
Alkene protection against acid using a bromide substituent: application in a total synthesis of (−)-6,7-dideoxysqualestatin H5†
Hasanain A. A.
Almohseni‡
,
Hamad H.
Al Mamari§
,
Anne
Valade¶
,
Herman O.
Sintim||
 and
David M.
Hodgson
*
and
David M.
Hodgson
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: david.hodgson@chem.ox.ac.uk
First published on 9th May 2018
Abstract
The presence of a bromide substituent, instead of a hydrogen or methyl group, on a carbon–carbon double bond, protects the alkene from addition reactions when exposed to trifluoroacetic acid. This concept is used to circumvent concomitant loss of unsaturation in a late-stage acid-catalysed 6,8- to 2,8-dioxabicyclo[3.2.1]octane rearrangement towards (−)-6,7-dideoxysqualestatin H5. The inertness of the alkenyl bromide functionality is demonstrated through several synthetic transformations in the assembly of the rearrangement substrate. Completion of the natural product synthesis is facilitated by post-rearrangement removal of the bromide substituent through stereoselective C–C cross-coupling in the presence of ester and hydroxyl functionalities.
(−)-6,7-Dideoxysqualestatin H5 (DDSQ) 5 (Scheme 1) is a member of the zaragozic acid/squalestatin family of natural products which display a variety of interesting bioactivities.1,2 Structurally, DDSQ possesses a synthetically challenging 2,8-dioxabicyclo[3.2.1]octane core bearing a hydroxyl and three carboxylic acid groups and an unsaturated hydrocarbon side-chain at C-1. In an earlier study, we attempted to access DDSQ through acid-catalysed transketalisation of the advanced intermediate 1.3 Rearrangement to the desired core could be induced, however concomitant loss of the unsaturation in the side-chain by protonation/Friedel–Crafts cyclisation to give tetralin 2 could not be avoided. Although this problem could be solved by introducing the olefin after the rearrangement (3 → 4 → 5),2b we became intrigued by the issue of how to directly protect this type of electron-rich carbon–carbon double bond from such unwanted reactivity. While masking the alkene could