
Iron transport in the kidney: implications for physiology and cadmium nephrotoxicity
Frank
Thévenod
* and
Natascha A.
Wolff
Institute of Physiology, Pathophysiology & Toxicology, Center for Biomedical Training and Research (ZBAF), University of Witten/Herdecke, Stockumer Str. 12, 58453 Witten, Germany. E-mail: frank.thevenod@uni-wh.de; natascha.wolff@uni-wh.de
First published on 12th October 2015
Abstract
The kidney has recently emerged as an organ with a significant role in systemic iron (Fe) homeostasis. Substantial amounts of Fe are filtered by the kidney, which have to be reabsorbed to prevent Fe deficiency. Accordingly Fe transporters and receptors for protein-bound Fe are expressed in the nephron that may also function as entry pathways for toxic metals, such as cadmium (Cd), by way of “ionic and molecular mimicry”. Similarities, but also differences in handling of Cd by these transport routes offer rationales for the propensity of the kidney to develop Cd toxicity. This critical review provides a comprehensive update on Fe transport by the kidney and its relevance for physiology and Cd nephrotoxicity. Based on quantitative considerations, we have also estimated the in vivo relevance of the described transport pathways for physiology and toxicology. Under physiological conditions all segments of the kidney tubules are likely to utilize Fe for cellular Fe requiring processes for metabolic purposes and also to contribute to reabsorption of free and bound forms of Fe into the circulation. But Cd entering tubule cells disrupts metabolic pathways and is unable to exit. Furthermore, our quantitative analyses contest established models linking chronic Cd nephrotoxicity to proximal tubular uptake of metallothionein-bound Cd. Hence, Fe transport by the kidney may be beneficial by preventing losses from the body. But increased uptake of Fe or Cd that cannot exit tubule cells may lead to kidney injury, and Fe deficiency may facilitate renal Cd uptake.
 Frank Thévenod | Frank Thévenod received his MD from Goethe University, Frankfurt, Germany and his PhD in cellular physiology and biophysics from Case Western Reserve University (CWRU), Cleveland, OH. After postdoctoral training at the Max-Planck-Institute of Biophysics, Frankfurt, Germany and CWRU, he became assistant professor at Saarland University, Homburg, Germany and subsequently senior lecturer at the University of Manchester, UK. Since 2002 he holds the Chair of Physiology, Pathophysiology and Toxicology in the Faculty of Health at Witten/Herdecke University, Germany. His work focuses on renal physiology and toxicology of transition metals, epithelial transport, signal transduction, and molecular mechanisms of cellular metal homeostasis. |
 Natascha A. Wolff | Natascha A. Wolff received her diploma in biology from the University of Hannover, Germany, and her PhD from the Ruhr-University Bochum on work carried out at the Max-Planck-Institute of Molecular Physiology (formerly Systems Physiology), Dortmund, Germany. After postdoctoral studies at the NIEHS/NIH, NC, she became research associate at the Georg-August University Göttingen, Germany, where she received her Venia legendi (Habilitation) for Physiology. Since 2005, she is research associate at the University of Witten/Herdecke, Germany. Her work focuses on epithelial transport of organic ions and metals, as well as cellular metal homeostasis and nephrotoxicity by transition metals. |
1. Introduction
Until recently, the kidney was thought to play no role in systemic iron (Fe) homeostasis.1 On the other hand, cadmium (Cd) has been known for decades to accumulate in the kidney (described in detail in the excellent review by G. F. Nordberg2). The major Fe compounds in biological systems are the redox pair ferrous (Fe2+) and ferric (Fe3+) iron whereas the major Cd compound is the divalent Cd ion (Cd2+). Due to their hydrophilicity Fe2+/Fe3+ and Cd2+ (and other metal ions) must cross cellular membranes via proteinous pathways, i.e. channels, transporters or receptors. In biological systems Fe2+/Fe3+ and Cd2+ are mostly found in bound form and are either complexed to small ligands, such as amino acids or peptides,3 or more or less specifically bound to proteins (e.g. ferritin, transferrin, metallothionein or albumin) whose affinity constants determine their residency as “free” or bound metal ions. As a non-essential metal in mammals, Cd2+ must exploit and compete for physiological entry pathways for essential metal ions, such as Fe2+, Cu2+, Zn2+ or Mn2+. To describe this process, the term “ionic and molecular mimicry” has been coined.4 In this context, molecular mimicry accounts for a condition in which a toxic metal ion forms a complex with an endogenous organic ligand (e.g. a peptide or a protein) and the resulting compound mimics the behavior of the endogenous ligand that binds to its receptor.Interestingly, epidemiological studies had shown an inverse relationship between the size of the Fe stores and the Cd burden of the body (and kidneys),5,6 thus hinting at a link between transport of Fe2+/Fe3+ and Cd2+ (reviewed in ref. 7). Mounting functional evidence for transport of Cd2+ by transporters and receptors for essential free and complexed metal ions in renal and other epithelia8,9 was then superseded by the discovery that the first cloned Fe2+ transporter, the divalent metal transporter 1 (DMT1/Nramp2/DCT1/SLC11A2), is equally well permeated by Cd2+.10,11 DMT1 (and other Fe2+/Cd2+ transporting proteins) is, however, highly expressed in the kidney.
This fact soon attracted the attention of Craig P. Smith from the University of Manchester (U.K.) who noticed the fact that the kidneys “…contain many if not all of the proteins that are central to iron balance, that in some cases are expressed in considerable amounts, implies that the kidney handles iron in some way that has demanded evolutionary conservation and therefore is likely to be of importance…”.12 In a series of pioneering studies, his group measured Fe reabsorption by the rat kidney in vivo.13 From their data they estimated that under physiological conditions ∼0.4 mg Fe is filtered daily by rat kidneys, but only 0.7% is excreted in the urine and also appears to depend on the renal expression of DMT1.14,15 Alterations of dietary Fe intake modulated renal DMT1 expression: iron restriction increased renal DMT1 whereas iron loading decreased renal DMT1 expression and DMT1 expression was inversely correlated with urinary Fe output.15 Therefore they concluded that long-term modulation of renal DMT1 expression may influence renal iron excretion rate. In addition, it soon became apparent that a certain proportion of filtered Fe is protein-bound and includes transferrin (Tf).16,17 Meanwhile, additional renal Fe transport pathways have been identified and characterized that now allow a better understanding of the role of the kidney in Fe handling and physiological Fe homeostasis. Thus, the notion that the kidney is involved in transport and excretion of Fe and other metal ions12 has gained recognition and has entered the fields of toxicology,18 iron biology19,20 and nephrology.21
2. Systemic iron homeostasis
For detailed accounts of systemic Fe homeostasis, the reader is referred to excellent recent reviews.1,22,23 Iron, with an amount of ∼2.5–4.5 g in adults, is the major transition metal in the body and is mostly localized in erythrocyte hemoglobin, amounting to roughly 60% of the human total body Fe. Yet, Fe is indispensable for other tissues as well, being an essential component of hundreds of proteins, including many enzymes. Thus, Fe is not only required for oxygen transport and storage with hemoglobin or myoglobin, but Fe-containing proteins are needed for a variety of additional functions, including, first and foremost, mitochondrial respiration (electron transport chain), but also metabolism and detoxification (cytochrome P450 enzymes), DNA synthesis (ribonucleotide reductase), antioxidant defense (catalase) and beneficial pro-oxidant functions, oxygen sensing (hypoxia-inducible factor (HIF) prolyl hydroxylases), and immune defense (myeloperoxidase).24 Yet, Fe is also toxic due to the production of cell-damaging radicals through Fenton-type reactions.25 Thus, body Fe homeostasis needs to be tightly regulated.Mammalian Fe homeostasis is unusual in that it is mainly controlled at the level of intestinal Fe absorption. To date, there is no known regulated short-term mechanism of Fe excretion (but see Section 1 for an example of long-term modulation in the kidney; reviewed in ref. 12) and the small daily Fe loss of about 1 mg in healthy adult males is closely balanced by duodenal Fe uptake. The daily Fe loss mainly (∼80%) occurs via shedding of Fe-laden duodenal enterocytes, complemented by much smaller losses via other pathways, including the kidneys26 (reviewed in ref. 27 and 28). The large fraction of Fe in erythrocyte hemoglobin is efficiently recycled, while excess Fe is stored in the liver (0.5–1 g) (reviewed in ref. 27 and 28). Under normal conditions, Fe loss is balanced via regulated intestinal absorption.29 Two forms of dietary Fe are taken up into duodenal enterocytes via different mechanisms: while heme Fe uptake occurs via not yet clearly defined pathways (see ref. 30–32), non-heme Fe absorption is mediated by the proton-coupled divalent metal transporter 1 (DMT1/Nramp2/DCT1/SLC11A2)10 (reviewed in ref. 33) after reduction of dietary Fe3+ to Fe2+ by duodenal cytochrome B with ascorbate as an electron donor (reviewed in ref. 34). An apical intestinal Na+/H+ exchanger appears to be responsible for generating the proton gradient necessary for DMT1-mediated Fe2+ uptake.35 Fe2+ is subsequently delivered to cytoplasmic ferritin for storage36 by chaperones, including the poly(rC)-binding protein 1 (PCBP1),37 or to the basolateral transporter ferroportin (FPN1/IREG1/MTP1/SLC40A1)38–41 for efflux into the plasma. Efficient export requires the presence of members of a family of copper-containing ferroxidases, e.g. hephaestin and/or ceruloplasmin,42–44 which convert effluxed Fe2+ to Fe3+ that mainly binds to the Fe-carrying serum protein Tf. Importantly, FPN1, to date the only known cellular Fe exporter,45 is regulated by Fe loading46 through homeostatically increased synthesis and release of the hepatic peptide hepcidin into the circulation that limits further absorption of dietary Fe and its release from stores (reviewed in ref. 47): hepcidin binds to FPN1, leading to its internalization and subsequent lysosomal degradation, hence preventing further Fe export into the plasma.48
Free, unbound Fe is incompatible with either plasma Fe transport (it would precipitate) or with cytosolic Fe trafficking (it would damage the cellular environment).49 Therefore, Fe must be complexed with appropriate ligands. The transport of Fe in plasma to its sites of use occurs predominantly as Tf-bound Fe50 (TBI), and to a lesser extent associated with several other serum proteins, including ferritin, albumin, neutrophil gelatinase associated lipocalin (NGAL/24p3/lipocalin-2), and possibly lactoferrin and hepcidin. Collectively, these latter forms of serum Fe – with the exception of ferritin – are termed non-Tf-bound Fe (NTBI).49,51,52 Under physiological conditions, NTBI is a minor entity within total serum Fe – although NTBI may become a relevant issue in patients with various pathological conditions in which Tf saturation is significantly elevated (reviewed in ref. 49). Ferritin, primarily an intracellular protein, is low in human serum under normal conditions, despite substantial inter-individual variations and substantial increases under Fe overload conditions53 where it may be secreted through a non-classical lysosomal secretory pathway by macrophages and renal proximal tubule (PT) cells.54 Additionally, serum Fe may exist in the form of holo-Tf bound to a soluble form of the Tf receptor.55
3. Cellular iron homeostasis
One major mechanism for Fe assimilation by erythrocyte precursors and non-erythroid cells is the internalization of serum Tf-bound Fe3+.56,57 Tf endocytosis is mediated by the ubiquitous Tf receptor 1 (TfR1)58,59 (a TfR2 has been cloned, but its expression is limited to the liver and erythropoietic progenitors,60 where it is thought to operate as an “Fe sensor”61). Endosomal acidification favors the release of iron from Tf, which itself remains bound to the receptor and is subsequently recycled to the cell surface, where the near neutral pH promotes dissociation of apo-Tf from the receptor and its release into the circulation.62,63 Endosomal Fe3+ is quickly reduced to Fe2+ by an oxidoreductase activity, now known to be represented by “Steap” (sixtransmembrane epithelial antigen of the prostate) family proteins, namely Steap2 to Steap4,64,65 (a reaction which may actually occur prior to dissociation from the Tf–TfR1 complex, especially since Fe3+ tightly binds to Tf, while Fe2+ does so only weakly66). Subsequent endosomal efflux of Fe2+ is mediated by DMT1.67,68 The transient receptor potential mucolipin 1 (TRPML1/ML1/MLN1/MCLN1) may function as another Fe2+ release channel in late endosomes and lysosomes.69The mechanisms of intracellular Fe trafficking to its sites of utilization are not well understood. In most cell types, it is agreed that Fe acquired during the Tf cycle is first released into the cytosol by entering a “labile cytosolic Fe pool” that is defined as a pool of chelatable and redox-active Fe2+ and represents a transition compartment for Fe sensing, metabolic utilization or storage.70,71 A variety of low molecular weight compounds have been suggested as Fe chelators in this readily accessible Fe reservoir, including organic anions like citrate and phosphate, oligopeptides such as glutathione (GSH),72 membrane phospholipids, as well as “mammalian siderophores”, namely 2,5-dihydroxybenzoic acid (2,5-DHBA),73 although an involvement of the latter has been recently challenged.74 Further, the conserved cytosolic glutaredoxins Grx3 and Grx4 could also play an essential role in intracellular Fe sensing and trafficking, as their depletion in yeast leads to impaired Fe transport to mitochondria and defects in Fe-dependent pathways.75
In contrast, in erythroid cells, kinetic and microscopy studies support a “kiss and run” hypothesis, which assumes the direct delivery of Tf-derived Fe to mitochondria through a transient contact with endosomes (reviewed in ref. 76): this concept was originally developed based on kinetic Fe release studies with 59Fe–Tf in reticulocytes at 4 °C that contain very little chelatable cytosolic Fe, thus preventing Fe mobilization from other compartments.77 Ponka and coworkers later observed direct, albeit transient inter-organellar contacts and a simultaneous increase in mitochondrial chelatable Fe at these sites by live confocal imaging.78
The major Fe-utilizing cellular organelles are mitochondria that require Fe for the synthesis of heme and Fe–sulfur clusters.79,80 Irrespective of whether Fe is delivered by cytosolic chaperones or direct endosome-mitochondria contacts, it has to cross two membranes to enter the mitochondrial matrix, where it is needed for synthetic processes. The outer mitochondrial membrane (OMM) has typically been assumed to be freely permeable to Fe due to the presence of “pores”81 represented by voltage-dependent anion channels (VDACs) that are regarded as the major permeability pathway of the OMM for small solutes.82 But this knowledge is based on in vitro studies that have been performed after reconstitution of VDAC in artificial membranes/planar lipid bilayers. Thus, in vivo the OMM may not be as freely permeable to inorganic cations as previously believed and VDAC function may be tightly regulated.83,84 We have recently identified DMT1 in several tissues as a possible mechanism for Fe2+ transfer across the OMM using a variety of experimental approaches85,86 (see Section 6.1.3), but additional pathways may also exist.
Entry of Fe into the mitochondrial matrix requires the SLC transporter mitoferrin-1 and -2 (also known as MFRN1/SLC25A37 and MFRN2/SLC25A28), which are found in the inner mitochondrial membrane (IMM).87 Mitoferrin-1 is highly enriched in erythroid cells and is stabilized during differentiation whereas mitoferrin-2 is ubiquitously expressed and its half-life is not regulated.88 The lack of functional mitoferrin-1 in the frascati zebrafish mutant is associated with severe defects in erythropoiesis, heme synthesis and Fe–sulfur clusters biogenesis.87 Some studies have also suggested that the mitochondrial calcium uniporter in the IMM represents an additional route of Fe entry into the matrix (e.g.ref. 89).
Cells may eliminate excess intracellular Fe by secretion of Fe2+via FPN1 or by secretion of heme through the putative heme exporter FLVCR (feline leukemia virus, subgroup C, receptor).90 Excess intracellular Fe may also be stored and detoxified in the cytosol by ferritin, which consists of 24 H (heavy) and L (light) subunits, encoded by two different genes.91 H-ferritin possesses ferroxidase activity, mediating conversion of ferrous Fe (Fe2+) to the ferric form (Fe3+), whereas L-ferritin chains provide a nucleation center. Ferritin assembles into a shell-like structure with a cavity of ∼80 nm that provides storage space for up to 4500 Fe3+ ions. Shuttling of Fe to ferritin appears to be mediated by the PCBP family chaperones37,92 (see Section 2). Both the lysosomal and the proteasomal pathways of degradation seem to be recruited to mobilize Fe from ferritin, probably depending on the cell type and the cellular conditions (reviewed in detail in ref. 91). Mitochondria contain a nuclear-encoded ferritin isoform93 whose expression is limited to a few organs, such as testis, neurons, heart and kidney, but not the liver or spleen.94 Mitochondrial ferritin may cooperate with cytosolic ferritin in the maintenance of intracellular Fe balance or protect mitochondria from Fe-dependent oxidative damage and increased production of reactive oxygen species (ROS) in cells with high metabolic rate.91
4. Function of the kidney
Detailed state-of-the-art descriptions of the morphology of the kidneys, the structure of the nephrons, i.e. the functional units of the kidneys, and their functions are beyond the scope of this review and can be found in standard handbooks of renal physiology.95–97 The aim of this very simple overview on kidney function is to introduce readers unfamiliar with renal physiology to basic principles that are required for a better understanding of the handling of Fe and Cd by the kidneys.The plasma permanently equilibrates with the interstitial fluid of the extracellular space and – through the interstitial fluid – with the intracellular space. Together with the lungs and the intestine, the kidneys keep the body fluid homeostasis of mammalian organisms constant by selectively excreting metabolic wastes, excess solutes and water as well as xenobiotics from the body into the urine. Blood is constantly pumped through the kidneys where plasma fluid is filtered through a capillary network called the glomerulus. The driving force for ultrafiltration is generated by the effective filtration pressure in the capillaries, which is set by the glomerular blood pressure. To fulfill the excretory function of the kidneys, large quantities of plasma amounting to >60× its total body volume are filtered daily in the renal glomeruli, complemented by secretory pathways along the renal tubule epithelium. Filtered water and solutes still of use for the body are efficiently recycled to the circulation by obligatory and regulated reabsorptive processes in the tubular sections of the nephrons. By these means, about 180 l of primary filtrate is generated every day to produce about 1–3 l final urine. This indicates that about 99% of the primary urine is reabsorbed along the more than 2 million nephrons.
At the glomerulus, a three-layer anatomical barrier allows fluid and solutes <10 kDa and/or 18 Å to cross that barrier, but permeation decreases with increasing molecular mass (cutoff of ∼80 kDa), molecular size (<42 Å), and also depends on charge (cationic > neutral > anionic) (see however ref. 98 for a critical discussion). Hence, the primary urine also contains essential nutrients and electrolytes that need to be actively reabsorbed to avoid critical losses and ensuing deficiencies. On the other hand, some metabolic wastes are actively secreted by the kidney since their rate of production exceeds their rate of glomerular filtration. All these selective processes are carried out by the nephrons, epithelial tubular structures that consist of several interconnected segments with characteristic morphological and functional properties, the PT with its convoluted segments S1 and S2 (PCT) and straight segment S3, the loop of Henle (LOH), the distal tubule (DT) with its convoluted segment (DCT) and connecting tubule, and finally the collecting duct (CD) (see Fig. 1). Glomeruli, convoluted segments of the PT, DT with connecting tubule, and cortical CD are localized in the kidney cortex. Parts of the straight segment of the PT, parts of the thin limb of the LOH, the thick ascending limb of the LOH and outer medullary CD are in the outer medulla. The remaining segments (most of the thin limbs of the LOH as well as initial and terminal inner medullary CD) are found in the inner medulla of the kidney.
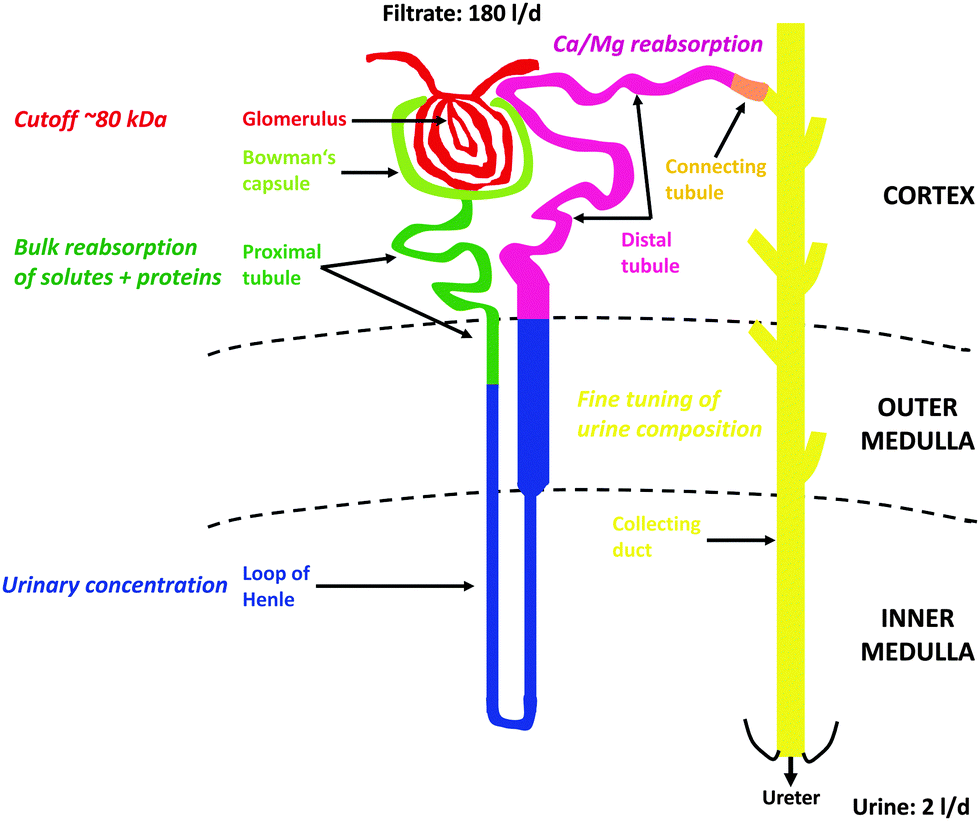 | ||
| Fig. 1 Structure and function of the nephron. For further details, see Section 4. | ||
In general, the PT is responsible for bulk reabsorption of the primary fluid that is filtrated into the lumen of that segment. About two-third of PT reabsorption occurs “paracellularly” at intercellular tight-junctions, through osmotically driven “solvent drag”. But amino acids, glucose, bicarbonate and several other essential molecules are also reabsorbed via luminal Na+-dependent transporters expressed in the luminal brush-border membranes (BBM) of PT cells and therefore require the energy of adenosine triphosphate (ATP) for activation of basolateral Na+/K+-ATPases to maintain these reabsorptive processes. The PT cells are also responsible for bulk reuptake of filtered proteins and peptides via a multi-ligand receptor complex expressed in the luminal BBM, megalin:cubilin:amnionless99 (see Section 6.1.1), that also binds metalloproteins, such as Tf (an Fe binding protein) or metallothionein (MT) (a Cd2+ binding protein).100 Finally, the PT is the major location for the secretion of xenobiotic and endogenous organic cations and anions. The LOH that follows the PT builds up the hyperosmotic interstitium surrounding the final segment of the nephron, the CD that is required for reabsorption of water to generate small volumes of concentrated urine (“antidiuresis”), thus preserving water for the body. Hyper-osmolarity of the kidney medulla is built up by several properties of the different segments of LOH, i.e. (1) active NaCl transport into the interstitium by the thick ascending limb of LOH; (2) high permeability of the descending LOH to water and low permeability of the ascending LOH; (3) increased permeability to urea in the medullary portions of LOH; and (4) magnification of the medullary hyper-osmolarity by the countercurrent flow within the descending and ascending limbs of LOH (“countercurrent multiplication”).95–97 A hypo-osmotic fluid reaches the DT where divalent metal ions are reabsorbed, such as Ca2+ and Mg2+ (and Fe2+) (see Section 6.3) and the luminal fluid is further depleted by active NaCl reabsorption. In the CD the final composition of the urine is adjusted by “fine-tuning” through hormonal regulation of the CD cells. The paracellular permeability of the CD epithelial layer to ions and water is very low; therefore the final content of the urine in NaCl and water must be controlled by hormonal regulation of CD cells via aldosterone and antidiuretic hormone. This occurs through regulated and temporary incorporation of epithelial Na+ channels (ENaC) and aquaporin-2 water channels (AQP2), respectively, into the apical membrane of principal (light) cells of the CD. Additional regulated functions of the CD include acid–base balance (type A- and type B-intercalated cells) and K+ homeostasis. Apart from the CD, PT and DT also represent nephron segments where hormones (i.e. parathyroid hormone, calcitonin, calcitriol) control Ca2+ and PO43− homeostasis. Fig. 1 summarizes the structure of the nephron with the major functions of the different nephron segments that are relevant to this review.
5. Plasma iron and renal glomerular filtration
Only recently has it been recognized that the kidney is also involved in systemic Fe homeostasis because certain Fe-containing complexes in plasma (e.g. Tf, NGAL/24p3/lipocalin-2, lactoferrin, albumin, hemoglobin, myoglobin and hepcidin) have the ability to cross the glomerular filter, even under physiological conditions101,102 (reviewed in ref. 12 and 103). There is also a rising interest in the role of Fe in both acute kidney injury and chronic kidney disease.21 Renal Fe losses are minimal under physiological conditions26 (reviewed in ref. 27). The lack of urinary Fe excretion has traditionally been attributed to binding of Fe (or, if erythrocytes are lysed within the blood, hemoglobin and free heme) to larger proteins that would ensure that little or no Fe is lost by glomerular filtration and entry into the urine because of the low protein permeability of the glomerular filter.28,104 That would also include the large 24-subunit serum ferritin complex that is unlikely to reach the ultrafiltrate. But NGAL is present in plasma as monomers of 25 kDa and dimers of 45 kDa that should permeate the glomerular filter.105 Similarly, the small molecule hepcidin (2–3 kDa) readily passes into the primary urine.47 Moreover, early micropuncture studies in animals indicated significant glomerular filtration of high-molecular weight proteins (HMWP), such as albumin (reviewed in ref. 98 and 106). In accordance with these observations, patients with renal Fanconi syndrome, i.e. with compromised renal PT function, including protein reabsorption (reviewed in ref. 107), display increased urinary excretion of proteins up to 160 kDa,17 suggesting that substantial amounts of TBI, i.e. Fe bound to Tf (80 kDa), as well as NTBI (see Section 2), such as Fe bound to albumin (66.5 kDa) and lactoferrin (80 kDa), reach the primary filtrate and must be reabsorbed by the PT (see Section 6.1.1).6. Iron transporters of the nephron
(See also Tables 1 and 2 for a summary).| Receptor/transporter | Nephron localization | Subcellular localization | Species | Ref. | Comments |
|---|---|---|---|---|---|
| a PT: proximal tubule; LOH: loop of Henle; DT: distal tubule; CD: collecting duct. b NGAL: neutrophil gelatinase-associated lipocalin. | |||||
| Megalin:cubilin:amnionless | PTa | Apical; subapical | Human; mouse; rat; rabbit | 108 | Immuno-fluorescence/-histochemistry/-gold; co-labeling with segment-/organelle-specific marker |
| Transferrin receptor 1 | PT; CD | Apical; subapical | Mouse | 103 | Immunofluorescence; poor resolution; specificity of antibody unclear |
| NGALb/24p3/lipocalin-2 receptor (SLC22A17) | DT; CD | Apical; subapical | Mouse; rat | 182 and 214 | Immunofluorescence/-histochemistry; co-labeling with segment-specific marker |
| DMT1 (SLC11A2) | PT | Intracellular (endosomes/lysosomes/mitochondria) | Mouse; rat | 12, 14, 15, 85, 86, 137 and 140 | Immuno-fluorescence/-histochemistry/-gold; co-labeling with segment-/organelle-specific marker |
| Apical? | Mouse | 138 | Immunohistochemistry; poor resolution; collapsed tubules; no co-labeling with segment-specific marker | ||
| LOH | Apical; intracellular | Rat | 14 | Immunofluorescence; co-labeling with segment-/cell-specific marker | |
| DT | Apical | Rat | 14 and 15 | Immunofluorescence; co-labeling with segment-/membrane-specific marker | |
| ∅? | Mouse | 138 and 140 | Immunohistochemistry; poor resolution; collapsed tubules; no co-labeling with segment-specific marker; specificity of antibody unclear | ||
| CD | Apical; intracellular; basolateral | Rat | 14, 15 and 145 | Immunofluorescence; co-labeling with segment-/cell-specific marker | |
| ZIP8 (SLC39A8) | PT | Apical?; subapical | Mouse | 153 | Immunofluorescence/-histochemistry; co-labeling with membrane-specific marker, yet poor resolution (discussed in ref. 141) |
| ZIP14 (SLC39A14) | PT? | ? | ∅ | ∅ | No staining of native tissue; staining in overexpressing cell lines only (discussed in ref. 141) |
| Ferroportin (FPN1/SLC40A1) | PT | Basolateral | Mouse; rat | 123, 160 and 161 | Immuno-fluorescence/-histochemistry/-gold; co-labeling with segment-/membrane-specific marker |
| Apical/basolateral? | Mouse | 140 and 162 | Immunohistochemistry; poor resolution; no co-labeling with segment-specific marker; specificity of antibody unclear | ||
| LOH | Basolateral | Mouse | 123 | Immunofluorescence; co-labeling with segment-/membrane-specific marker | |
| ∅? | Mouse | 140 | Immunohistochemistry; poor resolution; no co-labeling with segment-specific marker; specificity of antibody unclear | ||
| CD | Intracellular? | Mouse | 140 | Immunohistochemistry; poor resolution; no co-labeling with segment-specific marker; specificity of antibody unclear | |
| ∅ | Mouse | 123 | Immunofluorescence; co-labeling with segment-/membrane-specific marker | ||
| TRPV5 (ECaC1) | DT | Apical; subapical | Rat | 164 | Immunofluorescence; co-labeling with segment-specific markers |
| Cav3.1 (α1G) | DT; CD | Apical | Rat | 175 | Immunohistochemistry; co-labeling with segment-specific markers |
| Uptake pathway | Localization | Fe | Cd2+ | ||||
|---|---|---|---|---|---|---|---|
| Substrate/ligand | K D/K0.5 (nmol l−1) | Ref. | Substrate/ligand | K D/K0.5 (nmol l−1) | Ref. | ||
| a PT: proximal tubule; LOH: loop of Henle; DT: distal tubule; CD: collecting duct. b See Section 7.2 for detailed explanations. c NGAL: neutrophil gelatinase-associated lipocalin. | |||||||
| Megalin:cubilin:amnionless | PTa | Transferrin | 20 | 16 | ?b | 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
207 |
| NGAL | 60 | 112 | Cd2+-metallothionein | ||||
| Transferrin receptor 1 | PT; CD | Transferrin | 0.2–0.4 | 124 and 125 | ? | ∅ | ∅ |
| NGALc/24p3/lipocalin-2 receptor (SLC22A17) | DT; CD | Transferrin | 100 | 182 | Cd2+-metallothionein | 100 | 182 |
| NGAL | 0.090 | 197 | |||||
| DMT1 (SLC11A2) | PT; LOH; DT; CD | Fe2+ | 1000 | 11 | Cd2+ | 1000 | 11 |
| ZIP8 (SLC39A8) | PT | Fe2+ | 700 | 152 | Cd2+ | 620 | 148 |
| ZIP14 (SLC39A14) | PT | Fe2+ | 2300 | 151 | Cd2+ | 100–1100 | 149 |
| Ferroportin (FPN1/SLC40A1) | PT; LOH; CD | Fe2+ | <100 | 41 | ∅ | ∅ | 41 |
| TRPV5 (ECaC1) | DT | Fe2+ | <1000 (estimated) | 169 | Cd2+ | Micromolar (estimated) | 297 |
| Cav3.1 (α1G) | DT; CD | Fe2+ | Low micromolar (estimated) | 180 | Cd2+ | Low nanomolar (estimated) | 295 |
6.1. Iron transporters of the proximal tubule (PT)
| Megalin:cubilin:amnionless | |||||
|---|---|---|---|---|---|
| Ligand | K D (nmol l−1) | Ref. | Concentration in plasma (μmol l−1) | Ref. | Concentration in glomerular filtratea (nmol l−1) |
| a Calculations are based on estimated glomerular sieving coefficients of plasma proteins.17 | |||||
| Transferrin | 20 | 16 | 35 | 17 | 2 |
| NGAL [human]/siderocalin/24p3 [rodent]/lipocalin-2 | 60 | 112 | 7 | 116 | 650 |
| Albumin | 630 | 115 | 690 | 17 | 53 |
| Metallothionein | 100000 |
207 | 0.0005–0.005 | 208 and 209 | 0.5–5 |
| β2-Microglobulin | 420 | 278 | 0.11 | 17 | 100 |
| α1-Microglobulin | n.d. | 108 | 1 | 17 | 92 |
Reabsorption of filtered TBI in the renal PT has been mainly attributed to megalin-dependent cubilin-mediated endocytosis.16 Yet, cubilin-independent megalin-mediated uptake of Tf may also occur.111 Based on its plasma concentration and calculated glomerular sieving coefficient (GSC; derived from studies in patients with renal Fanconi syndrome),17 the Tf concentration in the primary filtrate has been estimated to ∼2 nM, which would allow its PT reabsorption via cubilin because this receptor binds Tf with a KD of ∼20 nM, as determined by surface plasmon resonance analysis.16 The same applies to other filtered Fe-binding proteins (see above) that are known substrates of cubilin and/or megalin, including NGAL,112 albumin,111,113 and hepcidin.114 Albumin requires cubilin for renal PT internalization, which is supported by experiments using cubilin-deficient mice.111 The concentration of albumin in the glomerular filtrate has been calculated to ∼53 nM17 and the KD of albumin to cubilin amounts to ∼0.63 μM.115 The plasma concentration of NGAL in healthy subjects amounts to ∼6.5 μM116 and should reach concentrations approximating ∼0.65 μM in the ultrafiltrate based on an estimated GSC of ∼0.1.17 Surface plasmon resonance analysis has demonstrated binding of apo-NGAL to megalin with a KD of ∼60 nM,112 which is about 10-fold lower than the estimated NGAL concentration in the primary filtrate (see Table 3). Although, both lactoferrin117 and hepcidin114 bind to megalin and are likely to be filtered by the glomerulus (the latter completely), their binding affinity to megalin has not be determined. Overall, significant amounts of both TBI and NTBI are filtered by the glomerulus and likely to be reabsorbed via megalin:cubilin:amnionless in the PT (but see also Section 6.1.2 for Tf reabsorption).
TBI and NTBI that has been reabsorbed by the PT can meet four possible and not mutually exclusive fates (see ref. 12 for a review): transcytosis; export back into the circulation via the Fe efflux transporter FPN1 aided by hephaestin (see Section 6.1.5); storage in ferritin (see Section 3); and utilization by PT cytosolic or mitochondrial Fe requiring processes (see Section 6.1.3). In vivo transcytosis of Fe transporting proteins has been recently reported by a number of groups. Thus, albumin transcytosis in the PT was inferred from intravital tracking of fluorescent albumin by two-photon microscopy118 as well as in a study showing the appearance of transgenic albumin specifically expressed in podocytes in the plasma where transcytosis was suggested to be mediated by a neonatal Fc (fragment crystallizable) receptor.119 However, the issue of whether transcytosis of intact albumin actually occurs in renal PT is highly controversial.120,121 Whether bound Fe may be retained on albumin (and possibly other proteins) during such transcellular transfer, or may rather be released in some intracellular transit compartment, has, to our knowledge, not yet been investigated. Although transcytosis has also been reported for ferritin infused into the renal PT122 this process is unlikely to play a role in vivo due to the large size and therefore poor glomerular filtration of ferritin (see Section 5).
At the tissue level, DMT1 is ubiquitously expressed, most notably in the proximal duodenum, red blood cells, macrophages, but also in the kidneys and the brain.10 DMT1 is expressed in the plasma membrane, typically in enterocytes, where it mediates Tf-independent Fe2+ absorption into the organism.10 Alternatively, when DMT1 is located intracellularly, it is involved in the TfR1 pathway of Fe acquisition (as demonstrated in erythrocyte precursors or macrophages67,134) (see Section 3). There, DMT1 is localized to intracellular endosomes and lysosomes that are formed during endocytosis of the Tf–TfR1 complex. Vacuolar-type ATPases acidify the endosomes and lysosomes which promotes dissociation of Fe3+. Fe3+ is reduced to Fe2+ by ferrireductase/oxidoreductase activity in the lumen of endosomes and lysosomes that is mediated by Steap proteins and that have also been found expressed at the mRNA level in epithelia, including the kidney.64,65 This, in turn, activates DMT1 in the lysosomal membrane to co-transport the metal ion along with H+ into the cytosol.67,134
The intracellular localization of DMT1 has also been demonstrated in epithelial cells,135e.g. in immunolocalization studies of human 1B/+IRE and 1B/−IRE isoforms overexpressed in HEp-2 human larynx carcinoma cells.136 In the kidney, the 1A/−IRE isoform was not detected by semiquantitative RT-PCR of total RNA from mouse kidney.130 In contrast, we have detected all four DMT1 transcripts in RNA from rat renal cortex and a rat renal proximal tubule cell line, albeit with different abundance.137 At the protein level, evidence could only be obtained for the presence of the +IRE isoforms in mouse kidney cortex that were expressed at the apical pole of PT cells.138 In another study, murine +IRE and −IRE DMT1 isoforms were transfected in LLC-PK1 cells: the +IRE isoform was associated with a higher surface expression and slower rate of internalization, as opposed to the −IRE isoform, which was efficiently sorted to recycling endosomes upon internalization, whereas the +IRE isoform was not efficiently recycled and rather targeted to lysosomes.139
Consistent with a major role of megalin:cubilin dependent endocytosis for Tf clearance from the ultrafiltrate, DMT1 has been detected in late endosomes and lysosomes of rat kidney PT cells by electron microscopy and also mainly co-localized with late endosomal and lysosomal markers in a renal PT cell line.137 Furthermore, a marked increase of punctate intracellular DMT1 immunostaining was observed in rat renal PT upon Fe deprivation, whereas DMT1 was decreased when animals were fed an Fe enriched diet.15 Free Fe2+ has previously been postulated to be reabsorbed via DMT1 residing in the luminal membrane of mouse PT cells.138 However, this localization contrasts with other reports indicating exclusive intracellular localization of DMT1, both in PT from rat14,15,137 and mouse12,140 (see Table 1 for an overview). Insufficient resolution of the immunohistochemical images in the study by Canonne-Hergaux and Gros138 that could not distinguish between apical staining and staining of subapical vesicles has been proposed as a reason for this discrepancy.12 Moreover, due to the high affinity of transferrin for Fe3+,141 a brush-border membrane DMT1 could only reabsorb Fe from NTBI as Fe3+ that would also require its reduction by a brush-border ferrireductase and that has not been described in the kidney so far (with the exception of anecdotal evidence for a ferrireductase activity of a prion protein expressed in the apical membrane of PT cells142). Moreover, Wareing et al.13 have performed tracer microinjections of 55FeCl3 in the early PCT of rat kidney in vivo to determine the percentage of Fe reabsorption in the PT. Since urinary 55Fe recovery was independent of the injection site (which varied between 1 and 6 mm from the glomerulus to the injection site) the authors concluded that free Fe is not reabsorbed across the surface convolutions of the PT. This further argues against a role for apical DMT1 (and other Fe transporters) in non-protein bound NTBI reabsorption by the PT.
