
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diguanidinocalix[4]arenes as effective and selective catalysts of the cleavage of diribonucleoside monophosphates†
Riccardo
Salvio
*a,
Roberta
Cacciapaglia
a,
Luigi
Mandolini
a,
Francesco
Sansone
b and
Alessandro
Casnati
 b
b
aDipartimento di Chimica and IMC – CNR Sezione Meccanismi di Reazione, Università La Sapienza, 00185 Roma, Italy. E-mail: riccardo.salvio@uniroma1.it
bDipartimento di Chimica, Università degli Studi di Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy
First published on 25th July 2014
Abstract
Calix[4]arenes derivatives 1 and 2, featuring two guanidine units at the upper rim, catalyze the transesterification of diribonucleoside monophosphates much more effectively than that of HPNP. Rate accelerations relative to the background range from 103 to 104-fold, and approach 105-fold with the most favorable substrate-catalyst combinations.
Introduction
The inertness of phosphodiesters towards hydrolysis1 has challenged many research groups to the design of artificial phosphodiesterases2–6 with the purpose of achieving efficient and selective cleavage of polynucleotides and polyribonucleotides. The potential application of these enzyme mimics to health related goals is an attractive target of investigation in this field.7Metal cations, notably copper(II) and zinc(II), are at the core of most artificial phosphodiesterases,2 but nonmetallic active units as well have received some attention.2p,3–6 In a recent work5 we found that upper rim diguanidino-cone-calix[4]arenes 1 and 2 catalyze the cleavage of the RNA model compound 2-hydroxypropyl-p-nitrophenyl phosphate (HPNP). It was shown that the catalysts are active in their protonated forms 1H+ and 2H+, in which the guanidine–guanidinium dyad combines the general base action of the neutral guanidine with the electrophilic/electrostatic activation of the protonated guanidine,4–6 (Fig. 1).
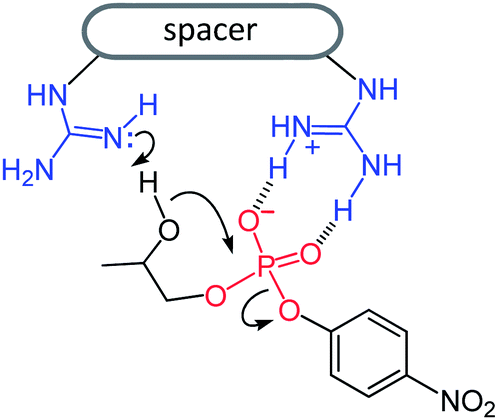 | ||
| Fig. 1 General-base/general-acid mechanism of HPNP cleavage catalyzed by monoprotonated diguanidino compounds. | ||
Since conclusions drawn from the cleavage of activated phosphodiesters do not necessarily apply to the cleavage of unactivated phoshodiesters,8 it seemed worthwhile to investigate the catalytic activity of 1 and 2 in the transesterification of a series of diribonucleoside 3′,5′-monophosphates NpN′, eqn (1), as more appropriate RNA models. The results of such an investigation are reported herein.
Results and discussion
Catalytic runs were carried out under the same conditions used for the cleavage of HPNP,5 namely, pH 10.4, 10 mM Me4NClO4, DMSO–H2O 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 (v/v), hereafter referred to as 80% DMSO. The sole difference is the higher temperature 50 °C rather than 25 °C, dictated by the lower reactivity of diribonucleoside monophosphates compared to HPNP. Solutions of precatalyst – 1·2HCl or 2·2HCl – were exactly half neutralized with 1 mol equiv. of Me4NOH to give buffer solutions at pH 10.4 (pOH 8.0)9 that were used for the catalytic experiments. According to the previously reported distribution diagrams of the species as a function of pH,5 these solutions contain the maximum concentration of monoprotonated species 1H+ or 2H+, amounting to a mole fraction of 0.86 in both cases. The kinetics were monitored by HPLC analysis of aliquots of the reaction mixture withdrawn at time intervals in the early stages of the reaction, as previously described.2d Initial rates of nucleoside N′ formation were translated into pseudo-first-order specific rates kobs.
:20 (v/v), hereafter referred to as 80% DMSO. The sole difference is the higher temperature 50 °C rather than 25 °C, dictated by the lower reactivity of diribonucleoside monophosphates compared to HPNP. Solutions of precatalyst – 1·2HCl or 2·2HCl – were exactly half neutralized with 1 mol equiv. of Me4NOH to give buffer solutions at pH 10.4 (pOH 8.0)9 that were used for the catalytic experiments. According to the previously reported distribution diagrams of the species as a function of pH,5 these solutions contain the maximum concentration of monoprotonated species 1H+ or 2H+, amounting to a mole fraction of 0.86 in both cases. The kinetics were monitored by HPLC analysis of aliquots of the reaction mixture withdrawn at time intervals in the early stages of the reaction, as previously described.2d Initial rates of nucleoside N′ formation were translated into pseudo-first-order specific rates kobs.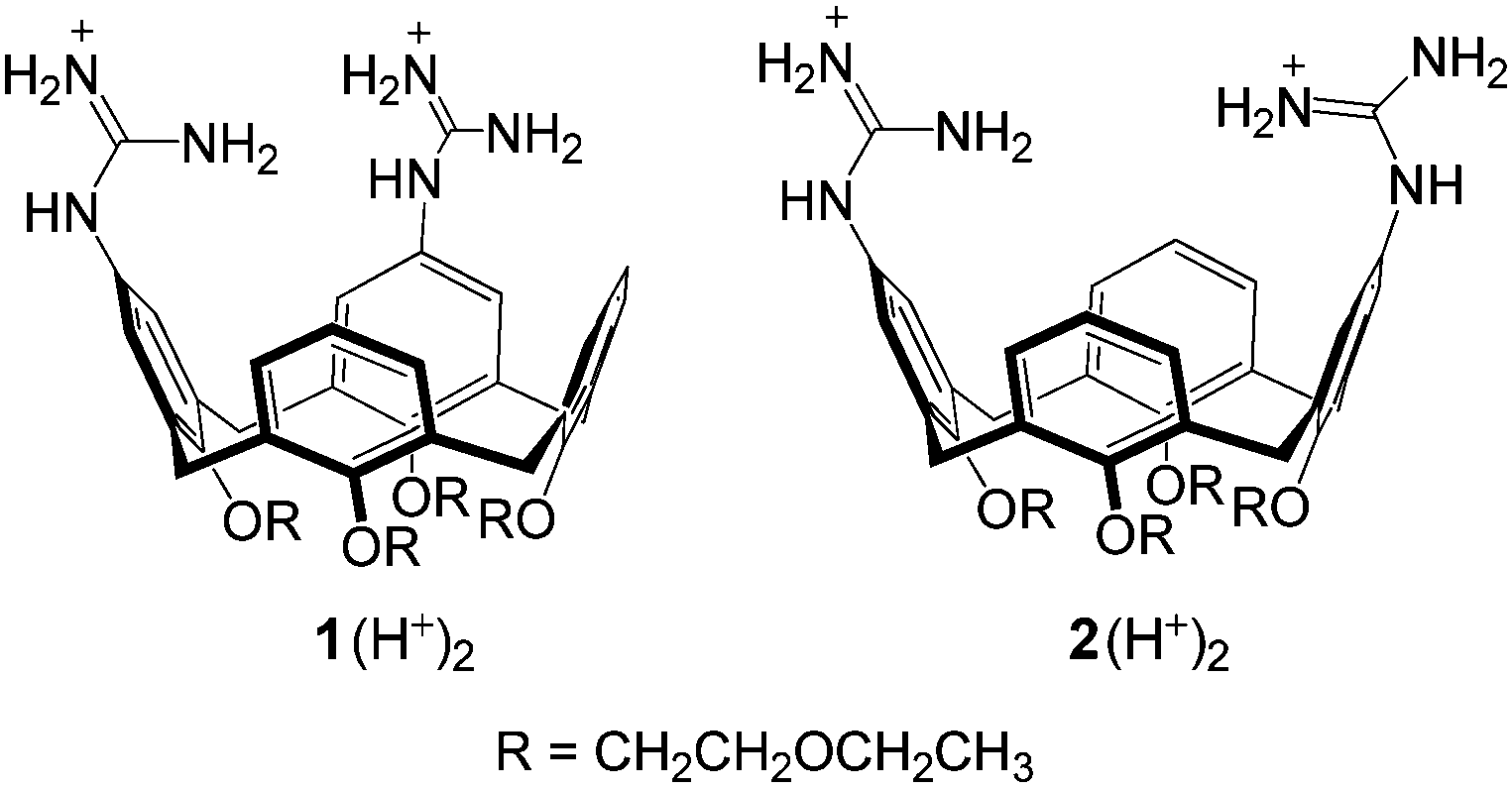
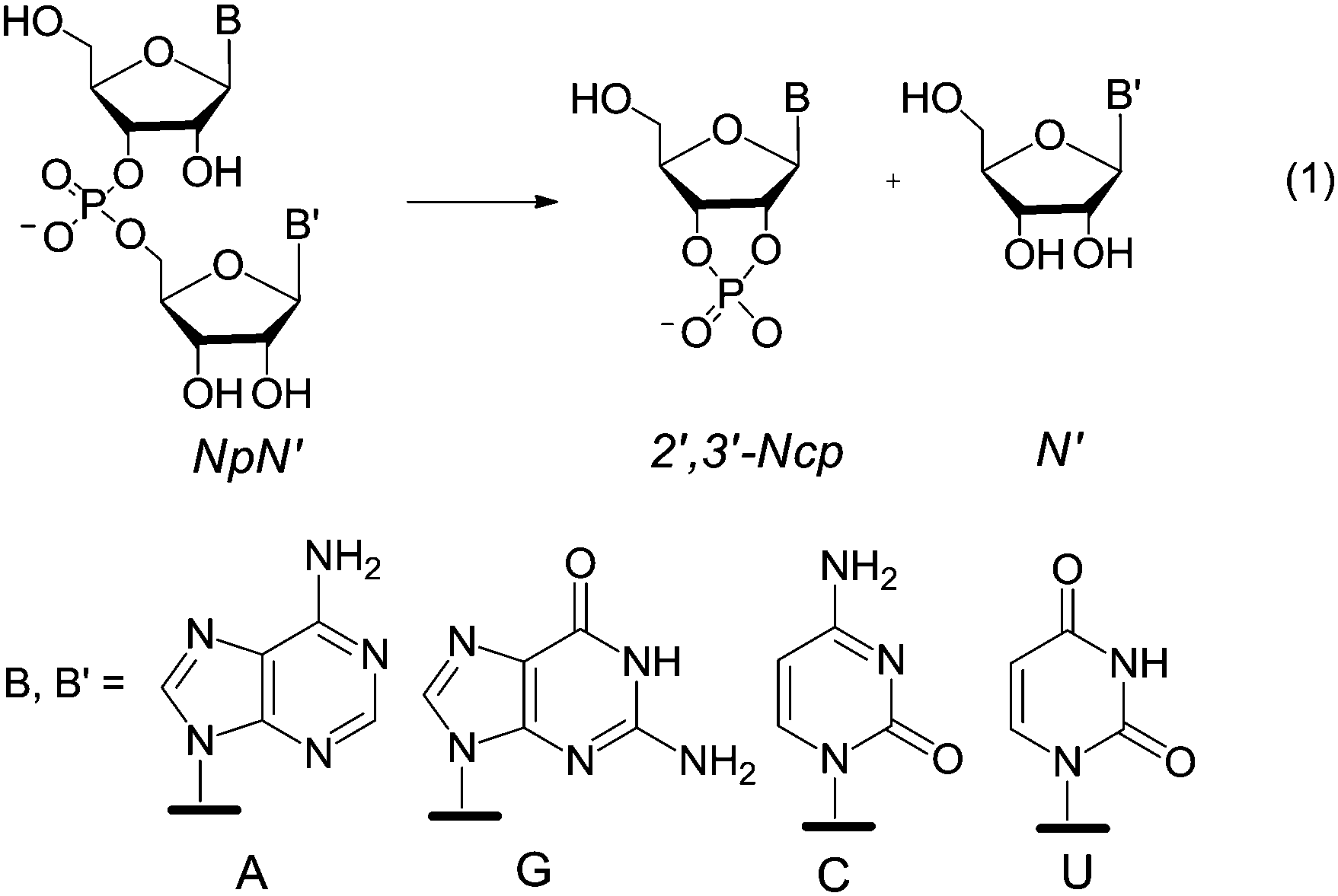
Typical plots of reaction percentage vs. time related to the cleavage of GpU catalyzed by 1 are shown in Fig. 2a. The diagram of kobsvs. buffer concentration (Fig. 2b) shows a good adherence of data points to a straight line with zero intercept, and this indicates (i) that the contribution of background hydrolysis to the overall rate is negligibly small and (ii) that the catalyst works under subsaturating conditions, i.e. binding of the catalyst to the substrate is too low to affect the kinetics in the investigated concentration range (K < 25 M−1). An analogous result was obtained in our previous studies of the cleavage of HPNP.5,6 This finding not only confirms that in the reactant state binding of guanidinium to the negatively charged phosphate moiety is insignificant, but also indicates that any possible stabilizing interaction between guanidine/guanidinium units and nucleobases B and B′ is negligibly small.
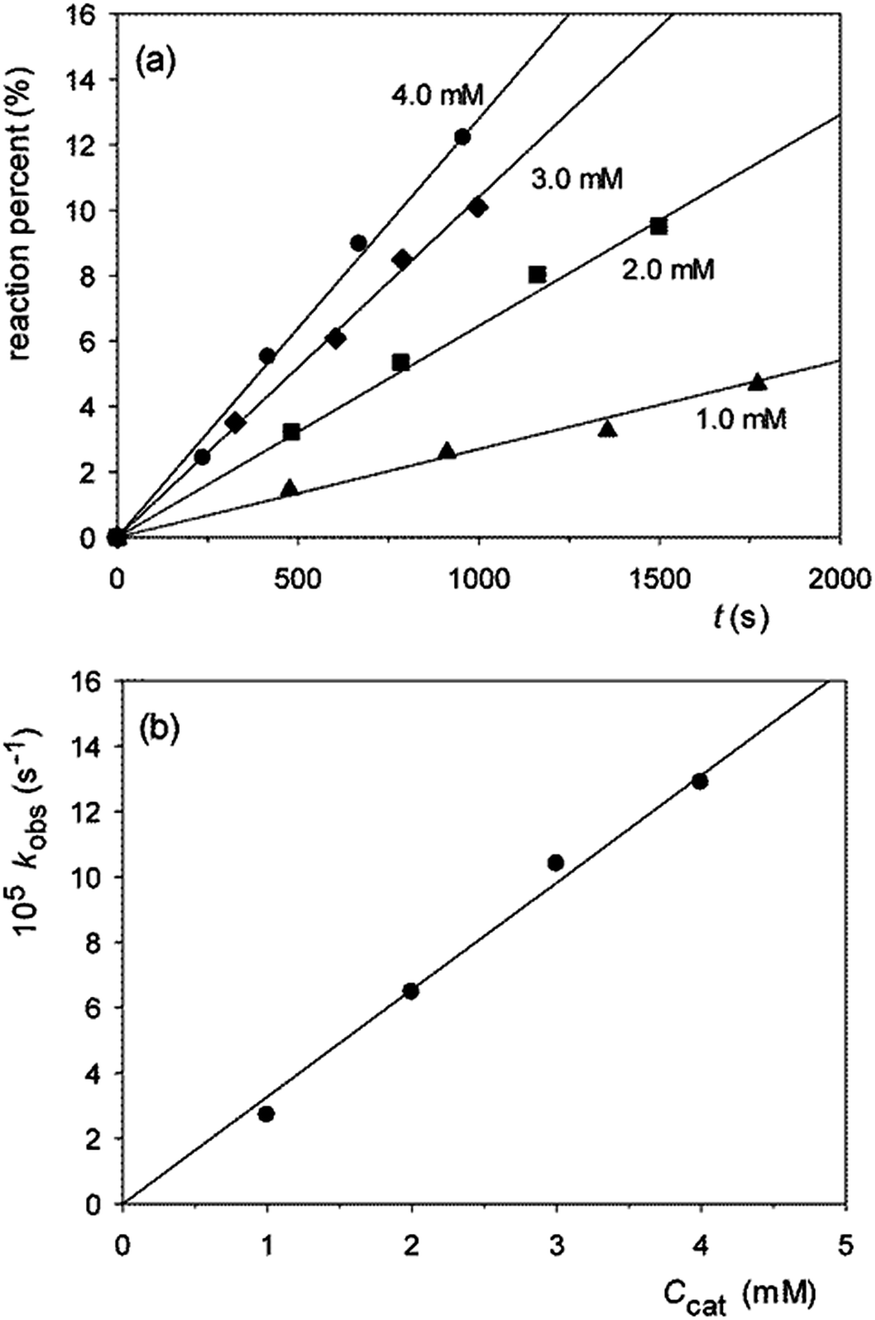 | ||
| Fig. 2 Cleavage of 0.10 mM GpU catalyzed by 1.0–4.0 mM 1 (80% DMSO, pH 10.4, 10 mM Me4NClO4, 50.0 °C). (a) Reaction percent vs. time, for experiments carried out at the given catalyst concentration. (b) Plot of kobsvs. total catalyst concentration ccat. | ||
Having established that the kinetics of the reaction of GpU catalyzed by 1 were not complicated by a pre-association equilibrium, the whole set of catalytic runs listed in Table 1 were carried out using a fixed precatalyst concentration of 2.0 mM, under the assumption that subsaturating conditions applies to all runs. Table 1 shows that both catalysts effectively cleave all of the investigated substrates, with a marked preference for GpU, GpG and UpU (entries 1–3), while the reactions of the remaining substrates experience a much lower nucleobase selectivity (entries 4–8). The finding that UpG is much less reactive than GpU, and that both ApG and GpA react slowly, clearly indicates that catalytic efficiency depends in a very critical way on the identity of both nucleobases B and B′. Interestingly, literature data on the cleavage of diribonucleoside monophosphates catalyzed by di- and trinuclear metal complexes2d,10 show that in many cases in which high nucleobase selectivity is observed, at least one of the two nucleobases is either uracyl or guanine.2d,10a,e–g In the latter cases enhanced rates of cleavage have been ascribed to a pre-association mechanism, supported by spectroscopic10f,g and kinetic10a,e–g data, in which the deprotonated amidic N–H of a uracyl or guanine serves as a site for anchoring to a ligated zinc(II) or copper(II) unit of the catalyst. In the present work GpU, GpG, and UpU turn out to be the most reactive substrates in the lot, but in the absence of metal ions, deprotonation of the amidic N–H bond is unlikely at the pH value of our measurements. No significant interaction between substrate and catalyst occurs in the reactant state, as clearly demonstrated by the kinetics (Fig. 2b). As suggested by CPK molecular models, interaction between the relatively large and conformationally mobile catalyst and substrate molecules generates a large number of potentially attractive and repulsive secondary interactions involving distal part structures of the reactants. In the lack of information of spectroscopic nature, a rational interpretation of the role played by uracyl and guanine bases appears to be out of reach.
| 1 (1,2-vicinal) | 2 (1,3-distal) | |||||
|---|---|---|---|---|---|---|
| Entry | NpN′ | 106 × kobs (s−1) | k rel | 106 × kobs (s−1) | k rel | k vicinal/kdistalc |
| a 2.0 mM precatalyst, 0.10 mM NpN′, 10 mM Me4NClO4; 80% DMSO, pH 10.4, 50.0 °C. b Pseudo-first order specific rates kobs calculated from initial rates of HPLC monitored nucleoside liberation. Error limits on the order of ±10%. c For HPNP kvicinal/kdistal = 0.5, see ref. 5. | ||||||
| 1 | GpU | 65 | 88 | 24 | 38 | 2.7 |
| 2 | GpG | 62 | 84 | 14 | 22 | 4.4 |
| 3 | UpU | 14 | 19 | 9.8 | 16 | 1.4 |
| 4 | ApG | 2.4 | 3.2 | 1.3 | 2.1 | 1.8 |
| 5 | GpA | 1.0 | 1.3 | 1.3 | 2.1 | 0.8 |
| 6 | CpC | 2.2 | 3.0 | 1.2 | 1.9 | 1.7 |
| 7 | CpA | 0.74 | 1.0 | 0.92 | 1.5 | 0.8 |
| 8 | UpG | 1.9 | 2.6 | 0.63 | 1.0 | 3.0 |
There is a definite tendency for the 1,2-vicinal regioisomer 1 to be a better catalyst than its 1,3-distal regioisomer 2 in the reaction of most substrates, but the range of kvicinal/kdistal ratios (Table 1) is within a factor of 5. Modest inversions are observed for the reaction of GpA and CpA (entries 5 and 7), for which the 1,3-distal regioisomer is a slightly better catalyst, as it is for the cleavage of HPNP,5 for which kvicinal/kdistal = 0.5.
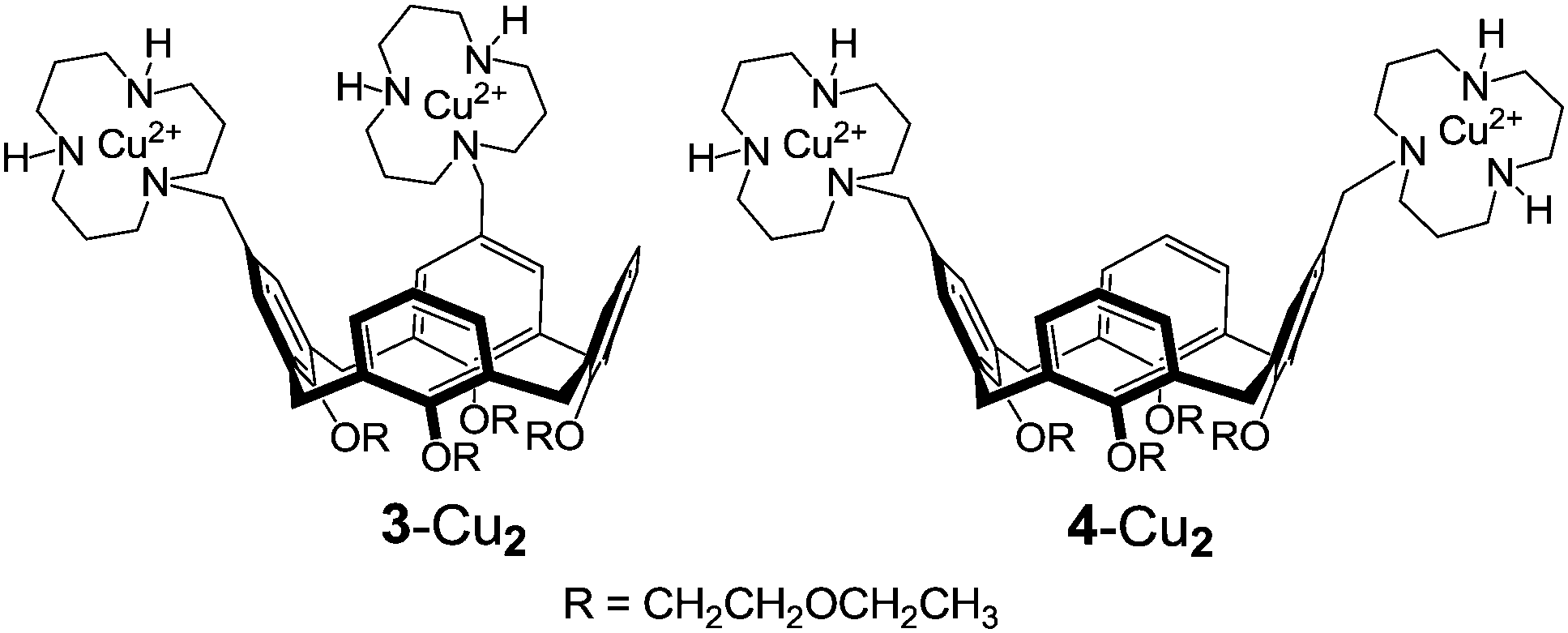
Interestingly, the kvicinal/kdistal ratio for the cleavage of CpA, UpU, and HPNP catalyzed by bimetallic complexes 3-Cu2 and 4-Cu2 (H2O, pH 7.0) are 2.8, 114, and 28, in the given order.2d Although the comparison is based on a limited set of substrates, it appears that the relative position of the two catalytic units in the diguanidinocalix[4]arenes has but a moderate influence on catalytic efficiency. This suggests the existence of a certain degree of flexibility in transition states in which contacts between catalyst and substrate are primarily provided by proton bridges (Fig. 1). In contrast, the marked preference exhibited by UpU and HPNP for the 1,2-vicinal catalyst 3-Cu2 is most likely ascribable to the more stringent geometrical requirements of coordinative bonds to copper(II) involved in the double Lewis acid activation.2d
In order to compare the catalytic efficiency of 1 and 2 in the cleavage of diribonucleoside monophosphates vs. HPNP, catalytic rates relative to background (kobs/kbg) are required. Initial rates of the hydroxide catalyzed cleavage of CpA and GpU, measured in the presence of 1.0 mM Me4NOH (pH 15.4), gave kbg values of 5.7 × 10−5 s−1 and 9.4 × 10−5 s−1, respectively. These values were extrapolated to pH 10.4 under the assumption that the reaction is specific base catalyzed, on the analogy of the corresponding reaction of HPNP, that was found to be strictly first order in hydroxide concentration in the pH range 9.3–13.0.5 The results listed in Table 2 show that both 1 and 2 are from 1 to 2 orders of magnitude more effective in the cleavage of CpA and GpU than in the cleavage of HPNP. Thus, replacement of a good leaving group with a bad leaving group has a favorable effect on catalytic efficiency.
Both an associative two step (AN + DN) mechanism involving a pentavalent phosphorane dianion intermediate, and a concerted (ANDN) mechanism in which no intermediate is involved, are likely possibilities for the hydrolysis of phosphate diesters.2o The reactions of phosphate diesters with good leaving groups are believed to proceed via a one step (ANDN) mechanism,11 also labeled as SN2(P).12 When the leaving group is poor the question of mechanism is still under debate, but there is little doubt that upon replacement of a good leaving group with a poor one the transition state becomes tighter, i.e. more associative in character and, consequently, bears a close resemblance to a pentavalent phosphorane dianion, independent of whether the mechanism is (AN + DN) or (ANDN). Accordingly, the larger rate enhancements experienced by the reactions of NpN′ substrates are understood as arising from a stronger electrophilic/electrostatic stabilization of the transition state by the guanidinium unit of the bifunctional catalysts.
