
Ultra-efficient and stable electro-optic dendrimers containing supramolecular homodimers of semifluorinated dipolar aromatics†
Jieyun
Wu
 a,
Bo
Wu
a,
Wen
Wang
a,
Kin Seng
Chiang
*ab,
Alex. K.-Y.
Jen
*cd and
Jingdong
Luo
*b
a,
Bo
Wu
a,
Wen
Wang
a,
Kin Seng
Chiang
*ab,
Alex. K.-Y.
Jen
*cd and
Jingdong
Luo
*b
aSchool of Optoelectronic Science and Engineering, Key Lab of Optical Fiber Sensing and Communication (Education Ministry of China), University of Electronic Science and Technology of China, China
bDepartment of Electronic Engineering, City University of Hong Kong, Hong Kong. E-mail: eeksc@cityu.edu.hk; jingdluo@cityu.edu.hk
cDepartment of Chemistry, City University of Hong Kong, Hong Kong. E-mail: alexjen@cityu.edu.hk
dDepartment of Materials Science & Engineering, University of Washington, USA
First published on 6th February 2018
Abstract
In organic electro-optic (EO) materials, strong dipole–dipole interactions hinder the highly efficient poling of nonlinear optical chromophores. Supramolecular self-assembly through π–π stacking of fluoroaromatics was proved to be one of the most effective strategies to simultaneously achieve high chromophore loading density and highly efficient poling. Herein, we demonstrated a new strategy of supramolecular homodimerization to self-assemble EO dendritic films, in which two dendritic units with semifluorinated dipolar 1,2,3-trifluorobenzene (TFB) moieties were attached to the donor end and the π-bridge centre of push–pull tetraene chromophores. In these new dendrimers, the use of monolithic and semifluorinated TFB rings to replace the heterodimers of phenyl and pentafluorophenyl moieties has greatly simplified the synthesis of dendrimers and their intermixing, and can further potentially enable more efficient and rapid intermixing of interacting moieties in the solid states than those in binary and ternary systems. Photophysical property analysis and DFT calculations were carried out to understand the macroscopic supramolecular self-assembly and microscopic polarizability of new TFB-based EO dendrimers. The poled films of these self-assembled dendritic EO films exhibited very large EO coefficients up to 248 pm V−1 at a wavelength of 1310 nm and excellent temporal stability at room temperature with a very minimal change of ∼5% for over 1000 hours. Our study therefore illustrates that homodimer stacking of TFB rings through dipole–dipole coupling provides stabilization energy similar to that of quadrupolar interaction of phenyl and pentafluorophenyl heterodimeric pairs. Due to the highly efficient poling and excellent temporal EO stability, TFB self-assembled EO dendrimers show great potential for application in photonic devices.
Introduction
The information era we are enjoying now is urgently expecting new photonic devices with low energy-consumption, high-speed and ultra-broad band for next-generation optical fiber communications.1 Organic electro-optic (EO) materials promise many applications in photonic devices, such as low driving-voltage EO modulators and ultra-fast EO switches.1–3 As the key active component in EO materials, nonlinear optical (NLO) dipolar chromophores are covalently or non-covalently incorporated into the polymer matrix to form the polymeric EO film.4–6 The intramolecular electron distribution of the dipolar chromophore is non-centro symmetrical, which allows the chromophore alignment under the electrical field-induced poling to translate the microscopic hyperpolarizability of dipolar chromophores into macroscopic EO coefficients (r33 values). Chromophores with a larger dipole moment are more likely to align in the direction of the electrical field. In this respect, highly electron-delocalized chromophores with suitable electron donors and acceptors are the primary requirement to achieve high r33 values. In other words, highly dipolar chromophores with a large dipole moment are essential.7 However, these dipolar chromophores easily aggregate to form antiparallel dimers during the high-voltage poling process, which is the stable state in thermodynamics.In molecular engineering of dipolar chromophores, several strategies have been proposed, such as the site-isolation strategy,8–10 the binary-chromophore systems, molecular optimization of spherical shaped chromophores,7,11 and self-assembled molecular glasses.12 One of the most noteworthy strategies is the self-assembly of dipolar chromophores, which can achieve high chromophore loading densities and high EO coefficients simultaneously.13 Through intermolecular interactions such as hydrogen-bonding, π–π stacking, or van der Waals interaction, neat chromophores can self-assemble into monolithic molecular glass films. In 2007, Kim et al. developed a new class of molecular glasses based on the π–π stacking of phenyl and pentafluoro-phenyl (ArH–ArF interactions) in dendritic chromophores.12 They reported r33 values higher than 100 pm V−1 with highly significant temporal stability. These self-assembled molecular glasses were further used in binary chromophore systems to achieve the highest r33 value of 327 pm V−1 at that time. Upon fine-tuning arene-pentafluoroarene interactions, Zhou et al. synthesized a series of polyene-based dendritic chromophores substituted with branched aryl- and pentafluorophenyl dendrons.14 The self-assembly was driven by the enhanced cohesive energy of heterodimers and structural rigidity through the π–π stacking of pentafluorophenyl with phenyl, naphthyl and anthracyl respectively. These self-assembled dendrimers exhibited large EO coefficients and excellent temporal EO stability. Besides the self-assembly of chromophores, Wu et al. investigated their self-assembly in polymers.4 A series of main-chain and side-chain self-assembled EO polymers with azo-dyes were synthesized and their nonlinear optical coefficients (d33) were as high as 257 pm V−1.15 These endeavors indicate that supramolecular self-assembly is an effective strategy to improve EO performance. Investigating the new self-assembly driving force for dipolar chromophores is attractive and promising in electro-optics.
Recently, Christopher has reported the stacked fluorinated aromatics, trifluorobenzyl (TFB) moieties, as supramolecular synthons for programming protein dimerization specificity.16 This revealed the surprisingly large contributions of dipole–dipole and dipole–induced-dipole interactions to the association of aromatic pairs in the stacked geometry. Due to the high electronegativity difference between carbon and fluorine, the C–F bond acted as a weak hydrogen bond acceptor. The weak C–H⋯F–C interactions arose from the electrostatic and dispersive forces between C(δ+)–F(δ−) and H(δ+)–C(δ−) fragments. Unlike the relatively nonpolar pentafluorophenyl moieties, the semifluorinated 1,2,3-trifluorobenzene ring contains a large dipole moment of 3.0 D.17 In crystal engineering of 1,2,3-trifluobenzene, antiparallel packing at three dimensions was furnished by dipole–dipole interactions and hydrogen-bonding.18 It has also been illustrated that the homodimer stacking of the 1,2,3-trifluorobenzene ring through dipole–dipole coupling provides very similar stabilization energy compared with that of the quadrupolar interaction of the phenyl and pentafluorophenyl heterodimeric pairs.
This work investigates the feasibility of supramolecular self-assembly of dipolar 1,2,3-trifluorobenzene in nonlinear optical chromophores. In general, the π–π stacking of fluoroaromatic rings is through electrostatic mechanisms. Using such motifs for the design and synthesis of organic π-functional materials requires an optimal consideration of many related parameters of molecular physics, such as the quadrupole moment or dipole moment, the dipole–induced-dipole interactions, and the hydrophobicity and the synthetic efficacy. Such research efforts will be very helpful to provide new guidelines and address the scope of this approach in designing new high performance self-assembled organic functional materials for photonics and electronics. The endeavour of this work follows our persistent research efforts in using supramolecular engineering of quadrupolar and dipolar interactions to improve the poling efficiency and thermal stability of organic EO materials. The use of a monolithic and semifluorinated TFB ring to replace the heterodimer of phenyl and pentafluorophenyl moieties has greatly simplified the synthesis of dendrimers and their intermixing, and can further potentially enable more efficient and rapid intermixing of interacting moieties in the solid states than those in binary and ternary systems. Photophysical property analysis and DFT calculations were carried out to understand their macroscopic supramolecular self-assembly and microscopic polarizability. The supramolecular homodimerization enabled the formation of high-quality EO thin films with a high chromophore loading density of 3.7 × 1020 cm−3. In EO activities, the r33 values of poled dendrimer-1 and dendrimer-2 films were as large as 237 and 248 pm V−1, respectively. Meanwhile, their EO activities only dropped by ∼5% after 1000 hours at ambient temperature. Given their exceptionally high poling efficiency, large EO coefficients and excellent temporal stability, our new self-assembly strategy of TFB-based dendritic NLO chromophores was very practical for photonic devices.
Results and discussion
Synthesis
Fig. 1 shows the synthetic route to dipolar branched RH and two dendritic chromophores. Trifluorobenzyl alcohol (B1) was first etherified with B2 by the Mitsunobu reaction to form the precursor branch B3. After hydrolysis in methanol, RH with a carboxyl group was obtained in a high yield. The synthesis of the chromophore backbone started from the substitution reaction of compound 1 and bromo-1-propanol. The esterified compound 3 was then formylated with POCl3 in DMF. The Knoevenagel condensation between compound 4 and 2-hydroxyethyl-substituted isophorone 5 was carried out with a strong base sodium ethoxide as the catalyst. The extension of the conjugated bridge of compound 6 was proceeded with Wittig reagent compound 7 and then the crude products were hydrolysed to form compound 8 with two hydroxyl groups in the donor and conjugated bridge.19 The synthesis of the intermediate compound 