
CO2 capture under humid conditions in metal–organic frameworks
Eduardo
González-Zamora
 *a and
Ilich A.
Ibarra
*b
*a and
Ilich A.
Ibarra
*b
aDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, San Rafael Atlixco 186, Col. Vicentina, Iztapalapa, C. P. 09340, Ciudad de México, Mexico. E-mail: egz@xanum.uam.mx; Fax: +52(55) 5622-4595
bInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacán, 04510, Ciudad de México, Mexico. E-mail: argel@unam.mx
First published on 18th January 2017
Abstract
Metal–organic frameworks (MOFs) are one of the most promising candidates for CO2 capture due to their sorption selectivity towards CO2. The optimisation of the physical–chemical interactions between MOFs and CO2 molecules is the key to further amplification of CO2 capture. An emerging technology for CO2 capture is the construction of hybrid adsorbent MOFs via confinement of water inside the pores. This review article describes the recent progress in this field. Indeed, the pre-adsorption of small quantities of water within the micropores of MOF materials (constructed with hydroxo functional groups, μ2-OH) provides a positive impact on the overall CO2 capture. We anticipate that the current review article can offer useful information on the significant developments made for the enhancement of CO2 capture by confining water (and other solvents) within the pores of MOFs, which is a very promising technology for real-world applications, where MOF materials could be capable of serving as next-generation CO2 capture systems.
 Eduardo González-Zamora | Eduardo González-Zamora, full Professor at the Metropolitan Autonomous University (UAM-I, Mexico), was born in 1961 in Mexico City. He received his MSc degree from UAM-I in 1988. In 1994, he moved to France and obtained his PhD (1998) from Paris XI University (France) under the supervision of Professor R. Beugelmans. After one year of a post-doctoral position with Professor Cruz at the Institute of Chemistry (UNAM, Mexico), he joined the “Institut de Chimie des Substances Naturelles”, CNRS (France) in 2000 as a post-doctoral fellow for two years. In 2011 he moved to UCLA (USA), visiting Professor, in Garcia-Garibay group's. |
 Ilich A. Ibarra | Ilich A. Ibarra was born in Mexico City in 1981. He completed his BSc in chemistry at Universidad Autónoma Metropolitana (UAM-I, Mexico) in 2005. Later, in 2010 he obtained his PhD in chemistry under the supervision of Professor Martin Schröder at Nottingham University (UK). He then took a postdoctoral position, 2010–2012, at The University of Texas at Austin (USA) under the supervision of Professor Simon Humphrey. Then in 2013 he was awarded as a Wenner-Gren researcher at Stockholm University (Sweden) under the supervision of Professor Xiaodong Zou. In 2014, he moved to UNAM (IIM, Mexico), working as an Assistant Professor. |
1. Introduction
The development of our modern world has many socioeconomic, political, technological and environmental implications. The global population is growing quickly and human consumption and energy demands are greater than our planet can tolerate. Unquestionably, in order to meet these demands it is necessary to increase chemical production and the combustion of large volumes of fossil fuels, leading to an indiscriminate release of carbon dioxide (CO2) into the atmosphere. These CO2 emissions generate the undesirable greenhouse effect,1 which is mainly responsible for global warming. A near-linear relationship between global temperature and cumulative CO2 emissions (from all sources: manufacturing processes, fossil fuel combustion and land-use change) has been previously demonstrated.2–4 Thus, a drastic reduction of these CO2 emissions is indispensable in order to limit the increase in global average temperature (below 2 °C).5 Very recently (December 2015), the United Nations Framework Convention on Climate Change (UNFCCC), convened in Paris, agreed to balance the anthropogenic greenhouse-gas budget by the vigorous removal of greenhouse gases from the atmosphere and immediate emission cuts.Different technologies for CO2 removal have been proposed. The success of each of them depends on viability, acceptability and costs. One step forward in these technologies is to capture the CO2 generated during combustion and store it in a suitable place. Carbon Capture and Storage (CCS) refers to a family of technologies and techniques to selectively remove CO2 from fuel combustion or industrial processes, compression of pure CO2 to a supercritical fluid, transportation and finally storage (either subterranean or submarine).6,7 One of the first-generation CCS technologies, which has been employed since 1930,8 is the absorption of CO2 (Lewis acid) by aqueous solutions of alkanolamine (Lewis base). Nevertheless, their main disadvantages, including corrosion of pipelines, heat instability and the substantial costs for their regeneration, considerably restrict the long-term use of these alkaline solutions.9 Alternatively, an emerging CCS technology is based on the use of porous solid materials for the capture of CO2 by adsorption processes and it is one of the most promising technologies to investigate.10 Poliakoff11 proposed ‘the twelve principles of CO2 chemistry’ where CO2 sequestration represents one of these principles (which maximises integration). Thus, the search for porous materials with high structural stability, adsorption capacity, solvent stability, fast sorption kinetics and mild regeneration conditions is a major challenge for scientists.
Metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) are among the most promising CCS candidates (as solid porous materials). Due to their sorption selectivity towards CO2, which can be directly tailored as a function of the topology and chemical composition of the pores,12–17 MOF materials provide a vast CCS research field.18 The most important aspect for CO2 capture in MOFs is the optimisation of the physical–chemical interactions between these materials and CO2 molecules. Interestingly, the search for different synthetic strategies in order to increase these interactions has afforded the discovery of new functional MOF materials with enhanced CO2 capture properties.19–22 Currently, the three foremost synthetic approaches to enhance CO2 interactions within MOF systems are: (i) the introduction of functional groups with high polarity (e.g. –OH, –CN, –NO2, –SH, etc.) which can induce polarisation of CO2 molecules (possessing a large quadrupole moment) resulting in an enhanced affinity,8,18 (ii) the generation of uncoordinated metal sites,23 and (iii) the chemical functionalisation of the pores with basic (Lewis) nitrogen-containing groups such as triazole,24,25 amine,19,26 and tetrazole.27 In particular, a cutting edge development was presented by Long et al.,26b where CO2 molecules were inserted into metal–amine bonds (in mmen-Mn2(dobpdc)), inducing a large CO2 separation capacity.
Although MOF materials have shown very promising capabilities for CO2 capture, their main drawback is their vulnerability towards H2O,28 since many CO2 separation processes in industry involve, unwittingly, contact with significant amounts of water vapour. For example, one of the products from the burning of fossil fuels is industrial flue gas, which commonly contains 5 to 7% of water.18 Indeed, H2O vapour molecules can compete with CO2 gas molecules for the active sites within MOF materials and thus drastically decrease the overall CO2 capture capacity. In addition, H2O can disrupt the coordination bonds between the organic ligands (e.g. carboxylate) and metal centres, resulting in the disintegration of the MOF structure.29
Therefore, water stability of MOFs is a fundamental requirement for any CO2 capture application. Water stable MOFs are commonly classified into three categories: (i) materials with high valence metal ions coordinated to carboxylate ligands; (ii) metal azolate frameworks; and (iii) functionalised MOFs with hydrophobic functional groups.30 Recently, a considerable amount of water-stable MOFs have been reported and some representative examples are: UiO-66,31 MIL-101,32 NOTT-400,33 Cu(bcppm)H2O,34 InOF-1,35 NOTT-40136 and Mg-CUK-1.37
Emerging and very interesting applications have been explored for some of these moisture-stable MOF materials.38 For example, a series of Zr(IV) MOFs39 showed very promising properties in water-storage technologies for the delivery of drinking water which could be applied in arid and remote areas. Yang40 reported a composite membrane based on ZIF-8/PMPS (PMPS = polymethylphenylsiloxane) for the recovery of furfural (furan-2-carbaldehyde) from water, showing a pervaporation performance of 0.9 kg m−2 h−1 total flux and a separation factor of 53.3. Bradshaw and co-workers used UiO-66-NH2 as a fluorescent pH sensor.41 Yan et al.42 incorporated Eu3+ ions within the channels of CPM-17-Zn to detect cations in aqueous solutions. The MOF material Al2(OH)2TCPP-Co (TCPP = 4,4′,4′′,4′′′-(porphyrin-5,10,15,20-tetrayl)tetrabenzoate) was shown to be very efficient for the reduction of CO2 to CO in aqueous electrolytes.43 Kitagawa et al.44 demonstrated high proton conductivity and high adsorption of water under low humidity conditions, in a novel class of MOFs, {NR3(CH2COOH)}[MCr(ox)3]·nH2O (R = methyl, ethyl or n-butyl and M = Mn or Fe). Finally, Janiak and Henninger have proposed the use of MOFs for low temperature heat transformation applications in thermally driven adsorption chillers (TDCs) or adsorption heat pumps (AHPs).45
Although these promising applications of water stable MOFs (H2O storage, membrane separation, sensing, catalysis, proton conductivity and heat-pump chillers) have been recently reviewed,38,45 an emerging technology for CO2 sequestration (based on the generation of hybrid adsorbent MOFs via confinement of water inside the pores) is presented in this review. Significant work on this new application is comprehensibly discussed with an eye on the near future for this developing technology. It is important to emphasise that the pre-adsorption of water within the pores of MOF materials is not an easy task to achieve. The disadvantages of pre-installing water in MOFs arise from their hydrophobic nature, low kinetic adoption rates, partial blocking of the pores and instability. At present, framework damage by water is a limitation. However, with the high diversity of synthetic strategies for the design of new MOF materials, it is realistic to expect many MOFs which will exhibit water stability.
The present contribution aims to provide a useful reference in the field of CO2 capture and to encourage more investigation groups to explore the exiting frontiers of knowledge in environmental applications for MOFs.
2. First evidence
In 2006, Llewellyn and co-workers46 reported how water inside the channels of material MIL-53(Cr) can significantly improve the adsorption selectivity of CO2 over methane (CH4). MIL-53(Cr) is constructed from infinite chains of corner-sharing CrO4(OH)2 octahedra which are linked by BDC (1,4-benzenedicarboxylate). The three dimensional structure of MIL-53(Cr) shows very particular diamond-shaped channels with a pore diameter of approximately 8.5 Å. Interestingly, in the presence of water these channels show a ‘breathing effect’ when the material is under ambient conditions (hydrated) and activated (dehydrated). This breathing phenomenon47 comprises two forms of MIL-53(Cr) channels; in the hydrated form (Fig. 1, left) the pores are slightly deformed due to hydrogen-bonding interactions between water molecules inside the channels and the carboxylic ligands and the μ2-OH groups of MIL-53(Cr). When the material is activated (heating under vacuum), water molecules can be removed from the inside of the pores affording a structure with a more regular diamond-shaped channels (Fig. 1, right), which, essentially, provides a more open porosity.47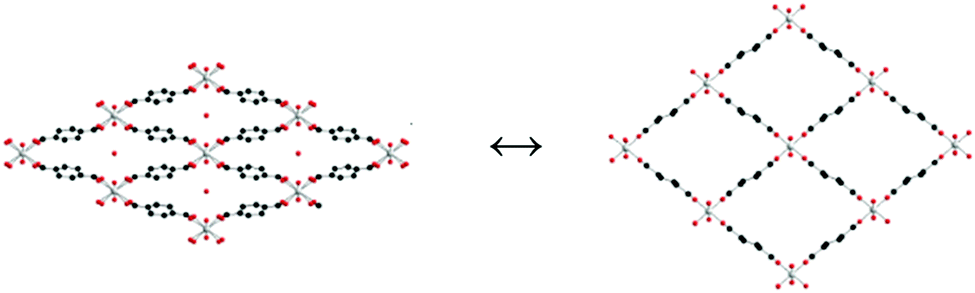 | ||
| Fig. 1 Views of the pore-shapes from: (left) hydrated MIL-53(Cr) and (right) dehydrated MIL-53(Cr). The hydration and dehydration is a reversible process. | ||
In this early study46 adsorptions of CO2 and CH4 were carried out on both forms of MIL-53(Cr): hydrated and dehydrated (Fig. 1). The final conclusion of these experiments was that the hydrated form was better for the recovery of CO2 from CH4. In Fig. 2 the CO2 and CH4 isotherms for both hydrated and dehydrated MIL-53(Cr) systems is presented. Clearly, the hydrated material showed almost no methane uptake (∼0.2 mmol g−1) even at 20 bar. The authors attributed this behaviour to be a result of the repulsive effect of water (polar molecules) towards nonpolar CH4 molecules. The overall consequence of this result was that the apparent selectivity drastically increased (CO2 over CH4) due to the presence of water. Although this selectivity was clearly demonstrated and led to potential applications for recovering CO2 in natural-gas feeds in which water contents are significant, the interesting behaviour of both CO2 adsorption isotherms in the two MIL-53(Cr) systems can be discussed in more detail. For example, in the dehydrated material there is a sharp CO2 uptake of 2 mmol g−1 from 0 to approximately 1 bar (Fig. 2). This suggests a very high affinity of the material for CO2, presumably, due to the relatively strong interactions between CO2 molecules and the μ2-OH groups as was recently demonstrated by Yang et al.48 On the other hand, the CO2 adsorption isotherm of hydrated MIL-53(Cr) did not show any CO2 uptake (at 1 bar). In fact, when the pressure was increased up to 10 bar, the CO2 uptake was approximately 0.5 mmol g−1 (Fig. 2), which indicates a considerably high difficulty for CO2 molecules to be adsorbed within the hydrated material. Later, while increasing the pressure, a sharp increase in the CO2 uptake takes place to finally reach a total CO2 uptake of nearly 8 mmol g−1 at 20 bar. This particular behaviour is associated with the breathing nature of the material, and remarkably when the total CO2 uptake is compared to the one from the dehydrated material (9 mmol g−1 at 20 bar) there is only a small drop in the CO2 capture. The precise amount of water is not described in this work, but it is clear that its presence did not drastically diminish the adsorption of CO2 and this result opened the possibility of studying the effect of pre-adsorbed water in MOFs for the capture of CO2.
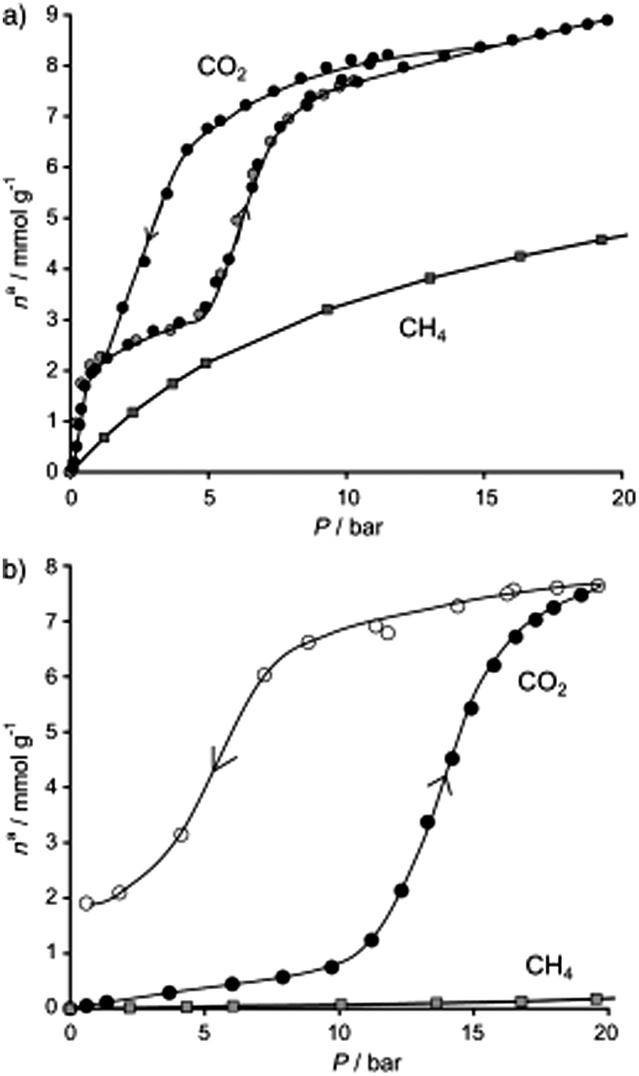 | ||
| Fig. 2 Comparison of the adsorption isotherms of CO2 and CH4 at 20 bar and 304 K on (a) dehydrated and (b) hydrated forms of MIL-53(Cr). (Reproduced with permission from ref. 46. Copyright© 2006 Wiley-VCH Verlag GmbH & Co.) | ||
Later in 2009, Snurr et al.49 demonstrated (predicted by GCMC molecular simulations and DFT calculations to finally be validated by adsorption experiments) that the pre-adsorption of small amounts of water in the material HKUST-1 increases the CO2 uptake and its selectivity over N2 and CH4. HKUST-1 (Cu3(BTC)2(H2O)3, BTC = 1,3,5-benzene-tricarboxylate)50 is perhaps one of the most well-studied MOFs which exhibits framework Cu(II) metal centres (coordinated to the BTC ligands and terminal water molecules, resembling paddle-wheels) from which H2O, under suitable activation conditions, can be removed to leave coordinatively unsaturated Cu(II) metal sites where the oxidation state of copper remains unchanged and the crystallinity of the material is maintained. Thus, the water loading that Snurr et al.49 proposed in their study was 4 wt%, which was estimated by simply deleting (derived from the single crystal X-ray diffraction structure) one of the coordinated H2O molecule from each paddle-wheel corner to leave only one Cu(II) metal centre coordinated to H2O (see Fig. 3).
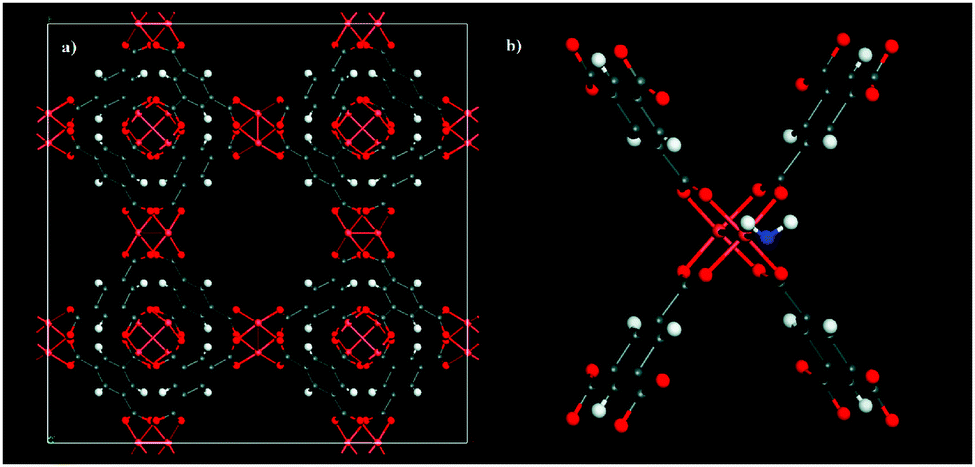 | ||
| Fig. 3 (a) Anhydrous HKUST-1 unit cell, (b) hydrated HKUST-1 (4 wt%) showing one coordinated water molecule to the Cu(II) paddle-wheel. (Reprinted with permission from ref. 49. Copyright© 2009, American Chemical Society.) | ||
Then, CO2 adsorption isotherm experiments were carried out from 0 to 5 bar and over the entire pressure range, the material HKUST-1 loaded with 4 wt% of water adsorbed more CO2 than the anhydrous sample (0 wt% of water), see Fig. 4. This was also confirmed by the computational simulations (GCMC and DFT) and finally corroborated by the very small adsorption of CO2 which was found for a fully hydrated (pores of the material completely occupied by water) HKUST-1 (Fig. 4). Interestingly, upon increasing the water loading on HKUST-1 to 8 wt%, the CO2 uptake was considerably lower than at 4 wt% (and even lower than the anhydrous material). Since at 8 wt% the material contains uncoordinated water molecules that block the free space inside the pores of HKUST-1 (similar to the water saturated HKUST-1), the CO2 adsorption is lower. For the first time the significance of having small amounts of pre-adsorbed water in order to obtain a higher CO2 uptake was established.
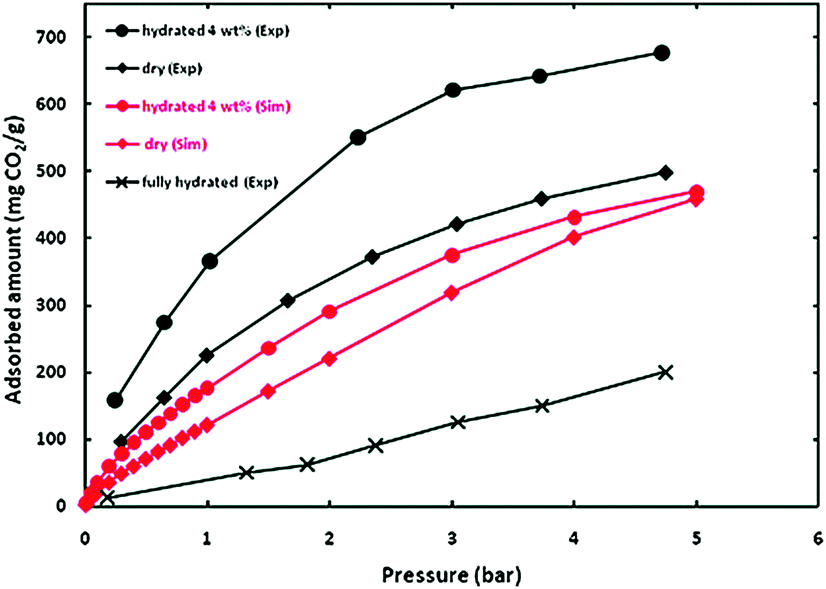 | ||
| Fig. 4 Simulated and experimental adsorption isotherms for CO2 at 298 K in HKUST-1 with different water contents. (Reprinted with permission from ref. 49. Copyright© 2009, American Chemical Society.) | ||
3. Current developments
After the previous finding presented in the last section, researchers focused their efforts on studying the effect of water on the CO2 capture in MOFs. Matzger et al.51 investigated the effect of humidity on a MOF series named M–DOBDC (M = Zn(II), Ni(II), Co(II) and Mg(II); DOBDC = 2,5-dioxidobenzene-1,4-dicarboxylate), by allowing a gaseous mixture of N2/CO2/H2O to flow through these materials until breakthrough was achieved for each component and was followed by the re-activation of the samples (thermal treatment) to finally evaluate the post-regeneration CO2 capacities. Then, the exposure to different relative humidities (RHs) (9, 36 and 70%) was carried out on these MOF materials followed by thermal regeneration to find that Co–DOBDC showed 85% of the initial CO2 capacity. Indeed, Matzger51 demonstrated that although CO2 capacity under dry conditions is an important parameter in the search for a good MOF candidate for CO2 capture, post-regeneration CO2 capacity retention after exposure to humidity is definitely a more important parameter for further CO2 sequestration developments. In the same context, Le Van and co-workers52 studied a couple of MOF systems: Ni–DOBDC and Mg–DOBDC. Their results showed that the material Ni–DOBDC kept its CO2 capacity almost intact after water vapour conditioning, while in the other material (Mg–DOBDC) the CO2 uptake was significantly reduced.52 In addition, Le Van et al.53 found that a small amount of adsorbed water did not decrease, and may actually increase, the CO2 capacity of the MOF material HKUST-1. Remarkably, Shekhah and Eddaoudi showed that CO2 sequestration by the material SIFSIX-3–Cu (a metal–organic framework based on pyrazine and Cu(II)) was not affected by the presence of water vapour (74% RH).54 Thus, these significant contributions emphasised the importance of water stability in MOFs for CO2 capture applications, since contact with water vapour is unavoidable in a more realistic CO2 capture scenario.Later, Liu and co-workers55 proposed a novel methodology to enhance the methane (CH4) storage capacity of the material ZIF-8 by forming a hydrate. The inclusion of different quantities of water within the MOF material (0.0%, 16.3%, 27.7%, 30.6% and 35.1%) was investigated under hydrate formation conditions, since it is well known that natural gas (CH4) and water can form a gas hydrate at a suitable temperature and pressure. Their results exhibited that hydrates can form in wet ZIF-8 pores and improve the net CH4 storage capacity up to 56%. For example, the CH4 uptake of dry ZIF-8 is 5.9 mmol g−1 at 296 K and 28.5 bar. When the water content in the sample was 35.1%, at the same temperature and pressure, the total CH4 uptake increased up to 9. 3 mmol g−1.55 In addition to this very important result, Liu provided the proof of concept of this methodology by investigating the adsorption behaviour of CH4 in the pores of ZIF-8 and finding that part of the volume is not used effectively. By pre-adsorbing a suitable amount of water within the pores of the material, the overall CH4 storage capacity is substantially improved due to the effective use of the pore volume.
