Synthetic methodologies of achiral diarylmethanols, diaryl and triarylmethanes (TRAMs) and medicinal properties of diaryl and triarylmethanes-an overview
Sankalan Mondal
and
Gautam Panda
*
Central Drug Research Institute, Medicinal and Process Chemistry Division, Jankipuram, Lucknow, India. E-mail: gautam.panda@gmail.com
First published on 30th June 2014
Abstract
The last decade has witnessed a high demand of various synthetic approaches towards bioactive achiral diarylmethanols, diaryl and triarylmethanes and the molecules derived thereof. Their biological and therapeutical relevancy in diverse areas such as antimicrobials, infectious, cardiovascular and nervous system disorders, genital tract diseases, estrogen related disorders and bone remodeling is quite well known. These small molecules have also been the starting materials for the development of a variety of pharmaceutically important compounds. Compounds belonging to this family have not only played a leading role in the development of small molecules as therapeutically useful compounds but also have become one of the mainstays for the development of organic synthesis. However, a comprehensive review which covers their synthesis as well as their biological activity is still lacking. (Two reviews cover the synthesis of chiral diarylmethanols through asymmetric aryl transfer, and three reviews cover the photochemical properties of triarylmethanes, bioconjugation, application of trityl ions and the use of triarylmethanes as dyes.) This review describes the synthesis as well as the biological activities of this group of molecules that came up in the last fifteen years (1995–2013). The current review will cover the various approaches followed for the synthesis of achiral diarylmethanols and the strategies followed for the synthesis of achiral diaryl as well as triarylmethanes. Finally, we will also cover the bioactivities of molecules containing the diaryl and triaryl methane core.
 Sankalan Mondal | Sankalan Mondal was born on 13th September, 1986. After receiving his B.Sc. and M.Sc. degree in chemistry in 2007 and 2009 respectively from University of Calcutta, Kolkata, India, he joined Medicinal and Process Chemistry Division of CSIR-Central Drug Research Institutes, Lucknow, India to work on his thesis title entitled “Design, Synthesis, Molecular Modeling Studies and Bioevaluation of Trisubstituted Methanes and Amino Acids Derived Heterocycles” under the supervision of Dr Gautam Panda. |
 Gautam Panda | Gautam Panda was born at Contai, Midnapore, West Bengal, India. After completing B.Sc. in Chemistry (Hons.) from Calcutta University, he obtained M.Sc. in Chemical Sciences from IIT Kharagpur in 1993. Later he completed Ph. D. in 1999 working with Prof. Goverdhan Mehta at University of Hyderabad on classical synthesis of bucky-bowls. He did postdoctoral work as NSERC visiting fellow in the lab of Prof. Howard Alper, University of Ottawa and Dr Prabhat Arya, National Research Council, Ottawa, Canada from 2000–2001. He later joined CSIR-Central Drug Research Institute, Lucknow in 2002 and he is currently holding Principal Scientist at CDRI and Associate Professor Position at Academy of Scientific and Innovative Research (AcSIR), New Delhi. His research interests are identification and chemical biology of new chemical entities in treating tuberculosis, malaria and breast cancer through synthesis of natural products and natural product-like privileged molecules from chiral amino acids and syn-2,3-dihydroxy esters. He is the recipient of Bronze medal from the Chemical Research Society of India for the year 2011–2012 as well as JSPS invitation fellowship from Japan. |
1. Introduction
The desired bioactivity, in principle, may be rationalized with a particular conformational structure of a small molecule and towards this perspective, the design of new chemical entities and synthetic routes for their assembly have typically focused on the quick assembly of diverse compounds with the correct conformations. Also for quickly accessing these pharmaceutically relevant molecules, small building blocks which can be readily obtained with very short synthetic endeavour are desired. In this regard, many small organic molecules containing diaryl and triaryl methyl cores have come up which has caused a high demand of various synthetic approaches towards diarylmethanols, diarylmethanes, triarylmethanes various trisubstituted methanes and the molecules derived thereof. Moreover, these molecules have not only been the cornerstone for the development of small molecules as therapeutic agents but also have been the pioneer for the development of asymmetric synthesis. For example the diaryl methanol core has formed an important structural subunit in many ligands (like TADDOL, CBS catalyst etc.) used in asymmetric synthesis of diverse array of compounds.Though there are excellent reviews on the asymmetric synthesis of the diarylmethanols,1 to the best of our knowledge there are no reviews that cover the synthesis of achiral diarylmethanols, which are also synthetically important fragment and has led to the development of synthetic organic and organometallic chemistry.
2. Diarylmethanols
The diaryl hydroxy methyl group has become an integral part in the development of the asymmetric synthesis of many compounds. Though itself not stereogenic, this structural unit has become the backbone of many chiral auxiliaries, ligands, as well as catalysts with the two flanking aryl groups playing a pivotal role in maintaining or enhancing the stereoselectivity. TADDOL (1) and CBS catalyst (2) exemplify the versatility and the importance of this “magic” group in the asymmetric synthesis (Fig. 1).2 Achiral diarylmethanols (3) and (4) are also used for the kinetic resolution of racemic α-aryl propanoic acids, α-aryl butanoic acids and β-substituted α-aryl propanoic acids which form an integral part of many pharmaceutically relevant molecules.3 Moreover Lewis acid or protic acid treatment of diarylmethanols, o-aryloxy diarylmethanols or chroman-aryl methanols followed by nucleophilic substitution yields various trisubstituted methanes, xanthenes or xanthenes like analogues which are important structural motifs/building blocks in organic chemistry as well as in pharmaceutical sciences.4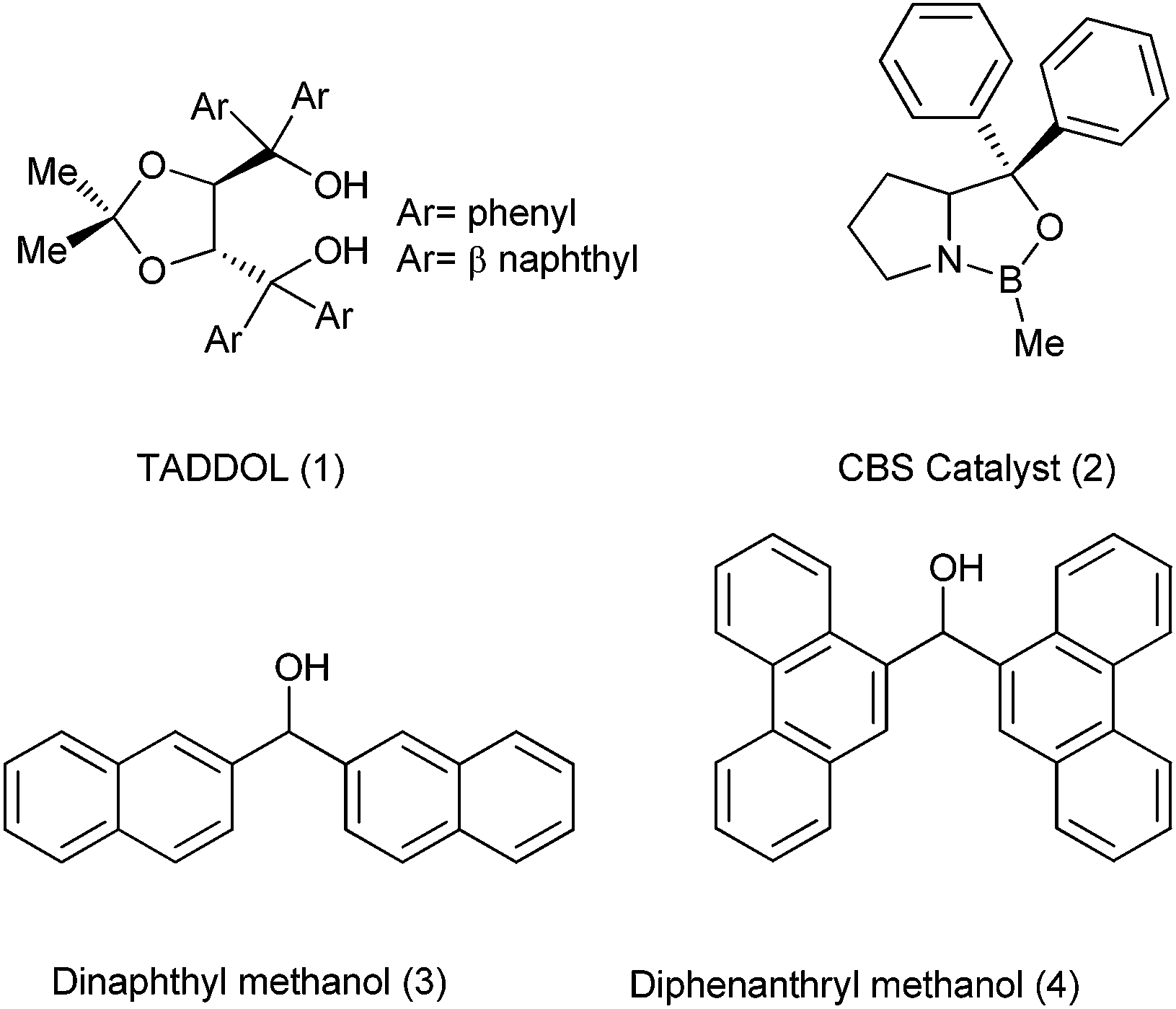 | ||
| Fig. 1 Some important molecules containing diarylmethanol motif used in the asymmetric synthesis. | ||
The most traditional way to achieve the synthesis of diarylmethanols is the treatment of the appropriate organo lithium, magnesium or zinc reagents with aromatic aldehydes. Preparation of such reagents requires usage of highly reactive metals like magnesium and lithium, leading to various competing side reactions, unstable in air or moisture making it difficult for the easy handling of these reagents. Moreover due to the high reactivity of these organometallic reagents, these reagents are incompatible with many functional groups. So, for the efficient synthesis of diarylmethanols as well as various related molecules, various synthetic protocols were developed which have caused a renaissance in the field of synthetic organic chemistry. Though these types of molecules are very much important from the synthetic as well as the pharmaceutical perspective, to the best of our knowledge there are no reviews which cover the synthesis of this benign group. We will cover the synthetic strategies followed for the preparation of achiral/racemic diarylmethanols in the last two decades (from 1995–2013).
3. Metal catalyzed synthesis of achiral or racemic diarylmethanols
3.1 Magnesium iodine/bromine exchange reaction to prepare functionalized Grignard reagents
Towards the preparation of highly functionalized organomagnesium reagents, P. Knochel and coworkers first reported the use of the concept of magnesium iodine exchange at low temperature to produce Grignard reagents which contain functional groups like ester, cyano and amide functional groups which are in general incompatible with Grignard reagents and used them in chemoselective arylation of the aldehydes (Scheme 1).5 Later, they could further extend this magnesium iodine exchange for the preparation of Grignard reagents in solid supports and for the preparation of polyfunctional organometallic reagents like those containing nitro or hydroxyl groups.6–8 Interestingly, for the preparation of ortho nitro Grignard reagent from 2,5-diiodo nitrobenzene (5) the iodine atom ortho to the nitro group was replaced leading to the chemoselective preparation of the corresponding Grignard reagent and this when quenched with benzaldehyde (6) yielded the diarylmethanol (7) in 89% yield (Scheme 2).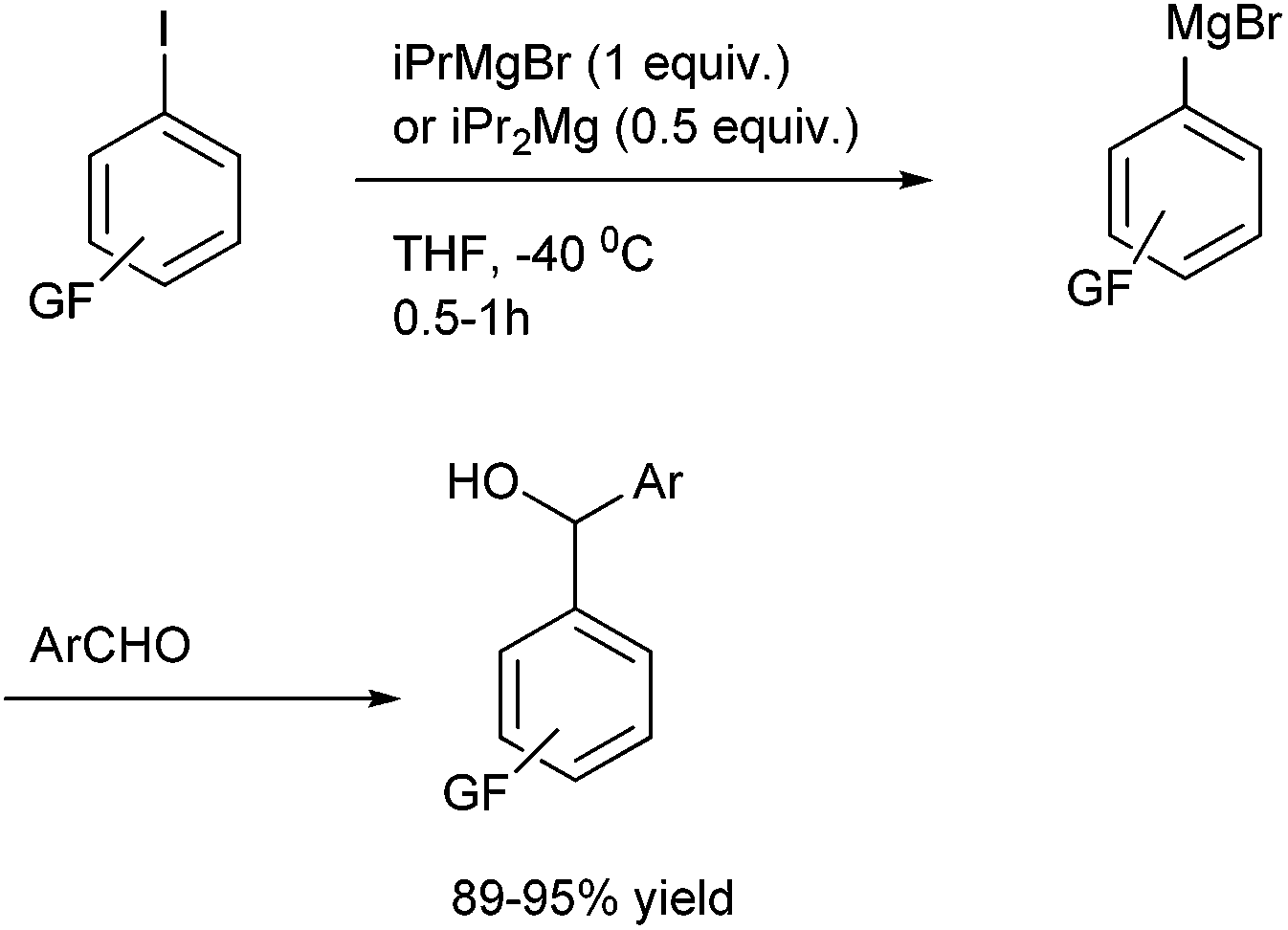 | ||
| Scheme 1 | ||
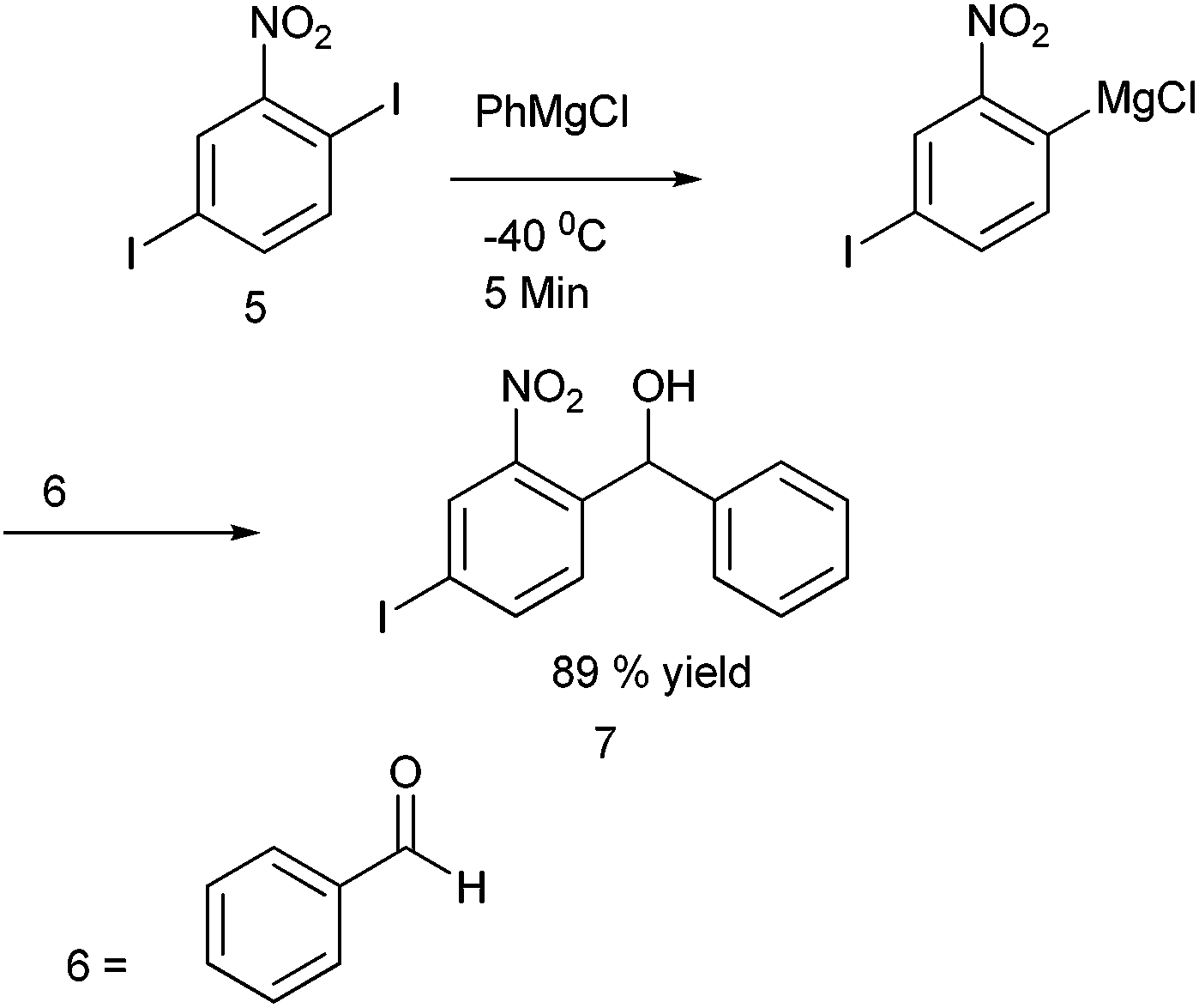 | ||
| Scheme 2 | ||
K. Oshima and coworkers were able to overcome the limitations of Knochel's protocol by using organomagnesium Ate complex for the magnesium halogen exchange reaction.9 This protocol was suitable for not only electron poor aryl halides but also for electron rich aryl halides and they noted that quenching the Grignard reagents with benzaldehyde furnishes an excellent yield of diarylmethanols (Scheme 3).
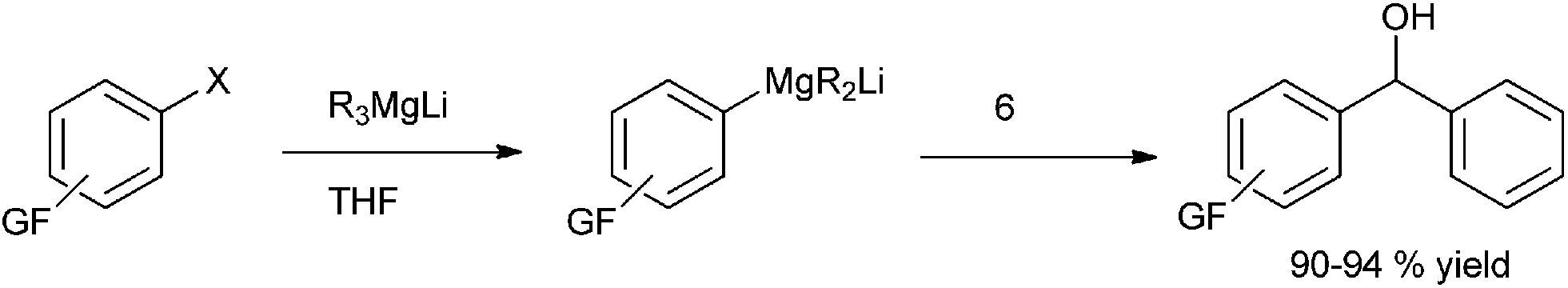 | ||
| Scheme 3 | ||
3.2 Electrochemical coupling of aryl halides with aromatic aldehydes
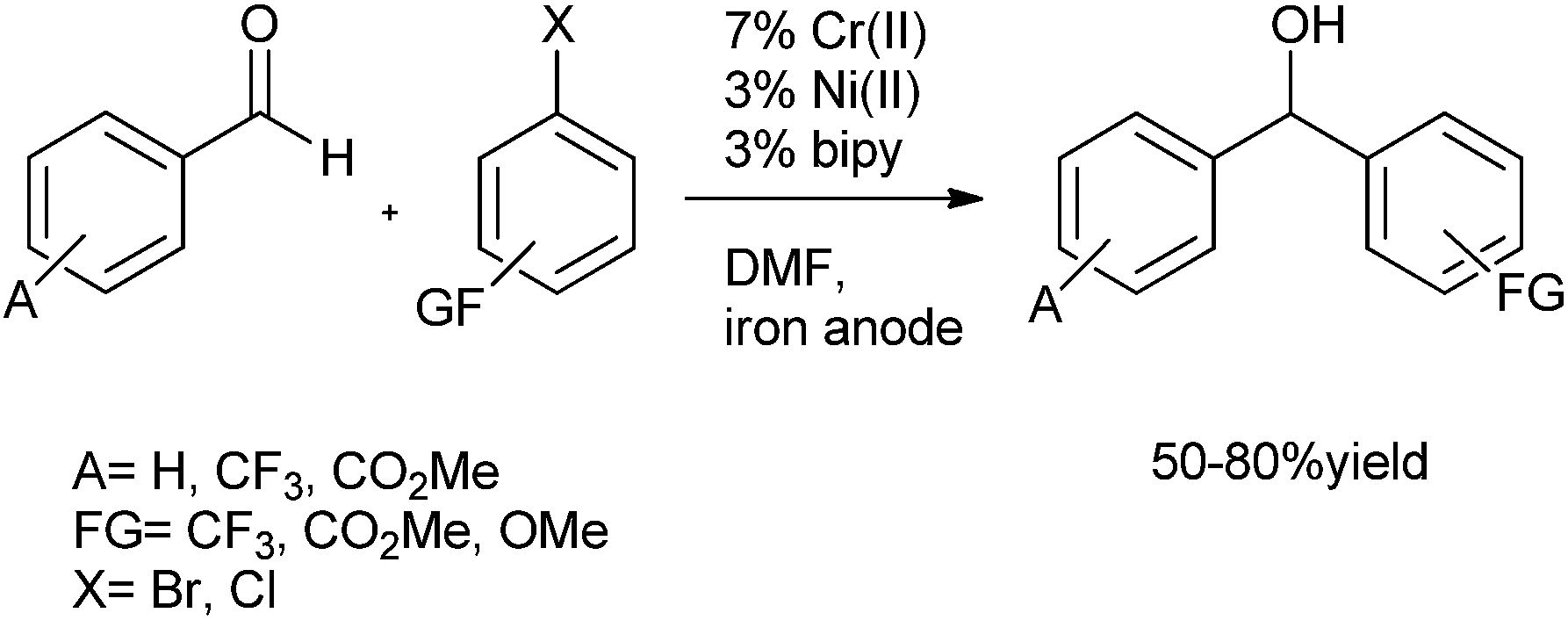 | ||
| Scheme 4 | ||
3.3 Nickel catalyzed arylation of aldehydes
C. H. Cheng et al. first reported the nickel catalyzed arylation of aromatic aldehydes using aryl halides as aryl source.12 Thus using 4-bromo anisole (8) as aryl halide, compound (6) as aldehyde, Ni(dppe)2Br2 as Nickel source, Zinc powder and THF, compound (9) was obtained in high yield (91%) (Scheme 5).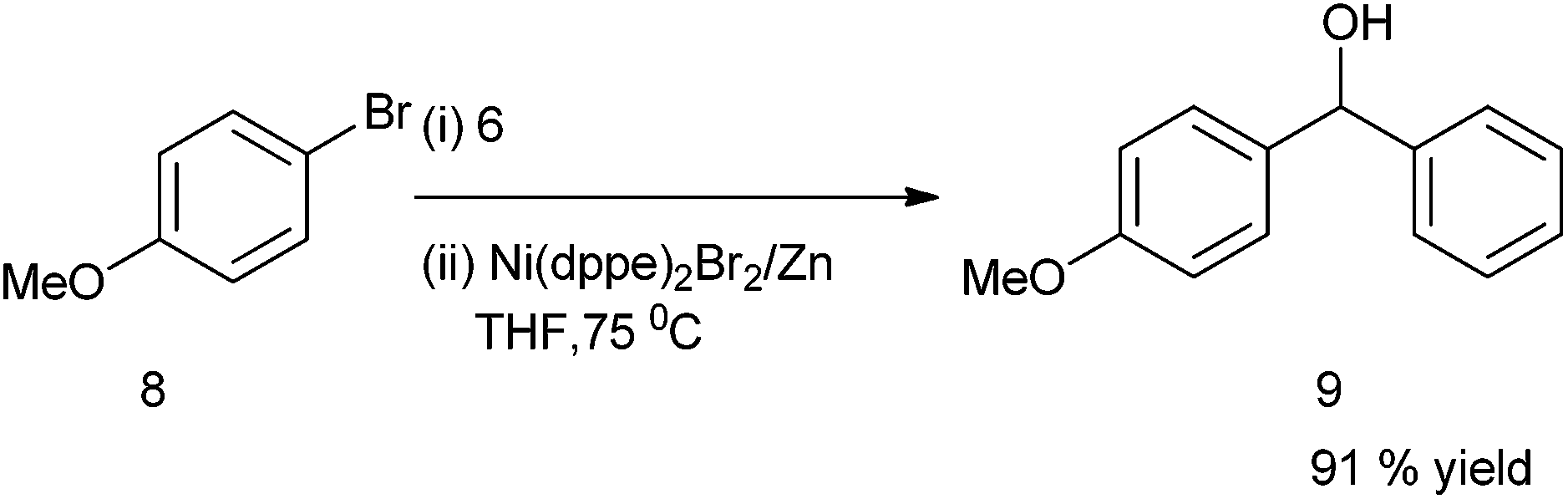 | ||
| Scheme 5 | ||
The authors noted that temperature of the reaction was crucial for maintaining yield and selectivity. At 66 °C (refluxing THF), using (8) and (6) as starting materials, they obtained only 52% of the diaryl methanol (9) along with good amount of the pinacol product (10) while at higher temperature (110 °C) the authors could get 67% of diaryl methanol (9) along with the ketone (11) (Scheme 6). The optimum temperature was found to be 75 °C.
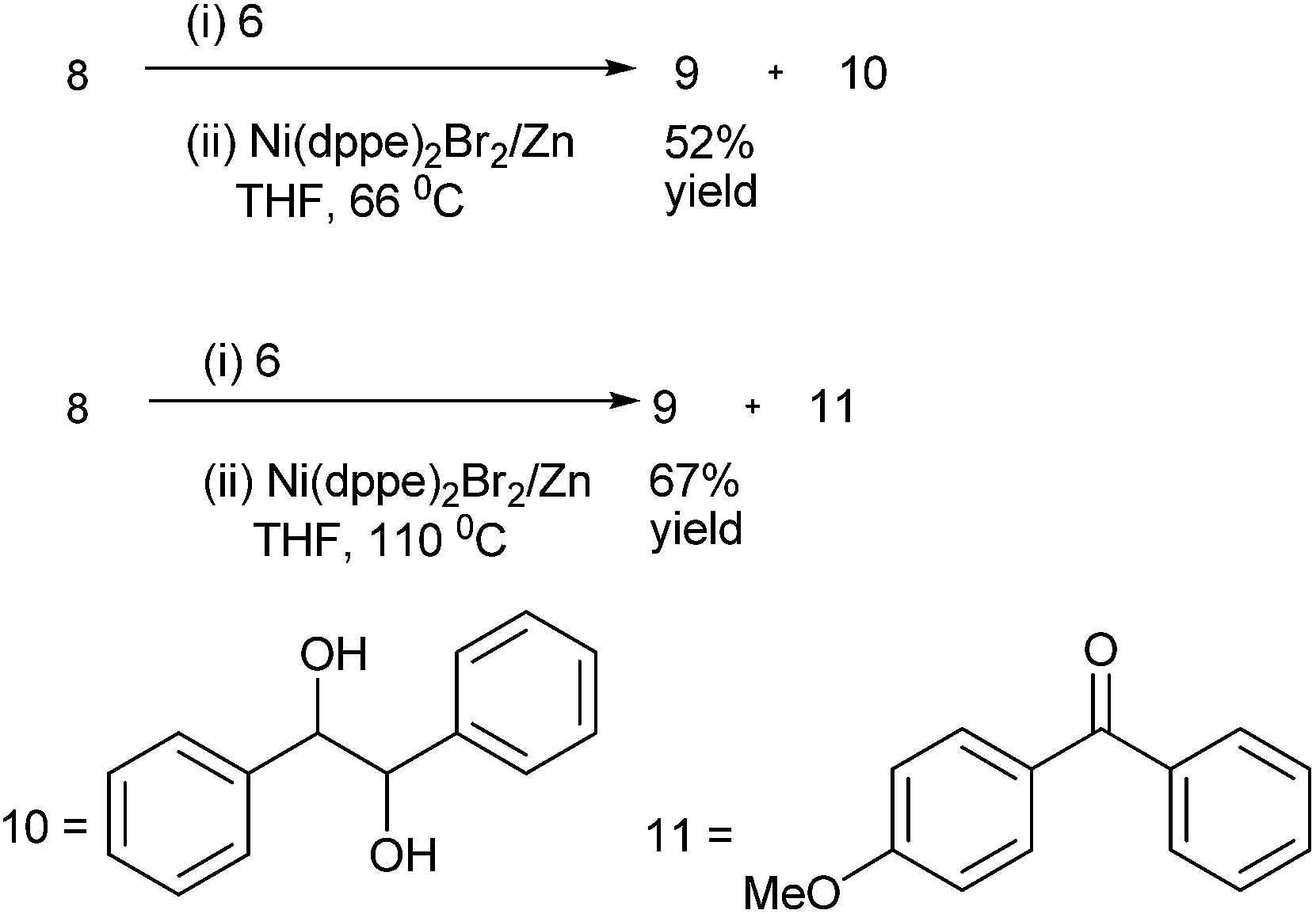 | ||
| Scheme 6 | ||
E. Shirakawa et al. in 2005 delineated that alkynes can act as co-catalyst in nickel catalyzed arylation of aldehydes with organoboronates as the aryl source (Scheme 7).13 The alkynes acted as the additives. Thus using Ni(cod)2/4-octyne as catalyst, 4-trifluoromethyl benzaldehyde (12) as the aldehyde and 2-p-toluyl-1,3,2-dioxaboronate (13) as the aryl source, they could prepare p-tolyl ((4-trifluoromethyl)phenyl) methanol (14) in 93% yield.
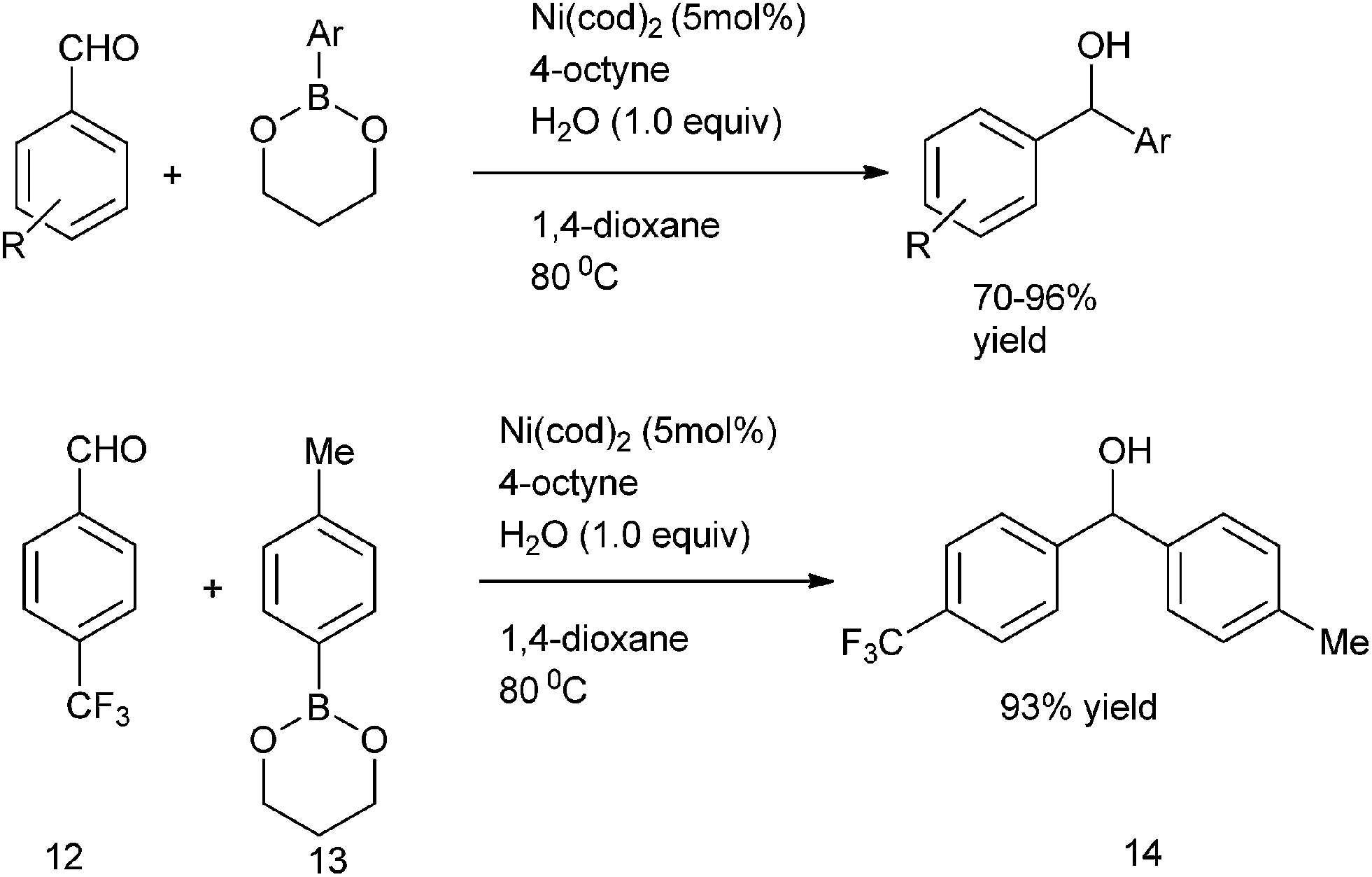 | ||
| Scheme 7 | ||
M. Bao et al. reported nickel catalyzed arylation of aromatic aldehydes using aryl boronic acids as the aryl source in IPA or toluene–IPA mixture in the ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in presence of potassium phosphate as the base to obtain the diarylmethanols in very high yields (Scheme 8).14
:1 in presence of potassium phosphate as the base to obtain the diarylmethanols in very high yields (Scheme 8).14
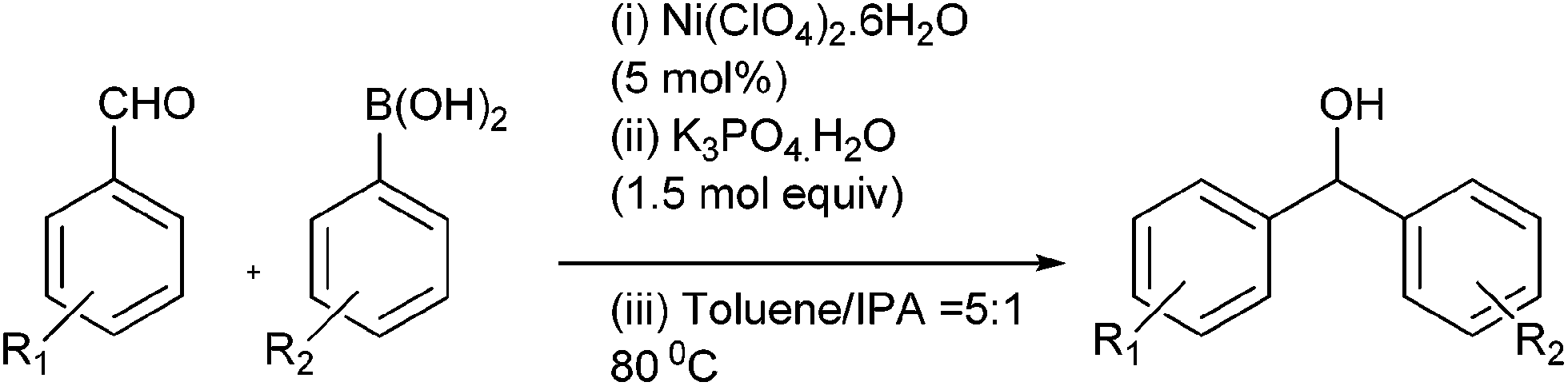 | ||
| Scheme 8 | ||
K. Itami et al. reported the use of 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) (15) ligated Ni(cod)2 catalyst for the addition of organoboronate esters to aldehydes and ketones (Scheme 9).15 Interestingly, the addition of organoboronate esters to ketones required caesium fluoride as an additive but for the aldehydes no such additives were required. This arylation procedure is highly chemoselective with the aldehyde group being arylated in preference to the aryl chlorides, ketones or nitriles. Interestingly, the authors noticed that when 1-(6-methoxynaphthalen-2-yl) ethanone (16) was used, change of the bulky ligand (15) to PCy3 results in the arylation of the methoxy group in preference to the carbonyl group furnishing compound (18) while when (15) was used, the carbonyl group was orthogonally arylated to compound (17) (Scheme 10).