potentially be achieved in numerous ways,4 we were attracted to the idea of reducing its propensity to protonation through the presence of a temporary halide substituent. Here, we communicate the realisation of this concept, and its application in a synthesis of DDSQ 5 by halide replacement of the alkenyl methyl group in rearrangement substrate 1 (italicised Me).
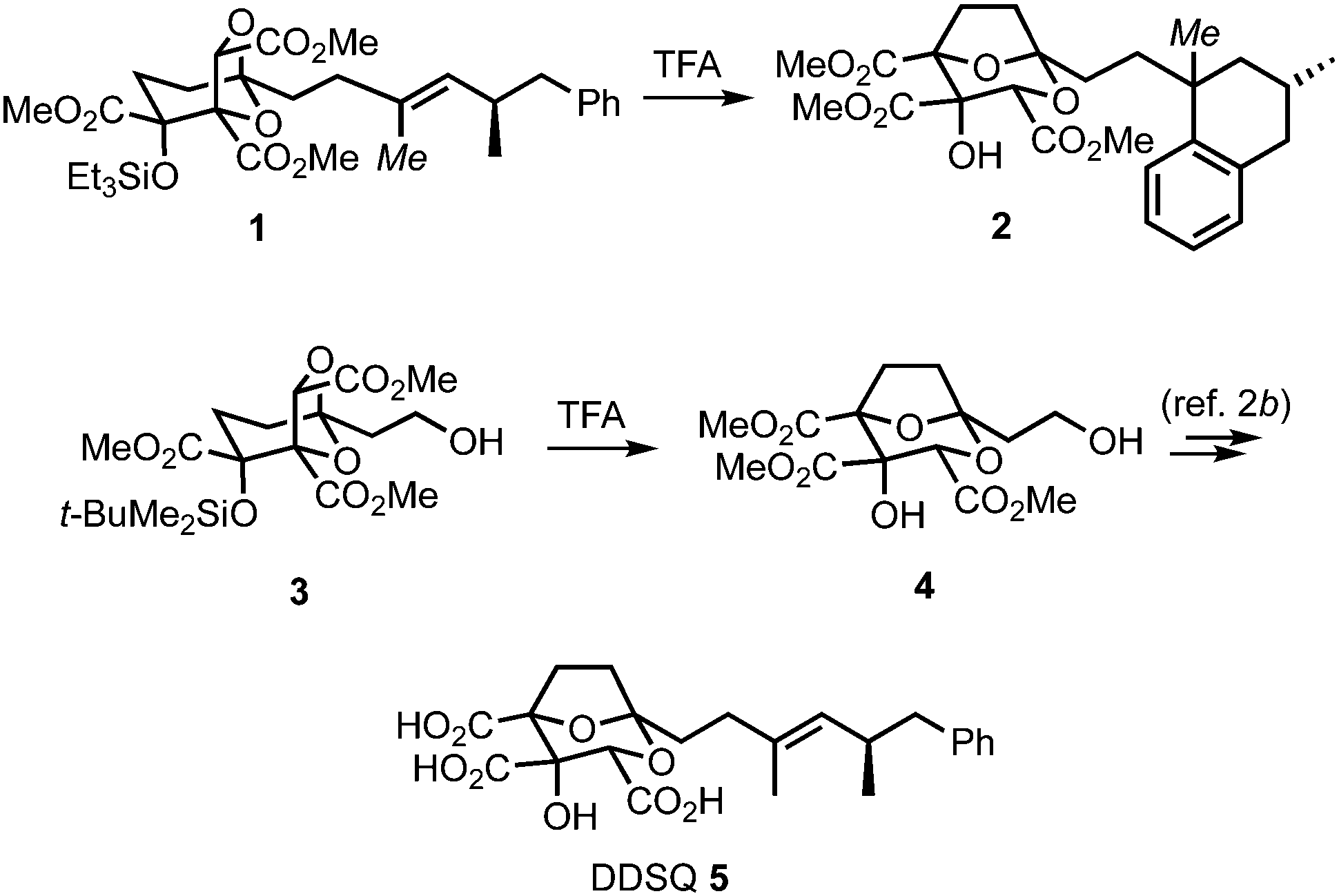 | ||
| Scheme 1 Transketalisation approaches to (−)-6,7-dideoxysqualestatin H5 5. | ||
The Z-alkenyl halide functionality envisaged in the rearrangement substrate for DDSQ synthesis discussed above would not only have to remain inert during transketalisation. It would also be required to pass unscathed through several synthetic steps in the construction of the substrate (vide infra), and yet would need enough reactivity to undergo stereoretentive (and chemoselective – in the presence of ester functionality) cross-coupling with a methyl donor;5 we therefore focused on bromide (rather than iodide or chloride) as the halide substituent. Before embarking on the target synthesis with this modification in place, the following observations provided initial support for the idea of alkene protection from an acid such as TFA, by a bromide substituent. 2-Methyl-1-octene (6a, Scheme 2) is known to be entirely converted to the corresponding tertiary trifluoroacetate 7 within 20 min using TFA (5 equiv.) in CH2Cl2 at 0 °C,6,7 whereas 2-bromo-1-octene (6b)8 was recovered unchanged from exposure to the acidic conditions typically necessary to induce the acid-catalysed 6,8- to 2,8-dioxabicyclo[3.2.1]octane rearrangement [TFA/CH2Cl2/H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:1), 40 °C, 48–68 h].2b,9 Also, 2-methyl-5-phenylpentene (8a) is known to undergo Friedel–Crafts cyclisation to 1,1-dimethyltetralin (9) with acid,10 and, similarly, we found 2,4-dimethyl-5-phenyl-2-pentene (10) with TFA gave the corresponding tetralin 11 in 95% yield (Scheme 2); in contrast, 2-bromo-5-phenylpentene (8b)11 was recovered quantitatively from exposure to the standard TFA rearrangement conditions.
