
Hexaalkylguanidinium salts as ionic liquids – applications in titanium and aluminium alcoholate assisted synthesis
Maria Arkhipova,
Svetlana Eichel and
Gerhard Maas*
Institute of Organic Chemistry I, University of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: gerhard.maas@uni-ulm.de; Fax: +49-731-5022803
First published on 27th October 2014
Abstract
The solubility of titanium and aluminium alcoholates and of titanium tetrakis(trimethylsilanolate) in several hexaalkylguanidinium-based room temperature ionic liquids was screened. The solvent/solute combinations which displayed the highest alcoholate solubility and stability were applied as Lewis-acidic catalytic media for several dehydrating cyclocondensations: lactamisation of ω-aminocarboxylic acids, direct amidation of carboxylic acids, synthesis of oxazolines from carboxylic acids and 2-aminoethanol, lactonisation of 6-hydroxyhexanoic acid, and Paal–Knorr synthesis of pyrroles.
Introduction
Over the past two decades ionic liquids (ILs) have received much attention as “green” replacements for volatile organic solvents. Reaction types successfully performed in ILs include Diels–Alder and Friedel–Crafts reactions, olefin hydrogenation, hydroformylation and oligomerization, and transition-metal catalysed C,C coupling reactions, to name just a few.1–10 In the domain of catalytic reactions, ILs may not only serve as solvents, but they can also act as catalysts, ligands and as stabilising agents for catalysts or reaction intermediates. They can influence chemo-, regio-, stereo- and enantioselectivity, enhance the reaction rate, increase the product yield, and facilitate the separation of products and catalyst recovery.11,12 Usually, extraction or distillation of the product is applied, so that the catalyst is retained in the ionic liquid phase. This allows then multiple use of the catalyst/solvent system, lowering the costs and disposal of chemicals. Because many transition metal complexes are well soluble in ionic liquids, the ILs are particularly suited for homogeneous catalysis and isolation of the catalyst in biphasic systems.The ionic liquids used as reaction media are usually based on 1,3-dialkylimidazolium, N-alkylpyridinium, tetraalkylammonium, N,N-dialkylpyrrolidinium, and tetraalkylphosphonium cations. Strangely enough, hexaalkylguanidinium-based ILs are often ignored in review articles and book chapters. As alternative reaction media, all aspects named above also apply to hexaalkylguanidinium-based ILs, and indeed some applications as reaction media in organic synthesis and catalysis have been reported. Guanidinium-based phosphotungstates were used for epoxidation of olefins,13 guanidinium acetates as media for the palladium-catalysed Heck reaction,14 and Lewis acidic guanidinium ILs for aminolysis of epoxides.15 Hexaalkylguanidinium ILs as reaction media have been reported for the oxidation of benzyl alcohols,16 nucleophilic substitution reactions,17 CO2 fixation,18 Sharpless dihydroxylation19,20 and asymmetric aldol reaction.21,22 Intramolecular carbenoid C–H insertion of an α-methoxycarbonyl-α-diazoacetamide was successfully performed with rhodium or ruthenium catalysts in hexaalkylguanidinium triflates.23
Hexaalkylguanidinium salts show high chemical stability, due to the superior charge delocalisation in the cation, and are thermally remarkably stable.24,25 Another advantage of guanidinium ionic liquids consists in the possibility to vary up to six substituents to adjust the properties of the IL to a specific task.24–27 In this paper we describe a new application of guanidinium-based ionic liquids as reaction media containing a titanium or aluminium alcoholate as a mild Lewis-acidic catalyst for several (cyclo)condensation reactions.
Titanium alkoxides (Ti(OR)4 and MeTi(OR)3) have been widely applied as Lewis acids, for example in aldol reactions,28,29 in Kulinkovich cyclopropanation,30 and in the formation of lactams. Titanium tetraisopropoxide is also a reagent for the Sharpless asymmetric epoxidation of allylic alcohols.31
Results and discussion
Synthesis of guanidinium-based ionic liquids and solubility of titanium and aluminium alcoholates
A number of guanidinium-based ionic liquids with different anions were synthesised (Table 1). Several methods of synthesis of guanidinium salts have already been described by Kantlehner,26 Afonso,24 Maas,25 and their coworkers. These protocols have been applied and adopted for the synthesis of new representatives of the guanidinium family.
|
|||||
|---|---|---|---|---|---|
| Compound | R1 = R2 | R3 = R4 | R5 = R6 | X | |
| 1a | [N11N22N44Gu]OTf | Me | Et | Bu | OSO2CF3 |
| 1b | [N11N22N1O21O2Gu]OTf | Me | Et | MeO(CH2)2 | OSO2CF3 |
| 2a | [N11N22N44Gu]NTf2 | Me | Et | Bu | N(SO2CF3)2 |
| 2b | [N22N44N66Gu]NTf2 | Et | Bu | Hex | N(SO2CF3)2 |
| 2c | [N11N22N1O21O2Gu]NTf2 | Me | Et | MeO(CH2)2 | N(SO2CF3)2 |
| 2d | [N11N11N1O21O2Gu]NTf2 | Me | Me | MeO(CH2)2 | N(SO2CF3)2 |
| 2e | [N11N11N66Gu]NTf2 | Me | Me | Hex | N(SO2CF3)2 |
| 2f | [N11N66N66Gu]NTf2 | Me | Hex | Hex | N(SO2CF3)2 |
| 3a | [N22N44N66Gu]N(CN)2 | Et | Bu | Hex | N(CN)2 |

Solubility tests (Table 2) showed that aluminium isopropoxide is not soluble in hexaalkylguanidinium ILs at room temperature and can be dissolved only at 120–130 °C, whereas titanium isopropoxide dissolves readily in liquid guanidinium bis(triflamides) with longer alkyl chains in the cation at room temperature. Interestingly, the metal alcoholates are only sparingly soluble in the ether-functionalised guanidinium salts, although it was expected that Lewis acid/Lewis base interactions would favour the dissolution. For comparison we also checked some commonly used salts, such as 1-butyl-3-methylimidazolium and hexyl-trimethylammonium bis(triflamides). It was noted that several of the guanidinium-based ILs are better solvents for titanium isopropoxide than the examined imidazolium and ammonium salts.
| IL | Ti(OiPr)4 | Al(OsecBu)3 | Al(OiPr)3 | |
|---|---|---|---|---|
| a Mass-to-volume ratio in those cases, where suspensions or two phases were formed. | ||||
| 1a | [N11N22N44Gu]OTf | 0.17 (turbid) | 0.01 (turbid) | |
| 1b | [N11N22N1O21O2Gu]OTf | 0.05 (turbid) | 0.12 (turbid) | 0.16 (turbid) |
| 2a | [N11N22N44Gu]NTf2 | 0.11 (clear) | 0.06 (turbid) | |
| 0.40 (turbid) | ||||
| 2b | [N22N44N66Gu]NTf2 | 0.84 (clear) | 0.37 (turbid) | 0.44 (turbid) |
| 3.00 (turbid) | ||||
| 2c | [N11N22N1O21O2Gu]NTf2 | 0.31 (turbid) | 0.01 (turbid) | |
| 2d | [N11N11N2O12O1Gu]NTf2 | 0.17 (turbid) | 0.24 (turbid) | 0.13 (turbid) |
| 2e | [N11N11N66Gu]NTf2 | 0.67 (clear) | ||
| 1.00 (turbid) | ||||
| 2f | [N11N66N66Gu]NTf2 | 0.53 (clear) | 0.14 (turbid) | 0.15 (turbid) |
| 3a | [N22N44N66Gu]N(CN)2 | 0.44 (clear) | 0.32 (turbid) | |
| [BMIm]NTf2 | 0.20 (turbid) | 0.06 (turbid) | ||
| [Me3HexN]NTf2 | 0.39 (turbid) | 0.04 (turbid) | ||
Solutions of titanium and aluminium isopropoxides in N,N-dibutyl-N′,N′-diethyl-N′′,N′′-dihexylguanidinium bis(trifluoromethylsulfonyl)imide (2b) or more easily obtainable N,N-dihexyl-N′,N′,N′′,N′′-tetramethylguanidinium bis(trifluoromethylsulfonyl)imide (2e), as the most concentrated and stable ones, were chosen for further investigations and applied as mild catalytic media for some typical dehydrating condensation reactions. Our attention was attracted mostly by bis(triflamide)-ILs, which are among the most hydrophobic ILs. This could have a positive impact on the stability and therefore activity of the catalyst and could facilitate the recovery and reuse of the IL.
Lactamisation reactions
Cyclocondensation of ω-amino acids in the presence of titanium-based Lewis acids is one of the ways to obtain macrocyclic lactams.32 P. Helquist and coworkers have investigated the lactamisation reaction of some amino acids (Scheme 1) and have applied the developed method to the synthesis of streptogramin A antibiotics.32,33 | ||
| Scheme 1 Lactamisation of ω-amino acids. | ||
We have studied the cyclisation of some ω-amino acids in guanidinium-based ILs with the goal to find a replacement for the less desirable solvent dichloroethane34 that was used in the reported procedure. Additionally we expected that the reaction could be run at higher temperature in the IL and the amount of the Ti-alkoxide catalyst could so be reduced considerably. We found that a complete conversion could be achieved within 3–5 h at 120 °C with only 5–10 mol% of catalyst.
Initial screening and optimisation reactions were carried out with 4-aminobutyric acid in a clear solution of Ti(OiPr)4 in [N22N44N66Gu]NTf2 (2b) (Table 3). The nature of guanidinium-based IL does not influence significantly the reaction time ([N22N44N66Gu]NTf2 (2b), [N11N22N44Gu]NTf2 (2a), [N11N11N66Gu]NTf2 (2e) and [N22N44N66Gu]N(CN)2 (3a) were also tested as solvents and showed similar results), and isolation of the product by bulb-to-bulb distillation was more effective than an extractive work-up procedure.
| Ti(OiPr)4, mol% | Temperature, °C | |||
|---|---|---|---|---|
| 40 | 80 | 100 | 120 | |
| 0 | — | |||
| 5 | 5 h | |||
| 10 | >10 h | 3 h | ||
| 20 | 4 h | 2 h | ||
| 50 | >8 h | 3 h | 2 h | |
The results obtained for the cyclisation of various ω-amino acids (4-aminobutyric acid, 4-(methylamino)butyric acid, 5-aminovaleric acid, 6-aminocaproic acid) under optimised IL conditions (10 mol% of catalyst, 120 °C, distillative work-up) and under “traditional” conditions (halogenated organic solvent) are compared in Table 4. For comparison, the experiments using Ti(OiPr)4 as a catalyst were also carried out with the same quantity of catalyst in an organic solvent (1,2-dichloroethane).
| Entry | n, R | Ti(OiPr)4 (50 mol%), DCEc, 84 °C | Ti(OiPr)4 (10 mol%), DCE, 84 °C | Ti(OiPr)4 (10 mol%), IL, 120 °C | Ti(OSiMe3)4 (10 mol%), IL, 120 °C | Al(OiPr)3 (10 mol%), IL, 120 °C |
|---|---|---|---|---|---|---|
| a DCE = 1,2-dichloroethane; IL: [N11N11N66]NTf2 (2e) or [N22N44N66]NTf2 (2b).b ILs 2e and 2b gave similar yields under the same conditions.c Ref. 32.d For comparison: a yield of 86% was achieved when the reaction was run in IL 2e at 84 °C for 3 h. | ||||||
| 1 | 2, H | 93% (3 h)d | 85% (3 h) | 85% (3 h) | 88% (3 h) | 89% (3 h) |
| 2 | 2, Me | 85% (5 h) | 41% (5 h) | 82% (3 h) | 88% (1 h) | 84% (1 h) |
| 3 | 3, H | 75% (4 h) | 87% (4 h) | 82% (2 h) | 94% (2 h) | 94% (3 h) |
| 4 | 4, H | 35% (26 h) | 9% (26 h) | 58% (26 h) | 62% (26 h) | 26% (26 h) |
In most cases the lactam yield was increased along with the expected reduction in reaction time. The system metal alcoholate/guanidinium IL was especially effective for the synthesis of the N-substituted lactam (the reaction duration was up to five times lower, entry 2) and for ε-caprolactam (during the same time the yield was almost doubled, entry 4).
Beside titanium isopropoxide other, somewhat less moisture sensitive, Lewis acids were tested. Titanium trimethylsilanolate turned out to be a bit more catalytically active in comparison to isopropanolate. Titanium 2-ethylhexanolate, which is more viscous than the isopropanolate and trimethylsilanolate, is somewhat less active than the others. This is why the reactions with titanium 2-ethylhexanolate were not extended. Aluminium isopropanolate did not show good solubility, but its catalytic activity proved to be quite similar to titanium trimethylsilanolate with one exception (synthesis of caprolactam). In order to accelerate the reaction we added molecular sieves 4 Å to adsorb the water produced in the condensation reaction and to prevent the hydrolysis of the catalyst. Notably, this had no impact on the reaction – for the reasons that became clear later (vide infra).
Another major advantage of our IL strategy is given by the fact that the IL/catalyst system could be reused in subsequent cycles with little or no reduction in yield (Table 5). To this end, the produced lactam was separated from the reaction mixture by vacuum distillation, and the IL/catalyst system, which contained a white precipitate, was used again for the next reaction cycle.
| n, R | Catalyst | Yields in four cycles, % |
|---|---|---|
| a For reaction conditions, see Table 4. | ||
| 2, H | Ti(OiPr)4 | 85 – 85 – 94 – 85 |
| 2, H | Ti(OSiMe3)4 | 88 – 91 – 89 – 85 |
| 2, H | Ti(O-2-ethylhexyl)4 | 75 – 75 – 54 – 50 |
| 2, H | Al(OiPr)3 | 89 – 89 – 89 – 95 |
| 2, Me | Ti(OiPr)4 | 82 – 92 – 94 – 77 |
| 2, Me | Ti(OSiMe3)4 | 88 – 90 – 87 – 94 |
| 3, H | Ti(OiPr)4 | 82 – 79 – 74 – 72 |
| 3, H | Ti(OSiMe3)4 | 94 – 98 – 96 – 96 |
| 3, H | Al(OiPr)3 | 94 – 96 – 95 – 95 |
Moreover, the possibility of phase separation into a product phase and catalyst/IL phase was investigated, as this would facilitate the work-up procedure. To this end, the concentration of the amino acid in the usual amount of IL was increased. Unfortunately, the formed lactam did not appear as a separate phase, and a distillative work-up was required. A three times higher concentration of 4-aminobutanoic acid in the ionic liquid increased the yield of butyrolactam from 85 to 96%.
To test the scope of the developed method, the cyclodehydration of 11-aminoundecanoic acid was tried. The experiment failed, perhaps mainly due to the low solubility of the amino acid in the IL. Cyclisation of the optically active L-lysine was also attempted, but the reaction with 10 mol% of Ti(OiPr)4 at 120 °C took almost two days and yielded less than 18% of α-amino-ε-caprolactam, not in pure form. The reaction with 100 mol% of the same catalyst gave 27% of impure product, so the racemisation of the product could not be checked.
Obviously, the lactamisation is catalysed by metal alcoholates or species derived therefrom. Without catalyst no reaction takes place, also not after 5 h at 120 °C (Table 3). NMR spectra of the reaction mixture before the second cycle no longer contained signals of the isopropyl substituent. The reason could be the hydrolysis of Ti(OiPr)4 with possible formation of TiO2. Nevertheless, the solid species formed (a precipitate appeared several minutes after the start of the reaction) is catalytically active in the subsequent cycles.
Some experiments were performed to understand the nature of the catalyst. To this end, several mixtures containing a white precipitate, which remained after the product isolation by bulb-to-bulb distillation or extraction, were diluted in dichloromethane, filtered, and the precipitate was dried. NMR and IR spectra indicated the presence of 4-aminobutanoic acid in admixture with TiO2. This powder was washed with water to remove the acid. X-ray diffraction (XRD) measurements of the solid so obtained showed that it was only amorphous material. After calcination at 550 °C for 4 h, the XRD spectrum of pure anatase was obtained (Fig. 1).
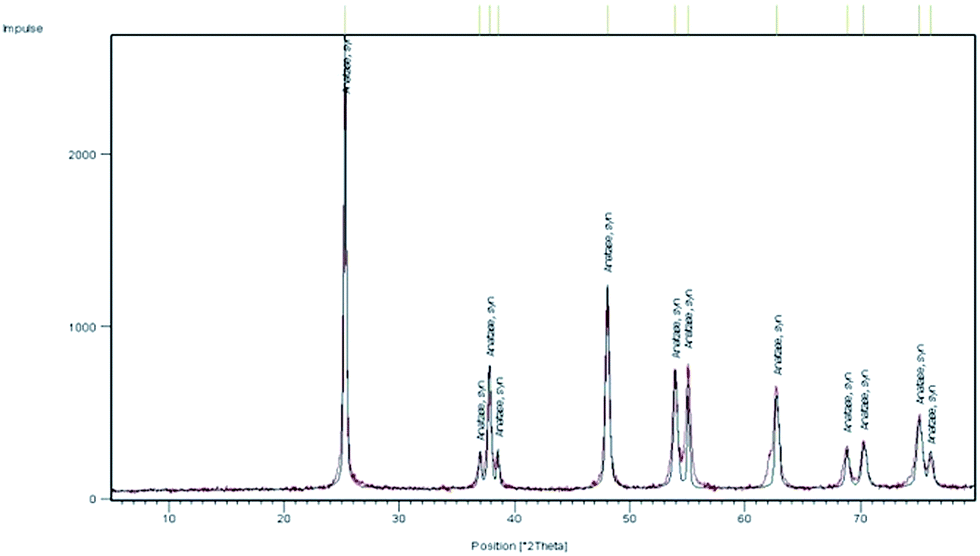 | ||
| Fig. 1 XRD spectrum of the calcinated catalytically active powder. | ||
Actually, it was not surprising to obtain anatase particles as a by-product of the reaction. It is well known that diverse ionic liquids are effective for the synthesis of TiO2 anatase nanoparticles from titanium alcoholates.35–38 Therefore, it was also interesting to get information on the size of particles obtained after the first reaction cycle. The TEM images of the anatase (obtained after calcination) are presented in Fig. 2 (left). Most particles are nanoparticles with a size of 30–60 nm. The image on the right shows the particles obtained after filtration from the catalyst/IL system. It is seen that they are bigger in size, the majority of them is conglomerated, and the single particles have a very porous structure.
 | ||
| Fig. 2 TEM images of anatase particles (left, length scale corresponds to 30 nm) and catalyst particles (right, length scale corresponds to 60 nm). | ||
Is the titanium dioxide the active catalytic species? To answer this question the experiment (lactamisation of 4-aminobutanoic acid in IL 2e) with 10 mol% of anatase as a catalyst was carried out, but no reaction was observed. Notably, after addition of some drops of water the reaction set in but proceeded very slowly. On the other hand, when the powder obtained from the reaction mixture by filtration (when Ti(OiPr)4 was used as catalyst) was applied one more time, the reaction was fast as usual. With the precipitate (TiO2) washed with water only a slow reaction was observed. There was no reaction in the IL sample remaining after filtration. So the possible catalyst could be (a) the freshly precipitated Ti(OH)4, its dimers or trimers39–41 or freshly obtained TiO2 (further written as TiO2*), (b) the mentioned Ti species complexed with amino acid. For clarification, the lactamisation of 4-aminobutanoic acid was carried out in the presence of a TiO2*/amino acid complex, which had been prepared separately from Ti(OiPr)4 and 4-aminobutanoic acid in isopropanol at reflux for 3 h with subsequent filtration of the catalyst. Under analogous reaction conditions (IL 2e, 10 mol% of catalyst, 120 °C, 3 h), a conversion of 58% was determined by 1H NMR spectroscopy with the TiO2*/amino acid catalyst, compared to 99% with Ti(OiPr)4 as catalyst. Further experiments with freshly precipitated TiO2* (from Ti(OiPr)4/H2O in isopropanol) and in situ precipitated TiO2* in IL (from Ti(OiPr)4/H2O in IL 2e) showed the same catalytic activity as in the original reaction.
Concluding the obtained results, it seems that in the first reaction cycle, the reaction starts with Lewis acidic Ti(OiPr)4 as the catalyst. Ti(OiPr)4, which is sensitive even to traces of water, is hydrolysed by water formed as a side product to yield Ti(OH)4 and finally titanium dioxide. It is also known that titanium alcoholates and titanium hydroxide are prone to form oligomers.39–41 So a number of species could act as a catalyst, but the most likely under our conditions (elevated temperature, vacuum distillation) is titanium dioxide in the form of nanoparticles with porous structure (Fig. 2, right). Water is known to adsorb both associatively and dissociatively (as HO−) on the surface of titania containing reduced Ti cations and to oxidise the surface,42,43 thus blocking the coordinatively unsaturated sites at the TiO2 surface. The amino acid could be adsorbed on the surface of titanium dioxide, where the reaction takes place. Moreover, TiO2 reveals Lewis acidic properties, as well as Lewis basic properties.44 In any case, the chemical behaviour of both Ti4+ (Lewis acid) and O2− (Lewis base) ions depends on the surface structure.45 It seems that in this reaction we deal with mildly Lewis acidic titanium dioxide as catalytic species. Moreover, Lewis acid activity of TiO2 nanoparticles has already been reported.46,47
With the application of hexaalkylguanidinium bis(triflamides) as solvents we managed: (a) to lower the amount of the catalyst significantly, (b) to reduce the reaction time (since the IL allows a higher reaction temperature), (c) to use an easier work-up procedure (vacuum distillation of the product from the reaction mixture), (d) to carry out several cycles without regeneration of IL and without extra addition of new catalyst before the next cycle, (e) to improve yields in some cases and (f) to avoid the use of organic solvents and some disadvantages associated with them (e.g. volatility, flammability, disposal of chlorinated solvents etc.).
Direct amidation of carboxylic acids
After successful intramolecular lactamisation we investigated intermolecular amidation in a guanidinium-IL reaction medium. Although various methods for the direct amide synthesis exist,48,49 still no procedures of this important reaction are known to be performed in an ionic liquid according to the principles of Green Chemistry.50For the optimisation purposes the reaction between phenylacetic acid and piperidine furnishing benzamide 4 (Scheme 2) was chosen. The same approach as for the lactamisation reaction was taken: the catalyst quantity and temperature were varied (Table 6), and the product was separated through bulb-to-bulb distillation. These experiments showed the suitability of this system for the investigated reaction, as very good yields could be achieved and only 10–20 mol% of the catalyst was required. For reactions with other substrates the conditions with 20 mol% of Ti(OiPr)4 and at 90 °C were selected. For the synthesis of phenylacetyl piperide, replacement of IL 2e with 1-butyl-3-methylimidazolium bis(triflamide) [BMIM]NTf2 as the solvent did not significantly affect reaction time and yield at several temperatures and catalyst loadings. The results of the direct amidation reactions of phenylacetic acid or benzoic acid with secondary and primary amines are presented in Table 7. For comparison, amide formation from phenylacetic acid and piperidine (at 100 °C), morpholine (at 100 °C) or benzylamine (at 70 °C) in dry THF, in the presence of Ti(OiPr)4 (20–10 mol%) and 4 Å molecular sieves, succeeded in yields of 69, 76 and 91% after chromatographic purification.49
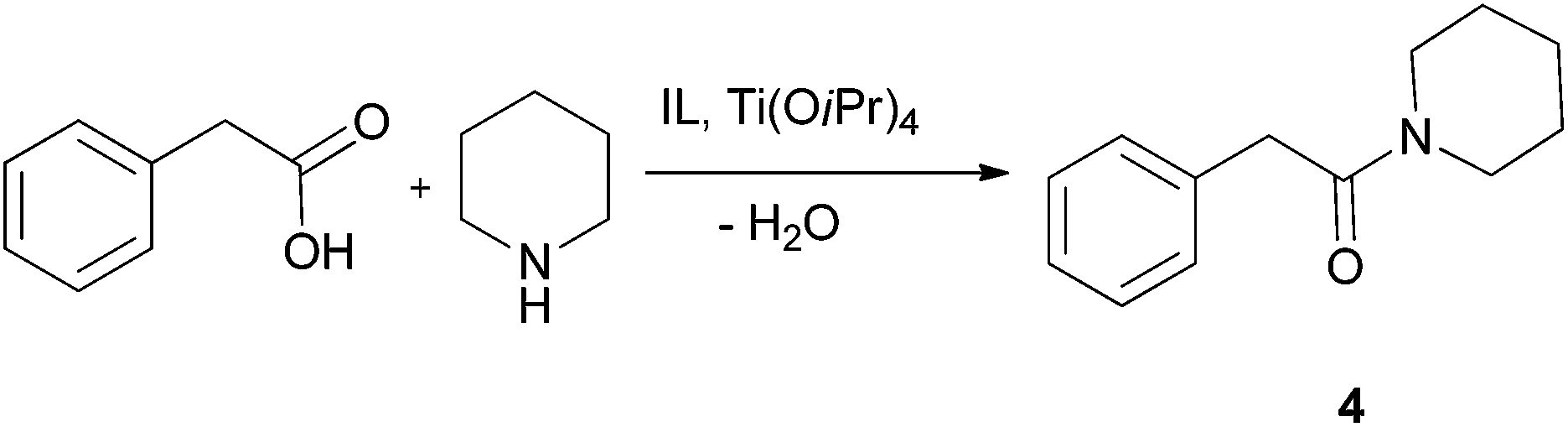 | ||
| Scheme 2 Reaction between phenylacetic acid and piperidine. | ||
| Catalyst, mol% | Temperature, °C | |||
|---|---|---|---|---|
| 77 | 90 | 120 | 200 | |
| 0 | 74% (6 h) | |||
| 10 | 90% (9 h) | 90% (7 h) | 90% (5 h) | 91% (4 h) |
| 20 | 91% (7 h) | 94% (4 h) | 93% (3 h) | 92% (2 h) |
| 50 | 91% (7 h) | 92% (4 h) | 93% (2.5 h) | 94% (2 h) |
| Acid | Amine | ||
|---|---|---|---|
| Piperidine | Morpholine | Benzylamine | |
| Phenylacetic acid | 94% (4 h) | 92% (4 h) | 92% (4 h) |
| Benzoic acid | 94% (5 h) | 92% (6 h) | 92% (7 h) |
As in the case of lactamisation it was desirable to develop a recyclable system for direct amidation in a guanidinium-based IL. The first experiments showed that the second cycle of the reaction required a longer time. To reduce this time, 3 Å molecular sieves were added in the second cycle. Reactions were also performed at higher temperatures than in the first cycle (Table 8). Under the optimal conditions (20 mol% of catalyst at 200 °C), four subsequent cycles could be performed in a short time with good yields (92% – 91% – 91% – 87%).
Although the recyclability of the direct amidation reaction of phenylacetic acid and piperidine in IL has been achieved, the procedure for reuse of the catalyst/IL system was not developed further for other substrates because of the high temperatures needed in the first case. To improve the reaction conditions, more effective Lewis acids may be required.51
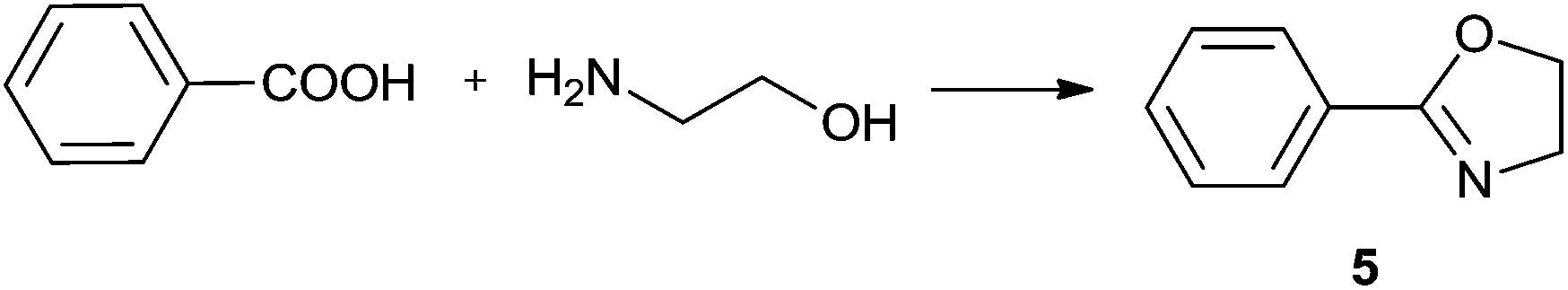
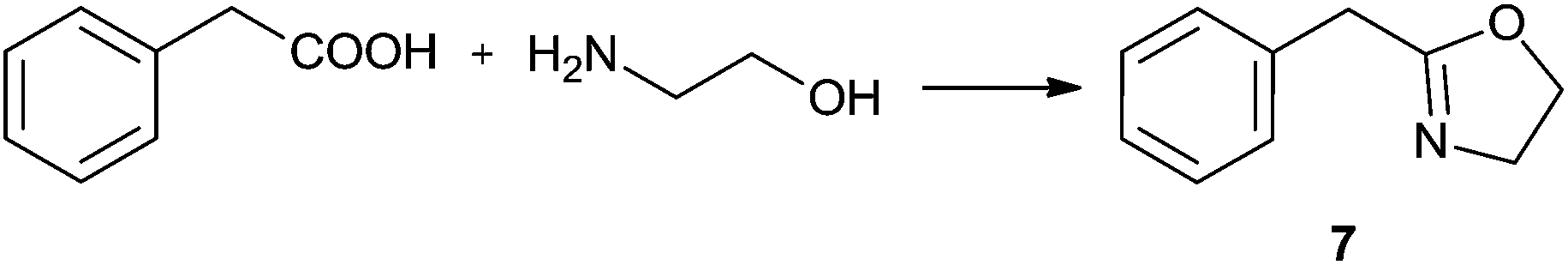
Synthesis of oxazolines
2-Oxazolines are an important class of heterocycles, widely present in natural products. 2-Oxazolines are structural entities in naturally occurring iron chelators, cytotoxic cyclic peptides, and in antiinflammatory, antimitotic and neuroprotective agents.52,53 They are intermediates in drug syntheses.54,55 Oxazolines are well known as protected forms of carboxylic acids as well as amino alcohols and are also widely utilised as chiral auxiliaries. Chiral bis(oxazoline) ligands have broad application in asymmetric hydrosilylation, carbenoid cyclopropanation reaction, Friedel–Crafts reaction, Diels–Alder reaction, aldol addition, Michael reaction, Henry reaction, allylic oxidation, 1,3-dipolar cycloaddition, and so on.56Methods of synthesis of 2-oxazolines have been explored extensively. Some of the cyclocondensations of carboxylic acids with 2-aminoalcohols suffer from using such harsh reagents as thionyl chloride and the necessity of high temperature conditions (up to 230 °C).57,58 Several milder approaches have also been developed, including preferable one-pot procedures. One of the most facile one-pot protocols utilises a tetranuclear zinc cluster in refluxing toluene,59 providing a number of oxazolines in good and excellent yields after chromatographic work-up. Another approach consists in the use of a [BMIm]Cl/InCl3 system, which affords 60–80% yields under mild conditions (60 °C, 3 h).60 Nevertheless, in order to obtain the pure products organic solvents were needed, and nothing was reported about the possibility of recycling the catalyst/IL system.
In order to develop an efficient procedure with a recyclable catalyst system, we explored Ti(OiPr)4/[N11N11N66Gu]NTf2 (2e) as a milieu for the direct dehydrating condensation. By analogy to the lactamisation and direct amidation reactions reported above, this system was expected to catalyse at least the first step in the oxazoline synthesis, namely amide formation (Scheme 3). The application of a thermally more stable guanidinium bis(triflamide) IL (instead of imidazolium chloride) would allow to work at higher temperatures.
 | ||
| Scheme 3 The stepwise formation of 2-phenyl-4,5-dihydro-1,3-oxazole. | ||
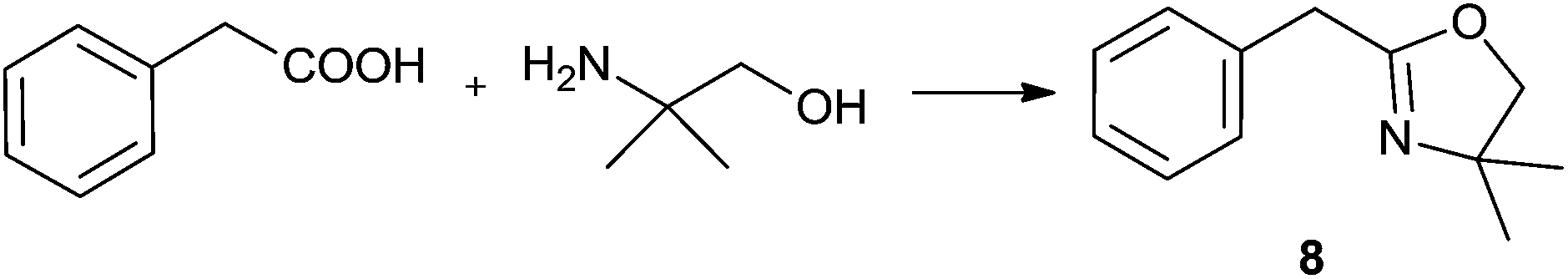
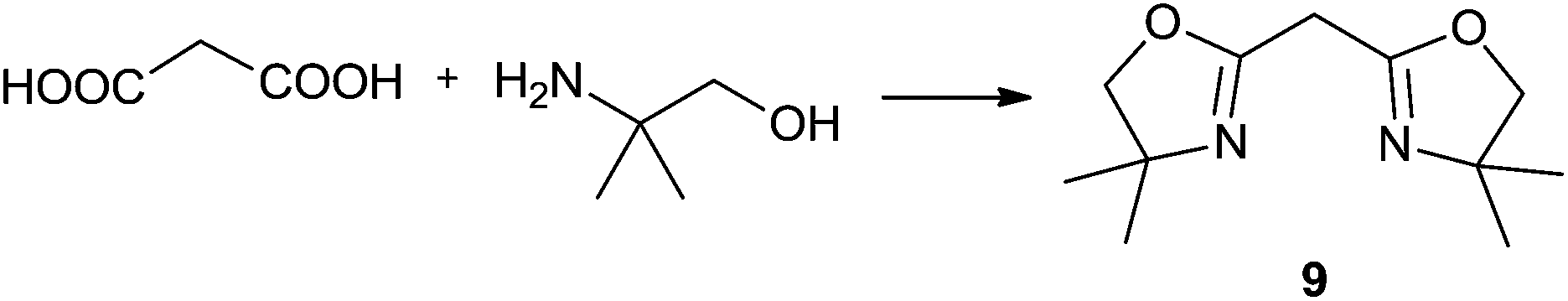
For the initial studies the reactions of benzoic acid with either monoethanolamine (MEA) or 2-amino-2-methyl-1-propanol were taken. In refluxing chlorobenzene the reactions needed 67 h and 120 h, respectively, to afford after chromatographic work-up the oxazolines in yields of only 40 and 50% (Table 9). The first experiments (benzoic acid with MEA) in IL showed that at 120 °C solely N-(2-hydroxyethyl)-benzamide is formed. However, the oxazolines could be obtained after bulb-to-bulb distillation at 200 °C instead of 120 °C during several hours. This means that the cyclic product was formed during distillation. When the whole reaction was conducted at 200 °C, formation of the oxazoline was observed in the NMR spectra, but the product could be isolated only in poor yields. To lower the required reaction time, the reaction mixture was kept at first at 120 °C for the amide formation, and then the temperature was increased to 200 °C. Unfortunately, all attempts led only to poor yields. A higher amount of catalyst (Ti(OiPr)4) accelerated the reaction, but high temperatures were still needed for the oxazoline formation. As a disadvantage, titanium tetraisopropoxide was distilled off together with the product; this means that the catalyst was not immobilised in the IL and therefore could not be used once more. In addition, the product had to be separated from the catalyst, and so the extraction with an organic solvent was unavoidable.
| Solvent | Cat.b, mol% | Conditions | Yield, % | Solvent | Cat.b, mol% | Conditions | Yield, % |
|---|---|---|---|---|---|---|---|
| a IL = [N11N11N66Gu]NTf2 (2e).b Catalyst = Ti(OiPr)4.c Not obtained in pure form; chromatographic purification or distillation was necessary. | |||||||
 |
 |
||||||
| PhCl | 50 | 140 °C/67 h | 40 | PhCl | 50 | 140 °C/120 h | 51 |
| ILa | 50 | 200 °C/20 h | 23c | IL | — | 200 °C/7 h | 18 |
| IL, MS 4 Å | — | 120 °C/1 h + 200 °C/15 h | 0 | IL, MS 4 Å | — | 200 °C/7 h | 49 |
| IL | 250 | 120 °C/2 h + 200 °C/7 h | 52 | IL, MS 4 Å | 50 | 120 °C/1 h + 200 °C/8.5 h | 67 |
 |
 |
||||||
| IL, MS 4 Å | 50 | 200 °C/6 h | 11 | IL, MS 4 Å | 50 | 120 °C/1 h + 200 °C/7 h | 63 |
 |
|||||||
| IL, MS 4 Å | 50 | 120 °C/4 h + 200 °C/7 h | 34 | ||||
This reaction suffers from poor yields and the necessity of high temperatures, likely because the Lewis acid used is not strong enough to activate the carboxylic acid. To improve the reaction conditions, an attempt with MW irradiation was made, but also after several hours only the benzamide was produced. Aluminium isopropoxide and titanium tetrakis(trimethylsilanolate) were found to be even less effective than titanium tetraisopropoxide.
As the reaction required a temperature of 200 °C in all cases, the question arose, whether it was catalysed or was simply promoted by the temperature and/or molecular sieves. In the case with monoethanolamine no reaction took place without the catalyst; in the case of 2-amino-2-methyl-propanol, unexpectedly, a 49% yield was obtained with molecular sieves and 18% without molecular sieves and catalyst. It can be concluded that the catalyst plays only a limited role in this reaction and that the nucleophilicity of the amine is a key factor for the reaction under these conditions.
Nevertheless, with the Ti(OiPr)4/IL system we were able to obtain 34% of bis-oxazoline 9 from dimethyl malonate after fractionating distillation (Table 9); this is still acceptable when considering the traditional multi-step synthesis of bis(oxazolines) involving corrosive acid chlorides or hazardous halogenating reagents.
For the synthesis of oxazolines 6 and 8, which gave the highest yields under optimised reaction conditions, we have also checked the reusability of the catalyst/IL system left after distillative separation of the product. In both cases, the yields of 6 and 8 decreased only slightly over three cycles and furthermore, we observed that the reaction was complete in a shorter time (Table 10).
| Oxazoline | Reaction conditions of the first cycle | Reaction conditions of subsequent cycles | Yields in three subsequent cycles, % |
|---|---|---|---|
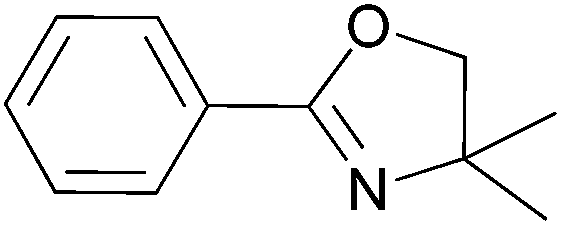 |
IL, 4 Å MS, 50 mol% of Ti(OiPr)4; 1 h at 120 °C, 8.5 h at 200 °C | Catalyst/IL system from the first cycle, 7 h at 200 °C | 67 – 63 – 63 |
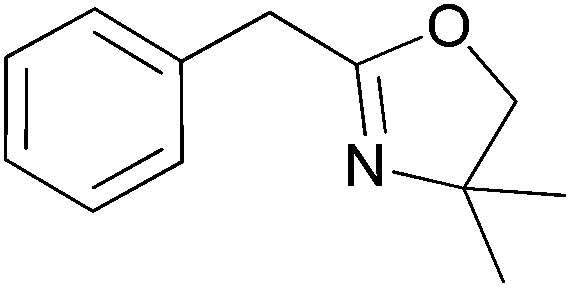 |
IL, 4 Å MS, 50 mol% of Ti(OiPr)4; 1 h at 120 °C, 7 h at 200 °C | Catalyst/IL system from the first cycle, 5 h at 200 °C | 63 – 60 – 59 |
To summarise, satisfactory conditions and yields of the 2-oxazoline synthesis were not achieved. Too high temperatures, low yields of oxazolines in many cases and the presence of the corresponding carboxamide as a side-product do not promote this procedure for a wide application, taking into account that some methods of oxazoline synthesis with high atom economy already exist. However, the low price of titanium tetraisopropoxide can in some cases be more favourable in comparison to more effective but also more expensive catalysts.
Lactonisation reactions
The procedure developed for intramolecular amidation was also applied to the intramolecular esterification of hydroxycarboxylic acids. For the cyclisation of 6-hydroxycaproic acid (Scheme 4) and 15-hydroxypentadecanoic acid it was found, that the reaction could not be completed under the chosen conditions even after several days. For example, treatment of 6-hydroxycaproic acid with 70 mol% of Ti(OiPr)4 at 120 °C gave after one day a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.4 ratio of acid to lactone 10. Unfortunately, increasing the catalyst amount (from 10 to 100 mol%) or the reaction temperature (22 → 120 °C), varying the catalyst (Ti(OiPr)4, Ti(OSiMe3)4, Al(OiPr)3), prolonging the reaction time (up to 5 days) and performing the reaction under MW conditions did not lead to the completion of the reaction, and unconsumed hydroxy acid was distilled together with the product. In all cases the yields were moderate. The methyl ester of 6-hydroxycaproic acid was also tried in this reaction at 40 and 120 °C but without success.
:0.4 ratio of acid to lactone 10. Unfortunately, increasing the catalyst amount (from 10 to 100 mol%) or the reaction temperature (22 → 120 °C), varying the catalyst (Ti(OiPr)4, Ti(OSiMe3)4, Al(OiPr)3), prolonging the reaction time (up to 5 days) and performing the reaction under MW conditions did not lead to the completion of the reaction, and unconsumed hydroxy acid was distilled together with the product. In all cases the yields were moderate. The methyl ester of 6-hydroxycaproic acid was also tried in this reaction at 40 and 120 °C but without success.
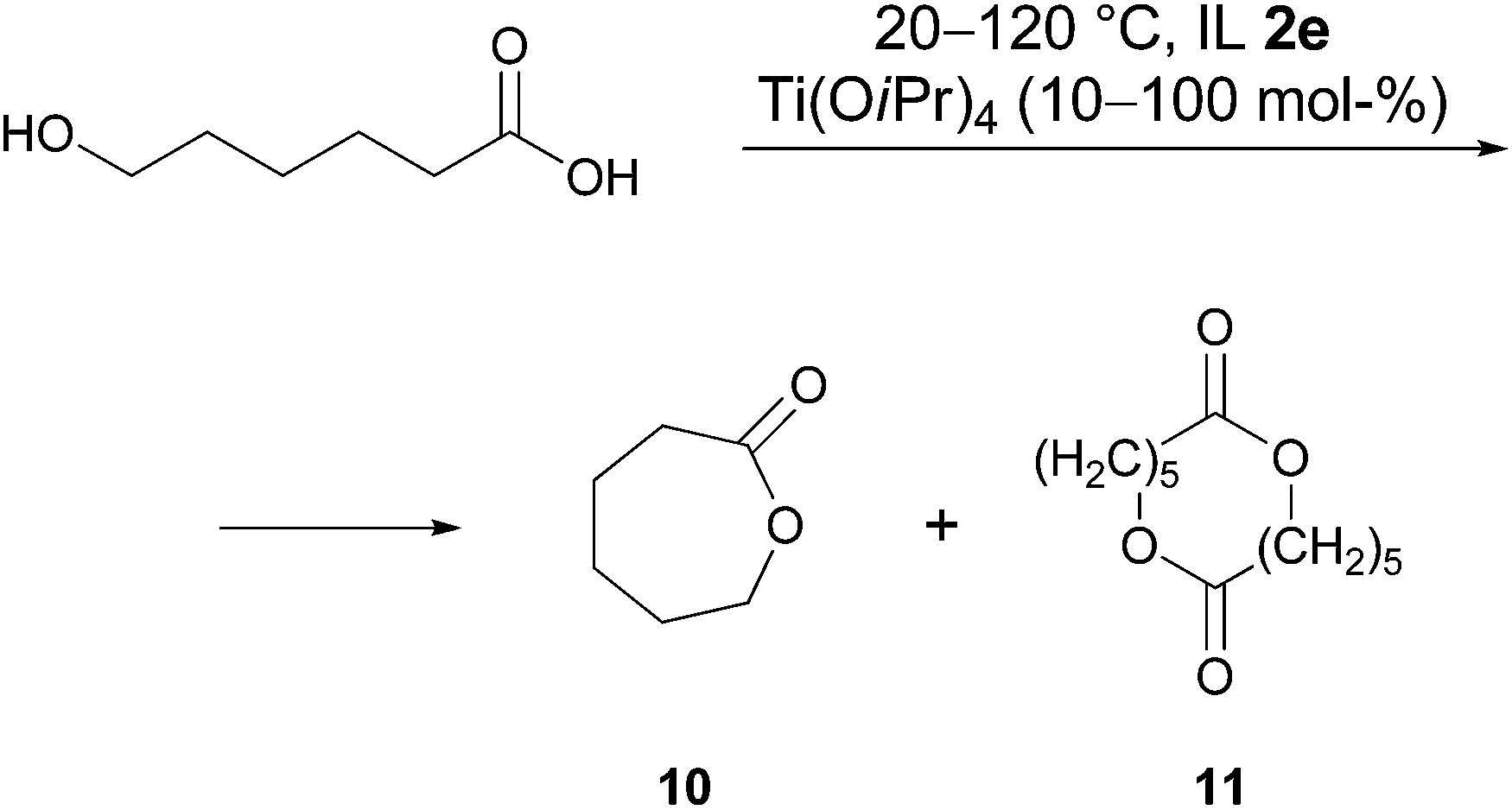 | ||
| Scheme 4 Cyclisation of 6-hydroxycaproic acid in IL 2e. | ||
Two reasons for the failure of lactonisation could be mentioned. Firstly, the competitive dimer formation, which was also observed in controlling NMR spectra. Along with signals of the ε-caprolactone in a 1H NMR spectrum taken in D2O at δ = 4.32 (m, CH2O) and 2.67 ppm (m, CH2C(O)), signals with smaller chemical shifts appear at δ = 4.08 (m, CH2O) 2.38 ppm (m, CH2C(O)), which were tentatively assigned to the dimer 11 shown in Scheme 4 (other signals of the product and by-product were overlapped by the signals of IL). This side reaction could perhaps be suppressed by carrying out the reaction with a low concentration of hydroxy acid, but in the case of ionic liquids this is not reasonable because of the high cost of the solvent. Another reason could be the relatively low Lewis acidity of the Ti(OiPr)4 in combination with the lower nucleophilicity of the oxygen of the hydroxy acid compared to nitrogen of the amino acid used for lactamisation.
Paal–Knorr synthesis
Pyrroles and their derivatives constitute one of the most important classes of heterocyclic compounds. They exhibit broad biological and pharmacological properties. Many pyrrole derivatives possess antibacterial, antiinflammatory, antioxidant, antitumor, antifungal and immune suppressant activities.61Up to day a great number of pyrrole syntheses exists, of which the Paal–Knorr condensation is the most simple, straightforward reaction, which consists in an acid-catalysed cyclisation of a 1,4-dicarbonyl compound and ammonia or a primary amine. Although this reaction was first reported concurrently by C. Paal62 and L. Knorr63 already in 1884, it still awakes the interest of researchers, who continue to develop new catalysts and solvents to render the reaction “green”. Noteworthy, a Paal–Knorr condensation is one of the steps in the synthesis of atorvastatin, a drug for lowering cholesterol levels.64,65
Both Brønsted and Lewis acids catalyse the Paal–Knorr reaction. The moderate Lewis acid Ti(OiPr)4 was employed in the synthesis of the alkaloid funebrine.66,67 It offered itself as a suitable catalyst, which would not catalyse polymerisation of pyrrole and decomposition of the aminolactone (which is a part of the funebrine structure), as in the case of stronger Lewis acids.67,68 Several papers have already reported the use of imidazolium-based ionic liquids as reaction media in the Paal–Knorr pyrrole synthesis; [BMIM]BF4 was used alone69 or in combination with catalytic bismuth(III) triflate,70 and [BMIM]HSO4 served as both a solvent and a Brønsted acid.71 We tried our reusable IL/catalyst system in this reaction type.
The condensation of 2,5-hexanedione and benzylamine to give pyrrole 12 (R1 = Bn) was investigated first (Scheme 5, Table 11). In the IL [N11N11N66Gu]NTf2 (2e), this reaction went to completion without catalyst at room temperature within 24 h; this is nearly twice as efficient than in the traditional solvent (benzene, Table 11), but distinctly slower than the reported reaction in the ionic liquids [BMIM]BF4 and [BMIM]I.69 The reaction could be accelerated not only at higher temperature, but also at ambient temperature by addition of 10 mol% of Ti(OiPr)4. Bulb-to-bulb distillation provided pure product without tedious work-up.
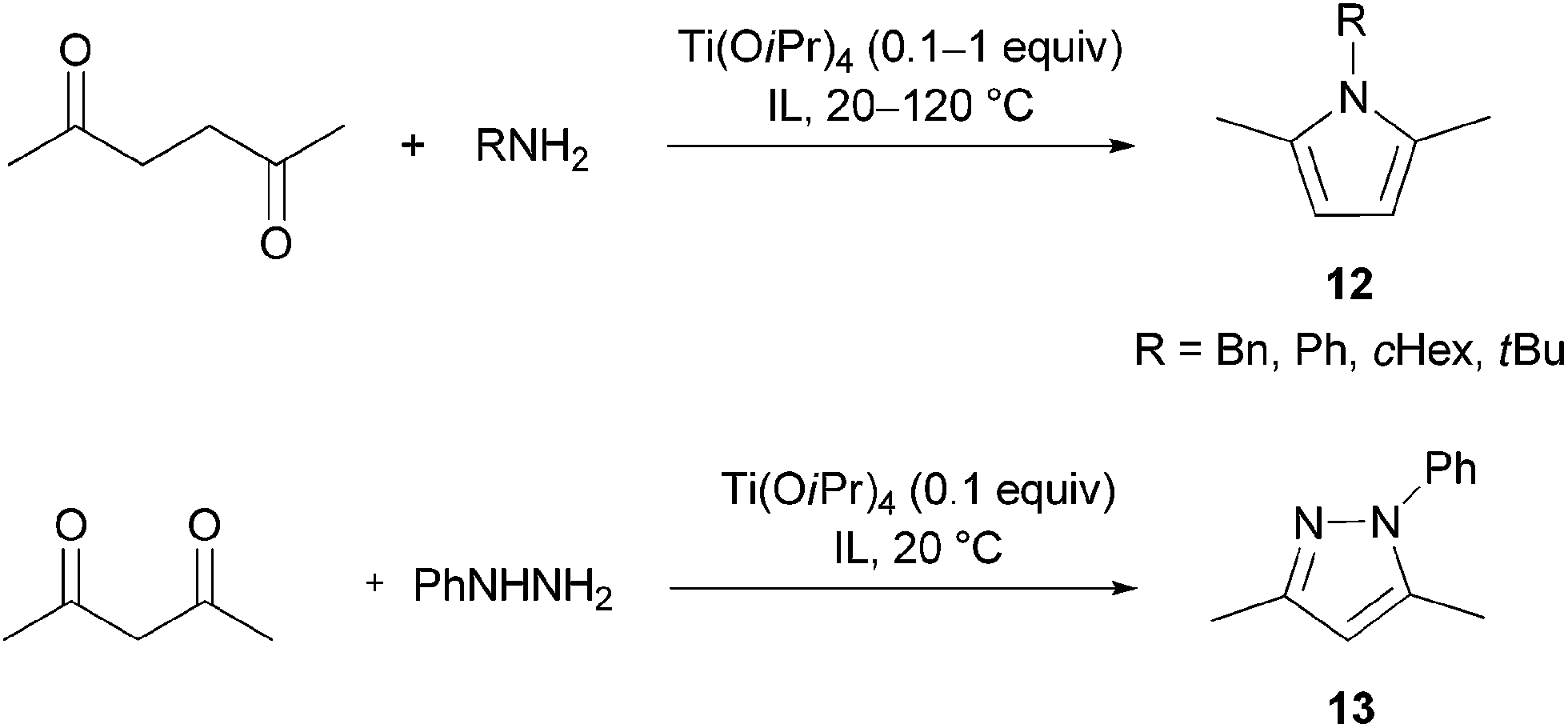 | ||
| Scheme 5 Investigated Paal–Knorr reactions. | ||
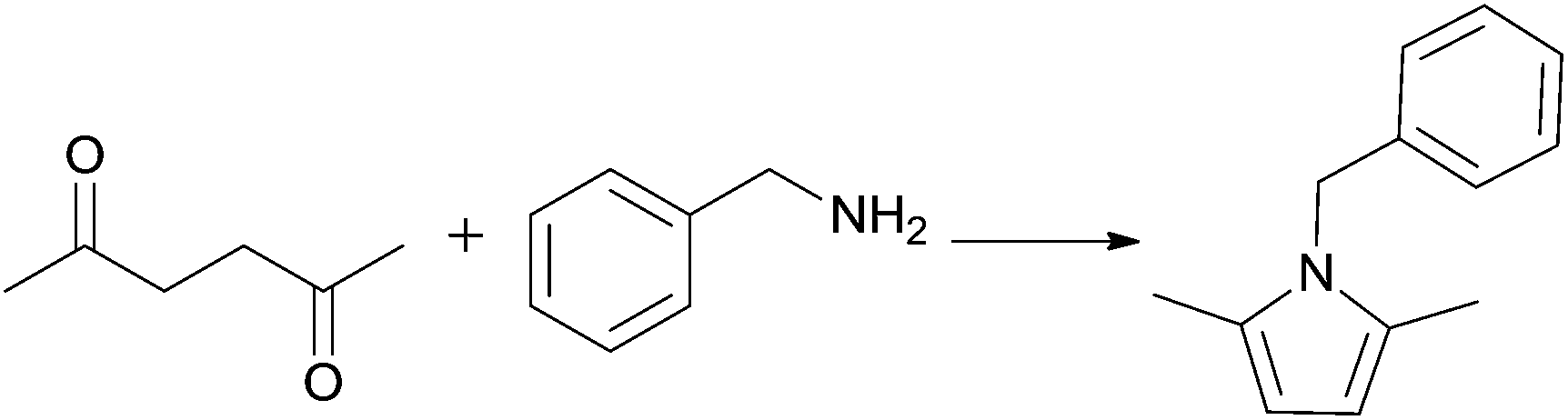
Further reactions were carried out with other, less nucleophilic (aniline) and more nucleophilic but sterically more hindered amines (tBuNH2, cHexNH2), in order to reveal the scope of the IL/catalyst system (Table 12). If the reaction was not complete after 24 h at room temperature, the catalyst amount was increased to 50 mol%. In some cases higher temperatures were needed. The reaction conditions were optimised for each case to improve the yields. Notably, the Paal–Knorr reaction failed completely, when tert-butylamine was used as an amine component. It is known that the Paal–Knorr reaction is a convenient synthesis of pyrroles from ammonia or primary amines, whereas sterically hindered tert-alkylamines are not prone to form pyrroles under mild conditions.72 The Paal–Knorr pyrrole synthesis was also successfully extended to the synthesis of 1-phenylpyrazole (13) from 2,4-pentanedione and phenylhydrazine (Scheme 5). In all cases, the catalyst/IL mixture remaining after product distillation could be reused several times with product yields remaining more or less unaltered (Table 13).
| Diketone | Amine | Cat.a, mol% | Temp., °C | Time, h | Yield, % |
|---|---|---|---|---|---|
| a Catalyst = Ti(OiPr)4.b Conversion of 1,4-diketone (determined by 1H NMR). | |||||
| CH3CO(CH2)2COCH3 | BnNH2 | 10 | 20 | 2 | 93 |
| PhNH2 | 10 | 20 | 22 | 56b | |
| PhNH2 | 50 | 20 | 48 | 94 | |
| cHexNH2 | 10 | 20 | 22 | 32b | |
| cHexNH2 | 50 | 20 | 20 | 45b | |
| cHexNH2 | 10 | 120 | 3 | 73 | |
| t-BuNH2 | 10 | 20 | 24 | 0 | |
| t-BuNH2 | 100 | 20 | 24 | 0 | |
| CH3COCH2COCH3 | PhNHNH2 | 10 | 20 | 24 | 95 |
The catalytic system of Ti(OiPr)4 in the hexa-alkylguanidinium bis(triflamide) 2e proved to be an effective and mild milieu for the Paal–Knorr reaction with all the advantages, which were also achieved when performing lactamisation reactions in guanidinium ILs. Pyrroles and pyrazoles can be obtained by the developed method easily and in high yields. In comparison to the procedure described by B. Wang et al.,69 who demonstrated the preparation of pyrroles in [BMIm]I at room temperature with an ensuing extraction of the product with diethylether, our procedure avoids the use of volatile and flammable organic solvents. On the other hand, the [BMIm]I system allows the synthesis of pyrroles from more sterically hindered amines, such as tert-butylamine and isopropylamine. Compared with the Bi(OTf)3/[BMIm]BF4 system proposed by Y. S. Yadav et al.,70 the developed guanidinium-based IL/Ti(OiPr)4 system has two advantages: the catalyst used is considerably cheaper and a bis(triflamide)-based IL is water-stable in contrast to an IL tetrafluoroborate.
Conclusions
We have found that the lactamisation of ω-aminocarboxylic acids is achieved effectively and conveniently with catalytic Ti(OiPr)4 in ionic liquids of the hexaalkylguanidinium type. The developed procedure ties in with principles of Green Chemistry, such as avoidance of volatile and hazardous organic solvents, significant reduction of the amount of catalyst compared to a reported procedure carried out in a traditional solvent, and reuse of the catalyst/IL system in several subsequent cycles, combined with a convenient distillative separation of the product. Moreover, the catalyst/IL system can be reused in several subsequent reaction cycles, although the catalytically active species after the first reaction cycle is no longer Ti(OiPr)4 but most likely an activated form of TiO2.The same catalyst/IL system is also suited to perform the direct amidation of carboxylic acids with benzylamine and cyclic secondary amines at moderately high temperatures (typically 90 °C), but its reuse in subsequent reaction cycles requires a much higher temperature (200 °C) to proceed in a short time and in high yield. A direct amidation is also included in the synthesis of 2-oxazolines from carboxylic acids and 1,2-aminoalcohols. It turned out that the crucial step of the reaction sequence is the final cyclisation of the initially formed N-(2-hydroxyethyl)carboxamide, and this step is no longer catalysed effectively by the Ti(OiPr)4/ionic liquid system.
On the other hand, good to excellent yields were obtained in the synthesis of pyrroles from a 1,4-diketone and sterically less encumbered primary amines (Paal–Knorr synthesis), and for the related pyrazole synthesis from acetylacetone and phenylhydrazine. Here again, the easy recovery and effective reusability of the Ti(OiPr)4/IL system deserve particular notice.
Experimental section
General information
NMR spectra were recorded on a Bruker DRX 400 spectrometer (1H: 400.13 MHz; 13C: 100.61 MHz; 19F: 376.46 MHz). 1H NMR spectra were referenced to the residual proton signal of the solvent [δ(CDCl3) = 7.26 ppm], 13C spectra to the solvent signal [δ(CDCl3) = 77.00 ppm] and 19F spectra to external C6F6 [δ(C6F6) = −162.9 ppm]. IR spectra were recorded with a Bruker Vector 22 FTIR spectrometer. The mass spectrometry measurements were performed with a Finnigan MAT instrument (SSQ-7000). Thermogravimetric analysis (TGA) was performed with a Mettler-Toledo TGA/SDTA 851 instrument and differential scanning calorimetry (DSC) with a Perkin Elmer DSC 7 instrument (Tdec = temperature of highest decomposition gradient, Tg = glass transition temperature). Microanalyses were obtained with an Elementar vario MICRO cube instrument. Transmission electron microscopy was performed with a TEM Zeiss EM10 (accelerating voltage 80 kW, CCD-camera (TVIPS 1K 1K)). X-ray diffraction (XRD) spectra were measured on a PANanalytical X'Pert MRD Pro diffractometer with Cu-Kα radiation.Rigorously dried organic solvents were used. All amines were dried with KOH pellets and distilled prior to use. Lithium bis(trifluoromethylsulfonyl)imide (99%) was purchased from Acros Organics or IoLiTec GmbH.
The guanidinium triflates 1a and 1b were obtained chloride-free from the corresponding ureas and amines according to the literature.25 Guanidinium bis(triflamides) 2a, 2b, 2c and 2f and guanidinium dicyanamide 3a were obtained from the corresponding hexaalkylguanidinium chlorides by anion exchange reactions as reported.24,73,74 The hexaalkylguanidinium chlorides in turn were prepared from N,N,N′,N′-tetraalkyl(chloro)formamidinium chlorides by an adaptation of the classical procedure.26,73,74 The corresponding chloride required for the synthesis of bis(triflamide) 2f was obtained from phosgene iminium chloride and dihexylamine.24 1-Butyl-3-methylimidazolium (BMIm) bis(triflamide) and n-hexyltrimethylammonium bis(triflamide) were obtained from the corresponding chlorides by anion exchange. N-Hexyltrimethylammonium iodide ([Me3HexN]I), used as a precursor for [Me3HexN]NTf2, was prepared according to MacFarlane et al.75
The NMR data of the synthesised lactams, amides, oxazolines, pyrroles, and pyrazoles were in agreement with data in the literature. The purity of the obtained products was confirmed by elemental analyses.76.77
General procedure for lactamisation
Dried IL 2b or 2e (2 g) was placed in a round-bottom flask equipped with a three-way Claisen adaptor, an argon inlet and a drying tube. Catalyst (10 mol%) and then 0.4 g of an amino acid were added. The mixture was stirred for some hours at 120 °C. The progress of the reaction was monitored by 1H NMR (in D2O). The product was then separated by bulb-to-bulb distillation at 120–140 °C/0.05 mbar. For the next cycle a new portion of the amino acid was added, and the procedure was repeated. The following substances were obtained: 2-pyrrolidone, 2-piperidinone, hexahydro-2-azepinone, N-methyl-2-pyrrolidone.General procedure for lactonisation
Dried IL 2b or 2e (2 g) was placed in a round-bottom flask equipped with a three-way Claisen adaptor, an argon inlet and a drying tube. The catalyst (10–100 mol%) and then 0.2 g of a hydroxy acid were added. The mixture was stirred for some hours at 20–120 °C. The reaction was controlled by NMR (in D2O). The product was then separated by bulb-to-bulb distillation at 120–130 °C/0.05 mbar. The obtained mixtures of lactone/hydroxy acid were not further separated.General procedure for direct amidation
Dried IL 2e (2 g) was placed in the round-bottom flask equipped with a three-way Claisen adaptor, an argon inlet and a drying tube. Carboxylic acid (0.3 g), dry amine (1 equiv.) and catalyst (20 mol%) were added. The mixture was stirred for some hours at the temperatures given in Tables 3 and 4. The reaction was controlled by NMR (in CDCl3). The product was then separated by bulb-to-bulb distillation at 100–120 °C/0.05 mbar. The following substances were obtained: 2-phenyl-1-(piperidin-1-yl)ethanone, 1-(morpholin-4-yl)-2-phenylethanone, N-benzyl-2-phenylacetamide, phenyl(piperidin-1-yl)methanone, morpholin-4-yl(phenyl)methanone, N-benzylbenzamide.General procedure for the synthesis of 2-oxazolines
Dried IL 2e (2 g) was placed in the round-bottom flask, equipped with a reflux condenser, a three-way Claisen adaptor, an argon inlet and a drying tube. Catalyst (50–250 mol%) and then acid (3 mmol), amino alcohol (3.6 mmol) and 1 g of molecular sieves 4 Å were added. The mixture was stirred for some hours at 120 °C, then at 200 °C. The reaction was controlled by NMR (in CDCl3 or CD3OD). The product was separated by bulb-to-bulb distillation at 120–140 °C/0.05 mbar. For the next cycle the new portions of the acid and amino alcohol were added, and the procedure was repeated. The following substances were obtained: 2-phenyl-4,5-dihydrooxazole, 4,4-dimethyl-2-phenyl-4,5-dihydrooxazole, 2-benzyl-4,5-dihydrooxazole, 4,4-dimethyl-2-benzyl-4,5-dihydrooxazole, bis(4,4-dimethyl-4,5-dihydrooxazol-2-yl)methane.General procedure for the Paal–Knorr synthesis
Dried IL 2e (2 g) was placed in a round-bottom flask equipped with a three-way Claisen adaptor, an argon inlet and a drying tube. Catalyst (10–100 mol%) and then 1,3- or 1,4-diketone (0.3 mL) and amine (1 equiv.) were added. The mixture was stirred for 2–48 h at 20–120 °C. The reaction was controlled by NMR (in CDCl3). The product was separated by bulb-to-bulb distillation at 100–130 °C/0.05 mbar. For the next cycle new portions of the 1,3- or 1,4-diketone and amine were added, and the procedure was repeated. The following substances were obtained: 1-benzyl-2,5-dimethyl-1H-pyrrole, 2,5-dimethyl-1-phenyl-1H-pyrrole, 1-cyclohexyl-2,5-dimethyl-1H-pyrrole, 3,5-dimethyl-1-phenyl-1H-pyrazole.N,N-Bis(2-methoxyethyl)-N′,N′,N′′,N′′-tetramethylguanidinium bis(trifluoromethylsulfonyl)imide (2d)
N,N-Bis(2-methoxyethyl)-N′,N′,N′′,N′′-tetramethylguanidinium chloride (2.0 g, 7.3 mmol) and lithium bis(trifluoromethylsulfonyl)imide (2.1 g, 7.3 mmol) were dissolved in deionised water (35 mL). The solutions were combined resulting in two phases. After vigorous stirring of the mixture at 70 °C for 30 min, it was cooled to room temperature and dichloromethane was added. The organic phase was separated and washed with several portions of deionised water, until a test on chloride ions in the rinsing water (AgNO3) was negative. The organic phase was dried with Na2SO4, stirred over charcoal for 15 min, and filtered. The solvent was removed in a rotary evaporator, and the product was dried for 8 h at 120 °C/0.05 mbar in the bulb-to-bulb apparatus. The product was obtained as a light yellow oil. Yield: 3.2 g (84%); m.p. = 7 °C, Tdec = 445 °C, Tg = −57 °C. 1H NMR (CDCl3): δ = 2.95 and 3.00 (2 s, each 6H, NCH3), 3.32 (s, 6H, OCH3), 3.32–3.65 (several m, 8H, NCH2CH2 and NCH2) ppm. 13C NMR (CDCl3): δ = 39.8 and 40.2 (NCH3), 49.3 (NCH2), 58.7 (CH3O), 68.7 (NCH2CH2), 119.8 (CF3), 164.4 (CN3) ppm. 19F NMR (CDCl3): δ = −75.2 ppm. IR (NaCl): ν = 2939 (m), 2902 (m), 2834 (m), 1602 (s), 1568 (s), 1456 (m), 1435 (m), 1411 (s), 1352 (s), 1190 (s), 1136 (s), 1058 (s), 1014 (m), 894 (m) cm−1. MS (CI): m/z = 232 (100%, [cation]+). Anal. calcd for C13H26F6N4O6S2 (512.49 g mol−1): C 30.47, H 5.11, N 10.93%; found: C 30.50, H 5.59, N 11.26%.N,N-Dihexyl-N′,N′,N′′,N′′-tetramethylguanidinium bis(trifluoromethylsulfonyl)imide (2e)
Prepared analogously to 2d from N,N-dihexyl-N′,N′,N′′,N′′-tetramethylguanidinium chloride (4.3 g, 13.3 mmol) and lithium bis(trifluoromethylsulfonyl)imide (3.8 g, 13.3 mmol) in deionised water (50 mL). The product was obtained as a colourless oil. Yield: 7.1 g (94%); Tdec = 460 °C, Tg = −82 °C. 1H NMR (CDCl3): δ = 0.87–0.91 (ψt, 6H, N(CH2)5CH3), 1.20–1.68 (several m, 16H, NCH2(CH2)4CH3), 2.97 and 3.01 (2 s, both 6H, NCH3), 3.04–3.20 (m, 4H, NCH2) ppm. 13C NMR (CDCl3): δ = 13.9 (N(CH2)5CH3), 22.4 (N(CH2)4CH2CH3), 26.4 (N(CH2)3CH2CH2CH3), 27.5 (N(CH2)2CH2(CH2)2CH3), 31.3 (NCH2CH2(CH2)3CH3), 40.31 and 40.34 (NCH3), 49.6 (NCH2), 119.9 (CF3), 163.2 (CN3) ppm. 19F NMR (CDCl3): δ = −75.2 ppm. IR (NaCl): ν = 2958 (m), 2935 (m), 2863 (m), 1594 (s), 1566 (s), 1411 (m), 1352 (s), 1333 (s), 1190 (s), 1138 (s), 1059 (s), 897 (m) cm−1. MS (CI): m/z = 284 (100%, [cation]+). Anal. calcd for C19H38F6N4O4S2 (564.65 g mol−1): C 40.42, H 6.78, N 9.92%; found: C 40.36, H 6.69, N 10.01%.N,N-Bis(2-methoxyethyl)-N′,N′,N′′,N′′-tetramethylguanidinium chloride
To a solution of N,N,N′,N′-tetramethyl(chloro)formamidinium chloride (4.0 g, 23.4 mmol) in dry acetonitrile (30 mL) was added a solution of bis(2-methoxyethyl)amine (3.4 mL, 23.4 mmol) and triethylamine (3.3 mL, 23.4 mmol) in dry diethylether (15 mL) under argon atmosphere at room temperature. The mixture was stirred overnight, the precipitated ammonium salt was filtered off, and the volatiles were evaporated at 40 °C/180 mbar. In order to destroy remaining ammonium salt(s), 0.1 M aqueous NaOH was added to the residual oil until the pH was slightly alkaline. To remove the coloured impurities, the aqueous solution was washed several times with diethyl ether. Water and triethylamine were distilled off on the rotary evaporator (40 °C/70 mbar), and the remaining residue was subsequently dried at 50 °C/0.05 mbar. Afterwards it was dissolved in dry acetonitrile/diethylether (2:1, v/v), and the remaining solid was filtered off. The organic solvents were evaporated, and the remaining oil was triturated with dry diethyl ether to remove coloured impurities and induce the crystallisation of the product. After drying for 8 h at 80 °C/0.05 mbar a colourless hygroscopic solid was obtained. Yield: 4.3 g (69%); m.p. = 83 °C, Tdec = 305 °C. 1H NMR (CDCl3): δ = 3.06 and 3.08 (2 s, each 6H, NCH3), 3.33 (s, 6H, OCH3), 3.42–3.55 (m, 6H, NCH2CH2 and NCH2), 3.65–3.75 (m, 2H, NCH2CH2) ppm. 13C NMR (CDCl3): δ = 40.2 and 40.6 (NCH3), 49.4 (NCH2), 58.9 (CH3O), 69.2 (NCH2CH2), 164.4 (CN3) ppm. IR (KBr): ν = 2937 (m), 2903 (m), 2829 (w), 1602 (vs), 1565 (vs), 1458 (m), 1432 (m), 1409 (s), 1115 (s), 1064 (w), 1012 (w), 893 (w) cm−1. MS (CI): m/z = 232 (20%, [cation]+). Anal. calcd for C11H26ClN3O2·0.5H2O (267.80 + 9.00 g mol−1): C 47.73, H 9.83, N 15.18%; found: C 47.75, H 10.07, N 15.21%.
N,N-Dihexyl-N′,N′,N′′,N′′-tetramethylguanidinium chloride
Prepared analogously to N,N-bis(2-methoxyethyl)-N′,N′,N′′,N′′-tetramethylguanidinium chloride from N,N,N′,N′-tetramethyl(chloro)formamidinium chloride (12.6 g, 73.8 mmol), di-n-hexylamine (17.1 mL, 73.8 mmol), triethylamine (10.3 mL, 73.8 mmol) in acetonitrile (100 mL) and diethyl ether (50 mL), recrystallised from dry cyclohexane/ethyl acetate (5:2, v/v). The product was obtained as a colourless hygroscopic solid. Yield: 16.3 g (69%); m.p. = 103 °C, Tdec = 299 °C. 1H NMR (CDCl3): δ = 0.87–0.91 (ψt, 6H, N(CH2)5CH3), 1.25–1.70 (several m, 16H, NCH2(CH2)4CH3), 3.06 and 3.21 (2 s, both 6H, NCH3), 3.10–3.20 (m, 4H, NCH2) ppm. 13C NMR (CDCl3): δ = 13.9 (N(CH2)5CH3), 22.5 (N(CH2)4CH2CH3), 26.5 (N(CH2)3CH2CH2CH3), 27.7 (N(CH2)2CH2(CH2)2CH3), 31.4 (NCH2CH2(CH2)3CH3), 40.7 and 41.0 (NCH3), 49.7 (NCH2), 163.2 (CN3) ppm. IR (ATR): ν = 2929 (m), 2856 (m), 1590 (s), 1559 (s), 1428 (m), 1403 (s), 1373 (m), 1271 (w), 1244 (w), 1121 (w), 1068 (m), 893 (m) cm−1. MS (CI): m/z = 284 (16%, [cation]+). Anal. calcd for C17H38ClN3 (319.92 g mol−1): C 63.82, H 11.97, N 13.13%; found: C 63.79, H 12.15, N 13.18%.
Acknowledgements
M. Arkhipova thanks the State of Baden-Württemberg for a postgraduate fellowship. S. Eichel thanks the Fonds der Chemischen Industrie for a fellowship. We are grateful to Prof. Dr W. Kantlehner (University of Applied Sciences Aalen) for the tetraalkylureas used in this study. We thank Prof. M. Lindén and S. Blessing, Institute of Inorganic Chemistry II of the University of Ulm, for the XRD measurements.Notes and references
- P. Wasserscheid and W. Keim, Angew. Chem., 2000, 112, 3926–3945 CrossRef.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS PubMed.
- N. Jain, A. Kumar, S. Chauhan and S. M. S. Chauhan, Tetrahedron, 2005, 61, 1015–1060 CrossRef CAS PubMed.
- Z. C. Zhang, Adv. Catal., 2006, 49, 153–237 CAS.
- V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS PubMed.
- H. Oliver-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef PubMed.
- J. P. Hallet and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef PubMed.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley–VCH, Weinheim, Germany, 2nd edn, 2008 Search PubMed.
- A. Mohammed and Inamuddin, Green Solvents II: Properties and Applications of Ionic Liquids, Springer, Dordrecht, 2012 Search PubMed.
- A. Kokorin, Ionic Liquids: Applications and Perspectives, InTech, Rijeka, Croatia, 2011 Search PubMed.
- For regio- and stereoselectivity, see also: V. Calò, A. Nacci and A. Monopoli, Eur. J. Org. Chem., 2006, 3791–3802 CrossRef.
- For solvent properties of ionic liquids relevant to catalysis, see: J. S. Wilkes, J. Mol. Catal. A: Chem., 2004, 214, 11–17 CrossRef CAS PubMed.
- L. Gharnati, O. Walter, U. Arnold and M. Döring, Eur. J. Inorg. Chem., 2011, 2756–2762 CrossRef CAS.
- S. Li, Y. Lin, H. Xie, S. Zhang and J. Xu, Org. Lett., 2006, 8, 391–394 CrossRef CAS PubMed.
- J. Cao, H. Cao, Y. Lin, D. Liang, H. Duan, S. Han and X. Zhao, Chem. Res. Chin. Univ., 2011, 27, 780–783 CAS.
- H. Xie, S. Zhang and H. Duan, Tetrahedron Lett., 2004, 45, 2013–2015 CrossRef CAS PubMed.
- Y. Lin, Z. Qiu, H. Duan, S. Li and S. Zhang, Chem. Res. Chin. Univ., 2004, 20, 46–49 CAS.
- H. Xie, H. Duan, S. Li and S. Zhang, New J. Chem., 2005, 29, 1199–1203 RSC.
- L. C. Branco and C. A. M. Afonso, J. Org. Chem., 2004, 69, 4381–4389 CrossRef CAS PubMed.
- L. C. Branco, P. M. P. Gois, N. M. T. Lourenço, V. B. Kurteva and C. A. M. Afonso, Chem. Commun., 2006, 2371–2372 RSC.
- J. Shah, H. Blumenthal, Z. Yacob and J. Liebscher, Adv. Synth. Catal., 2008, 350, 1267–1270 CrossRef CAS.
- J. Shah and J. Liebscher, Z. Naturforsch., B: J. Chem. Sci., 2011, 66, 88–94 CrossRef CAS PubMed.
- T. Large, T. Müller, H. Kunkel, S. Buck and G. Maas, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 347–353 CrossRef CAS PubMed.
- N. M. M. Mateus, L. C. Branco, N. M. T. Lourenço and C. A. M. Afonso, Green Chem., 2003, 5, 347–352 RSC.
- H. Kunkel and G. Maas, Eur. J. Org. Chem., 2007, 3746–3757 CrossRef CAS.
- W. Kantlehner, E. Haug, W. Mergen, P. Speh, T. Maier, J. Kapassakalidis, H.-J. Bräuner and H. Hagen, Liebigs Ann. Chem., 1984, 108–126 CrossRef CAS.
- M. G. Bogdanov, I. Svinyarov, H. Kunkel, C. Steinle, M. Arkhipova, W. Kantlehner and G. Maas, Z. Naturforsch., B: J. Chem. Sci., 2010, 65, 791–797 CAS.
- K. Yachi, H. Shinokubo and K. Oshima, J. Am. Chem. Soc., 1999, 121, 9465–9466 CrossRef CAS.
- R. Mahrwald and H. Schick, Synthesis, 1990, 592–595 CrossRef CAS.
- O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597–2632 CrossRef CAS PubMed.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- M. Mader and P. Helquist, Tetrahedron Lett., 1988, 29, 3049–3052 CrossRef CAS.
- P. Helquist, M. Bergdahl, R. Hett, A. R. Gangloff, M. Demillequand, M. Cottard, M. M. Mader, T. Friebe, J. Iqbal, Y. Wu, B. Akermark, T. Rein and N. Kann, Pure Appl. Chem., 1994, 66, 2063–2066 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- H. Lin, P. W. de Oliveira, V. Huch and M. Veith, Chem. Mater., 2010, 22, 6518–6523 CrossRef CAS.
- H. Lin, P. W. de Oliveira, I. Grobelsek, A. Haettich and M. Veith, Z. Anorg. Allg. Chem., 2010, 636, 1947–1954 CrossRef CAS.
- H. Choi, Y. J. Kim, R. S. Varma and D. D. Dionysiou, Chem. Mater., 2006, 18, 5377–5384 CrossRef CAS.
- T. Alammar, A. Birkner, O. Sheknah and A.-V. Mudring, Mater. Chem. Phys., 2010, 120, 109–113 CrossRef CAS PubMed.
- I. S. Ignatyev, M. Montejo and J. J. L. Gonzalez, J. Phys. Chem. A, 2007, 111, 7973–7979 CrossRef CAS PubMed.
- Y. Hu, H. Liu, Q. Rao, X. Kong, W. Sun and X. Guo, J. Nanosci. Nanotechnol., 2011, 11, 3434–3444 CrossRef CAS PubMed.
- L. Y. Shteinberg, S. A. Kondratov, S. M. Shein and V. V. Marshalova, Kinet. Catal., 2007, 48, 632–635 CrossRef CAS.
- G. B. Raupp and J. A. Dumesic, J. Phys. Chem., 1985, 89, 5240–5246 CrossRef CAS.
- M. Olea, M. Tada and Y. Iwasawa, J. Catal., 2007, 248, 60–67 CrossRef CAS PubMed.
- K. Tanabe, M. Misono, Y. Ono and H. Hattori, New solid acids and bases, Elsevier, Tokyo, 1989 Search PubMed.
- G. Martra, Appl. Catal., A, 2000, 200, 275–285 CrossRef CAS.
- S. Abdolmohammadi, M. Mohammadnejad and F. Shafaei, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 362–366 CrossRef CAS.
- M. Z. Kassaee, R. Mohammadi, H. Masrouri and F. Movahedi, Chin. Chem. Lett., 2011, 22, 1203–1206 CAS.
- H. Lundberg, F. Tinnis and H. Adolfsson, Chem.–Eur. J., 2012, 18, 3822–3826 CrossRef CAS PubMed and ref. cited therein.
- H. Lundberg, F. Tinnis and H. Adolfsson, Synlett, 2012, 23, 2201–2204 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, USA, 1998 Search PubMed.
- J. D. Wilson and H. Weingarten, Can. J. Chem., 1970, 48, 983–986 CrossRef CAS PubMed.
- A. R. Katritzky, C. Cai, K. Suzuki and S. K. Singh, J. Org. Chem., 2004, 69, 811–814 CrossRef CAS PubMed and ref. cited therein.
- S. Crosignani and D. Swinnen, J. Comb. Chem., 2005, 7, 688–696 CrossRef CAS PubMed and ref. cited therein.
- A. I. Meyers and J. J. Willemsen, Tetrahedron, 1998, 54, 10493–10511 CrossRef CAS.
- S. Ghosh, A. S. Kumar, R. Soundararajan and G. N. Mehta, Synth. Commun., 2009, 39, 3880–3887 CrossRef CAS.
- W. J. Li and S. X. Qiu, J. Heterocycl. Chem., 2010, 47, 1340–1343 CrossRef CAS and ref. cited therein.
- J. A. Frump, Chem. Rev., 1971, 71, 483–505 CrossRef CAS.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., 1966, 20, 913–952 CrossRef.
- T. Ohshima, T. Iwasaki and K. Mashima, Chem. Commun., 2006, 2711–2713 RSC.
- R. Kamakshi and B. S. R. Reddy, Aust. J. Chem., 2006, 59, 463–467 CrossRef CAS.
- P. B. S. Dawadi and J. Lugtenburg, Global Journal of Science Frontier Research Chemistry, 2012, 12, 23–36 Search PubMed and ref. cited therein.
- C. Paal, Ber. Dtsch. Chem. Ges., 1884, 17, 2756–2767 CrossRef.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 2863–2870 CrossRef.
- J. J. Li, D. S. Johnson, D. R. Sliskovic and B. D. Roth, Contemporary Drug Synthesis, Wiley–VCH, Weinheim, Germany, 2004 Search PubMed.
- K. L. Baumann, D. E. Butler, C. F. Deering, K. E. Mennen, A. Millar, T. N. Nanninga, C. W. Palmer and B. D. Roth, Tetrahedron Lett., 1992, 33, 2283–2284 CrossRef CAS.
- S.-X. Yu and P. W. Le Quesne, Tetrahedron Lett., 1995, 36, 6205–6620 CrossRef CAS.
- Y. Dong, N. N. Pai, S. L. Ablaza, S.-X. Yu, S. Bolvig, D. A. Forsyth and P. W. Le Quesne, J. Org. Chem., 1999, 64, 2657–2666 CrossRef CAS PubMed.
- O. Tamura, N. Iyama and H. Ishibashi, J. Org. Chem., 2004, 69, 1475–1480 CrossRef CAS PubMed.
- B. Wang, Y. Gu, C. Luo, T. Yang and J. Suo, Tetrahedron Lett., 2004, 45, 3417–3419 CrossRef CAS PubMed.
- J. S. Yadav, B. V. S. Reddy, B. Eeshwaraiah and M. K. Gupta, Tetrahedron Lett., 2004, 45, 5873–5876 CrossRef CAS PubMed.
- Y.-H. He, G.-Q. Wang, K.-L. Xu and Z. Guan, Z. Naturforsch., B: J. Chem. Sci., 2011, 66, 191–196 CrossRef CAS PubMed.
- R. G. Kostyanovsky, G. K. Kadorkina, A. G. Mkhitaryan, I. I. Chervin and A. E. Aliev, Mendeleev Commun., 1993, 21–23 CrossRef CAS PubMed.
- M. Gnahm, C. Berger, M. Arkhipova, H. Kunkel, T. Pajkossy, G. Maas and D. M. Kolb, Phys. Chem. Chem. Phys., 2012, 14, 10647–10652 RSC.
- H. Kunkel, Dissertation, University of Ulm, Ulm, Germany, 2008.
- J. Sun, D. R. MacFarlane and M. Forsyth, Ionics, 1997, 3, 356–362 CrossRef CAS.
- S. Eichel, Zulassungsarbeit, University of Ulm, Ulm, Germany, 2013 Search PubMed.
- M. Arkhipova, Dissertation, University of Ulm, Ulm, Germany, 2014.
| This journal is © The Royal Society of Chemistry 2014 |