
Enantioselective total synthesis of sagittacin E and related natural products†
Hideki
Abe
 *ab,
Mitsuru
Fujimaki
a,
Eri
Nakagawa
a,
Toyoharu
Kobayashi
a and
Hisanaka
Ito
*a
*ab,
Mitsuru
Fujimaki
a,
Eri
Nakagawa
a,
Toyoharu
Kobayashi
a and
Hisanaka
Ito
*a
aSchool of Life Sciences, Tokyo University of Pharmacy and Life Sciences, 1432-1 Horinouchi, Hachioji, Tokyo 192-0392, Japan
bDepartment of Chemical and Biological Sciences, Faculty of Science, Japan Women's University, 2-8-1 Mejirodai, Bunkyo-ku, Tokyo 112-8681, Japan
First published on 29th May 2018
Abstract
The first enantioselective total synthesis of eremophilane-type sesquiterpenoids, sagittacin E and related natural products, was achieved. This synthesis features an asymmetric desymmetrization by Shi asymmetric epoxidation, intramolecular aldol-type cyclization, allylic oxidation of a 1,4-diene compound, and stereoselective epoxidation.
The genus Ligularia is an important member of the family Compositae, which is a rich source of biologically active natural products. This genus produces mainly eremophilane-type sesquiterpenoids and pyrrolizidine alkaloids as secondary metabolites. Sagittacin E (1), isolated from Ligularia sagitta by Gao and co-workers in 2014, is a highly oxygenated eremophilane-type sesquiterpenoid that possesses mild cytotoxic activities against three human tumor cell lines, HL-60, SMMC-7721, and HeLa cells, and moderates antibacterial activities against E. coli and E. carotovora (Fig. 1).1 Three structurally similar sesquiterpenoids 2–4 were isolated from Ligularia sagitta,1,2Ligularia veitchana,2 and Senecio nemorensis.3 Although the antibacterial activity of 3 against E. coli was reported, biological tests of 2 and 4 have not yet been performed. These four eremophilane-type natural products have very simple bicyclic skeletons, but highly oxygenated frameworks. Consequently, there are few total syntheses or synthetic studies on them to date. Very recently, Liu reported the efficient total synthesis of five eremophilane-type natural products in racemic form, including 3 and 4, by using Robinson annulation and Suzuki coupling.4