Mitochondria heavily rely on Fe-dependent metabolism and are therefore intracellular targets for Fe trafficking, which is particularly relevant in the kidney PT and thick ascending limb of LOH where mitochondria provide ATP for active reabsorption and secretion of solutes. Recently, we have obtained evidence for expression of the four major DMT1 isoforms in the OMM in several cell lines and tissues from multiple origin, including the kidney PT, and proposed that mitochondrial DMT1 represents a possible entry pathway for Fe and other metal ions utilized by mitochondria.85,86 We used a variety of methods, including (1) cryo-immunogold electron microscopy to detect DMT1 co-localization with the OMM protein VDAC1; (2) confocal immunofluorescence microscopy to visualize partial co-localization of DMT1 with the mitochondrial markers VDAC1 and Tom6 (translocase of outer membrane 6); (3) immunoblotting of OMM and IMM fractions to demonstrate co-purification with the OMM marker VDAC1, but not with the IMM marker adenine nucleotide translocase; (4) a split ubiquitin yeast-two hybrid screen where the mitochondrial protein cytochrome C oxidase subunit II (COXII) was identified as an interaction partner of DMT1; (5) co-immunoprecipitation of COXII with DMT1 from cell lysates.85 Most importantly, preliminary studies indicate that mitochondria isolated from stably DMT1-transfected HEK293 cells exhibit substantially higher uptake of the known DMT1 substrate 54Mn2+ when the cells had been pretreated with doxycycline to induce the DMT1 promoter.143 Moreover, 54Mn2+ uptake into mitochondria from induced cells was sensitive to a specific DMT1 inhibitor.143 Taken together, these data suggest that DMT1 not only exports Fe2+ (and Mn2+) from endosomes and lysosomes, but also serves to import metal ions, including Mn2+ and Fe2+, for mitochondrial utilization in the kidney PT and other tissues and cells.
Homozygous Belgrade rats (b/b) have a G185R mutation of DMT1 that diminishes transport and results in significantly increased serum Fe levels due to the inability of the tissues to utilize Fe.68,128 These animals have been investigated to estimate the role of renal DMT1 in reabsorption of Fe, however with conflicting results. Belgrade rats showed significantly reduced renal kidney 59Fe3+ 2 hours after intravenous injection of Fe–Tf, compared to wild-type or heterozygous animals, suggesting that DMT1 is involved in Fe uptake by renal tissue.144 In contrast, another study showed urinary iron excretion rates that were unchanged in b/b compared to heterozygous animals.145 This study may cast doubts on a functional role of DMT1 in reabsorption of Fe in the kidney, but alternate path(s) for Fe reabsorption by renal cells may also compensate for the lack of DMT1 protein. Indeed, significantly increased urinary Ca2+ excretion was measured in Belgrade rats that did not show DMT1 dependence of urinary Fe excretion rates,145 which may be explained by increased competition of Fe2+ with Ca2+ for renal reabsorption by Ca2+ channels in the DT of DMT1-deficient Belgrade rats (see Sections 6.3.1 and 6.3.2 for a further discussion). Another aspect needs also to be considered: a recent study with Belgrade rats has hinted to the fact that urinary Fe excretion increases with increasing age of the animals,146 suggesting a subtle but cumulative impact of DMT1 (dys)function on Fe handling by the kidney. This study may provide another explanation for the negative results described previously where young animals had been used.145 Consequently, we investigated renal Fe handling in >25 weeks old Belgrade rats and their heterozygous litter mates and measured ∼2-fold increased urinary Fe excretion (184 ± 40 versus 108 ± 9 μg l−1 kg−1 b.w.; n = 3) as well as ∼2-fold decreased kidney Fe concentrations (0.39 ± 0.11 versus 0.21 ± 0.03 mg g−1 kidney tissue; n = 5) in Belgrade animals compared with heterozygous controls (F. Thévenod, A. R. Nair, W.-K. Lee & M. D. Garrick; unpublished), which is in agreement with the studies in Belgrade rats demonstrating the importance of DMT1 for renal Fe reabsorption.144,146
Using thoroughly characterized affinity-purified rabbit polyclonal antibodies against rat FPN1, we have previously reported that FPN1 is expressed in rat PT (S2 > S1 > S3) where it is mainly localized in the basolateral plasma membrane (and some intracellular vesicles), as evidenced by immunohistochemistry and immunogold electron microscopy at high magnification.160 Interestingly, FPN1 was absent from glomeruli and DT. Iron loading resulted in increased surface expression of FPN1 in a rat renal PT cell line, as detected by immunofluorescence labeling of non-permeabilized cells as well as surface biotinylation experiments, but with no change in total cellular FPN1 expression, suggesting that FPN1 redistributes to the cell surface and that increased insertion of FPN1 into the plasma membrane may play a role in protecting PT cells from Fe overload.160 The basolateral localization of FPN1 in PT was subsequently confirmed in hepcidin(−/−)123 and heme oxygenase 1(−/−) mice161 using commercial antibodies, but FPN1 expression was much weaker in control animals. In contrast to those studies, Veuthey et al. showed both apical and basolateral FPN1 distribution in the mouse PT,140 and FPN1 was found only at the apex of PT cells in another mouse study.162 In addition to the poor resolution of the images shown in these mouse studies, the specificity and quality of the antibodies used is difficult to assess as they were either from commercial sources and/or poorly characterized (Dr B. Galy, European Molecular Biology Laboratory, Germany; personal communication) (information summarized in Table 1). There is also evidence to suggest that renal FPN1 expression is regulated by hepcidin: intraperitoneal hepcidin pre-injection (24 h) prevents FPN1 upregulation induced by ischemia-reperfusion injury, as demonstrated in whole membranes of mouse kidney.163 Furthermore, intraperitoneal hepcidin injection in mice induces a rapid (1 h) degradation of FPN1 in kidney homogenates (Drs R. P. L. van Swelm & D. W. Swinkels, Department of Laboratory Medicine, RUMC, Nijmegen, The Netherlands, personal communication; manuscript submitted).
6.2. Iron transporters of the loop of Henle (LOH)
6.3. Iron transporters of the distal tubule (DT)
Using a calcein-AM fluorescence assay to detect Fe in the cytosol under various Fe loading conditions, T-type calcium channels have been implicated in Fe2+ uptake by cardiomyocytes through the use of selective blockers.179 In a more detailed study, Lopin et al.180 examined the effects of extracellular Fe2+ on permeation and gating of Cav3.1 channels stably transfected in HEK293 cells, using whole-cell patch-clamp electrophysiology recording. In the absence of extracellular Ca2+, Fe2+ carried detectable, whole-cell, inward currents at millimolar concentrations (73 ± 7 pA at −60 mV with 10 mM extracellular Fe2+). With a two-site/three-barrier Eyring model for permeation of Cav3.1 channels,181 the authors estimated a transport rate for Fe2+ of ∼20 ions per s for each open channel at −60 mV, with 1 μM extracellular Fe2+ and in the presence of physiological Ca2+ concentrations (2 mM extracellular Ca2+). Reversal potentials indicated a Fe2+/Ca2+ permeability ratio of 0.06–0.18. Because Cav3.1 channels exhibit a significant “window current” at resting membrane voltage (open probability, ∼1%), the authors concluded that Cav3.1 channels represent a likely pathway for Fe2+ entry into cells at resting membrane potentials and possibly during the course of action potentials in excitable cells with clinically relevant concentrations of extracellular Fe2+180 (see Table 2).
6.4. Iron transporters of the collecting duct (CD)
The mounting relevance of Lip2 in the medical field has increased the interest in identifying putative receptors of this ligand. Megalin, the epithelial multi-ligand receptor expressed in renal PT (see Section 6.1.1) binds Lip2 with high affinity.112 In addition, a receptor for murine Lip2, Lip2-R, has also been cloned196 whose mRNA encodes 520 amino acids (molecular mass ∼60 kDa and 11 or 12 transmembrane domains depending on the predicted topology) and whose affinity for Lip2 is ∼1000× higher (KD ∼ 90 pM)197 than that of megalin (KD ∼ 60 nM).112 According to the SLC (solute carrier) nomenclature system this receptor is also named SLC22A17 or BOCT (brain organic cation transporter).198 However, classical substrates of organic cation transporters are not transported by SLC22A17 (ref. 199 and N. A. Wolff & F. Thévenod; unpublished). Interestingly, several short N- and C-terminal splicing variants (22 kDa and ∼30 kDa, respectively) of the Lip2-R have been described in humans and rodents, respectively,196,200 but their function in health or disease is unknown. Although Lip2-R protein is expressed in the kidney196 its localization and functions in that organ were unknown until recently. Using two affinity-purified polyclonal rabbit antibodies directed against the N- and C-terminal domains of Lip2-R, we showed apical expression of Lip2-R in rodent kidney DCT (where it co-localized with calbindin, Lip2-R being expressed apically and calbindin intracellularly) and CD (mainly inner medullary CD), but not in PT (where it was found weakly expressed intracellularly). In DCT, some cells were Lip2-R-positive but calbindin-negative, suggesting that Lip2-R is expressed in both early and late DCT (see Table 1). Lip2-R was also found expressed in respective mouse cell lines (mDCT209; mIMCD3, mCCDcl1), but not in PT cell lines (WKPT-0293 Cl.2) (ref. 182 and unpublished). We also confirmed the expression of several immunoreactive protein bands in purified plasma membranes by immunoblotting (MM ∼35 kDa, ∼45 kDa, ∼60 kDa and ∼130 kDa), thus confirming the presence of “short” and “long” forms of the protein that may represent splicing variants or dimers of the receptor, respectively (ref. 182 and unpublished). Chinese hamster ovary (CHO) overexpressing Lip2-R or mDCT209 cells expressing Lip2-R endogenously internalized submicromolar concentrations of fluorescence-labelled Tf, albumin, or MT and their uptake was blocked by 500 pM Lip2,182 which confirms that the uptake of these proteins is mediated by the Lip2-R. Using microscale thermophoresis, a powerful technique to quantify biomolecular interactions,201 we showed that MT binds to Lip2-R with a KD of ∼100 nM.182 Hence, Lip2-R seems to bind proteins filtered by the kidney, including Tf and MT, with high affinity and may contribute to receptor-mediated endocytosis of these proteins as well as of Lip2 in the distal nephron (see Table 2).
Is the uptake of metalloproteins, such as Tf, Lip2 or MT, by Lip2-R physiologically and pathophysiologically relevant when bulk protein reabsorption is thought to take place in the PT? Experimental evidence has demonstrated that physiologically a small but significant proportion of filtered proteins is reabsorbed by the distal segments of the nephron.202–205 Although megalin:cubilin:amnionless is a high-capacity receptor complex for endocytotic reabsorption of filtered proteins,206 some proteins/metalloproteins may bypass reabsorption in the PT, either as the consequence of their low affinity to megalin and low concentration in the ultrafiltrate (e.g. MT with a KD of ∼5–100 μM207 but a plasma concentration of ∼0.5–5 nM208,209) (in this context see Table 3) or due to limited reabsorptive capacity of the system (e.g. following glomerular or PT damage and ensuing proteinuria).17,210,211 A high-affinity protein receptor in the distal nephron such as Lip2-R could contribute to exhaustive protein/metalloprotein reabsorption and deplete the final urine from protein-bound Fe (and other metals) under physiological conditions, or limit losses associated with renal diseases, including various forms of inherited or acquired Fanconi syndrome.17 Indeed, two in vivo studies have demonstrated Fe uptake into the distal nephron of nephrotic rats212 or following glomerular damage induced by acute Fe overload.213 Interestingly, Fe deposits were found in lysosomes of DT by electron microscopy212 and kidney medullary tubule cells by histochemistry.213 Furthermore, in hepcidin(−/−) mice, a model of the Fe overload disease hemochromatosis, Fe deposits were also found in the distal nephron.123 Hence, increased uptake of proteins/metalloproteins by Lip2-R in the distal nephron could initiate or enhance kidney injury. Along these lines, a recent in vivo study has implicated the Lip2-R in the CD in contributing to initiation and/or aggravation of renal inflammation and fibrosis in response to proteinuria.214
Correnti et al.74 have recently questioned a role of Lip2 in cellular Fe metabolism based on their observation that gentisic acid (a putative mammalian siderophore) could not form a stable ternary complex with Lip2 and Fe and on their inability to demonstrate any physical interaction between Lip2 and N-(NTD) or C-terminal domains (CTD) of mouse Lip2-R by surface plasmon resonance analyses. However, using the 105 residue NTD of human Lip2-R and analysis of its interaction by microscale thermophoresis, isothermal titration calorimetry and nuclear magnetic resonance, we could demonstrate binding of human Lip2 to its cellular receptor NTD (A.-I. Cabedo Martinez et al.; submitted). Although the affinity we measured between human Lip2-R-NTD and human Lip2, i.e. ∼7 μM for apo-Lip2 and ∼20 μM for holo-Lip2 (Lip2 bound to the bacterial siderophore enterobactin) suggests that the N-terminus alone cannot account for the internalization of Lip2 by Lip2-R and that other parts of the receptor must contribute to the interaction, our results are in contradiction with the conclusions of Correnti et al.74 We suspect that their failure to observe any direct interaction between Lip2 and mouse Lip2-R results from (1) their inability to control the state of their recombinant Lip2 (apo- or holo-) and (2) a lack of proper formation of the disulfide bridges of their mouse Lip2-R-NTD preparation, as the formation of aberrant disulfides would probably lead to forms of mouse Lip2-R-NTD that are unable to bind to Lip2 (A.-I. Cabedo Martinez et al.; submitted). Either or both of these points could explain their inability to observe an interaction between Lip2 and mouse Lip2-R-NTD. Overall, our data suggest that Lip2-R represents a high-affinity multiligand receptor for apical endocytosis of proteins and/or metalloproteins (such as Tf or Cd2+–MT) in renal epithelia. Increased endocytosis subsequent to glomerular and/or PT damage may promote renal epithelial damage by death, inflammation and fibrosis.
7. Cadmium toxicity
7.1. General considerations and link to iron transport
Pollution by cadmium (Cd) is rising worldwide because of intensified industrial activities that have increased its availability and because Cd cannot be degraded further.7,215 Chronic exposure to low Cd concentrations is a significant health hazard for ∼10% of the world population that increases morbidity and mortality.216 Indeed, Cd damages multiple organs in humans and other mammalian organisms by causing nephrotoxicity, osteoporosis, neurotoxicity, genotoxicity, teratogenicity, or endocrine and reproductive defects.217In mammalian organisms, Cd is a toxic-only element with no known role in physiological processes: as a non-essential metal ion Cd2+ competes with essential metal ions in cells where it disrupts cellular functions and leads to disease. Although Cd2+ is not capable of catalyzing Fenton chemistry in biological systems, it may initiate free radical chain reactions by depleting endogenous redox scavengers, inhibiting anti-oxidative enzymes, blocking the mitochondrial electron transport chain, and/or displacing redox active metals, such as Fe2+ or Cu2+ from their carrier proteins218 and thereby trigger cell death by apoptosis (reviewed in ref. 219). Cd2+ can also substitute for Ca2+ in cellular signaling or for Zn2+ in many enzymes and transcription factors which may account for some of the biological effects of Cd2+.219,220 In order for toxicity to occur Cd2+ must first enter cells by utilizing transport pathways for essential metals, such as Fe2+, Zn2+, Cu2+, Ca2+ or Mn2+, that are present in biological systems mostly as complexes with small organic molecules or as metalloproteins. These metal ion compounds are hydrophilic and must permeate lipophilic cellular membranes through intrinsic proteinous pathways. Hence, free or small complexed metal ions may be transported via ion channels or carrier proteins whereas metalloproteins are taken up by receptor-mediated endocytosis (RME). Cd2+ has similar physico-chemical properties as essential metal ions (for a detailed account see ref. 18 and references therein) and Cd2+ complexes are analogous to endogenous biological molecules, therefore this attribute has been termed “ionic and molecular mimicry”.4,221 Hence, transport (and toxicity) of Cd2+ can only occur if cells possess pertinent transport pathways for essential metals or biological molecules. A number of pathways has been suggested to allow Cd2+ entry in excitable and non-excitable cells9 and the most likely candidates have been recently reviewed.18,215
Chronic exposure to Cd2+ involves very low concentrations of Cd2+ that originate from environmental pollution and mainly results from dietary sources and cigarette smoking. Hence Cd2+ enters the body primarily through the lungs and the gastrointestinal (GI) tract: the absorption of Cd2+ from the lungs is much more effective than that from the gut; however, Cd2+ absorption from the GI tract is the main route of Cd2+ exposure in humans.215 Following absorption in the lungs and/or intestine, Cd2+ in the blood at first largely binds to albumin and other thiol-containing HMWP and low molecular weight proteins (LMWP) in the plasma, including MT, as well as to blood cells. But Cd2+ tends to concentrate in blood cells (mainly erythrocytes) and <10% remains in the plasma.222 Since intravenously injected MT-bound Cd2+ in mice is quickly cleared from the plasma by the kidneys223 this protein fraction in the circulation – that is assumed to originate from Cd2+ stored in liver cells as Cd2+–MT and is released from damaged cells (see below) – has been thought to be of great importance for the transport of Cd2+ to the kidney during long-term exposure224–226 (although the plasma Cd2+–MT concentrations following experimental injections exceeded physiological MT concentrations by >2000-fold;208,209 see Section 7.2 for a critical discussion). The blood level of Cd2+ largely reflects recent Cd2+ exposure with a half-life of 75–128 days.227 It ranges between 0.03 and 0.5 μg l−1 (∼0.3–5 nM) depending on the preparation method and the populations studied (reviewed in ref. 228) and its concentration in plasma will be at least ten-fold lower.222
Cd2+ reaching the plasma is thought to be initially transported to the liver where intracellular Cd2+ induces the synthesis of the endogenous detoxicant MT, which binds, sequesters and detoxifies Cd2+ because its affinity to Cd2+ is very high with a KD of ∼10−14 M (reviewed in ref. 229). Yet, a small proportion of liver (Cd2+–)MT is assumed to be slowly released into blood plasma as the hepatocytes in which Cd2+ is sequestered die off, either through normal turnover or as a result of Cd2+ injury.224,230,231 Several studies have demonstrated that following long-term exposure to Cd2+ and even at long time intervals after a single exposure, the level of Cd2+ is initially highest in the liver and then gradually increases in the kidneys.232,233 The strongest evidence for the concept that the major source of renal Cd2+ during chronic Cd2+ exposure is derived from hepatic Cd2+, which is transported in the form of Cd2+–MT in blood plasma, was derived from studies with transplanted livers of Cd2+-exposed rats to normal rats.234 Cd2+ and MT in the liver of recipient rats decreased over time after surgery whereas renal Cd2+ and MT levels increased and most of the Cd2+ in the kidney was bound to MT.234 Although none of these data proved that redistribution of Cd2+ from the liver to the kidney is mediated by circulating Cd2+–MT, this hypothesis still prevails in the literature (see Section 7.2 for a critical discussion).
Once absorbed Cd2+ is stored in various organs, including the kidneys and liver, with a half-life of several decades.2,7,215 This happens because Cd2+ induces the expression of detoxifying molecules that form a complex with the metal ion and thereby alleviate its toxic effects. But this apparently beneficial effect is a two edged-sword because these seemingly harmless Cd2+ complexes represent an endogenous source of high concentrations of potentially toxic Cd2+. The major detoxifying tool of the cell for Cd2+ complexation is MT. MTs are low-molecular weight (MM ranging from 3.5–14 kDa), cysteine-rich metal-binding proteins that have the capacity to bind both physiological Zn2+ ions and toxic Cd2+ ions through the thiol group of their cysteine residues that represent nearly 30% of its amino acidic residues.100,235,236
7.2. Cadmium handling by the kidney
As a consequence of its storage in tissues Cd2+ is very poorly excreted, mainly in urine and feces. With low, or even moderate, levels of exposure, little or no Cd2+ is excreted in the urine,237 which indicates that Cd2+ is reabsorbed and stored by the kidney. In humans, the amount of Cd2+ excreted daily in urine represents only about 0.005–0.015% of the total body burden237 and amounts to 0.05–0.2 μg l−1 (reviewed in ref. 228). Most of the Cd2+ in urine is bound to MT238,239 and it is assumed that urinary Cd2+ and MT stem from filtered Cd2+–MT and normal turnover and shedding of epithelial cells, or – perhaps – from exosomes derived from Cd2+–MT containing tubule epithelia. This supposition is based on chronic studies in several mammalian species showing that urinary excretion of Cd2+ increases slowly for a considerable time as a reflection of the level of Cd2+ exposure and the body burden of the toxic metal ion, which correlates with an increase of Cd2+ in the renal cortex (reviewed in ref. 240). But when the concentration of Cd2+ in the renal epithelial cells reaches a threshold value of ∼150–200 μg g−1 wet weight Cd2+ disrupts tubular reabsorptive processes and the excretion of Cd2+ and MT begins to increase more strongly in a linear manner, which is associated with the onset of polyuria and proteinuria241,242 (reviewed in ref. 240). When kidney dysfunction aggravates and a sharp increase in excretion of Cd2+ and MT occurs, a decrease in renal and liver Cd2+ concentrations is also observed.243,244 Hence, the early, slow linear phases of Cd2+ and MT excretion likely mirror the level of chronic Cd2+ exposure whereas the later sharp increases in excretion reflect Cd2+-induced tubular injury.In the previous paragraphs it has been emphasized that chronic exposure to low environmental or dietary Cd2+ concentrations results in accumulation of the metal ion in the kidney with a biological half-life of ∼20 years or more2,7,215 where it may cause damage, fibrosis or failure,245,246 or – with Cd2+ being a class 1 human carcinogen – cancer.247 In contrast, acute or subchronic Cd2+ nephrotoxicity is associated with a general transport defect of the PT that mimics the de Toni-Debré-Fanconi-syndrome248,249 with proteinuria, aminoaciduria, glucosuria and phosphaturia (for review, see ref. 250).
For several decades the following scenario has prevailed to account for acute or chronic Cd2+ toxicity in the kidney: it has been presumed that Cd2+ in the circulation is filtered by the glomerulus because of the small molecular mass of most circulating Cd2+ forms: in the plasma, Cd2+ is thought to be loosely associated with molecules, such as LMWP – e.g. β-2 microglobulin, α-1 microglobulin, retinol-binding protein, insulin or parathyroid hormone – with amino acids or the sulfhydryl compounds GSH or cysteine, or tightly bound to specific metal-binding proteins such as the LMWP MT.229 Several HMWP, e.g. albumin, bind Cd2+ with low affinity,251 also show some degree of glomerular filtration17 and may therefore carry Cd2+ into the ultrafiltrate. Furthermore, the Fe-binding protein Tf that is filtered by the glomerulus and ferritin (see Section 5) may also bind Cd2+ in plasma.252–254 The PT largely contributes to the reabsorption of Cd2+ because as the first segment of the nephron it is responsible for bulk reabsorption of primary urine, which mainly takes place by solvent drag via paracellular routes (see Section 4). But PT cells also possess apical transporters (as proposed for ZIP8 and ZIP14 transporters that may carry both Fe2+ and Cd2+;148,149 however see Section 6.1.4 and ref. 147 for a note of caution), amino acid transporters, metabolizing brush-border enzymes (such as γ-glutamyl transpeptidase that degrades GSH), and the receptor for protein endocytosis megalin:cubilin:amnionless108 that may mediate apical uptake of Cd2+ ions and Cd2+ complexes (see Section 6.1.1, and ref. 9 and 18 for reviews). There is also evidence that Cd2+ is taken up at the basolateral surface of PT cells255,256 and it has recently been shown to take place via the organic cation transporter 2 (OCT2).257,258 Although it is mechanistically remarkable that an organic cation transporter is able to carry a divalent metal ion as a substrate, a Km value of ∼54 μM for Cd2+258 suggests that the in vivo toxicological relevance of this transporter is questionable.
Like other LMWP, Cd2+–MT/MT is thought to be reabsorbed from primary urine into PT cells of the kidneys by megalin:cubilin:amnionless receptor-mediated endocytosis206,259–261 (see Table 3). Studies with cultured PT cells have provided evidence that Cd2+–MT/MT is trafficked to acidic late endosomes and lysosomes262,263 where MT may be degraded by lysosomal proteases whereas Cd2+ may exit the endosomal/lysosomal compartment by DMT1-mediated efflux into the cytosol.137,264 This may cause acute PT toxicity in cases where PT cells would have to handle high concentrations of endocytosed Cd2+–MT.264 However, if the Cd2+–MT stress is low PT cells may adapt by inducing the upregulation of detoxifying proteins, including MT265 that inactivate and complex Cd2+ released from lysosomes into the cytosol for long-term storage.266 Cd2+ accumulation in the PT (and storage as Cd2+–MT) may be likely further promoted by the absence of an efflux pathway for cytosolic Cd2+ into the extracellular fluid or blood plasma because FPN1 that is expressed at the basolateral cell side of PT cells160 does not transport Cd2+ (as opposed to Fe2+)41 (see also Section 6.1.5).
The concept that endocytosis of filtered Cd2+–MT by megalin:cubilin:amnionless is mainly responsible for accumulation of Cd2+ in the PT was based on studies demonstrating redistribution of hepatic Cd2+ to the kidney that was supposed to be Cd2+–MT,232–234 on in vivo animal studies with intravenously injected Cd2+–MT223,267–272 as well as on microinjections of Cd2+–MT in isolated PT.273 It was confirmed and elaborated in cell culture studies260–263 (reviewed in ref. 274). However, all in vivo and cell culture studies applied Cd2+–MT at micromolar concentrations. Meanwhile surface plasmon resonance analyses, cell culture and in vivo studies have established that the binding affinity of megalin for MT is ∼100 μM,207,260,272 which is compatible with the observations from the in vivo animal studies. But considering that plasma concentrations of MT are in the range of ∼0.5–5 nM208,209 in humans (and healthy laboratory animals), the concept that filtered (Cd2+)–MT is also taken up by PT via megalin:cubilin:amnionless-dependent endocytosis under physiological conditions275 is unfounded, and thus the current models of Cd2+ accumulation (and chronic toxicity) in the PT should be revised (nevertheless Cd2+–MT may still be useful as a model compound to study RME of Cd2+–protein complexes in cell culture or in vivo as the high binding affinity of MT to Cd2+ precludes dissociation of toxic Cd2+ ions). It is more likely that other LMWP (e.g. α1- or β2-microglobulin) and albumin, which also bind Cd2+ and reach submicromolar concentrations in the ultrafiltrate,276,277 are more relevant ligands (e.g. β2-microglobulin binds to megalin with a KD of ∼0.42 μM278) (see also Table 3) that are endocytosed by megalin:cubilin:amnionless to induce Cd2+ accumulation and eventually PT toxicity. It could be argued that these proteins exhibit relatively low affinities to Cd2+ compared to MT (reliable KD values of ∼10−6 M for Cd2+ and other divalent metal ions are only available for albumin and β2-microglobulin251,279), indicating that at steady-state maximally 1% of these proteins in the circulation will form complexes with blood Cd2+ (with a concentration of 0.3–5 nM228) whereas MT in the circulation will be Cd2+-saturated (based on equivalent low nM concentrations of MT and Cd2+ and a KD of ∼10−14 M229). Yet the relatively high concentration of microglobulins and albumin in the primary filtrate and their high binding affinity to megalin (see Table 3) combined with the multiplicative effect of their continuous glomerular filtration makes them more prone to accumulate in the PT and contribute to chronic renal PT toxicity than Cd2+–MT whose concentration in the primary filtrate is at least 105-times lower208,209 than its KD for megalin binding.207
Because only 0.02–03% of filtered proteins, including MT (based on measured values for plasma and urinary MT 99.7% of filtered MT must be reabsorbed by the kidney208,209), are excreted with the urine280 (reviewed in ref. 281) additional uptake pathways for proteins and protein–Cd2+ complexes must exist in the distal nephron (see below).
Cd2+ may not be only toxic to PT cells, but also to glomeruli and the distal nephron.282 Glomerular damage with a decreased GFR has been observed in occupationally exposed workers283 and in environmentally exposed populations where it may occur at similar Cd2+ dose levels as the tubular damage.246,284 But overall, the pathogenesis of the glomerular lesion in Cd2+ nephropathy is not well understood.285 Downstream segments of the nephron, both in the cortex and medulla, also exhibit a high permeability to Fe2+ and other metal ions (see Sections 6.2–6.4) and could hence contribute to uptake of Cd2+. This would be particularly the case when proximal segments of the nephron are defective or “overwhelmed” as a consequence of increased filtration (e.g. due to glomerular damage). Under those circumstances, later segments should become more relevant for uptake. As an example, a chronic in vivo study in ducks demonstrated substantial damage to glomerular podocytes following exposure to a combination of lead, methylmercury and cadmium that was associated with enhancement of degenerative changes in PT and CD; in contrast exposure to cadmium alone showed no podocyte damage and tubular damage was restricted to PT whereas the CD was not affected.286 Sporadic evidence for chronic Cd2+ toxicity of the distal portions of the nephron induced by Cd2+ exposure has also been obtained, both in experimental animals287,288 and in Cd2+-exposed workers,289 but the mechanisms of distal nephron damage remain unclear.
In agreement with studies demonstrating in vivo55Fe transport in the rat LOH13 that may be mediated by apical DMT114 (see Section 6.2.1), Barbier et al.290 performed 109Cd2+ tracer microinjections into the late PCT and early DCT of rat kidney and 109Cd2+ reabsorption in the LOH was obtained by calculating the difference between 109Cd2+ recovery after early DCT and late PCT. The authors obtained 46.8% unidirectional 109Cd2+ fluxes that were reduced to 25.4% in the presence of 100 μM Fe2+, suggesting that DMT1 in the LOH is involved in 109Cd2+ uptake (and competes with Fe2+ for reabsorption).290 This report is unique for its exhaustive characterization of Cd2+ transport by the nephron, but unfortunately no additional studies have been published to confirm its conclusions. Despite variable and partly questionable results of FPN1 expression and localization in the rodent LOH123,140,160 (see Section 6.2.2), even if FPN1 were expressed in the LOH Cd2+ would remain trapped within the cells of the LOH because it is not transported by FPN141 (see Section 6.1.5).
Given the large number of Ca2+ channels expressed throughout the body, the importance of Ca2+ signaling, and the large number of ions a channel can transport (∼105 ions per s), even slight permeability of a Ca2+ channel to Cd2+ might lead to significant Cd2+ entry. Indeed, several Ca2+ channels that are expressed in the apical membrane of DCT164,175 are known to transport Cd2+. T-type Ca2+ channels are blocked by Cd2+,291,292 but their role in Cd2+ transport had not been investigated until recently. Cav3.1, also known as α1G, is a T type Ca2+ channel and is expressed in the DT (see Section 6.3.2). Cav3.1 channels may be suitable for Cd2+ transport, because they have a well-defined and substantial window current at negative membrane potentials at which the driving force for divalent cation entry is high293 and they are ∼2-fold less selective for Ca2+ than are L-type Ca2+ channels,170 which suggests that Cd2+ may have an increased chance of permeating the channel in the presence of competing Ca2+. Furthermore, development of resistance to Cd2+ in cell culture has been linked to down-regulation of Cav3.1, which suggested the involvement of this channel in Cd2+ toxicity.294 Consequently, Lopin et al.295 examined the effects of extracellular Cd2+ on permeation and gating of Cav3.1 channels stably transfected in HEK293 cells, by using whole-cell recording. In the absence of other permeant ions (Ca2+ and Na+ were replaced by N-methyl-D-glucamine), Cd2+ carried sizable inward currents through Cav3.1 channels (210 ± 20 pA at −60 mV with 2 mM Cd2+). Incubation with radiolabeled 109Cd2+ confirmed uptake of Cd2+ into cells with Cav3.1 channels. With a two-site/three-barrier Eyring model for permeation of Cav3.1 channels,181 a transport rate for Cd2+ of ∼1 ion per s was estimated for each open channel at −60 mV, with 3–10 nM extracellular Cd2+ and in the presence of 2 mM extracellular Ca2+. On the basis of the Goldman–Hodgkin–Katz theory,296 a Cd2+/Ca2+ permeability ratio of 0.66 was calculated, with Cd2+ being only slightly less permeable than Ca2+.295 Blood Cd2+ concentrations range between 0.3 and 5 nM (reviewed in ref. 228). Following glomerular filtration, the concentration of the “free” ionic form of Cd2+ in the primary urine of the nephron may increase up to 15-fold in the lumen of the DT (see below). In addition, luminal ionic Cd2+ may be further increased by its release from small peptides that are degraded by brush-border enzymes (such as γ-glutamyl transpeptidase that degrades GSH). Hence, in view of the significant “window current” at negative voltages and the high permeability of Cav3.1 channels for Cd2+ at low nanomolar concentrations (in the presence of physiological Ca2+ concentrations), these channels are a likely candidate pathway for Cd2+ entry into cells expressing Cav3.1 channels, including the kidney DT. Thus, Cav3.1 channels could significantly contribute to the in vivo renal toxicity of Cd2+ (see Table 2). In another study, the human TRPV5 (ECaC1) of the vanilloid family of the transient receptor channel (TRP) superfamily was transiently expressed in the plasma membrane of human embryonic kidney (HEK293) cells297 (see also Section 6.3.1). Cd2+ (and less well Zn2+) permeated hTRPV5 in ion imaging experiments using Fura-2 or Newport Green DCF (dichlorofluorescein) with an EC50 of ∼10 respective ∼100 μM Cd2+ depending on the absence or presence of 1 mM Ca2+ in the extracellular medium. The results were further confirmed using whole-cell patch clamp technique. Transient overexpression of hTRPV5 sensitized cells to Cd2+ toxicity. Hence, although micromolar concentrations of Cd2+ appeared to be required for permeation the results suggest that TRPV5 may also play a role in Cd2+ uptake by the DCT, especially under low Ca2+ dietary conditions, when these channels are maximally upregulated. Functional studies in vivo support these cell culture studies. Using both, 109Cd2+ and 45Ca2+ tracer microinjections into the early and late DCT of rat kidney and recovery in the urine, Barbier et al. showed about 20–25% unidirectional reabsorption of either of the tracers in the DCT that were almost completely abolished in the presence of 100 μM Fe2+ or 20 μM Cd2+, respectively (which, of course, seems too high from a viewpoint of physiological relevance).290 This suggests that Cd2+ is taken up by Fe2+ and/or Ca2+ transporters in the DCT, possibly DMT1 that is expressed in the luminal membrane of this nephron segment14,15 (see Section 6.3.3), but also TRPV5/Cav3.1 Ca2+ channels (see above). However, the experimental design of this tracer microinjection study could not exclude that DMT1 expressed in the CD may also mediate 109Cd2+ reabsorption290 (see below). Finally, we have previously shown apical expression of the Lip2-R in rodent kidney DT (see Sections 6.3.4 and 6.4.2) and cultured mDCT209 cells expressing Lip2-R at their surface internalized submicromolar concentrations of fluorescence-labelled MT that was blocked by 500 pM of the endogenous ligand Lip2.182 And Cd2+–MT caused cell death in mDCT209 cells that could be rescued by 500 pM Lip2.182 Hence, it is possible that Lip2-R contributes to receptor-mediated endocytosis of toxic Cd2+–MT and other Cd2+–protein complexes in the DT.
Cd2+ reabsorption by terminal nephron segments, i.e. CD, has been investigated by Barbier et al.290 using 109Cd2+ and 45Ca2+ tracer microinjections. Unidirectional 45Ca2+ fluxes in the terminal segments of the nephron were not affected by 20 μM Cd2+, which suggests that Cd2+ permeating Ca2+ channels are less likely expressed in the CD. In contrast, they showed that 50–100 μM Fe2+, Co2+ and Zn2+ increased 109Cd recovery in the urine after microinjection in the early DCT. This suggests involvement of DMT1 in nephron segments downstream of the early DCT, i.e. late DCT and CD (see Section 6.4.1). Mouse IMCD3 cells are sensitive to CdCl2 (LC50 ∼ 40 μM), indicating uptake of Cd2+ by these CD cells298 although the toxic concentrations of Cd2+ in these cells were much higher than the Km of DMT1 for Cd2+ transport of ∼1 μM.11,132 The likely Fe transport pathways of the distal nephron segments (LOH, DT, CD) that compete with Cd2+ for uptake are summarized in Table 2. Although Lip2-R is expressed in CD (see Section 6.4.2) and mediates uptake and toxicity of Cd2+–MT in various cultured cells,182,299 its role in Cd2+–MT transport and cell damage in the CD has not been investigated so far.
A difficulty in the attempt to estimate the role of distal nephron segments (LOH, DT, CD) in Cd2+ uptake – and given the binding affinity of putative transport pathways for Cd2+ – is the inability to determine accurately the actual concentrations of ionic and complexed forms of Cd2+ in nephron segments downstream of the PT. Nevertheless, an approximation can be obtained from the ratio of inulin concentration in the tubule fluid over plasma (TF/P). Inulin is an indicator of the GFR, i.e. a molecule that is only filtered by the glomerulus and neither reabsorbed nor secreted by the nephron. The TF/P ratio of inulin therefore reflects fluid reabsorption by the nephron.300 The TF/P ratio of inulin increases from 1 to 3 at about 2/3 of the PT length. It reaches a value of 7 at the beginning of the DT and increases up to ∼15 towards its end to reach a final value of 10–200 in the final urine depending on the diuresis condition (water- and anti-diuresis, respectively). In other words, the concentration of non-reabsorbed solutes increases by a factor of 3 along the PT, varies between 7 and 15 along the DT and can increase up to 200-fold in the CD. Consequently, the concentrations of Cd2+ and MT/Cd2+–MT may increase up to 10–15-fold in the DT and up to 200-fold in the CD, suggesting that these nephron segments may be more relevant segments of the kidney cortex for Cd2+ and MT/Cd2+–MT uptake and accumulation under conditions of chronic low Cd2+ exposure than previously thought. DMT1 and the Lip2-R expressed in DT (cortex) and CD segments (cortex and medulla) are more likely to efficiently reabsorb Cd2+ and Cd2+–MT, respectively because of their high affinity to these Cd2+ compounds (Km of DMT1 for Cd2+ ∼ 1 μM;11,132KD of Lip2-R for MT ∼ 120 nM182). The renal medulla also accumulates significant amounts of both Cd2+ and (Cd2+–)MT in humans and concentrations of both Cd2+ compounds can reach ∼50% of the levels found in the cortex.301,302 Indeed, MT has been detected by immunohistochemistry in distal segments of the nephron using cortical and medullary sections of rodent and human kidney303–305 (although no co-localization with nephron segment-specific markers was performed) and its expression was increased by exposure to Cd2+.304,305 But why then is Cd2+ nephrotoxicity less apparent in the DT and kidney medulla? The relative resistance of the DT and kidney medulla to Cd2+ toxicity may result from their lower sensitivity to oxidative stress,306,307 their increased potential for adaptive responses, and stress-induced factors (e.g. hypoxia-inducible factor-1α (HIF-1α), hepcidin, neutrophil gelatinase-associated lipocalin (NGAL) to name a few),21,308,309 and their metabolic profile (largely anaerobic glycolysis due to a low partial pressure for O2 in the medullary segments).310 All these issues are known to account for the resistance of the DT and kidney medulla to acute tubular damage (e.g. involving mitochondria)311 and necrosis (outer medulla (straight segment of PT, medullary thick ascending limb of LOH) > cortex (PT, DT) ≫ inner medulla) elicited by various inducers of AKI (reviewed in ref. 312).
8. Determinants of the fate of the kidney exposed to iron or cadmium
Irrespective of the nature of the Cd2+ compound that is taken up by tubule cells, its impact on cell viability differs from the effect of Fe. Both metal ions appear to be partly taken up as protein–metal complexes via RME. Moreover, both metal ions may accumulate intracellularly and be detoxified by binding to high-affinity chaperone proteins: for instance, the intracellular Fe storage protein ferritin is induced by overload of the PT with Fe in vivo.54,123,313,314 Similarly, following Cd2+ exposure in vivo PT cells upregulate the scavenger protein MT for Cd2+ storage.234,235 Both proteins even share some degree of overlapping specificity for Cd2+ and Fe2+: apart from binding Fe with high affinity91 ferritin also binds Cd2+.253,254,315 Conversely, although MT binds Cd2+ with very high affinity316 it has the ability to form complexes with Fe2+ as well.317 Yet, it is surprising that acute or chronic Fe overload generally does not cause manifest renal damage213,314 whereas nephrotoxicity is not an unusual sequel of acute or chronic Cd2+ exposure.245,246,250 Transport (TfR1, megalin:cubilin:amnionless, Lip2-R, DMT1, Cav3.1, etc.) (compare Sections 6 and 7 and Table 2) and detoxification/storage mechanisms (MT, ferritin) are shared by Fe2+ and Cd2+. Hence, the differential toxicity of Fe2+ and Cd2+ in PT cells could be attributed to disparate expression and/or upregulation of the intracellular metal scavenger proteins ferritin and MT,318–320 which are both regulated by antioxidant response elements in their gene promoter region (reviewed in ref. 91 and 235). In addition, the differences in binding characteristics of these chaperone proteins for Fe and Cd2+ could be accountable: both, the binding capacity (maximally ∼4500 for ferritin91versus 7 metal ion binding sites for MT321) and/or in the binding affinities of ferritin or MT to Fe and Cd2+317,322 are at variance. However, currently there is no stringent evidence for the relative contribution of both chaperone proteins in determining the extent of Fe and Cd2+ toxicity in PT cells, therefore further work is needed to clarify these issues.
In contrast, obvious differences concern the cellular utilization or non-utilization (i.e. toxicity) of both metal ions, and the efflux pathway for Fe2+ and Cd2+. Fe2+ enters mitochondria possibly via DMT1 in the OMM85 and mitoferrins in the IMM87 for synthesis of heme and Fe–sulfur clusters.79,80 In contrast, after crossing the OMM (possibly via DMT1 in the OMM85) and entering the mitochondrial matrix through the mitochondrial Ca2+ uniporter in the IMM323 Cd2+ disrupts mitochondrial function,324,325 which leads to increased formation of ROS and death through apoptosis and/or necrosis (reviewed in ref. 219 and 326). Another principle difference is the inability of FPN1 at the basolateral side of PT cells to transport Cd2+ into the circulation and thereby to clear it from the cell, which is in contrast to FPN1 handling of Fe2+.41 Hence, these differences may underlie – or at least contribute to – the mode of damage likely developing after acute exposure to high concentrations of Cd2+ (necrosis) or to nephrotoxicity induced by chronic accumulation of low concentrations of Cd2+ (apoptosis, cancer development).
Considering the competition between Fe2+ and Cd2+ for transport at renal entry pathways (“ionic and molecular mimicry”),4,221 it should also be deduced that Fe deficiency may not only augment body and kidney Cd2+ burden5 by increased gastrointestinal absorption of Cd2+327 but also facilitate renal Cd2+ reabsorption and thereby elicit a higher likelihood of renal tubule damage.
9. Outlook
Despite a wealth of novel data suggesting a contribution of the kidney to systemic Fe homeostasis and good evidence for uptake and reabsorption of Fe by specific transporters in various nephron segments as well as their involvement in Cd2+ uptake and nephrotoxicity, there is still not enough in vivo data available. Presently, a detailed characterization of Fe transporters has been performed in cell lines and heterologous expression systems and these studies have unambiguously demonstrated which transporters are Fe2+ and Cd2+ selective and which are not (see Table 2). In contrast, only one study has described in vivo transport of Fe by the nephron and this report is already 15 years old.13 Similarly, only one study has investigated the role of different nephron segments in uptake of Cd2+ and other divalent metal ions in vivo.290 These studies – as exhaustive and thorough as they are – would need to be confirmed and extended, in particular by using nephron specific transporter knockout models. Indeed, this approach has been successfully used to clarify the role of megalin:cubilin:amnionless in reabsorption of proteins by the PT,115,278,328 including reabsorption of the Fe-binding protein Tf.16 Unfortunately, no study has attempted to investigate systemic Fe homeostasis in nephron specific megalin or megalin:cubilin deficient animals or whether the PT of these animals is protected against Cd2+ nephrotoxicity. Studies are underway that aim to investigate the role of renal Lip2-R in the uptake and toxicity of metalloproteins, including Cd2+–MT and Tf, in nephron specific lip2-R knockout mice, (F. Thévenod & S. de Seigneux; in preparation).Another weakness of this area of research is the inconsistent characterization of the renal localization of renal Fe transporters that has resulted in contradictory results (see Table 1). These conflicting data have even inspired some authors to indiscriminately adopt models of renal Fe transport that are not compatible with renal epithelial physiology and membrane transport. A critical analysis of the relevant experimental reports identifies four principle methodological problems that may also be linked: the tissue sections used for immunostaining were often of poor quality (e.g. tubule lumina were collapsed); authors failed to properly identify nephron segments, e.g. by co-localization studies with nephron specific markers; the images showed poor resolution; and last not least, antibodies used were often of doubtful origin or their specificity had not been proven (see Table 1). This lack of methodological rigor (obviously immunostaining data were not important enough and therefore did not seem to concern the authors) has, at least in part, weakened this field of research. Once more, the use of renal Fe transporter knockout models will hopefully shed light on these confusing data.
Nevertheless, this review has clearly demonstrated that the kidney plays a previously unsuspected role in systemic iron balance and that renal Fe transporters are crucial for the accumulation of Cd2+ in the kidney and the development of nephrotoxicity. Future studies, as suggested above, should be able to verify the significance of Fe transporters described in this review and possibly identify additional relevant uptake pathways for renal Fe transport. Last not least, we think that the contribution of circulating Cd2+–MT (originating from the liver or not) to chronic Cd2+ accumulation in the PT and its toxicity may not be as important as previously suggested and that other hypotheses should be envisaged and experimentally tested.
Abbreviations
| 2,5-DHBA | 2,5-Dihydroxybenzoic acid |
| AKI | Acute kidney injury |
| AQP2 | Aquaporin 2 |
| ATP | Adenosine triphosphate |
| BBM | Brush-border membrane |
| BOCT | Brain organic cation transporter (Lip2-R/Lipocalin-2 receptor) |
| calcein-AM | Calcein acetoxymethyl (AM) ester |
| CaV3.1 | Calcium channel, voltage-dependent (T-type, α1G subunit) |
| CCD | Cortical collecting duct |
| CD | Collecting duct |
| CHO | Chinese hamster ovary (cell line) |
| COXII | Cytochrome C oxidase subunit II |
| CTD | Carboxy-terminal domain |
| DCF | Dichlorofluorescein |
| DCT | Distal convoluted tubule |
| DMT1/Nramp2/DCT1/SLC11A2 | Proton-coupled divalent metal transporter 1 |
| DT | Distal tubule |
| EC50 | Half maximal effective concentration |
| ECaC | Epithelial calcium channel |
| ENaC | Epithelial sodium channel |
| Fc | Fragment crystallizable (region of an antibody) |
| FLVCR1 | Feline leukemia virus, subgroup C, receptor (heme exporter) |
| FPN1/IREG1/MTP1/SLC40A1 | Ferroportin |
| GFR | Glomerular filtration rate |
| GI | Gastrointestinal |
| Grx3_4 | Glutaredoxin 3_4 |
| GSC | Glomerular sieving coefficient |
| GSH | Glutathione |
| H-_L-ferritin | Heavy-_light ferritin subunit |
| HEK | Human embryonic kidney (cell line) |
| HIF-1α | Hypoxia-inducible factor-1α |
| HMWP | High-molecular weight protein |
| IMM | Inner mitochondrial membrane |
| IRE | Iron response element |
| K M0.5/Km | Substrate/metal concentration at which velocity is half-maximal |
| K D | Equilibrium dissociation constant |
| LC50 | Half maximal lethal concentration |
| Lip2 | Lipocalin-2 (NGAL/24p3) |
| Lip2-R | Lipocalin-2 receptor |
| LMWP | Low-molecular weight proteins |
| LOH | Loop of Henle |
| MDCK | Madin-Darby canine kidney (cell line) |
| MFRN1/SLC25A37 | Mitoferrin-1 |
| MFRN2/SLC25A28 | Mitoferrin-2 |
| MM | Molecular mass |
| MT | Metallothionein |
| NCCT | NaCl cotransporter |
| NGAL/24p3 | Neutrophil gelatinase-associated lipocalin |
| NTBI | Non-transferrin-bound iron |
| NTD | Amino-terminal domain |
| OCT2 | Organic cation transporter 2 |
| OMCD | Outer medullary collecting duct |
| OMM | Outer mitochondrial membrane |
| pA | Picoampere |
| PCBP1 | Poly(rC)-binding protein 1 |
| PCT | Proximal convoluted tubule |
| pS | Picosiemens |
| PT | Proximal tubule |
| RME | Receptor-mediated endocytosis |
| ROS | Reactive oxygen species |
| RT-PCR | Reverse transcription polymerase chain reaction |
| S1_2_3 | Segment 1_2_3 (PT) |
| SLC | Solute carrier |
| Steap | Sixtransmembrane epithelial antigen of the prostate (oxidoreductase) |
| T_L-type | Transient opening_long lasting-type (calcium channel) |
| TAL | Thick ascending limb |
| TBI | Transferrin-bound iron |
| Tf | Transferrin |
| TF/P | Tubule fluid over plasma |
| TfR1_2 | Transferrin receptor 1_2 |
| TOM6 | Translocase of outer membrane 6 |
| TRPML1/ML1/MLN1/MCLN1 | Transient receptor potential mucolipin 1 |
| TRPV | Vanilloid (V) family of the transient receptor potential channel (TRP) superfamily |
| VDAC | Voltage-dependent anion channel/porin |
| ZIP8_14/SLC39A8_14 | Zrt, Irt-related proteins 8 (ZIP8/SLC39A8) and 14 |
Acknowledgements
Financial support was obtained by the Deutsche Forschungsgemeinschaft TH345 and the Center for Biomedical Training and Research ZBAF from the University of Witten/Herdecke. We thank our long-time collaborators Drs Craig P. Smith (University of Manchester, U.K.), Robert A. Fenton (Aarhus University, Denmark) and Michael D. Garrick (SUNY, Buffalo, U.S.A.) for valuable discussions and support.References
- T. Ganz, Physiol. Rev., 2013, 93, 1721–1741 CrossRef CAS PubMed.
- G. F. Nordberg, Toxicol. Appl. Pharmacol., 2009, 238, 192–200 CrossRef CAS PubMed.
- I. Sovago and K. Varnagy, Met. Ions Life Sci., 2013, 11, 275–302 CAS.
- T. W. Clarkson, Annu. Rev. Pharmacol. Toxicol., 1993, 33, 545–571 CrossRef CAS PubMed.
- L. Jarup, M. Berglund, C. G. Elinder, G. Nordberg and M. Vahter, Scand. J. Work, Environ. Health, 1998, 24(Suppl 1), 1–51 Search PubMed.
- M. Berglund, A. Akesson, B. Nermell and M. Vahter, Environ. Health Perspect., 1994, 102, 1058–1066 CrossRef CAS PubMed.
- L. Jarup and A. Akesson, Toxicol. Appl. Pharmacol., 2009, 238, 201–208 CrossRef PubMed.
- G. L. Diamond, in Molecular biology and toxicology of metals, ed. R. K. Zalups and J. Koropatnick, Taylor & Francis, London and New York, 2000, ch. 11, pp. 300–345 Search PubMed.
- R. K. Zalups and S. Ahmad, Toxicol. Appl. Pharmacol., 2003, 186, 163–188 CrossRef CAS PubMed.
- H. Gunshin, B. Mackenzie, U. V. Berger, Y. Gunshin, M. F. Romero, W. F. Boron, S. Nussberger, J. L. Gollan and M. A. Hediger, Nature, 1997, 388, 482–488 CrossRef CAS PubMed.
- A. C. Illing, A. Shawki, C. L. Cunningham and B. Mackenzie, J. Biol. Chem., 2012, 287, 30485–30496 CrossRef CAS PubMed.
- C. P. Smith and F. Thévenod, Biochim. Biophys. Acta, 2009, 1790, 724–730 CrossRef CAS PubMed.
- M. Wareing, C. J. Ferguson, R. Green, D. Riccardi and C. P. Smith, J. Physiol., 2000, 524(pt 2), 581–586 CrossRef CAS.
- C. J. Ferguson, M. Wareing, D. T. Ward, R. Green, C. P. Smith and D. Riccardi, Am. J. Physiol. Renal Physiol., 2001, 280, F803–F814 CAS.
- M. Wareing, C. J. Ferguson, M. Delannoy, A. G. Cox, R. F. McMahon, R. Green, D. Riccardi and C. P. Smith, Am. J. Physiol. Renal Physiol., 2003, 285, F1050–F1059 CrossRef CAS PubMed.
- R. Kozyraki, J. Fyfe, P. J. Verroust, C. Jacobsen, A. Dautry-Varsat, J. Gburek, T. E. Willnow, E. I. Christensen and S. K. Moestrup, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12491–12496 CrossRef CAS PubMed.
- A. G. Norden, M. Lapsley, P. J. Lee, C. D. Pusey, S. J. Scheinman, F. W. Tam, R. V. Thakker, R. J. Unwin and O. Wrong, Kidney Int., 2001, 60, 1885–1892 CrossRef CAS PubMed.
- F. Thévenod, BioMetals, 2010, 23, 857–875 CrossRef PubMed.
- J. Wang and K. Pantopoulos, Biochem. J., 2011, 434, 365–381 CrossRef CAS PubMed.
- K. Gkouvatsos, G. Papanikolaou and K. Pantopoulos, Biochim. Biophys. Acta, 2012, 1820, 188–202 CrossRef CAS PubMed.
- A. M. Martines, R. Masereeuw, H. Tjalsma, J. G. Hoenderop, J. F. Wetzels and D. W. Swinkels, Nat. Rev. Nephrol., 2013, 9, 385–398 CrossRef CAS PubMed.
- I. De Domenico, D. McVey Ward and J. Kaplan, Nat. Rev. Mol. Cell Biol., 2008, 9, 72–81 CrossRef CAS PubMed.
- K. Pantopoulos, S. K. Porwal, A. Tartakoff and L. Devireddy, Biochemistry, 2012, 51, 5705–5724 CrossRef CAS PubMed.
- Iron in biochemistry and medicine, II., ed. A. Jacobs and M. Worwood, Academic Press, London, 1980, pp. 1–706 Search PubMed.
- J. Prousek, Pure Appl. Chem., 2007, 79, 2325–2338 CrossRef CAS.
- R. Green, R. Charlton, H. Seftel, T. Bothwell, F. Mayet, B. Adams, C. Finch and M. Layrisse, Am. J. Med., 1968, 45, 336–353 CrossRef CAS PubMed.
- M. Hynes, J. Clin. Pathol., 1948, 1, 57–67 CrossRef CAS PubMed.
- M. C. Linder, Nutrients, 2013, 5, 4022–4050 CrossRef PubMed.
- C. Finch, Blood, 1994, 84, 1697–1702 CAS.
- M. Shayeghi, G. O. Latunde-Dada, J. S. Oakhill, A. H. Laftah, K. Takeuchi, N. Halliday, Y. Khan, A. Warley, F. E. McCann, R. C. Hider, D. M. Frazer, G. J. Anderson, C. D. Vulpe, R. J. Simpson and A. T. McKie, Cell, 2005, 122, 789–801 CrossRef CAS PubMed.
- A. Qiu, M. Jansen, A. Sakaris, S. H. Min, S. Chattopadhyay, E. Tsai, C. Sandoval, R. Zhao, M. H. Akabas and I. D. Goldman, Cell, 2006, 127, 917–928 CrossRef CAS PubMed.
