu, R. Shi, Y. X. Zhao, T. Bian, Y. F. Zhao, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. R. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- L. Lin, C. Hou, X. Zhang, Y. Wang, Y. Chen and T. He, Appl. Catal., B, 2018, 221, 312–319 CrossRef.
- K. Wada, C. S. K. Ranasinghe, R. Kuriki, A. Yamakata, O. Ishitani and K. Maeda, ACS Appl. Mater. Interfaces, 2017, 9, 23869–23877 CrossRef PubMed.
- S. Aoi, K. Mase, K. Ohkubo, T. Suenobu and S. Fukuzumi, ACS Energy Lett., 2017, 2, 532–536 CrossRef.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- K.-L. Bae, J. Kim, C. K. Lim, K. M. Nam and H. Song, Nat. Commun., 2017, 8, 1156 CrossRef PubMed.
- M. Xing, Y. Zhou, C. Dong, L. Cai, L. Zeng, B. Shen, L. Pan, C. Dong, Y. Chai, J. Zhang and Y. Yin, Nano Lett., 2018, 18, 3384–3390 CrossRef PubMed.
- Y. Zhaoa, Y. Weia, X. Wua, H. Zhenga, Z. Zhao, J. Liu and J. Li, Appl. Catal., B, 2018, 226, 360–372 CrossRef.
- S. Wanga, Y. Guan, L. Lu, Z. Shi, S. Yan and Z. Zou, Appl. Catal., B, 2018, 224, 10–16 CrossRef.
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi, L. Song, J. Jiang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2017, 139, 4486–4492 CrossRef PubMed.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef.
- S. Neaţu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef PubMed.
- J.-C. Wang, L. Zhang, W.-X. Fang, J. Ren, Y.-Y. Li, H.-C. Yao, J.-S. Wang and Z.-J. Li, ACS Appl. Mater. Interfaces, 2015, 7, 8631–8639 CrossRef PubMed.
- S. Das, S. Kumar, S. Garai, R. Pochamoni, S. Paul and S. Roy, ACS Appl. Mater. Interfaces, 2017, 9, 35086–35094 CrossRef PubMed.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- A. Iwase, S. Yoshino, T. Takayama, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2016, 138, 10260–10264 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |