
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double quick, double click reversible peptide “stapling”†
Claire M.
Grison
 ab,
George M.
Burslem
ab,
Jennifer A.
Miles
ab,
Ludwig K. A.
Pilsl
ab,
David J.
Yeo
ab,
Zeynab
Imani
ab,
Stuart L.
Warriner
ab,
Michael E.
Webb
ab and
Andrew J.
Wilson
*ab
ab,
George M.
Burslem
ab,
Jennifer A.
Miles
ab,
Ludwig K. A.
Pilsl
ab,
David J.
Yeo
ab,
Zeynab
Imani
ab,
Stuart L.
Warriner
ab,
Michael E.
Webb
ab and
Andrew J.
Wilson
*ab
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK. E-mail: A.J.Wilson@leeds.ac.uk
bAstbury Centre For Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK
First published on 31st May 2017
Abstract
The development of constrained peptides for inhibition of protein–protein interactions is an emerging strategy in chemical biology and drug discovery. This manuscript introduces a versatile, rapid and reversible approach to constrain peptides in a bioactive helical conformation using BID and RNase S peptides as models. Dibromomaleimide is used to constrain BID and RNase S peptide sequence variants bearing cysteine (Cys) or homocysteine (hCys) amino acids spaced at i and i + 4 positions by double substitution. The constraint can be readily removed by displacement of the maleimide using excess thiol. This new constraining methodology results in enhanced α-helical conformation (BID and RNase S peptide) as demonstrated by circular dichroism and molecular dynamics simulations, resistance to proteolysis (BID) as demonstrated by trypsin proteolysis experiments and retained or enhanced potency of inhibition for Bcl-2 family protein–protein interactions (BID), or greater capability to restore the hydrolytic activity of the RNAse S protein (RNase S peptide). Finally, use of a dibromomaleimide functionalized with an alkyne permits further divergent functionalization through alkyne–azide cycloaddition chemistry on the constrained peptide with fluorescein, oligoethylene glycol or biotin groups to facilitate biophysical and cellular analyses. Hence this methodology may extend the scope and accessibility of peptide stapling.
Introduction
Inhibition of α-helix mediated protein–protein interactions (PPIs)1–4 represents an area of intense interest, due to their role in intracellular signalling processes5 and demonstrated tractability for drug discovery.6,7 The last decade has seen an increasing emphasis on the development of constrained peptides8–10 for PPI inhibition. Constraint of a peptide in a bioactive conformation is proposed to improve target binding affinity (by preorganization),11,12 increase stability (e.g. by protecting against proteolysis)13–15 and enhance cell-uptake,16,17 although a number of recent studies have provided conflicting evidence for the enhanced cell permeability and cellular potency that is conferred through stapling.18–20 Available strategies9,13,21–25 to constrain a peptide in a helical conformation (Fig. 1a–e), include hydrocarbon “staples”,26–28 lactam bridges,29–31 hydrogen-bond surrogates,32 photoswitches11,33,34 and triazoles introduced by “click” chemistry.35–38 The nature of the linker, including any stereocenters, plays a role in regulating peptide conformation and target protein affinity.31,39 Introduction of constraints may be achieved either by intramolecular reaction (Fig. 1a)28,31 or by chemoselective intermolecular reaction (Fig. 1b) between suitably disposed residues within the peptide sequence and an appropriate coupling partner.36,40 Introduction of a constraint by intermolecular reaction, whilst synthetically more demanding, may be advantageous in that it provides access to functionalized constraints36 and thus to peptides tailored for biophysical, cellular and chemical proteomics analyses. Herein, we use BID and RNase S variant peptides (1a–d) which respectively bind Bcl-xL proteins41 and RNase S protein42 (Fig. 1f and g), to demonstrate that dibromomaleimides43–51 serve as versatile constraints that can be introduced and removed.52–57 These reagents have not previously been studied for their ability to control peptide helicity. Introduction of an alkyne onto the dibromomaleimide permits further divergent functionalization by “click” chemistry.58 This approach may therefore complement recently reported methods to constrain peptides by reaction of dichloroacetone59 with side chain thiols and photo-reversible constraints using S,S′-tetrazine.60 | ||
| Fig. 1 Overview of peptide stapling strategies and linkers for constraining peptides in an α-helical conformation together with structures for the model protein–protein interactions used in this study; (a) schematic for intramolecular crosslinking; (b) schematic for intermolecular crosslinking; (c) S,S-α,α′-disubstituted i, i + 4 hydrocarbon “staple”; (d) S,S-des-methyl i, i + 4 hydrocarbon constraint; (e) i, i + 4 S,S′-maleimide staple reported in this work; (f) and (g) model peptide sequences 1a–d for BID (1a–b) and RNase S (1c–d) and protein–protein interactions in this study (Bcl-xL (green)/BID (blue) PDB ID: 4QVE; RNase S protein (green)/S peptide (blue) PDB ID: 1CJQ; hotspot residues (red)). | ||
Previously, we14 and others61 introduced α-pentenylglycine as an alternative to the widely used α,α′-pentenylalanine for cross-linking the i and i + 4 residues within a peptide (Fig. 1c and d).26 Production of this des-methyl constraint requires a more easily synthesized amino acid which is less sterically hindered for peptide coupling, and, as shown by us, has no deficiencies in terms of biophysical properties when applied to BH3 peptides BID and BIM.62 We hypothesized that this all hydrocarbon 8-atom constraint could be replaced with an isoatomic S,S′-maleimide crosslink by reaction of suitably disposed homocysteine (hCys) residues with dibromomaleimide (or for cysteine (Cys), a 6-atom constraint) (Fig. 1e).