- A. Rajagopal, A. U. Rao, J. Amigo, M. Tian, S. K. Upadhyay, C. Hall, S. Uhm, M. K. Mathew, M. D. Fleming, B. H. Paw, M. Krause and I. Hamza, Nature, 2008, 453, 1127–1131 CrossRef CAS PubMed.
- A. Shawki, P. B. Knight, B. D. Maliken, E. J. Niespodzany and B. Mackenzie, Curr. Top. Membr., 2012, 70, 169–214 CAS.
- D. J. Lane, D. H. Bae, A. M. Merlot, S. Sahni and D. R. Richardson, Nutrients, 2015, 7, 2274–2296 CrossRef CAS PubMed.
- B. Mackenzie, A. Shawki, R. Kim, S. R. Anthony, P. B. Knight, E. M. Bradford and G. E. Shull, FASEB J., 2011, 25, 238.231 Search PubMed (abstract).
- L. Vanoaica, D. Darshan, L. Richman, K. Schumann and L. C. Kuhn, Cell Metab., 2010, 12, 273–282 CrossRef CAS PubMed.
- H. Shi, K. Z. Bencze, T. L. Stemmler and C. C. Philpott, Science, 2008, 320, 1207–1210 CrossRef CAS PubMed.
- S. Abboud and D. J. Haile, J. Biol. Chem., 2000, 275, 19906–19912 CrossRef CAS PubMed.
- A. Donovan, A. Brownlie, Y. Zhou, J. Shepard, S. J. Pratt, J. Moynihan, B. H. Paw, A. Drejer, B. Barut, A. Zapata, T. C. Law, C. Brugnara, S. E. Lux, G. S. Pinkus, J. L. Pinkus, P. D. Kingsley, J. Palis, M. D. Fleming, N. C. Andrews and L. I. Zon, Nature, 2000, 403, 776–781 CrossRef CAS PubMed.
- A. T. McKie, P. Marciani, A. Rolfs, K. Brennan, K. Wehr, D. Barrow, S. Miret, A. Bomford, T. J. Peters, F. Farzaneh, M. A. Hediger, M. W. Hentze and R. J. Simpson, Mol. Cell, 2000, 5, 299–309 CrossRef CAS PubMed.
- C. J. Mitchell, A. Shawki, T. Ganz, E. Nemeth and B. Mackenzie, Am. J. Physiol.: Cell Physiol., 2014, 306, C450–C459 CrossRef CAS PubMed.
- Z. L. Harris, A. P. Durley, T. K. Man and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10812–10817 CrossRef CAS.
- C. D. Vulpe, Y. M. Kuo, T. L. Murphy, L. Cowley, C. Askwith, N. Libina, J. Gitschier and G. J. Anderson, Nat. Genet., 1999, 21, 195–199 CrossRef CAS PubMed.
- S. Cherukuri, R. Potla, J. Sarkar, S. Nurko, Z. L. Harris and P. L. Fox, Cell Metab., 2005, 2, 309–319 CrossRef CAS PubMed.
- N. Montalbetti, A. Simonin, G. Kovacs and M. A. Hediger, Mol. Aspects Med., 2013, 34, 270–287 CrossRef CAS PubMed.
- E. Ramos, L. Kautz, R. Rodriguez, M. Hansen, V. Gabayan, Y. Ginzburg, M. P. Roth, E. Nemeth and T. Ganz, Hepatology, 2011, 53, 1333–1341 CrossRef CAS PubMed.
- T. Ganz and E. Nemeth, Biochim. Biophys. Acta, 2012, 1823, 1434–1443 CrossRef CAS PubMed.
- E. Nemeth, M. S. Tuttle, J. Powelson, M. B. Vaughn, A. Donovan, D. M. Ward, T. Ganz and J. Kaplan, Science, 2004, 306, 2090–2093 CrossRef CAS PubMed.
- P. Brissot, M. Ropert, C. Le Lan and O. Loreal, Biochim. Biophys. Acta, 2012, 1820, 403–410 CrossRef CAS PubMed.
- B. J. Scott and A. R. Bradwell, Clin. Chem., 1983, 29, 629–633 CAS.
- C. Hershko and T. E. Peto, Br. J. Haematol., 1987, 66, 149–151 CrossRef CAS PubMed.
- R. C. Hider, Eur. J. Clin. Invest., 2002, 32(suppl 1), 50–54 CrossRef CAS PubMed.
- C. A. Finch, V. Bellotti, S. Stray, D. A. Lipschitz, J. D. Cook, M. J. Pippard and H. A. Huebers, West. J. Med., 1986, 145, 657–663 CAS.
- L. A. Cohen, L. Gutierrez, A. Weiss, Y. Leichtmann-Bardoogo, D. L. Zhang, D. R. Crooks, R. Sougrat, A. Morgenstern, B. Galy, M. W. Hentze, F. J. Lazaro, T. A. Rouault and E. G. Meyron-Holtz, Blood, 2010, 116, 1574–1584 CrossRef CAS PubMed.
- B. S. Skikne, Am. J. Hematol., 2008, 83, 872–875 CrossRef CAS PubMed.
- D. M. Frazer and G. J. Anderson, BioFactors, 2014, 40, 206–214 CrossRef CAS PubMed.
- A. N. Luck and A. B. Mason, Curr. Top. Membr., 2012, 69, 3–35 CAS.
- P. Aisen, Int. J. Biochem. Cell Biol., 2004, 36, 2137–2143 CrossRef CAS PubMed.
- J. E. Levy, O. Jin, Y. Fujiwara, F. Kuo and N. C. Andrews, Nat. Genet., 1999, 21, 396–399 CrossRef CAS PubMed.
- H. Kawabata, R. Yang, T. Hirama, P. T. Vuong, S. Kawano, A. F. Gombart and H. P. Koeffler, J. Biol. Chem., 1999, 274, 20826–20832 CrossRef CAS PubMed.
- J. Chen and C. A. Enns, Biochim. Biophys. Acta, 2012, 1820, 256–263 CrossRef CAS PubMed.
- B. D. Grant and J. G. Donaldson, Nat. Rev. Mol. Cell Biol., 2009, 10, 597–608 CrossRef CAS PubMed.
- P. Ponka, C. Beaumont and D. R. Richardson, Semin. Hematol., 1998, 35, 35–54 CAS.
- R. S. Ohgami, D. R. Campagna, E. L. Greer, B. Antiochos, A. McDonald, J. Chen, J. J. Sharp, Y. Fujiwara, J. E. Barker and M. D. Fleming, Nat. Genet., 2005, 37, 1264–1269 CrossRef CAS PubMed.
- R. S. Ohgami, D. R. Campagna, A. McDonald and M. D. Fleming, Blood, 2006, 108, 1388–1394 CrossRef CAS PubMed.
- S. Dhungana, C. H. Taboy, O. Zak, M. Larvie, A. L. Crumbliss and P. Aisen, Biochemistry, 2004, 43, 205–209 CrossRef CAS PubMed.
- S. Gruenheid, F. Canonne-Hergaux, S. Gauthier, D. J. Hackam, S. Grinstein and P. Gros, J. Exp. Med., 1999, 189, 831–841 CrossRef CAS PubMed.
- M. D. Fleming, M. A. Romano, M. A. Su, L. M. Garrick, M. D. Garrick and N. C. Andrews, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1148–1153 CrossRef CAS.
- X. P. Dong, X. Cheng, E. Mills, M. Delling, F. Wang, T. Kurz and H. Xu, Nature, 2008, 455, 992–996 CrossRef CAS PubMed.
- W. Breuer, M. Shvartsman and Z. I. Cabantchik, Int. J. Biochem. Cell Biol., 2008, 40, 350–354 CrossRef CAS PubMed.
- F. Petrat, H. de Groot, R. Sustmann and U. Rauen, Biol. Chem., 2002, 383, 489–502 CrossRef CAS PubMed.
- R. C. Hider and X. L. Kong, BioMetals, 2011, 24, 1179–1187 CrossRef CAS PubMed.
- L. R. Devireddy, D. O. Hart, D. H. Goetz and M. R. Green, Cell, 2010, 141, 1006–1017 CrossRef CAS PubMed.
- C. Correnti, V. Richardson, A. K. Sia, A. D. Bandaranayake, M. Ruiz, Y. Suryo Rahmanto, Z. Kovacevic, M. C. Clifton, M. A. Holmes, B. K. Kaiser, J. Barasch, K. N. Raymond, D. R. Richardson and R. K. Strong, PLoS One, 2012, 7, e43696 CAS.
- U. Muhlenhoff, S. Molik, J. R. Godoy, M. A. Uzarska, N. Richter, A. Seubert, Y. Zhang, J. Stubbe, F. Pierrel, E. Herrero, C. H. Lillig and R. Lill, Cell Metab., 2010, 12, 373–385 CrossRef PubMed.
- D. R. Richardson, D. J. Lane, E. M. Becker, M. L. Huang, M. Whitnall, Y. Suryo Rahmanto, A. D. Sheftel and P. Ponka, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10775–10782 CrossRef CAS PubMed.
- A. S. Zhang, A. D. Sheftel and P. Ponka, Blood, 2005, 105, 368–375 CrossRef CAS PubMed.
- A. D. Sheftel, A. S. Zhang, C. Brown, O. S. Shirihai and P. Ponka, Blood, 2007, 110, 125–132 CrossRef CAS PubMed.
- R. S. Ajioka, J. D. Phillips and J. P. Kushner, Biochim. Biophys. Acta, 2006, 1763, 723–736 CrossRef CAS PubMed.
- R. Lill, B. Hoffmann, S. Molik, A. J. Pierik, N. Rietzschel, O. Stehling, M. A. Uzarska, H. Webert, C. Wilbrecht and U. Muhlenhoff, Biochim. Biophys. Acta, 2012, 1823, 1491–1508 CrossRef CAS PubMed.
- W. Xu, T. Barrientos and N. C. Andrews, Cell Metab., 2013, 17, 319–328 CrossRef CAS PubMed.
- R. Benz, Biochim. Biophys. Acta, 1994, 1197, 167–196 CrossRef CAS.
- W. Tan and M. Colombini, Biochim. Biophys. Acta, 2007, 1768, 2510–2515 CrossRef CAS PubMed.
- M. Y. Liu and M. Colombini, J. Bioenerg. Biomembr., 1992, 24, 41–46 CrossRef CAS PubMed.
- N. A. Wolff, A. J. Ghio, L. M. Garrick, M. D. Garrick, L. Zhao, R. A. Fenton and F. Thévenod, FASEB J., 2014, 28, 2134–2145 CrossRef CAS PubMed.
- N. A. Wolff, L. M. Garrick, L. Zhao, M. D. Garrick and F. Thévenod, Channels, 2014, 8, 458–466 CrossRef PubMed.
- G. C. Shaw, J. J. Cope, L. Li, K. Corson, C. Hersey, G. E. Ackermann, B. Gwynn, A. J. Lambert, R. A. Wingert, D. Traver, N. S. Trede, B. A. Barut, Y. Zhou, E. Minet, A. Donovan, A. Brownlie, R. Balzan, M. J. Weiss, L. L. Peters, J. Kaplan, L. I. Zon and B. H. Paw, Nature, 2006, 440, 96–100 CrossRef CAS PubMed.
- P. N. Paradkar, K. B. Zumbrennen, B. H. Paw, D. M. Ward and J. Kaplan, Mol. Cell. Biol., 2009, 29, 1007–1016 CrossRef CAS PubMed.
- J. Sripetchwandee, S. B. KenKnight, J. Sanit, S. Chattipakorn and N. Chattipakorn, Acta Physiol., 2014, 210, 330–341 CrossRef CAS PubMed.
- S. B. Keel, R. T. Doty, Z. Yang, J. G. Quigley, J. Chen, S. Knoblaugh, P. D. Kingsley, I. De Domenico, M. B. Vaughn, J. Kaplan, J. Palis and J. L. Abkowitz, Science, 2008, 319, 825–828 CrossRef CAS PubMed.
- D. Finazzi and P. Arosio, Arch. Toxicol., 2014, 88, 1787–1802 CrossRef CAS PubMed.
- S. Leidgens, K. Z. Bullough, H. Shi, F. Li, M. Shakoury-Elizeh, T. Yabe, P. Subramanian, E. Hsu, N. Natarajan, A. Nandal, T. L. Stemmler and C. C. Philpott, J. Biol. Chem., 2013, 288, 17791–17802 CrossRef CAS PubMed.
- S. Levi, B. Corsi, M. Bosisio, R. Invernizzi, A. Volz, D. Sanford, P. Arosio and J. Drysdale, J. Biol. Chem., 2001, 276, 24437–24440 CrossRef CAS PubMed.
- P. Santambrogio, G. Biasiotto, F. Sanvito, S. Olivieri, P. Arosio and S. Levi, J. Histochem. Cytochem., 2007, 55, 1129–1137 CrossRef CAS PubMed.
- Brenner & Rector's The Kidney, ed. M. W. Taal, G. M. Chertow, P. A. Marsden, K. Skorecki, A. S. L. Yu and B. M. Brenner, Saunders, Philadelphia, 9th edn, 2012, pp. 1–3064 Search PubMed.
- Seldin and Giebisch's The Kidney: Physiology & Pathophysiology, ed. R. J. Alpern, M. J. Caplan and O. W. Moe, Academic Press, London, 5th edn, 2013, pp. 1–3352 Search PubMed.
- Handbook of Physiology, ed. E. E. Windhager, Oxford University Press, Oxford, 1992, pp. 1–2544 Search PubMed.
- W. D. Comper, B. Haraldsson and W. M. Deen, J. Am. Soc. Nephrol., 2008, 19, 427–432 CrossRef PubMed.
- E. I. Christensen and H. Birn, Nat. Rev. Mol. Cell Biol., 2002, 3, 256–266 CrossRef CAS PubMed.
- C. D. Klaassen, J. Liu and B. A. Diwan, Toxicol. Appl. Pharmacol., 2009, 238, 215–220 CrossRef CAS PubMed.
- M. J. Burne, T. M. Osicka and W. D. Comper, Kidney Int., 1999, 55, 261–270 CrossRef CAS PubMed.
- P. R. Cutillas, R. J. Chalkley, K. C. Hansen, R. Cramer, A. G. Norden, M. D. Waterfield, A. L. Burlingame and R. J. Unwin, Am. J. Physiol. Renal Physiol., 2004, 287, F353–F364 CrossRef CAS PubMed.
- D. Zhang, E. Meyron-Holtz and T. A. Rouault, J. Am. Soc. Nephrol., 2007, 18, 401–406 CrossRef CAS PubMed.
- P. Aisen, in Iron metabolism in health and disease, ed. J. H. Brock, J. W. Halliday, M. J. Pippard, L. W. Powell and W. B. Saunders, London, 1994, pp. 1–30 Search PubMed.
- L. Kjeldsen, A. H. Johnsen, H. Sengelov and N. Borregaard, J. Biol. Chem., 1993, 268, 10425–10432 CAS.
- A. Tojo and S. Kinugasa, Internet J. Nephrol., 2012, 2012, 481520 Search PubMed.
- E. D. Klootwijk, M. Reichold, R. J. Unwin, R. Kleta, R. Warth and D. Bockenhauer, Nephrol., Dial., Transplant., 2015, 30, 1456–1460 CrossRef PubMed.
- E. I. Christensen, H. Birn, T. Storm, K. Weyer and R. Nielsen, Physiology, 2012, 27, 223–236 CrossRef CAS PubMed.
- B. Seetharam, E. I. Christensen, S. K. Moestrup, T. G. Hammond and P. J. Verroust, J. Clin. Invest., 1997, 99, 2317–2322 CrossRef CAS PubMed.
- J. C. Fyfe, M. Madsen, P. Hojrup, E. I. Christensen, S. M. Tanner, A. de la Chapelle, Q. He and S. K. Moestrup, Blood, 2004, 103, 1573–1579 CrossRef CAS PubMed.
- S. Amsellem, J. Gburek, G. Hamard, R. Nielsen, T. E. Willnow, O. Devuyst, E. Nexo, P. J. Verroust, E. I. Christensen and R. Kozyraki, J. Am. Soc. Nephrol., 2010, 21, 1859–1867 CrossRef CAS PubMed.
- V. Hvidberg, C. Jacobsen, R. K. Strong, J. B. Cowland, S. K. Moestrup and N. Borregaard, FEBS Lett., 2005, 579, 773–777 CrossRef CAS PubMed.
- S. Cui, P. J. Verroust, S. K. Moestrup and E. I. Christensen, Am. J. Physiol., 1996, 271, F900–F907 CAS.
- H. P. Peters, C. M. Laarakkers, P. Pickkers, R. Masereeuw, O. C. Boerman, A. Eek, E. A. Cornelissen, D. W. Swinkels and J. F. Wetzels, BMC Nephrol., 2013, 14, 70 CrossRef CAS PubMed.
- H. Birn, J. C. Fyfe, C. Jacobsen, F. Mounier, P. J. Verroust, H. Orskov, T. E. Willnow, S. K. Moestrup and E. I. Christensen, J. Clin. Invest., 2000, 105, 1353–1361 CrossRef CAS PubMed.
- N. E. Magnusson, M. Hornum, K. A. Jorgensen, J. M. Hansen, C. Bistrup, B. Feldt-Rasmussen and A. Flyvbjerg, BMC Nephrol., 2012, 13, 8 CrossRef CAS PubMed.
- R. A. Orlando, M. Exner, R. P. Czekay, H. Yamazaki, A. Saito, R. Ullrich, D. Kerjaschki and M. G. Farquhar, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2368–2373 CrossRef CAS.
- R. M. Sandoval, M. C. Wagner, M. Patel, S. B. Campos-Bilderback, G. J. Rhodes, E. Wang, S. E. Wean, S. S. Clendenon and B. A. Molitoris, J. Am. Soc. Nephrol., 2012, 23, 447–457 CrossRef CAS PubMed.
- V. Tenten, S. Menzel, U. Kunter, E. M. Sicking, C. R. van Roeyen, S. K. Sanden, M. Kaldenbach, P. Boor, A. Fuss, S. Uhlig, R. Lanzmich, B. Willemsen, H. Dijkman, M. Grepl, K. Wild, W. Kriz, B. Smeets, J. Floege and M. J. Moeller, J. Am. Soc. Nephrol., 2013, 24, 1966–1980 CrossRef CAS PubMed.
- E. I. Christensen and H. Birn, Nat. Rev. Nephrol., 2013, 9, 700–702 CrossRef CAS PubMed.
- L. E. Dickson, M. C. Wagner, R. M. Sandoval and B. A. Molitoris, J. Am. Soc. Nephrol., 2014, 25, 443–453 CrossRef CAS PubMed.
- S. Cui, S. Nielsen, T. Bjerke and E. I. Christensen, Exp. Nephrol., 1993, 1, 309–318 CAS.
- B. Moulouel, D. Houamel, C. Delaby, D. Tchernitchko, S. Vaulont, P. Letteron, O. Thibaudeau, H. Puy, L. Gouya, C. Beaumont and Z. Karim, Kidney Int., 2013, 84, 756–766 CrossRef CAS PubMed.
- H. Tsunoo and H. H. Sussman, J. Biol. Chem., 1983, 258, 4118–4122 CAS.
- H. Fuchs and R. Gessner, Biochim. Biophys. Acta, 2002, 1570, 19–26 CrossRef CAS.
