
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli-responsive protection of optically excited triplet ensembles against deactivation by molecular oxygen†
N. V.
Nazarova
 a,
Yu. S.
Avlasevich
a,
K.
Landfester
a and
S.
Baluschev
*ab
a,
Yu. S.
Avlasevich
a,
K.
Landfester
a and
S.
Baluschev
*ab
aMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: balouche@mpip-mainz.mpg.de
bOptics and Spectroscopy Department, Faculty of Physics, Sofia University, “St. Kliment Ochridski”, 5 James Bourchier, 1164 Sofia, Bulgaria
First published on 23rd January 2018
Abstract
Herein we demonstrate temperature-dependent sacrificial singlet oxygen scavenging properties of N-butyl-2-pyridone, ensuring efficient stimuli-responsive protection of densely populated excited triplet state ensembles against deactivation by molecular oxygen. As an acting external stimulus the temperature was chosen: it will be shown that at low temperature the concentration of singlet oxygen will be substantially lowered; in contrast, at elevated temperatures singlet oxygen will not be captured, and thus the optically excited densely populated triplet ensembles will be effectively depopulated. The singlet oxygen scavenging ability of N-butyl-2-pyridone demonstrates long-term protection of a triplet–triplet annihilation upconversion process against photooxidation.
Introduction
The triplet–triplet annihilation photon energy upconversion process (TTA-UC) relies on optically created densely populated organic triplet ensembles, in which the inter-molecular energy transfer depends strongly on a variety of environmental parameters, such as temperature or contamination with molecular oxygen. The TTA-UC takes place in multi-chromophore systems consisting of energetically optimized pairs of emitter molecules (typically aromatic hydrocarbons) and sensitizer molecules (metallated macrocycles,1 such as porphyrins and phthalocyanines).Metallated porphyrins are widely used for thermal- or oxygen sensing,2 where the dependence of integral phosphorescence emission or the decay time of the phosphorescence is registered. These sensing processes involve optical excitation of the metallated macrocycle molecule and registration of the decreased phosphorescence or phosphorescence decay time as a function of the increased sample temperature or oxygen content.3
Oxygen sensors for detecting extremely low levels of oxygen i.e. physiologically relevant O2 concentrations (ranging from nM–200 μM) deserve special attention.4 For example, one can mention the transient state imaging technique,5 single-nanoparticles with a ratiometric O2-response6 or phosphorescent metalloporphyrins encapsulated in hydrophobic dendrimers.7,8 The main experimental drawback of these sensing techniques is that phosphorescence emission is an integral parameter, which depends simultaneously on the local temperature and local oxygen contamination.
The triplet–triplet annihilation photon energy upconversion process represents a thorough solution for the problem of oxygen sensing: instead of a single material response on the acting parameter (variation of the oxygen concentration), the TTA-UC provides a ratiometric material response. The use of the mutual dependence of the residual sensitizer phosphorescence and emitter delayed fluorescence on the acting parameter ensures inherent/instantaneous compensation regarding other unwanted local changes in the sample parameters.9 Another advantage of the TTA-UC as a sensing technique is its extremely low excitation intensity (less than 1–10 mW cm−2): efficient TTA-UC was observed even with non-coherent light (sunlight10). Application potential of the TTA-UC process ranges from the fields of molecular sensing11 and bioimaging,12 solid state optical technologies13 up to renewable energy sources.14
A simplified representation of the TTA-UC process is shown in Fig. 1. The chemical structures of the used sensitizer – meso-tetraphenyl-tetrabenzo[2,3]porphyrin palladium(II) (PdTBP) and the emitter – 3,9(10)-bis(3,3-dimethylbutyn-1-yl)perylene (BDP) are shown as insets in Fig. 1. The photon energy is absorbed by the sensitizer (the green arrow, Fig. 1). The long-lived triplet state of the sensitizer is formed due to the efficient inter-system crossing (ISC) enhanced by the spin–orbital coupling of the heavy metal centre. Furthermore, this energy is transferred to an emitter triplet state via the triplet–triplet transfer (TTT) process. Then two excited emitter molecules undergo triplet–triplet annihilation (TTA), in which one emitter molecule returns to its singlet ground state and the other molecule gains the energy of both triplet states and is excited to the higher singlet state. As the emitter singlet state decays radiatively back to the ground state, a delayed fluorescence photon (dark blue line, Fig. 1) bearing higher energy than that of the excitation photons is emitted. It is important to note that the sensitizer excited triplet state is not completely depopulated via the TTT process; therefore residual sensitizer phosphorescence is observed too (Fig. 1, red arrow).