Peptides 1a–d bearing Cys or hCys amino acids in positions used previously26,62 for i → i + 4 constraints were independently oxidised to the constrained disulfide 2a–d or cross-linked with dibromomaleimide 4 to generate 3a–d with rapid and complete conversion. Alternatively, this sequence 1a–d to 3a–d could be performed in one pot and peptides 1a–d fully regenerated by addition of an excess of thiol (Fig. 2 and S4–7†). It is noteworthy that conversion of the Cys-derived sequences 1a and 1c to their constrained derivatives 3a and 3c was more rapid than for the corresponding hCys derivatives 1b and 1d. It should be noted that these studies were carried out in unbuffered aqueous solution and future studies will focus on a more detailed analysis of the reaction rates, their pH dependence and comparison to other rapid constraining reactions.63 Of further note is the fact that a range of thiols were used including a 10 mM aqueous solution of glutathione, which corresponds to the average concentration present in cancer cells.64,65 It may thus be feasible to harness this approach for controlled delivery of a peptide to the diseased cell; a focus of future studies. Interestingly, removal of the maleimide cross-link could be abrogated through addition of an excess of the reducing agent TCEP, resulting in a permanent succinimide cross-link (Fig. S9†). Succinimide linkages of a similar nature have been shown to undergo thiol mediated cleavage;48 future efforts to “lock” the constraint may therefore be more appropriately served by hydrolysis of the imide in the maleimide crosslink.66
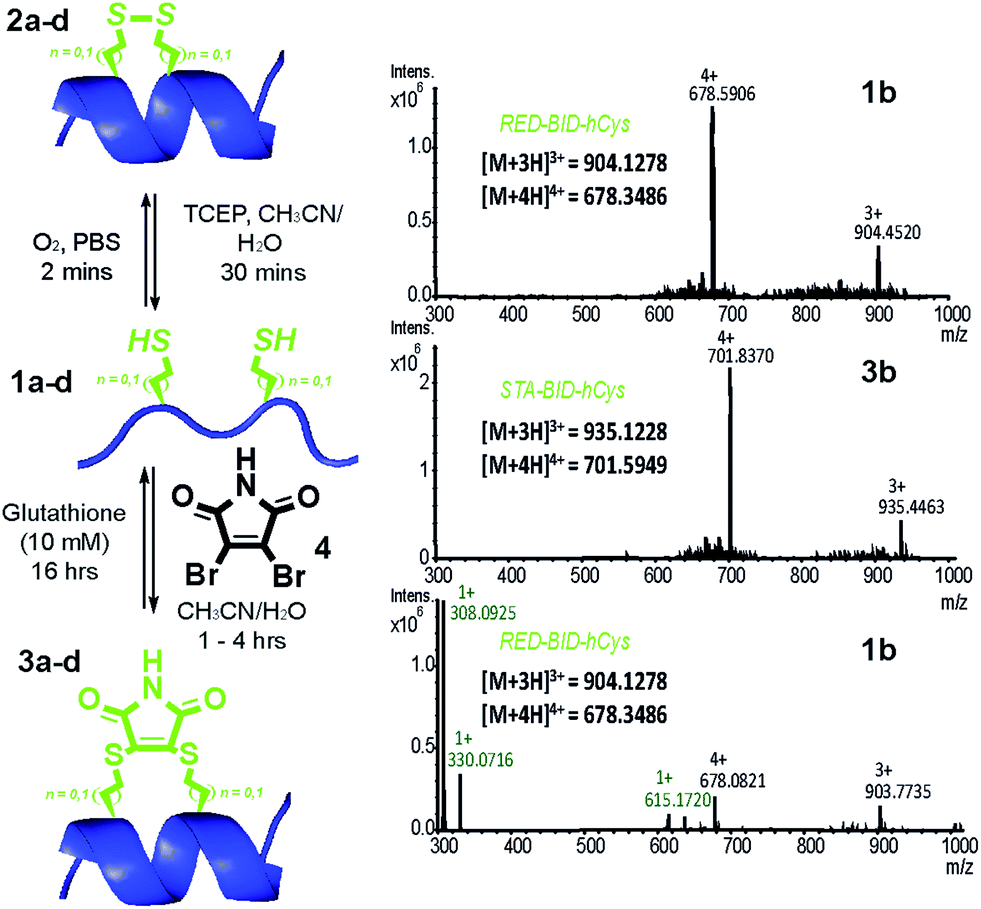 | ||
| Fig. 2 Reversible constraining reaction sequence for peptides 1–3a–d: conditions for conversion from the oxidised state (OX) through the intermediate reduced state (RED) to the constrained peptide (STA) and vice versa (LHS); HRMS analyses of reaction mixture for reversible interconversion between 1b and 3b (relevant masses are indicated in black for the peptide and in green for excess glutathione used during unconstraining) (RHS). | ||