- M. Haley, M. Lowe and C. P. Smith, Proc. Physiol. Soc., 2012, 27, 24P Search PubMed (abstract C41).
- B. Mackenzie, M. L. Ujwal, M. H. Chang, M. F. Romero and M. A. Hediger, Pfluegers Arch., 2006, 451, 544–558 CrossRef CAS PubMed.
- M. A. Su, C. C. Trenor, J. C. Fleming, M. D. Fleming and N. C. Andrews, Blood, 1998, 92, 2157–2163 CAS.
- P. L. Lee, T. Gelbart, C. West, C. Halloran and E. Beutler, Blood Cells, Mol., Dis., 1998, 24, 199–215 CrossRef CAS PubMed.
- N. Hubert and M. W. Hentze, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12345–12350 CrossRef CAS PubMed.
- B. Mackenzie, H. Takanaga, N. Hubert, A. Rolfs and M. A. Hediger, Biochem. J., 2007, 403, 59–69 CrossRef CAS PubMed.
- M. Okubo, K. Yamada, M. Hosoyamada, T. Shibasaki and H. Endou, Toxicol. Appl. Pharmacol., 2003, 187, 162–167 CrossRef CAS PubMed.
- M. Arredondo, M. J. Mendiburo, S. Flores, S. T. Singleton and M. D. Garrick, BioMetals, 2014, 27, 115–123 CrossRef CAS PubMed.
- N. Jabado, F. Canonne-Hergaux, S. Gruenheid, V. Picard and P. Gros, Blood, 2002, 100, 2617–2622 CrossRef CAS PubMed.
- M. Tabuchi, T. Yoshimori, K. Yamaguchi, T. Yoshida and F. Kishi, J. Biol. Chem., 2000, 275, 22220–22228 CrossRef CAS PubMed.
- M. Tabuchi, N. Tanaka, J. Nishida-Kitayama, H. Ohno and F. Kishi, Mol. Biol. Cell, 2002, 13, 4371–4387 CrossRef CAS PubMed.
- M. Abouhamed, J. Gburek, W. Liu, B. Torchalski, A. Wilhelm, N. A. Wolff, E. I. Christensen, F. Thévenod and C. P. Smith, Am. J. Physiol. Renal Physiol., 2006, 290, F1525–F1533 CrossRef CAS PubMed.
- F. Canonne-Hergaux and P. Gros, Kidney Int., 2002, 62, 147–156 CrossRef CAS PubMed.
- S. Lam-Yuk-Tseung and P. Gros, Biochemistry, 2006, 45, 2294–2301 CrossRef CAS PubMed.
- T. Veuthey, M. C. D'Anna and M. E. Roque, Am. J. Physiol. Renal Physiol., 2008, 295, F1213–F1221 CrossRef CAS PubMed.
- P. Aisen, A. Leibman and J. Zweier, J. Biol. Chem., 1978, 253, 1930–1937 CAS.
- S. Haldar, A. Tripathi, J. Qian, A. Beserra, S. Suda, M. McElwee, J. Turner, U. Hopfer and N. Singh, J. Biol. Chem., 2015, 290, 5512–5522 CrossRef CAS PubMed.
- N. A. Wolff, M. D. Garrick, L. M. Garrick and F. Thévenod, Acta Physiol., 2015, 213(suppl 699), 83(P039) Search PubMed.
- A. C. Chua and E. H. Morgan, J. Comp. Physiol., B, 1997, 167, 361–369 CrossRef CAS.
- C. J. Ferguson, M. Wareing, M. Delannoy, R. Fenton, S. J. McLarnon, N. Ashton, A. G. Cox, R. F. McMahon, L. M. Garrick, R. Green, C. P. Smith and D. Riccardi, Kidney Int., 2003, 64, 1755–1764 CrossRef CAS PubMed.
- T. Veuthey, D. Hoffmann, V. S. Vaidya and M. Wessling-Resnick, Am. J. Physiol. Renal Physiol., 2014, 306, F333–F343 CrossRef CAS PubMed.
- S. Jenkitkasemwong, C. Y. Wang, B. Mackenzie and M. D. Knutson, BioMetals, 2012, 25, 643–655 CrossRef CAS PubMed.
- L. He, K. Girijashanker, T. P. Dalton, J. Reed, H. Li, M. Soleimani and D. W. Nebert, Mol. Pharmacol., 2006, 70, 171–180 CAS.
- K. Girijashanker, L. He, M. Soleimani, J. M. Reed, H. Li, Z. Liu, B. Wang, T. P. Dalton and D. W. Nebert, Mol. Pharmacol., 2008, 73, 1413–1423 CrossRef CAS PubMed.
- J. P. Liuzzi, F. Aydemir, H. Nam, M. D. Knutson and R. J. Cousins, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13612–13617 CrossRef CAS PubMed.
- J. J. Pinilla-Tenas, B. K. Sparkman, A. Shawki, A. C. Illing, C. J. Mitchell, N. Zhao, J. P. Liuzzi, R. J. Cousins, M. D. Knutson and B. Mackenzie, Am. J. Physiol.: Cell Physiol., 2011, 301, C862–C871 CrossRef CAS PubMed.
- C. Y. Wang, S. Jenkitkasemwong, S. Duarte, B. K. Sparkman, A. Shawki, B. Mackenzie and M. D. Knutson, J. Biol. Chem., 2012, 287, 34032–34043 CrossRef CAS PubMed.
- B. Wang, S. N. Schneider, N. Dragin, K. Girijashanker, T. P. Dalton, L. He, M. L. Miller, K. F. Stringer, M. Soleimani, D. D. Richardson and D. W. Nebert, Am. J. Physiol.: Cell Physiol., 2007, 292, C1523–C1535 CrossRef CAS PubMed.
- A. Lymboussaki, E. Pignatti, G. Montosi, C. Garuti, D. J. Haile and A. Pietrangelo, J. Hepatol., 2003, 39, 710–715 CrossRef CAS PubMed.
- N. Wilkinson and K. Pantopoulos, Front. Pharmacol., 2014, 5, 176 Search PubMed.
- D. L. Zhang, R. M. Hughes, H. Ollivierre-Wilson, M. C. Ghosh and T. A. Rouault, Cell Metab., 2009, 9, 461–473 CrossRef CAS PubMed.
- B. K. Fuqua, Y. Lu, D. Darshan, D. M. Frazer, S. J. Wilkins, N. Wolkow, A. G. Bell, J. Hsu, C. C. Yu, H. Chen, J. L. Dunaief, G. J. Anderson and C. D. Vulpe, PLoS One, 2014, 9, e98792 Search PubMed.
- I. De Domenico, D. M. Ward, M. C. di Patti, S. Y. Jeong, S. David, G. Musci and J. Kaplan, EMBO J., 2007, 26, 2823–2831 CrossRef CAS PubMed.
- D. M. Ward and J. Kaplan, Biochim. Biophys. Acta, 2012, 1823, 1426–1433 CrossRef CAS PubMed.
- N. A. Wolff, W. Liu, R. A. Fenton, W. K. Lee, F. Thévenod and C. P. Smith, J. Cell. Mol. Med., 2011, 15, 209–219 CrossRef CAS PubMed.
- R. R. Starzynski, F. Canonne-Hergaux, M. Lenartowicz, W. Krzeptowski, A. Willemetz, A. Stys, J. Bierla, P. Pietrzak, T. Dziaman and P. Lipinski, Biochem. J., 2013, 449, 69–78 CrossRef CAS PubMed.
- A. Zarjou, S. Bolisetty, R. Joseph, A. Traylor, E. O. Apostolov, P. Arosio, J. Balla, J. Verlander, D. Darshan, L. C. Kuhn and A. Agarwal, J. Clin. Invest., 2013, 123, 4423–4434 CAS.
- Y. Scindia, P. Dey, A. Thirunagari, H. Liping, D. L. Rosin, M. Floris, M. D. Okusa and S. Swaminathan, J. Am. Soc. Nephrol., 2015 DOI:10.1681/ASN.2014101037.
- J. G. Hoenderop, D. Muller, A. W. Van Der Kemp, A. Hartog, M. Suzuki, K. Ishibashi, M. Imai, F. Sweep, P. H. Willems, C. H. Van Os and R. J. Bindels, J. Am. Soc. Nephrol., 2001, 12, 1342–1349 CAS.
- R. Vennekens, J. G. Hoenderop, J. Prenen, M. Stuiver, P. H. Willems, G. Droogmans, B. Nilius and R. J. Bindels, J. Biol. Chem., 2000, 275, 3963–3969 CrossRef CAS PubMed.
- B. Nilius, R. Vennekens, J. Prenen, J. G. Hoenderop, R. J. Bindels and G. Droogmans, J. Physiol., 2000, 527(Pt 2), 239–248 CrossRef CAS.
- M. van Abel, J. G. Hoenderop, A. W. van der Kemp, M. M. Friedlaender, J. P. van Leeuwen and R. J. Bindels, Kidney Int., 2005, 68, 1708–1721 CrossRef CAS PubMed.
- T. de Groot, R. J. Bindels and J. G. Hoenderop, Kidney Int., 2008, 74, 1241–1246 CrossRef CAS PubMed.
- A. Qiu and C. Hogstrand, Gene, 2004, 342, 113–123 CrossRef CAS PubMed.
- E. Perez-Reyes, Physiol. Rev., 2003, 83, 117–161 CrossRef CAS PubMed.
- W. A. Catterall, Cold Spring Harbor Perspect. Biol., 2011, 3, a003947 Search PubMed.
- D. M. Rodman, K. Reese, J. Harral, B. Fouty, S. Wu, J. West, M. Hoedt-Miller, Y. Tada, K. X. Li, C. Cool, K. Fagan and L. Cribbs, Circ. Res., 2005, 96, 864–872 CrossRef CAS PubMed.
- A. M. Yunker and M. W. McEnery, J. Bioenerg. Biomembr., 2003, 35, 533–575 CrossRef CAS PubMed.
- P. B. Hansen, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2015, 308, R227–R237 CrossRef CAS PubMed.
- D. Andreasen, B. L. Jensen, P. B. Hansen, T. H. Kwon, S. Nielsen and O. Skott, Am. J. Physiol. Renal Physiol., 2000, 279, F997–F1005 CAS.
- M. Leclerc, M. G. Brunette and D. Couchourel, Kidney Int., 2004, 66, 242–250 CrossRef CAS PubMed.
- M. G. Brunette, M. Leclerc, D. Couchourel, J. Mailloux and Y. Bourgeois, Can. J. Physiol. Pharmacol., 2004, 82, 30–37 CrossRef CAS PubMed.
- D. Couchourel, M. Leclerc, J. Filep and M. G. Brunette, Mol. Cell. Endocrinol., 2004, 222, 71–81 CrossRef CAS PubMed.
- S. Kumfu, S. Chattipakorn, S. Srichairatanakool, J. Settakorn, S. Fucharoen and N. Chattipakorn, Eur. J. Haematol., 2011, 86, 156–166 CrossRef CAS PubMed.
- K. V. Lopin, I. P. Gray, C. A. Obejero-Paz, F. Thévenod and S. W. Jones, Mol. Pharmacol., 2012, 82, 1194–1204 CrossRef CAS PubMed.
- K. V. Lopin, C. A. Obejero-Paz and S. W. Jones, J. Membr. Biol., 2010, 235, 131–143 CrossRef CAS PubMed.
- C. Langelueddecke, E. Roussa, R. A. Fenton, N. A. Wolff, W. K. Lee and F. Thévenod, J. Biol. Chem., 2012, 287, 159–169 CrossRef CAS PubMed.
- N. Paragas, R. Kulkarni, M. Werth, K. M. Schmidt-Ott, C. Forster, R. Deng, Q. Zhang, E. Singer, A. D. Klose, T. H. Shen, K. P. Francis, S. Ray, S. Vijayakumar, S. Seward, M. E. Bovino, K. Xu, Y. Takabe, F. E. Amaral, S. Mohan, R. Wax, K. Corbin, S. Sanna-Cherchi, K. Mori, L. Johnson, T. Nickolas, V. D'Agati, C. S. Lin, A. Qiu, Q. Al-Awqati, A. J. Ratner and J. Barasch, J. Clin. Invest., 2014, 124, 2963–2976 CAS.
- B. S. Nielsen, N. Borregaard, J. R. Bundgaard, S. Timshel, M. Sehested and L. Kjeldsen, Gut, 1996, 38, 414–420 CrossRef CAS PubMed.
- D. H. Goetz, M. A. Holmes, N. Borregaard, M. E. Bluhm, K. N. Raymond and R. K. Strong, Mol. Cell, 2002, 10, 1033–1043 CrossRef CAS PubMed.
- G. Bao, M. Clifton, T. M. Hoette, K. Mori, S. X. Deng, A. Qiu, M. Viltard, D. Williams, N. Paragas, T. Leete, R. Kulkarni, X. Li, B. Lee, A. Kalandadze, A. J. Ratner, J. C. Pizarro, K. M. Schmidt-Ott, D. W. Landry, K. N. Raymond, R. K. Strong and J. Barasch, Nat. Chem. Biol., 2010, 6, 602–609 CrossRef CAS PubMed.
- T. H. Flo, K. D. Smith, S. Sato, D. J. Rodriguez, M. A. Holmes, R. K. Strong, S. Akira and A. Aderem, Nature, 2004, 432, 917–921 CrossRef CAS PubMed.
- T. Berger, A. Togawa, G. S. Duncan, A. J. Elia, A. You-Ten, A. Wakeham, H. E. Fong, C. C. Cheung and T. W. Mak, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1834–1839 CrossRef CAS PubMed.
- R. J. Abergel, M. C. Clifton, J. C. Pizarro, J. A. Warner, D. K. Shuh, R. K. Strong and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 11524–11534 CrossRef CAS PubMed.
- J. Yang, K. Mori, J. Y. Li and J. Barasch, Am. J. Physiol. Renal Physiol., 2003, 285, F9–F18 CrossRef CAS PubMed.
- K. M. Schmidt-Ott, K. Mori, J. Y. Li, A. Kalandadze, D. J. Cohen, P. Devarajan and J. Barasch, J. Am. Soc. Nephrol., 2007, 18, 407–413 CrossRef CAS PubMed.
- A. Haase-Fielitz, M. Haase and P. Devarajan, Ann. Clin. Biochem., 2014, 51, 335–351 CrossRef CAS PubMed.
- W. F. t. Peacock, A. Maisel, J. Kim and C. Ronco, Postgrad. Med., 2013, 125, 82–93 Search PubMed.
- N. Paragas, A. Qiu, Q. Zhang, B. Samstein, S. X. Deng, K. M. Schmidt-Ott, M. Viltard, W. Yu, C. S. Forster, G. Gong, Y. Liu, R. Kulkarni, K. Mori, A. Kalandadze, A. J. Ratner, P. Devarajan, D. W. Landry, V. D'Agati, C. S. Lin and J. Barasch, Nat. Med., 2011, 17, 216–222 CrossRef CAS PubMed.
- J. Mishra, Q. Ma, A. Prada, M. Mitsnefes, K. Zahedi, J. Yang, J. Barasch and P. Devarajan, J. Am. Soc. Nephrol., 2003, 14, 2534–2543 CrossRef CAS PubMed.
- L. R. Devireddy, C. Gazin, X. Zhu and M. R. Green, Cell, 2005, 123, 1293–1305 CrossRef CAS PubMed.
- L. R. Devireddy, J. G. Teodoro, F. A. Richard and M. R. Green, Science, 2001, 293, 829–834 CrossRef CAS PubMed.
- H. Koepsell and H. Endou, Pfluegers Arch., 2004, 447, 666–676 CrossRef CAS PubMed.
- K. M. Bennett, J. Liu, C. Hoelting and J. Stoll, Mol. Cell. Biochem., 2011, 352, 143–154 CrossRef CAS PubMed.
- W. K. Fang, L. Y. Xu, X. F. Lu, L. D. Liao, W. J. Cai, Z. Y. Shen and E. M. Li, Biochem. J., 2007, 403, 297–303 CrossRef CAS PubMed.
- C. J. Wienken, P. Baaske, U. Rothbauer, D. Braun and S. Duhr, Nat. Commun., 2010, 1, 100 CrossRef PubMed.
- A. Tojo and H. Endou, Am. J. Physiol., 1992, 263, F601–F606 CAS.
- P. D. Leber and D. J. Marsh, Am. J. Physiol., 1970, 219, 358–363 CAS.
- R. G. Galaske, J. B. Van Liew and L. G. Feld, Kidney Int., 1979, 16, 394–403 CrossRef CAS PubMed.
- K. M. Madsen, R. H. Harris and C. C. Tisher, Kidney Int., 1982, 21, 354–361 CrossRef CAS PubMed.
- E. I. Christensen, P. J. Verroust and R. Nielsen, Pfluegers Arch., 2009, 458, 1039–1048 CrossRef CAS PubMed.
- R. B. Klassen, K. Crenshaw, R. Kozyraki, P. J. Verroust, L. Tio, S. Atrian, P. L. Allen and T. G. Hammond, Am. J. Physiol. Renal Physiol., 2004, 287, F393–F403 CrossRef CAS PubMed.
- D. F. Akintola, B. Sampson and A. Fleck, J. Lab. Clin. Med., 1995, 126, 119–127 CAS.
- H. Milnerowicz and A. Bizon, Acta Biochim. Pol., 2010, 57, 99–104 CAS.
- R. Nielsen and E. I. Christensen, Pediatr. Nephrol., 2010, 25, 813–822 CrossRef PubMed.
- C. Kastner, M. Pohl, M. Sendeski, G. Stange, C. A. Wagner, B. Jensen, A. Patzak, S. Bachmann and F. Theilig, Am. J. Physiol. Renal Physiol., 2009, 296, F902–F911 CrossRef CAS PubMed.
- A. C. Alfrey and W. S. Hammond, Kidney Int., 1990, 37, 1409–1413 CrossRef CAS PubMed.
- N. Ishizaka, K. Saito, E. Noiri, M. Sata, I. Mori, M. Ohno and R. Nagai, Nephron Physiol., 2004, 98, p107–p113 CrossRef CAS PubMed.
- E. Dizin, U. Hasler, S. Nlandu-Khodo, M. Fila, I. Roth, T. Ernandez, A. Doucet, P. Y. Martin, E. Feraille and S. de Seigneux, Am. J. Physiol. Renal Physiol., 2013, 305, F1053–F1063 CrossRef CAS PubMed.
- F. Thévenod and W. K. Lee, Met. Ions Life Sci., 2013, 11, 415–490 Search PubMed.
- J. M. Moulis, BioMetals, 2010, 23, 877–896 CrossRef CAS PubMed.
- T. S. Nawrot, J. A. Staessen, H. A. Roels, E. Munters, A. Cuypers, T. Richart, A. Ruttens, K. Smeets, H. Clijsters and J. Vangronsveld, BioMetals, 2010, 23, 769–782 CrossRef CAS PubMed.
- A. Cuypers, M. Plusquin, T. Remans, M. Jozefczak, E. Keunen, H. Gielen, K. Opdenakker, A. R. Nair, E. Munters, T. J. Artois, T. Nawrot, J. Vangronsveld and K. Smeets, BioMetals, 2010, 23, 927–940 CrossRef CAS PubMed.