10, containing the tetrahydroquinolinyl donor, was similar to that of the dendritic tetraene-based chromophore, which can be found elsewhere.20 RH was introduced into the donor-bridge aldehyde 8 and 10 by esterification using DIPC as a catalyst to facilitate dendritic donor-bridges 9 and 11. The condensation of dendritic donor-bridges and the strong acceptor Ph-CF3-TCF was carried out to finally obtain the dendritic chromophores dendrimer-1 and dendrimer-2. Dendrimer-2 was obtained in a higher yield than Dendrimer-1 (67% versus 48%), which might be attributed to the stronger electron-donation of the tetrahydroquinolinyl donor than the trimethylindolinyl donor. Both chromophores were well soluble in common solvents such as hexane, toluene, chloroform, and acetone. Comparing the EO dendrimers with the heterodimers, the homodimer-based dendrimers required relatively facile synthetic efforts.19–21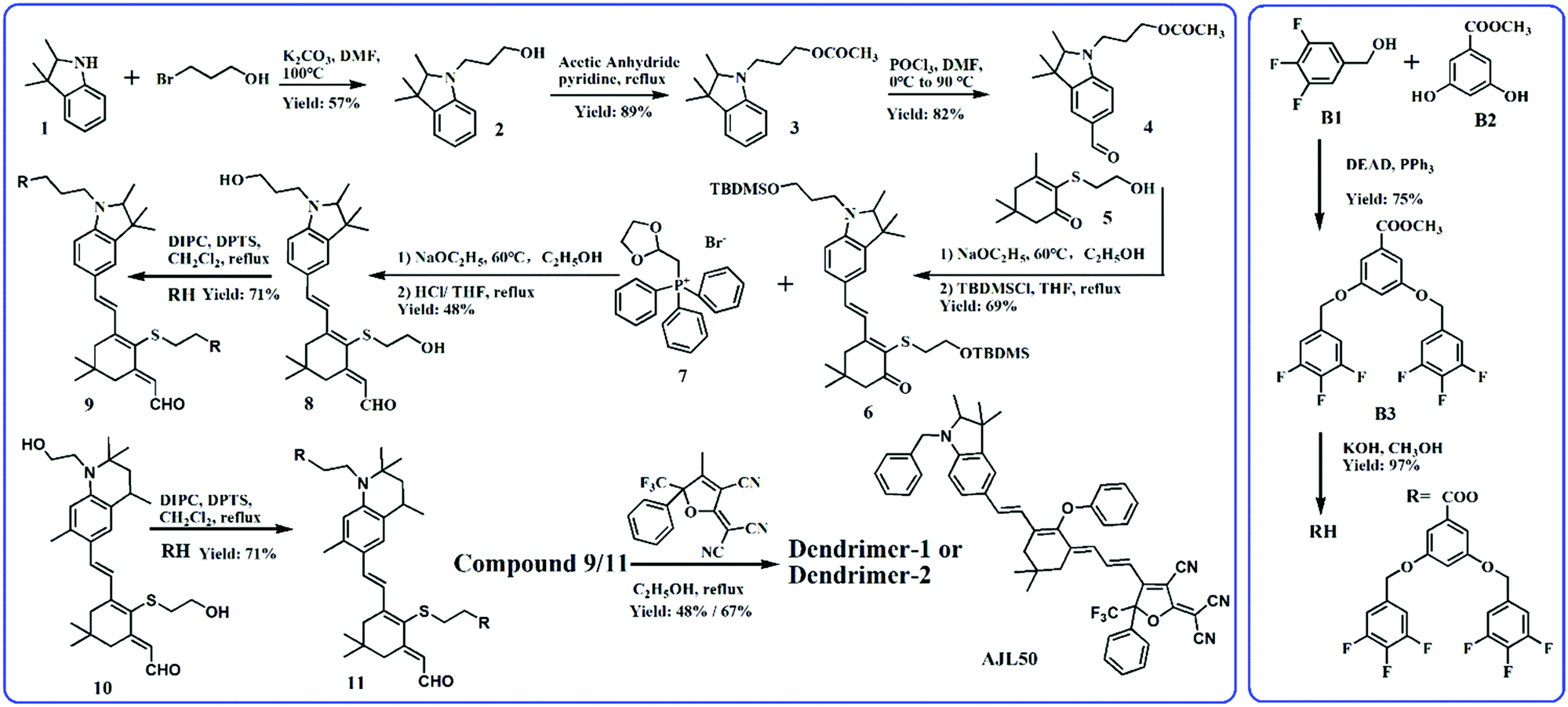 | ||
| Fig. 1 Synthesis of dendrimers and chemical structure of reference chromophore AJL50. | ||
Structure analysis
The chemical structures of dendrimer-1 and dendrimer-2 are shown in Fig. 2. Both dendrimers had a typical tetraene-conjugated core chromophore structure. They contained the same acceptor and tetraene-conjugated electron-bridge, but different donor structures. The donor of dendrimer-1 was a derivate of trimethylindoline, while that of dendrimer-2 was a tetrahydroquinolinyl derivate. The bond angle of N–C in the excited state (sp2) in the penta-cyclic donor of dendrimer-1 was less coplanar than that in the hexa-cyclic donor (close to 120°) of dendrimer-2. This structural variation should lead to a higher energy gap of transition in the intramolecular charge-transfer of dendrimer-1 and also different glass transition temperatures (Tg). Both donors used are relatively rigid compared to the regular aniline donor, which showed better chemical stability and resistance. The flexible alkyl chains ethyl and propyl groups, which respectively connected the donor and the dendritic TFB moiety (TFBD), also affected the Tg of both dendrimers. In differential scanning calorimeter (DSC) analysis, dendrimer-2 exhibits a higher Tg than dendrimer-1 (85 °C versus 54 °C, see the ESI†). And there was no glass transition temperature for the reference chromophore AJL50 (Fig. 1) without TFB. It was also found that TFB self-assembled dendrimers exhibited a lower Tg than ArH–ArF self-assembled dendrimers.14 | ||
| Fig. 2 Chemical structures (a) and the illustration of dipole–dipole homodimerization of TFB (b). Red: acceptor; blue: donor; green: conjugated bridge. | ||
The TFB moieties were located in the donor and the centre of the conjugated bridge, which served two functions in this self-assembly system. In terms of site-isolation, TFB played the role of a site-isolator to isolate the antiparallel stacking between core chromophores. More significantly, TFB, with the donor–acceptor structure (μ = 3.0 D), dispersed around the core chromophores, which allowed the formation of antiparallel TFB homodimers via dipole–dipole interactions. This effect has been proved previously by mass spectrometry and crystal analyses.16 The powder XRD, mass and infrared spectrometry (see ESI†) were not able to characterize the motion of self-assembly. These are in accordance with the conclusion pointed out by Kirchner, M. T et al. that the self-assembly of TFB was poorly directional and can be easily deformed by other predominant interactions. As the structure analysis showed, the core chromophore exhibited a much stronger dipole moment than trifluorobenzene (TFB). The dipole–dipole interactions between chromophore cores, core–TFB and TFB–TFB interactions, show a synergistic effect in the formation of self-assembled dendritic films. However, the effects of heterodimer self-assembly on the highly efficient poling and excellent temporal stability of EO activities have been successfully demonstrated. Hence, the homodimer self-assembly in this work was expected to provide a significant improvement in the EO performance, which might be the strongest evidence to prove the homodimer self-assembly motion in our EO dendrimers.
Photophysical properties
Fig. 3 shows the measured absorption spectra of two dendrimers in CH2Cl2 solution and in spin-coated thin films. The key features of the absorption spectra are listed in Table 1. Two dendrimers showed strong absorption bands with similar shapes. In CH2Cl2 solution, the λmax of dendrimer-1 was 776 nm and a strong bathochromic shift to 799 nm was observed in dendrimer-2. This was attributed to the fact that the hexa-cyclic tetrahydroquinolinyl donor of dendrimer-2 was stronger in electron-donation than the penta-cyclic trimethylindolinyl donor of dendrimer-1. The absorption bandwidths of dendrimer-1 and dendrimer-2, defined as the full-width at half-maximum (FWHM), were 277 nm and 288 nm, respectively. Their FWHMs were similar to those of the previously reported tetraene-based chromophores with ArH–ArF dendritic moieties.14 | ||
| Fig. 3 Normalized absorption spectra of dendrimer-1 and dendrimer-2 in CH2Cl2 solution and in self-assembled films. | ||
In the case of the solid state of self-assembled films, they showed significant differences in absorption features. As shown in Fig. 3 and Table 1, the absorption peaks respectively bathochromic-shifted to longer wavelengths of 798 nm and 828 nm for dendrimer-1 and dendrimer-2. Meanwhile, the absorption bands significantly extended to the near-infrared regime with a broad FWHM bandwidth greater than 400 nm and the onset wavelength around 1150 nm. These distinct changes in the absorption bands were also observed in self-assembled dendrimers through π–π stacking between arene and pentafluoroarene rings (ArH–ArF interactions). This phenomenon in both self-assembled systems can be attributed to the inhomogeneous broadening and solvatochromism of electronic absorption characteristics associated with chromophore–chromophore and chromophore–dendron interactions in the self-assembled thin films.14
Due to their different style of self-assembly, namely ArH–ArF of π–π stacking heterodimerization versus the TFB–TFB of dipole–dipole interacting homodimerization, there were some fundamental differences in the photophysical properties in the solid state. Firstly, self-assembled dendrimer-1 and dendrimer-2 based on TFB homodimerization displayed a stronger shoulder absorption peak than self-assembled dendrimers TED 1–3 (see the chemical structure in Fig. 5 and the absorption spectrum in ref. 14) based on ArH–ArF π–π stacking. Secondly, the FWHMs of dendrimer-1 and dendrimer-2 were more than 400 nm, which were also considerably broadened compared to 330–350 nm of TED 1–3. The differences might be due to their different nanoscale environment of dipole–dipole interactions and different donating power of donor structures for the chromophoric core. For dendrimer-1 and dendrimer-2, the surrounding TFBs create a more polarizable environment for the tetraene chromophore core through the dipole–induced-dipole interaction, and the ring-locked structures of the donors also increase the donating power of chromophores and the rigidity of dendrimers for more efficient intramolecular charge transfer. In contrast, for TED 1–3, the ArH–ArF π–π stacking motif is less polar and the dialkylaminophenyl donor gives less donating power for the chromophore core. Such structural differences in both the dendritic units and chromophore cores explain why the TFB-functionalized dendrimers in the present study of Dendrimer-1 and Dendrimer-2 showed much stronger shoulder peaks, more bathochromic shifts and wider FWHMs than those of TED 1–3.
DFT calculations
Since TFB is a dipolar moiety, it is necessary to evaluate the effects of TFB on the chromophore's microscopic dipole moment and ground-state polarizability. DFT calculations using Gaussian 03 were carried out at the hybrid B3LYP level by employing the split valence 6-31g(d) basis set.21 Chromophore AJL50 with a similar push–pull structure (see Fig. 1) was chosen as the reference for comparison. Their simulated configurations are shown in Fig. 4. In terms of steric hindrance, two dendritic TFBs in the donor and the conjugated bridge acted as site-isolators to attenuate the dipole–dipole interaction between strongly dipolar cores of the conjugated chromophore structures with a dipole moment greater than 20 D.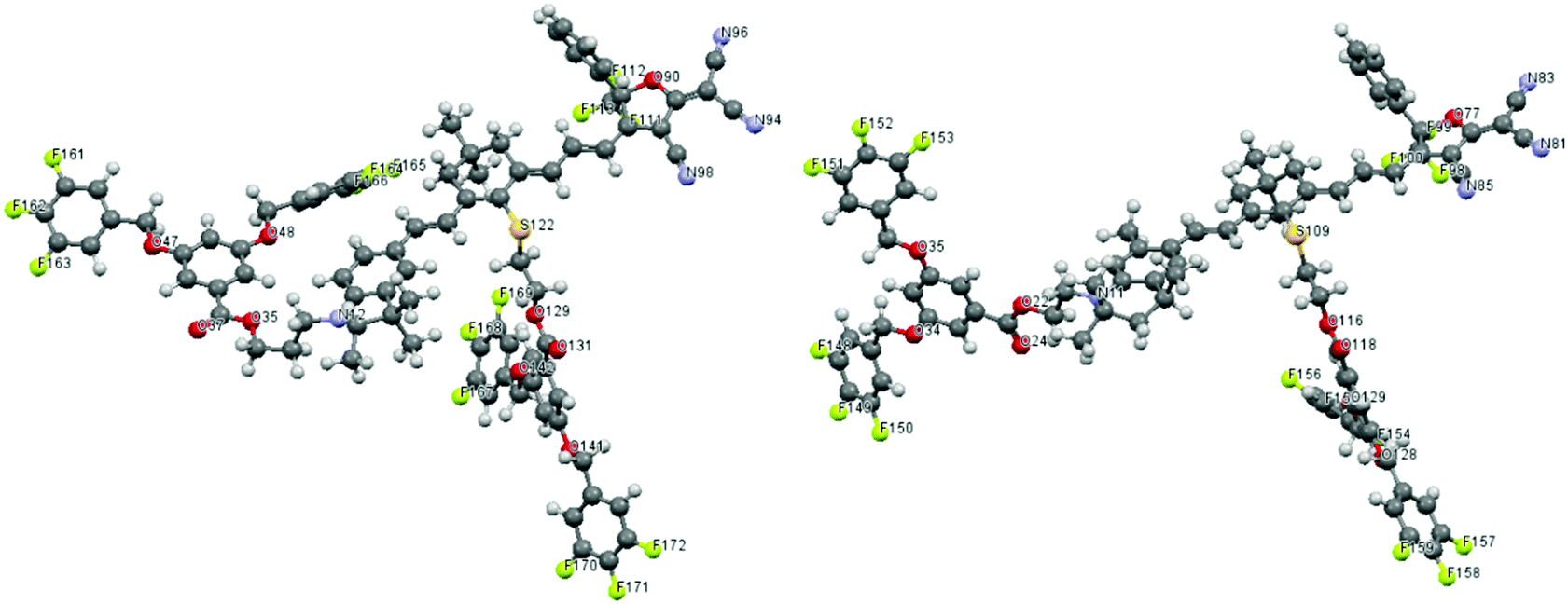 | ||
| Fig. 4 Theoretically optimized configuration of dendrimers-1 (left) and dendrimer-2 (right). C and H atom labels are omitted. | ||
As shown in Table 2, the dipole moments of AJL50 and dendrimer-1 were almost the same, and dendrimers-2 exhibited a slightly larger dipole moment. The main dipole component was along the direction of the core conjugated system (x axis). It was interesting that μy and μz were weaker after the introduction of TFB moieties. Although the introduction of small dipolar moieties TFB (μ = 3.0 D) had a weak influence on μtotal, their influence on molecular dipole derivatives and polarizability was significant. Recent theoretical investigations showed that atomic polar tensor charges can be obtained from dipole moment derivatives. The dipole moment derivatives are obtained from the derivative of the dipole moment with respect to internal coordinates or ordinary coordinates. In periodic table of elements, the fluorine atom is the strongest in electron negativity. In every case of the resultant vector for the fluorine atom, the dipole is directed along the A–F bond axis, which is also named “bond dipole”.22–24 In the case of TFB with highly polar C–F bonds, three C–F bond dipoles associated with the aromatic ring enabled the formation of TFB with a dipole moment of 3.0 D. This configuration led to the combination of five dipolar centres consisting of four TFB dipolar and one tetraene dipolar moieties. This molecular variety resulted in very different dipole moment derivative values with respect to five individual ordinal coordinates. Chromophore AJL50 without TFB had 954 dipole moment derivatives, which mainly originated from heteroatoms N, O, F, double bonds and triple bonds, whereas 1620 and 1647 dipole moment derivatives (see the ESI†) were obtained in dendrimer-1 and dendrimer-2 with TFBs, respectively. A higher density of dipole moment derivatives indicates strong atomic polar tensors, leading to enhanced polarizabilities for dendrimers. Hence the calculated polarizabilities of dendrimer-1 and dendrimer-2 were 368.25 and 381.52, much higher than 293.04 of AJL50. In general, theoretical calculations showed that the strong atom polar tensor of TFB dendrons created a much more polarized environment, which might be of great significance for the chromophore alignment in electrical field-induced poling.
| μ x | μ y | μ z | μ total | Nμb | α x | α y | α z | α total | |
|---|---|---|---|---|---|---|---|---|---|
| a μ total = (μx2 + μy2 + μz2)1/2, unit: Debye. b number of dipole moment derivatives. c α total = (αx2 + αy2 + αz2)1/2, unit: esu. | |||||||||
| Dendrimer-1 | 23.20 | 4.72 | 3.25 | 23.91 | 1620 | 303.42 | 182.54 | 129.40 | 368.25 |
| Dendrimer-2 | 25.67 | 2.47 | 1.65 | 25.82 | 1647 | 320.41 | 160.53 | 142.79 | 381.52 |
| AJL50 | 22.20 | 6.51 | 5.45 | 23.62 | 954 | 273.35 | 90.55 | 60.30 | 293.04 |
Electro-optic properties
Dendrimer-1 and dendrimer-2 were respectively dissolved in dibromomethane at a concentration of 6 wt% for thin film fabrication. The solutions were filtered through a 0.2 μm PTFE-syringe filter and then spin-coated onto indium tin oxide (ITO) glass substrates. The obtained optical-quality thin films had a thickness of about 800 nm and contained a high chromophore loading density of 3.7 × 1020 cm−3. The films were then baked in a vacuum at 50 °C overnight to ensure the removal of any residual solvent. A thin layer of gold was sputtered onto each film as a top electrode for contact poling. The optimal poling conditions and the measured r33 values as well as the poling efficiency for our two dendrimers are summarized in Table 3.| Mna | Nb | Epc | r 33 | r 33/Epe | r 33/Ep/Nf | |
|---|---|---|---|---|---|---|
| a Unit: g mol−1. b Core chromophore moiety, unit: 1020 cm−3 (formula of dendrimer-1, -2 and AJL33: C38H33F3N4O, molecular weight: 618.7); AJL50: 13 wt% chromophore in PMMA; AJL33: 25 wt% chromophore in PMMA. c Poling voltage, unit: V μm−1. d Measured at 1310 nm,25 unit: pm V−1. e Poling efficiency, unit: (nm V−1)2. f Unit: 10−20 nm2 V−2 cm3. | ||||||
| Dendrimer-1 | 1618 | 3.7 | 87 | 237 | 2.72 | 0.74 |
| Dendrimer-2 | 1628 | 3.7 | 71 | 248 | 3.49 | 0.94 |
| AJL50 | 814 | 0.95 | 100 | 90 | 0.90 | 0.95 |
| AJL33 | 781 | 1.9 | 100 | 168 | 1.68 | 0.88 |
Because of their different glass transition temperatures and different chromophore structures, the two dendrimers require different poling voltages and temperatures for poling. To achieve a high poling efficiency, the applied voltage as the driving force to induce the dipolar chromophore to align in the direction of the electrical field is expected to be as large as possible. However, high poling field could cause the damage of the EO thin film due to high leakage current and possible dielectric breakdown. Due to this restriction, the typical guest–host or self-assembled EO materials were usually poled with an electric field of about 100 V μm−1. The other key point for highly efficient poling is that the highly polarized dipolar chromophore is easily poled under the electrical field. In DFT analysis, it was shown that our TFB self-assembled dendrimers showed much more dipole moment derivatives and higher polarizability than benchmark chromophore AJL50, indicating the possibility of higher poling efficiency. Finally, the poling results turned out to be as expected. Both dendrimers required relatively low electric fields (87 V μm−1 for dendrimer-1 and 71 V μm−1 for dendrimer-2), yet very large r33 values, 237 pm V−1 for dendrimer-1 and 248 pm V−1 for dendrimer-2 respectively. It is not common to achieve such large EO coefficients under relatively low poling fields in guest–host systems or self-assembled dendrimers. This highly efficient poling can be attributed to the more polarizable microscopic environment. In dendrimer-1 and dendrimer-2, the dipole–dipole interactions between the core chromophores are attenuated and interfered by the dipole–dipole interactions of the core chromophore–TFB and the TFB homodimers, leading to a good poling efficiency of the poled films.
Fig. 5 and Table 3 summarize the poling efficiency of typical guest–host polymer films (AJL50 and AJL33) and ArH–ArF self-assembled dendrimers (TED 1–3). The poling efficiency r33/Ep is defined as the contribution of the poling field to the chromophore alignment parameter r33. In the TFB-containing self-assembled EO dendrimers, the r33/Ep was much higher than ArH–ArF self-assembled dendrimers (TED 1–3), demonstrating a higher poling efficiency of homodimer-based dendrimers than heterodimer-based dendrimers. Meanwhile, the r33/Ep/N of dendrimer-2 with a high chromophore loading density of 3.7 × 1020 cm−3 was nearly identical to that of simple guest–host AJL50 with a much lower loading density of 0.95 × 1020 cm−3. The purpose of the comparison of self-assembled dendrimers and efficiently poled guest–host films with a low loading density is to illustrate that even at a high chromophore loading density, dendrimer-2 and dendrimer-2 still showed a high poling efficiency. These comparisons indicate that new supramolecular dendrimers can achieve a high poling efficiency that exceeds the performance of early generation dendrimers and simple guest–host polymers.
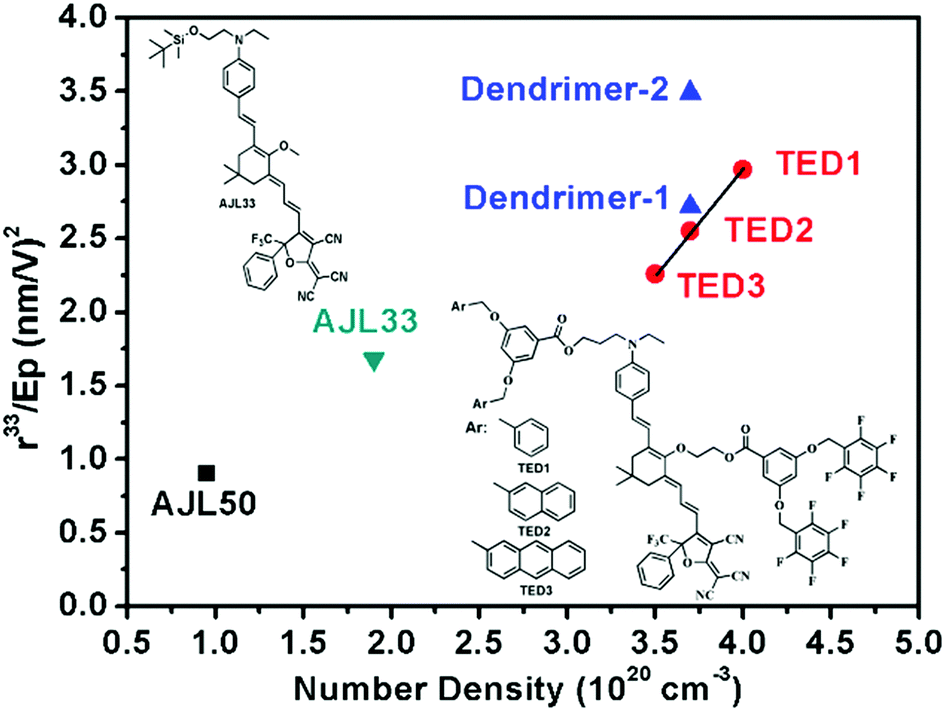 | ||
| Fig. 5 Comparison of poling efficiency in relation to chromophore loading density in our work and ref. 14. The formulas for AJL33 and AJL50 were the same as presented in Table 3. | ||
An EO material with a higher Tg usually has a more stable r33 value. Since the glass transition temperatures of both dendrimers were not high, one might worry about the temporal stability of EO performance. EO materials with such high chromophore loading densities and low Tg values usually exhibited poor temporal stability of EO coefficients. But our study on the stability of the poled self-assembled dendrimers showed a surprisingly good result. As illustrated in Fig. 6, 94% and 96% of their original EO coefficient values were maintained at ambient temperature for over 1000 h after an initial fast decay. Considering the low Tgs of the dendrimers, especially for dendrimer-1 (54 °C), the stability of TFB self-assembled dendrimers was really excellent. Although the Tg of dendrimer-2 is 31 °C higher than that of dendrimer-1, their temporal stabilities of EO coefficients were almost the same. This finding suggested that the glass transition temperature may not necessarily be the determining factor for maintaining the long-term stability of EO activities. Rather the self-assembled interactions should play a more predominant role in obstructing the motion of dipole relaxation in these dendrimers.
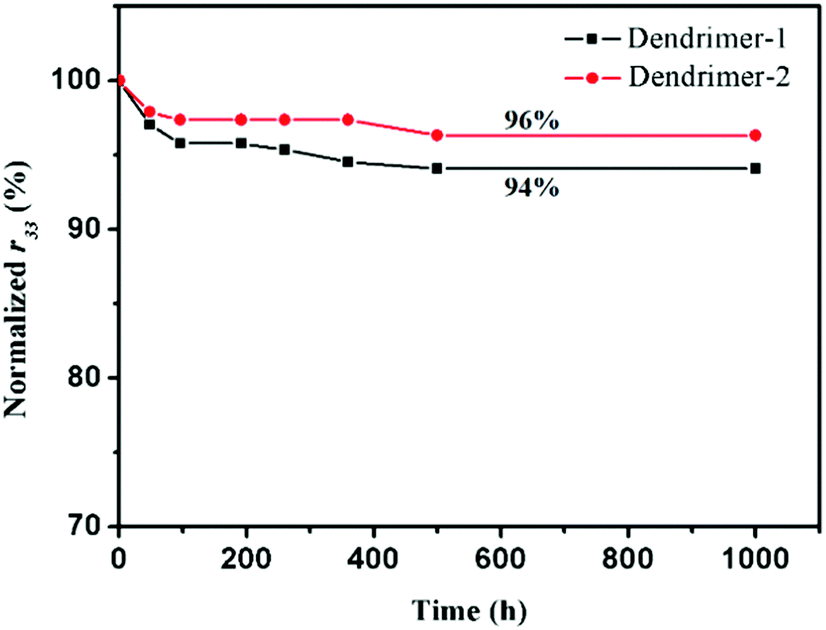 | ||
| Fig. 6 EO coefficients r33 of the two dendrimers, measured over a period of 1000 hours at room temperature. | ||
The analysis of the thermally stimulated discharge (TSD) current of poled films in orientation relaxation can provide useful information for the understanding of dipole relaxation. In our experiments, poled EO films were connected to an electrometer in a short circuit configuration and placed into a programmable heater. The temperature was first held at 30 °C for 10 min and then ramped up at a rate of 1 °C min−1. The TSD current was recorded and plotted as a function of temperature, as shown in Fig. 7. In the TSD spectrum, a notable α peak around the glass transition was observed. Previous studies have shown that the α peak of an EO polymer is a signal of the dipole relaxation process. Integration in the time domain of the curves in Fig. 7 yielded a total charge release of 51 nC for dendrimer-1 and 59 nC for dendrimer-2, which confirmed the higher poling efficiency and r33 value of dendrimer-2 compared to dendrimer-1. For dendrimer-1, the dipole relaxation starts at around 50 °C and the α peak appears at 60 °C, while for dendrimer-2, the α peak appears at 80 °C. Routinely, a higher dipole relaxation temperature indicates the higher temporal stability of EO activity. However, two dendrimers showed quite similar stability. This again indicated that the self-assembly force of dipole–dipole interactions of TFB in dendrimers accounted for the outstanding temporal stability of EO coefficients at room temperature.