:20:1), 40 °C, 48–68 h].2b,9 Also, 2-methyl-5-phenylpentene (8a) is known to undergo Friedel–Crafts cyclisation to 1,1-dimethyltetralin (9) with acid,10 and, similarly, we found 2,4-dimethyl-5-phenyl-2-pentene (10) with TFA gave the corresponding tetralin 11 in 95% yield (Scheme 2); in contrast, 2-bromo-5-phenylpentene (8b)11 was recovered quantitatively from exposure to the standard TFA rearrangement conditions.
 | ||
| Scheme 2 Addition and cyclisation reactions of alkenes with acids. | ||
For the prospective synthesis of (−)-DDSQ 5, the Z-alkenyl bromide-bearing rearrangement substrate 12 was anticipated to be available through a Rh(II)-catalysed tandem carbonyl ylide formation and cycloaddition of a diazoketone 13 with methyl glyoxylate (Scheme 3).12 In turn, diazoketone 13 was considered as being accessible from substituted tartrate 14, with the latter expected to arise through application of Seebach's tartrate alkylation methodology2b,13 using the enolate of tartrate 15 and iodide 16.
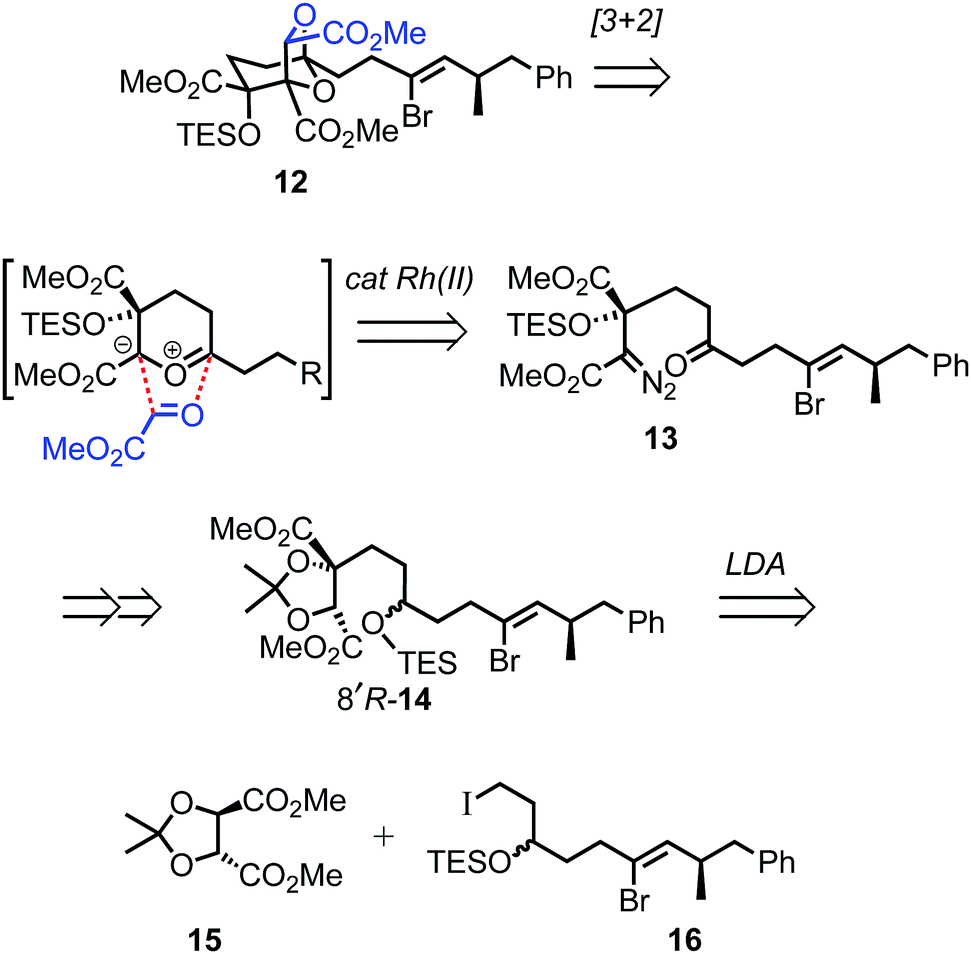 | ||
| Scheme 3 Outline of projected route to Z-alkenyl bromide-bearing rearrangement substrate 12. | ||
Access to the Z-alkenyl bromide-containing iodide 16 for tartrate alkylation was achieved from R-aldehyde 1714 (Scheme 4). Wittig olefination (known to proceed without erosion of enantiointegrity with such an ester-stabilised ylide on a related α-methyl aldehyde15) to give E-enoate 18, was followed by bromination–dehydrobromination15 to give the Z-α-bromoenoate 19. The dianion of ethyl acetoacetate16 was efficiently alkylated with the derived allylic bromide 20, and the resulting β-ketoester 21 was reduced to the 1,3-diol 22 using NaBH4 in MeOH/THF.17 Iodination of the primary alcohol and TES protection of the secondary alcohol gave the iodide 16.