- F. Thévenod and W. K. Lee, Arch. Toxicol., 2013, 87, 1743–1786 CrossRef PubMed.
- W. Maret and J. M. Moulis, Met. Ions Life Sci., 2013, 11, 1–29 CAS.
- C. C. Bridges and R. K. Zalups, Toxicol. Appl. Pharmacol., 2005, 204, 274–308 CrossRef CAS PubMed.
- R. Cornelis, B. Heinzow, R. F. Herber, J. M. Christensen, O. M. Poulsen, E. Sabbioni, D. M. Templeton, Y. Thomassen, M. Vahter and O. Vesterberg, J. Trace Elem. Med. Biol., 1996, 10, 103–127 CAS.
- M. Nordberg and G. F. Nordberg, Environ. Health Perspect., 1975, 12, 103–108 CrossRef CAS PubMed.
- R. E. Dudley, L. M. Gammal and C. D. Klaassen, Toxicol. Appl. Pharmacol., 1985, 77, 414–426 CrossRef CAS PubMed.
- K. Nomiyama, H. Nomiyama and N. Kameda, Toxicology, 1998, 129, 157–168 CrossRef CAS PubMed.
- S. Thijssen, J. Maringwa, C. Faes, I. Lambrichts and E. Van Kerkhove, Toxicology, 2007, 229, 145–156 CrossRef CAS PubMed.
- L. Jarup, A. Rogenfelt, C. G. Elinder, K. Nogawa and T. Kjellstrom, Scand. J. Work, Environ. Health, 1983, 9, 327–331 CAS.
- K. Klotz, W. Weistenhofer and H. Drexler, Met. Ions Life Sci., 2013, 11, 85–98 CAS.
- E. Freisinger and M. Vasak, Met. Ions Life Sci., 2013, 11, 339–371 CAS.
- H. M. Chan, Y. Tamura, M. G. Cherian and R. A. Goyer, Proc. Soc. Exp. Biol. Med., 1993, 202, 420–427 CrossRef CAS PubMed.
- K. Tanaka, Dev. Toxicol. Environ. Sci., 1982, 9, 237–249 CAS.
- S. A. Gunn and T. C. Gould, Proc. Soc. Exp. Biol. Med., 1957, 96, 820–823 CrossRef CAS PubMed.
- G. F. Nordberg and K. Nishiyama, Arch. Environ. Health, 1972, 24, 209–214 CrossRef CAS PubMed.
- H. M. Chan, L. F. Zhu, R. Zhong, D. Grant, R. A. Goyer and M. G. Cherian, Toxicol. Appl. Pharmacol., 1993, 123, 89–96 CrossRef CAS PubMed.
- I. Sabolic, D. Breljak, M. Skarica and C. M. Herak-Kramberger, BioMetals, 2010, 23, 897–926 CrossRef CAS PubMed.
- W. Maret, J. Biol. Inorg. Chem., 2011, 16, 1079–1086 CrossRef CAS PubMed.
- T. Kjellstrom, Environ. Health Perspect., 1979, 28, 169–197 CrossRef CAS PubMed.
- H. Roels, R. Lauwerys, J. P. Buchet, A. Bernard, J. S. Garvey and H. J. Linton, Int. Arch. Occup. Environ. Health, 1983, 52, 159–166 CrossRef CAS PubMed.
- C. Tohyama, Z. A. Shaikh, K. Nogawa, E. Kobayashi and R. Honda, Toxicology, 1981, 20, 289–297 CrossRef CAS PubMed.
- W. C. Prozialeck and J. R. Edwards, BioMetals, 2010, 23, 793–809 CrossRef CAS PubMed.
- L. Jarup, Nephrol., Dial., Transplant., 2002, 17(suppl 2), 35–39 CrossRef CAS.
- H. Roels, A. Bernard, J. P. Buchet, A. Goret, R. Lauwerys, D. R. Chettle, T. C. Harvey and I. A. Haddad, Lancet, 1979, 1, 221 CrossRef CAS.
- WHO, Cadmium, World Health Organization, 1992 Search PubMed.
- Y. Suzuki, J. Toxicol. Environ. Health, 1980, 6, 469–482 CrossRef CAS PubMed.
- P. M. Ferraro, S. Costanzi, A. Naticchia, A. Sturniolo and G. Gambaro, BMC Public Health, 2010, 10, 304 CrossRef PubMed.
- A. Navas-Acien, M. Tellez-Plaza, E. Guallar, P. Muntner, E. Silbergeld, B. Jaar and V. Weaver, Am. J. Epidemiol., 2009, 170, 1156–1164 CrossRef PubMed.
- A. Hartwig, Met. Ions Life Sci., 2013, 11, 491–507 CAS.
- A. Bernard, J. P. Buchet, H. Roels, P. Masson and R. Lauwerys, Eur. J. Clin. Invest., 1979, 9, 11–22 CrossRef CAS PubMed.
- K. Nogawa, A. Ishizaki, M. Fukushima, I. Shibata and N. Hagino, Environ. Res., 1975, 10, 280–307 CrossRef CAS PubMed.
- N. Johri, G. Jacquillet and R. Unwin, BioMetals, 2010, 23, 783–792 CrossRef CAS PubMed.
- W. Goumakos, J. P. Laussac and B. Sarkar, Biochem. Cell Biol., 1991, 69, 809–820 CrossRef CAS PubMed.
- W. R. Harris and L. J. Madsen, Biochemistry, 1988, 27, 284–288 CrossRef CAS PubMed.
- D. J. Price and J. G. Joshi, J. Biol. Chem., 1983, 258, 10873–10880 CAS.
- T. Granier, G. Comberton, B. Gallois, B. L. d'Estaintot, A. Dautant, R. R. Crichton and G. Precigoux, Proteins, 1998, 31, 477–485 CrossRef CAS.
- W. C. Prozialeck and P. C. Lamar, Arch. Toxicol., 1993, 67, 113–119 CrossRef CAS PubMed.
- R. K. Zalups, Toxicol. Appl. Pharmacol., 2000, 164, 15–23 CrossRef CAS PubMed.
- S. Soodvilai, J. Nantavishit, C. Muanprasat and V. Chatsudthipong, Toxicol. Lett., 2011, 204, 38–42 CrossRef CAS PubMed.
- F. Thévenod, G. Ciarimboli, M. Leistner, N. A. Wolff, W. K. Lee, I. Schatz, T. Keller, R. Al-Monajjed, V. Gorboulev and H. Koepsell, Mol. Pharmacol., 2013, 10, 3045–3056 CrossRef PubMed.
- C. D. Klaassen, J. Liu and S. Choudhuri, Annu. Rev. Pharmacol. Toxicol., 1999, 39, 267–294 CrossRef CAS PubMed.
- N. A. Wolff, M. Abouhamed, P. J. Verroust and F. Thévenod, J. Pharmacol. Exp. Ther., 2006, 318, 782–791 CrossRef CAS PubMed.
- N. A. Wolff, W. K. Lee, M. Abouhamed and F. Thévenod, Toxicol. Appl. Pharmacol., 2008, 230, 78–85 CrossRef CAS PubMed.
- C. Erfurt, E. Roussa and F. Thévenod, Am. J. Physiol.: Cell Physiol., 2003, 285, C1367–C1376 CrossRef CAS PubMed.
- N. A. Wolff, W. K. Lee and F. Thévenod, Toxicol. Lett., 2011, 203, 210–218 CrossRef CAS PubMed.
- M. Abouhamed, N. A. Wolff, W. K. Lee, C. P. Smith and F. Thévenod, Am. J. Physiol. Renal Physiol., 2007, 293, F705–F712 CrossRef CAS PubMed.
- R. D. Palmiter, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1219–1223 CrossRef CAS.
- Y. Liu, J. Liu, S. M. Habeebu, M. P. Waalkes and C. D. Klaassen, Toxicol. Sci., 2000, 57, 167–176 CrossRef CAS PubMed.
- C. Dorian, V. H. Gattone II and C. D. Klaassen, Toxicol. Appl. Pharmacol., 1992, 114, 173–181 CrossRef CAS PubMed.
- C. Dorian, V. H. Gattone II and C. D. Klaassen, Toxicol. Appl. Pharmacol., 1992, 117, 242–248 CrossRef CAS PubMed.
- W. Tang and Z. A. Shaikh, J. Toxicol. Environ. Health, Part A, 2001, 63, 221–235 CrossRef CAS PubMed.
- M. Nordberg, Environ. Health Perspect., 1984, 54, 13–20 CrossRef CAS PubMed.
- J. Sudo, T. Hayashi, J. Terui, M. Soyama, M. Fukata and K. Kakuno, Eur. J. Pharmacol., 1994, 270, 229–235 CAS.
- A. Onodera, M. Tani, T. Michigami, M. Yamagata, K. S. Min, K. Tanaka, T. Nakanishi, T. Kimura and N. Itoh, Toxicol. Lett., 2012, 212, 91–96 CrossRef CAS PubMed.
- E. Felley-Bosco and J. Diezi, Toxicol. Appl. Pharmacol., 1987, 91, 204–211 CrossRef CAS PubMed.
- F. Thévenod, Nephron Physiol., 2003, 93, 87–93 CrossRef.
- A. Chaumont, M. Nickmilder, X. Dumont, T. Lundh, S. Skerfving and A. Bernard, Toxicol. Lett., 2012, 210, 345–352 CrossRef CAS PubMed.
- P. E. Evrin and L. Wibell, Scand. J. Clin. Lab. Invest., 1972, 29, 69–74 CrossRef CAS PubMed.
- K. Takagi, K. Kin, Y. Itoh, H. Enomoto and T. Kawai, J. Clin. Pathol., 1980, 33, 786–791 CrossRef CAS PubMed.
- J. R. Leheste, B. Rolinski, H. Vorum, J. Hilpert, A. Nykjaer, C. Jacobsen, P. Aucouturier, J. O. Moskaug, A. Otto, E. I. Christensen and T. E. Willnow, Am. J. Pathol., 1999, 155, 1361–1370 CrossRef CAS PubMed.
- C. M. Eakin, J. D. Knight, C. J. Morgan, M. A. Gelfand and A. D. Miranker, Biochemistry, 2002, 41, 10646–10656 CrossRef CAS PubMed.
- A. G. Norden, P. Sharratt, P. R. Cutillas, R. Cramer, S. C. Gardner and R. J. Unwin, Kidney Int., 2004, 66, 1994–2003 CrossRef CAS PubMed.
- H. Birn and E. I. Christensen, Kidney Int., 2006, 69, 440–449 CrossRef CAS PubMed.
- J. Liu, R. A. Goyer and M. P. Waalkes, in Casarett and Doull's Toxicology, ed. C. D. Klaassen, McGraw-Hill, New York, NY, 5th edn, 2008, pp. 931–979 Search PubMed.
- L. Jarup, B. Persson and C. G. Elinder, Occup. Environ. Med., 1995, 52, 818–822 CrossRef CAS PubMed.
- A. Akesson, T. Lundh, M. Vahter, P. Bjellerup, J. Lidfeldt, C. Nerbrand, G. Samsioe, U. Stromberg and S. Skerfving, Environ. Health Perspect., 2005, 113, 1627–1631 CrossRef PubMed.
- W. Xiao, Y. Liu and D. M. Templeton, Toxicol. Appl. Pharmacol., 2009, 238, 315–326 CrossRef CAS PubMed.
- P. V. Rao, S. A. Jordan and M. K. Bhatnagar, J. Environ. Pathol., Toxicol. Oncol., 1989, 9, 19–44 CAS.
- J. P. Girolami, J. L. Bascands, C. Pecher, G. Cabos, J. P. Moatti, J. F. Mercier, J. M. Haguenoer and Y. Manuel, Toxicology, 1989, 55, 117–129 CrossRef CAS PubMed.
- G. Oner, U. K. Senturk and N. Izgut-Uysal, Biol. Trace Elem. Res., 1995, 48, 111–117 CrossRef CAS PubMed.
- R. Lauwerys and A. Bernard, Toxicol. Lett., 1989, 46, 13–29 CrossRef CAS PubMed.
- O. Barbier, G. Jacquillet, M. Tauc, P. Poujeol and M. Cougnon, Am. J. Physiol. Renal Physiol., 2004, 287, F1067–F1075 CrossRef CAS PubMed.
- L. Lacinova, N. Klugbauer and F. Hofmann, Neuropharmacology, 2000, 39, 1254–1266 CrossRef CAS PubMed.
- D. Diaz, R. Bartolo, D. M. Delgadillo, F. Higueldo and J. C. Gomora, J. Membr. Biol., 2005, 207, 91–105 CrossRef CAS PubMed.
- J. R. Serrano, E. Perez-Reyes and S. W. Jones, J. Gen. Physiol., 1999, 114, 185–201 CrossRef CAS PubMed.
- E. M. Leslie, J. Liu, C. D. Klaassen and M. P. Waalkes, Mol. Pharmacol., 2006, 69, 629–639 CrossRef CAS PubMed.
- K. V. Lopin, F. Thévenod, J. C. Page and S. W. Jones, Mol. Pharmacol., 2012, 82, 1183–1193 CrossRef CAS PubMed.
- C. J. Frazier, E. G. George and S. W. Jones, Biophys. J., 2000, 78, 1872–1880 CrossRef CAS PubMed.
- G. Kovacs, N. Montalbetti, M. C. Franz, S. Graeter, A. Simonin and M. A. Hediger, Cell Calcium, 2013, 54, 276–286 CrossRef CAS PubMed.
- E. K. Park, S. K. Mak and B. D. Hammock, Interdiscip. Toxicol., 2013, 6, 157–158 Search PubMed.
- C. Langelueddecke, W. K. Lee and F. Thévenod, Toxicol. Lett., 2014, 226, 228–235 CrossRef CAS PubMed.
- K. J. Ullrich, in Kurzgefaßtes Lehrbuch der Physiologie, ed. W. D. Keidel, Georg Thieme Verlag, Stuttgart, 1975, ch. 10 Search PubMed.
- M. Torra, J. To-Figueras, M. Brunet, M. Rodamilans and J. Corbella, Bull. Environ. Contam. Toxicol., 1994, 53, 509–515 CrossRef CAS PubMed.
- M. Yoshida, H. Ohta, Y. Yamauchi, Y. Seki, M. Sagi, K. Yamazaki and Y. Sumi, Biol. Trace Elem. Res., 1998, 63, 167–175 CrossRef CAS PubMed.
- S. H. Garrett, M. A. Sens, J. H. Todd, S. Somji and D. A. Sens, Toxicol. Lett., 1999, 105, 207–214 CrossRef CAS PubMed.
- T. Nagamine, K. Nakazato, K. Suzuki, T. Kusakabe, T. Sakai, M. Oikawa, T. Satoh, T. Kamiya and K. Arakawa, Biol. Trace Elem. Res., 2007, 117, 115–126 CrossRef CAS PubMed.
- L. Wang, D. Chen, H. Wang and Z. Liu, Biol. Trace Elem. Res., 2009, 129, 190–199 CrossRef CAS PubMed.
- R. Campos, F. Maureira, A. Garrido and A. Valenzuela, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1993, 105, 157–163 CrossRef CAS.
- B. Gonzalez-Flecha, P. Evelson, N. Sterin-Speziale and A. Boveris, Biochim. Biophys. Acta, 1993, 1157, 155–161 CrossRef CAS.
- A. P. Zou and A. W. Cowley, Jr., Acta Physiol. Scand., 2003, 179, 233–241 CrossRef CAS PubMed.
- R. A. Zager, A. C. Johnson and K. B. Frostad, Am. J. Physiol. Renal Physiol., 2014, 307, F856–F868 CrossRef CAS PubMed.
- A. A. McDonough and S. C. Thompson, in Brenner & Rector's The Kidney, ed. M. W. Taal, G. M. Chertow, P. A. Marsden, K. Skorecki, A. S. L. Yu and B. M. Brenner, Saunders, Philadelphia, 9th edn, 2012, ch. 4, vol. 1, pp. 138–157 Search PubMed.
- A. M. Hall, G. J. Rhodes, R. M. Sandoval, P. R. Corridon and B. A. Molitoris, Kidney Int., 2013, 83, 72–83 CrossRef CAS PubMed.
- A. Sharfuddin, S. D. Weisbord, P. M. Palevsky and B. A. Molitoris, in Brenner & Rector's The Kidney, ed. M. W. Taal, G. M. Chertow, P. A. Marsden, K. Skorecki, A. S. L. Yu and B. M. Brenner, Saunders, Philadelphia, 9th edn, 2012, ch. 30, vol. 1, pp. 1044–1099 Search PubMed.
- G. W. Richter, J. Exp. Med., 1957, 106, 203–218 CrossRef CAS PubMed.
- S. Fagoonee, J. Gburek, E. Hirsch, S. Marro, S. K. Moestrup, J. M. Laurberg, E. I. Christensen, L. Silengo, F. Altruda and E. Tolosano, Am. J. Pathol., 2005, 166, 973–983 CrossRef CAS PubMed.
- J. G. Wardeska, B. Viglione and N. D. Chasteen, J. Biol. Chem., 1986, 261, 6677–6683 CAS.
- J. H. Kagi and B. L. Vallee, J. Biol. Chem., 1961, 236, 2435–2442 CAS.
- X. Ding, E. Bill, M. Good, A. X. Trautwein and M. Vasak, Eur. J. Biochem., 1988, 171, 711–714 CrossRef CAS PubMed.
- J. P. Muller, M. Vedel, M. J. Monnot, N. Touzet and M. Wegnez, DNA Cell Biol., 1991, 10, 571–579 CrossRef CAS PubMed.
- D. M. Durnam and R. D. Palmiter, Mol. Cell. Biol., 1984, 4, 484–491 CrossRef CAS PubMed.
- J. C. Fleet, G. K. Andrews and C. C. McCormick, J. Nutr., 1990, 120, 1214–1222 CAS.
- J. H. Kagi and A. Schaffer, Biochemistry, 1988, 27, 8509–8515 CrossRef CAS PubMed.
- D. Price and J. G. Joshi, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 3116–3119 CrossRef CAS.
- W. K. Lee, U. Bork, F. Gholamrezaei and F. Thévenod, Am. J. Physiol. Renal Physiol., 2005, 288, F27–F39 CrossRef CAS PubMed.
- Y. Wang, J. Fang, S. S. Leonard and K. M. Rao, Free Radical Biol. Med., 2004, 36, 1434–1443 CrossRef CAS PubMed.
- W. K. Lee, M. Spielmann, U. Bork and F. Thévenod, Am. J. Physiol.: Cell Physiol., 2005, 289, C656–C664 CrossRef CAS PubMed.
- W. K. Lee and F. Thévenod, Am. J. Physiol.: Cell Physiol., 2006, 291, C195–C202 CrossRef CAS PubMed.
- M. Kippler, W. Goessler, B. Nermell, E. C. Ekstrom, B. Lonnerdal, S. El Arifeen and M. Vahter, Environ. Res., 2009, 109, 914–921 CrossRef CAS PubMed.
- K. Weyer, T. Storm, J. Shan, S. Vainio, R. Kozyraki, P. J. Verroust, E. I. Christensen and R. Nielsen, Nephrol., Dial., Transplant., 2011, 26, 3446–3451 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |