
Enzymatically crosslinked alginate hydrogels with improved adhesion properties†‡
Junxia
Hou
a,
Chong
Li
a,
Ying
Guan
*a,
Yongjun
Zhang
*a and
X. X.
Zhu
*b
aKey Laboratory of Functional Polymer Materials, Institute of Polymer Chemistry, College of Chemistry, Nankai University, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China. E-mail: yingguan@nankai.edu.cn; yongjunzhang@nankai.edu.cn
bDepartment of Chemistry, Université de Montréal, C. P. 6128, Succursale Centre-ville, Montreal, QC H3C 3J7, Canada. E-mail: julian.zhu@umontreal.ca
First published on 13th January 2015
Abstract
Enzymatic crosslinking of polymer–phenol conjugates in the presence of horseradish peroxidase (HRP) and H2O2 has emerged as an important method to synthesize in situ-forming, injectable hydrogels. Here we show that the alginate–dopamine (Alg–DA) conjugate, a polymer with catechol side groups instead of phenol groups, also gels in situ in the presence of HRP and H2O2. The effects of various factors, including the concentration of HRP, H2O2, and the polymer, and the degree of substitution of the polymer, on the gelation rate and the mechanical strength of the resulting gels, were studied by rheology. The influence of these factors on the gelling of catechol-functionalized polymer is similar to their influence on phenol-functionalized polymers, suggesting that they have a similar crosslinking mechanism. Compared to the phenol-functionalized alginate–tyramine (Alg–TA) hydrogel, catechol-functionalized Alg–DA gels exhibit significantly improved adhesion properties. When both the polymers have a degree of substitution of 10%, the adhesion strength of the latter is about 10-fold of the former. Replacement of phenol groups with catechol groups also results in very different cell behaviour. While the cells seeded on the Alg–TA gels do not attach onto the substratum, they attach onto the Alg–DA gels and exhibit a spread morphology. The significantly enhanced adhesion properties of the Alg–DA hydrogels are attributed to the catechol moiety, a structure found in the adhesive proteins of blue mussels.
Introduction
Hydrogels are crosslinked three-dimensional networks of hydrophilic polymers. Because of their unique properties, such as high water content, biocompatibility, low inflammatory response and mechanical properties similar to real tissues, hydrogels have found a wide range of biomedical applications, including drug release, biosensing and tissue engineering.1–3 In the recent years in situ-forming hydrogels, or injectable hydrogels, are receiving more and more attention. Unlike the preformed hydrogels, homogeneous encapsulation of bioactive molecules and/or cells in the in situ-forming hydrogels can be achieved by a simple mixing. They can be implanted facilely by injection. In addition, they can fill the defects with any shape. Therefore, from a clinical point of view, in situ-forming hydrogels are more desirable for many biomedical applications.4–7A lot of in situ gelling systems, either gelled via physical interactions or chemical reactions, have been designed in the last few years.6 An interesting gelling system is based on enzyme-catalyzed crosslinking reactions. In this system, crosslinking was achieved via the oxidative coupling of phenol or aniline moieties in the presence of H2O2. The reaction was catalyzed by an enzyme, usually horseradish peroxidase (HRP), a single-chain β-type hemoprotein.4,8 Kaplan et al.9 reported the in situ formation of hydrogels from poly(aspartic acid) functionalized with tyramine (TA), tyrosine or aminophenol in the presence of H2O2 and HRP. Kurisawa et al.10 reported the enzymatic crosslinking of hyaluronic acid–TA conjugates. Jin et al.7 synthesized dextran hydrogels via the enzymatic crosslinking of dextran–tyramine conjugates. These hydrogels are chemically crosslinked and therefore superior to physically crosslinked ones in terms of stability and mechanical strength.4 Unlike many chemical gelling reactions, enzyme-catalyzed crosslinking occurs under mild conditions. In addition, this approach allows for an independent tuning of the gelation rate and mechanical strength of the resulting gels.4,11 These in situ-forming gels were widely used for cell immobilization,10 drug delivery,4,10,12 tissue engineering,4,8,10,13–16 and bone cement.17
In most of the previous approaches, phenol groups were first conjugated onto a polymer, such as hyaluronic acid10,11,17 and dextran.7 These groups were then coupled under the catalysis of HRP to form a 3D network. In this work, however, we used catechol groups instead of phenol groups. Considering their similar structures, it is expected that polymers with catechol side groups can also be gelled by the HRP-catalyzed crosslinking. In addition, the resulting hydrogels are expected to exhibit an improved adhesion property, which is a valuable property that can be exploited clinically.18 Previously, the extraordinary ability of blue mussels to attach to virtually any type of surface was attributed to the high level of 3,4-dihydroxy-L-phenylalanine (DOPA) of the proteins found in their plaques.19 Inspired by this finding, various adhesive biomaterials were designed by the incorporation of DOPA and DOPA-mimetic catechols into hydrogels.20–25 Alginate is a linear polysaccharide with homopolymeric blocks of (1,4)-linked β-D-mannuronate and α-L-guluronate. Because of its biocompatibility, low toxicity and a relatively low cost, it has been widely exploited in biomedical applications.2 Here the catechol-functionalized alginate was synthesized by conjugation with dopamine. We demonstrate that, like the phenol-functionalized polymers, the alginate–dopamine conjugate gels in situ in the presence of HRP and H2O2. Compared to their phenol-functionalized analogues, the bulk adhesion strength of the resulting hydrogels improved significantly. Unlike ordinary hydrogels, cells can attach onto the gel surface. These in situ-forming hydrogels with improved adhesion properties may find many biomedical applications including wound closure and healing, drug delivery, tissue engineering, and dental and bone applications.26
Experimental section
Materials
Sodium alginate (Alg, low viscosity) was purchased from Alfa Aesar. Dopamine hydrochloride (DA), tyramine hydrochloride (TA), hydrogen peroxide (H2O2, 30 wt% aqueous solution), horseradish peroxidase (HRP), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (NHS) were purchased from Heowns. Acridine orange (AO), ethidium bromide (EB) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazoliumbromide (MTT) were purchased from Tianjin Junyao Biological Company. All the chemicals were of analytical grade and were used without further purification.Synthesis of alginate–dopamine (Alg–DA) and alginate–tyramine (Alg–TA) conjugates
Alg–DA conjugates were synthesized by the modification of Alg with DA under the catalysis of EDC/NHS (Scheme 1).23 Briefly Alg (1.08 g, 5 mmol in terms of repeating unit) was dissolved in 100 mL of distilled water. EDC and NHS (both 5 mmol) were added and the solution was purged with nitrogen for 45 min, after which a predetermined amount of DA was added. The reaction mixture was stirred overnight at room temperature. During the reaction, pH of the mixture was maintained at 4.5–5.5 with 0.1 M Na2CO3 or HCl. After being dialyzed against deionized water for 3 days (cutoff: 3500 Da), the products were lyophilized.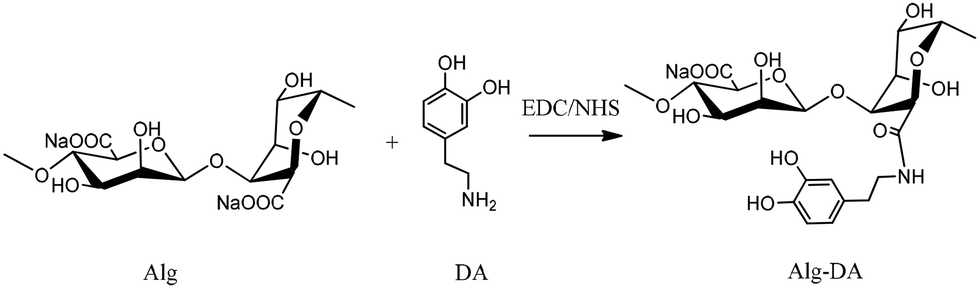 | ||
| Scheme 1 Synthesis of Alg–DA conjugates by coupling Alg with DA under the catalysis of EDC and NHS. | ||
To determine the degree of substitution (DS) of the products, i.e., percentage of carboxylic acid groups in Alg coupled with DA, Alg–DA solutions with known concentrations were prepared. The absorbance at 280 nm was measured, from which the amount of DA moieties was calculated with the help of a calibration curve, which was plotted by measuring the absorbance of a series of dopamine hydrochloride solution with different concentrations at the same wavelength.23 DSs of the products and their synthesis conditions are reported in Table 1. Alg–TA conjugates were synthesized from Alg and TA in the same way.
| Entry no. | Feeding molar ratio | DS (%) | |
|---|---|---|---|
Alg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EDC:NHS :EDC:NHS |
Alg:DA |
||
| 1 | 1:1:1 |
3:1 |
10 |
| 2 | 1:1:1 |
2:1 |
16 |
| 3 | 1:1:1 |
1:1 |
21 |
Enzymatic crosslinking of Alg–DA conjugates
As a typical example, Alg–DA solution was prepared by dissolving Alg–DA in pH 7.4, 0.01 M phosphate buffer. Predetermined amounts of HRP and H2O2 solutions were added and mixed. The final concentration of Alg–DA, HRP and H2O2 was 4.0 wt%, 23.04 U mL−1, and 52.0 mM, respectively. The mixture was incubated at 37 °C and gelled quickly (Scheme 2).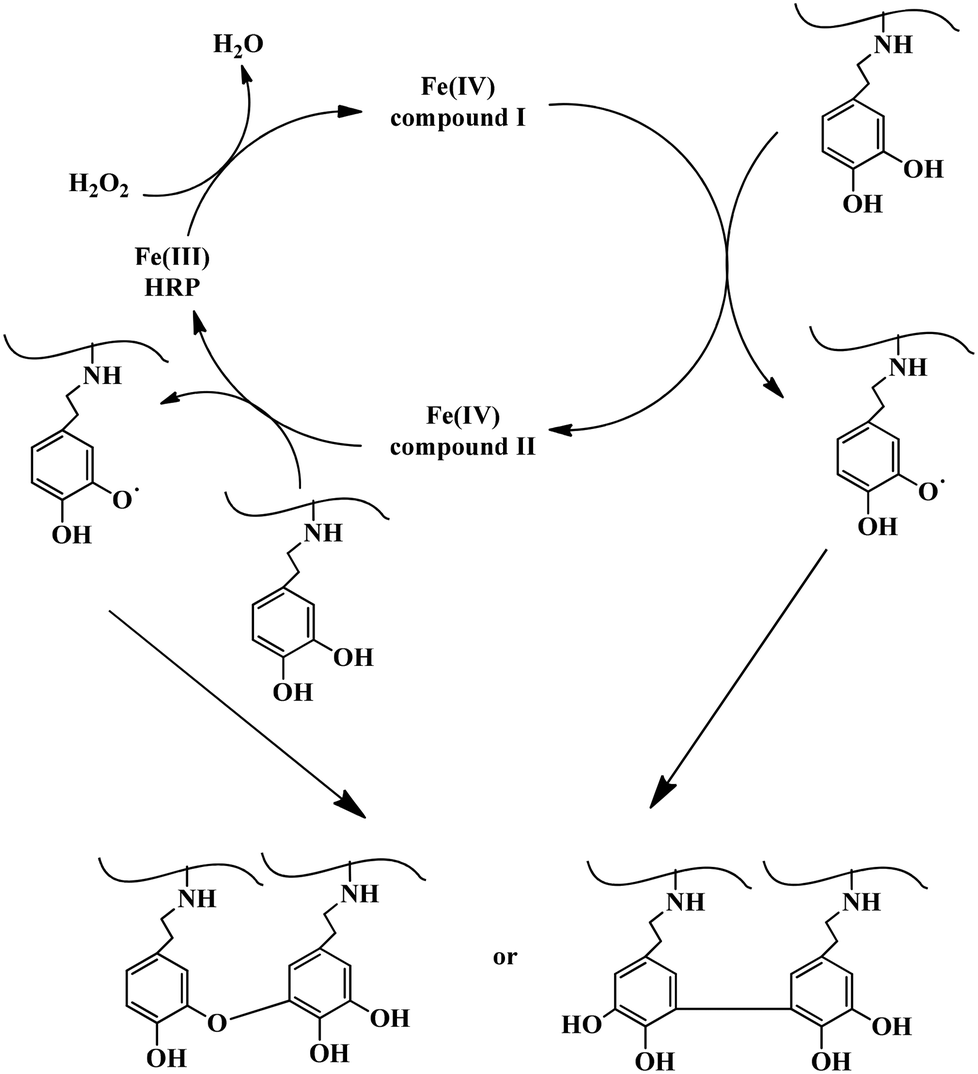 | ||
| Scheme 2 Possible mechanism of the enzymatic crosslinking of Alg–DA conjugates.4 | ||
Rheological measurements
Rheological measurements were performed using an AR2000ex rheometer (TA Instruments). Parallel plate geometry with a diameter of 40 mm was used. The sample gap was set to be 1.0 mm. The temperature was controlled by a Peltier system in the bottom plate connected with a water bath. For the kinetic study, the pre-gel solution containing Alg–DA, HRP and H2O2 was quickly prepared and loaded. Silicon oil was placed around the rim to prevent water evaporation. The change in storage modulus (G′) and loss modulus (G′′) during the gelation process was monitored as a function of time. All the experiments were performed within the linear viscoelastic region.Swelling ratio
The hydrogel samples were prepared by enzymatic crosslinking at 37 °C for 2 h to ensure complete crosslinking. Dry gels were obtained by lyophilization. To study the swelling kinetics, the dry gels were immersed in pH 7.4, 0.01 M phosphate buffer at 37 °C. At predetermined time intervals, the swollen gels were taken out. They were weighed after blotting with tissue paper to remove the adhering water. Swelling ratio of the gels was calculated using the following equation:| Swelling ratio = (Ws − W0)/W0 |
Adhesion strength
Bulk adhesion property of the hydrogels was studied by lap shear tests.21 Commercial glass slides with a size 75 mm × 25 mm × 1 mm were used as adherends. The gel samples were prepared in situ between the glass slides. For this purpose, the pre-gel solutions were placed between the glass slides and allowed to gel at 37 °C for 2 h. The overlapped area was 25 mm × 25 mm × 1 mm. Tests were performed at room temperature on a universal testing machine (Instron 5848) with a crosshead of 1.3 mm min−1 and a 100 N load cell. Adhesion strength was calculated by dividing the maximum load (force) by overlapping contact area. Each measurement was repeated at least 3 times.Cell culture
To each well of 48-well cell culture plates, 200 μL of the pre-gel solution ([polymer] = 4 wt%, [HRP] = 23.04 U mL−1, [H2O2] = 52.0 mM) was added. After gelling at 37 °C for 4 h, the gels were washed with PBS. NIH 3T3 cells were seeded at a density of 5.0 × 104 cells per well. After culturing for 24 h, the cell viability was measured using MTT assay. For this purpose, 100 μL of MTT (5.0 mg mL−1) was added into each well and the cells were incubated for 4 h at 37 °C. After washing with PBS three times, 10 mL of DMSO was added to dissolve the resulting formazan crystals. OD values at 490 nm were then measured using a microplate reader (Infinite F50, TECAN).The appearance of the cells was examined using an Olympus LX70-140 inverted fluorescence microscope. The cells were stained with AO and EB before imaging. The excitation wavelength used for AO was 450 ± 20 nm.
Other characterization
UV-vis spectra were recorded on a Varian Cary100 spectrophotometer. NMR spectra were recorded on a Bruker spectrometer (AVANCE III).Statistical analysis
All the experiments were repeated at least three times. A comparison of the means was performed by one-way analysis of variance. Differences were considered significant if p < 0.05.Results and discussion
Synthesis and characterization of Alg–DA conjugates
The Alg–DA conjugates were synthesized by coupling Alg with DA under the catalysis of EDC and NHS, as shown in Scheme 1.23,27 To protect DA from oxidation and self-polymerization, the reaction mixture was purged with N2 throughout the reaction.27 Compared to the pristine Alg, the Alg–DA conjugate exhibits an absorption peak at ∼280 nm in its UV-vis absorption spectra, as shown in Fig. 1A, confirming the successful introduction of DA moieties.23,27 The successful introduction of DA moieties was also confirmed by the appearance of peaks from 6.5 to 7.0 ppm in its 1H NMR spectra (Fig. 1B).23 By varying the feeding molar ratio of DA to Alg, a series of Alg–DA conjugates with different degree of substitutions (DSs) were synthesized23,27 (Table 1).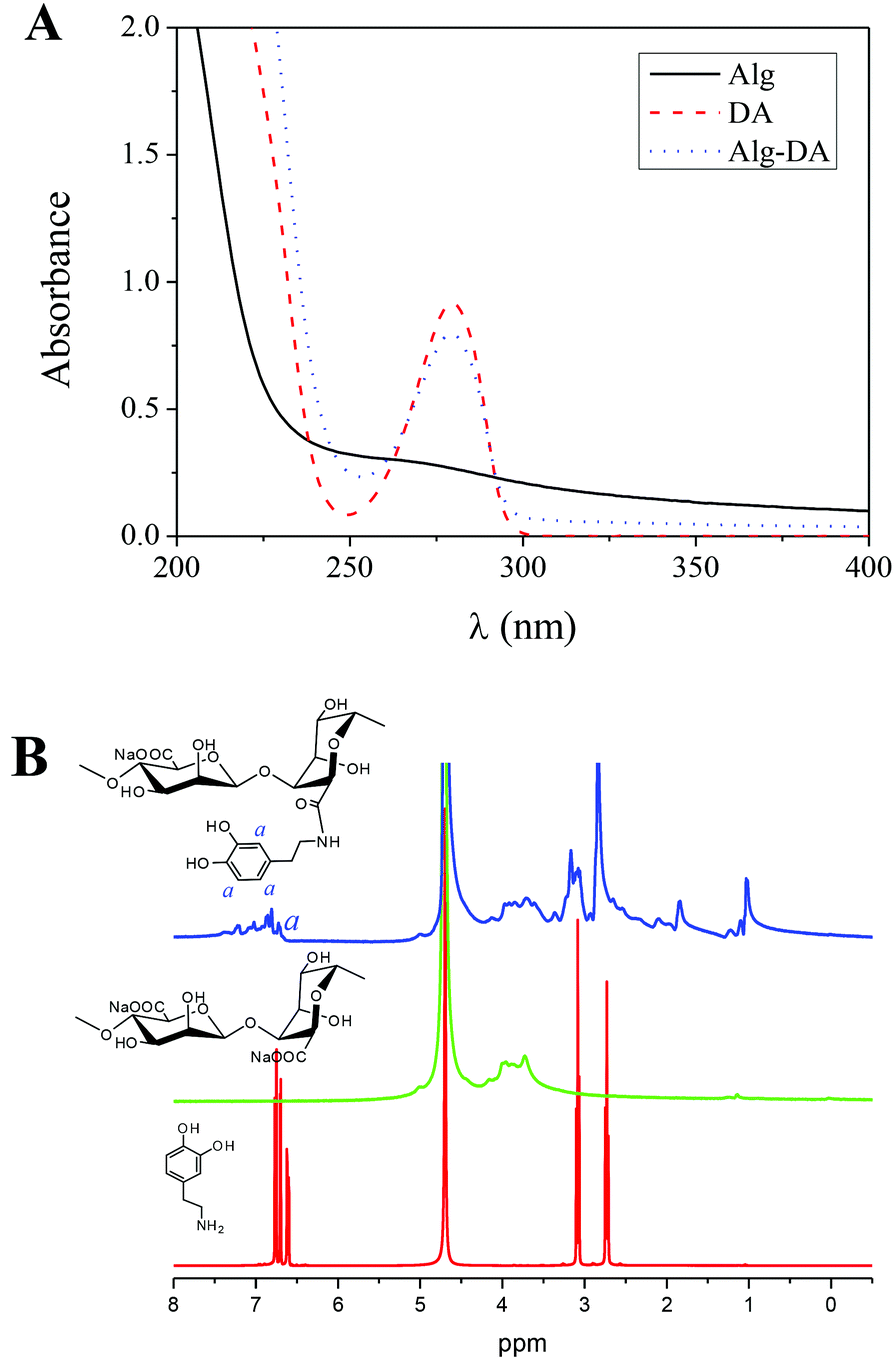 | ||
| Fig. 1 (A) UV-vis spectra of Alg, DA and Alg–DA conjugate. (B) 1H NMR spectra of DA, Alg and Alg–DA conjugate. Solvent: D2O. | ||
In situ gelation of Alg–DA via enzymatic crosslinking
Similar to polymers with phenol functionalities, Alg–DA conjugate, a polymer with catechol functionalities, can also be crosslinked by horseradish peroxidase (HRP) and H2O2. As an example, Fig. 2 shows the photograph of an Alg–DA solution before and after the in situ gelation. In the absence of HRP and H2O2, the solution remains in a liquid state and flows when tilted. After a brief incubation with HRP and H2O2, the solution gelled and could not flow upon tilting. | ||
| Fig. 2 Photographs of an Alg–DA solution before and after gelation. [Polymer] = 4.0 wt%. DS = 16%, [HRP] = 23.04 U mL−1, [H2O2] = 52.0 mM. | ||
The mechanism for the enzymatic crosslinking of catechol-modified polymers is expected to be similar to the enzymatic crosslinking of phenol-modified polymers as shown in Scheme 2.4 First HRP is converted to compound I by reacting with a H2O2 molecule. Then it reacts with a catechol molecule and is converted to compound II. As a result, a catechol radical forms. By reacting with another catechol molecule to form a second catechol radical, compound II is reduced back to its native state. Therefore, in a complete catalytic cycle, one H2O2 molecule is consumed and two catechol radicals are produced. The catechol radicals then couple with each other, either at the C–C or C–O positions, and form crosslinks between the polymer chains4,11,17,28 (Scheme 2). Previously, crosslinking of catechol-modified polymers has been accomplished by reactions between the catechol moiety and metal ions such as Fe3+ (ref. 22) and Mn3+.29,30 They can also be crosslinked by the oxidation of the catechol groups using oxidants (e.g., NaIO4) under basic conditions.20,31–33 Very recently Lee et al.34 reported the hematin-catalyzed gelation of chitosan–catechol conjugate in the presence of H2O2, probably via a similar mechanism proposed above. Like the gel synthesized via NaIO4 oxidation,23 the gels synthesized here are also brown coloured, which may have originated from the coupled catechol structures.19
The in situ gelation of Alg–DA in the presence of HRP and H2O2 was also studied using small-deformation oscillatory rheological measurements. Unless otherwise specified, all measurements were performed at a frequency of 1 Hz and a strain of 1% which is within the linear viscoelastic region. Fig. 3 shows a typical result. At the beginning, the loss modulus G′′ dominates the storage modulus G′. In addition, the phase angle is ∼90°. These results indicate that the system is in a liquid state. As time elapses, both G′ and G′′ increase gradually. Because G′ increases faster than G′′, they crossover at a reaction time of ∼125 s. At this point, the phase angle is 45°. Beyond this point, G′ remains larger than G′′, indicating the formation of 3D networks in the system. G′ continues to increase with time and eventually reaches a plateau at which the phase angle is close to zero, indicating the formation of a solid-like elastic material. Here, the point where G′ and G′′ crossover is defined as the gel point.23,35 The time required for the system to reach the gel point is defined as the gelation time (tgel), which is used as a measure of the gelation rate.7,10,36,37 The plateau G′ is used to represent the mechanical strength of the resulting gel.
 | ||
| Fig. 3 Evolution of the storage modulus G′ (■), loss modulus G′′ (□) and phase angle δ (△) of a 4.0 wt% Alg–DA solution (DS = 21%) with time in the presence of HRP (23.04 U mL−1) and H2O2 (52.0 mM). T = 37 °C. | ||
To study the effect of H2O2, the gelation of Alg–DA in the presence of various concentrations of H2O2 was studied. The polymer concentration remains constant at 4.0 wt%, while the HRP concentration remains constant at 23.04 U mL−1. As shown in Fig. 4A, for the solution of Alg–DA with a DS of 16%, on increasing [H2O2] from 10.4 to 52.0 mM, its gelation time decreases from ∼597 s to ∼140 s. It is expected that the HRP-catalyzed crosslinking reaction takes place at a faster rate with increasing [H2O2]. Therefore the gelation rate of the system increases with increasing [H2O2]. However, on further increasing [H2O2], a gradual increase in gelation time was observed. A similar phenomenon was previously observed while studying the HRP-catalyzed gelling of phenol-modified polymers, such as dextran–tyramine conjugates.7,38 This phenomenon was explained by the inactivation of HRP upon exposure to a high concentration of H2O2.7 Here, the reduced gelling rate in the high H2O2 concentration range may be explained similarly. For Alg–DA with a DS of 21%, the gelation rate also first increases with increasing [H2O2], reaches a maximum at [H2O2] = 52.0 mM, and then decreases with increasing [H2O2]. The optimal H2O2 concentration for the gelation of Alg–DA is higher than that for phenol-functionalized polymers. Previously Kurisawa et al.10 reported that hyaluronic acid–TA gels quickly in the presence of 2.4 mM H2O2. Jin et al.7 reported that, for the gelation of dextran-TA, a concentration of H2O2 from 7 to 70 mM is needed, depending on the DS and concentration of the polymer. For the gelling of Alg–DA, it is likely that the steric hindrance makes the catechol radicals more difficult to couple with each other, therefore a higher H2O2 concentration is needed.
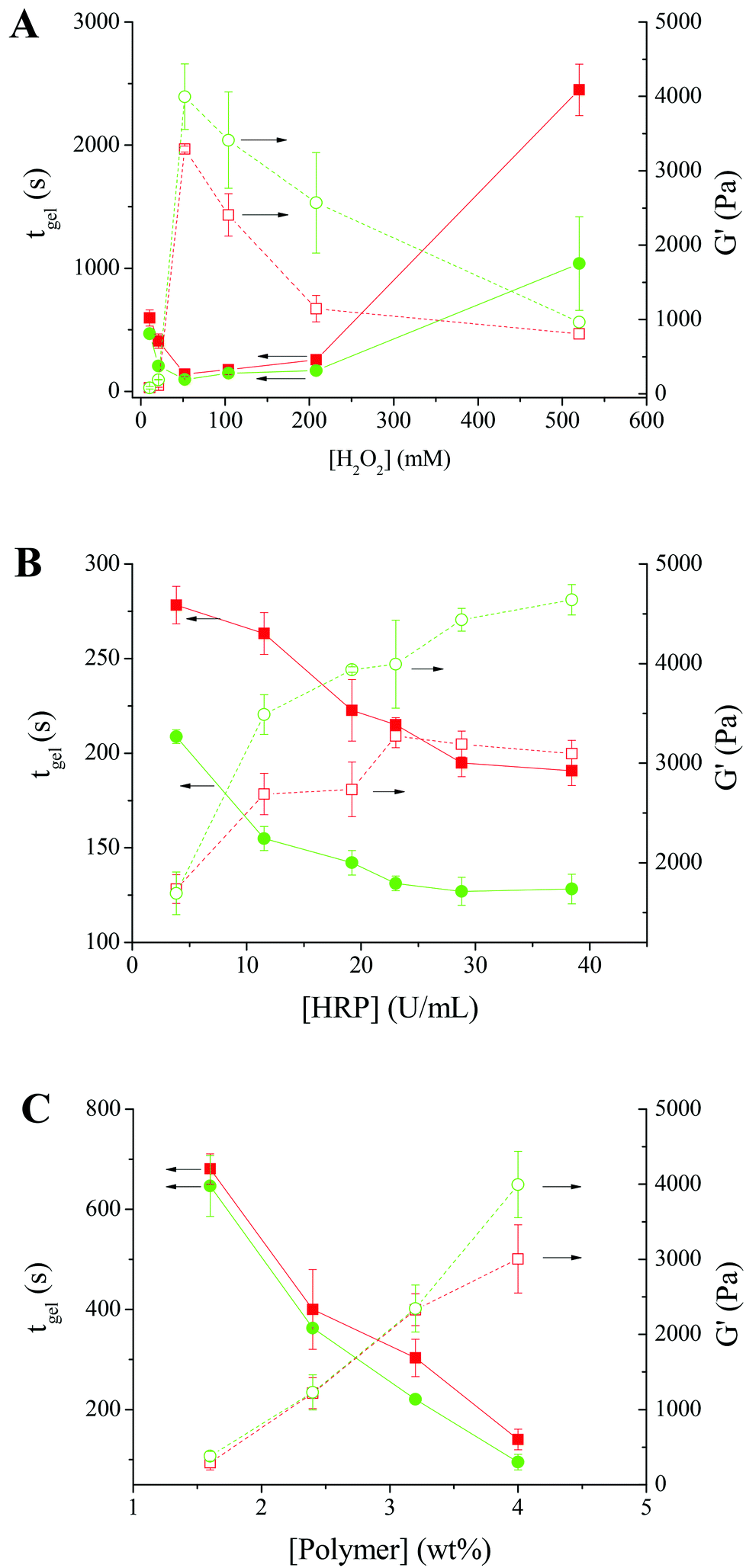 | ||
| Fig. 4 (A) Influence of H2O2 concentration on the gelation time (solid symbols) and plateau G′ (open symbols). [HRP] = 23.04 U mL−1. [Polymer] = 4 wt%. DS = 16% (■/□) or 21% (●/○). T = 37 °C. (B) Influence of HRP concentration on the gelation time (solid symbols) and plateau G′ (open symbols). [H2O2] = 52.0 mM. [Polymer] = 4 wt%. DS = 16% (■/□) or 21% (●/○). T = 37 °C. (C) Influence of polymer concentration on the gelation time (solid symbols) and plateau G′ (open symbols). [H2O2] = 52.0 mM. [HRP] = 23.04 U mL−1. DS = 16% (■/□) or 21% (●/○). T = 37 °C. The lines are a guide to the eye. | ||
For the mechanical strength of the resulting hydrogels, an opposite trend was observed. The behaviour of the two Alg–DA conjugates is similar. For Alg–DA with a DS of 16%, on increasing [H2O2] from 10.4 to 52.0 mM, the plateau G′ increases from ∼80 to ∼3300 Pa. As mentioned above, in this [H2O2] range, more catechol radicals will be produced at a higher [H2O2], which will result in a higher crosslinking density and therefore a higher strength of the gel. However, when further increasing [H2O2] from 52.0 to 520.0 mM, the plateau G′ decreases gradually from ∼3300 to ∼800 Pa. The result suggests that with the deactivation of HRP by excess H2O2, less catechol radicals will form. Therefore, the resulting gel has a lower crosslinking density and lower mechanical strength. When studying the enzymatic crosslinking of hyaluronic acid-tyramine and dextran–tyramine conjugates,7,11 a similar decline in the gel strength was reported when H2O2 concentration is higher than a critical concentration.
Fig. 4B shows the effect of HRP concentration on the gelation of Alg–DA. A constant H2O2 concentration of 52.0 mM was chosen for this study because a faster gelation rate and a higher gel strength were obtained at this concentration in the above study (Fig. 4A). The polymer concentration used was still 4.0 wt%. Again the two Alg–DA conjugates behave similarly. Their gelation rates increase with increasing [HRP]. For the Alg–DA with a DS of 16%, when [HRP] increases from 3.84 to 38.42 U mL−1, its gelation time decreases from ∼280 s to ∼190 s. Meanwhile, the strength of the resulting hydrogel increases with increasing [HRP]. For the hydrogels from Alg–DA with a DS of 16%, a plateau G′ of ∼1700 Pa was obtained when catalyzed with 3.84 U mL−1 HRP, while it increases to ∼3100 Pa when catalyzed with 38.42 U mL−1 HRP. As shown in Scheme 2, HRP acts as a catalyst in the reaction. A higher concentration of HRP should catalyze the reaction more effectively, resulting in a faster gelation rate. Higher concentration of catechol radicals will be produced at a higher [HRP]. Therefore, the polymer chains will be crosslinked more effectively, producing a hydrogel with a higher mechanical strength. It is noteworthy that when crosslinking phenol-modified polymers, such as dextran–tyramine conjugates, the gelation rate and gel strength also increase with increasing HRP concentrations.7
The effect of polymer concentration was studied at a constant [H2O2] of 52.0 mM and a constant [HRP] of 23.04 U mL−1 because the above studies show Alg–DA can be gelled effectively under these conditions. As shown in Fig. 4C, the gelation time decreases with increasing polymer concentration. For Alg–DA with a DS of 16%, it takes ∼680 s to gel a 1.6 wt% solution, while only ∼140 s is required for a 4.0 wt% solution. The decrease in gelation time may be attributed to the increased possibility for the formation of crosslinks at a high polymer concentration.39Fig. 4C also shows that the mechanical strength of the resulting hydrogels increases with increasing polymer concentration. In this perspective, the behaviour of the catechol-modified polymer is again similar to the phenol-modified polymers.7
The degree of substitution of catechol groups in the polymers should also influence their gelation. In the above studies, polymers with two different DSs were used. Generally, the polymer with a higher DS gels faster and produces a hydrogel with a higher mechanical strength (Fig. 4A–C). It is understandable that a higher DS means a higher concentration of catechol groups. Therefore, the reaction rate increases with increasing DS. Also for polymers with a higher DS, more crosslinks form, resulting in a hydrogel with a higher mechanical strength.7
Swelling behaviour
Swelling in aqueous solutions maybe the most important property for hydrogels. The swelling properties of the in situ formed Alg–DA gels were studied by measuring the weight change of the gels in phosphate buffer. The gels reach their swelling equilibrium in about 10 h as shown in Fig. 5A. As for the equilibrium swelling ratio, the gel with a higher DS usually has a lower swelling degree than the gel with a lower DS, because the former has a higher crosslinking density (Fig. 5B). For the gels synthesized at the same [HRP] but different [H2O2], the equilibrium swelling degree of the gel gelled at [H2O2] = 52.0 mM is the lowest because its crosslinking density is the highest (Fig. 5C). For the gels synthesized at the same [H2O2] but different [HRP], the equilibrium swelling degree of the gel decreases with increasing [HRP] (Fig. 5D). These observations are generally in agreement with the results from the kinetic studies.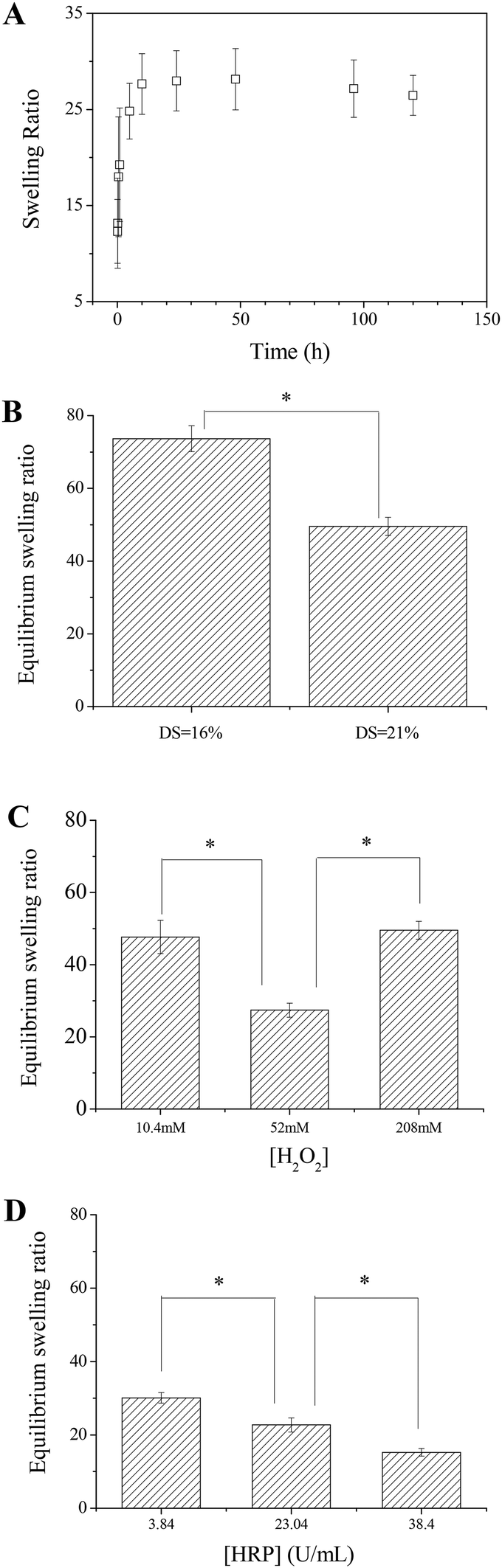 | ||
| Fig. 5 (A) Swelling kinetics of an Alg–DA hydrogel in PBS. T = 37 °C. Gel parameters: [polymer] = 4 wt%, DS = 21%, [HRP] = 23.04 U mL−1, [H2O2] = 52 mM. (B) Equilibrium swelling ratios of Alg–DA hydrogels as a function of DS. Other parameters: [polymer] = 4 wt%, [HRP] = 23.04 U mL−1, [H2O2] = 208 mM. (C) Equilibrium swelling ratios of Alg–DA hydrogels as a function of [H2O2]. Other parameters: [polymer] = 4 wt%, DS = 21%, [HRP] = 23.04 U mL−1. (D) Equilibrium swelling ratios of Alg–DA hydrogels as a function of [HRP]. Other parameters: [polymer] = 4.0 wt%, DS = 21%, [H2O2] = 52.0 mM. * indicates statistical significance at the 0.05 level. | ||
Adhesion strength
A major objective of the study is to improve the bioadhesion property of the injectable alginate hydrogels. For comparison, Alg–TA hydrogels were also prepared via enzymatic crosslinking of Alg–TA. We first tested if porcine skins can be bonded with Alg–DA or Alg–TA hydrogels. When the skins were bonded with the Alg–DA gel, they were bonded so firmly that they could not be pulled apart by 300 g weight (Fig. 6). In contrast, when they were bonded with the Alg–TA hydrogel, they could not even support their own weight (data not shown). These results suggest that the Alg–DA hydrogels exhibit much better bioadhesion property than the Alg–TA hydrogel.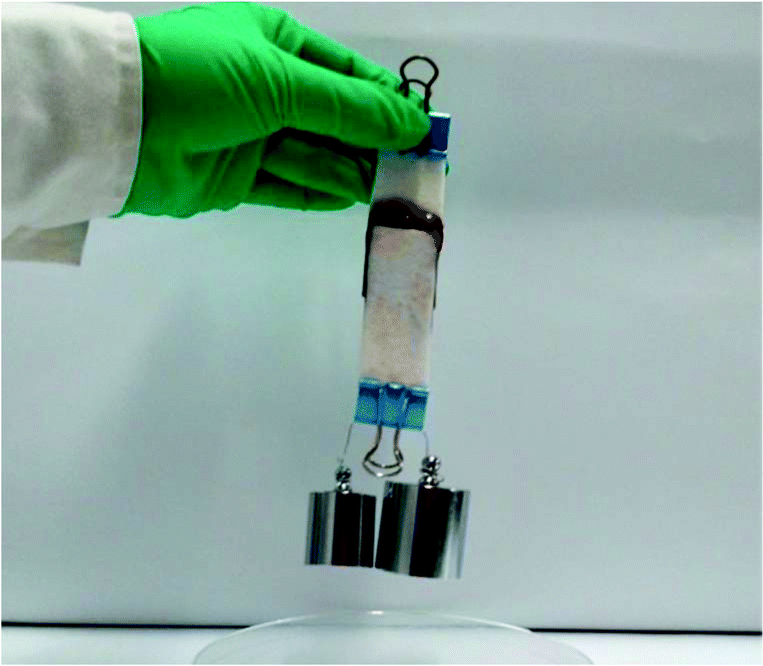 | ||
| Fig. 6 Photograph of two pieces of porcine skin bonded together with an Alg–DA hydrogel showing that they are able to support 300 g weight. Overlapped area: 25 mm × 25 mm × 1 mm. | ||
To compare their adhesion property quantitatively, the bulk adhesion strength of the hydrogels was measured by lap shear tests, the most widely used method for quantifying adhesion.18,25,40,41 For convenience, we used commercial glass slides as adherends. The hydrogel samples were prepared in situ by placing the pre-gel solutions between the glass slides and allowing them to gel at 37 °C for 2 h.
Fig. 7A compares the adhesion strength of Alg–DA hydrogels to an Alg–TA hydrogel. The polymer content was 4.0 wt% for all the hydrogels. They were all crosslinked in the presence of 23.04 U mL−1 HRP and 52 mM H2O2. The adhesion strength of the Alg–TA hydrogel is only 0.38 kPa, indicating a weak interaction between the gel and glass slides. For the Alg–DA hydrogel with the same DS (10%), the adhesion strength is 3.5 kPa. The result indicates that replacing of the phenol groups in the Alg–TA hydrogel with catechol groups significantly increases the adhesion strength of the hydrogel by about 10 folds. An improvement of adhesion properties by the introduction of catechol functionalities have been widely reported in the literature. Previously Ryu et al.25 have reported that the adhesion force between chitosan/thiolated Pluronic hydrogels and subcutaneous tissue layers increases by about twofold when chitosan is functionalized with catechol groups. Matos-Pérez and Wilker42 reported that the adhesion strength of polystyrene increases by 5 folds when 10% catechol was introduced. In another example, Chung and Grubbs21 synthesized terpolymers with a poly(acrylic acid) backbone. The adhesion strength of the one containing a DOPA moiety is 190% over the one without a DOPA moiety. The mechanism for the extraordinary adhesion of DOPA-containing proteins and catechol-containing polymers has been studied in the literature. It is believed that the oxidative crosslinking43 and metal chelation30,44 within the bulk result in strong cohesive forces. These materials could also interact with a surface via hydrogen bonding,45,46 metal chelation,44,47 and radical–surface coupling.30,44 S. A. Mian et al.45 recently calculated that the binding energy between the catechol and silica surface with adsorbed water molecules is 23 kcal mol−1. The unique bonding properties of catechol-containing polymers are a combined result of these interactions. It is noteworthy that the exact mechanism has not been precisely described.42
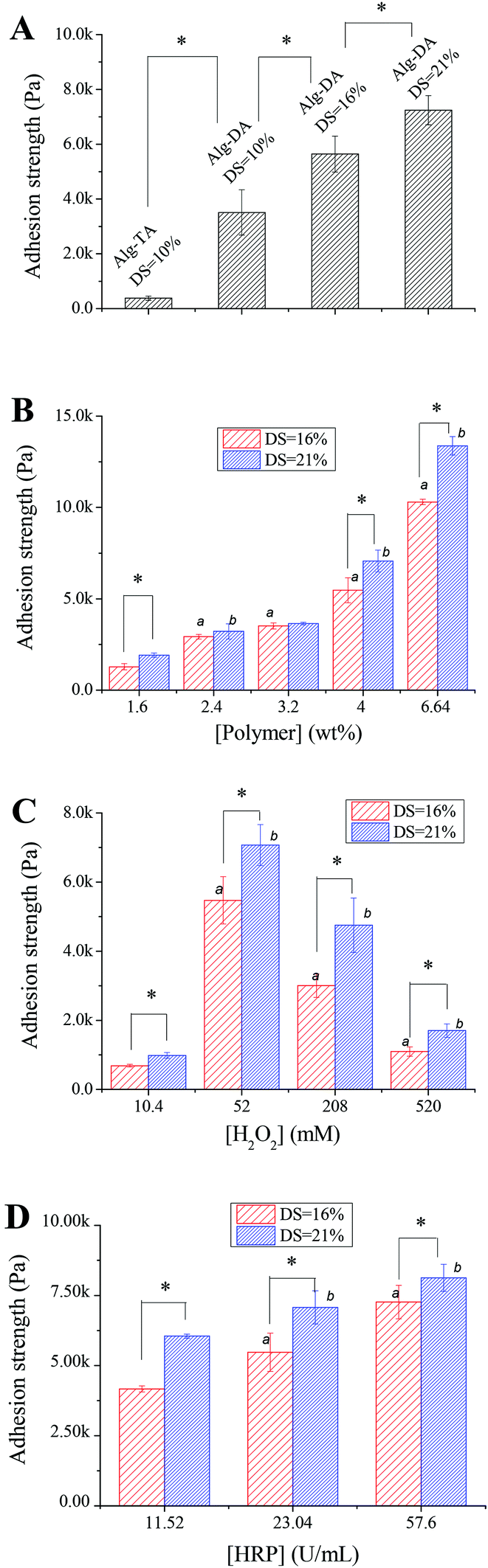 | ||
| Fig. 7 (A) Lap shear strength of Alg–TA hydrogels and Alg–DA hydrogels with various DSs. Other parameters for the hydrogels: [polymer] = 4.0 wt%, [HRP] = 23.04 U mL−1, [H2O2] = 52.0 mM. (B) Lap shear strength of the Alg–DA hydrogel as a function of the polymer concentration. Other parameters for the hydrogels: [HRP] = 23.04 U mL−1, [H2O2] = 52.0 mM. (C) Lap shear strength of Alg–DA hydrogels crosslinked at various concentrations of H2O2. Other parameters for the hydrogels: [polymer] = 4.0 wt%, [HRP] = 23.04 U mL−1. (D) Lap shear strength of Alg–DA hydrogels crosslinked at various concentrations of HRP. Other parameters for the hydrogels: [polymer] = 4.0 wt%, [H2O2] = 52.0 mM. The superscript letters and * indicate statistical significance at the 0.05 level. a and b: compared to the previous datum in the same group. a: DS 16% group, b: DS 21% group. | ||
Fig. 7A also shows that the adhesion strength of Alg–DA hydrogels can be further improved by increasing the DA content in the hydrogels. When DS of the polymer increases to 16% and 21%, the adhesion strength of the gel increases further to be 5.6 and 7.2 kPa. Previously Wilker et al.41 studied the effect of catechol content on the properties of the mussel mimetic polymers. For their model polymer with a polystyrene backbone, they found that the adhesion strength of the polymer increases with increasing catechol content up to the point of ∼33%. Further addition of catechol did not enhance the adhesion. Interestingly, this value is close to the DOPA content in the mussel foot proteins Mfp-3 (∼25% DOPA) and Mfp-5 (∼30% DOPA). In light of these results, we expect that the adhesion properties of Alg–DA hydrogel may be further improved by introducing more catechol moieties.
Fig. 7B shows the adhesion properties of Alg–DA hydrogel as a function of polymer concentration. For both DS 21% hydrogels and DS 16% hydrogels, their adhesion strength increases with increasing polymer concentration. A hydrogel with a higher polymer concentration can provide more catechol moieties to interact with the adherend (glass slide in this case), therefore it can stick to the adherend more firmly. Generally at the same concentration, the adhesion strength of a DS 21% hydrogel is higher than a DS 16% hydrogel, which is in agreement with the observation shown in Fig. 7A.
Besides the catechol content and polymer concentration, the gelling conditions also influence the adhesion properties of the hydrogel. Fig. 7C shows the adhesion properties of hydrogels gelled with 23.04 U mL−1 HRP and different concentrations of H2O2. For hydrogels gelled with 10.4 mM H2O2, they adhere to the glass slides weakly. In contrast, hydrogels gelled with 52.0 mM H2O2 exhibit a strong adhesion property. Further increasing [H2O2] deteriorates the adhesion properties of the hydrogels. The different adhesion properties may originate from different network structures of the hydrogels. Previous studies show that crosslinking can often enhance the adhesion properties of polymers.41 It is expected that the adhesion strength will increase with increasing crosslinking density because a gel network with a higher crosslinking density can provide stronger cohesive interactions to help resist bond failure. It can also dissipate energy more effectively when the gel is under strain.48 As revealed above, the hydrogels gelled with 10.4 mM H2O2 have a low crosslinking density, while the ones gelled with 52 mM H2O2 are effectively crosslinked (Fig. 4A). Therefore, the former shows a weak adhesion while the latter adheres firmly. On further increasing [H2O2], the crosslinking density decreases again, so does the adhesion strength (Fig. 4A). It is noteworthy that according to previous studies,41,48 too much crosslinking may reduce the wettability of the polymer and thus decrease its adhesion.
The adhesion properties of hydrogels gelled with the same concentration of H2O2 but different concentrations of HRP are shown in Fig. 7D. The adhesion of the hydrogels increases with increasing HRP concentrations, because higher HRP concentration produces hydrogels with a higher crosslinking density as indicated by their increasing mechanical strength as shown in Fig. 4C.
Cell culture
To test the biocompatibility of the new hydrogels, they were synthesized directly on the bottom of the wells of cell culture plates. NIH 3T3 cells were then seeded and the viability of the cells was assessed using the MTT assay. For a comparison, bare cell culture plate surface (TCPS) was used as a control. As shown in Fig. 8, the percentage viability of the cells cultured on the hydrogel surface is more than 80%. In addition, the viability of the cells cultured on Alg–DA hydrogels is comparable to the cells cultured on Alg–TA hydrogels. These results indicate that the Alg–DA hydrogels, similar to the Alg–TA hydrogels, are non-toxic to the cells. The result is not surprising because it is well-known that alginate is highly biocompatible.2 | ||
| Fig. 8 Percentage viability of cells seeded on hydrogel surfaces and cultured for 24 h. | ||
To study the cell morphology, the cells were first double stained with AO/EB. In this way, the live cells were stained green and dead ones were stained red. As shown in Fig. 9, most of the cells are alive, confirming again a high viability of the cells grown on the gel surfaces. One can see that the cells grown on Alg–DA gels exhibit a very different morphology compared to the cells grown on Alg–TA gels. The cells on Alg–TA gels are round in shape, indicating that the cells do not attach the substratum. The Alg–TA gels are highly hydrophilic and highly swollen, therefore the interaction of the cells with the gel is weak. The weak interaction between the cells and the substratum also results in the aggregation of the cells, especially on the gel with a DS = 21%. A similar cell behaviour has been widely observed from other hydrogels.49–51 In contrast, many of the cells grown on Alg–DA gels present a spread, irregular shape with protrusions. The result indicates that the cells attach on the gel surface, just like the cells grown on bare cell culture plates. The attachment and spreading of cells on Alg–DA gels result in a larger cell spreading area (Table S1 in ESI‡). The cell area on the DS 21% Alg–DA surface was calculated to be 1160 ± 238 μm2, while it is 549 ± 81 μm2 for cells on Alg–TA with the same DS. The structure of Alg–DA gels is similar to Alg–TA gels. Like the latter, the Alg–DA gels are highly hydrophilic and highly swollen too. The very different behaviour of the cells on the two kinds of hydrogels could only be explained by the replacement of phenol groups with catechol groups, which results in a significantly increased interaction between the cells and the gels. Fig. 9 also shows that when DS of the Alg–DA gel increases from 10% to 21%, the cells present a more prolonged morphology. This observation may imply that the interaction between the cells and the gel increases with increasing catechol content.
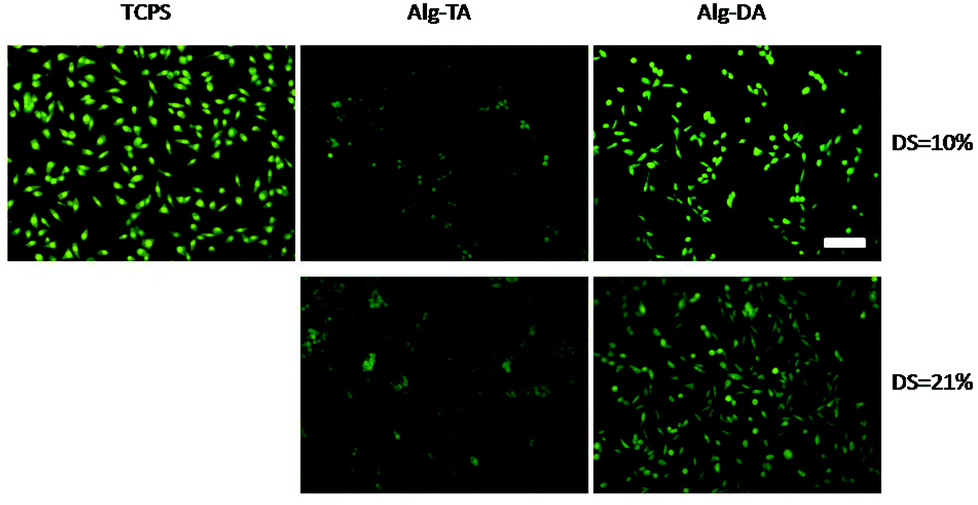 | ||
| Fig. 9 Fluorescence image of NIH 3T3 cells cultured on hydrogel surfaces for 24 h. The cells were double stained with AO/EB. Scale bar: 200 μm. | ||
Conclusions
In conclusion, we show that polymers with catechol side groups, similar to polymers with phenol side groups, can gel in the presence of HRP and H2O2. As an example, the in situ gelling of Alg–DA conjugates was studied in detail using rheological measurements. Various factors, including the concentration of HRP, H2O2, and the polymer, and the degree of substitution of the polymer, can influence the kinetics of the enzyme-catalyzed crosslinking reaction, thereby influencing the mechanical strength and swelling behaviour of the resulting hydrogels. Replacement of phenyl side groups with catechol groups results in significantly improved adhesion properties. For Alg–DA and Alg–TA hydrogels with the same DS of 10%, the adhesion strength of the former is about 10-fold more than the latter. While both the hydrogels are biocompatible, cells seeded on the gel surface behave very differently. They do not attach on Alg–TA gels, but attach on Alg–DA gels. The in situ-forming hydrogels with unusual adhesion properties are expected to find use in important biomedical applications such as drug delivery and wound closure. A further improvement of the gelling system is needed to reduce H2O2 concentration to avoid its cytotoxicity.Acknowledgements
We acknowledge the financial support received for this work from the National Natural Science Foundation of China (Grants Nos 21174070, 21274068, 21228401 and 21374048), the Tianjin Public Health Bureau (13KG110), Program for New Century Excellent Talents in University (NCET-11-0264), and the PCSIRT program (IRT1257).Notes and references
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- Y. Guan and Y. Zhang, Chem. Soc. Rev., 2013, 42, 8106–8121 RSC.
- M. Kurisawa, F. Lee, L. Wang and J. E. Chung, J. Mater. Chem., 2010, 20, 5371–5375 RSC.
- S. R. Van Tomme, G. Storm and W. E. Hennink, Int. J. Pharm., 2008, 355, 1–18 CrossRef CAS PubMed.
- Y. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- R. Jin, C. Hiemstra, Z. Y. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS PubMed.
- L. S. Moreira Teixeira, J. Feijen, C. A. van Blitterswijk, P. J. Dijkstra and M. Karperien, Biomaterials, 2012, 33, 1281–1290 CrossRef CAS PubMed.
- S. J. Sofia, A. Singh and D. L. Kaplan, J. Macromol. Sci., Part A:Pure Appl. Chem., 2002, 39, 1151–1181 CrossRef PubMed.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312–4314 RSC.
- F. Lee, J. E. Chung and M. Kurisawa, Soft Matter, 2008, 4, 880–887 RSC.
- F. Lee, J. E. Chung and M. Kurisawa, J. Controlled Release, 2009, 134, 186–193 CrossRef CAS PubMed.
- F. Yu, X. Cao, Y. Li, L. Zeng, B. Yuan and X. Chen, Polym. Chem., 2014, 5, 1082–1090 RSC.
- R. Jin, L. Teixeira, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Biomaterials, 2010, 31, 3103–3113 CrossRef CAS PubMed.
- R. Jin, L. S. M. Teixeira, P. J. Dijkstra, Z. Zhong, C. A. van Blitterswijk, M. Karperien and J. Feijen, Tissue Eng., Part A, 2010, 16, 2429–2440 CrossRef CAS PubMed.
- M. K. McHale, L. A. Setton and A. Chilkoti, Tissue Eng., 2005, 11, 1768–1779 CrossRef CAS PubMed.
- Y. S. Pek, M. Kurisawa, S. Gao, J. E. Chung and J. Y. Ying, Biomaterials, 2009, 30, 822–828 CrossRef PubMed.
- O. Jeon, J. E. Samorezov and E. Alsberg, Acta Biomater., 2014, 10, 47–55 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- C. E. Brubaker and P. B. Messersmith, Biomacromolecules, 2011, 12, 4326–4334 CrossRef CAS PubMed.
- H. Chung and R. H. Grubbs, Macromolecules, 2012, 45, 9666–9673 CrossRef CAS.
- B. J. Kim, D. X. Oh, S. Kim, J. H. Seo, D. S. Hwang, A. Masic, D. K. Han and H. J. Cha, Biomacromolecules, 2014, 15, 1579–1585 CrossRef CAS PubMed.
- C. Lee, J. Shin, J. S. Lee, E. Byun, J. H. Ryu, S. H. Um, D. Kim, H. Lee and S. Cho, Biomacromolecules, 2013, 14, 2004–2013 CrossRef CAS PubMed.
- J. D. White and J. J. Wilker, Macromolecules, 2011, 44, 5085–5088 CrossRef CAS.
- J. H. Ryu, Y. Lee, W. H. Kong, T. G. Kim, T. G. Park and H. Lee, Biomacromolecules, 2011, 12, 2653–2659 CrossRef CAS PubMed.
- M. L. B. Palacio and B. Bhushan, Phil. Trans. R. Soc. A, 2012, 370, 2321–2347 CrossRef CAS PubMed.
- X. Wang, Z. Jiang, J. Shi, Y. Liang, C. Zhang and H. Wu, ACS Appl. Mater. Interfaces, 2012, 4, 3476–3483 CAS.
- H. Zhang, L. P. Bré, T. Zhao, Y. Zheng, B. Newland and W. Wang, Biomaterials, 2014, 35, 711–719 CrossRef CAS PubMed.
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS PubMed.
- M. J. Sever, J. T. Weisser, J. Monahan, S. Srinivasan and J. J. Wilker, Angew. Chem., Int. Ed., 2004, 43, 448–450 CrossRef CAS PubMed.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS PubMed.
- C. E. Brubaker, H. Kissler, L. Wang, D. B. Kaufman and P. B. Messersmith, Biomaterials, 2010, 31, 420–427 CrossRef CAS PubMed.
- M. Cencer, Y. Liu, A. Winter, M. Murley, H. Meng and B. P. Lee, Biomacromolecules, 2014, 15, 2861–2869 CrossRef CAS PubMed.
- E. Byun, J. H. Ryu and H. Lee, Chem. Commun., 2014, 50, 2869–2872 RSC.
- C. M. Tung and P. J. Dynes, J. Appl. Polym. Sci., 1982, 27, 569–574 CrossRef CAS.
- A. Tobitani and S. B. Ross-Murphy, Macromolecules, 1997, 30, 4845–4854 CrossRef CAS.
- W. Liao, Y. Zhang, Y. Guan and X. X. Zhu, Macromol. Chem. Phys., 2011, 212, 2052–2060 CrossRef CAS.
- Y. Sun, Z. Deng, Y. Tian and C. Lin, J. Appl. Polym. Sci., 2013, 127, 40–48 CrossRef CAS.
- J. He, A. Zhang, Y. Zhang and Y. Guan, Macromolecules, 2011, 44, 2245–2252 CrossRef CAS.
- L. Ninan, J. Monahan, R. L. Stroshine, J. J. Wilker and R. Shi, Biomaterials, 2003, 24, 4091–4099 CrossRef CAS.
- C. R. Matos-Pérez, J. D. White and J. J. Wilker, J. Am. Chem. Soc., 2012, 134, 9498–9505 CrossRef PubMed.
- C. R. Matos-Pérez and J. J. Wilker, Macromolecules, 2012, 45, 6634–6639 CrossRef PubMed.
- J. Yu, W. Wei, E. Danner, R. K. Ashley, J. N. Israelachvili and J. H. Waite, Nat. Chem. Biol., 2011, 7, 588–590 CrossRef CAS PubMed.
- J. J. Wilker, Angew. Chem., Int. Ed., 2010, 49, 8076–8078 CrossRef CAS PubMed.
- S. A. Mian, L. Yang, L. C. Saha, E. Ahmed, M. Ajmal and E. Ganz, Langmuir, 2014, 30, 6906–6914 CrossRef CAS PubMed.
- S. A. Mian, L. C. Saha, J. Jang, L. Wang, X. Gao and S. Nagase, J. Phys. Chem. C, 2010, 114, 20793–20800 CAS.
- A. Akemi Ooka and R. L. Garrell, Biopolymers, 2000, 57, 92–102 CrossRef CAS.
- C. L. Jenkins, H. J. Meredith and J. J. Wilker, ACS Appl. Mater. Interfaces, 2013, 5, 5091–5096 CAS.
- J. Lee, M. J. Cuddihy, G. M. Cater and N. A. Kotov, Biomaterials, 2009, 30, 4687–4694 CrossRef CAS PubMed.
- A. Ivascu and M. Kubbies, J. Biomol. Screening, 2006, 11, 922–932 CrossRef CAS PubMed.
- Z. Zhao, J. Gu, Y. Zhao, Y. Guan, X. X. Zhu and Y. Zhang, Biomacromolecules, 2014, 15, 3306–3312 CrossRef CAS PubMed.
Footnotes |
| † This work is dedicated to Professor Weixiao Cao, Peking University, China, for his 80th birthday in November 2014. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py01757a |
| This journal is © The Royal Society of Chemistry 2015 |