Peptides 1a,b and 3a,b were analysed by CD spectroscopy and subjected to proteolysis assays and fluorescence anisotropy competition assays for inhibition of the BID/Mcl-1, BAK/Bcl-xL and NOXA-B/Mcl-1 interactions (Fig. 3, S19–23 and S29–30†). Constrained peptides 3a,b showed no significant difference in helicity (42% and 43%) to each other as a result of the different cross-link length but both were more helical than the corresponding reduced peptide 1a or 1b, (24% and 29%). Molecular dynamics calculations were conducted on 1b and 3b – these further support the conclusion that the helical conformation predominates when constrained by the maleimide linker (Fig. S25 and S26†). Tryptic proteolysis as monitored by LC-MS14 established that the conformationally constrained peptides 3a,b exhibit improved proteolytic resistance in comparison to the reduced peptides 1a,b; proteolysis was retarded up to 7 residues removed from either side of the thiol residues (Fig. S19–23†). Taken together these data indicate that the maleimide constraint confers comparable enhancement of helicity and proteolytic resistance to the des-methyl hydrocarbon constraint reported by us earlier.62 Peptides 1a,b and 3a,b inhibited the BID/Mcl-1 interaction with comparable potency to the native WT-BID 8 sequence (IC50 ∼ 15–25 μM) except for the constrained peptide 3b which was slightly more potent (IC50 ∼ 4.2 μM), thus the constraining/unconstraining reagents have either a weak or no effect on the inhibitory potency for this PPI. Although inconsistent with the concept that preorganization should promote higher binding affinity, the result is consistent with our own recent studies on hydrocarbon-constrained peptides, which illustrated enthalpy-entropy compensation operates for these peptides which bind through an induced-fit mechanism.62 For inhibition of BAK/Bcl-xL both constrained peptides 3a,b were shown to be significantly superior inhibitors compared to the reduced peptides 1a,b. The differential effects upon potency for the same peptide against Bcl-xL and Mcl-1 may arise as a consequence of favourable non-covalent contacts between the maleimide and Bcl-xL; it is anticipated that future structural analyses will shed light on these differences.