 | ||
| Scheme 4 Synthesis of iodide 16. | ||
Initial availability of the 1,3-diol 22 (from rac-17) provided an early opportunity to test the stability of the Z-alkenyl bromide motif under the TFA rearrangement conditions. Pleasingly, reaction of 1,3-diol 22 gave the bistrifluoroacetate 25 (quant., Scheme 5), in which the alkenyl bromide was unaffected. Moreover, stereoretentive Suzuki reactions between the 1,3-diol-derived mono- and bis-TBS ethers 23 and 24 and the borinate complex 26 (from MeLi and 9-MeO-9-BBN)5 gave E-alkenes 27 and 28 in 75% and 76% yields, respectively.18
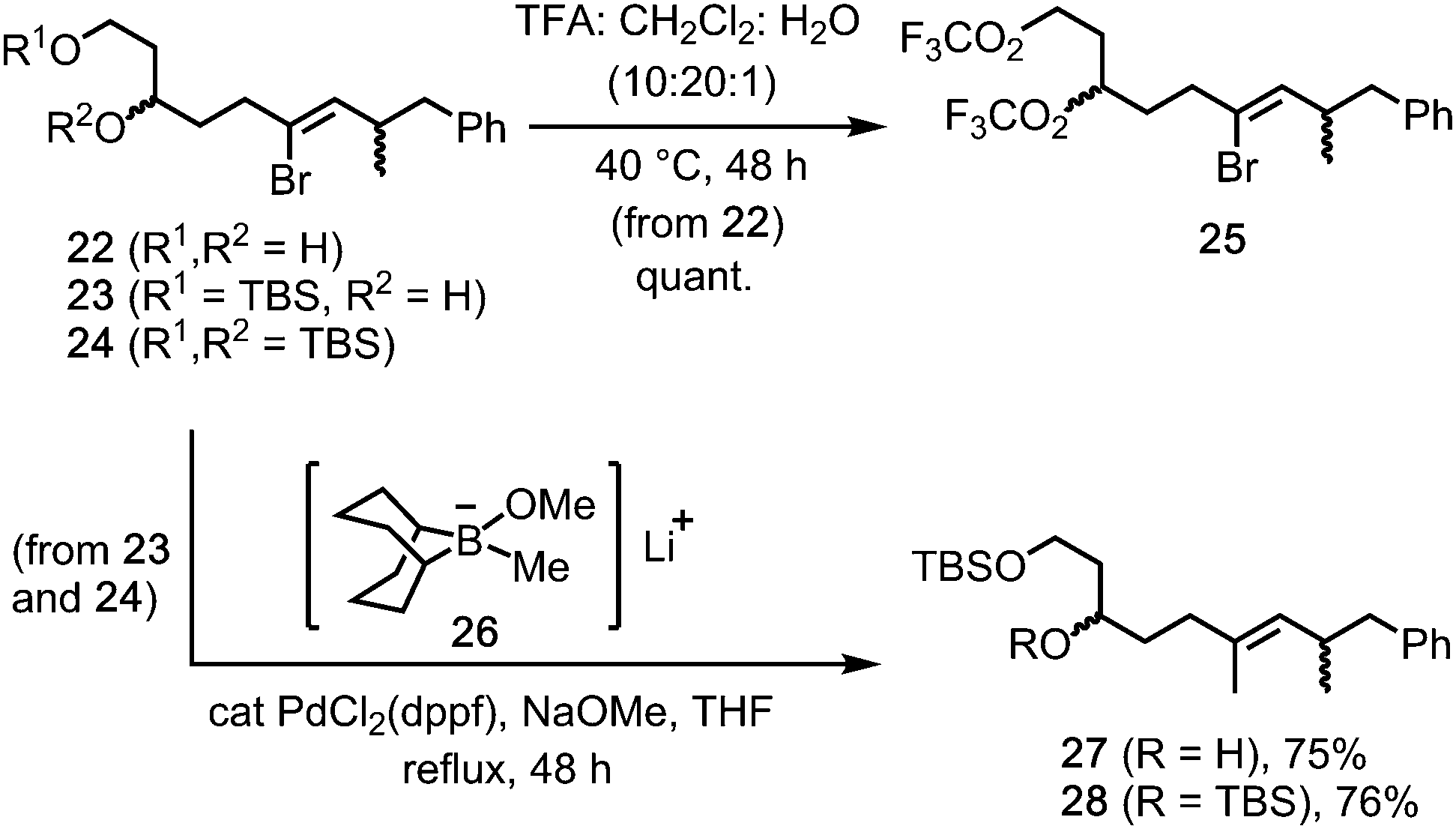 | ||
| Scheme 5 Stability of alkenyl bromide functionality to TFA, and stereoretentive methylation. | ||
The chemistry in Scheme 5 encouraged progression of iodide 16 towards (−)-DDSQ 5. Thus, tartrate alkylation,2b,13 followed by oxidation19 of the resulting alkylated tartrate 14 gave hydroxy acetonide 29 (Scheme 6). The acidic conditions necessary to remove acetone from 29 resulted in concomitant desilylation, but the silyl group was reinstalled during formation of the bis-TES ether 30 from the intermediate lactol. This bis-TES ether 30 underwent condensation with tosylhydrazide, followed by Et3N-induced generation of diazo functionality;20 under the basic reaction conditions partial desilylation of the secondary TES ether occurred and this was completed by addition of aqueous acetic acid. The lability of the secondary TES ether under mild conditions was crucial, as it, along with mild Dess–Martin oxidation of the resulting intermediate secondary alcohol, allowed access to ketone 13 in which both the tertiary TES ether and the diazo functionality were retained.9
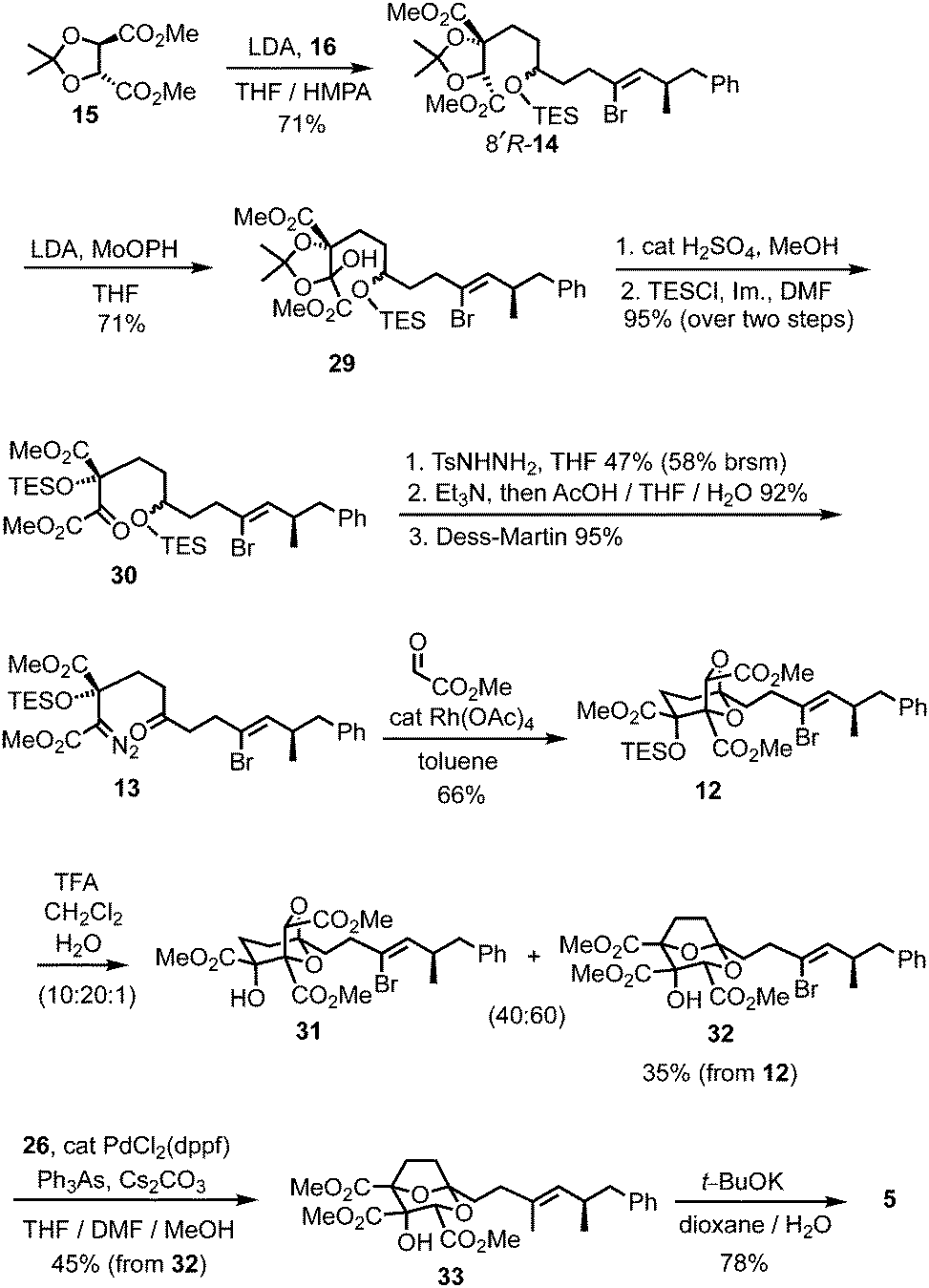 | ||
| Scheme 6 Completion of the synthesis of (−)-6,7-dideoxysqualestatin H5 5. | ||
Following cycloaddition of diazoketone 13 with methyl glyoxylate, we were pleased to observe that, in line with the model study (Scheme 5), rearrangement of the resulting cycloadduct 12 could be induced with preservation of the alkenyl bromide, to give the 2,8-dioxabicyclo[3.2.1]octane 32.21 Further Suzui methylation studies with alkylated tartrate 14 (from rac-17) as a closer (ester-containing) model system to 32, led to the identification of Cs2CO3 (in MeOH) as the preferred base and Ph3As as additive in DMF.† 22 Application of these conditions to alkenyl bromide 32 gave the squalestatin trimethyl ester 33, which underwent global saponification using anhydrous KOH23 to give 5.2
The present work exemplifies prevention of electrophilic attack (protonation/Friedel–Crafts cyclisation) of an alkene by using a vinylic bromine substituent. Conceptually, this differs from earlier approaches, that mask a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond by transient addition of elements across it, which are then subsequently eliminated.4 During the sequence to prepare 5, the alkenyl bromide functionality is also robust enough to survive a variety of other chemistry (Schemes 4 and 6), before being selectively replaced in a C–C bond-forming event at the penultimate step. The strategy should find application in other synthetic endeavours.