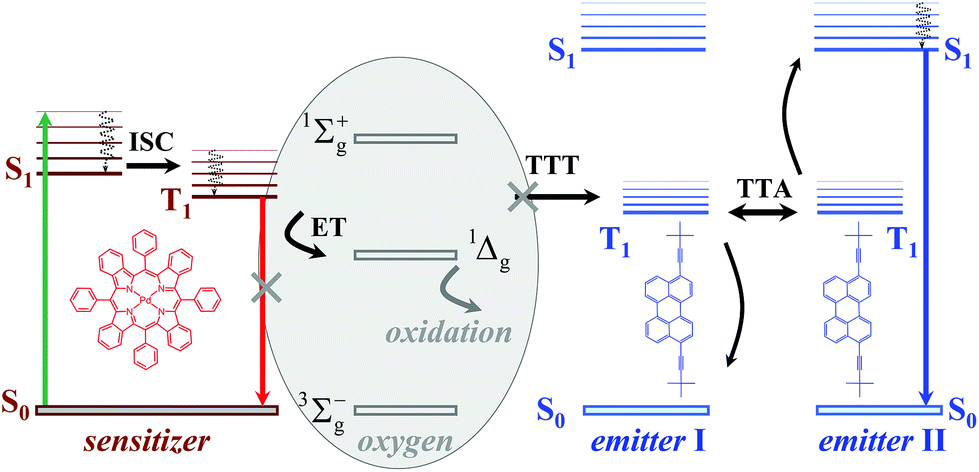 | ||
| Fig. 1 Simplified energetic scheme of the triplet–triplet annihilation upconversion process in an oxygen contaminated environment, containing singlet oxygen scavenging moieties. Insets: Structure of the sensitizer (PdTBP) and emitter (BDP); SOS – singlet oxygen scavenger. | ||
The above described excitation energy transfers occur in an oxygen-free environment. In an environment contaminated with molecular oxygen15 even at the ppm-level, the TTA-UC process becomes more complicated. In the presence of molecular oxygen, the energy stored in the excited states of the triplet ensembles is actively dissipated, competing with emissive (phosphorescence) or non-emissive (triplet to triplet) energy transfer processes. The reason for this is the process of energy transfer between the excited organic triplet state and the ground state of molecular oxygen, followed by the generation of singlet oxygen (1Δg, Fig. 1). Singlet oxygen is a highly reactive species, leading to the oxidation of the photoactive molecules, followed by further loss of efficiency. An extensive review16 describing the oxygen protection strategies of organic ensembles, undergoing triplet–triplet annihilation, has been recently published. Earlier reversible photoswitching and chemical-responsive TTA-UC were demonstrated.17
Herein the sensing portfolio of the TTA-UC process will be enriched – we demonstrate for the first time stimuli responsive protection of the upconverting organic ensemble. As an acting external stimulus we chose temperature: it will be shown that at low temperature the concentration of singlet oxygen will be substantially lowered; in contrast, at elevated temperatures the singlet oxygen will be not captured; thus the optically excited densely populated triplet ensembles will be effectively depopulated.
Polycyclic aromatic hydrocarbons are able to bind singlet oxygen forming the corresponding endoperoxides (EPOs).18 Recently, reversible singlet oxygen binding for optical and photo-medical applications was reviewed.19 The endoperoxide formation is strongly temperature dependent. Furthermore, some EPOs exhibit the exceptional feature of releasing oxygen, frequently in the excited singlet state,20 under heating21 or UV irradiation.18 Thus the endoperoxide formation allows for keeping the singlet O2 concentration in the UC-ensemble at a certain level, co-regulated by the sample temperature. The fractional photodynamic therapy22 needs a sensitive technology, measuring the concentration of the singlet oxygen released during the “dark”-periods in an all-optical manner and under a change in the sample temperature: since the TTA-UC process demonstrates the necessary sensitivity and after the corresponding calibration, the TTA-UC can be used as such a sensing tool. The reported stimuli-responsive UC-process could be used as real-time control of the concentration of singlet oxygen, generated during the post-illumination period for the purpose of fractional photodynamic therapy.22 Even more, the proposed TTA-UC process is much more sensitive to the presence of singlet oxygen than the commonly used sensing process – phosphorescence.
The mechanism of the cycloaddition of 1O2 to the aromatic compounds is very similar to the classical Diels Alder type reactions. The reactivity of aromatic hydrocarbons toward 1O2 increases with the electron density of the aromatic substrate, reflecting the electrophilic nature of 1O2.18 The easiness of oxygen release from these systems depends on the polycyclic aromatic system and the nature of the substituents at the meso-positions.20
As the next step, it will be shown that N-butyl-2-pyridone (NBP) is capable of binding 1O2 dissolved in an organic solvent in a temperature dependent manner. 2-Pyridone itself exists in two forms; the proton bound to nitrogen can migrate to oxygen, giving the second tautomer, 2-hydroxypyridine.23 The first form can be stabilized by N-alkylation; a long alkyl chain in such compounds facilitates their miscibility with nonpolar organic solvents. N-Alkyl-2-pyridones represent a stable diene structure, which can undergo [4 + 2]-cycloaddition (Diels–Alder reaction) with different dienophiles, like alkenes24 or molecular oxygen in the singlet excited state.25,26 It is known that 2-pyridones bind singlet oxygen efficiently at lower temperatures and release it upon heating.25 Therefore, 2-pyridones were used for catalytic purposes,25,26 as well as prominent biological antifungal and antiviral agents.27
Irradiation of palladium benzoporphyrin (PdTBP) dissolved in a mixture of toluene (80% vol) and NBP solution (20% vol) leads to the formation of N-butyl-2-pyridone EPO. The reaction scheme is shown in Fig. 2. This reaction is strongly temperature dependent; therefore the endoperoxides decompose thermally and regenerate oxygen (probably in a singlet state, we did not study this process explicitly) and the parent hydrocarbon. The lifetime of the EPO depends strongly on the temperature.19 At low temperature singlet oxygen can be stored for prolonged periods of time and then released by decomposition upon heating; thus the binding–release cycle can be repeated.
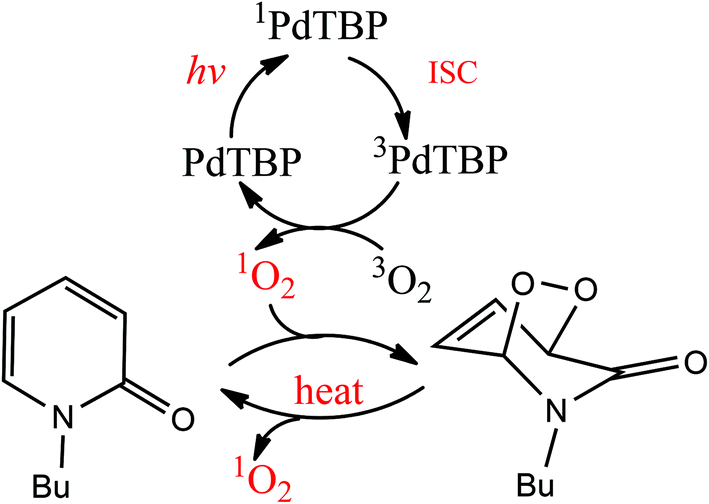 | ||
| Fig. 2 Reversible addition of singlet oxygen to N-butyl-2-pyridone. 1PdTBP represents the first excited singlet state of the sensitizer (corresponds to excitation at the Q-band); 3PdTBP represents the first excited triplet state of the sensitizer. | ||
Experimental
Materials
The sensitizer – meso-tetraphenyl-tetrabenzo[2,3]porphyrin palladium(II) (PdTBP)12 and the emitter – 3,10-bis(3,3-dimethylbutyn-1-yl)perylene (BDP)28 were synthesized as described elsewhere. N-Butyl-2-pyridone was synthesized by alkylation of 2-hydroxypyridine with 1-bromobutane29 and purified via vacuum distillation.For the samples prepared in a glove-box commercial solvents (ultra-dry, packed under argon, septum sealed) were used as received. NBP solutions were placed into a glove-box (4 ppm of O2) and stirred in an open bottle for 2 h; thus the residual oxygen contamination at the NBP corresponds to the glove-box oxygen content. Sample solutions were prepared and sealed in the glove-box. For all optical measurements, the molar concentrations of the sensitizer (PdTBP) and emitter (BDP) were the same, 1 × 10−5 M/2 × 10−4 M, respectively.
Method
Luminescence spectra were recorded by a home-made setup. Typically, sample was located in a temperature-controlled cuvette holder. The cuvette holder temperature was controlled with a Peltier element by means of the T-app computer program from Electron Dynamics Ltd (Southampton, UK). The sample temperature additionally was measured through a thermistor (PT100) attached on the top of the cuvette. The sample was excited with wavelength λexc = 633 nm light beam from a HeNe laser. The excitation power was controlled using a power meter PM 100D (Thorlabs, USA). For regular measurements, the excitation intensity was set to 12 mW cm−2 and the laser spot diameter was d = 2 × 10−3 m.Results
Active protection of the densely populated triplet ensembles from deactivation by molecular oxygen is based on the application of oxygen scavenging species which react with singlet oxygen to minimize the amount of oxygen available for deteriorative reactions, leading to the degradation of the photoactive molecules.16 An evident drawback of this method is the possible unwanted interaction between the scavenger and the UC-active moieties. Therefore, to start with the influence of the NBP on the efficiency of the TTA-UC process was studied. The luminescence spectra of the studied UC-couple under glove-box conditions were compared (Fig. 3). Experimental conditions: samples prepared and sealed in a nitrogen filled glove-box (4 ppm O2); temperature – 25 °C; excitation intensity – 12 mW cm−2; HeNe laser; concentrations of the sensitizer (PdTBP) and emitter (BDP), 1 × 10−5 M/2 × 10−4 M, respectively.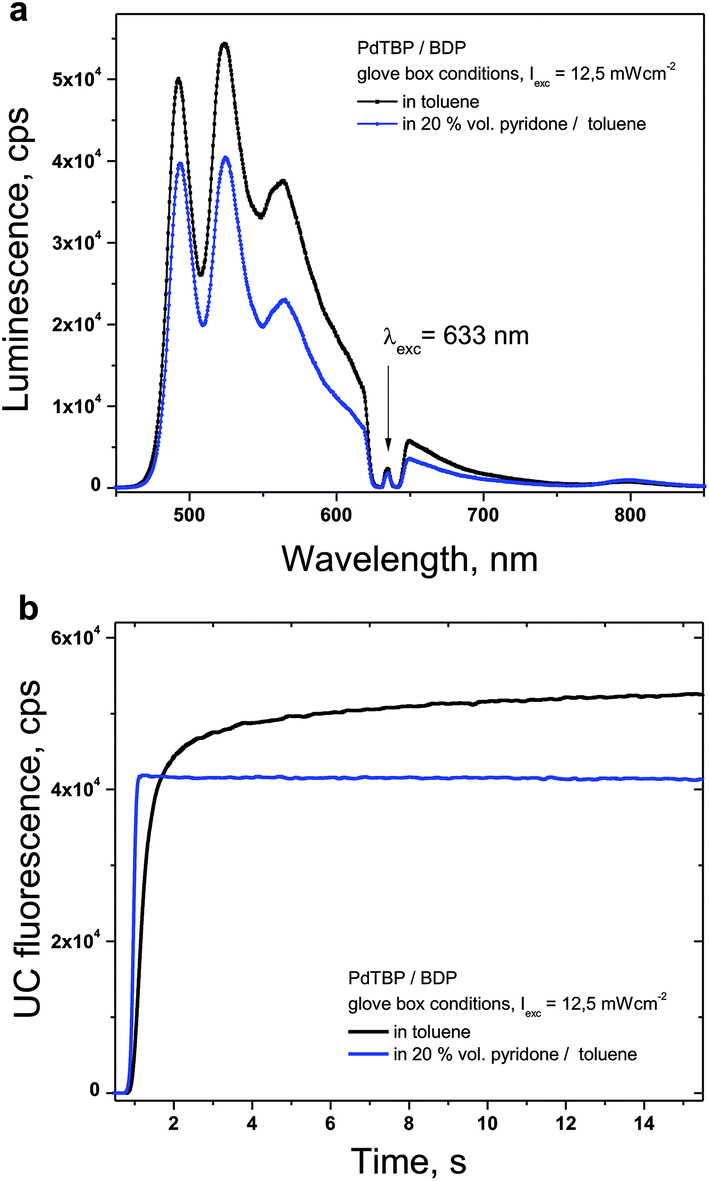 | ||
| Fig. 3 (a) Luminescence spectra for the samples containing the UC-couple PdTBP/BDP, dissolved in toluene (black line); in a mixture of N-butyl-2-pyridone/toluene (blue line). (b) Temporal dependences of the UC-signal at the fluorescence maximum (λ = 526 nm). | ||
As shown in Fig. 3a, the steady-state UC-signal in a solvent mixture containing 20% vol NBP is slightly lower (about 15% less efficiency) than the UC-signal in toluene. The temporal dependence of the UC-signal for the pure toluene sample shows typical behaviour (Fig. 3b, black line) for the UC-samples in an environment with residual oxygen contamination (4 ppm O2): the UC-fluorescence rises during the first 10 seconds of optical excitation, until the complete oxygen content is consumed (for instance, by oxidation of the BDP emitter molecule30). In contrast, the UC-sample containing 20% vol NBP demonstrates fast growth (the integration time of the registration device is set to 100 ms) because the concentration of the oxygen dissolved in the toluene/NBP solution is very low.
Temperature effect
The densely populated organic triplet ensemble participating in the TTA-UC process is created by optical excitation performed in the continuous wave (cw) regime. Therefore the delayed fluorescence of the emitter molecule (the UC-signal) – the emission with a central wavelength around 526 nm (Fig. 4) and the residual phosphorescence of the sensitizer – the emission with a central wavelength around 800 nm (Fig. 4) can be observed simultaneously. In the environment contaminated with molecular oxygen an additional lost channel is present – it is the generation of singlet oxygen (Fig. 2) leading to the modulation and loss of efficiency of both optical signals – delayed fluorescence as well the residual phosphorescence. If a sacrificial singlet oxygen scavenger such as NBP is present, it is possible to bind a large amount of the present molecular oxygen, thus excluding it as a loss mechanism for the TTA-UC and phosphorescence processes. The usage of N-butyl-2-pyridone allows implementation of an additional acting parameter – since the probability of the creation of EPOs is temperature dependent, the rate in which the local amount of molecular oxygen will be bound will also be temperature dependent. In Fig. 4 the temperature dependence of the UC-fluorescence and the residual phosphorescence for samples protected by NBP is demonstrated. Experimental conditions: UC-couple PdTBP/BDP, dissolved in a mixture of 20% vol N-butyl-2-pyridone/toluene; excitation intensity – 12 mW cm−2; HeNe laser; concentration of the sensitizer (PdTBP) and emitter (BDP), 1 × 10−5 M/2 × 10−4 M, respectively; ambient environment (oxygen saturated solvents). The sample is continuously illuminated; the luminescence spectra were obtained at the 100th second after the start. For each temperature measurement a new sample point, with a large lateral displacement, was chosen.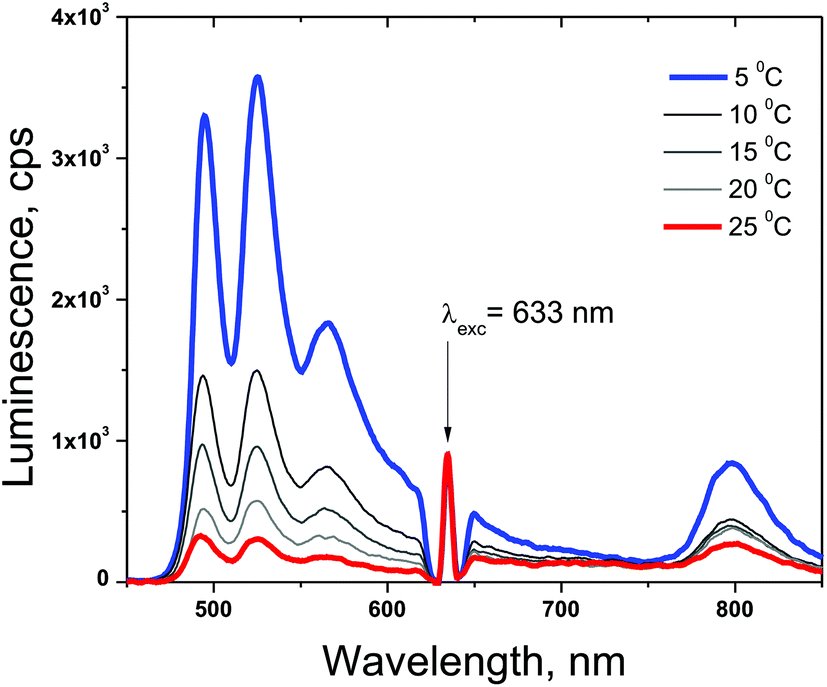 | ||
| Fig. 4 Dependence of the delayed emitter fluorescence and residual sensitizer phosphorescence on the sample temperature. UC-couple PdTBP/BDP, dissolved in a mixture of 20% vol N-butyl-2-pyridone/toluene. | ||
In Fig. 5 the temporal dependences of the UC-signal and residual sensitizer phosphorescence in an ambient environment for different sample temperatures are shown. It is clearly demonstrated that the ability of NBP to bind singlet oxygen decreases with a rise in the sample temperature. It is also evident that the delayed emitter fluorescence is much more sensitive to the local changes in the oxygen concentration than the residual phosphorescence: if the sample temperature is lower, this leads to an increase probability to bind and trap singlet oxygen, which cause a local decrease in the molecular oxygen concentration.