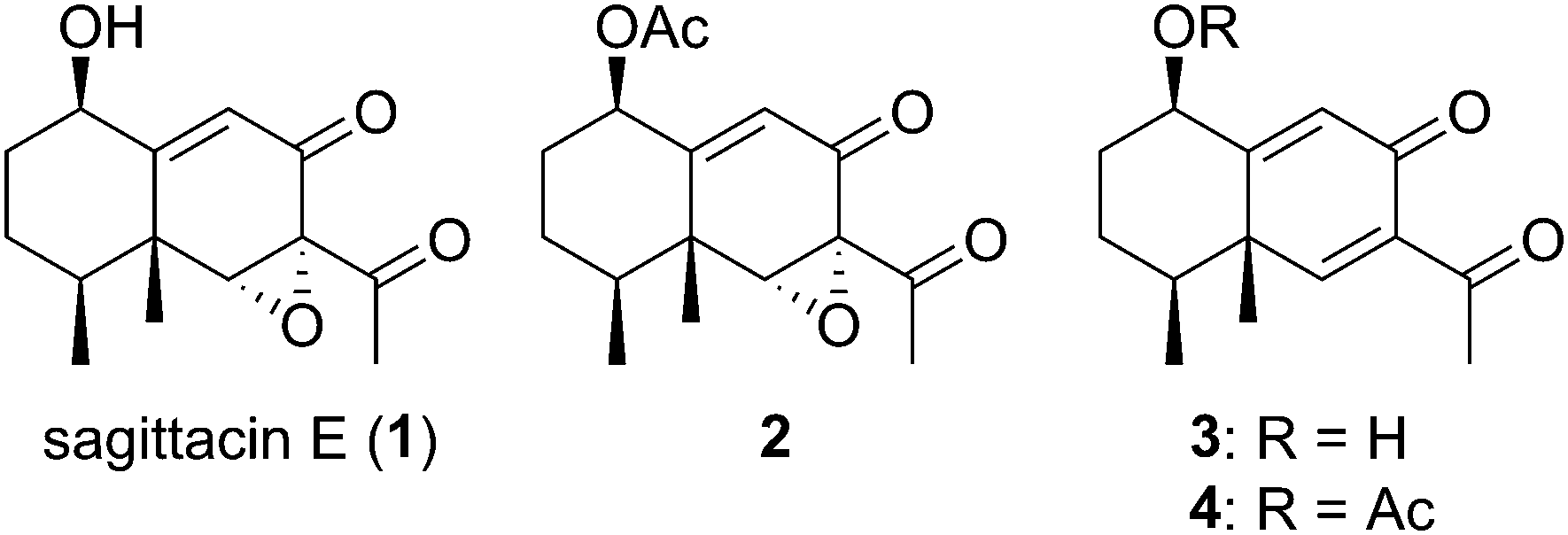 | ||
| Fig. 1 Structures of sagittacin E (1) and related natural products 2–4. | ||
We previously reported an asymmetric synthetic study of briarane-type diterpenoid pachyclavulide B using asymmetric epoxidation.5 Our synthetic strategy featured desymmetrization of symmetric 1,4-cyclohexadiene derivatives by Shi asymmetric epoxidation. This desymmetrization technique is useful for the asymmetric total syntheses of natural products. Thus, we planned the asymmetric total synthesis of highly oxygenated eremophilane-type natural product sagittacin E and related compounds based on our developed desymmetrization reaction.
Herein, we describe the enantioselective total synthesis of eremophilane-type sesquiterpenoids, sagittacin E and structurally similar natural products, based on our asymmetric desymmetrization strategy.
Our synthetic strategy for the target natural products is outlined in Scheme 1. Sagittacin E (1) would easily be obtained by deacetylation of 2. Likewise, 3 would be obtained by deacetylation of 4. The acetyl sagittacin E (2) would be synthesized from the bicyclic diene 9via allylic oxidation and stereoselective epoxidation. On the other hand, natural compound 4 would be derived from 9 only by allylic oxidation. The common intermediate 9 would be constructed by intramolecular aldol type condensation of nitrile 7 with a tethered aldehyde, followed by transformation of the nitrile group of the resulting bicyclic compound 8 to a methyl ketone. The precursor 7 would be synthesized in a multi-step operation from the optically active epoxide 6, which would be obtained by asymmetric desymmetrization of 1,1,2,6-tetrasubstituted cyclohexadiene 5.