C bond by transient addition of elements across it, which are then subsequently eliminated.4 During the sequence to prepare 5, the alkenyl bromide functionality is also robust enough to survive a variety of other chemistry (Schemes 4 and 6), before being selectively replaced in a C–C bond-forming event at the penultimate step. The strategy should find application in other synthetic endeavours.
We thank the Higher Committee for Education Development in Iraq, the Sultanate of Oman and the University of Oxford for studentship support (to H. A. A. A., H. H. A. M., and H. O. S., respectively), and the European Union for a Marie Curie Fellowship (MEIF-CT-2004-515366 to A. V.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For a review, see: A. Armstrong and T. J. Blench, Tetrahedron, 2002, 58, 9321 CrossRef CAS.
- For previous syntheses of 5, see: (a) S. Naito, M. Escobar, P. R. Kym, S. Liras and S. F. Martin, J. Org. Chem., 2002, 67, 4200 CrossRef CAS PubMed; (b) Y. Fegheh-Hassanpour, T. Arif, H. O. Sintim, H. A. Al-Mamari and D. M. Hodgson, Org. Lett., 2017, 19, 3540 CrossRef CAS PubMed.
- H. O. Sintim, DPhil thesis, University of Oxford, 2002.
- For a review on protection of CC bonds, see: J. Svoboda and J. Paleček, Chem. Listy, 1994, 88, 86 CAS.