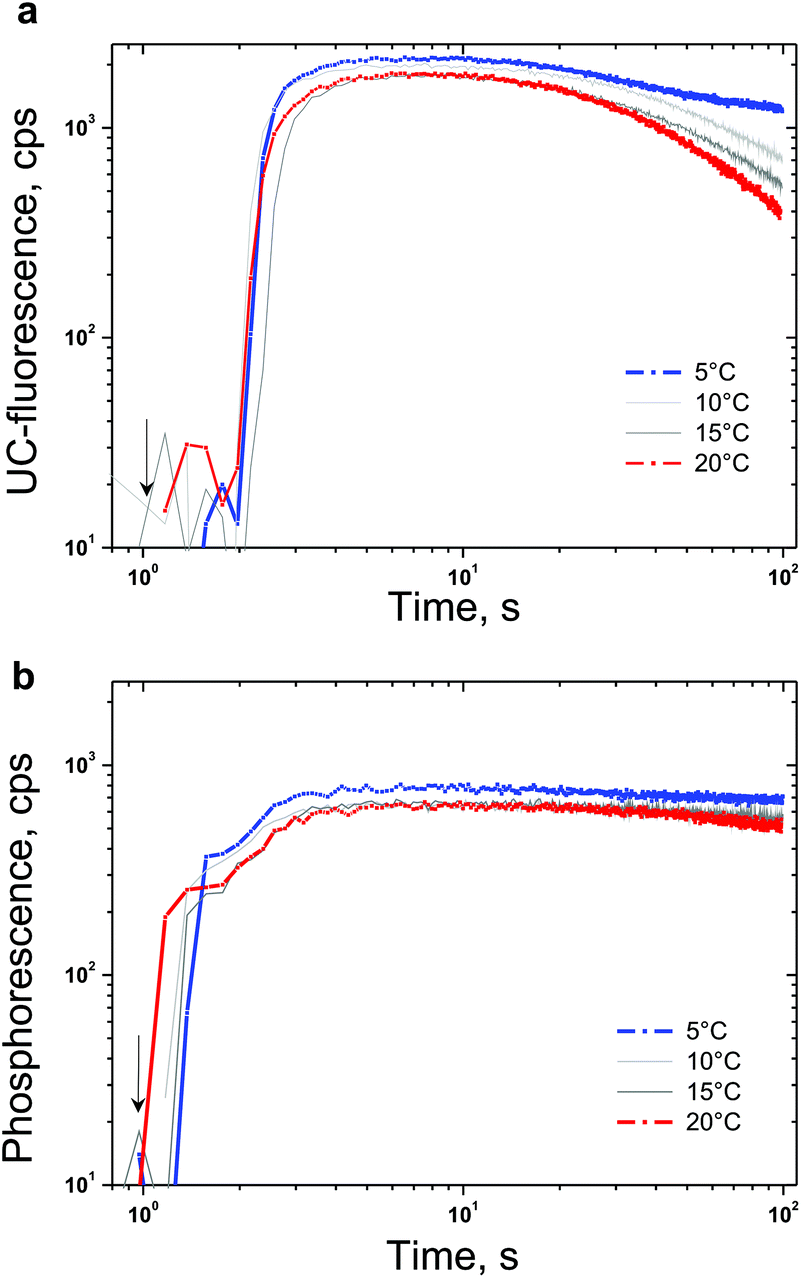 | ||
| Fig. 5 (a) Temporal dependence of the UC-signal at the fluorescence maximum (λ = 526 nm) for different sample temperatures. (b) Temporal dependence of the residual sensitizer phosphorescence at the maximum (λ = 800 nm) for different sample temperatures. Experimental conditions: the same as in Fig. 4. The start moment of optical excitation is marked by an arrow. | ||
Despite the unique property to deliver temperature dependent protection of the excited triplet ensembles against singlet oxygen damage, NBP also demonstrates constraint potential to bind large amounts of singlet oxygen. Fig. 5 reveals the efficiency limitations of the NBP as a sacrificial singlet oxygen scavenger – even at a relatively large concentration (20% vol), the total amount of the bound molecular oxygen can be estimated to be at least 200 μM (saturation concentration of the oxygen in toluene31). Furthermore, in a bulk sample, after binding the present local amount of molecular oxygen (coinciding with the optically accessed spot) a strong increase in the UC-fluorescence is observed, since the optically accessed spot is a subject of increased molecular oxygen diffusion (from the non-excited regions) the newly arrived molecular oxygen will not be bound and a gradual loss of efficiency is observed first for UC-fluorescence, and afterwards for sensitizer residual phosphorescence (Fig. 5a and b, large observation times).