In 2012, Llewellyn et al.56 reported a comprehensive study on the enhancement of CO2 capture, in three different topical MOF materials, by pre-adsorbing water. In fact, this study represents a benchmark work for the present review since, in the context of post-combustion CO2 capture, they built a flow system in which a carrier gas (N2), CO2 and different quantities of H2O vapour (3, 10, 20 and 40% relative humidity (RH)) were allowed to flow through different MOF materials (HKUST-1, UiO-66 and MIL-100(Fe)).56 The UiO-66 MOF material (Zr6O4(OH)4(BDC)6, BDC = 1,4-benzenedicarboxylate) is constructed from zirconium oxoclusters, which are connected via 12 BDC ligands.31 HKUST-1 is built from Cu(II) paddlewheels connected together through 4 BTC ligands,50 and MIL-100(Fe), (Fe3O(H2O)2X(BTC)2 (X = F or OH)),57 is a mesoporous MOF material that shows two types of mesoporous cages of 25 and 29 Å, respectively, which are accessible through microporous windows of dimension 4.7 × 5.5 and 8.6 Å.57
First, water isotherm cycles were performed in all three MOF materials in order to investigate their water vapour stability (Fig. 5). Then, water adsorption isotherms from 0 to 100% RH at 298 K were cycled six times, without any further activation, on each sample. For material UiO-66, a recyclable Type-V isotherm, was observed with a significant water uptake around 20% RH. On the other hand, HKUST-1 showed a Type-I isotherm, but without recyclability, because this is followed by a decrease in water uptake, to lead to more than 50% loss in mass at the sixth run (Fig. 5). In addition, this loss of water capacity was corroborated by N2 adsorption and PXRD experiments. In the case of MIL-100(Fe), a Type-V isotherm was observed with a marked hysteresis loop where the water uptake started at approximately 25% RH and stabilised at 50% RH. Interestingly, the recyclability of this sample is remarkable since each of the six water isotherms overlapped, confirming no sample degradation after water cycling.
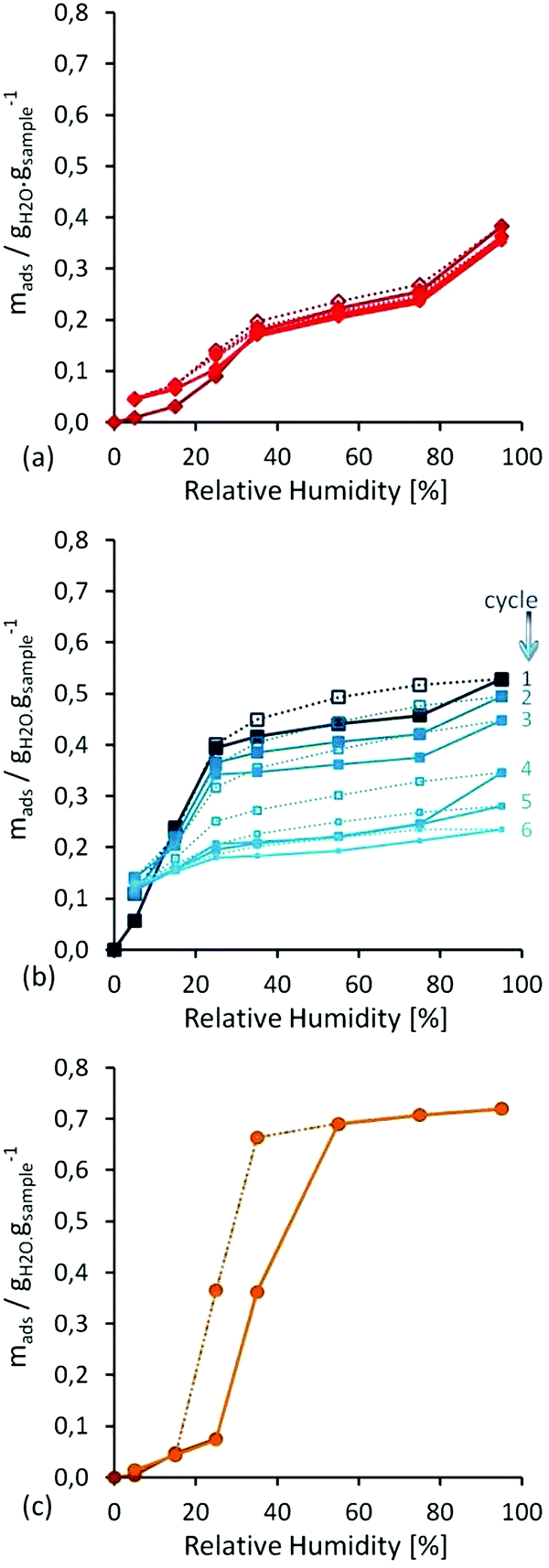 | ||
| Fig. 5 Water adsorption–isotherm cycles performed at 298 K on (a) UiO-66, (b) HKUST-1 and (c) MIL-100(Fe). (Reprinted with permission from ref. 56. Copyright© 2012, American Chemical Society.) | ||
After the evaluation of the water stability for these materials, CO2 adsorption experiments in the presence of water were carried out on HKUST-1, UiO-66 and MIL-100(Fe) by using a built laboratory scale dynamic system (including a Tian-Calvet microcalorimeter to directly measure heat changes) at 1 bar with nitrogen as a carrier gas. Thus, CO2 uptake (estimated from breakthrough curves) was performed at a partial pressure of 0.2 (characteristic partial pressure from CO2 emissions) and the RH varied (3, 10, 20 and 40%) at this fixed CO2 partial pressure. The CO2 uptake in the UiO-66 material showed only minor differences under different amounts of water vapour, which are calculated to be within experimental error (Fig. 6). The enthalpy of adsorption signals observed during the CO2 adsorption experiments were very similar, from −21 to −26 kJ mol−1, for all the RHs and the initial enthalpy of adsorption of pure CO2 (anhydrous) on this MOF material was reported earlier58 with a value of around −25 to −26 kJ mol−1. Thus, the CO2 uptake in UiO-66 was barely modified by the presence of water vapour.
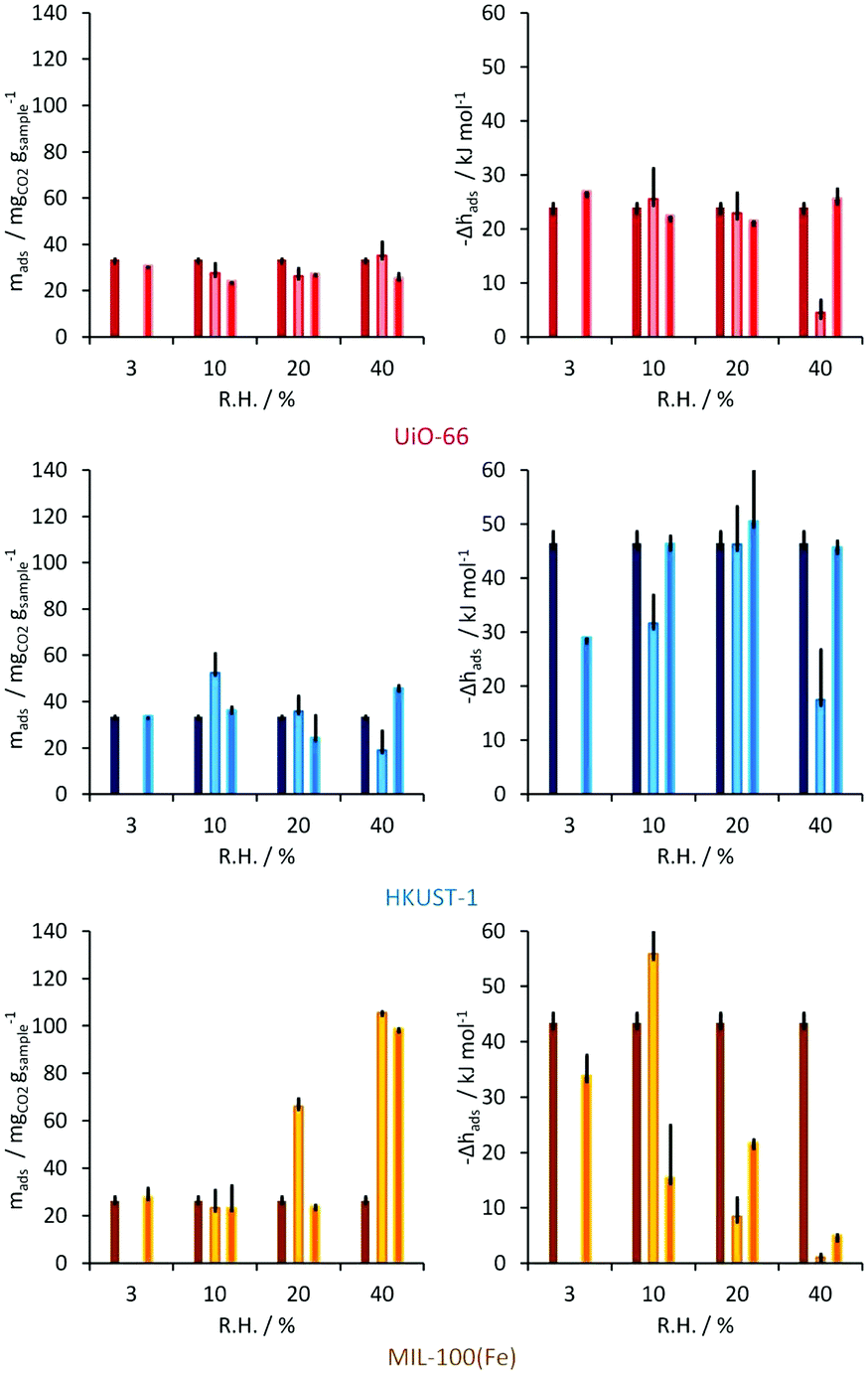 | ||
| Fig. 6 Dynamic CO2 adsorption for UiO-66 (top), HKUST-1 (middle) and MIL-100(Fe) (bottom). Left: Amounts of CO2 adsorbed for various relative humidities. Right: Corresponding adsorption enthalpies. Black bars show standard deviations. (Reprinted with permission from ref. 56. Copyright© 2012, American Chemical Society.) | ||
In the case of the material HKUST-1, the behaviour is different; at 3% RH the CO2 uptake is slightly lower (35 mg g−1) than under anhydrous conditions (0% RH, 40 mg g−1)59 but at 10% RH there is an increase in the CO2 uptake up to 52 mg g−1 (Fig. 6). When the RH was increased to 20 and 40% RH, the total CO2 capture was considerably decreased to 41 and 20 mg g−1, respectively. This drop in the CO2 adsorption capacity was demonstrated to be due to sample degradation as shown by a lower N2 adsorption (77 K) uptake than the anhydrous HKUST-1 sample. The enthalpy of adsorption for CO2 at 3% RH was measured to be between −43 and −44 kJ mol−1, which was considerably higher than for the anhydrous material (−29 kJ mol−1).59 Thus, the overall results on HKUST-1 are in good agreement with those of Snurr et al.49 and Le Van53 in which a small quantity of water appears to increase the CO2 uptake and conversely, at higher water amounts the uptake of CO2 decreases.
MIL-100(Fe) showed very interesting and promising results. At 3% RH, CO2 uptake reached 26 mg g−1 with corresponding adsorption enthalpies of −43 and −34 kJ mol−1 for the repeated second run (Fig. 6). These values can be compared with those previously reported, by Férey and co-workers,60 for anhydrous CO2. At 10% RH, a small decrease in the amount of CO2 captured on MIL-100(Fe) is observed with a corresponding enthalpy of adsorption of approximately −56 kJ mol−1. When the amount of pre-adsorbed water is increased to 20% RH the overall CO2 uptake is considerably augmented to approximately 66 mg g−1 and remarkably upon increasing to 40% RH, the total CO2 capture reached 105 mg g−1 (Fig. 6). Thus, at 40% RH, five times more CO2 is adsorbed than under anhydrous conditions.
These results suggested that the pre-adsorbed water, within the mesopores of the material MIL-100(Fe), forms microporous pockets (Fig. 7a), which are filled with CO2 (Fig. 7b) at lower pressures. However, it seems that CO2 molecules are able to relocate some of the water molecules (Fig. 7c). On the other hand, the microporous material UiO-66 did not show any enhanced CO2 capture at low pressures in the presence of water molecules. This phenomenon was attributed to the absence of accessible microporosity (Fig. 7d), since pre-adsorbed water occupies that free space, even though CO2 is capable of displacing some water molecules (Fig. 7e).
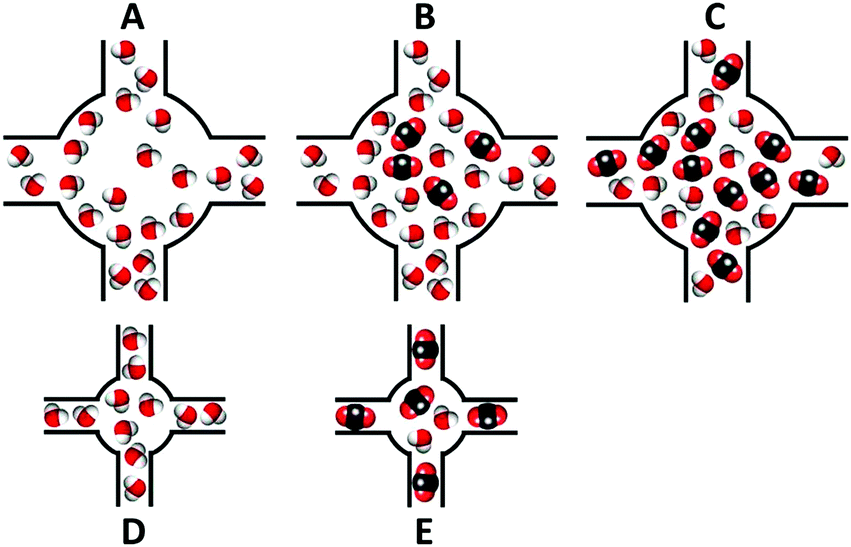 | ||
| Fig. 7 Schematic representation of possible mechanisms of CO2 adsorption in the presence of humidity. (A) Pre-equilibrated water in the mesoporous MIL-100(Fe) forms microporous pockets; (B) these microporous pockets are filled with CO2; (C) CO2 displacing some water molecules; (D) microporous materials (zeolites and UiO-66) saturated with water and (E) pre-adsorbed water in microporous materials do not produce any enhanced CO2 uptake. (Reprinted with permission from ref. 56. Copyright© 2012, American Chemical Society.) | ||
According to these outstanding results from Llewellyn,56 the pre-adsorption of water molecules can considerably enhance the CO2 capture in mesoporous MOF materials (e.g. MIL-100(Fe)) and conversely cannot work for microporous materials (UiO-66). Interestingly, Walton et al.61 functionalised this microporous MOF material (UiO-66) with nonpolar methyl groups (–CH3) to obtain UiO-66-MM. UiO-66-MM is less hydrophilic than the parent UiO-66 and thus adsorbs almost 30% less water than UiO-66. UiO-66-MM showed a much higher CO2/CH4 selectivity (under dry conditions) due to enhanced van der Waals interactions with CO2 molecules and since this material adsorbed less water than UiO-66, UiO-66-MM was expected to show a much better performance (over UiO-66) in humid gas separations. Although in this work there is no explicit enhancement of the CO2 capture derived from pre-adsorbing water, the incorporation of functional groups (such as –CH3) within the pores of microporous MOF materials opened the door to a general strategy for tuning the water vapour and CO2 adsorption properties.
In the search for effective and efficient methods for functionalisation of MOF materials, Zhang and co-workers62 proposed the incorporation of monodentate hydroxide as an active site for the adsorption of CO2. Thus, the framework material MAX-X25ox (Mn(II)Mn(III)(OH)Cl2(bbta), bbta = 1H,5H-benzo(1,2-d:4,5-d′′)bistriazole) showed very high CO2 capture, CO2/N2 selectivity, good recycling stability, very fast sorption kinetics and ultrahigh working capacity for sequestering CO2 under flue gas conditions, even in the presence of water. Although water was not used for enhancing CO2 capture, the incorporation of particular functional monodentate hydroxide groups within MAX-X25ox modified the CO2 and water adsorption properties considerably since CO2 and monodentate hydroxide react to form a reversible bicarbonate.62
Long et al.63 functionalised the framework material Mg2(dobpdc) (dobpdc = 4,4′-dioxido-3,3′-biphenyldicarboxylate) with N,N′-dimethylethylenediamine (mmen) and outstandingly, it exhibited a record CO2 capacity of 4.2 mmol g−1 at 0.1 bar and 40 °C in the presence of a high amount of water (partial pressure = 19 mbar).
Our group focused CO2 capture research on water-stable MOF materials that exhibited hydroxo functional groups (μ2-OH). First, the framework material named InOF-1,64 which is constructed from a flexible BPTC (biphenyl-3,3′,5,5′-tetracarboxylate) and based on a binuclear [In2(μ2-OH)] building block, was employed to study its CO2 capture properties in the presence of water vapour.65 Remarkably, when this material was exposed to water (relative humidity, RH) there was a significant 2-fold increase (approximately 11 wt%) in CO2 capture under 20% RH and 30 °C (Fig. 8).
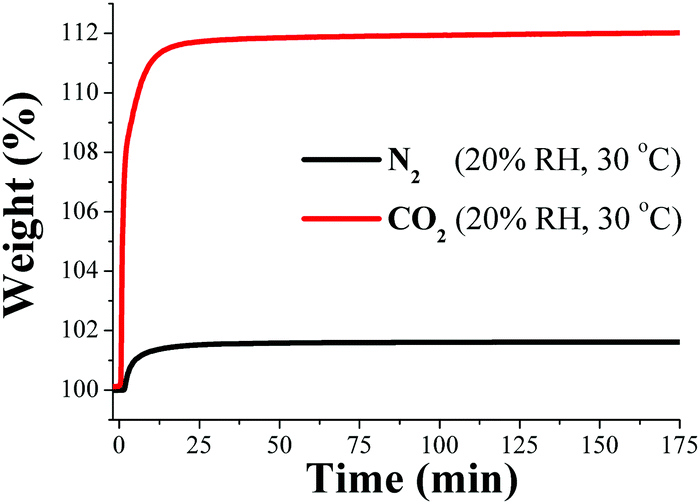 | ||
| Fig. 8 Kinetic uptake experiments carried out at 30 °C and 20% RH with CO2 (red line) and N2 (black line) flow rates of 60 mL min−1, respectively. (Reproduced from ref. 65 with permission from the Chinese Chemical Society (CCS), Peking University (PKU), and the Royal Society of Chemistry.) | ||
This enhancement of CO2 capture in the presence of water was explained by CO2 confinement effects induced by bulky molecules (H2O),66 since functional groups (e.g. hydroxo functional groups inside the pores of InOF-1) act as a directing agent for water in the pores allowing more efficient packing, as demonstrated by Walton and co-workers.67
When the RH was increased to 40%, the CO2 capture (40% RH and 30 °C) was considerably reduced in comparison with that under anhydrous conditions (from 5.24 to 1.00 wt%). Presumably, at 40% RH the saturation of the micropores in InOF-1, with H2O molecules, was completed and therefore, the inclusion of CO2 molecules, into the micropores, was not possible.65 Finally, it was also demonstrated that this MOF material captured CO2 under humid conditions (20% RH) and at moderately high temperatures (40 and 50 °C) which are necessary in a more realistic CO2 capturing state, like flue gas.
Later, the water adsorption properties and the CO2 capture were investigated, in the presence of water, on an isostructural water stable MOF material named NOTT-40068 (also known as MFM-40069) to corroborate this hypothesis. This material is also constructed with BPTC and Sc(III) metal centres which are arranged on a binuclear [Sc2(μ2-OH)] building block (such as in InOF-1). First, water isotherms were obtained on an activated NOTT-400 sample at 30 °C and in Fig. 9 the water adsorption isotherm is shown.70
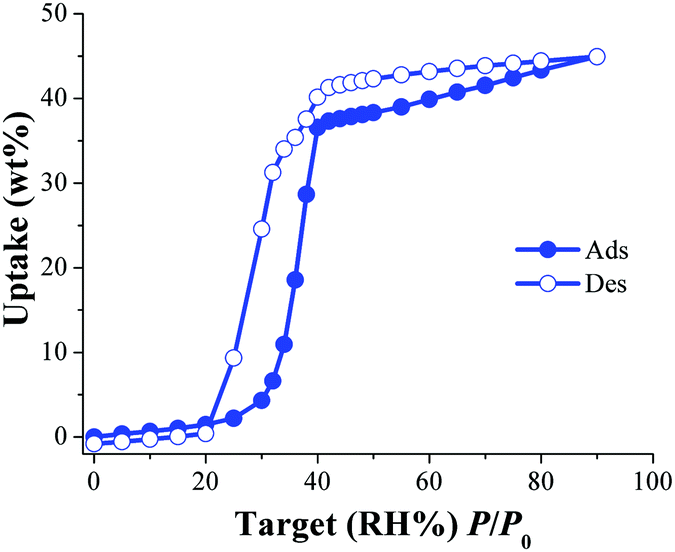 | ||
| Fig. 9 Water adsorption isotherm at 30 °C of NOTT-400. Solid circles represent adsorption, and open circles show desorption. (Reproduced from ref. 70 with permission from the Chinese Chemical Society (CCS), Peking University (PKU), and the Royal Society of Chemistry.) | ||
Dynamic and isothermal CO2 experiments were performed on an activated sample of NOTT-400 and the maximum amount of CO2 captured, under anhydrous conditions, was 4.4 wt% at 30 °C. Then, dynamic CO2 experiments at 30 °C under different relative humidities (10, 20, 35 and 60% RH) were performed on activated NOTT-400 samples. At 20% RH the total CO2 uptake was equal to 10.2 wt% representing a 2.5-fold CO2 capture increase (from anhydrous conditions to 20% RH), Fig. 10.
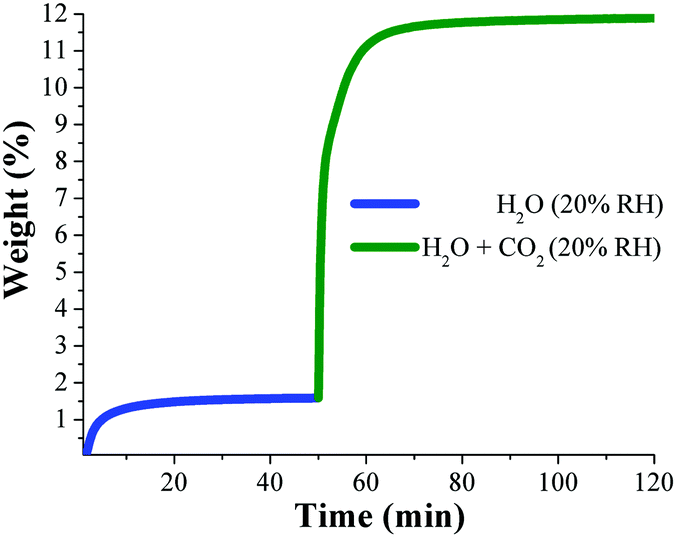 | ||
| Fig. 10 Kinetic uptake experiments performed at 20% RH (at 30 °C); H2O (blue line) and H2O + CO2 (green line). (Reproduced from ref. 70 with permission from the Chinese Chemical Society (CCS), Peking University (PKU), and the Royal Society of Chemistry.) | ||
It was then demonstrated in these two isostructural MOF materials (InOF-1 and NOTT-400) that when small quantities of water are pre-adsorbed into the micropores, they could considerably enhance the CO2 capture. Experimental evidence presented in these two examples concluded that water molecules could strongly interact with the hydroxo functional groups (μ2-OH) of these MOF materials, by hydrogen bonding, generating a reduction of the micropores (confinement effect), which permits a more efficient packing of CO2 molecules.
Then, a different water-stable MOF material which exhibited the same hydroxo functional groups (μ2-OH) and also belongs to the microporous system (pore diameter of 6.3 Å) was tested for the CO2 capture under humid conditions. NOTT-40168 (also known as MFM-40169) is based on a binuclear [Sc2(μ2-OH)] building block and 2,5-thiophenedicarboxylate. Thus, Fig. 11 shows the water adsorption isotherm of activated NOTT-401.71
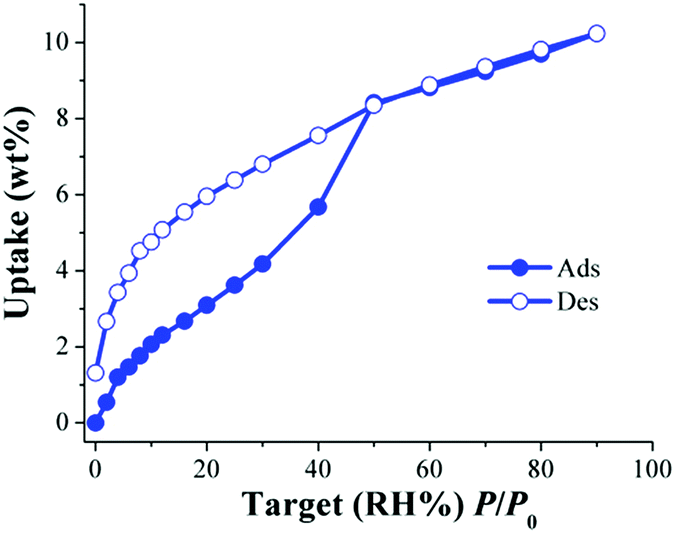 | ||
| Fig. 11 Water adsorption isotherm at 30 °C of NOTT-401. Solid circles represent adsorption, and open circles show desorption. (Reprinted with permission from ref. 71. Copyright© 2016, American Chemical Society.) | ||
Dynamic and isothermal (30 °C) CO2 capture experiments (anhydrous) gave a total CO2 uptake of 1.2 wt%. In agreement with the information obtained from the water adsorption isotherm (see Fig. 11), kinetic CO2 isotherm experiments at 30 °C and under different RHs (5, 20, and 30% RH) were performed. These three RHs corresponded to the low uptake region where the H2O uptake slowly increased with increasing pressure (see Fig. 11). The total CO2 uptakes at 5, 20 and 30% RH were 3.9, 1.9 and 0.4 wt%, respectively (Fig. 12).71 Thus, the CO2 capture augmented from 1.2 wt% (anhydrous conditions) to 3.9 wt% under 5% RH at 30 °C, signifying a 3.2-fold improvement.
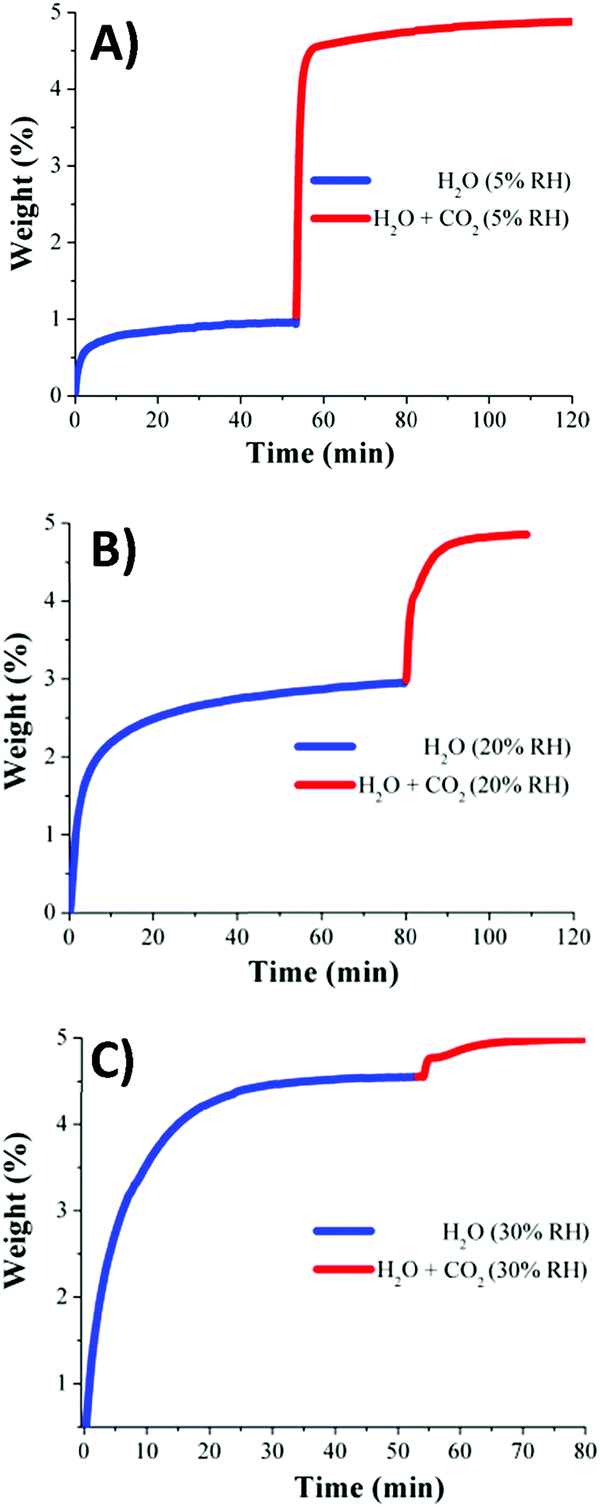 | ||
| Fig. 12 Kinetic CO2 uptake experiments performed at 30 °C and a series of relative humidities: (A) 5, (B) 20, and (C) 30% RH; H2O (blue line) and H2O + CO2 (red line). (Reprinted with permission from ref. 71. Copyright© 2016, American Chemical Society.) | ||
In the last three examples (InOF-1, NOTT-400 and NOTT-401) it was experimentally demonstrated that by pre-adsorbing small amounts of water in these microporous materials the CO2 capture was considerably enhanced. When these MOF materials adsorbed small quantities of water it was hypothesised that a first adsorption domain was created where these H2O molecules interact relatively strongly with the hydroxo functional groups (μ2-OH) by hydrogen bonding and this was mainly observed in the hysteresis of the water adsorption isotherms and the isosteric heat of adsorption for H2O. Thus, once these H2O molecules are “pinned” to the μ2-OH groups, a reduction of the micropores takes place (confinement effect), which allows a better packing of CO2 molecules and therefore, this provides a positive impact on the overall CO2 capture. Later, when the amounts of pre-adsorbed water were increased, a second adsorption domain was reached and there was no CO2 capture improvement, due to H2O saturation within the micropores of the MOF materials. Interestingly, these experimental water adsorption domain findings were previously described using computational spectroscopy.72
Paesani et al.72 investigated using computational infrared spectroscopy the behaviour of water confined in a microporous MOF material MIL-53(Cr).73 MIL-53(Cr) is constructed of one-dimensional chains of corner-sharing CrO4(μ2-OH) octahedra, linked by 1,4-benzenedicarboxylate (BDC) ligands. Although, MIL-53(Cr) is not an isostructural MOF material to any of the examples described before (vide supra, InOF-1, NOTT-400 and NOTT-401) it also exhibits the same hydroxo functional groups (μ2-OH). Thus, they demonstrated72 that H2O molecules (at low water loadings) interact strongly with the framework, via hydrogen bonding between the μ2-OH functional group and H2O molecules, corresponding to the first adsorption domain. Water molecules bound to the μ2-OH groups in the MIL-53(Cr) framework were also corroborated by molecular dynamics (MD)74 and Grand Canonical Monte Carlo (GCMC) by Maurin and co-workers.75 Upon increasing the amount of adsorbed water, the second adsorption domain occurs and intermolecular interactions between H2O molecules become considerably stronger (Fig. 13). Thus, confinement effects resulted in two distinct chemical environments (first and second water adsorption domains) depending on the amount of water within the micropores of MIL-53(Cr).
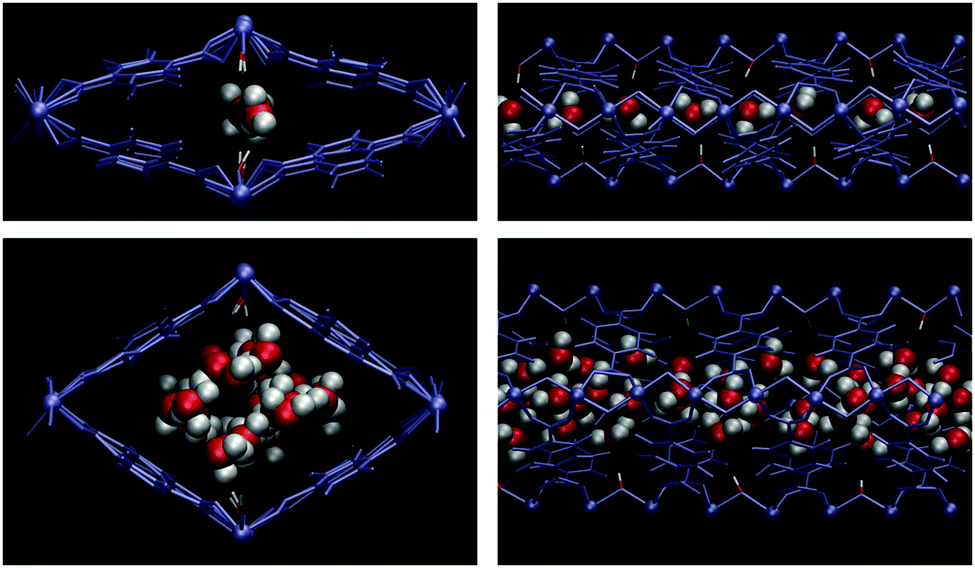 | ||
| Fig. 13 MD snapshots of water adsorbed in MIL-53(Cr). The top panels correspond to a loading of four molecules per unit cell, while the bottom panels correspond to a loading of 20. The μ2-OH moiety (the red/white rod coordinating the chromium ions) strongly influences the water structure for NW = 4, but its effect is diminished at higher loadings. The left panels depict a cross section of the XY plane, while the right panels show the YZ plane. (Reprinted with permission from ref. 72. Copyright© 2014, American Chemical Society.) | ||
These computational spectroscopy studies provided a better understanding of the experimental evidence, which can be summarised as follows: at low water loadings in InOF-1, NOTT-400 and NOTT-401 the channels (of these MOF materials) provide a template (via the μ2-OH groups) for a more efficient packing of the H2O molecules and therefore, these H2O molecules produce a reduction of the micropores which allows a better storage of CO2 molecules (water can donate a hydrogen bond to the CO2 molecules) to finally enhance the total CO2 uptake.