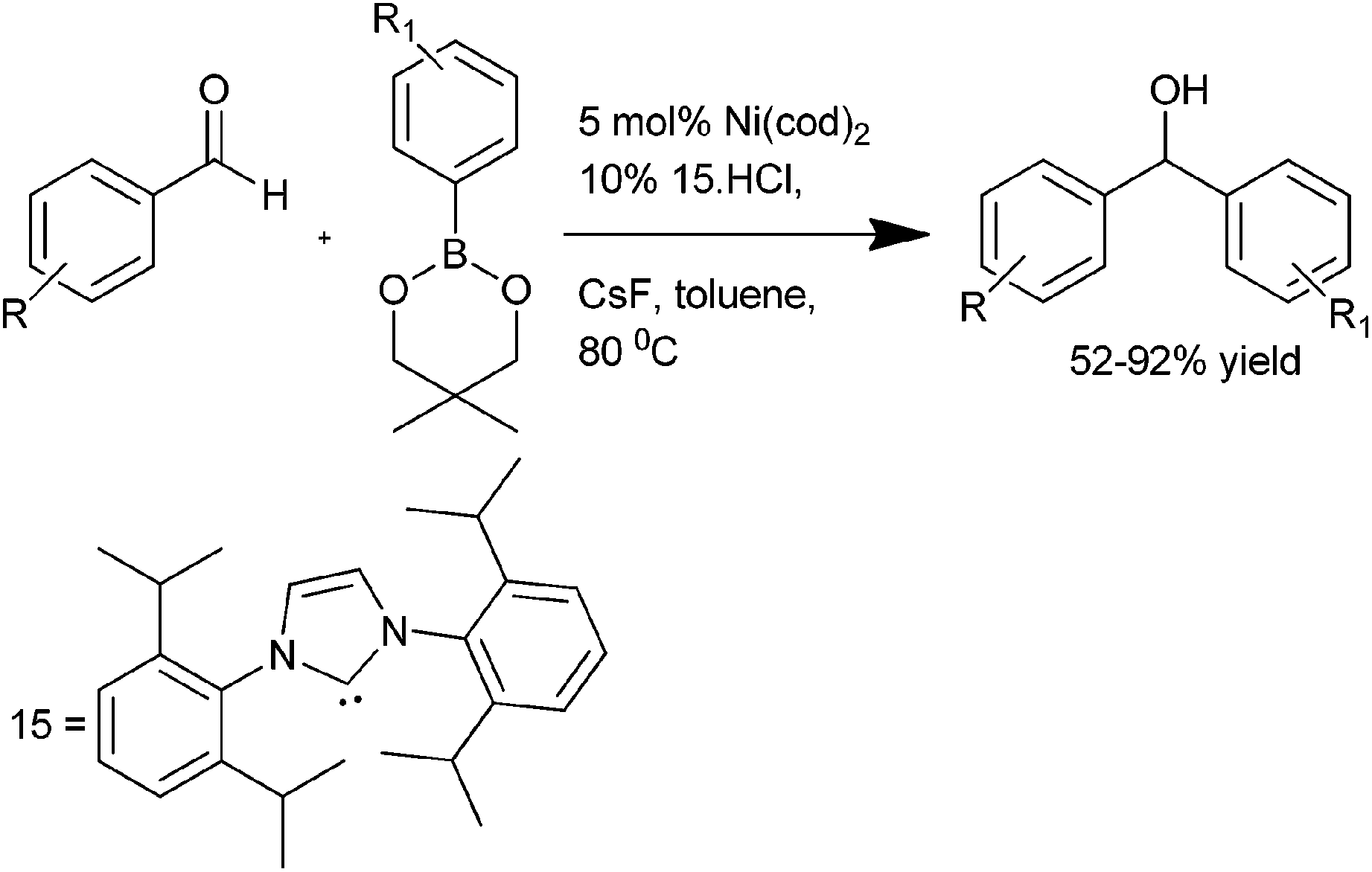 | ||
| Scheme 9 | ||
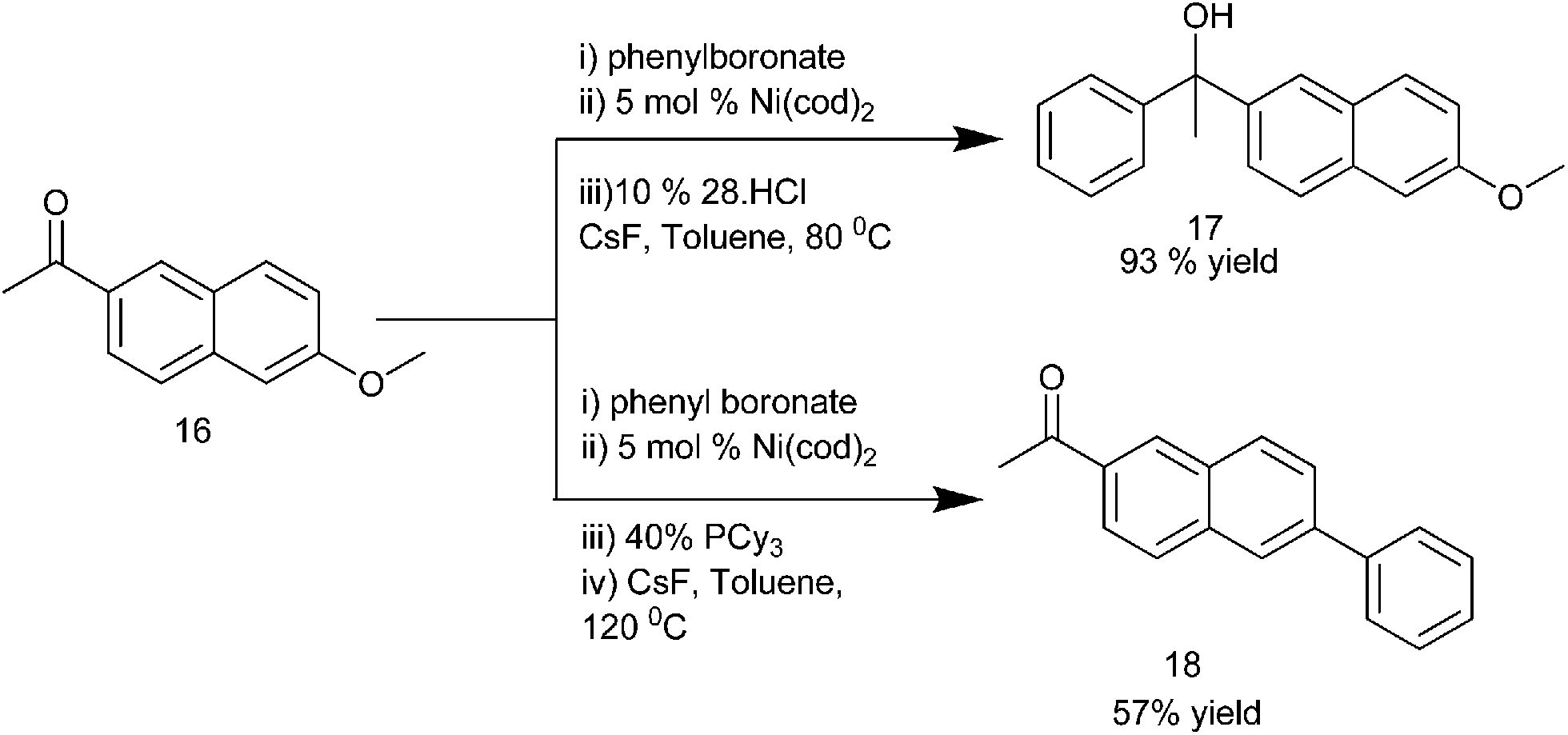 | ||
| Scheme 10 | ||
Q. S. Hu and co-workers reported 4-chloro benzaldehyde, 4-chloro acetophenones and 4-chloro benzophenones ligated Ni(cod)2 to be excellent catalytic system for the arylation of aldehydes using aryl boroxines as the arylating agent to obtain the diarylmethanols in excellent yields (Scheme 11).16 4-chloro acetophenones or 4-chloro benzophenones acted as ligands to the nickel catalyst.
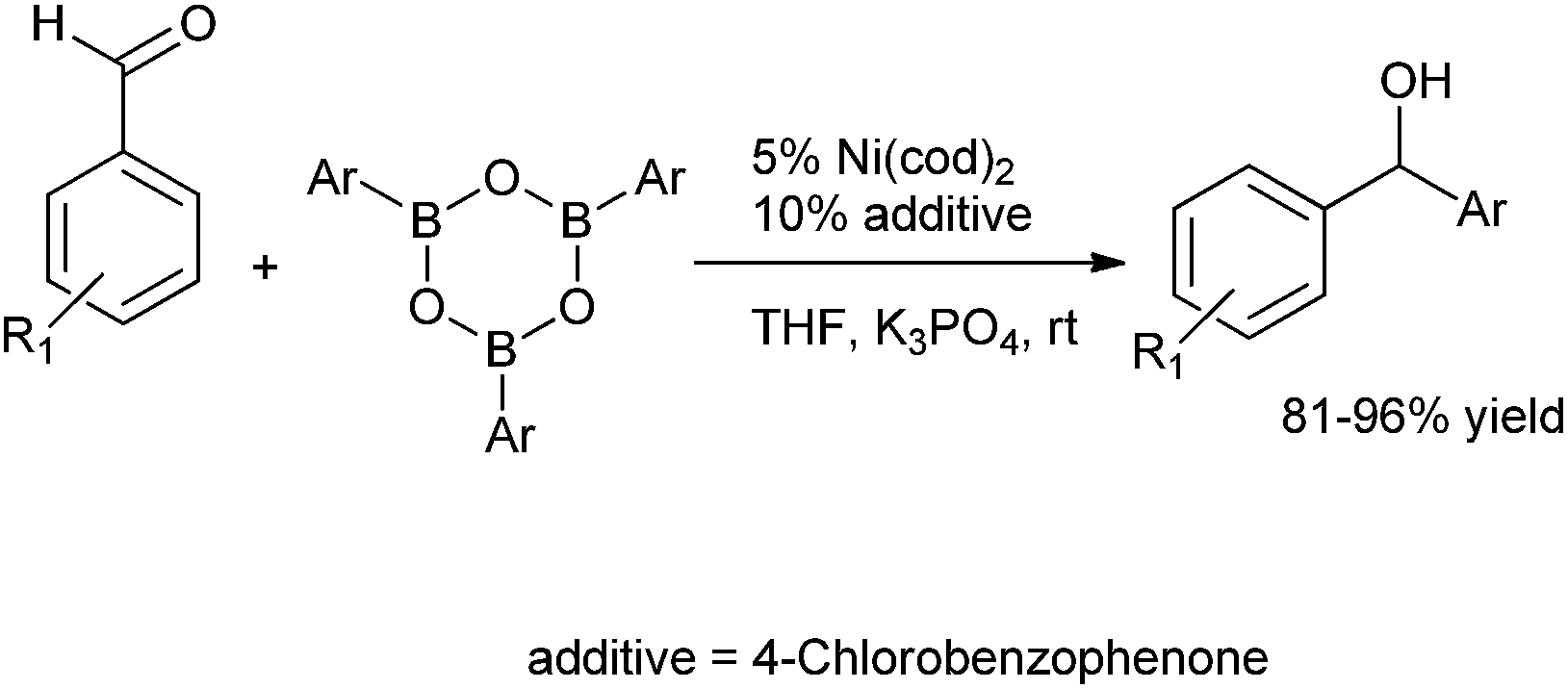 | ||
| Scheme 11 | ||
3.4 Rhodium catalyzed arylation of aromatic aldehydes
C. J. Li and coworkers reported Grignard type phenylation of aldehydes in water under air using a catalytic amount of Rh(COD)2BF4 and phenyltrimethyl tin or phenyltributyl tin as arylating agent (Scheme 12).17 Using this procedure, various aromatic aldehydes could be arylated to give the respective diarylmethanols in good yields. They noted that presence of sodium fluoride as an additive increased the yield of the product. The current protocol was compatible with several reactive functional groups that were incompatible with Grignard reagents. Interestingly, aryl halides were inert under the present conditions, thereby giving rise to chemoselective arylation of aldehydes.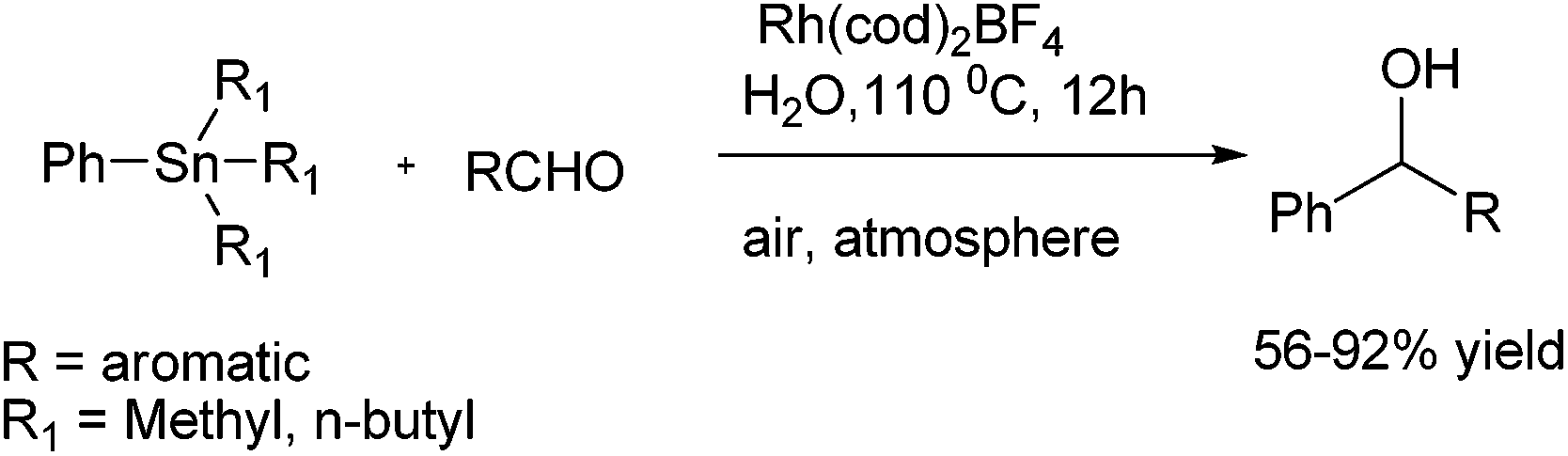 | ||
| Scheme 12 | ||
D. A. Sweigart and coworkers reported the use of redox non-innocent hydroquinone as a ligand in rhodium complexes for the arylation of aromatic aldehydes (Scheme 13).18 However the parent hydroquinone complex was incapable in the catalysis but treatment with potassium tertiary butoxide yielded a quinonoid complex that acted as a heterobimetallic catalyst. Further the fact that the anionic quinonoid ligand acts as a ligand for the boronic acid as well as a Lewis acid receptor site highlights that it plays a bifunctional role (Fig. 2).
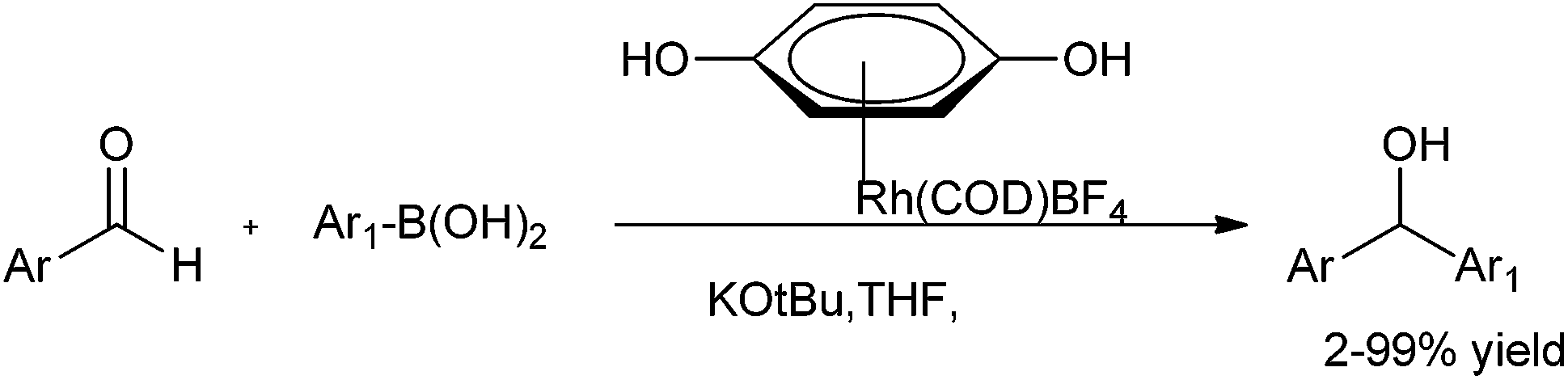 | ||
| Scheme 13 | ||
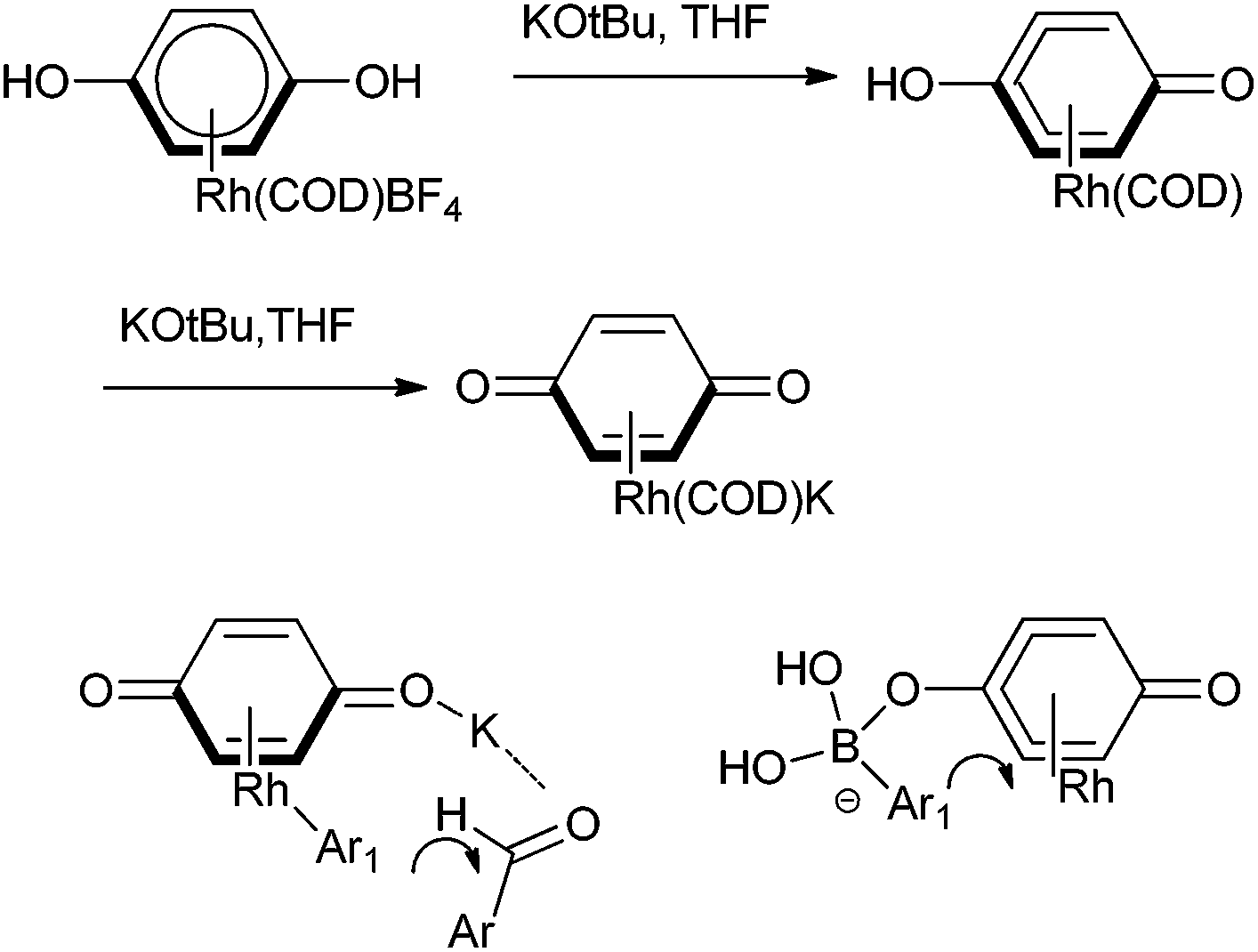 | ||
| Fig. 2 Preparation of rhodium quinoid complexes and its dual role in arylation of aldehydes. | ||
C. G. Frost et al. reported rhodium catalyzed arylation of aromatic aldehydes and ketones using a water soluble sulfonated biaryl phosphine ligand (19) (Scheme 14).19 The water soluble ligand allows for the catalyst recycling.