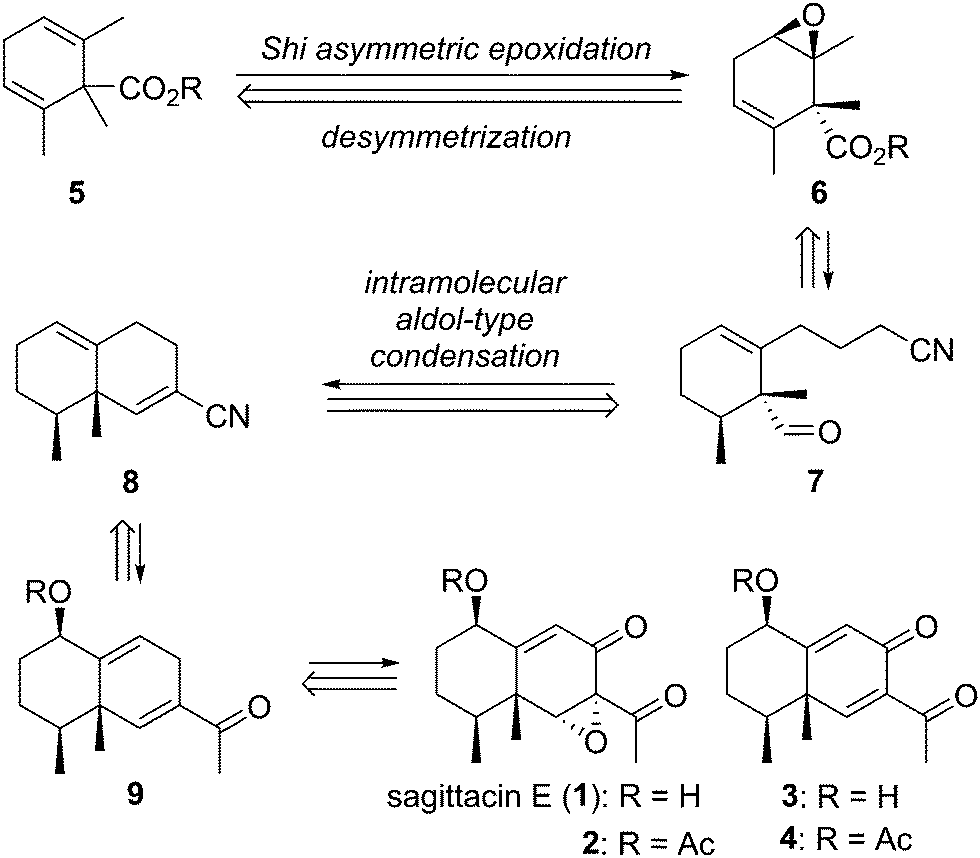 | ||
| Scheme 1 Synthetic plan for sagittacin E (1) and related compounds. | ||
Our synthetic project started with the construction of an asymmetric carbon via asymmetric desymmetrization with the Shi epoxidation protocol as shown in Scheme 2. Symmetric 1,4-diene derivative 12 having a quaternary carbon atom was synthesized from 2,6-dimethylbenzoic acid in a two-step procedure, i.e., reductive methylation of 10 under Birch reduction conditions (78% yield), followed by esterification of the resulting symmetric diene derivative 11 (99% yield). Shi asymmetric epoxidation6,7 of 12 with Shi ketone 13 prepared from D-fructose, and Oxone® as the oxidant afforded the desymmetrized epoxide 14 in 49% yield, 89% de, and 85% ee.8 Reduction of the benzyl ester of 14 with DIBALH at −78 °C gave the alcohol 15 in 94% yield as a crystalline compound. After recrystallization, the primary alcohol 15 was obtained in enantiomerically pure form (99% ee).9
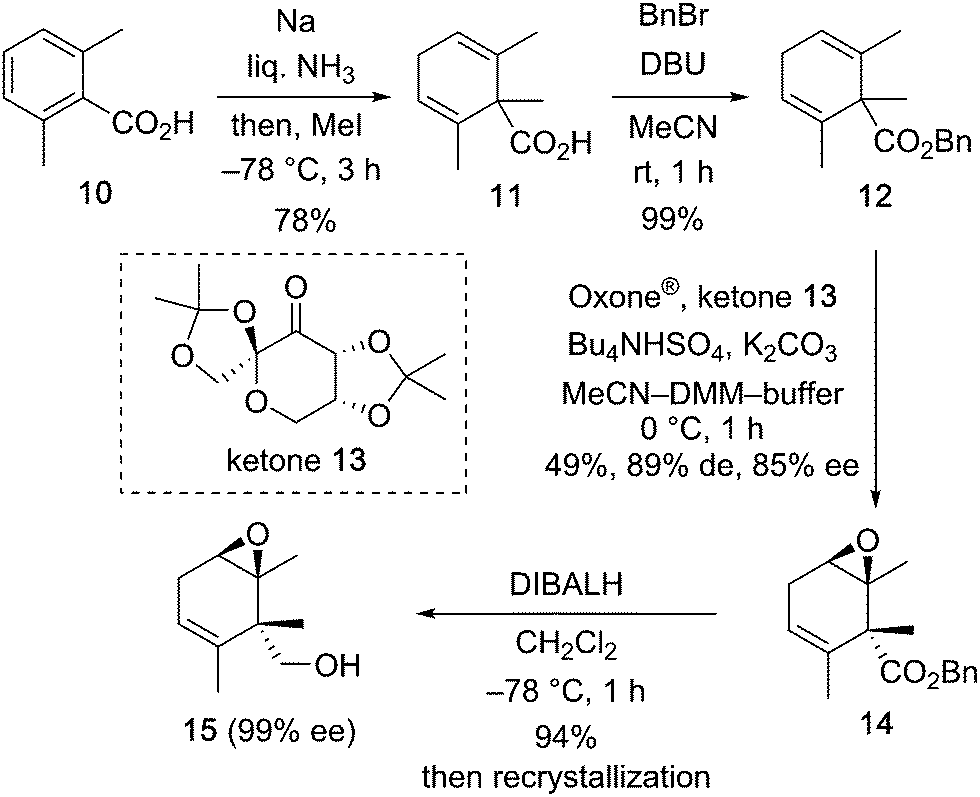 | ||
| Scheme 2 Asymmetric desymmetrization of the symmetric 1,4-diene derivative 12. | ||
With the desymmetrization of the symmetric 1,4-diene derivative achieved by Shi asymmetric epoxidation, we focused our efforts on the construction of the eremophilane skeleton (Scheme 3). After many attempts to hydrogenate the double bond without opening the epoxide of 15, the use of platinum on carbon as a heterogeneous catalyst in AcOEt under hydrogen gave the best result to afford hydrogenated compound 16 in 75% yield. The hydrogenation of 15 with platinum catalyst proceeded from the same side of the primary alcohol, thus the relative configuration of the two vicinal methyl groups was syn. Regioselective epoxide ring opening of 16 was achieved with DATMP (diethylaluminum 2,2,6,6-tetramethylpiperidine)10 developed by Yamamoto as a strong base to produce the exo-methylene compound 17 in 76% yield. Extension of the side chain at the C1 position was carried out in two steps: Johnson–Claisen rearrangement of allyl alcohol 17, followed by reduction of the resulting ester compound, to give the diol 18 in 78% yield for 2 steps. Transformation of the primary alcohol of 18 to a cyano group took place as a two-step operation, selective toluenesulfonylation of the sterically less hindered primary alcohol group of diol 18, followed by nucleophilic substitution of the resulting monotoluenesulfonate 19 with sodium cyanide, to afford the nitrile 20 in 91% yield (2 steps). Parikh–Doering oxidation11 of 20 gave the aldehyde 7, the precursor of the planned aldol-type condensation to construct the bicyclic framework, in high yield. Treatment of 7 with LHMDS in THF at −60 °C furnished the bicyclic compound 21 as a separable mixture in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in quantitative yield. These compounds were diastereomers related to the cyano group at the C7 position. Dehydration of 21 was executed in a single operation composed of a two-step reaction, acetylation of the hydroxyl group with acetic anhydride, followed by deacetoxylation via deprotonation of the α proton of the cyano group with DBU, to afford the α,β-unsaturated nitrile derivative 8 in 96% yield.
:1 ratio in quantitative yield. These compounds were diastereomers related to the cyano group at the C7 position. Dehydration of 21 was executed in a single operation composed of a two-step reaction, acetylation of the hydroxyl group with acetic anhydride, followed by deacetoxylation via deprotonation of the α proton of the cyano group with DBU, to afford the α,β-unsaturated nitrile derivative 8 in 96% yield.
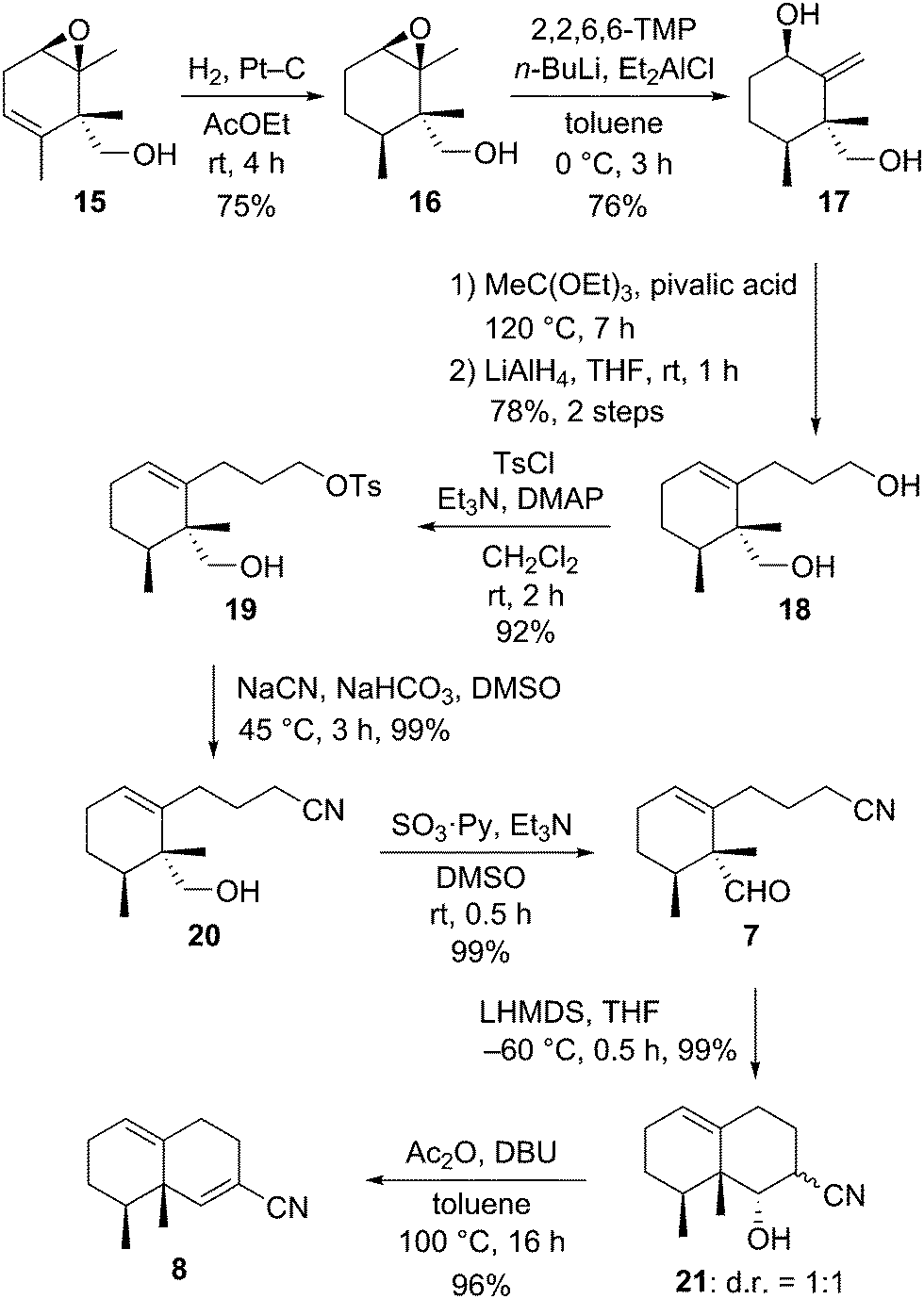 | ||
| Scheme 3 Synthesis of the nitrile derivative 8. | ||
After construction of the bicyclic framework, we transformed the nitrile to a methyl ketone group and performed the stereoselective introduction of the allyl alcohol unit on the bicyclic skeleton (Scheme 4). Nucleophilic addition of methyl lithium to the carbon atom of the nitrile group of 8, followed by treatment with a Brønsted acid, afforded methyl ketone derivative 22 in 94% yield. Stereo- and chemoselective epoxidation of 22 with mCPBA gave the epoxide 23 and its diastereoisomer 24 in 68% and 16% yields, respectively. Many reaction conditions for transformation of the epoxide to the allyl alcohol via epoxide ring opening of 23 were attempted. As a result, use of p-toluenesulfonic acid as a Brønsted acid and sec-butyl alcohol as a solvent afforded the desired allyl alcohol 25 in 62% yield. The stereochemistry of 25 was confirmed by X-ray crystallographic analysis of p-nitrobenzoate derivative 26,12 prepared from 25 with p-nitrobenzoyl chloride and base. This result indicated that the stereoselective epoxidation of 22 occurred at the more electron-rich olefin from the same face as the two methyl groups.
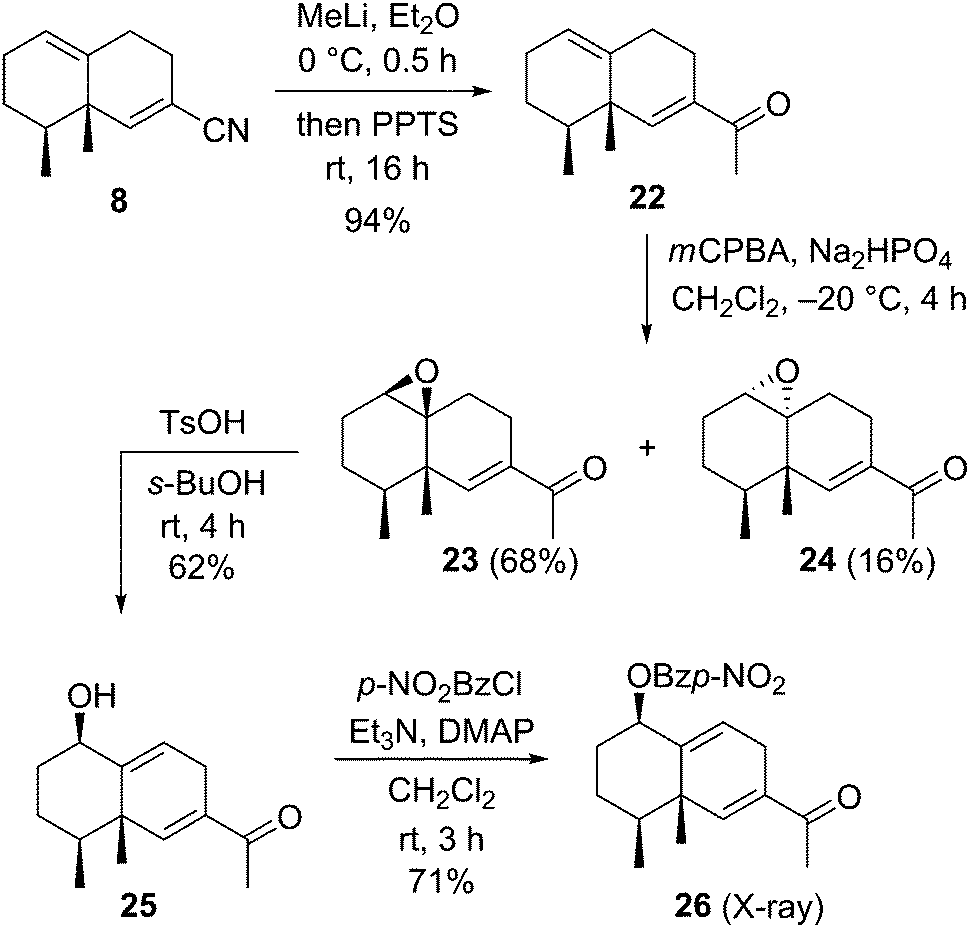 | ||
| Scheme 4 Synthesis of the allyl alcohol 25 and its benzoate 26. | ||
With the desired allyl alcohol in hand, we were on track to achieve our goal for the synthesis of the eremophilane-type target molecules (Scheme 5). After acetylation of 25, many conditions for allylic oxidation of the resulting 27 were examined. Although manganese acetate-catalyzed,13 or palladium-catalyzed14 allylic oxidations failed, giving a complex mixture or recovered 27, respectively, allylic oxidation using 3,5-dimethylpyrazole–chromium trioxide complex15 in dichloromethane at 0 °C afforded the oxidized product 416 in 52% yield. Alternatively, the combination of tert-butyl hydroperoxide and copper iodide17 caused sequential allylic oxidation and stereoselective epoxidation of 27 to give the epoxide 2 in 44% yield. Finally, removal of the acetoxy group of the resulting oxidized products 4 and 2 quantitatively produced the corresponding alcohols 3 and 1, respectively. Both 1H and 13C NMR spectra of the synthetic compounds 1–4 were identical with those of natural sagittasin E (1) and related natural products 2–4.1,2 The optical rotation of the synthetic 1 had the same rotation as that reported for the natural product [synthetic 1: [α]D +104.5 (c 0.5, MeOH); natural product 1: [α]D +90.0 (c 0.5, MeOH)1]. Therefore, we determined the absolute configuration of naturally occurring sagittasin E as 1R,4S,5R,6R and 7S (natural product numbering). Optical rotations of synthetic alcohol 3 and its acetate 4 also had the same rotations as those reported [synthetic 3: [α]D −43.3(c 1.0, CHCl3); natural product 3: [α]D −36.0 (c 0.6, CHCl3)2] and [synthetic 4: [α]D −33.3 (c 0.5, CHCl3); natural product 4: [α]D −34.6 (c 0.5, CDCl3)2]. The absolute configurations of natural products 3 and 4 were determined as 1R,4S and 5S, respectively. However, interestingly, the optical rotation of the synthetic epoxide 2 was different from the reported value of the natural product [synthetic 2: [α]D +52.1 (c 0.4, CHCl3); natural product 2: [α]D −11.6 (c 0.4, CHCl3)2]. Fortunately, we were able to obtain a single crystal of 2 by recrystallization from hexane. The stereochemistry of 2 was confirmed by the X-ray crystallographic analysis of 218 to be the same configuration as that of sagittasin E (1). Since natural product 2 was isolated along with 3 and 4,2 the optical rotation value of our synthetic sample 2 must be the correct value for natural product 2.
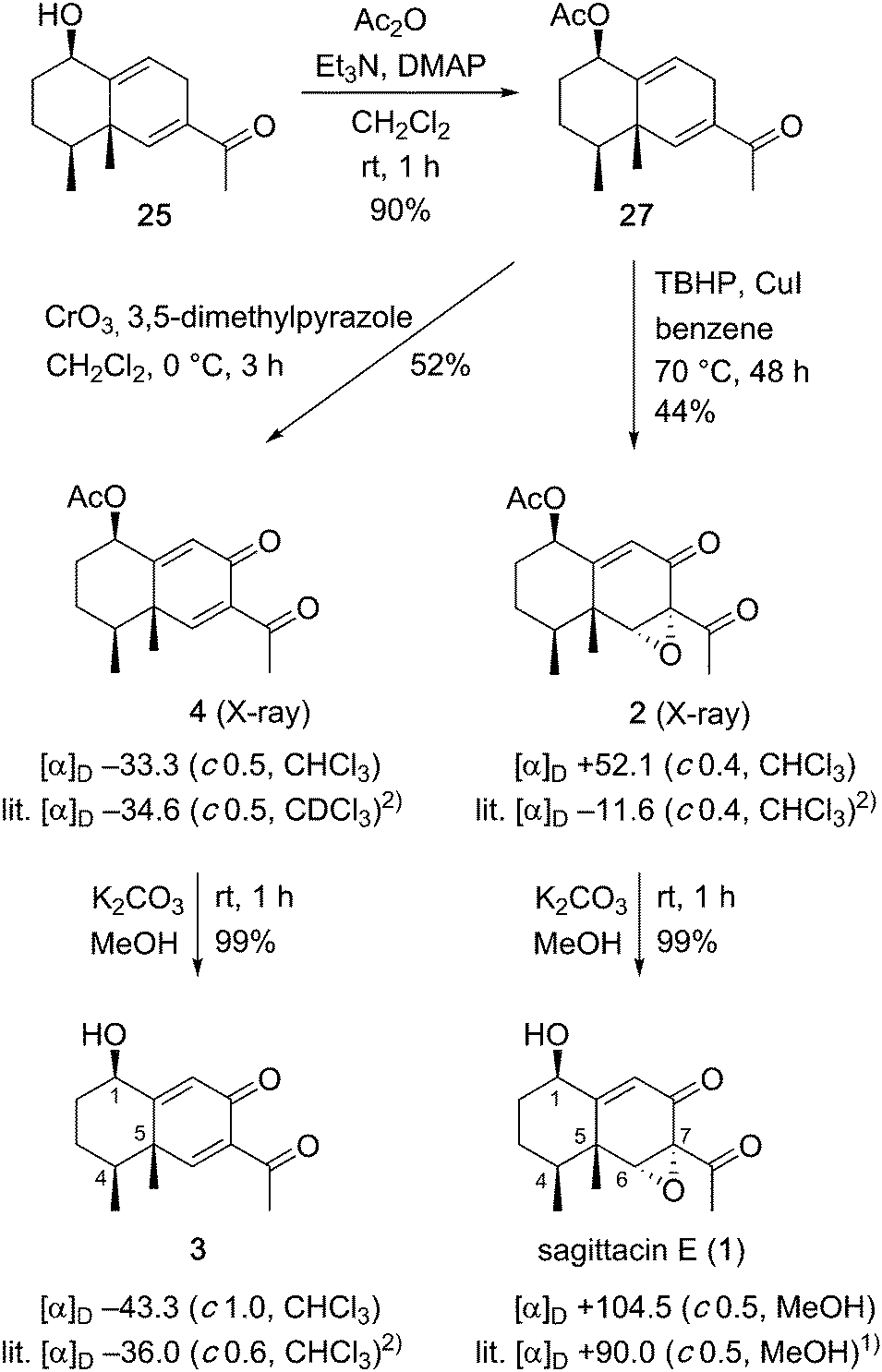 | ||
| Scheme 5 Asymmetric synthesis of sagittacin E (1) and related natural products. | ||
The first enantioselective total synthesis of (+)-sagittacin E and three related natural products was achieved. This synthesis features an asymmetric desymmetrization of a symmetric 1,4-cyclohexadiene derivative having a quaternary carbon by Shi asymmetric epoxidation, intramolecular aldol-type cyclization of a nitrile compound to construct the bicyclic skeleton, allylic oxidation of a 1,4-diene compound, and stereoselective epoxidation.
This work was supported by the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-J. Chen, C.-J. Chen, X.-J. Yao, X.-J. Jin and K. Gao, J. Nat. Prod., 2014, 77, 1329–1335 CrossRef PubMed.
- Y. Zhao, H. Peng and Z. J. Jia, J. Nat. Prod., 1994, 57, 1626–1630 CrossRef.
- D.-Q. Fei, Z.-X. Zhang, J.-J. Chen and K. Gao, Plants Med., 2007, 73, 1292–1297 CrossRef PubMed.
- Z. Meng and B. Liu, Org. Biomol. Chem., 2018, 16, 957–962 Search PubMed.
- J. Iwasaki, H. Ito, M. Nakamura and K. Iguchi, Tetrahedron Lett., 2006, 47, 1483–1486 CrossRef.
- Y. Shi, Acc. Chem. Res., 2004, 37, 488–496 CrossRef PubMed.
- O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958–3987 CrossRef PubMed.
- The enantiomeric excess of compound 14 was determined by chiral HPLC [AD-H column, hexane–isopropanol (150:1), 8.8 min for the first eluted isomer (minor) and 9.7 min for the second eluted isomer (major)].
- The enantiomeric excess of 15 was determined for the benzoate derivative prepared from 15 with benzoyl chloride by chiral HPLC. See ESI†.
- A. Yasuda, S. Tanaka, K. Oshima, H. Yamamoto and H. Nozaki, J. Am. Chem. Soc., 1974, 96, 6513–6514 CrossRef.
- J. R. Parikh and W. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505–5507 CrossRef.
- CCDC 1835801 (26)†.
- T. K. M. Shing, Y.-Y. Yeung and P. L. Su, Org. Lett., 2006, 8, 3149–3151 CrossRef PubMed.
- J.-Q. Yu and E. J. Corey, Org. Lett., 2002, 4, 2727–2730 CrossRef PubMed.
- W. G. Salmond, M. A. Barta and J. L. Havens, J. Org. Chem., 1978, 43, 2057–2059 CrossRef.
- CCDC 1843993 (4)†.
- J. A. R. Salvador, M. L. Sá e Melo and A. S. Campos Neves, Tetrahedron Lett., 1997, 38, 119–122 CrossRef.
- CCDC 1835813 (2)†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1835801, 1835813 and 1843993. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc03438a |
| This journal is © The Royal Society of Chemistry 2018 |