Since the computational study was performed on MIL-53(Cr), accordingly, it was decided to test the isostructural MOF material MIL-53(Al) to investigate its CO2 capture properties in the presence of water.76 By following the methodology previously described (see above) for NOTT-400 and NOTT-401, a total CO2 uptake of 3.5 wt% at 30 °C was achieved. For this study, only two relative humidities were explored: 20 and 40% RH. At 40% RH and 30 °C (pre-adsorbed water of 3 wt%) the CO2 capture was approximately 4.8 wt%, representing a 1.3-fold increase in the total CO2 capture (in comparison to the anhydrous value).
When the relative humidity was reduced to 20% RH, the pre-adsorbed water in MIL-53(Al) was approximately 1 wt% and a considerable 1.5-fold increase in the CO2 capture up to 5.2 wt% was observed (Fig. 14). Thus, in this work it was proposed that at higher water loadings (40% RH, 3 wt%) the directing effect of the μ2-OH groups (over H2O molecules) is considerably diminished due to an increase in the water disorder (within the pore) caused by thermal agitation, therefore reducing the CO2 capture.76
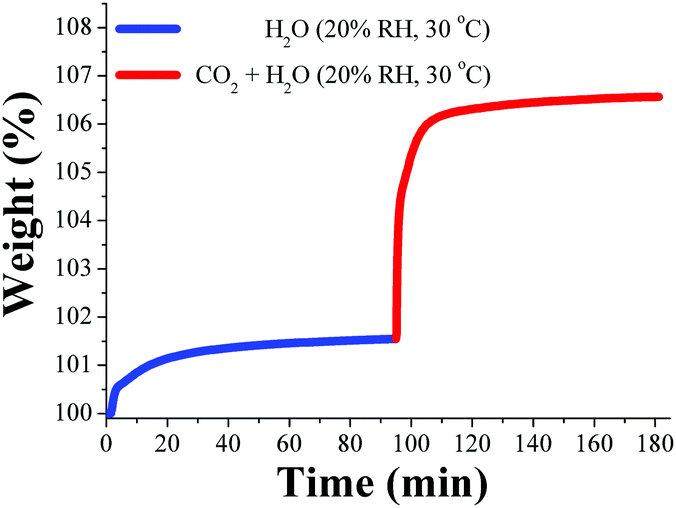 | ||
| Fig. 14 Kinetic uptake experiment carried out at 30 °C and 20% RH with H2O (blue line) and CO2 (red line). (Reproduced from ref. 76 with permission from the Centre National de la Recherche Scientifique (CNRS) and the Royal Society of Chemistry.) | ||
Later, the CO2 capture under humid conditions was investigated on the amine functionalised MIL-53(Al), NH2-MIL-53(Al), where the influence on the incorporation of an additional hydrophilic functional group (NH2) was presented.77
NH2-MIL-53(Al), reported by Blom et al.,78 is constructed by infinite trans chains of corner-sharing (via OH groups, μ2-OH) AlO4(OH)2 octahedra interconnected by NH2-BDC ligands (2-amino-terephthalate). Dynamic and isothermal (30 °C) CO2 experiments were performed on NH2-MIL-53(Al) showing a maximum CO2 uptake of 4.9 wt%. Interestingly, the amine functionalised NH2-MIL-53(Al) showed an enhancement of CO2 capture over the non-functionalised material (MIL-53(Al)).76 In fact, Gascon et al.79 showed that this CO2 enhancement was not due to the direct interaction of the amine functional (NH2) group with adsorbed CO2; instead, the amine group modulates the ‘breathing’ behaviour of NH2-MIL-53(Al), in other words, the flexibility of this MOF material. Therefore, the amine functional groups (within the pores) favour the narrow-pore structure conformation in which the interactions of CO2 molecules with the pore walls are stronger than for the parent MIL-53(Al), resulting in a higher CO2 uptake.
Kinetic CO2 isotherm experiments (30 °C) at different relative humidities (5, 10 and 30% RH) were performed on both MOF materials (MIL-53 and NH2-MIL-53(Al)). Then, an activated MIL-53(Al) sample, ht form,80 was stabilised at 30 °C and at 5% RH the CO2 capture increased 1.7-fold (from 3.5 wt% to 6.0 wt%) in comparison to that under anhydrous conditions (see Fig. 15, left).
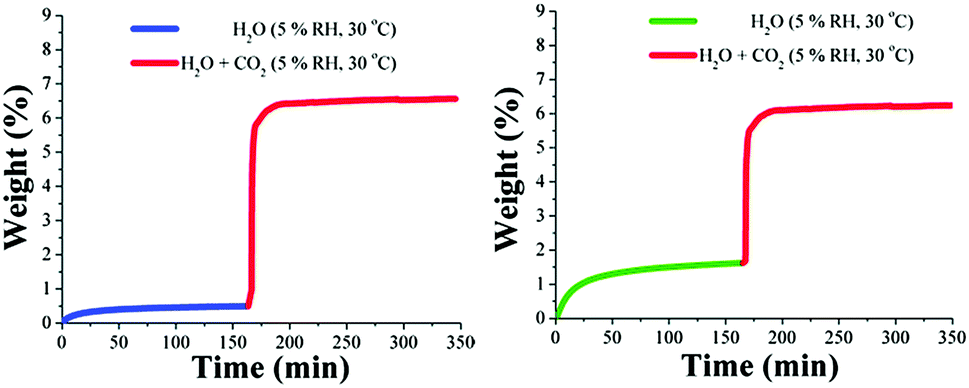 | ||
| Fig. 15 Kinetic uptake experiments carried out at 30 °C and 5% RH for (left) MIL-53(Al), with H2O (blue line) and H2O + CO2 (red line); (right) NH2-MIL-53(Al), with H2O (green line) and H2O + CO2 (red line). (Reproduced from ref. 77 with permission from the Royal Society of Chemistry.) | ||
On an activated sample (ht form)80 of NH2-MIL-53(Al), kinetic CO2 uptake experiments (at 30 °C and 5% RH) showed a total CO2 capture of 4.6 wt% (see Fig. 15, right). This CO2 capture, under relative humidity conditions, was (definitely) lower than under anhydrous conditions (4.9 wt%) representing a 0.3 wt% decrease. Clearly, the MOF material with the amine functional group (NH2-MIL-53(Al)) adsorbs more water than the non-functionalised (MIL-53(Al)), 1.6 and 0.5 wt%, respectively, suggesting that the affinity for H2O of the material NH2-MIL-53(Al) is significantly higher than for MIL-53(Al).
The water uptakes showed by MIL-53(Al) and NH2-MIL-53(Al) were in good agreement with the water adsorption isotherms and water uptakes previously presented by Farrusseng and co-workers.81 In this comprehensive work,81 a set of 15 MOFs (including MIL-53 series) was studied in detail underlying the mechanism of water adsorption. Water adsorption mechanisms for these MOF materials were rationalised by the Henry constant, the pressure at which pore filling occurs, and the maximum water adsorption capacity.
Upon increasing the relative humidity on the kinetic CO2 uptake experiments, in both MIL-53(Al) and NH2-MIL-53(Al) MOF materials, their capability to capture CO2 was certainly reduced. Thus, the NH2-MIL-53(Al) material exhibited a considerable stronger affinity for water than the non-functionalised material (MIL-53(Al)) and as a result, at 30 RH%, the material was practically saturated with water leaving no room for the CO2 molecules, resulting in a very small capability to capture CO2 (only 0.5 wt%) in comparison to anhydrous conditions (4.9 wt%). Additionally, due to the preference for the contracted structure (narrow-pore) that the amine functional group (NH2) enforces79,82 on the MOF material NH2-MIL-53(Al), the accessibility to the pores is much lower than in the non-functionalised material (MIL-53(Al)), resulting in lower H2O + CO2 captures at different RH.77
In situ Fourier transform infrared (FTIR) spectroscopy experiments (Fig. 16) showed how the hydrophobicity of the pores within a MOF material can actually have dramatic effects on the overall CO2 capture under humid conditions. Precisely, they demonstrated how a hydrophobic pore might help to enhance and sustain CO2 sequestration capabilities of a MOF material under more realistic CO2 capture conditions.
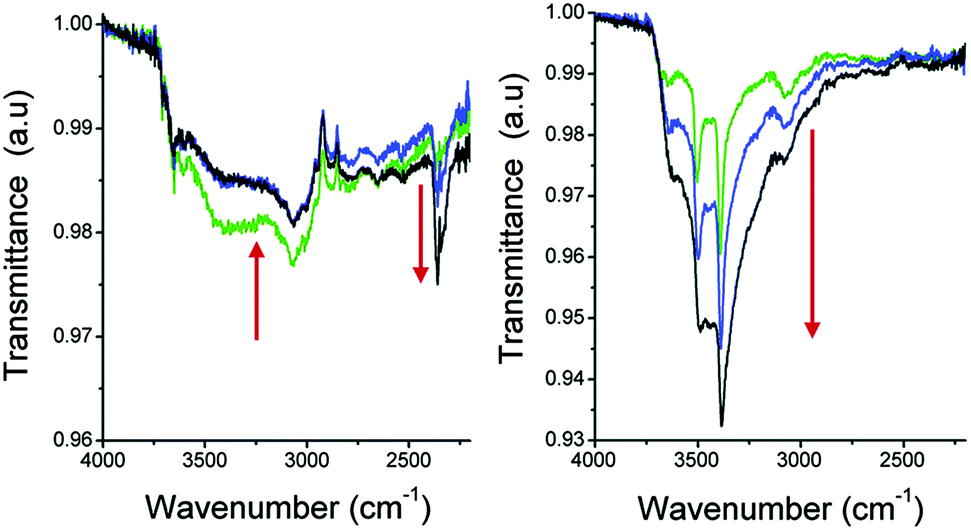 | ||
| Fig. 16 FTIR spectra of activated samples of (left) MIL-53(Al) and (right) NH2-MIL-53(Al) under atmospheric conditions (20 °C and 50% RH). The green lines show the T = 0 spectra, blue is at T = 10 min and the black one shows the spectra at T = 30 min (saturated). (Reproduced from ref. 77 with permission from the Royal Society of Chemistry.) | ||
Remarkable contributions by Yaghi et al.83 were recently reported. First, a series of ZIF MOFs83a were shown to be very effective at the dynamic separation of CO2 from N2 under humid conditions without any loss of performance over three cycles. Later, a MOF family (IRMOF-74-III) was covalently functionalised with primary amines and used for the selective capture of CO2 in 65% relative humidity.83b Certainly, in these two examples water did not enhance the overall CO2 capture but the outstanding performance and selectivity towards CO2 molecules (over water) that these MOF materials exhibited are significantly important to be mentioned in this review. In this context, Wang and Feng84 confined polynaphthylene (PN, hydrophobic polymer) inside MOF-5 (PN@MOF-5) which exhibited a doubled CO2 capacity, higher CO2/N2 selectivity and significantly improved moisture stability in comparison with pristine MOF-5.