- G. Seidel and A. Fürstner, Chem. Commun., 2012, 48, 2055 RSC.
- M. Julia and C. Schmitz, Bull. Soc. Chim. Fr., 1986, 630 CAS.
- Simple terminal alkenes (e.g., 1-hexene) and prenyl-type systems also undergo addition of TFA, see: (a) P. A. Peterson, J. Am. Chem. Soc., 1960, 82, 5834 CrossRef CAS; (b) L. Streinza, Z. Wimmera, G. K. Roshkab, R. I. Ishchenkob, M. Romaňuka and B. G. Kovalev, Collect. Czech. Chem. Commun., 1985, 50, 2174 CrossRef.
- M. Ahmed, A. G. M. Barrett, J. C. Beall, D. C. Braddock, K. Falck, V. C. Gibson, P. A. Procopiou and M. M. Salter, Tetrahedron, 1999, 55, 3219 CrossRef CAS.
- D. M. Hodgson, C. Villalonga-Barber, J. M. Goodman and S. C. Pellegrinet, Org. Biomol. Chem., 2010, 8, 3975 CAS.
- R. A. Bunce and A. N. Cox, Org. Prep. Proced. Int., 2010, 42, 83 CrossRef CAS.
- Z. Li, R. Ebule, J. Kostyo, G. B. Hammond and B. Xu, Chem. – Eur. J., 2017, 23, 12739 CrossRef CAS PubMed.
- D. M. Hodgson, A. H. Labande and S. Muthusamy, Org. React., 2013, 80, 133 CAS.
- (a) R. Naef and D. Seebach, Angew. Chem., Int. Ed. Engl., 1981, 20, 1030 CrossRef; (b) D. Seebach, J. D. Aebi, M. Gander-Coquoz and R. Naef, Helv. Chim. Acta, 1987, 70, 1194 CrossRef CAS.
- A. G. Myers, B. H. Yang and H. Chen, Org. Synth., 2004, X, 509 Search PubMed.
- (a) B. M. Trost, G. D. Probst and A. Schoop, J. Am. Chem. Soc., 1998, 120, 9228 CrossRef CAS; (b) T. Müller, M. Göhl, I. Lusebrink, K. Dettner and K. Seifert, Eur. J. Org. Chem., 2012, 2323 CrossRef.
- S. N. Huckin and L. Weiler, J. Am. Chem. Soc., 1974, 96, 1082 CrossRef CAS.
- K. Soai and H. Oyamada, Synthesis, 1984, 605 CrossRef CAS.
- In the absence of NaOMe, E-alkene 27 was obtained from mono TBS ether 23 in 37% yield.
- E. Vedejs and S. Larsen, Org. Synth., 1990, VII, 277 Search PubMed.
- Y. Fegheh-Hassanpour, F. Ebrahim, T. Arif, H. O. Sintim, T. D. W. Claridge, N. T. Amin and D. M. Hodgson, Org. Biomol. Chem., 2018, 16, 2876 CAS.
- The desilylated unrearranged cycloadduct 31 could be resubmitted to the rearrangement conditions to provide more 2,8-dioxabicyclo[3.2.1]octane 32.
- P. Gersbach, A. Jantsch, F. Feyen, N. Scherr, J.-P. Dangy, G. Pluschke and K.-H. Altmann, Chem. – Eur. J., 2011, 17, 13017 CrossRef CAS PubMed.
- (a) P. G. Gassman and W. N. Schenk, J. Org. Chem., 1977, 42, 918 CrossRef CAS; (b) A. Krief and A. Kremer, Chem. Rev., 2010, 110, 4772 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, model studies and copies of 1H NMR and 13C NMR spectra of all new compounds. See DOI: 10.1039/c8cc02690d |
| ‡ Permanent address: University of Kufa, Najaf Governorate, Iraq. |
| § Present address: Department of Chemistry, College of Science, Sultan Qaboos University, P.O. Box 36, Al Khoud 123, Muscat, Sultanate of Oman. |
| ¶ Present address: New Medicines, UCB Biopharma, Avenue de l’Industrie, 1420 Braine-l’Alleud, Belgium. |
| || Present address: Department of Chemistry, Purdue University, West Lafayette, IN 47907–2112, USA. |
| This journal is © The Royal Society of Chemistry 2018 |