 | ||
Fig. 3 Analyses on BID derived peptides 1a,b and 3a,b. (a) Circular dichroism spectra (water/30% acetonitrile); (b) trypsin proteolysis in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10000 enzyme/substrate ratio (50 mM phosphate buffer, 200 mM NaCl, pH 7.50); (c) fluorescence anisotropy competition assay for inhibition of the FITC-BID/Mcl-1 interaction (25 nM tracer, 150 nM protein, 50 mM Tris buffer, 150 mM NaCl, 0.61% of Triton X-100, pH 7.40); (d) fluorescence anisotropy competition assay for inhibition of the BODIPY-BAK/Bcl-xL interaction (25 nM tracer, 150 nM protein, 50 mM phosphate buffer, 200 mM NaCl, 0.02 mg mL−1 BSA, pH 7.50). :10000 enzyme/substrate ratio (50 mM phosphate buffer, 200 mM NaCl, pH 7.50); (c) fluorescence anisotropy competition assay for inhibition of the FITC-BID/Mcl-1 interaction (25 nM tracer, 150 nM protein, 50 mM Tris buffer, 150 mM NaCl, 0.61% of Triton X-100, pH 7.40); (d) fluorescence anisotropy competition assay for inhibition of the BODIPY-BAK/Bcl-xL interaction (25 nM tracer, 150 nM protein, 50 mM phosphate buffer, 200 mM NaCl, 0.02 mg mL−1 BSA, pH 7.50). | ||
The synthetic method was further adapted to allow the functionalization of the BID–Cys peptides 7a–c. Using Mitsunobu alkylation,67 alkyne–dibromomaleimide 5 was prepared and conjugated to peptide 1a to generate the constrained peptide 6. Without isolation or purification, subsequent “click” reaction with biotin, fluorescein and PEG azides38 afforded complete conversion to click-conjugated constrained peptides 7a–c (Fig. 4a and b). Related “clickable maleimides” have previously been used to conjugate small molecule drugs to polymers,68 whilst “clicked” dibromomaleimides have been used to conjugate polymers to oxytocin so as to improve solubility.51 Here we have expanded this methodology, demonstrating that this functionality can be used to constrain peptide conformation and as a divergent method for peptide functionalization. The utility of these modifications was demonstrated through a direct fluorescence anisotropy titration with the fluorescein linked peptide 7b (Fig. 4c). The observed potencies are consistent with the EC50 values determined in the competition assays for 3a,b indicating no detrimental consequence from the introduction of a fluorophore in this position. Such a modification could thus facilitate cellular studies whilst leaving the N-terminus of the peptide free to allow further orthogonal derivatization.
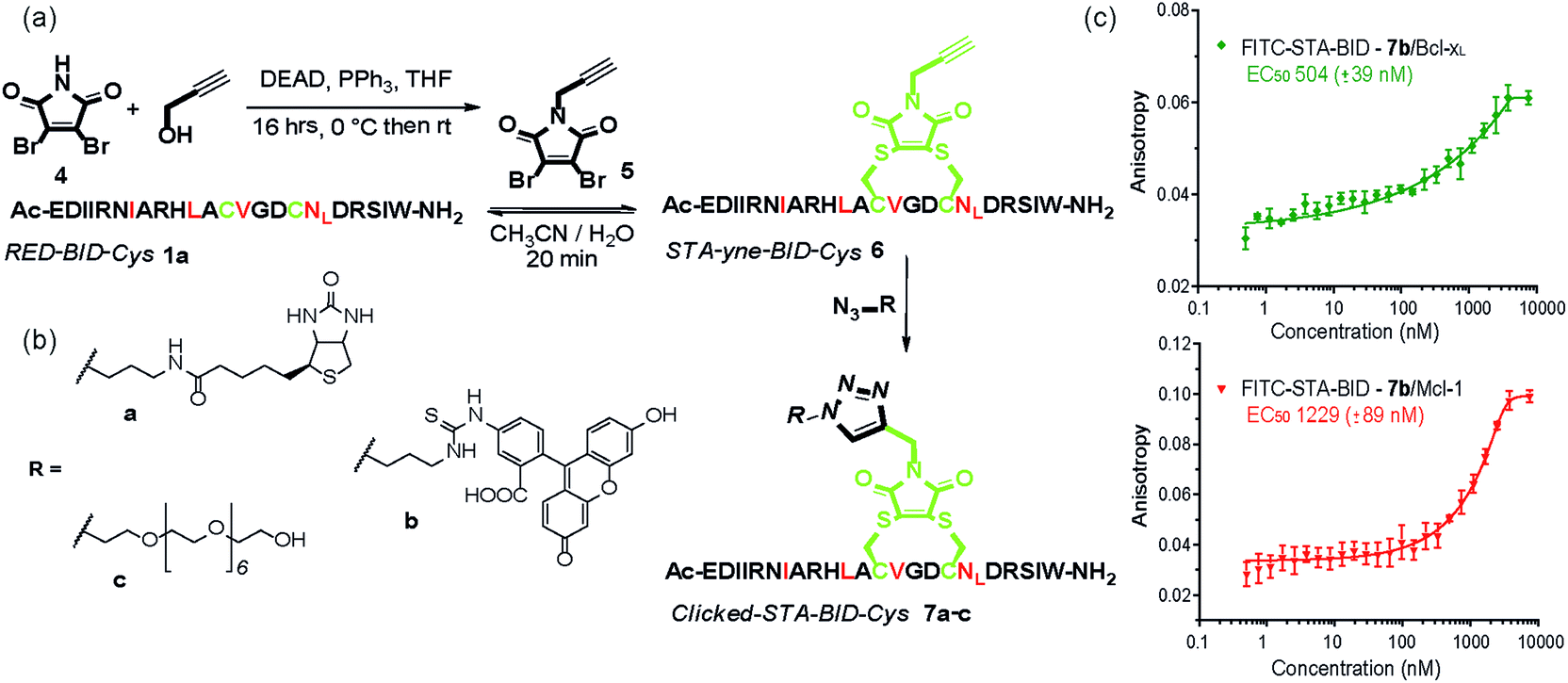 | ||
| Fig. 4 (a) Scheme illustrating double click chemistry for generating functionalized constrained peptides 7a–c. An alkyne-maleimide was used to generate yne-STA-BID-Cys 6, which was further functionalized by click reaction with (b) various azide derivatives. (c) Fluorescence anisotropy direct binding assay for interaction of FITC-STA-BID 7b with Mcl-1 and Bcl-xL (75 nM tracer, 15 μM protein, 50 mM Tris buffer, 150 mM NaCl, 0.61% of Triton X-100, pH 7.40). | ||