4. New alternatives
The pre-adsorption of water in MOFs was shown to be a very effective strategy to enhance the total CO2 capture. Naturally, the first restriction for this purpose is the use of water stable MOF materials. Microporous MOFs that show hydroxo (μ2-OH) functional groups (e.g. InOF-1, NOTT-400, NOTT-401 and MIL-53(Al)) can interact relatively strongly (via hydrogen bonding) with a small amount of pre-adsorbed H2O molecules and thus resulting in an augmented CO2 sequestration. Thus, the exploration of more and new water-stable MOFs that exhibit different hydrophilic functional groups is, unquestionably, an exciting research field which can provide more interesting CO2 capture results.On the other hand, the incorporation of different solvents, within the pores of MOF materials, opens a new research pathway. For example, if the fundamental step for the overall enhanced CO2 capture is hydrogen bond formation between μ2-OH and H2O, what would the effect of incorporating another polar solvent be (such as an alcohol) that can form hydrogen bonds with the hydroxo (μ2-OH) functional groups? Thus, InOF-164 was chosen to investigate the pre-adsorption of ethanol (EtOH), within the micropores of this MOF material, and study its CO2 capture properties.85 Then, a sample of InOF-1 was completely outgassed (activated) and the pore volume (of InOF-1) was partially filled with EtOH via a solvent wet impregnation method. In order words, a small amount of EtOH (2.63 wt%) was pre-adsorbed in InOF-1 and the CO2 capture was enhanced 2.7-fold, by the pre-adsorption of EtOH in InOF-1, in comparison to the fully activated InOF-1 (from 5.24 wt% to 14.14 wt%, Fig. 17).
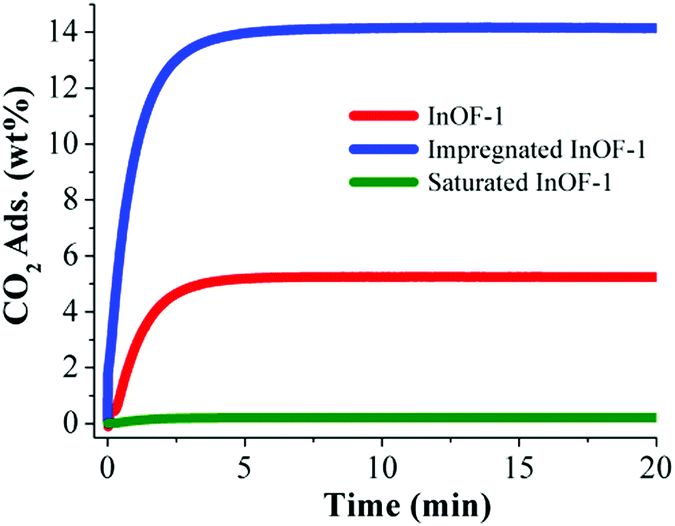 | ||
| Fig. 17 Kinetic CO2 uptake experiments performed at 30 °C with a CO2 flow of 60 mL min−1 in InOF-1 (red curve), impregnated InOF-1 (10% of the pore volume filled with EtOH, 2.63 wt% EtOH, blue curve) and saturated InOF-1 (85% of the pore volume filled with EtOH, 22.3 wt% EtOH, green curve). (Reproduced from ref. 85 with permission from the Royal Society of Chemistry.) | ||
In order to understand the role of ethanol in the adsorption of CO2 by InOF-1, the crystal structure of InOF-1 completely saturated with EtOH was determined. The pre-adsorption of 2.63 wt% with EtOH used in the adsorption studies means that if all EtOH molecules were adsorbed and uniformly distributed in the bulk MOF material, there would be approximately 1.35 molecules of EtOH in the unit cell and in this case all ethanol would be involved in hydrogen bonds with the μ2-OH groups (Fig. 18). Thus, this even distribution signifies two molecules of EtOH in each 37 Å of the length of the channel dividing it efficiently into wide sections separated by a “bottleneck” caused by the hydrogen bonded EtOH molecule. As Fig. 19 shows, this “bottleneck” effect is not strong enough to inhibit the CO2 molecules to pass this point, but will force them to “stop” or “slow down”, facilitating the adsorption of CO2 in the material since the MOF material InOF-1 possess a large number of free μ2-OH groups available on the surface of the channels, which can bind the CO2 molecules through the formation of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO(δ−)⋯H(δ+) hydrogen bonds, as previously demonstrated by Yang and co-workers, in a isostructural material to InOF-1 named NOTT-300 (Table 1).86
CO(δ−)⋯H(δ+) hydrogen bonds, as previously demonstrated by Yang and co-workers, in a isostructural material to InOF-1 named NOTT-300 (Table 1).86
 | ||
| Fig. 18 Crystal structure of InOF-1 emphasising the formation of a hydrogen bond between one ethanol molecule and the In2(μ2-OH) hydroxo group. | ||
 | ||
| Fig. 19 Crystal structures of InOF-1A (top) and InOF-1B (bottom) and the effect of the amount of ethanol confined in the pores via the formation of a hydrogen bond to the In2(μ2-OH) hydroxo groups on the pore diameter and shape. View along the crystallographic 001 direction with and without the channel surface (left) and along the 110 direction rotated additionally by 35° around 010 (right) to demonstrate the change from almost cylindrical to helicoidal shape. (Reproduced from ref. 85 with permission from the Royal Society of Chemistry.) | ||
| Name | Structure | Synthesis | Solvent/amount | CO2 capture performance | Ref. |
|---|---|---|---|---|---|
| a CO2 uptake under static conditions. The rest of the CO2 uptakes were recorded under dynamic conditions. | |||||
| MIL-100 (Fe) | Fe3O(H2O)2X(BTC)2 (X = F or OH) | Ref. 57 | H2O/40% RH | 105 mg g−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
56 |
| InOF-1 | In2(OH)2(BPTC) | Ref. 64 | H2O/20% RH | 11.0 wt% | 65 |
| EtOH/2.6 wt% | 14.1 wt% | 85 | |||
| NOTT-400 | Sc2(OH)2(BPTC) | Ref. 68 | H2O/20% RH | 10.2 wt% | 70 |
| NOTT-401 | Sc(OH)(TDA) | Ref. 68 | H2O/5% RH | 3.9 wt% | 71 |
| MIL-53(Al) | Al(OH)(BDC) | Ref. 87 | H2O/20% RH | 5.2 wt% | 76 |
| NH2-MIL-53(Al) | Al(OH)(NH2-BDC) | Ref. 87 | H2O/5% RH | 4.6 wt% | 77 |
5. Conclusions and perspectives
In the search for new alternatives for capturing CO2, metal–organic frameworks (MOFs) are one of the most promising candidates due to their sorption selectivity towards CO2, which can be synthetically modified as a function of the topology and chemical composition of the pores. Recently, the assembly of hybrid adsorbent MOFs via confining water inside the pores (a rising technology for CO2 capture) has been successfully proposed and the previous sections of this review article have described the recent progress made in the research of water-stable MOFs as water pre-adsorbent materials for the enhancement of CO2 capture.This rising CO2 capture technology was remarkably demonstrated on the mesoporous material MIL-100(Fe), where a remarkable 5-fold increase in CO2 capture was achieved. For this mesoporous MOF system the pre-adsorption of relatively high quantities of water considerably enhanced the overall CO2 capture. Conversely, for microporous MOF systems, it was demonstrated that only small amounts of pre-adsorbed water can enhance CO2 capture. The experimental evidence presented in three microporous examples (InOF-1, NOTT-400 and NOTT-401) concluded that water molecules can strongly interact with the hydroxo functional groups (μ2-OH) of these MOF materials, via hydrogen bonding, generating a reduction of the micropores (confinement effect) which permits a more efficient packing of CO2 molecules. Thus, when these MOF materials pre-adsorbed small quantities of H2O, a first adsorption domain was created where H2O molecules are “pinned” to the μ2-OH groups. Later, when the quantities of pre-adsorbed H2O were increased, a second adsorption domain was constructed and there was no detectable improvement in the CO2 capture, due to H2O saturation within the micropores of the MOF materials. These experimental water adsorption domain findings were corroborated by computational infrared spectroscopy in the microporous MOF material MIL-53(Cr), which is also constructed with μ2-OH groups.
Furthermore, the influence on the incorporation of an additional hydrophilic functional group (NH2) was investigated on the amine functionalised MIL-53(Al), NH2-MIL-53(Al). NH2-MIL-53(Al) demonstrated a considerably stronger affinity for water than the non-functionalised material (MIL-53(Al)), and therefore its ability to capture CO2 under humid conditions was significantly reduced.
Thus, the incorporation of the hydroxo (μ2-OH) functional group, within microporous MOF materials, is the key to pre-adsorbing small amounts of water which can enhance the overall CO2 capture. Nevertheless, more experimental and computational research is needed in order to understand better the mechanism of CO2 capture under humid conditions.
For example, the experimental evidence has shown how small amounts of water can improve the CO2 adsorption but it is not clear why. For instance, in the bulk phase, it is very well known that CO2 and water participate in hydration reactions to form unstable carbonic acid and eventually dissociate into more stable ions (CO2 + H2O ↔ H2CO3 ↔ HCO3− + H+). Therefore, can we find these hydration reactions in the confined environment that microporous materials provide? Additionally, the adsorption of the binary mixture (CO2 and H2O) is known to be a very non-ideal system where highly complex interactions between these two species and a pore surface take place. For example, CO2 can interact with field gradients generated by H2O molecules. It is also important to considerer the hydrophobicity of the pores within MOF materials, which can have considerable effects on the overall CO2 capture under humid conditions.
Certainly, the confinement of water within the pores of MOF materials reduces the application opportunities to those MOFs which are water-stable or can be synthetically modified to have moisture stability. Therefore, new and exciting perspectives on the confinement of different solvents within the pores of MOF materials can illuminate a new and vast research field. On one hand, the use of other solvent (e.g. alcohols) opens up a great window to work with MOFs which are water unstable, and on the other hand, many interesting CO2 capture properties may be discovered. For example, the confinement of EtOH molecules in a microporous MOF material (InOF-1) showed a considerable CO2 capture enhancement and provided a better explanation of this improvement. By visualising (single crystal X-ray diffraction experiments) the EtOH molecules hydrogen-bonded to the μ2-OH groups, it was proposed that these create a ‘‘bottleneck effect’’ which could accommodate CO2 molecules more efficiently by partially blocking the pores of InOF-1.
Clearly, incorporating different functional groups (hydrophilic or hydrophobic) into MOFs along with the design of new pore architectures (where sizes and different functionalities are integrated) and the confinement of different solvents (polar and non-polar) provide a huge research perspective for the field of CO2 capture. The principal tasks to be investigated will be the interactions of the solvent with the functional groups and the interactions of the CO2 molecules with the solvent in order to properly explain the CO2 capture mechanism.
Finally, we anticipate that this review article can provide useful information on the significant progress of the enhancement of CO2 capture by confining water (and other solvents) within the pores of MOF materials, which is very promising for real-world applications where MOF materials could be capable of serving as next-generation CO2 capture systems.