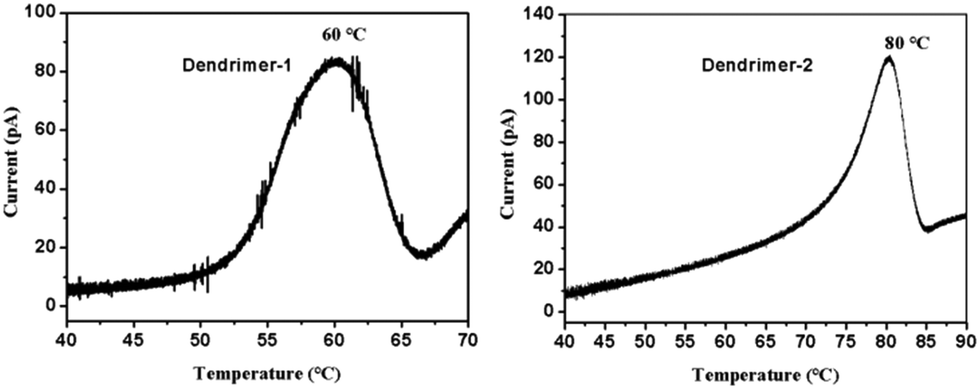 | ||
| Fig. 7 TSD current curves for dendrimer-1 and dendrimer-2 as a function of temperature. | ||
As a result, highly efficient poling and ultra-large EO activity with good temporal stability were achieved simultaneously in this new class of dendrimers containing homodimers of semifluorinated aromatic moieties.
Conclusions
In conclusion, novel self-assembled EO dendrimers containing supramolecular homodimers of semifluorinated aromatic moieties for ultra-efficient and stable EO performance were successfully demonstrated. Our homodimer-based self-assembled dendrimers required more facile synthesis than previously reported heterodimer-based dendrimers. The dipole–dipole interactions of the TFB–TFB pairs and the chromophore–TFB moieties could effectively attenuate the strong intermolecular interaction of a large dipole tetraene chromophore. As a result, dendrimer-1 and dendrimer-2 can be efficiently poled under relatively modest poling field, which is lower than the required field strengths of typical guest–host and heterodimer-based self-assembled EO materials. Under such poling conditions, they exhibited large EO coefficients (r33) up to 248 pm V−1 and excellent long-term temporal stability at ambient temperature. These results indicated that the self-assembled force of TFB dipole–dipole interactions can greatly improve the poling efficiency and effectively restrict dipole relaxation. This new self-assembled strategy showed attractive potential in EO devices with high-speed, low energy consumption and long-term stability.Conflicts of interest
The authors declare no commercial conflict on this manuscript.Acknowledgements
The authors would like to thank Dr Huajun Xu (University of Washington) for his help on DFT calculations. This work was supported by the Air Force Office of Scientific Research (FA8650-14-C-5005), the National Natural Science Foundation of China (No. U1533121), the New Faculty Start-up Grant of the City University of Hong Kong (7200550), the China Postdoctoral Science Foundation (2017M610596) and the 111 Project (B14039). J. Wu is thankful to the support from the National Postdoctoral Program for Innovative Talents (BX201600027).Notes and references
- M. Lee, H. E. Katz, C. Erben, D. M. Gill, P. Gopalan, J. D. Heber and D. J. McGee, Broadband modulation of light by using an electro-optic polymer, Science, 2002, 298, 1401–1403 CrossRef CAS PubMed.
- Y. Enami, C. T. Derose, D. Mathine, C. Loychik, C. Greenlee, R. A. Norwood, T. D. Kim, J. Luo, Y. Tian, A. K. Y. Jen and N. Peyghambarian, Hybrid polymer/sol–gel waveguide modulations with exceptionally large electro-optic coefficients, Nat. Photonics, 2007, 1, 423 CrossRef CAS.
- M. Hochberg, T. Baehr-Jones, G. X. Wang, M. Shearn, K. Harvard, J. D. Luo, B. Q. Chen, Z. W. Shi, R. Lawson, P. Sullivan, A. K. Y. Jen, L. Dalton and A. Scherer, Terahertz all-optical modulation in a silicon-polymer hybrid system, Nat. Mater., 2006, 5, 703–709 CrossRef CAS PubMed.
- Z. Li, Q. Li and J. Qin, Some new design strategies for second-order nonlinear optical polymers and dendrimers, Polym. Chem., 2011, 2, 2723 RSC.
- L. R. Dalton, D. Lao, B. C. Olbricht, S. Benight, D. H. Bale, J. A. Davies, T. Ewy, S. R. Hammond and P. A. Sullivan, Theory-inspired development of new nonlinear optical materials and their integration into silicon photonic circuits and devices, Opt. Mater., 2010, 32, 658–668 CrossRef CAS.