Conclusions
The temperature-dependent sacrificial singlet oxygen scavenging ability of N-butyl-2-pyridone, ensuring efficient stimuli-responsive protection of densely populated excited triplet state ensembles against deactivation by molecular oxygen, was demonstrated. As an acting external stimulus the temperature was chosen: it was shown that at low temperature the concentration of singlet oxygen will be substantially lowered; in contrast, at elevated temperatures singlet oxygen will be not captured; thus the optically excited densely populated triplet ensembles will be effectively depopulated. The singlet oxygen scavenging ability of N-butyl-2-pyridone demonstrates long-term protection of a triplet–triplet annihilation upconversion process against photooxidation. Additionally, the reported stimuli-responsive UC-process could be used as a real-time control for the concentration of singlet oxygen, generated during the post-illumination period for the purpose of fractional photodynamic therapy.Conflicts of interest
The authors declare no competing financial interest or other potential conflict of interest.Acknowledgements
This work is performed under European Horizon 2020 research and innovation programme under grant agreement no. 732794 – project HYPOSENS. S. Baluschev also acknowledges the DFNI E 02/11 – SunStore of the Bulgarian National Science Fund. Open Access funding provided by the Max Planck Society.Notes and references
- Y. Y. Cheng, B. Fueckel, T. Khoury, R. G. C. R. Clady, M. J. Y. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, J. Phys. Chem. Lett., 2010, 1, 1795–1799 CrossRef CAS; S. K. Sugunan, C. Greenwald, M. F. Paige and R. P. Steer, J. Phys. Chem. A, 2013, 117, 5419–5427 CrossRef PubMed; X. Cao, B. Hu and P. Zhang, J. Phys. Chem. Lett., 2013, 4, 2334–2338 CrossRef; J. S. Lissau, D. Nauroozi, M. P. Santoni, S. Ott, J. M. Gardner and A. Morandeira, J. Phys. Chem. C, 2013, 117, 14493–14501 Search PubMed.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- X. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- A. Fercher, S. M. Borisov, A. V. Zhdanov, I. Klimant and D. B. Papkovsky, ACS Nano, 2011, 5, 5499–5508 CrossRef CAS PubMed.
- T. Spielmann, L. Xu, A. K. B. Gad, S. Johansson and J. Widengren, FEBS J., 2014, 281, 1317–1332 CrossRef CAS PubMed.
- C. Wu, B. Bull, K. Christensen and J. McNeill, Angew. Chem., Int. Ed., 2009, 48, 2741–2745 CrossRef CAS PubMed.
- T. V. Esipova, A. Karagodov, J. Miller, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Anal. Chem., 2011, 83, 8756–8765 CrossRef CAS PubMed.
- J. A. Spencer, F. Ferraro, E. Roussakis, A. Klein, J. Wu, J. M. Runnels, W. Zaher, L. J. Mortensen, C. Alt, R. Turcotte, R. Yusuf, S. A. Vinogradov, D. T. Scadden and C. P. Lin, Nature, 2014, 508, 269–273 CrossRef CAS PubMed.
- K. Landfester, A. Svagan, J. Risbo and S. Balouchev, WO2015/059179A1, EP 2865443A1, 2015 Search PubMed; K. Landfester, M. Filatov, Yu. Avlasevich, D. Busko, A. Turshatov, F. Wurm, F. Marsico and S. Balouchev, EP2851407A1, WO 2015/044129A1, 2015 Search PubMed.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed; S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Mullen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693–7696 CrossRef PubMed; S. Hoseinkhani, R. Tubino, F. Meinardi and A. Monguzzi, Phys. Chem. Chem. Phys., 2015, 17, 4020–4024 RSC.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- S. H. C. Askes, N. López Mora, R. Harkes, R. I. Koning, B. Koster, T. Schmidt, A. Kros and S. Bonnet, Chem. Commun., 2015, 51, 9137–9140 RSC; Ch. Wohnhaas, V. Mailänder, M. Dröge, M. A. Filatov, D. Busko, Y. Avlasevich, S. Baluschev, T. Miteva, K. Landfester and A. Turshatov, Macromol. Biosci., 2013, 13, 1422–1430 CrossRef CAS PubMed; J. H. Kim and J. H. Kim, J. Am. Chem. Soc., 2012, 134, 17478–17481 CrossRef PubMed; Q. Liu, T. S. Yang, W. Feng and F. Y. Li, J. Am. Chem. Soc., 2012, 134, 5390–5397 CrossRef PubMed; C. Wohnhaas, A. Turshatov, V. Mailander, S. Lorenz, S. Baluschev, T. Miteva and K. Landfester, Macromol. Biosci., 2011, 11, 772–778 CrossRef PubMed; S. M. Borisov, R. Saf, R. Fischer and I. Klimant, Inorg. Chem., 2013, 52, 1206–1216 CrossRef PubMed; G. Massaro, J. Hernando, D. Ruiz-Molina, C. Roscini and L. Latterini, Chem. Mater., 2016, 28, 738–745 CrossRef.
