
A seed-engineering approach toward a hollow nanoreactor suitable for the confined synthesis of less-noble Ni-based nanocrystals†
Kwanyong
Jeong
,
Soo Min
Kim
and
In Su
Lee
*
Department of Chemistry, Pohang University of Science and Technology (POSTECH), Gyeongbuk, 790-784, Korea. E-mail: insulee97@postech.ac.kr
First published on 10th November 2014
Abstract
A hollow nanoreactor suitable for the cultivation of Ni-nanocrystals was developed through a distinct seed-engineering stratagem, which involved the assembly of a catalytically active Au/Pd-heterojunction-nanocrystal inside the hollow silica nanoshell. The resulting hollow nanoreactor demonstrated a targeted performance in the cavity-confined growth of the catalytic Ni nanocrystal.
Yolk@shell nanoparticles, which carry a functional core inside a hollow and porous nanoshell, are attractive candidates for nanoreactors that allow spatially confined reactions for selectively internalized molecules.1 Most research efforts to date have focused on developing highly durable and recyclable nanocatalyst systems that efficiently and selectively catalyze the transformation of organic molecules.2,3 Very recently, a new angle of approach has been suggested by a few researchers to utilize hollow nanoreactors for synthesizing and decorating nanoparticles within the confines of the protected interior space.4 For instance, we recently devised the nanoreactor approach for templating the synthesis of noble metal nanocrystals, such as Pt, Ag, Au and their alloys, through exploitation of the seed-mediated growth inside the cavity of Au-seed containing hollow silica nanospheres, Au@h-SiO2.5 The hollow nanoreactor-based approach has enabled the production of ligand-free noble-metal nanocrystals with well-controlled morphologies from a highly concentrated suspension. In this context, the current study was performed with the aim of further extending the utility of the hollow nanoreactors toward the production of less confinable non-noble metal nanocrystals beyond the noble metals. Unlike noble metals that can be easily restricted to grow on Au-seeds using mild reducing agents, the growth of less reducible non-noble metals is more complicated as it requires severe reaction conditions, which results in its homogenous nucleation in the bulk solution. In particular, this study focuses on devising a hollow nanoreactor suitable for the cultivation of Ni-nanocrystals, which plays important roles as versatile and representative non-noble metal catalysts in many applications.6 Herein, we report our strategy for engineering a seeding core toward the Ni-growing nanoreactor, which includes an assembly of a catalytically active Au/Pd-heterojunction-nanocrystal inside the hollow silica nanoshell. The nanoreactor-synthesized Ni-nanocrystals exhibited an active and recyclable performance in catalyzing hydrogen generation from NH3BH3 and the chemoselective reduction of nitroarenes in aqueous solutions at room temperature. Moreover, we demonstrate the usefulness of these hollow nanoreactors in the synthesis and post-modification of Ni-based hybrid nanocrystals.
Firstly, the employability of the previously developed Au@h-SiO2 was investigated in growing Ni nanocrystals. NiCl2 in a suspension of Au@h-SiO2 was treated with a range of mild reducing agents, including ascorbic acid, H2O2, and hydrazine. Although these reducing agents had previously allowed the Au-seed-mediated growth of Au,5a Pt,5b and Ag,5c respectively, it did not lead to the reduction of Ni2+. An alternative trial with a stronger reducing agent, NaBH4, caused Ni deposition on the Au seed. However, this process also formed undesirably large Ni particles on the outer surface of the Au@h-SiO2 nanoreactor (Fig. S1, ESI†). This preliminary study suggested that a more powerful seed needs to be engineered for restricting the Ni growth to the interior of the hollow nanoreactor. Therefore, a strategy was planned to introduce Pt, Pd, or Ni, which is known to catalyze the reductive decomposition of Ni2+–hydrazine complex,7 as a seeding material by incorporating Pt2+, Pd2+, or Ni2+ ions into the reductive dissolution process of the Fe3O4, which had been adopted previously for hollowing the (Fe3O4/Au)@SiO2 into Au@h-SiO2.5c It was hypothesized that in situ reduced Pt, Ni, or Pd would be deposited on the Au-nanocrystal inside the cavity during the hollowing process (Scheme 1).
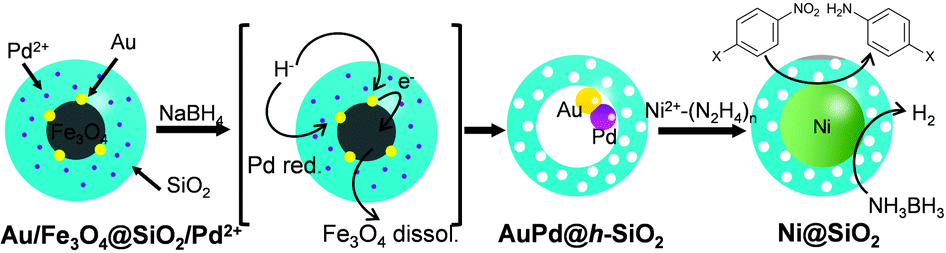 | ||
| Scheme 1 The synthetic strategy of the Ni-growing (Au/Pd)@h-SiO2 nanoreactor. | ||
The incorporation of Ni2+, Pt2+, and Pd2+ ions was carried out by adding aliquots of Ni(NO3)2, Na2PtCl4, and Na2PdCl4 solutions, respectively, into the microemulsion suspension which had previously been used to prepare (Fe3O4/Au)@SiO2 (for the detailed procedure, refer to the experimental section in ESI†). The transmission electron microscopy (TEM) images showed that all reactions afforded similar core@shell structures, which contain an embedded hybrid nanocrystal, composed of a Fe3O4 nanocrystal and surrounding satellite nanocrystals, at the core of the silica nanosphere (Fig. 1a–c). The control reactions without tetraethyl orthosilicate (TEOS) revealed that a comparable amount of Pt and Au were present around the Fe3O4 nanoparticle in isolated nanoparticles. Conversely, only a small amount of Ni and Pd were detected around the Fe3O4 nanoparticle (Fig. S2, ESI†). Additional control experiments, excluding both HAuCl4 and TEOS, showed a sparse population of Pt element on the isolated nanoparticles (Fig. S3, ESI†). From these observations, it can be deduced that, in the resultant core@shell nanospheres, the Ni2+ and Pd2+ ions were dispersed throughout the silica shell, generating (Fe3O4/Au)@(SiO2/Ni2+) and (Fe3O4/Au)@(SiO2/Pd2+) nanospheres, respectively. In contrast, the Pt2+ ions were co-included as the components of satellite nanocrystals together with Au surrounding the Fe3O4 nanocrystal, resulting in the formation of a (Fe3O4/AuPt2+)@SiO2 nanosphere. When the resultant nanospheres were exposed to a solution of NaBH4, which is expected to reductively dissolve the Fe3O4 and simultaneously reduce the metal ions, they were found to undergo different reactions depending on the incorporated ions, Pt2+, Ni2+, and Pd2+. In the case of (Fe3O4/AuPt2+)@SiO2, it was found that the Fe3O4 core was not subject to the expected reductive dissolution. This was presumably due to the reduced catalytic activity of the attached Au nanocrystal, which alloyed with Pt. The NaBH4 treatment therefore afforded an unexpected nanostructure, (Fe3O4/AuPt)@h-SiO2, in which a satellite-type nanoparticle, consisting of a 9(±1) nm-sized Fe3O4 core and surrounding Au/Pt bimetallic nanocrystals of 2.1(±0.5) nm size, is placed inside the 19(±2) nm-sized internal cavity, which was developed through silica etching (Fig. 1e). The interior cavity of the (Fe3O4/AuPt)@h-SiO2, which was partially occupied by the undissolved Fe3O4 core, was regarded as unsuitable for metal growth. In comparison, when (Fe3O4/Au)@(SiO2/Ni2+) was immersed in a NaBH4 solution, the Fe3O4 core dissolved rapidly due to the reductive dissolution process, facilitated by the attached Au-nanocrystals, while the Ni2+ ions slip away from the silica through in situ generated small pores. Therefore, regardless of the content of the initially incorporated Ni2+ ions in the silica shell, the reaction yielded a nanorattle-like structure containing a spherical Au core, Au@h-SiO2, which was found to be almost identical to the one previously prepared from (Fe3O4/Au)@SiO2 (Fig. 1d and Fig. S4, ESI†). When Fe3O4@SiO2/M2+ (M = Ni, Pd, Pt) nanospheres, which were prepared without containing Au nanocrystals, were treated with NaBH4 solution for the purpose of control, the removal of the Fe3O4 core was not observed from any nanosphere, which indicates little effect of metal ions other than the Au nanoparticle on the reductive dissolution process (Fig. S5, ESI†).
 | ||
| Fig. 1 TEM and HRTEM (insets) images of (a) (Fe3O4/Au)@(SiO2/Ni2+), (b) (Fe3O4/AuPt2+)@SiO2 and (c) (Fe3O4/Au)@(SiO2/Pd2+) core@shell nanospheres and (d) Au@h-SiO2 (e) (Fe3O4/AuPt)@h-SiO2 (insets: EDX elementary maps, Au (red), Pt (green)) and (f) (Au/Pd)@h-SiO2 yolk@shell nanospheres, which were produced by treating the core@shell nanospehres with NaBH4. | ||
The NaBH4 treatment of the (Fe3O4/Au)@(SiO2/Pd2+) also generated a similar nanorattle-like structure. This consisted of a 21(±2) nm-sized cavity and an encaged tiny core nanocrystal which appeared ellipsoidal in TEM images rather than spherical (Fig. 1f). Further investigation using high resolution-TEM (HR-TEM), STEM, and energy-dispersive X-ray spectroscopy (EDX) identified the ellipsoidal core to be an Au/Pd-heterodimer-nanocrystal with a dumbbell-like structure that includes spherical Au and Pd grains of a similar size (ca. 2.5 nm) joined by a small interfacial area (Fig. 2d). The above-identified structure of the (Au/Pd)@h-SiO2 represents the successful introduction of the reduced Pd into the cavity of the hollow silica nanoshell, as outlined in the initial synthetic strategy. Time-course TEM and HR-TEM studies revealed that the Fe3O4 at the core of the (Fe3O4/Au)@(SiO2/Pd2+) was dissolved very early in the reaction (within 10 min), leaving a spherical Au-nanocrystal in the newly formed interior cavity (Fig. 2a). Whilst the cavity expanded gradually over the reaction period, from 12(±1) nm at 10 min to 21(±2) nm at 1 h, the Pd, as seen in the STEM images, continuously and gradually migrated inward toward the Au-seed. This nucleated a Pd grain on the Au seed at 20 min; the grain eventually proliferated to a size of 4.9(±0.8) nm (Fig. 2 and Fig. S6, ESI†). It can be deduced that the reduced Pd was gradually released inward from the hollowed silica, which was continuously bored from within, and deposited on the pre-existing Au nanocrystal in the internal cavity. When the preparation of (Au/Pd)@h-SiO2 was attempted using an increased content of silica-incorporated Pd2+ ions, the Pd grain of the resulting Au/Pd-core-nanocrystal was found to increase in size, while maintaining the size of the Au grain (Fig. S7, ESI†).
 | ||
| Fig. 2 TEM (upper) and HRTEM (lower) images of samples isolated during the reaction of the (Fe3O4/Au)@(SiO2/Pd2+) with NaBH4 at (a) 10, (b) 20, (c) 30, and (d) 60 min, respectively. Insets: EDX elementary maps for showing the distribution of Au (red) and Pd (yellow) elements. | ||
The nanorattle-like structure of the (Au/Pd)@h-SiO2, bearing a catalytically active Pd grain as part of the internal core, signifies the formation of the targeted nanoreactor framework which guides the reduction of the Ni2+–hydrazine complex to occur inside the cavity, thus spatially confining the synthesis of Ni nanocrystals. A pre-mixed aliquot of NiCl2 and hydrazine was added to an aqueous suspension containing the (Au/Pd)@h-SiO2 in a capped vessel with constant stirring at 45 °C.7a The color of the suspension slowly changed from violet to light brown during the initial 20 min of the reaction, and then instantly changed to black with effervescence. The TEM and STEM images of the isolated black powder after a reaction time of 1.5 h revealed the confined growth of a 23(±3) nm Ni nanocrystal exclusively on the inside of the hollow cavity, creating a Ni@SiO2 core@shell structure in which a reduced Ni grown on the Au/Pd core was compacted within the porous silica nanoshell (Fig. 3a). HRTEM and XRD analyses confirmed the formation of a polycrystalline Ni nanoparticle with a face-centered-cubic (fcc) crystalline phase (Fig. 3b and c). Field dependent magnetization measurements show Ni@SiO2 exhibited superparamagnetic and ferromagnetic behaviors at 300 K and 5 K, respectively (Fig. S8, ESI†). The time-course TEM images showed that 24% of the hollow nanoreactors were filled by the Ni growth at 20 min, and as the reaction proceeded, the number of (Au/Pd)@h-SiO2 that transformed into Ni@SiO2 increased, reaching a conversion yield of 94% at 1.5 h (Fig. S9, ESI†). These observations suggest that the nucleation of the Ni was initiated on the surface of the Pd grain, which catalyzed the decomposition of the Ni2+−hydrazine complexes, and the subsequent crystal growth was significantly boosted on the Ni nuclei.
 | ||
| Fig. 3 (a) TEM, (b) HRTEM and STEM-HAADF (inset) images and (c) XRD pattern of the Ni@SiO2. The red line in (c) shows the position of the reflections corresponding to the fcc Ni phase (JCPDS Card No. 01-1260). | ||
To prove the effectiveness of the synthesized Ni@SiO2 in catalysis, we examined its catalytic performance in the hydrolytic dehydrogenation of NH3BH3 (ammonia borane, AB), which is required in high-performance hydrogen storage and generation systems.8Fig. 4a displays time-course curves of the hydrogen released of AB (2 M, 2 mL) during hydrolysis at room temperature by the Ni@SiO2 catalyst. The experiment was conducted with various Ni/AB molar ratios from 0.064 to 0.032, which generated stoichiometric amounts of hydrogen (67 mL, 3 mmol) in less than 15 min. The reaction rate of each curve was almost linearly dependent on the Ni/AB molar ratio, resulting in consistent turnover frequency (TOF) values among the curves. The average TOF of the Ni@SiO2 catalyst (8.0 min−1), calculated on the basis of the total moles of Ni, is somewhat lower than the reported highest value (30.7 min−1) for 6.5 nm-sized surfactant-free Ni nanoparticles.8b However, when defined as the moles of hydrogen generated per unit surface area of Ni nanocrystals, the TOF of the Ni@SiO2 catalyst (4.6 × 10−18 mol min−1 nm−2) is considered to be comparable to the highest reported value (5.02 × 10−18 mol min−1 nm−2), which implies high catalytic activity and well-exposedness of the surfactant-free surface of the Ni nanocrystal grown inside the porous silica nanoshell. Reuse of the magnetically recovered Ni@SiO2 did not result in a significant decline in the catalytic activity over five consecutive runs (Fig. 4b). The usefulness of the Ni@SiO2 catalyst was investigated further in the industrially important catalytic reduction of nitroarenes, which was recently carried out by using a Ni nanocatalyst in an aqueous solution at room temperature and using environment-friendly hydrazine as a reducing agent (Table S1, ESI†).9 A trial reaction of p-nitroaniline (0.1 M) and hydrazine (4 M) with 15 mol% of Ni@SiO2 was carried out in water at room temperature. The product, p-diaminobenzene, was afforded in a quantitative yield within 30 min, and this showed a higher TOF (13 h−1) than that of the reported ca. 10 nm-sized Ni nanoparticles (10 h−1). The Ni@SiO2 catalysts were well dispersed in the stirred reaction suspension, and after the reaction was completed, they could be readily retrieved using a small magnet (Fig. S10, ESI†). The reusability tests showed that the magnetically recovered catalyst retained most of its initial activity in terms of conversion yield and selectivity over three consecutive reactions. The same reaction performed using various functionalized nitroarenes also led to the complete conversion of substrates in less than 4 h. More importantly, while catalyzing the transformation of the nitro groups to the amino groups with a high TOF, the Ni@SiO2 did not much affect other functional groups (halogen, ether, carbonyl-, or alkenyl-groups) and predominantly afforded the corresponding substituted anilines with a selectivity higher than 83%. The remarkable chemoselectivity of the Ni@SiO2 toward the reduction of nitro groups may be attributed to the mild reaction conditions, at room temperature and using a using weak reducing agent, which is allowed by the surfactant-free catalytic surface of the Ni nanocrystals protected by the porous silica shell.
 | ||
| Fig. 4 Plots of volume of hydrogen generated vs. time for catalytic hydrolysis of AB (a) at various concentrations of the Ni@SiO2 catalyst and (b) during the recycling experiment. | ||
In addition to the synthesis of the Ni nanocrystals, the hollow nanoreactor engineered in this study was also applicable to the fabrication of alloy nanocrystals.10 Therefore, an additional injection of a mixture of CoCl2 and hydrazine into the Ni-growing suspension containing the (Au/Pd)@h-SiO2 and Ni2+–hydrazine complex led to the co-reduction of Co2+ together with Ni2+. This led to the consequent growth of a Ni/Co alloy nanocrystal inside the cavity, producing a (Ni/Co)@SiO2 nanosphere with a core@shell-type structure (Fig. S11, ESI†). The control reaction with Co2+– or Fe2+–hydrazine complexes and the (Au/Pd)@h-SiO2 suspension did not generate any reduced Co or Fe species, implying that the pre-grown Ni surface is responsible for reducing the Co2+ complex and forming the Ni/Co alloy. Meanwhile, the successive addition of CuCl2 solution and hydrazine to the (Au/Pd)@h-SiO2 suspension led to the homogeneous nucleation and the generation of large Cu particles at the outside solution of the hollow nonreactor. The annealing process of the synthesized (Ni/Co)@SiO2 at temperatures as high as 700 °C led to an alteration in the morphology and crystallinity of the Ni/Co nanocrystal coated by the thermally stable silica shells, hence preserving their nanocrystalline nature (Fig. S11, ESI†). This allowed the post-synthetic modification of the hollow nanoreactor-synthesized nanocrystals.
In summary, a hollow nanoreactor suitable for templating the synthesis of less reducible and, therefore, less-confinable Ni nanocrystals was developed through a seed-engineering strategy which involved introducing a catalytically active material on the seed entrapped within a hollow silica nanoshell. The Au/Pd-heterojunction-nanocrystal-bearing nanoreactor demonstrated targeted reduction of Ni2+–hydrazine complexes and consequent growth of the Ni nanocrystals within the confines of its cavity. The usefulness of the nanoreactor-synthesized Ni nanocrystals was verified by catalyzing the hydrogen generation of NH3BH3 and chemoselective reduction of nitroarenes in the aqueous phase and at room temperature. Moreover, we demonstrated the extendable utility of the hollow nanoreactors in the synthesis and post-modification of alloy nanocrystals.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (2011-0017377).
Notes and references
- For general reviews on yolk@shell-type nanoparticles, (a) J. Liu, S. Z. Qiao, J. S. Chen, X. W. Lou, X. Xing and G. Q. Lu, Chem. Commun., 2011, 47, 12578–12591 RSC; (b) X.-J. Wu and D. Xu, Adv. Mater., 2010, 22, 1516–1520 CrossRef CAS; (c) Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 3621–3638 CrossRef CAS.
- For reviews on hollow nanoreactor-based catalytic systems, (a) Y. Li and J. Shi, Adv. Mater., 2014, 28, 3176–3205 CrossRef PubMed; (b) X. Li, Y. Yang and Q. Yang, J. Mater. Chem. A, 2013, 1, 1525–1535 RSC; (c) M. Pérez-Lorenzo, B. Vaz, V. Salgueiriño and M. A. Correa-Duarte, Chem. – Eur. J., 2013, 19, 12196–12211 CrossRef PubMed; (d) Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816–1824 CrossRef CAS PubMed.
- Recent examples of hollow nanoreactor-based catalytic systems, (a) G.-H. Wang, J. Hilgert, F. H. Richter, F. Wang, H.-J. Bongard, B. Spliethoff, C. Weidenthaler and F. Schüth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS PubMed; (b) S. M. Kim, M. Jeon, K. W. Kim, J. Park and I. S. Lee, J. Am. Chem. Soc., 2013, 135, 15714–15717 CrossRef CAS PubMed; (c) X. Fang, Z. Liu, M.-F. Hsieh, M. Chen, P. Liu, C. Chen and N. Zheng, ACS Nano, 2012, 6, 4434–4444 CrossRef CAS PubMed; (d) Y. Yang, X. Liu, X. Li, J. Zhao, S. Bai, J. Liu and Q. Yang, Angew. Chem., Int. Ed., 2012, 51, 9164–9168 CrossRef CAS PubMed.
- (a) M. Xiao, C. Zhao, H. Chen, B. Yang and J. Wang, Adv. Funct. Mater., 2012, 22, 4526–4532 CrossRef CAS; (b) M. Sanlés-Sobrido, M. Pérez-Lorenzo, B. Rodríguez-González, V. Salgueiriño and M. A. Correa-Duarte, Angew. Chem., Int. Ed., 2012, 51, 3877–3882 CrossRef; (c) J. Lian, Y. Xu, M. Lin and Y. Chan, J. Am. Chem. Soc., 2012, 134, 8754–8757 CrossRef CAS PubMed; (d) S. Ding, J. S. Chen, G. Qi, X. Duan, Z. Wang, E. P. Giannelis, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 21–23 CrossRef CAS PubMed; (e) L. Tan, D. Chen, H. Liu and F. Tang, Adv. Mater., 2010, 22, 4885–4889 CrossRef CAS PubMed.
- (a) T.-L. Ha, J. Shin, C. W. Lim and I. S. Lee, Chem. – Asian J., 2012, 7, 36–39 CrossRef CAS PubMed; (b) K. M. Yeo, S. H. Choi, J. W. Kim and I. S. Lee, Angew. Chem., Int. Ed., 2011, 50, 745–748 CrossRef CAS PubMed; (c) K. M. Yeo, J. Shin and I. S. Lee, Chem. Commun., 2010, 46, 64–66 RSC.
- (a) C.-J. Liu, J. Ye, J. Jiang and Y. Pan, ChemCatChem, 2011, 3, 529–541 CrossRef CAS; (b) H.-L. Jiang and Q. Xu, Catal. Today, 2011, 170, 56–63 CrossRef CAS PubMed; (c) F. Alonso, P. Riente and M. Yus, Acc. Chem. Res., 2011, 44, 379–391 CrossRef CAS PubMed.
- (a) M. Grzelczak, J. Pérez-Juste, B. Rodríguez-González, M. Spasova, I. Barsukov, M. Farle and L. M. Liz-Marzán, Chem. Mater., 2008, 20, 5399–5405 CrossRef CAS; (b) M. Grzelczak, B. Rodríguez-González, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2007, 19, 2262–2266 CrossRef CAS; (c) S.-H. Wu and D.-H. Chen, J. Colloid Interface Sci., 2003, 259, 282–286 CrossRef CAS; (d) M. S. Hegde, D. Larcher, L. Dupont, B. Beaudoin, K. Tekaia-Elhsissen and J. M. Tarascon, Solid State Ionics, 1997, 93, 33–50 CrossRef.
- (a) M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 RSC; (b) P.-Z. Li, A. Aijaz and Q. Xu, Angew. Chem., Int. Ed., 2012, 51, 6753–6756 CrossRef CAS PubMed; (c) Ö. Metin, V. Mazumder, S. Özkar and S. Sun, J. Am. Chem. Soc., 2010, 132, 1468–1469 CrossRef PubMed.
- (a) R. K. Rai, A. Mahata, S. Mukhopadhyay, S. Gupta, P.-Z. Li, K. T. Nguyen, Y. Zhao, B. Pathak and S. K. Singh, Inorg. Chem., 2014, 53, 2904–2909 CrossRef CAS PubMed; (b) H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- (a) A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652–676 CrossRef CAS; (b) D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: More experimental data including TEM images. See DOI: 10.1039/c4cc07306a |
| This journal is © The Royal Society of Chemistry 2015 |