- S. R. Marder, B. Kippelen, A. K. Y. Jen and N. Peyghambarian, Design and synthesis of chromophores and polymers for electro-optic and photorefractive applications, Nature, 1997, 388, 845–851 CrossRef CAS.
- R. L. Tang, S. M. Zhou, Z. Y. Cheng, G. Yu, Q. Peng, H. Y. Zeng, G. C. Guo, Q. Q. Li and Z. Li, Janus second-order nonlinear optical dendrimers: their controllable molecular topology and corresponding largely enhanced performance, Chem. Sci., 2017, 8, 340–347 RSC.
- W. Wu, Q. Huang, G. Xu, C. Wang, C. Ye, J. Qin and Z. Li, Using an isolation chromophore to further improve the comprehensive performance of nonlinear optical (NLO) dendrimers, J. Mater. Chem. C, 2013, 1, 3226 RSC.
- W. Wu, L. Huang, L. Xiao, Q. Huang, R. Tang, C. Ye, J. Qin and Z. Li, New second-order nonlinear optical (NLO) hyperbranched polymers containing isolation chromophore moieties derived from one-pot “A2 + B4” approach via Suzuki coupling reaction, RSC Adv., 2012, 2, 6520 RSC.
- J. Y. Wu, J. D. Luo, N. Cernetic, K. X. Chen, K. S. Chiang and A. K. Y. Jen, PCBM-doped electro-optic materials: investigation of dielectric, optical and electro-optic properties for highly efficient poling, J. Mater. Chem. C, 2016, 4, 10286–10292 RSC.
- W. Wu, L. Huang, C. Song, G. Yu, C. Ye, Y. Liu, J. Qin, Q. Li and Z. Li, Novel global-like second-order nonlinear optical dendrimers: convenient synthesis through powerful click chemistry and large NLO effects achieved by using simple azo chromophore, Chem. Sci., 2012, 3, 1256 RSC.
- T. D. Kim, J. W. Kang, J. D. Luo, S. H. Jang, J. W. Ka, N. Tucker, J. B. Benedict, L. R. Dalton, T. Gray, R. M. Overney, D. H. Park, W. N. Herman and A. K. Y. Jen, Ultralarge and thermally stable electro-optic activities from supramolecular self-assembled molecular glasses, J. Am. Chem. Soc., 2007, 129, 488–489 CrossRef CAS PubMed.
- J. D. Luo, X. H. Zhou and A. K. Y. Jen, Rational molecular design and supramolecular assembly of highly efficient organic electro-optic materials, J. Mater. Chem., 2009, 19, 7410–7424 RSC.
- X. H. Zhou, J. D. Luo, S. Huang, T. D. Kim, Z. W. Shi, Y. J. Cheng, S. H. Jang, D. B. Knorr, R. M. Overney and A. K. Y. Jen, Supramolecular Self-Assembled Dendritic Nonlinear Optical Chromophores: Fine-Tuning of Arene-Perfluoroarene Interactions for Ultralarge Electro-Optic Activity and Enhanced Thermal Stability, Adv. Mater., 2009, 21, 1976–1981 CrossRef CAS.
- W. Wu, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, Using two simple methods of Ar-Ar(F) self-assembly and isolation chromophores to further improve the comprehensive performance of NLO dendrimers, Chemistry, 2013, 19, 630–641 CrossRef CAS PubMed.
- C. J. Pace, H. Zheng, R. Mylvaganam, D. Kim and J. M. Gao, Stacked Fluoroaromatics as Supramolecular Synthons for Programming Protein Dimerization Specificity, Angew. Chem., Int. Ed., 2012, 51, 103–107 CrossRef CAS PubMed.
- M. Onda, H. Mukaida, H. Akiba, M. Mori, H. Miyazaki and I. Yamaguchi, Microwave-Spectrum of 1,2,3-Trifluorobenzene, J. Mol. Spectrosc., 1995, 169, 480–483 CrossRef CAS.
- M. T. Kirchner, D. Bläser, R. Boese, T. S. Thakur and G. R. Desiraju, 1,2,3-Trifluorobenzene, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o2670 CAS.
- C. L. Hu, Z. Chen, H. Y. Xiao, Z. Zhen, X. H. Liu and S. H. Bo, Synthesis and characterization of a novel indoline based nonlinear optical chromophore with excellent electro-optic activity and high thermal stability by modifying the pi-conjugated bridges, J. Mater. Chem. C, 2017, 5, 5111–5118 RSC.
- Y. J. Cheng, J. D. Luo, S. Hau, D. H. Bale, T. D. Kim, Z. W. Shi, D. B. Lao, N. M. Tucker, Y. Q. Tian, L. R. Dalton, P. J. Reid and A. K. Y. Jen, Large electro-optic activity and enhanced thermal stability from diarylaminophenyl-containing high-beta nonlinear optical chromophores, Chem. Mater., 2007, 19, 1154–1163 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul; S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford, CT, 2004.
- R. F. W. Bader and C. F. Matta, Atoms in molecules as non-overlapping, bounded, space-filling open quantum systems, Found. Chem., 2012, 15, 253–276 CrossRef.
- R. F. W. Bader, A. Larouche, C. Gatti, M. T. Carroll, P. J. MacDougall and K. B. Wiberg, Properties of atoms in molecules: Dipole moments and transferability of properties, J. Chem. Phys., 1987, 87, 1142–1152 CrossRef CAS.
- A. E. de Oliveira, R. L. A. Haiduke and R. E. Bruns, Atomic mean dipole moment derivatives and GAPT charges, J. Phys. Chem. A, 2000, 104, 5320–5327 CrossRef CAS.
- C. C. Teng and H. T. Man, Simple Reflection Technique for Measuring the Electrooptic Coefficient of Poled Polymers, Appl. Phys. Lett., 1990, 56, 1734–1736 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, DSC and DFT results. See DOI: 10.1039/c8qm00006a |
| This journal is © the Partner Organisations 2018 |