The use of dibromomaleimide-based constraints for promotion of α-helicity was then further demonstrated using the RNase S-protein/S-peptide interaction with the choice of amino acid positions at which to introduce the constraint informed by previous studies by the Verdine,26 Hamachi69 and Dawson59 groups. S peptides 1c and 1d bearing Cys and hCys residues at the i and i + 4 positions were synthesized using standard solid-phase synthesis in good yield and purified by HPLC (Fig. S2 and S3†). Peptides 1c and 1d could not readily be isolated as reduced bis-thiols; one-pot reduction followed by the reaction with dibromomaleimide 4 afforded peptides 3c and 3d (Fig. 2). Both peptides were then analysed by CD spectroscopy and used in an RNA degradation assay to determine the restoration of enzymatic activity to S-protein in place of the S-peptide (Fig. 5a and b). In this instance, different and more pronounced behaviour for the Cys and hCys variants was observed, highlighting that the effects of introducing a constraint are context-dependent. The Cys-derived RNase S peptides 2–3c exhibit poor helicity (e.g.3c = 9% versus wild-type 8 = 30%) and corresponding low restoration of enzymatic activity (Fig. S31†). Molecular modelling suggests that the introduction of a disulfide or a maleimide bridge does not support a helical conformation (Fig. S27 and S28†). In contrast, for the hCys 1–3d series introduction of a disulfide or a maleimide crosslink supports a helical structure comparable to the WT sequence and consistent with the moderate effects on helicity for an 8-carbon constraint in these positions. (Furthermore, and fully consistent with the work of Hamachi and co-workers,69 upon addition of trifluoroethanol (TFE), the constrained peptides reach a greater overall α-helical content). Here, introduction of a disulfide (2d) or a maleimide (3d) crosslink results in enhanced helicity in comparison to the WT sequence (2d = 56%, 3d = 52% and wild-type 8 = 30%). As a result 3d is much better able to restore RNase S protein activity than is 3c.
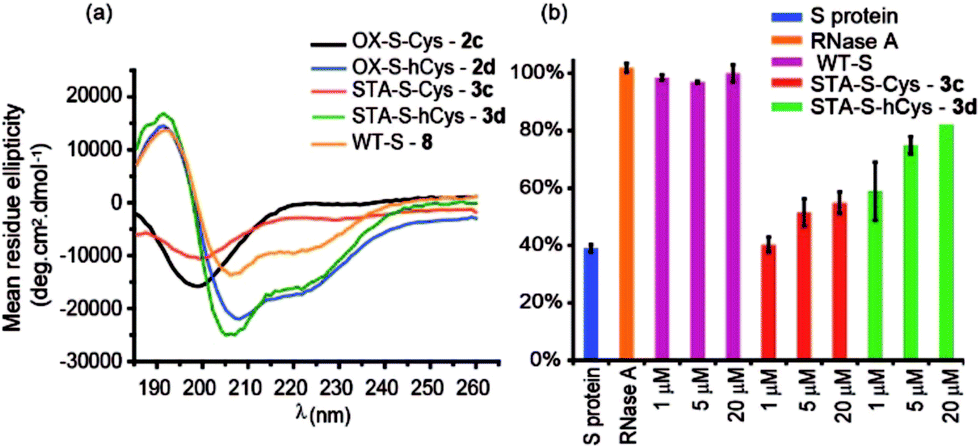 | ||
| Fig. 5 Biophysical and bioactivity analyses of WT-S and S derived peptides 3–4a,c. (a) Circular dichroism spectra recorded in water (30% TFE); (b) RNA degradation assay showing the restoration of S protein activity. S protein was used as a negative control, and RNase A as a positive control. (50 mM Tris, 100 mM NaCl, pH 7.50). | ||
Conclusions
In conclusion, the attractiveness of thiol-based “stapling” approaches has been recognized for some time,24 but no general approach has been widely adopted; we have developed a new method to rapidly and reversibly introduce a functionalized constraint into two model peptides that enhances a range of biophysical and biochemical properties. Moreover, the constraint may be further functionalized in a divergent manner with motifs useful in chemical biology and proteomics applications. The approach can be carried out using natural readily available amino acid side chains on unprotected peptides, potentially bringing peptide stapling to a larger community than hitherto have had access and extends the applications of dibromomaleimide and related reagents. Future efforts will apply this approach to a wider range of helix mediated PPIs with the aim of establishing the generality of the method. The approach may also be useful