Acknowledgements
The authors thank CONACyT Mexico (212318), PAPIIT UNAM Mexico (IN100415 and IN101517), and CONACyT Mexico (236879) for financial support. Thanks to U. Winnberg (ITAM) for scientific discussions.References
- J. T. Litynski, S. M. Klara, H. G. McIlvried and R. D. Srivastava, Environ. Int., 2006, 32, 128–144 CrossRef CAS PubMed.
- M. R. Allen, D. J. Frame, C. Huntingford, C. D. Jones, J. A. Lowe, M. Meinshausen and N. Meinshausen, Nature, 2009, 458, 1163–1166 CrossRef CAS PubMed.
- H. D. Matthews, N. P. Gillett, P. A. Stott and K. Zickfeld, Nature, 2009, 459, 829–832 CrossRef CAS PubMed.
- R. J. Andres, T. A. Boden and D. Higdon, Tellus, Ser. B, 2014, 66, 23616 CrossRef.
- S. Fuss, J. G. Canadell, G. P. Peters, M. Tavoni, R. M. Andrew, P. Ciais, R. B. Jackson, C. D. Jones, F. Kraxner, N. Nakicenovic, C. Le Quéré, M. R. Raupach, A. Sharifi, P. Smith and Y. Yamagata, Nat. Clim. Change, 2014, 4, 850–853 CrossRef CAS.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- (a) Special Report on Carbon Dioxide Capture and Storage, ed. B. Metz, O. Davidson, H. de Coninck, M. Loos and L. Meyer, Cambridge Univ. Press, Cambridge, 2005 Search PubMed; (b) E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed; (c) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS; (d) L. Joos, J. M. Huck, V. Van Speybroeck and B. Smit, Faraday Discuss., 2016, 192, 391–414 RSC.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- M. Poliakoff, W. Leitner and E. S. Streng, Faraday Discuss., 2015, 183, 9–17 RSC.
- S. Yang, X. Lin, A. J. Blake, G. S. Walker, P. Hubberstey, N. R. Champness and M. Schröder, Nat. Chem., 2009, 1, 487–493 CrossRef CAS PubMed.
- A. J. Nuñez, L. N. Shear, N. Dahal, I. A. Ibarra, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 11855–11857 RSC.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- K. Okada, R. Ricco, Y. Tokudome, M. J. Styles, A. J. Hill, M. Takahashi and P. Falcaro, Adv. Funct. Mater., 2014, 24, 1969–1977 CrossRef CAS.
- K. Sumida, N. Moitra, J. Reboul, S. Fukumoto, K. Nakanishi, K. Kanamori, S. Furukawa and S. Kitagawa, Chem. Sci., 2015, 6, 5938–5946 RSC.
- W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906–912 CrossRef CAS PubMed.
- (a) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, Z. T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed; (b) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zho, Coord. Chem. Rev., 2011, 255, 1791–1823 CrossRef CAS.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed.
- R. Babarao, C. J. Coghlan, D. Rankine, W. M. Bloch, G. K. Gransbury, H. Sato, S. Kitagawa, C. J. Sumby, M. R. Hill and C. J. Doonan, Chem. Commun., 2014, 50, 3238–3241 RSC.
- A. M. Bohnsack, I. A. Ibarra, P. W. Hatfield, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 52, 8514–8517 Search PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- A. Demessence, D. M. D’Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed.
- J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2009, 131, 5516–5521 CrossRef CAS PubMed.
- (a) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed; (b) T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2016, 519, 303–308 CrossRef PubMed.
- P. Pachfule and R. Banerjee, Cryst. Growth Des., 2011, 11, 5176–5181 CAS.
- J. J. Low, A. I. Benin, P. Jakubezak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- M. Bosch, M. Zhang and H.-C. Zhou, Adv. Chem., 2014, 2014, 1–8 CrossRef.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- M. R. Gonzalez, J. H. González-Estefan, H. A. Lara-García, P. Sánchez-Camacho, E. I. Basaldella, H. Pfeiffer and I. A. Ibarra, New J. Chem., 2015, 39, 2400–2403 RSC.
- W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed.
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696–9698 RSC.
- H. A. Lara-García, M. R. Gonzalez, J. H. González-Estefan, P. Sánchez-Camacho, E. Lima and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 442–447 RSC.
- B. Saccoccia, A. M. Bohnsack, N. W. Waggoner, K. H. Cho, J. S. Lee, D.-Y. Hong, V. M. Lynch, J.-S. Chang and S. M. Humphrey, Angew. Chem., Int. Ed., 2015, 54, 5394–5398 CrossRef CAS PubMed.
- C. Wang, X. Liu, N. K. Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- X. Liu, H. Jin, Y. Li, H. Bux, Z. Hu, Y. Ban and W. Yang, J. Membr. Sci., 2013, 428, 498–506 CrossRef CAS.
- J. Aguilera-Sigalat and D. Bradshaw, Chem. Commun., 2014, 50, 4711–4713 RSC.
- C. Liu and B. Yan, J. Colloid Interface Sci., 2015, 459, 206–211 CrossRef CAS PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- M. Sadakiyo, H. Ōkawa, A. Shigematsu, M. Ohba, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 5472–5475 CrossRef CAS PubMed.
- C. Janiak and S. K. Henninger, Chimia, 2013, 67, 419–424 CrossRef CAS PubMed.
- P. L. Llewellyn, S. Bourrelly, C. Serre, Y. Filinchu and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 7751–7754 CrossRef CAS PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed.
- A. Ö. Yazaydın, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425–1430 CrossRef.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. Le Van, Langmuir, 2011, 27, 11451–11456 CrossRef CAS PubMed.
- J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. Le Van, Langmuir, 2010, 26, 14301–14307 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- L. Mu, B. Liu, H. Liu, Y. Yang, C. Sun and G. Chen, J. Mater. Chem., 2012, 22, 12246–12252 RSC.
- E. Soubeyrand-Lenoir, C. Vagner, J. W. Yoon, P. Bazin, F. Ragon, Y. K. Hwang, C. Serre, J.-S. Chang and P. L. Llewellyn, J. Am. Chem. Soc., 2012, 134, 10174–10181 CrossRef CAS PubMed.
- P. Horcajada, S. Surblé, C. Serre, D.-Y. Hong, Y.-K. Seo, J.-S. Chang, J.-M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820–2822 RSC.
- A. D. Wiersum, E. Soubeyrand-Lenoir, Q. Yang, B. Moulin, V. Guillerm, M. B. Yahia, S. Bourrelly, A. Vimont, S. Miller, C. Vagner, M. Daturi, G. Clet, C. Serre, G. Maurin and P. L. Llewellyn, Chem. – Asian J., 2011, 6, 3270–3280 CrossRef CAS PubMed.
- L. S. Grajciar, A. D. Wiersum, P. L. Llewellyn, J.-S. Chang and P. Nachtigall, J. Phys. Chem. C, 2011, 115, 17925–17933 CAS.
- J. W. Yoon, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, E. Bloch, P. L. Llewellyn, C. Serre, P. Horcajada, J.-M. Grenèche, A. E. Rodrigues and G. Férey, Angew. Chem., Int. Ed., 2010, 49, 5949–5952 CrossRef CAS PubMed.
- H. Jasuja, J. Zang, D. S. Sholl and K. S. Walton, J. Phys. Chem. C, 2012, 116, 23526–23532 CAS.
- P.-Q. Liao, H. Chen, D.-D. Zhou, S.-Y. Liu, C.-T. He, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chena, Energy Environ. Sci., 2015, 8, 1011–1016 CAS.
- J. A. Mason, T. M. McDonald, T.-H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787–4803 CrossRef CAS PubMed.
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696–9698 RSC.
- R. A. Peralta, B. Alcántar-Vázquez, M. Sánchez-Serratos, E. González-Zamora and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 898–903 RSC.
- N. L. Ho, F. Porcheron and R. J.-M. Pellenq, Langmuir, 2010, 26, 13287–13296 CrossRef CAS PubMed.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606–15613 CrossRef CAS PubMed.
- I. A. Ibarra, S. Yang, X. Lin, A. J. Blake, P. J. Rizkallan, H. Nowell, D. R. Allan, N. R. Champness, P. Hubberstey and M. Schröder, Chem. Commun., 2011, 47, 8304–8306 RSC.
- I. A. Ibarra, A. Mace, S. Yang, J. Sun, S. Lee, J.-S. Chang, A. Laaksonen, M. Schröder and X. Zou, Inorg. Chem., 2016, 55, 7219–7228 CrossRef CAS PubMed.
- J. R. Álvarez, R. A. Peralta, J. Balmaseda, E. González-Zamora and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 1080–1084 RSC.
- E. Sánchez-González, J. R. Álvarez, R. A. Peralta, A. Campos-Reales-Pineda, A. Tejeda-Cruz, E. Lima, J. Balmaseda, E. González-Zamora and I. A. Ibarra, ACS Omega, 2016, 1, 305–310 CrossRef.
- G. R. Medders and F. Paesani, J. Phys. Chem. Lett., 2014, 5, 2897–2902 CrossRef CAS PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louer and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- V. Haigis, F.-X. Coudert, R. Vuilleumier and A. Boutin, Phys. Chem. Chem. Phys., 2013, 15, 19049–19056 RSC.
- F. Salles, S. Bourrelly, H. Jobic, T. Devic, V. Guillerm, P. Llewellyn, C. Serre, G. Férey and G. Maurin, J. Phys. Chem. C, 2011, 115, 10764–10776 CAS.
- M. Sánchez-Serratos, P. A. Bayliss, R. A. Peralta, E. González-Zamora, E. Lima and I. A. Ibarra, New J. Chem., 2016, 40, 68–72 RSC.
- A. Zárate, R. A. Peralta, P. A. Bayliss, R. Howie, M. Sánchez-Serratos, P. Carmona-Monroy, D. Solis-Ibarra, E. González-Zamora and I. A. Ibarra, RSC Adv., 2016, 6, 9978–9983 RSC.
- B. Arstad, H. Fjellvåg, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755–762 CrossRef CAS.
- E. Stavitski, E. A. Pidko, S. Couck, T. Remy, E. J. M. Hensen, B. M. Weckhuysen, J. Denayer, J. Gascon and F. Kapteijn, Langmuir, 2011, 27, 3970–3976 CrossRef CAS PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- J. Canivet, J. Bonnefoy, C. Daniel, A. Legrand, B. Coasne and D. Farrusseng, New J. Chem., 2014, 38, 3102–3111 RSC.
- P. Serra-Crespo, E. Gobechiya, E. V. Ramos-Fernadez, J. Juan-Alcañiz, A. Martinez-Joaristi, E. Stavitski, C. E. A. Kirschhock, J. A. Martens, F. Kapteijn and J. Gascon, Langmuir, 2012, 28, 12916–12922 CrossRef CAS PubMed.
- (a) N. T. T. Nguyen, H. Furukawa, F. Gándara, H. T. Nguyen, K. E. Cordova and O. M. Yaghi, Angew. Chem., Int. Ed., 2014, 53, 10645–10648 CrossRef CAS PubMed; (b) A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed.
- N. Ding, H. Li, X. Feng, Q. Wang, S. Wang, L. Ma, J. Zhou and B. Wang, J. Am. Chem. Soc., 2016, 138, 10100–10103 CrossRef CAS PubMed.
- R. A. Peralta, A. Campos-Reales-Pineda, H. Pfeiffer, J. R. Álvarez, J. A. Zárate, J. Balmaseda, E. González-Zamora, A. Martínez, D. Martínez-Otero, V. Jancik and I. A. Ibarra, Chem. Commun., 2016, 52, 10273–10276 RSC.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed.
- P. A. Bayliss, I. A. Ibarra, E. Pérez, S. Yang, C. C. Tang, M. Poliakoff and M. Schröder, Green Chem., 2014, 16, 3796 RSC.
| This journal is © the Partner Organisations 2017 |