As a final comment, the close similarity of kbg values measured for CpA and GpU is consistent with the fact that rates of background cleavage of the phosphodiester bond of diribonucleoside monophosphates are affected by nucleobase identity to a moderate extent.8a,13 It seems therefore reasonable to assume that the kbg values actually measured for two members of the series are representative of the whole series and, consequently, that rate enhancements as high as 103–105 characterize the performances of catalysts 1 and 2 in the cleavage of the eight NpN′ investigated substrates. These values are comparable in magnitude with rate accelerations reported for artificial di- and trimetallic phosphodiesterases.2d,2j,10,14
Conclusions
To sum up, we have shown that diguanidinocalix[4]arenes 1 and 2 catalyze the cleavage of diribonucleoside monophosphates much more efficiently than the cleavage of HPNP, most likely on account of a greater negative charge development on the nonbridging oxygen atoms in the transition state of the reaction of phosphodiesters with bad leaving groups. To the best of our knowledge, reactivity data related to the cleavage of a series of diribonucleoside monophosphates catalyzed by nonmetallic synthetic catalysts are here presented for the first time. When combined with data from previous studies, the data reported in this work reinforce the notion that cone-calix[4]arenes are useful platforms for the design of efficient bifunctional catalysts.15 The good catalytic efficiency and selectivity of the diguanidino derivatives described in this work encourages further studies in the cleavage of RNA polynucleotides.Experimental
Instruments and general methods
Warning! Care was taken when handling tetramethylammonium perchlorate because it is potentially explosive. No accident occurred in the course of the present work.
:0 to 85:15 in 25 min, flow 0.9 mL min−1. The pseudo-first-order rate constant for the hydroxide catalyzed cleavage of CpA and GpU was measured at 50.0 °C in the presence of 1.0 mM Me4NOH (pOH 3.0), 10 mM Me4NClO4, by HPLC monitoring of the nucleoside liberation (initial rate method).
Acknowledgements
Thanks are due to the Ministero dell'Istruzione e dell'Università e della Ricerca (MIUR, PRIN 2010 JMAZML-006) and the Consiglio Nazionale delle Ricerche (CNR) for financial support.Notes and references
- R. Wolfenden, Chem. Rev., 2006, 106, 3379 CrossRef CAS PubMed.
- (a) P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, Chem. Soc. Rev., 2000, 29, 75 RSC; (b) J. R. Morrow and O. Iranzo, Curr. Opin. Chem. Biol., 2004, 8, 192 CrossRef CAS PubMed; (c) R. Cacciapaglia, A. Casnati, L. Mandolini, D. N. Reinhoudt, R. Salvio, A. Sartori and R. Ungaro, J. Org. Chem., 2005, 70, 624 CrossRef CAS PubMed; (d) R. Cacciapaglia, A. Casnati, L. Mandolini, D. N. Reinhoudt, R. Salvio, A. Sartori and R. Ungaro, J. Am. Chem. Soc., 2006, 128, 12322 CrossRef CAS PubMed; (e) A. Scarso, G. Zaupa, F. B. Houillon, L. J. Prins and P. Scrimin, J. Org. Chem., 2007, 72, 376 CrossRef CAS PubMed; (f) R. Cacciapaglia, A. Casnati, L. Mandolini, A. Peracchi, D. N. Reinhoudt, R. Salvio, A. Sartori and R. Ungaro, J. Am. Chem. Soc., 2007, 129, 12512 CrossRef CAS; (g) T.-S. A. Tseng and J. N. Burstyn, Chem. Commun., 2008, 6209 RSC; (h) R. Bonomi, F. Selvestrel, V. Lombardo, C. Sissi, S. Polizzi, F. Mancin, U. Tonellato and P. Scrimin, J. Am. Chem. Soc., 2008, 130, 15744 CrossRef CAS PubMed; (i) C. Bazzicalupi, A. Bencini, C. Bonaccini, C. Giorgi, P. Gratteri, S. Moro, M. Palumbo, A. Simionato, J. Sgrignani, C. Sissi and B. Valtancoli, Inorg. Chem., 2008, 47, 5473 CrossRef CAS; (j) K. Nwe, C. M. Andolina and J. R. Morrow, J. Am. Chem. Soc., 2008, 130, 14861 CrossRef CAS PubMed; (k) H. Katada and M. Komiyama, ChemBioChem, 2009, 10, 1279 CrossRef CAS PubMed; (l) M. F. Mohamed and R. S. Brown, J. Org. Chem., 2010, 75, 8471 CrossRef CAS PubMed; (m) R. Salvio, R. Cacciapaglia and L. Mandolini, J. Org. Chem., 2011, 76, 5438 CrossRef CAS PubMed; (n) H. Lönnberg, Org. Biomol. Chem., 2011, 9, 1687 RSC; (o) F. Mancin, P. Scrimin and P. Tecilla, Chem. Commun., 2012, 48, 5545 RSC; (p) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734 RSC.
- (a) A. M. Piatek, M. Gray and E. V. Anslyn, J. Am. Chem. Soc., 2004, 126, 9878 CrossRef CAS; (b) U. Scheffer, A. Strick, V. Ludwig, S. Peter, E. Kalden and M. W. Göbel, J. Am. Chem. Soc., 2005, 127, 2211 CrossRef CAS PubMed; (c) C. Gnaccarini, S. Peter, U. Scheffer, S. Vonhoff, S. Klussmann and M. W. Göbel, J. Am. Chem. Soc., 2006, 128, 8063 CrossRef CAS PubMed; (d) N. J. V. Lindgren, J. R. Lars Geiger, C. Schmuck and L. Baltzer, Angew. Chem., Int. Ed., 2009, 48, 6722 CrossRef CAS PubMed; (e) J. M. Thomas, J.-K. Yoon and D. M. Perrin, J. Am. Chem. Soc., 2009, 131, 5648 CrossRef CAS PubMed; (f) M. Hollenstein, C. J. Hipolito, C. H. Lam and D. M. Perrin, ChemBioChem, 2009, 10, 1988 CrossRef CAS; (g) R. Salvio, A. Casnati, L. Mandolini, F. Sansone and R. Ungaro, Org. Biomol. Chem., 2012, 10, 8941 RSC; (h) R. Salvio and A. Cincotti, RSC Adv., 2014, 4, 28678 RSC.
- D. O. Corona-Martinez, O. Taran and A. K. Yatsimirsky, Org. Biomol. Chem., 2010, 8, 873 CAS.
- L. Baldini, R. Cacciapaglia, A. Casnati, L. Mandolini, R. Salvio, F. Sansone and R. Ungaro, J. Org. Chem., 2012, 77, 3381 CrossRef CAS PubMed.
- R. Salvio, L. Mandolini and C. Savelli, J. Org. Chem., 2013, 78, 7259 CrossRef CAS PubMed.
- (a) A. Demesmaeker, R. Haner, P. Martin and H. E. Moser, Acc. Chem. Res., 1995, 28, 366 CrossRef CAS; (b) B. N. Trawick, A. T. Daniher and J. K. Bashkin, Chem. Rev., 1998, 98, 939 CrossRef CAS PubMed.
- (a) M. Oivanen, S. Kuusela and H. Lönnberg, Chem. Rev., 1998, 98, 961 CrossRef CAS PubMed; (b) K. Worm, F. Chu, K. Matsumoto, M. D. Best, V. Lynch and E. V. Anslyn, Chem.– Eur. J., 2003, 9, 741 CrossRef CAS.
- The pKw for water autoprotolysis in 80% DMSO is 18.4 ( M. M. Kreevoy and E. H. Baughman, J. Phys. Chem., 1974, 78, 421 CrossRef ). This implies that the pH value of a neutral solution is 9.2.
- (a) P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, Angew. Chem., Int. Ed., 1999, 38, 3189 CrossRef CAS; (b) S. Liu and A. D. Hamilton, Chem. Commun., 1999, 587 RSC; (c) M. Komiyama, S. Kina, K. Matsumara, J. Sumaoka, S. Tobey, V. M. Lynch and E. Anslyn, J. Am. Chem. Soc., 2002, 124, 13731 CrossRef CAS PubMed; (d) M. Yashiro, H. Kaneiwa, K. Onaka and M. Komiyama, Dalton Trans., 2004, 605 RSC; (e) Q. Wang, S. Mikkola and H. Lönnberg, Chem. Biodiversity, 2004, 1, 1316 CrossRef CAS PubMed; (f) Q. Wang and H. Lönnberg, J. Am. Chem. Soc., 2006, 128, 10716 CrossRef CAS PubMed; (g) Q. Wang, E. Leino, A. Jancsó, I. Szilágyi, T. Gajda, E. Hietamäki and H. Lönnberg, ChemBioChem, 2008, 9, 1739 CrossRef CAS PubMed.
- W. W. Cleland and A. C. Hengge, Chem. Rev., 2006, 106, 3252 CrossRef CAS PubMed.
- A. J. Kirby, M. Medeiros, J. R. Mora, P. S. M. Oliveira, A. Amer, N. H. Williams and F. Nome, J. Org. Chem., 2013, 78, 1343 CrossRef CAS PubMed.
- M. Komiyama, Carbohydr. Res., 1989, 192, 97 CrossRef CAS.
- (a) N. H. Williams, B. Takasaki, M. Wall and J. Chin, Acc. Chem. Res., 1999, 32, 485 CrossRef CAS; (b) H. Linjalahti, G. Feng, J. C. Mareque-Rivas, S. Mikkola and N. H. Williams, J. Am. Chem. Soc., 2008, 130, 4232 CrossRef CAS PubMed.
- R. Cacciapaglia, S. Di Stefano, L. Mandolini and R. Salvio, Supramol. Chem., 2013, 25, 537 CrossRef CAS , and references cited therein.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC chromatograms for the monitoring of NpN′ cleavage. See DOI: 10.1039/c4ra05751a |
| This journal is © The Royal Society of Chemistry 2014 |