to identify transient binding pockets proximal to a native ligand binding site using the template directed approach introduced by Sharpless.70 Similarly, the reversibility of the constraint, may permit more facile delivery of a peptide-based reagent to the cell, where it can become unconstrained and less easily transported back out of the cell. Together with the additional functional handle introduced on the maleimide constraint (which is orthogonal in terms of its reactivity), this could facilitate detailed cellular and biochemical analyses e.g. identification of additional targets of the constrained peptide by pull-down. These objectives will represent the focus of our future studies.Acknowledgements
This work was supported by the European Research Council [ERC-StG-240324] and [ERC-PoC-632207], the Leverhulme Trust [RPG-2013-065], the EPSRC [EP/P505593/1], [EP/KO39292/1], for funding NMR facilities and The Wellcome Trust [094232/Z/10/Z] for funding CD facilities. We thank Dr Nasir Khan for assistance with CD. Ms Kirstin Spence for help with protein expression and Dr Thomas A Edwards for useful discussions.References
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- L.-G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Chem. Rev., 2014, 114, 4695–4748 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- A. Vinayagam, U. Stelzl, R. Foulle, S. Plassmann, M. Zenkner, J. Timm, H. E. Assmus, M. A. Andrade-Navarro and E. E. Wanker, Sci. Signaling, 2011, 4, rs8 CrossRef PubMed.
- W. H. Wilson, O. A. O'Connor, M. S. Czuczman, A. S. LaCasce, J. F. Gerecitano, J. P. Leonard, A. Tulpule, K. Dunleavy, H. Xiong, Y.-L. Chiu, Y. Cui, T. Busman, S. W. Elmore, S. H. Rosenberg, A. P. Krivoshik, S. H. Enschede and R. A. Humerickhouse, Lancet Oncol., 2010, 11, 1149–1159 CrossRef CAS PubMed.
- I. Ray-Coquard, J.-Y. Blay, A. Italiano, A. Le Cesne, N. Penel, J. Zhi, F. Heil, R. Rueger, B. Graves, M. Ding, D. Geho, S. A. Middleton, L. T. Vassilev, G. L. Nichols and B. N. Bui, Lancet Oncol., 2012, 13, 1133–1140 CrossRef CAS PubMed.
- P. M. Cromm, J. Spiegel and T. N. Grossmann, ACS Chem. Biol., 2015, 10, 1362–1375 CrossRef CAS PubMed.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302–3315 RSC.
- P. Wysoczanski, R. J. Mart, E. J. Loveridge, C. Williams, S. B. M. Whittaker, M. P. Crump and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 7644–7647 CrossRef CAS PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K.-H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS PubMed.
- D. Wang, W. Liao and P. S. Arora, Angew. Chem., Int. Ed., 2005, 44, 6525–6529 CrossRef CAS PubMed.
- D. J. Yeo, S. L. Warriner and A. J. Wilson, Chem. Commun., 2013, 49, 9131–9133 RSC.
- P. M. Cromm, J. Spiegel, P. Küchler, L. Dietrich, J. Kriegesmann, M. Wendt, R. S. Goody, H. Waldmann and T. N. Grossmann, ACS Chem. Biol., 2016, 11, 2375–2382 CrossRef CAS PubMed.
- Q. Chu, R. E. Moellering, G. J. Hilinski, Y.-W. Kim, T. N. Grossmann, J. T. H. Yeh and G. L. Verdine, MedChemComm, 2015, 6, 111–119 RSC.
- G. H. Bird, E. Mazzola, K. Opoku-Nsiah, M. A. Lammert, M. Godes, D. S. Neuberg and L. D. Walensky, Nat. Chem. Biol., 2016, 12, 845–852 CrossRef CAS PubMed.