- K. Börjesson, P. Rudquist, V. Gray and K. Moth-Poulsen, Nat. Commun., 2016, 7, 12689 CrossRef CAS PubMed; Z. Huang, X. Li, M. Mahboub, K. M. Hanson, V. M. Nichols, H. Le, M. L. Tang and C. J. Bardeen, Nano Lett., 2015, 15, 5552–5557 CrossRef PubMed; C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef PubMed.
- T. F. Schulze, J. Czolk, Y. Y. Cheng, B. Fuckel, R. W. MacQueen, T. Khoury, M. J. Crossley, B. Stannowski, K. Lips and U. Lemmer, J. Phys. Chem. C, 2012, 116, 22794–22801 Search PubMed; W. Q. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560–564 CrossRef CAS; P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef PubMed; T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 Search PubMed; R. E. Keivanidis, S. Baluschev, G. Lieser and G. Wegner, ChemPhysChem, 2009, 10, 2316–2326 CrossRef PubMed.
- M. I. J. Lorenz, S. H. Lorenz and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102 RSC.
- M. A. Filatov, S. Baluschev and K. Landfester, Chem. Soc. Rev., 2016, 45, 4631–4852 RSC.
- X. Cui, J. Zhao, Y. Zhou, J. Ma and Y. Zhao, J. Am. Chem. Soc., 2014, 136(26), 9256–9259 CrossRef CAS PubMed; R. Tao, J. Zhao, F. Zhong, C. Zhang, W. Yang and K. Xu, Chem. Commun., 2015, 51, 12403–12406 RSC.
- J. M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS PubMed.
- M. A. Filatov and M. O. Senge, Mol. Syst. Des. Eng., 2016, 1, 219–346 Search PubMed.
- H. H. Wasserman and J. R. Scheffer, J. Am. Chem. Soc., 1967, 89, 3073–3075 CrossRef CAS PubMed.
- M. A. Filatov, E. Heinrich, D. Busko, I. Z. Ilieva, K. Landfester and S. Baluschev, Phys. Chem. Chem. Phys., 2015, 17, 6501–6510 RSC.
- I. S. Turan, D. Yildiz, A. Turksoy, G. Gunaydin and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 2875–2878 CrossRef CAS PubMed; S. Kolemen, T. Ozdemir, D. Lee, G. M. Kim, T. Karatas, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 3606–3610 CrossRef PubMed; A. M. Asadirad, Z. Arno and N. R. Branda, Chem. Commun., 2013, 49, 5639–5641 RSC.
- S. Mata, V. Cortijo, W. Caminati, J. L. Alonso, M. Sanz, J. C. Lopez and S. Blanco, J. Phys. Chem. A, 2010, 114, 11393–11398 CrossRef CAS PubMed.
- H. Tomisawa, H. Hongo, T. Hatano, H. Nakano and R. Fujita, Chem. Pharm. Bull., 1987, 35, 530–534 CrossRef CAS.
- S. Benz, S. Nötzli, J. S. Siegel, D. Eberli and H. J. Jessen, J. Med. Chem., 2013, 56, 10171–10182 CrossRef CAS PubMed.
- C. Changtong, D. W. Carney, L. Luo, C. A. Zoto, J. L. Lombardi and R. E. Connors, J. Photochem. Photobiol., A, 2013, 260, 9–13 CrossRef CAS.
- D. Saini, N. K. Rangra and P. C. Behra, Exploring the Biological Potential of Pyridones, Lambert Academic Publishing, 2015, ISBN-13:978-3-659-39505-5 Search PubMed.
- A. J. Svagan, D. Busko, Yu. Avlasevich, G. Glasser, S. Baluschev and K. Landfester, ACS Nano, 2014, 8, 8198–8820 CrossRef CAS PubMed.
- S. McN. Sieburth, C. H. Lin and D. Rucando, J. Org. Chem., 1999, 64, 950–953 CrossRef CAS PubMed.
- K. Katta, D. Busko, Yu. Avlasevich, R. Muñoz-Espí, S. Baluschev and K. Landfester, Macromol. Rapid Commun., 2015, 36, 1084–1088 CrossRef CAS PubMed.
- Z. A. Li, S. Tang, P. Tan, C. Liu and B. Liang, J. Chem. Eng. Data, 2007, 52, 2339–2344 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt03698a |
| This journal is © The Royal Society of Chemistry 2018 |