- T. Okamoto, K. Zobel, A. Fedorova, C. Quan, H. Yang, W. J. Fairbrother, D. C. S. Huang, B. J. Smith, K. Deshayes and P. E. Czabotar, ACS Chem. Biol., 2012, 8, 297–302 CrossRef PubMed.
- Y.-C. Li, L. W. Rodewald, C. Hoppmann, E. T. Wong, S. Lebreton, P. Safar, M. Patek, L. Wang, K. F. Wertman and G. M. Wahl, Cell Rep., 2014, 9, 1946–1958 CrossRef CAS PubMed.
- T. Okamoto, D. Segal, K. Zobel, A. Fedorova, H. Yang, W. J. Fairbrother, D. C. S. Huang, B. J. Smith, K. Deshayes and P. E. Czabotar, ACS Chem. Biol., 2014, 9, 838–839 CrossRef CAS PubMed.
- M. M. Madden, A. Muppidi, Z. Li, X. Li, J. Chen and Q. Lin, Bioorg. Med. Chem. Lett., 2011, 21, 1472–1475 CrossRef CAS PubMed.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed.
- A. Muppidi, K. Doi, S. Edwardraja, E. J. Drake, A. M. Gulick, H.-G. Wang and Q. Lin, J. Am. Chem. Soc., 2012, 134, 14734–14737 CrossRef CAS PubMed.
- D. P. Fairlie and A. Dantas de Araujo, Pept. Sci., 2016, 106, 843–852 CrossRef CAS PubMed.
- A. J. Rojas, C. Zhang, E. V. Vinogradova, N. Buchwald, J. Reilly, B. Pentelute and S. Buchwald, Chem. Sci., 2017 10.1039/C6SC05454D.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- Y.-W. Kim, P. S. Kutchukian and G. L. Verdine, Org. Lett., 2010, 12, 3046–3049 CrossRef CAS PubMed.
- Y.-W. Kim, T. N. Grossmann and G. L. Verdine, Nat. Protoc., 2011, 6, 761–771 CrossRef CAS PubMed.
- C. Bracken, J. Gulyás, J. W. Taylor and J. Baum, J. Am. Chem. Soc., 1994, 116, 6431–6432 CrossRef CAS.
- R. S. Harrison, G. Ruiz-Gómez, T. A. Hill, S. Y. Chow, N. E. Shepherd, R.-J. Lohman, G. Abbenante, H. N. Hoang and D. P. Fairlie, J. Med. Chem., 2010, 53, 8400–8408 CrossRef CAS PubMed.
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS PubMed.
- A. Patgiri, A. L. Jochim and P. S. Arora, Acc. Chem. Res., 2008, 41, 1289–1300 CrossRef CAS PubMed.
- S. Kneissl, E. J. Loveridge, C. Williams, M. P. Crump and R. K. Allemann, ChemBioChem, 2008, 9, 3046–3054 CrossRef CAS PubMed.
- L. Nevola, A. Martín-Quirós, K. Eckelt, N. Camarero, S. Tosi, A. Llobet, E. Giralt and P. Gorostiza, Angew. Chem., Int. Ed., 2013, 52, 7704–7708 CrossRef CAS PubMed.
- O. Torres, D. Yüksel, M. Bernardina, K. Kumar and D. Bong, ChemBioChem, 2008, 9, 1701–1705 CAS.
- Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Skold, T. J. Sum, P. J. E. Rowling, T. L. Joseph, C. Verma, M. Hyvonen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- Y. H. Lau, P. de Andrade, N. Skold, G. J. McKenzie, A. R. Venkitaraman, C. Verma, D. P. Lane and D. R. Spring, Org. Biomol. Chem., 2014, 12, 4074–4077 CAS.
- P. T. Tran, C. Ø. Larsen, T. Røndbjerg, M. De Foresta, M. B. A. Kunze, A. Marek, J. H. Løper, L.-E. Boyhus, A. Knuhtsen, K. Lindorff-Larsen and D. S. Pedersen, Chem.–Eur. J., 2017, 23, 3490–3495 CrossRef CAS PubMed.
- K. Hu, H. Geng, Q. Zhang, Q. Liu, M. Xie, C. Sun, W. Li, H. Lin, F. Jiang, T. Wang, Y.-D. Wu and Z. Li, Angew. Chem., Int. Ed., 2016, 55, 8013–8017 CrossRef CAS PubMed.
- S. K. Sia, P. A. Carr, A. G. Cochran, V. N. Malashkevich and P. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14664–14669 CrossRef CAS PubMed.
- M. Kvansakul and M. Hinds, Apoptosis, 2015, 20, 136–150 CrossRef CAS PubMed.
- R. T. Raines, Chem. Rev., 1998, 98, 1045–1066 CrossRef CAS PubMed.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- P. Moody, M. E. B. Smith, C. P. Ryan, V. Chudasama, J. R. Baker, J. Molloy and S. Caddick, ChemBioChem, 2012, 13, 39–41 CrossRef CAS PubMed.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS PubMed.
- F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1525 CrossRef PubMed.
- M. P. Robin, P. Wilson, A. B. Mabire, J. K. Kiviaho, J. E. Raymond, D. M. Haddleton and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 2875–2878 CrossRef CAS PubMed.
- C. Marculescu, H. Kossen, R. E. Morgan, P. Mayer, S. A. Fletcher, B. Tolner, K. A. Chester, L. H. Jones and J. R. Baker, Chem. Commun., 2014, 50, 7139–7142 RSC.
- J. Youziel, A. R. Akhbar, Q. Aziz, M. E. B. Smith, S. Caddick, A. Tinker and J. R. Baker, Org. Biomol. Chem., 2014, 12, 557–560 CAS.
- M. P. Robin and R. K. O'Reilly, Chem. Sci., 2014, 5, 2717–2723 RSC.
- J. Collins, J. Tanaka, P. Wilson, K. Kempe, T. P. Davis, M. P. McIntosh, M. R. Whittaker and D. M. Haddleton, Bioconjugate Chem., 2015, 26, 633–638 CrossRef CAS PubMed.
- Oxime and Hydrazone constraints employing non-natural amino acids introduced by Horne and co-workers [ref. 53 and 54] have been used to stabilize a helical conformation but not applied to PPI inhibition.
- C. M. Haney, M. T. Loch and W. S. Horne, Chem. Commun., 2011, 47, 10915–10917 RSC.
- C. M. Haney and W. S. Horne, J. Pept. Sci., 2014, 20, 108–114 CrossRef CAS PubMed.
- Both iminoboronate formation56 and reaction of Meldrums acid with an amine and thiol side chain57 have recently shown to be amenable to reversible peptide cyclization and might be applied to stabilization of peptides for inhibiton of PPIs.
- A. Bandyopadhyay and J. Gao, J. Am. Chem. Soc., 2016, 138, 2098–2101 CrossRef CAS PubMed.
- A. M. Johnson and E. V. Anslyn, Org. Lett., 2017, 19, 1654–1657 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef CAS PubMed.
- S. P. Brown and A. B. Smith, J. Am. Chem. Soc., 2015, 137, 4034–4037 CrossRef CAS PubMed.
- W. Nomura, H. Aikawa, N. Ohashi, E. Urano, M. Métifiot, M. Fujino, K. Maddali, T. Ozaki, A. Nozue, T. Narumi, C. Hashimoto, T. Tanaka, Y. Pommier, N. Yamamoto, J. A. Komano, T. Murakami and H. Tamamura, ACS Chem. Biol., 2013, 8, 2235–2244 CrossRef CAS PubMed.
- J. A. Miles, D. J. Yeo, P. Rowell, S. Rodriguez-Marin, C. M. Pask, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Sci., 2016, 7, 3694–3702 RSC.
- P. Diderich, D. Bertoldo, P. Dessen, M. M. Khan, I. Pizzitola, W. Held, J. Huelsken and C. Heinis, ACS Chem. Biol., 2016, 11, 1422–1427 CrossRef CAS PubMed.
- J. M. Estrela, A. Ortega and E. Obrador, Crit. Rev. Clin. Lab. Sci., 2006, 43, 143–181 CrossRef CAS PubMed.
- P. Monostori, G. Wittmann, E. Karg and S. Túri, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3331–3346 CrossRef CAS PubMed.
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 CAS.
- F. F. Schumacher, M. Nobles, C. P. Ryan, M. E. B. Smith, A. Tinker, S. Caddick and J. R. Baker, Bioconjugate Chem., 2011, 22, 132–136 CrossRef CAS PubMed.
- H. Wang, M. Xu, M. Xiong and J. Cheng, Chem. Commun., 2015, 51, 4807–4810 RSC.
- I. Hamachi, Y. Yamada, T. Matsugi and S. Shinkai, Chem.–Eur. J., 1999, 5, 1503–1511 CrossRef CAS.
- W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radić, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, additional biophysical and biochemical analyses. See DOI: 10.1039/c7sc01342f |
| This journal is © The Royal Society of Chemistry 2017 |