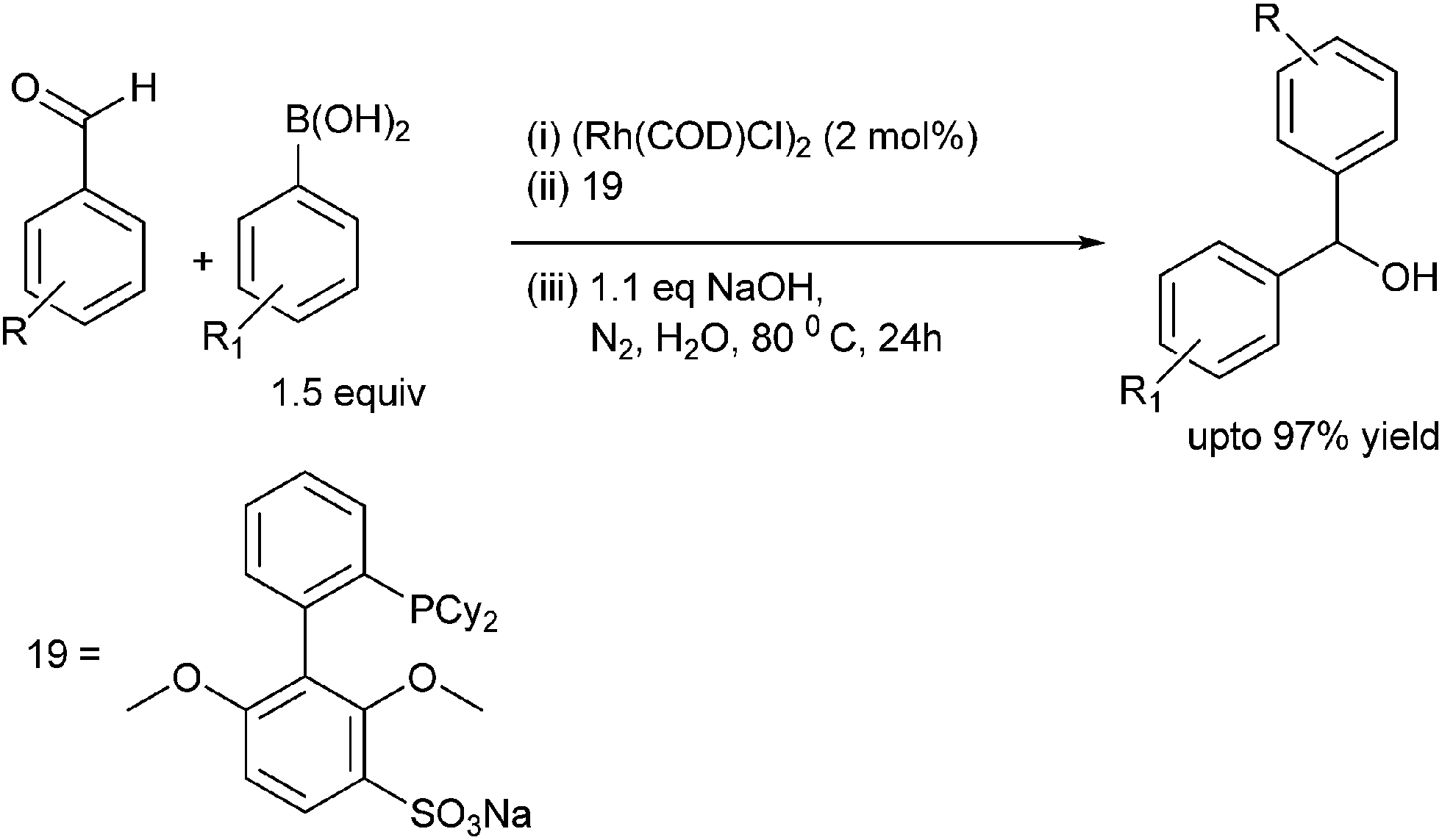 | ||
| Scheme 14 | ||
3.5 Palladium catalyzed arylation of aromatic aldehydes
T. Ohta et al. reported palladium catalyzed arylation of aldehydes using arylboronic acid as the arylating agent and triphenyl phosphine ligated palladium(0) complex as the catalyst.20 They observed addition of chloroform was necessary for the reaction. Later K. Kondo and T. Aoyama improved the protocol by using sodium tertiary butoxide as the base and DME–H2O (5:1) as the solvent and (±)-tol-BINAP as the ligand (Scheme 15).21
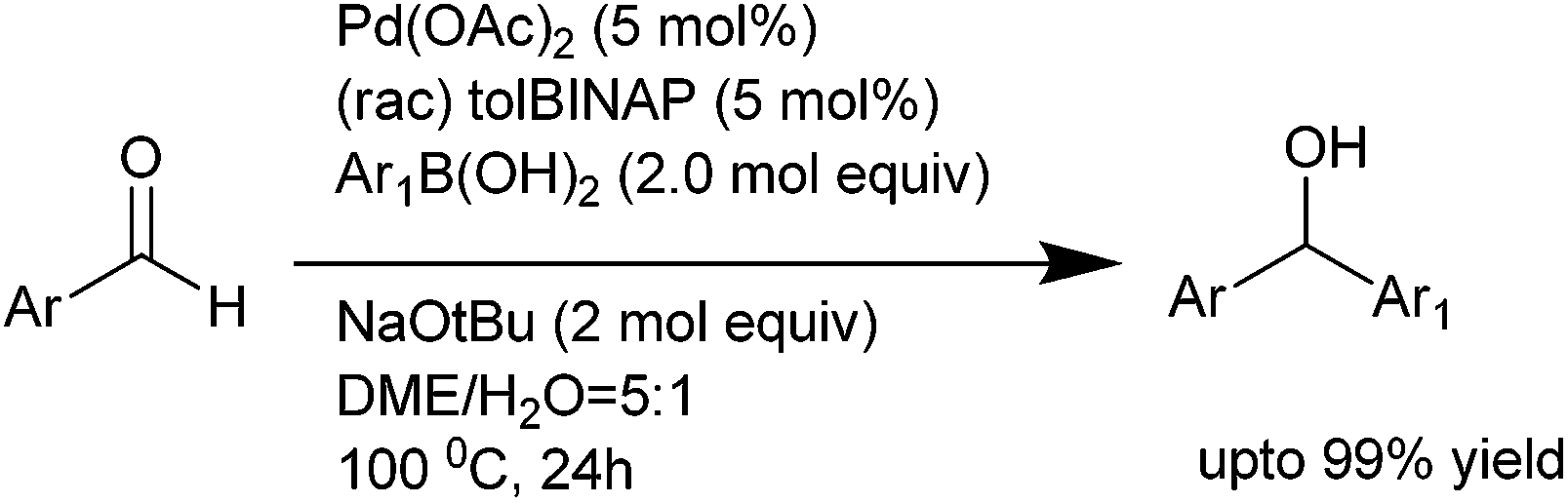 | ||
| Scheme 15 | ||
Q. S. Hu and co-workers in 2007 first reported the use of anionic 4 electron donor based palladacycles (20a–d) (Fig. 3) for the arylation of aromatic aldehydes using aryl boronic acids as the arylating agents.22,23 Thus using the ferrocenyl based palladacycle (20d), they could prepare the respective diarylmethanols in excellent yields (up to 99%) at room temperature (Scheme 16). Later on they reported a more diverse method by changing the ligand to aromatic phosphites and phosphinites.
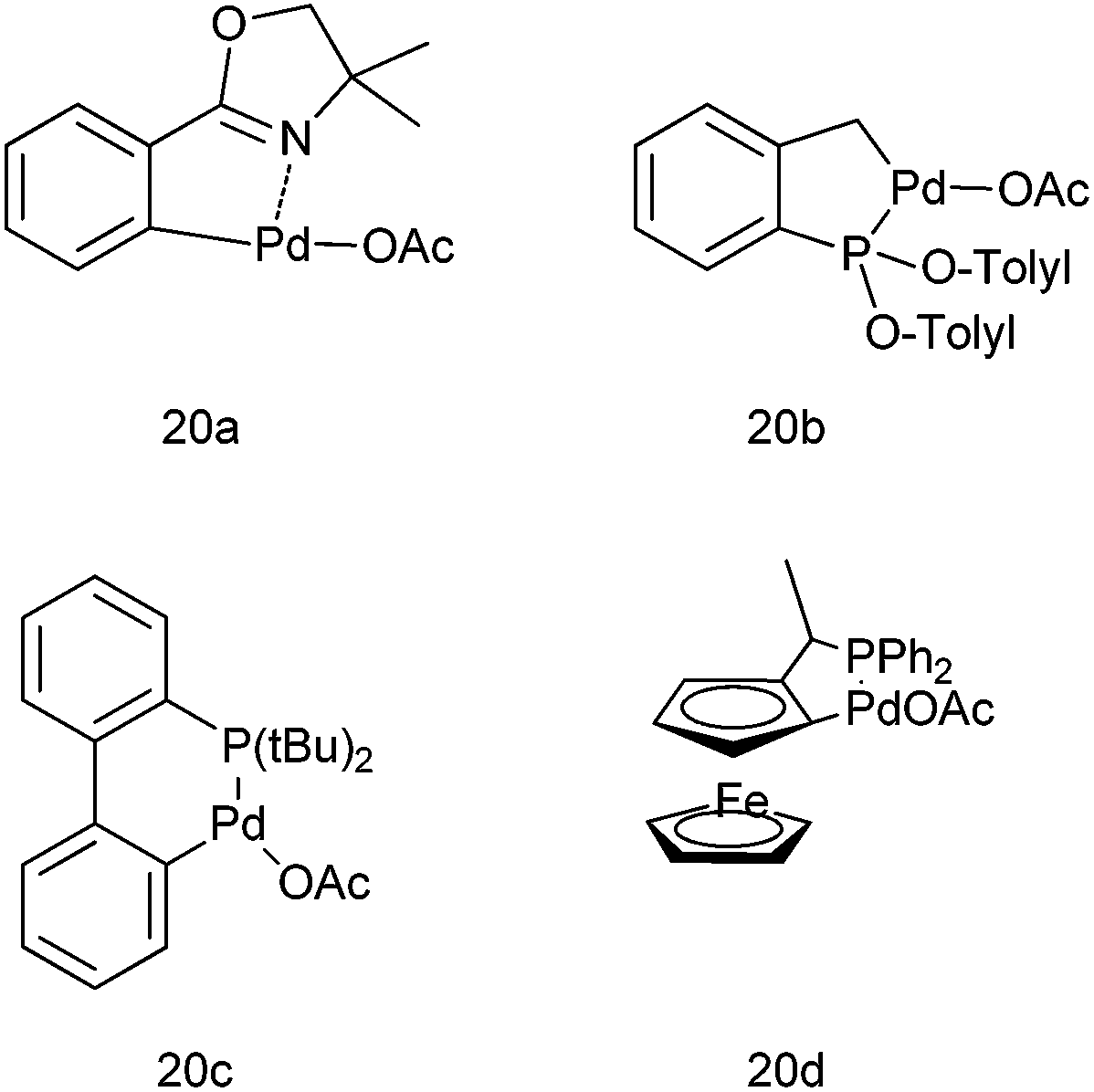 | ||
| Fig. 3 Palladacycles used in Scheme 16. | ||
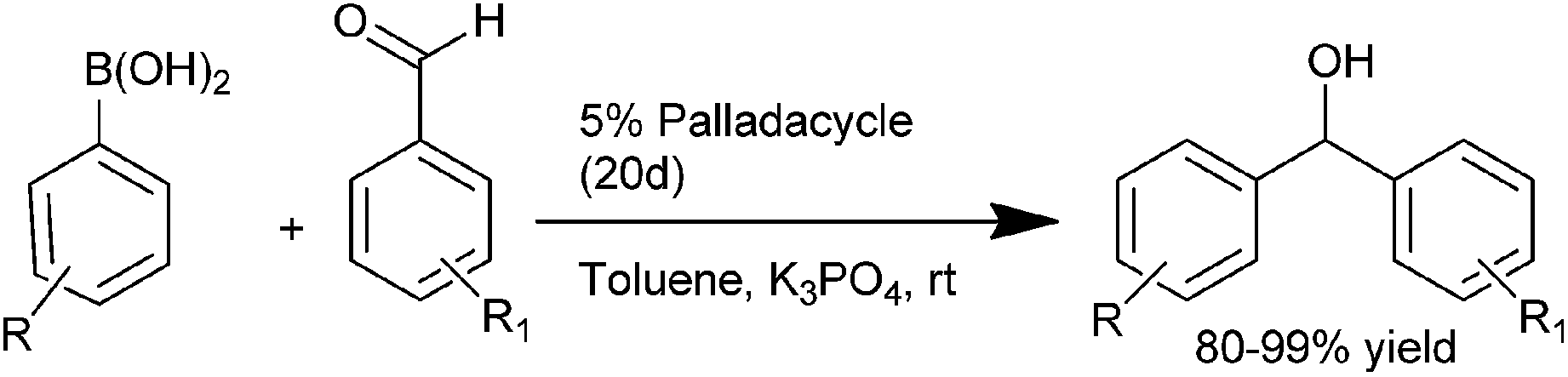 | ||
| Scheme 16 | ||
H. Wu et al. in 2007 reported a general efficient protocol for the palladium catalyzed arylation of aldehydes which was compatible with a wide range of functional groups. They used palladium chloride as the palladium source and trinaphthyl phosphine as the ligand (Scheme 17).24 Using this protocol, a wide range of diarylmethanols with sterically demanding groups could be synthesized in high yields. As arylating agents, aryl boronic acids with electron withdrawing substituents as well as heteroaryl boronic acids could be used.
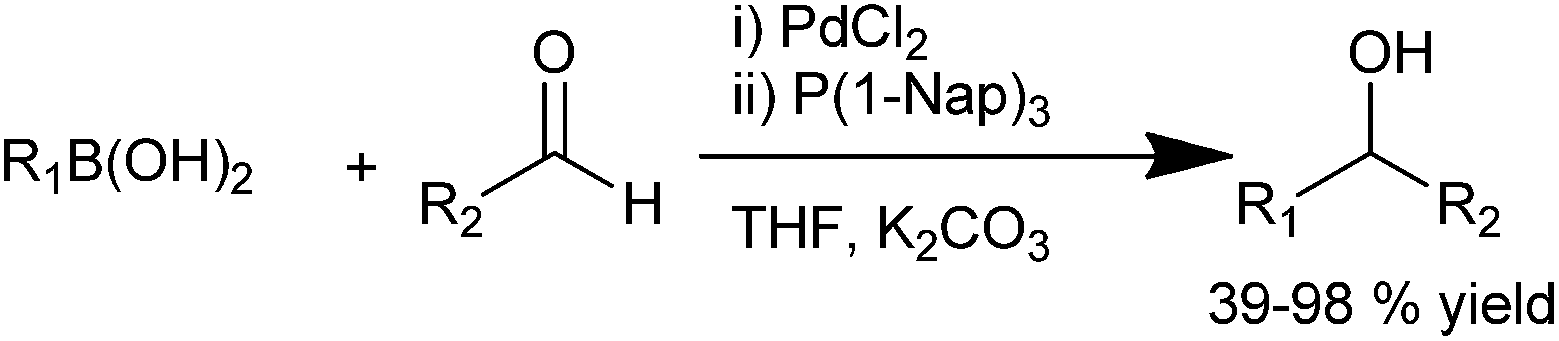 | ||
| Scheme 17 | ||
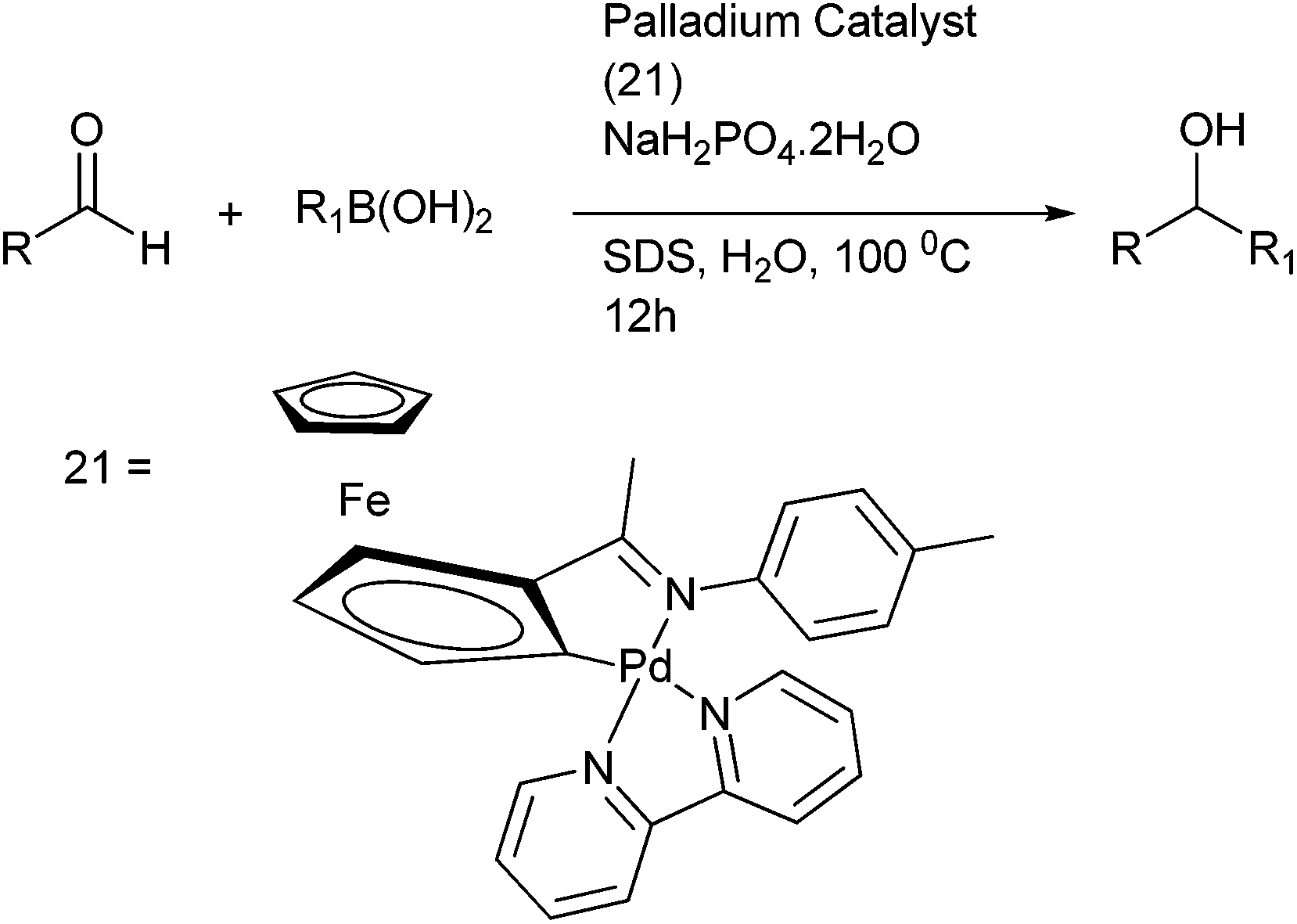
Y. Wu and co-workers developed a protocol for arylation of aldehydes under environmentally benign conditions using cyclopalladated ferrocenyl imine complex (21) in neat water and a weak acid sodium dihydrogen phosphate as an additive (Scheme 18).25 They could prepare both diarylmethanols containing electron rich as well as electron poor substituents as well as diarylmethanols containing sterically demanding groups. They noted that with electron rich aldehydes, quantitative yield of the diarylmethanols could be obtained if the additive was changed to potassium fluoride.
 | ||
| Scheme 18 | ||
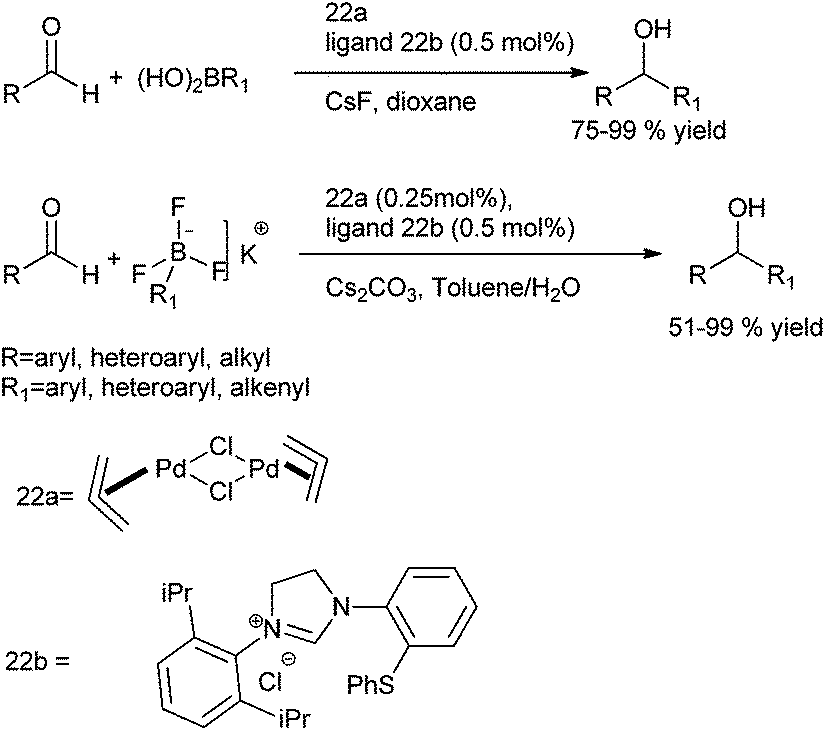
M. Kuriyama et al. & R. Shirai et al. in 2008 reported novel N-heterocyclic carbene (22b) ligated palladium catalyzed arylation of aldehydes using arylboronic acids and aryl trifluoro borates as the aryl source (Scheme 19). Thus using [Pd(allyl)Cl]2 as the palladium source, they could prepare the respective diarylmethanols in excellent yields (Scheme 19).26
 | ||
| Scheme 19 | ||
In 2010 they delineated only water was enough for the successful achievement of the reaction and no further additives or reagents were necessary.27,28
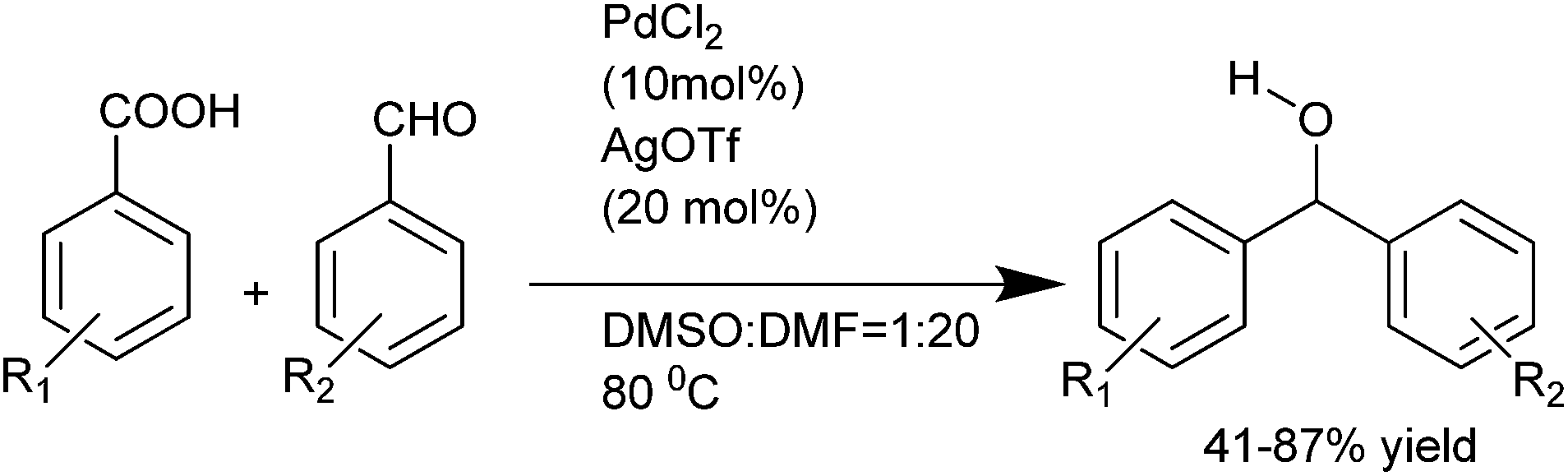
J. Wu et al. first reported the palladium catalyzed decarboxylative addition of aryl carboxylic acids with the aryl aldehydes thus avoiding the use of stoichiometric organometallic reagents (Scheme 20).29 However, silver triflate was used as an additive.
 | ||
| Scheme 20 | ||
3.6 Platinum catalyzed arylation of aromatic aldehydes
Q. S. Hu et al. reported the use of readily available, air and moisture stable ortho platinated triaryl phosphite (23) for the addition of aryl boronic acids to aromatic aldehydes. They could obtain a large variety of diaryl methanol with various activating and deactivating functional groups in high yields by using low catalyst loading (as low as 0.01%) (Scheme 21).30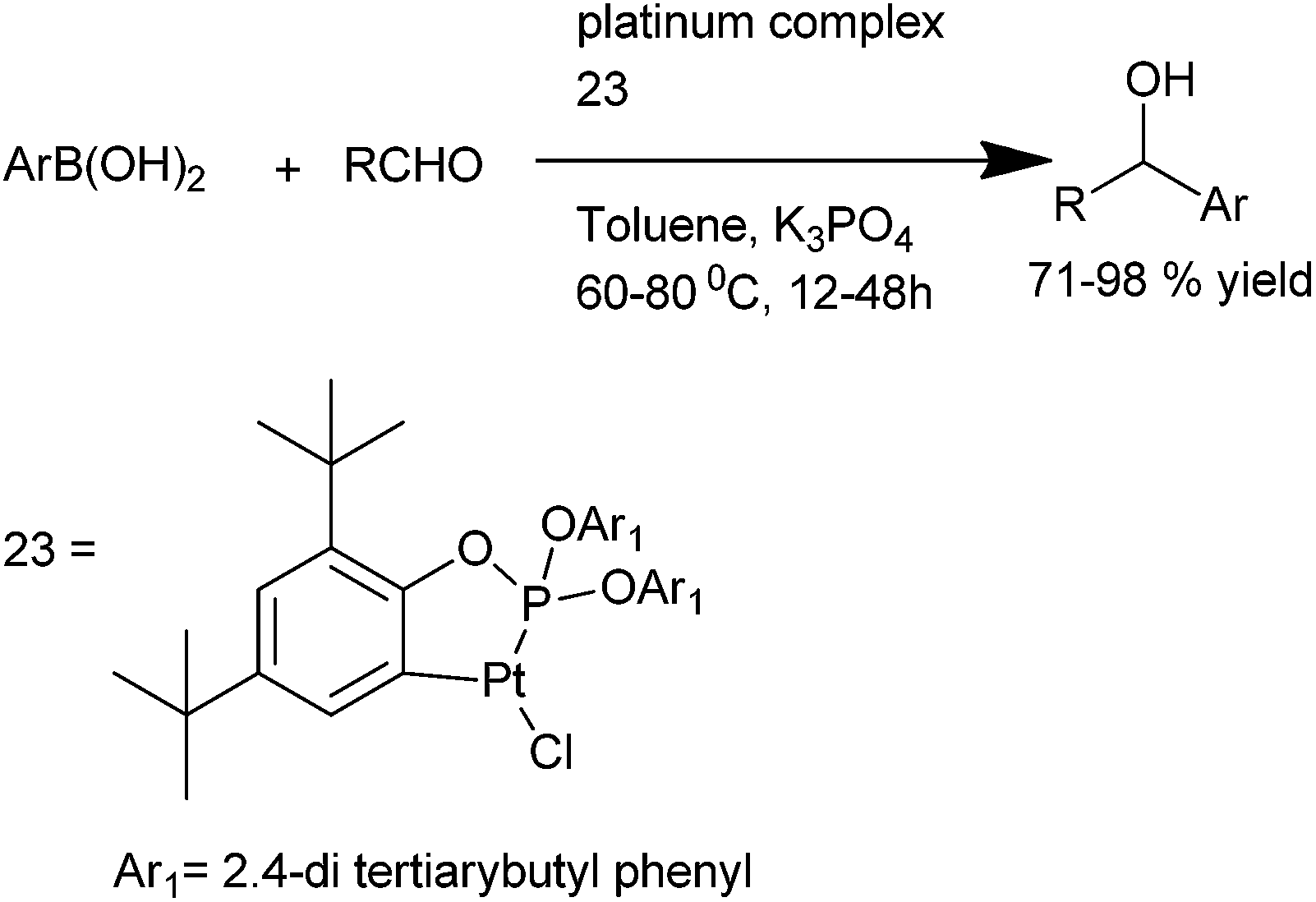 | ||
| Scheme 21 | ||
3.7 Copper catalyzed arylation of aromatic aldehydes
J. Ding et al. in 2009 reported the use of copper catalyzed arylation of aldehydes. They used copper(II) acetate as the copper source and aryl boronic acid as the aryl source (Scheme 22).31 When monodented phosphine ligands and sterically demanding ligands were used, the yields of the diarylmethanols were low to moderate; while when bidentate ligands were used, the yields were high. Among the bases and solvents screened, sodium acetate and toluene were found to be the best. When boronic acids with electron withdrawing groups were used, the yields were low with many side products while electro neutral and electron donating groups gave the diarylmethanols in high yield. The authors proposed that electron withdrawing groups decrease the nucleophilicity of the boronic acids while electron donating groups increase the nucleophilicity of the boronic acids. Putting electron withdrawing groups on the aldehyde moiety gave the expected diarylmethanols but electron releasing and even electroneutral groups did not yield the diarylmethanols probably due to the diminished electrophilicity of the aldehydes. Particularly interesting was the fact that with phthalaldehyde, isophthalaldehyde or terephthalaldehyde, only single arylated product could be obtained. The reaction depends on the electronic nature of the aldehyde and that the electrophilicity of the product hydroxy methyl benzaldehydes being less than the starting aldehydes the product does not react further with the boronic acid.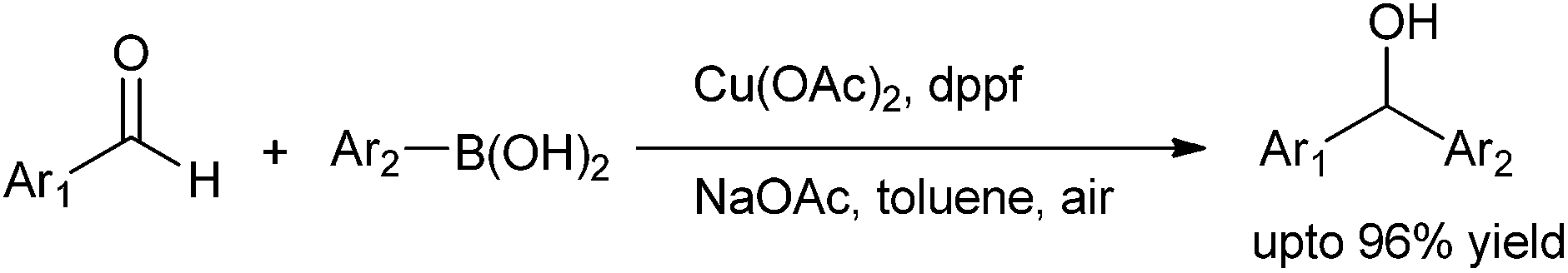 | ||
| Scheme 22 | ||
Q. S. Hu et al. in 2011, reported bipyridine ligated copper(I) chloride catalyzed addition of aryl boroxines to aldehydes to obtain the diarylmethanols with various electron deficient as well as electron rich substituents in very good yields (up to 92%) (Scheme 23).32
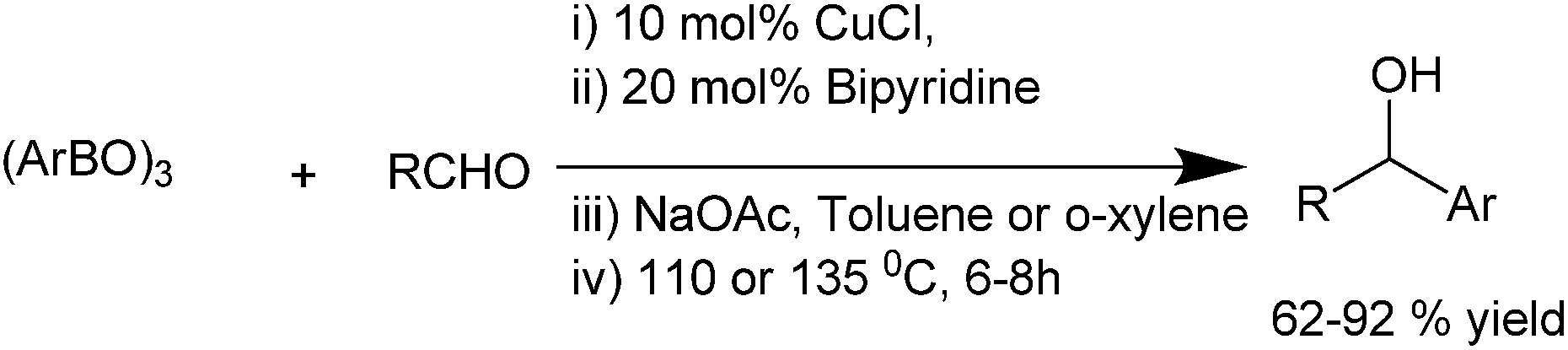 | ||
| Scheme 23 | ||
3.8 Iron catalyzed arylation of aromatic aldehydes
J. H. Li et al. was the first to report the use of environmentally friendly iron catalyzed arylation of aromatic aldehydes (Scheme 24).33 They reported the use of 2-(di-tert-butylphosphino) biphenyl (24) ligated FeCl3 catalyzed addition of aryl boronic acid to aromatic aldehydes. However this protocol is limited for electron withdrawing aldehydes as electro neutral and electron donating aldehydes being less electrophilic failed to give any product. For electron rich and electro neutral boronic acids, the product was obtained in good yield. However electron poor boronic acids, being poor nucleophiles, gave only low yield of the product.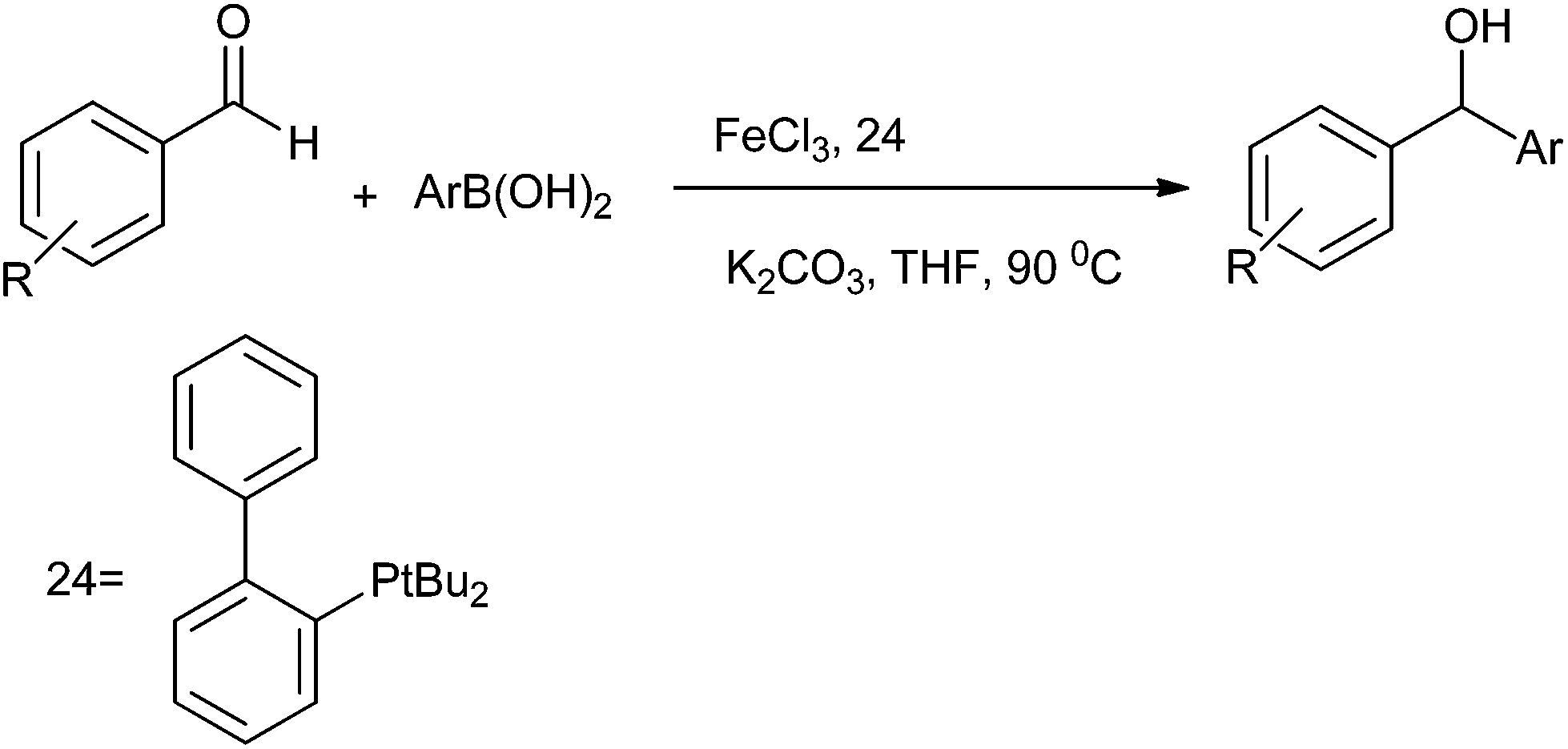 | ||
| Scheme 24 | ||
3.9 Arylation of aldehydes using ArB(OH)2–GaMe3 as the aryl source
C. Zhu and coworkers used a mixture of trimethyl gallium aryl boronic acid as the arylating agent in the room temperature arylation of aromatic aldehydes to obtain the respective diarylmethanols in high yields (up to 98%) (Scheme 25).34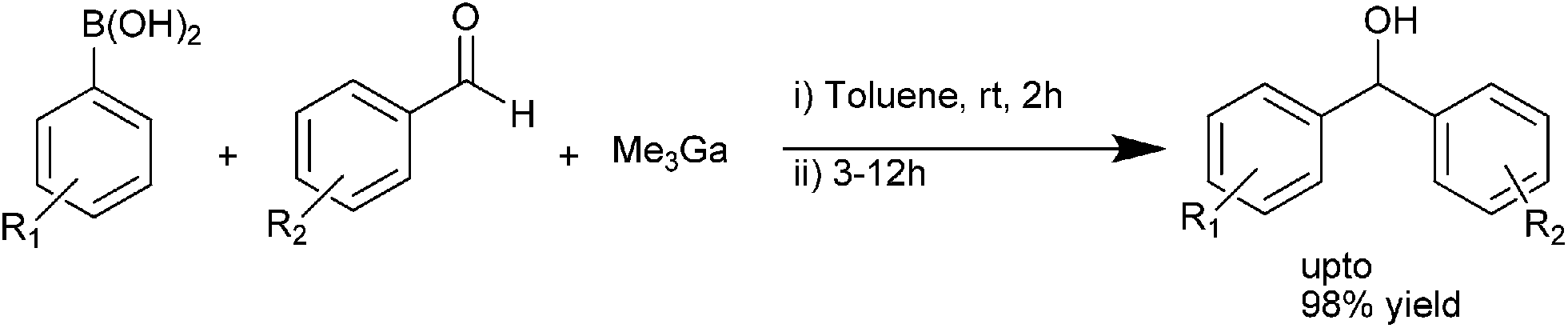 | ||
| Scheme 25 | ||
4. Diarylmethanes-overview
Diarylmethanes based/derived architectures have played a pivotal role in the development of supramolecular chemistry. This molecular entity has been the part and parcel of much supramolecular architecture like calixarenes, pillararenes and many synthetic receptors. Not only this, these macromolecular architectures have also played a pivotal role in the understanding of various molecular self assemblies and in the understanding of various molecular recognition processes.35,36,37aFrom the pharmaceutical perspective the importance of the diarylalkanes are well documented,37b–f with various diaryl methane based molecules like podophyllotoxin,37g peperomin B,37h tolterodine37i and lasofoxifene37j finding use in different types of diseases. The last two decades have witnessed an exponential growth in the synthesis of this benign group. However to the best of our knowledge, there has been no review covering the synthesis of this group. This review will cover the synthetic approaches and the biomedical applications of this rewarding group that have come up in the last 20 years.
5. Synthetic approaches towards diarylmethanes
This molecular scaffold has mostly been accessed through Friedel–Crafts alkylation of the corresponding benzyl alcohols with another arene, metal catalyzed cross coupling of aryl halides with benzyl nucleophiles, metal catalyzed cross coupling of benzyl halides with aryl nucleophiles and C–C bond formation between tosyl hydrazones and aryl boronic acids.6. Friedel–Crafts reaction for the synthesis of diarylmethanes
P. E. Georghiou et al. in 1995 reported the synthesis of all the isomers of calix[4] naphthalenes derived from 1-naphthols using TFA or TiCl4 in the cyclization of the hydroxymethyl naphthols.38 S. Fukuzawa in 1997 reported trifluoromethane sulfonic acid catalyzed reductive Friedel–Crafts alkylation of benzaldehyde acetals for the preparation of diarylmethanes (Scheme 26).39 When low boiling arenes were used, the arene served as the solvent as well as the reagent while for high boiling arenes 1,2-dichloroethane or nitromethane was used as a solvent. However, interestingly, common Friedel–Crafts solvent like carbon disulfide, dichloromethane or chloroform proved unsuitable for this reaction. Though both electron rich as well as electron poor aldehyde acetals responded to this reaction, the reaction rate was faster with the electron rich aldehyde acetals with the ratio of ortho:meta:para ratio being same as that observed for general Lewis acid catalyzed Friedel–Crafts alkylation reaction. The authors observed that except for anisole, the reaction was very clean with electron rich arenes yielding a high percentage of diarylmethanes. Anisole under similar conditions yielded a complex mixture of polybenzylated product. Later on, the authors reported Sc(OTf)3 as a mild, moisture tolerant, recyclable Lewis acid catalyst for the Friedel–Crafts alkylation of benzyl alcohols in nitromethane as the solvent.40 Though the reaction yield was quantitative in nitromethane, the reaction was low yielding in refluxing carbon disulfide or 1,2-dichloroethane.
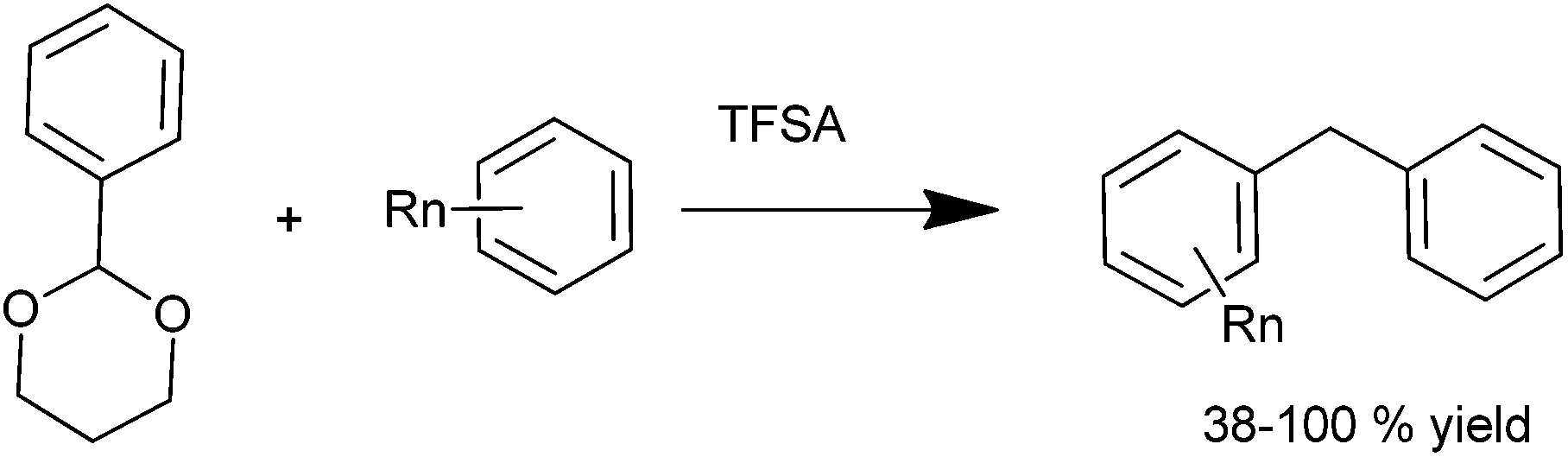 | ||
| Scheme 26 | ||
When the authors applied this reductive Friedel–Crafts alkylation on aldehydes interestingly, they found that the reaction was successful when 1,3-propane diol was used but yielded complex reaction mixture when 2,4-dimethyl pentane-2,4-diol was used and the reaction was unsuccessful without the propane diol.41 The authors proposed the mechanism of the reaction (Scheme 27). At first the diol reacts with the aldehyde to form the acetal. The Lewis acid then coordinates with the acetal thereby increasing the electrophilicity of the acetal carbon. A Friedel–Crafts alkylation reaction with the arene leads to the formation of the compound (25a). The Lewis acid then again coordinates the acetal oxygen and a 1,3-hydride shift or a 1,5-hydride shift (the marked hydrogens) generated the diarylmethane. A deuterium labelling study with deuteriated propane diol yielded deuterium labelled diaryl methane proving the hydrogen atom was obtained from the diol.
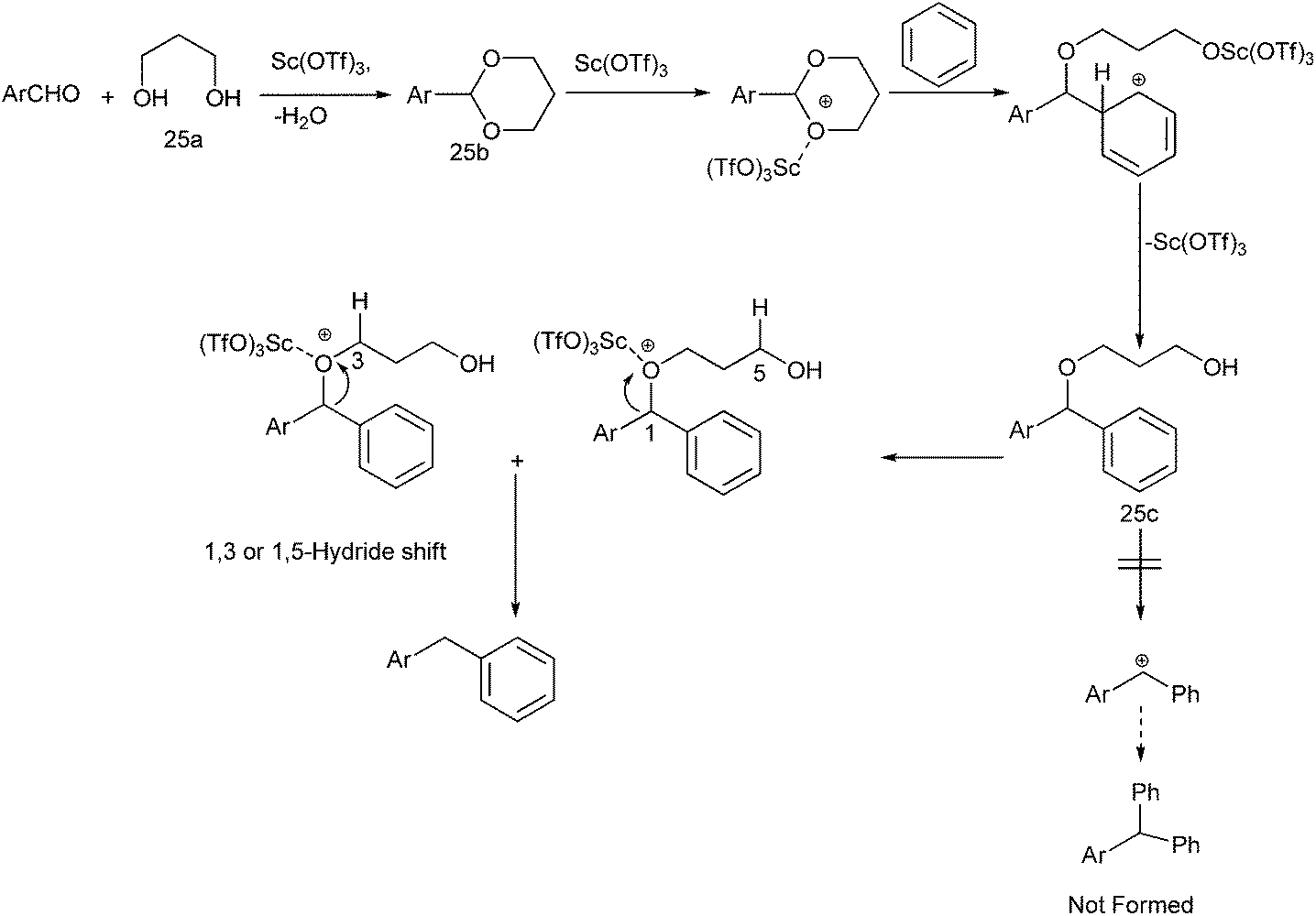 | ||
| Scheme 27 | ||
R. Hua et al. in 2006 reported indium(III) chloride catalyzed electrophilic substitution of trioxanes with arenes for the preparation of diarylmethanes.42 The authors found that the reaction was high yielding only with electron rich arenes while electronically poor ones gave low yield. Later in 2007, the authors reported indium trichloride acetyl acetone to be an efficient Lewis catalyst for the Friedel–Crafts arylation of benzyl alcohols for the efficient preparation of diarylmethanes (Scheme 28).43
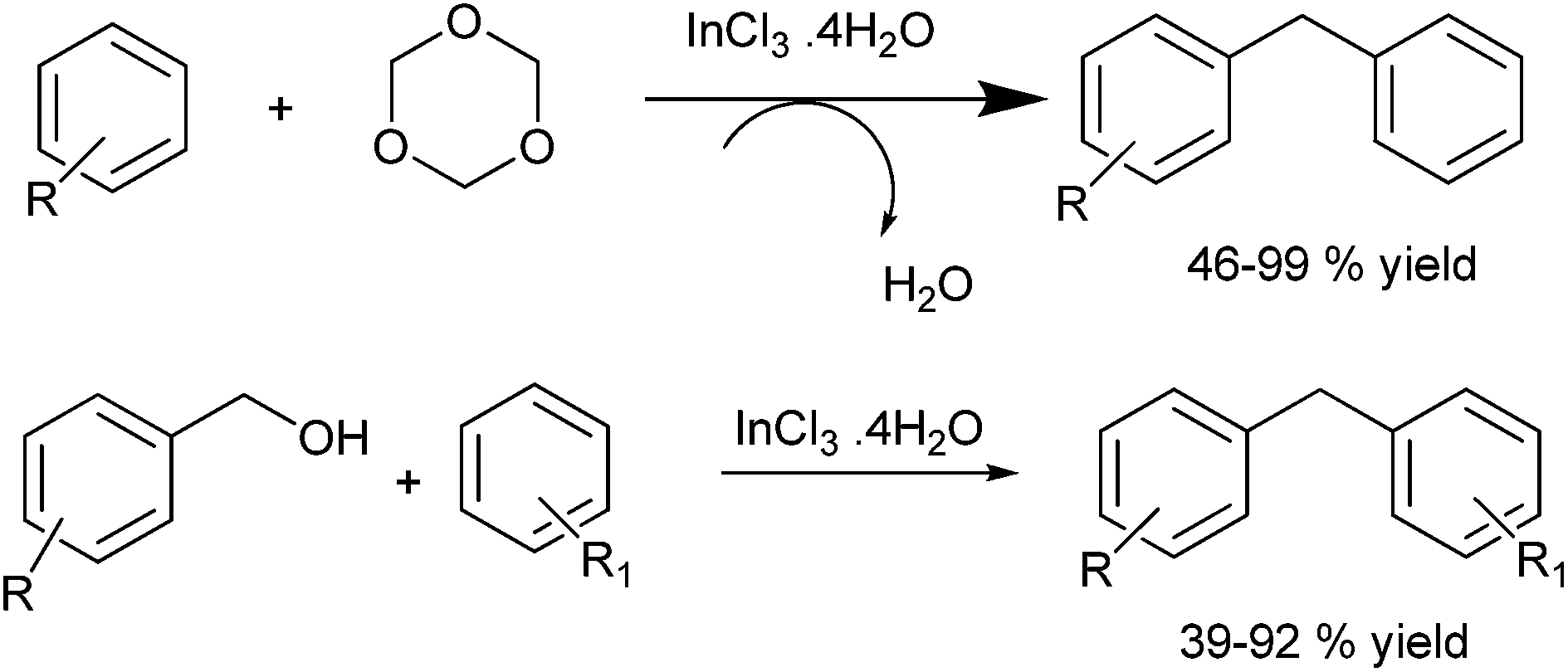 | ||
| Scheme 28 | ||
B. Myrboh et al. reported AlCl3 catalyzed Friedel–Crafts alkylation of benzalazines with polynuclear hydrocarbons to form diarylmethanes in aprotic solvents.44 The corresponding azines were accessed from the respective carbonyl compounds through condensation with aqueous solution of hydrazines.
L. Ghosez et al. in 2011 reported Brønsted acid catalyzed arylation of benzyl acetates and anisyl acetates with arenes for the preparation of diarylmethanes (Scheme 29).45 Trifluoromethane sulfonic acid and triflimide respectively were used as the Bronsted acid catalyst. Except for the triflimide, the reactions were carried out without the use of solvent. The use of either phosphoric acid or triflouro acetic acid did not lead any reaction. The authors observed that this protocol was highly successful for electron rich arenes. However with anisyl acetates and para xylene, the reaction led to the decomposition of the starting materials.
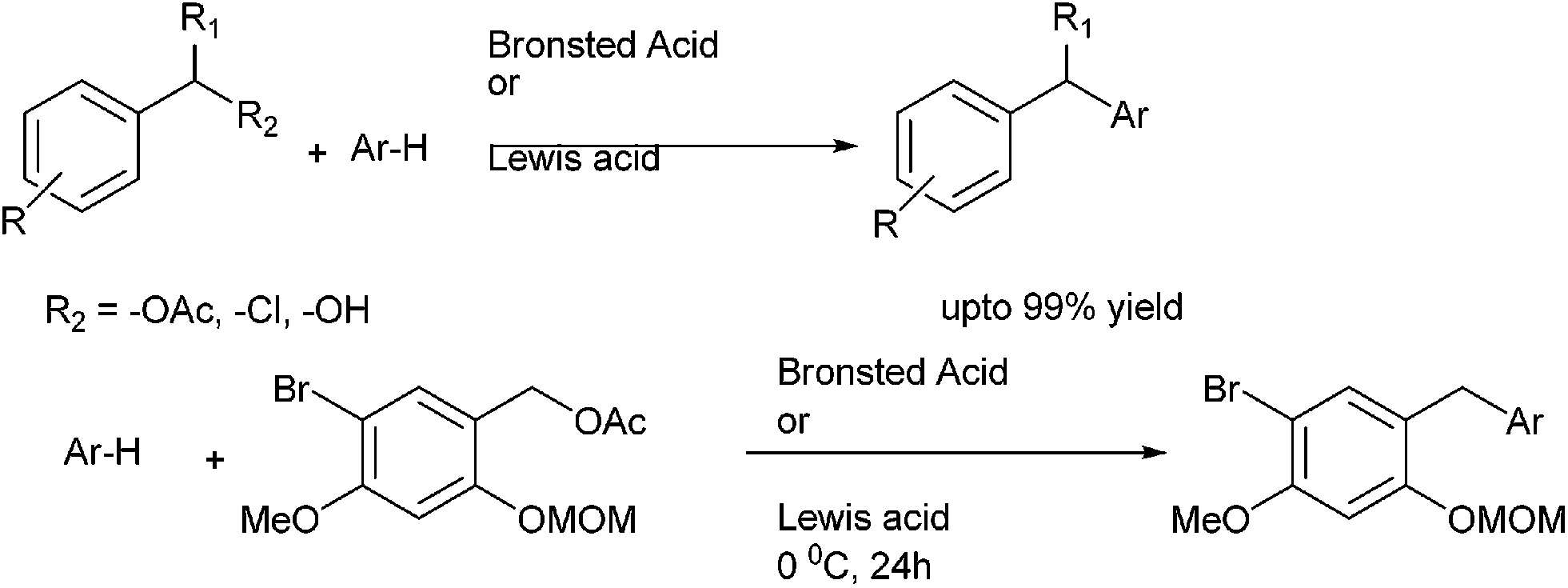 | ||
| Scheme 29 | ||
In 2011, L. N. He et al. improved this protocol by reporting an environmentally benign synthesis of diarylmethanes.46 They used ferric ion based ionic liquids for the Friedel–Crafts alkylation of benzyl alcohols and acetates with electron rich arenes and heteroarenes (Scheme 29).
W. Phakodee et al. in 2013 reported acid catalyzed generation of o-quinone methides followed by nucleophilic substitution with electron rich aromatic systems for the synthesis of diarylmethanes (Scheme 29).47
7. Metal free C–C bond formation
J. Barluenga in 2009 reported metal free C–C bond formation for the preparation of diarylmethanes from tosyl hydrazones (Scheme 30).48 The authors used aryl boronic acids as the aryl source. They found that the reaction can be carried out in one pot starting directly from the carbonyl compound without further isolation of the hydrazones. The authors proposed heating the tosyl hydrazone salts in presence of a base generates the diazo intermediate (26a) (Scheme 30a). This intermediate reacts with aryl boronic acids to form the benzyl boronic acid. The formation of benzyl boronic acid can be explained either through the intermediate formation of a boronate (26b) or through the formation of a carbene (through the loss of nitrogen molecule) (26c). Further reaction of the carbene with the boronic acid forms the benzyl boronic acid. Further protodeboronation of the benzyl boronic acid led to the formation of the product. In 2012, Gang Zou improved this metal free reductive cross coupling of N-tosyl hydrazones by using diaryl borinic acid or its anhydride as the aryl source (Scheme 30b).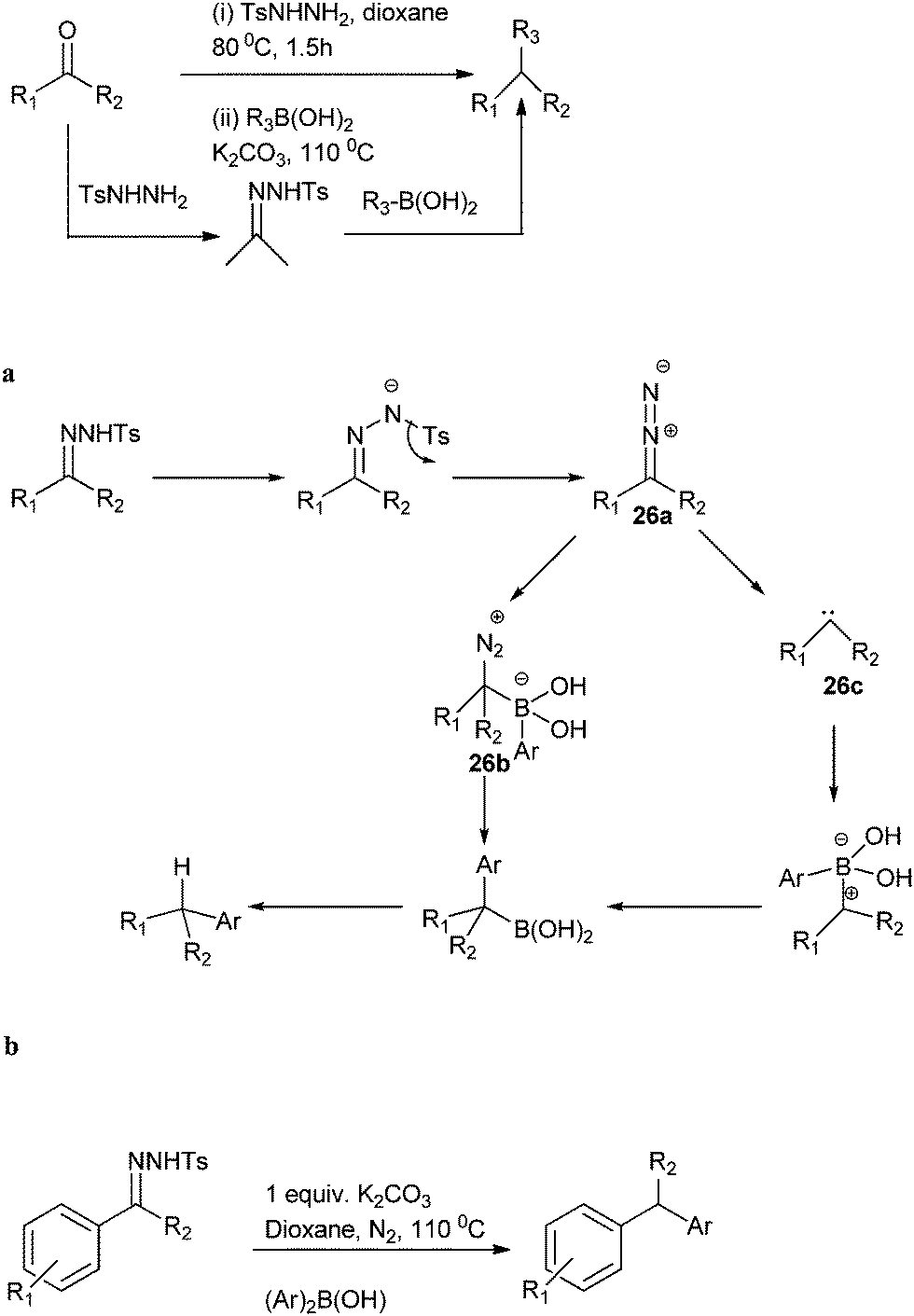 | ||
| Scheme 30 | ||
M. M. Khodaei et al. reported triphenyl phosphine ditriflate mediated Friedel–Crafts alkylation of benzyl alcohols with arenes for the facile preparation of diarylmethanes (Scheme 31).49
 | ||
| Scheme 31 | ||
8. Synthesis of diarylmethanes through metal catalyzed cross-coupling reactions
8.1 Copper catalyzed cross coupling of benzyl halides with Grignard reagents
Y. Y. Ku et al. in 1996 reported copper catalyzed cross coupling of aryl Grignard reagents with benzyl halides for the preparation of diarylmethanes (Scheme 32).50 This protocol was high yielding and was highly suitable not only for laboratory scale preparation of diarylmethanes but also for the multi kilogram industrial scale. | ||
| Scheme 32 | ||
L. S. Liebeskind et al. in 1997 and in 1999 reported metal catalyzed cross coupling reactions of heterobenzylic sulfonium salts with organostannanes, organoboronic acids as well as organozinc reagents (Scheme 33).51,52 For the cross coupling with organostannanes and organoboronic acids, palladium was used while for organozinc halides, nickel was used. For improving the efficiency of the organostannanes Ph2P(O)O−Bu4N+ was used as Bu3Sn scavenger. The use of highly nucleophilic phosphine ligands in order to stabilize the metal catalyst and the electrophilic sulfonium salts leads to the competing side reactions which were overcomed by using essentially non-nucleophilic triaryl phosphites as ligands.
 | ||
| Scheme 33 | ||
8.2 Suzuki–Miyaura coupling
P. E. Georghiou et al. in 1999 reported the Suzuki–Miyaura coupling of benzyl halides with aryl and naphthyl boronic acids to form the respective diarylmethanes (Scheme 34).53 The reactions were high yielding and were rather inert towards change in the electronic properties of the benzyl bromides. The authors noticed that the change of the coupling partner from benzyl bromide to the benzyl iodide caused a minor increase in the reaction yield. Moreover for compounds having two bromomethyl groups, higher catalyst loading had to be done and also the reactions took longer time to reach completion. Further using this methodology the authors could achieve a convergent synthesis of mixed endo and exo-functionalised calix[4]naphthalenes.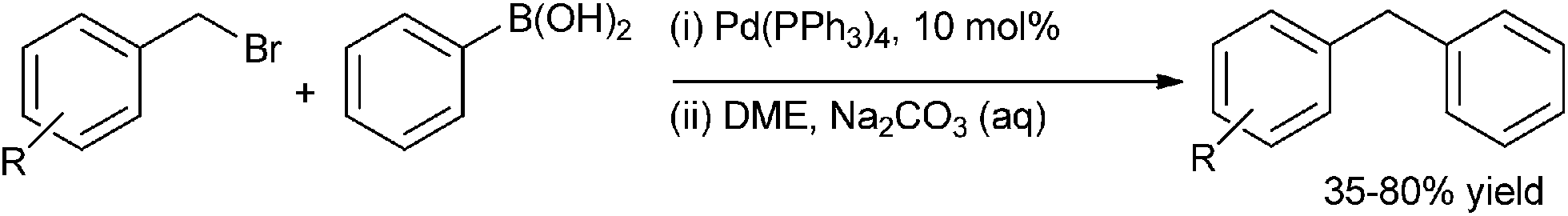 | ||
| Scheme 34 | ||
L. A. Sarandeses et al. in 2001 reported palladium catalyzed cross coupling of Triorgano indium reagents with aryl halides and pseudo halides (triflates), vinyl triflates, benzyl bromides and acid chlorides.54 The authors noticed this transformation to be highly chemoselective and atom efficient transferring all the attached organic groups effectively. The reaction of triphenyl indium with benzyl halide under palladium catalysis yielded diphenyl methane almost quantitatively.
C. Nájera et al. in 2002 reported the use of oxime derived palladacycles (27) for the Suzuki–Miyaura cross coupling of the aryl and benzylic halides with aryl boronic acids (Scheme 35).55 Tetrabutyl ammonium bromide was used as an additive. Acetone and water in the ratio of 3:2 were used as the solvent. However the authors found that in refluxing water, significant amount of alcohol was formed. Thus treatment of 3-Methoxy benzyl chloride (28) with phenyl boronic acid (29) under the optimized condition yielded the corresponding diarylmethane (30) in 75% yield (Scheme 35).
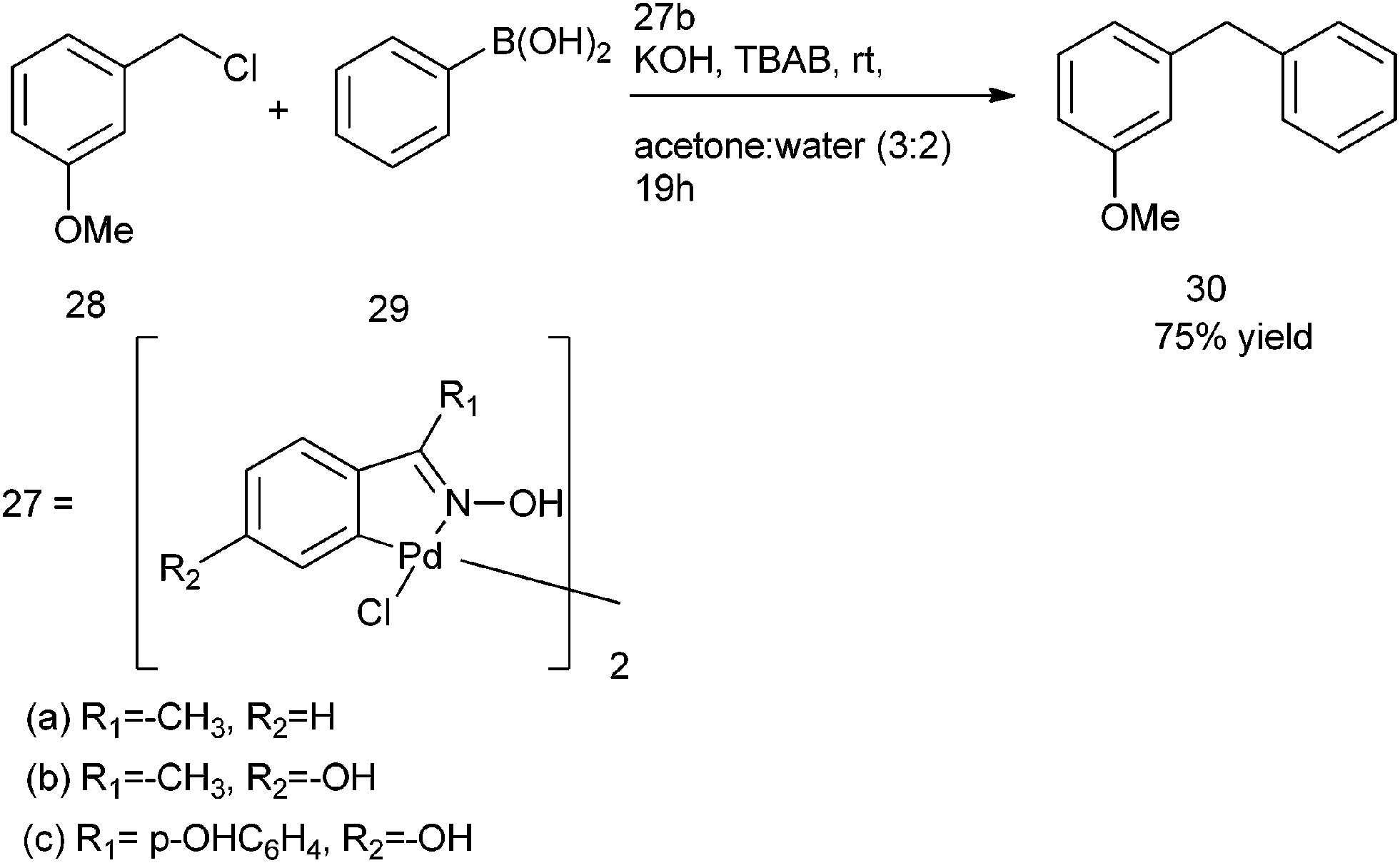 | ||
| Scheme 35 | ||
A. Duchêne et al. in 2003 reported tetrakis(triphenyl phosphine) palladium(0) (31) catalyzed chemoselective Suzuki cross coupling between ortho and para bromo benzyl bromides with aryl boronic acids for the synthesis of unsymmetrical diarylmethanes (Scheme 36).56 For the arylation of the aryl bromide, 2.1 equivalents of aryl boronic acid had to be used.
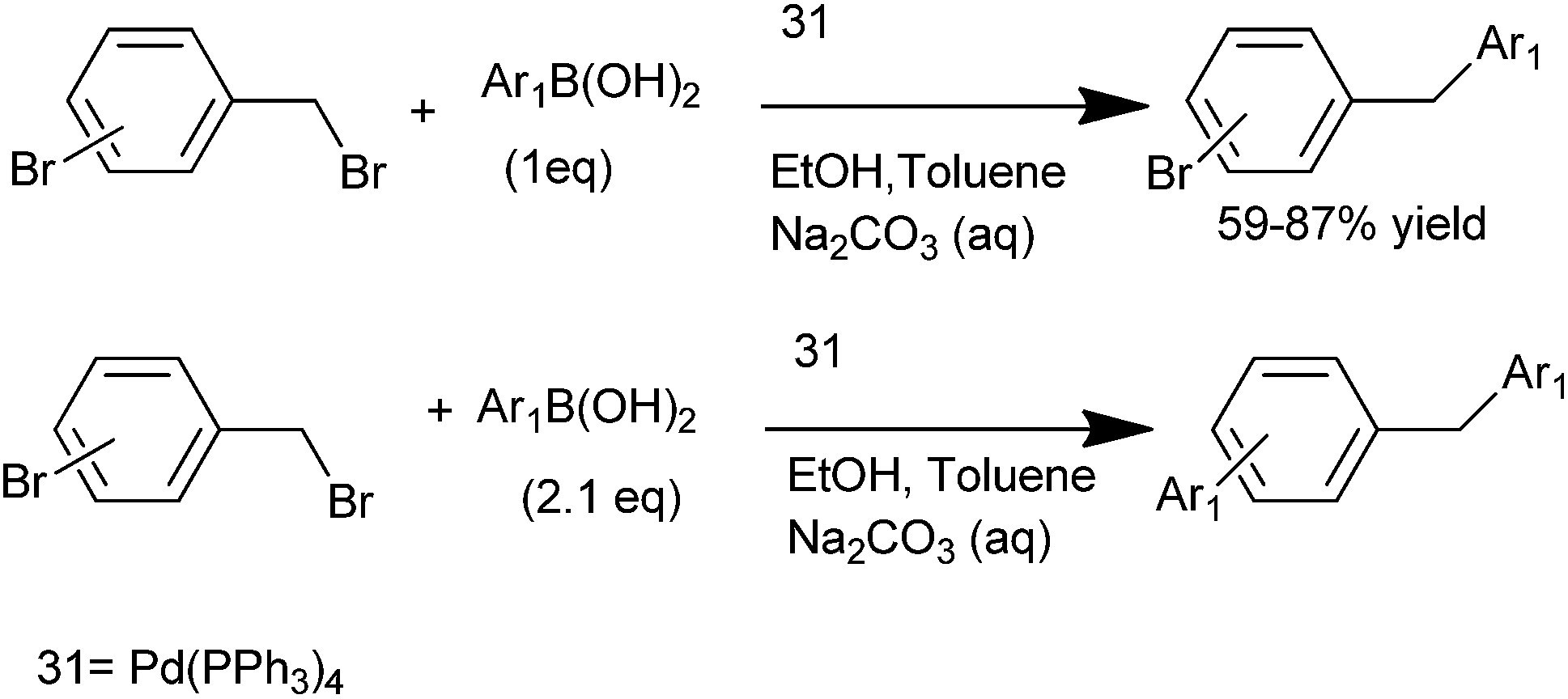 | ||
| Scheme 36 | ||
A. L. Monteiro et al. in 2004 reported an efficient protocol for the recycling of the palladium catalyst for the Suzuki–Miyaura coupling of the aryl and the benzyl halides in polyethylene oxide/MeOH as the solvent system.57 The authors reported extraction of the reaction mixture with heptane led to the separation of the nonpolar diarylmethanes in the heptane layer and the polar solvent containing the palladium catalyst could be reused for the next coupling reaction.
B. P. Bandgar et al. reported in 2004 the ligand free Suzuki–Miyaura coupling of benzylic halides with aryl boronic acids to form the diarylmethanes in high yields with palladium chloride as the catalyst and acetone:water (3:1) as the solvent system (Scheme 37).58 Thus using benzyl iodides and aryl boronic acids the diarylmethanes were obtained within 0.8 h in high yields. However the authors observed that the benzyl chlorides were inert to this coupling methodology. When this cross coupling procedure was extended to the o- and p-xylene dibromides, the authors found that this cross coupling reaction was smooth but took longer time to reach completion. Interestingly, the authors observed for benzyl bromides with fluoro or chloro substituents, the cross coupling chemoselectively occurred with the benzyl bromides but for the bromo or iodo substituted benzyl bromides, the cross coupling occurred with aryl halides.
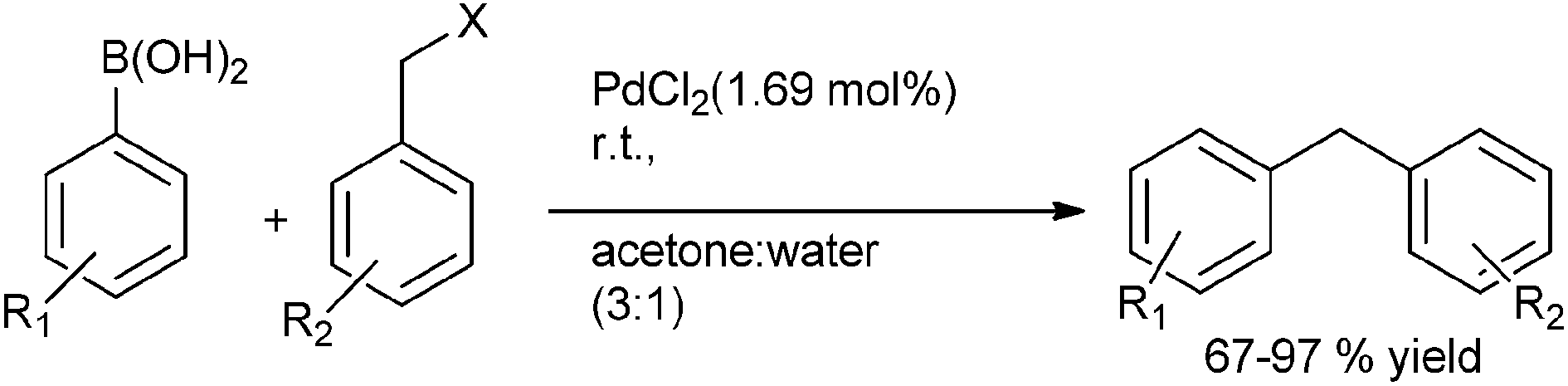 | ||
| Scheme 37 | ||
A. L. Monteiro et al., in 2004 reported triphenyl phosphine ligated Pd(OAc)2 catalyzed Suzuki–Miyaura coupling of benzylic halides with aryl boronic acid to produce diarylmethanes in high yields under mild reaction conditions and with low catalyst loadings (Scheme 38).59 The authors found that the benzyl chlorides could also be used for the cross coupling reactions and the reactivity pattern was aryl iodide > benzyl bromide > benzyl chloride > aryl bromide.
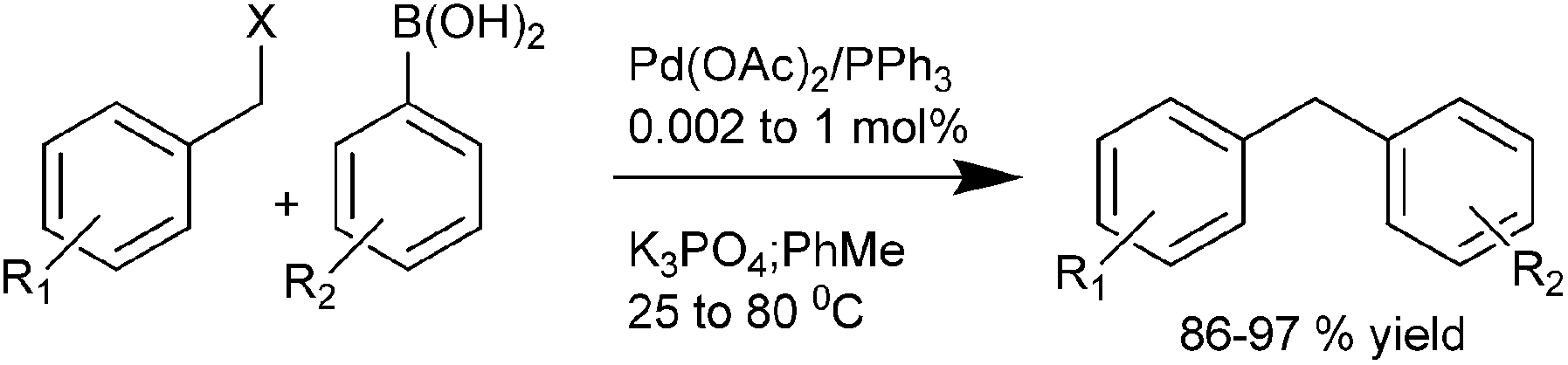 | ||
| Scheme 38 | ||
R. Kuwano et al. in 2005 reported Suzuki–Miyaura coupling of benzylic carbonates with aryl boronic acids for the preparation of diarylmethanes in high yields (Scheme 39).60 (22a) and 1,5-bis-diphenyl phosphino pentane (32) were used as palladium source and ligand respectively. The authors observed that the yield of the product and the rate of the reaction were highly depended on the nature as well as the bite angle of the phosphine ligands. They found that monodented ligands exhibited poor catalytic activity. The authors also found that the bidentate ligands with small bite angles were ineffective for this cross coupling reaction.
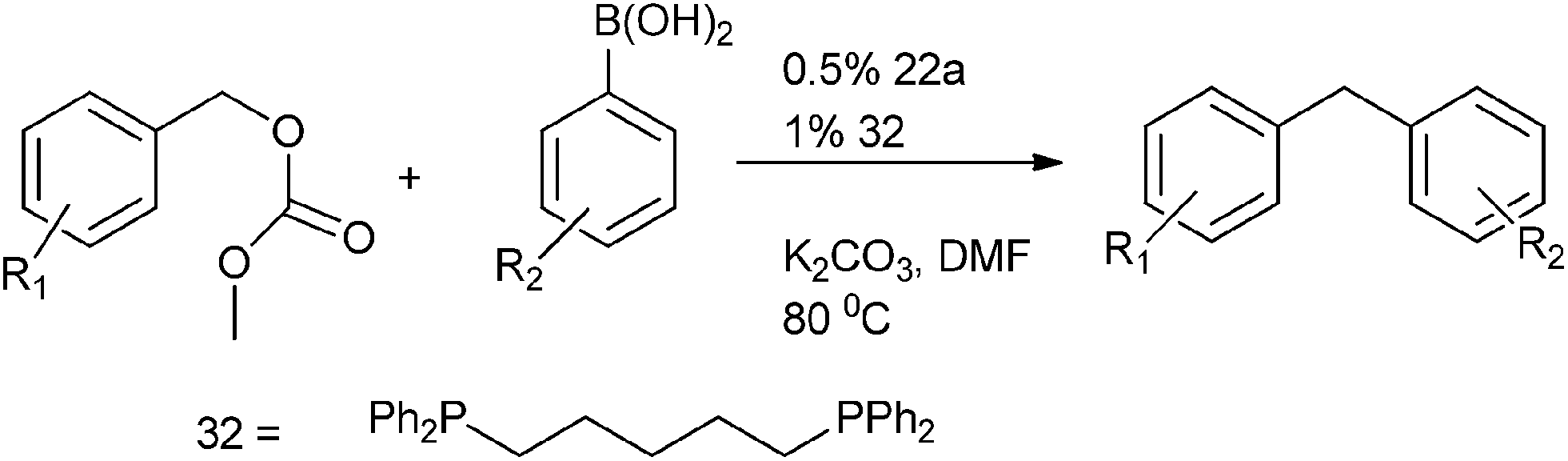 | ||
| Scheme 39 | ||
The authors described the reaction pathway (Scheme 40) by stating that at first the benzylic C–O bond is cleaved by the metal catalyst to form the intermediate (32b) and decarboxylation of the cleaved carbonate group generates the intermediate (32c). Transmetallation of this intermediate with the aryl boronic acids generates (32d). Reductive elimination of the alkyl benzyl palladium (32d) regenerates the active catalyst while delivering the product diaryl methane.
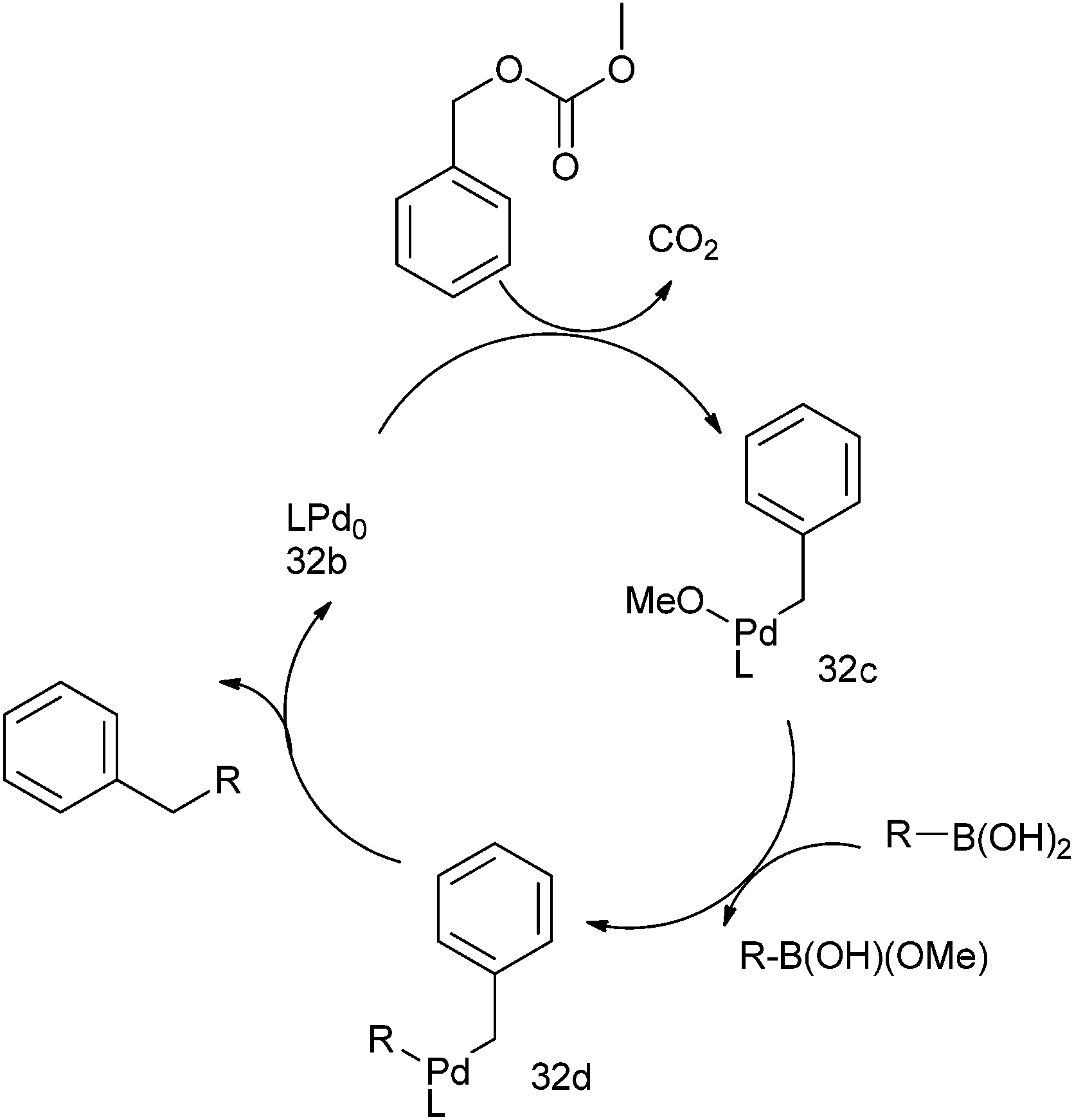 | ||
| Scheme 40 | ||
When the authors tried to extend this protocol for the Suzuki–Miyaura coupling with the acetates the reaction failed. Later on, the authors hypothesized that the alkoxide ligand formed from the oxidative addition of the carbonates to the palladium complex helped in the transmetallation step with the aryl boronic acids. But for the (acetato) benzyl palladium generated from the oxidative addition of the benzyl acetates with the palladium complex were ineffective for the transmetallation with aryl boronic acids thereby failing to give any cross coupled product. When the authors carried out the cross coupling reaction of the benzylic acetates with the aryl boronic acids using methanol or isopropyl alcohol as the solvent, the reaction did not proceed. In methanol the solvolysis of the acetates took place while isopropyl alcohol caused the decomposition of the catalyst. However the authors found that in tertiary amyl alcohol the cross coupling reaction was successful yielding high percentage of diarylmethanes.61 The cross coupled product was scarcely observed in aprotic solvents. The authors observed benzyl acetates having electron rich as well as poor substrate could be well tolerated in this coupling reaction.
M. McLaughlin et al. in 2005 reported Suzuki–Miyaura coupling of benzylic phosphates for the preparation of diarylmethanes (Scheme 41)62 with palladium acetate as palladium source and triphenyl phosphine as ligand. Interestingly, the author found that the ratio of the palladium catalyst to the ligand was important as with higher metal to ligand ratio higher yield of the product was obtained. Interestingly, the author found that the secondary aryl alkyl phosphate did not give the expected diaryl methane with the aryl boronic acid but yielded styrene as the sole product through β-hydride elimination of the palladium alkyl aryl complex.
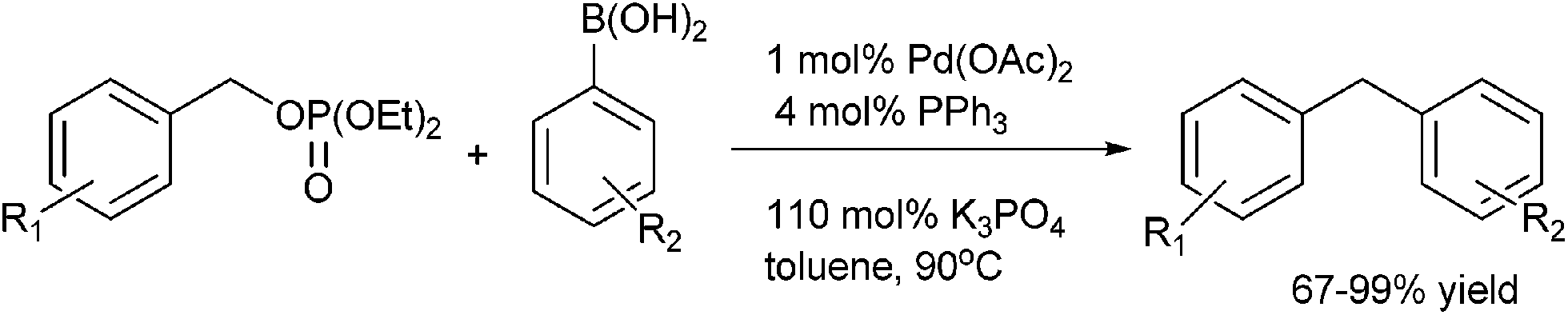 | ||
| Scheme 41 | ||
S. P. Nolan et al. in 2005 reported N-heterocyclic carbenes (15) and (33) ligated palladium(II) acetate complexes in Suzuki–Miyaura coupling of the alkyl chlorides with β-hydrogens for the respective synthesis of diarylmethanes, biaryls etc. (Scheme 42).63
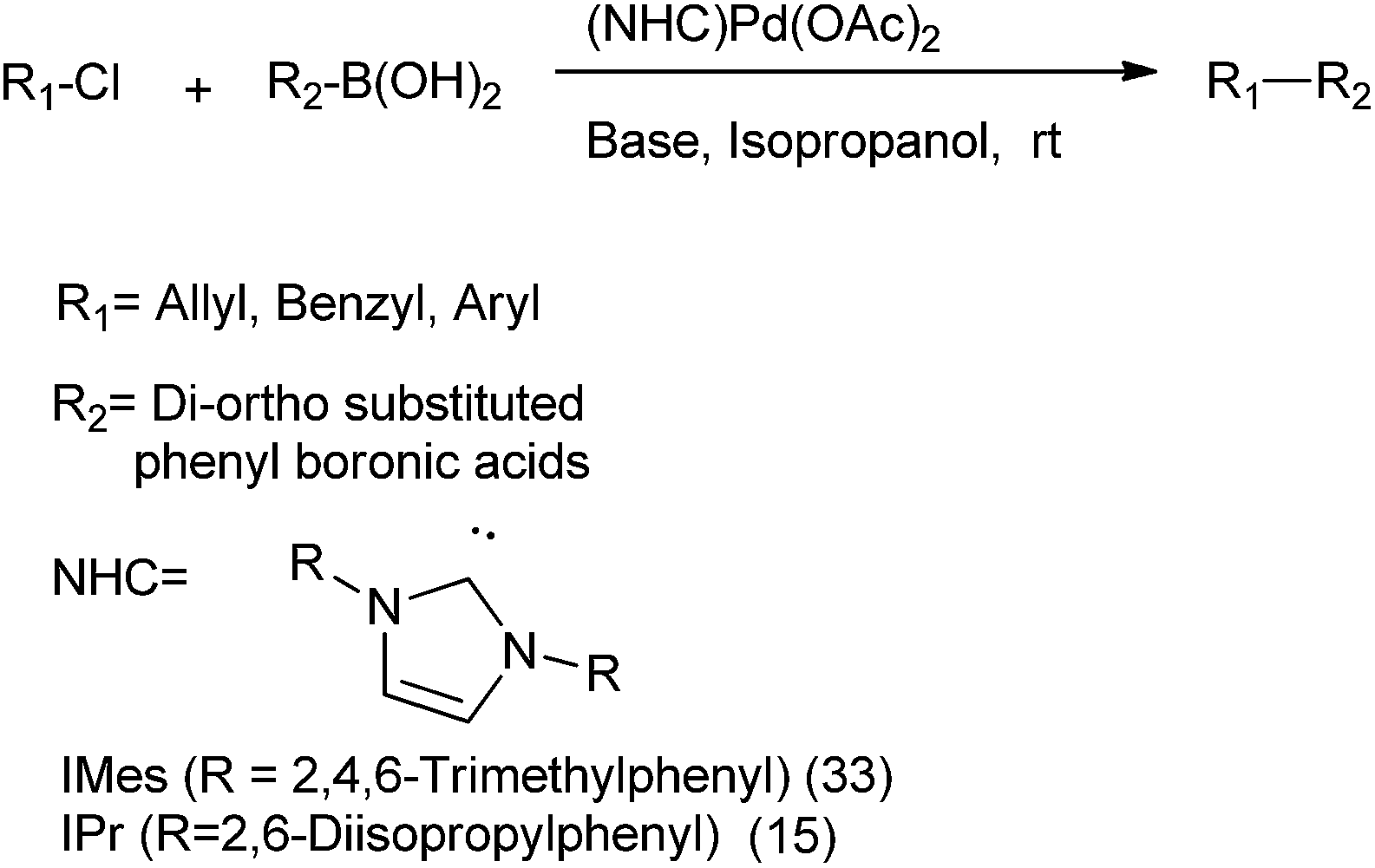 | ||
| Scheme 42 | ||
Y. Ma et al. in 2005 reported bulky phenanthryl N-heterocyclic carbene (34) ligated Palladium complexes for the Suzuki–Miyaura coupling of the vinyl, aryl and benzylic chlorides with organoboron compounds (Scheme 43).64 Later on 2013, J. M. Lu et al. reported NHC ligated palladium(II)-1-methyl imidazole complex for the Suzuki–Miyaura cross coupling of aryl boronic acids with benzyl chlorides in neat water for the preparation of diarylmethanes.65
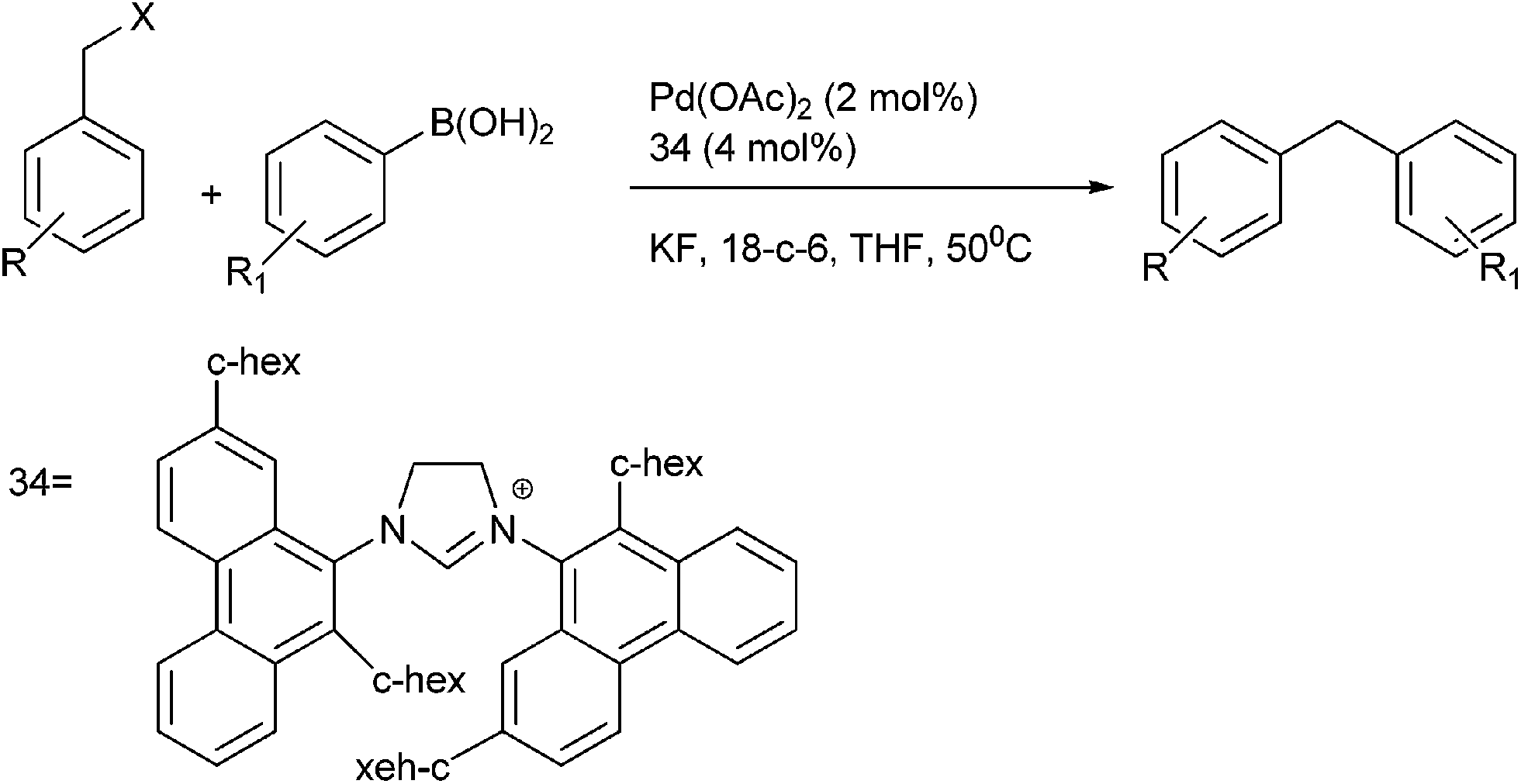 | ||
| Scheme 43 | ||
Z. Hell et al. in 2005 reported Pd/Mg La mixed oxide as an efficient catalyst for the Suzuki–Miyaura coupling of the aryl halides, triflates and for benzylic bromides.66 The authors observed that though longer reaction time was required for the cross coupling reaction, the yields of the diphenylmethanes were high.
I. J. S. Fairlamb et al. in 2006 reported binuclear anionic palladacyclopentadienyl catalysts (35) in the Stille coupling of benzyl halides with vinyl and aryl stannanes (Scheme 44).67 The authors observed that the halides and pseudo halides remarkably affected the palladium catalysts. Moreover the authors also observed a Baldwin type cooperative effect with CsF and CuI in DMF but not in toluene. The authors proposed that copper iodide probably activates the organostannanes towards the transmetallation.
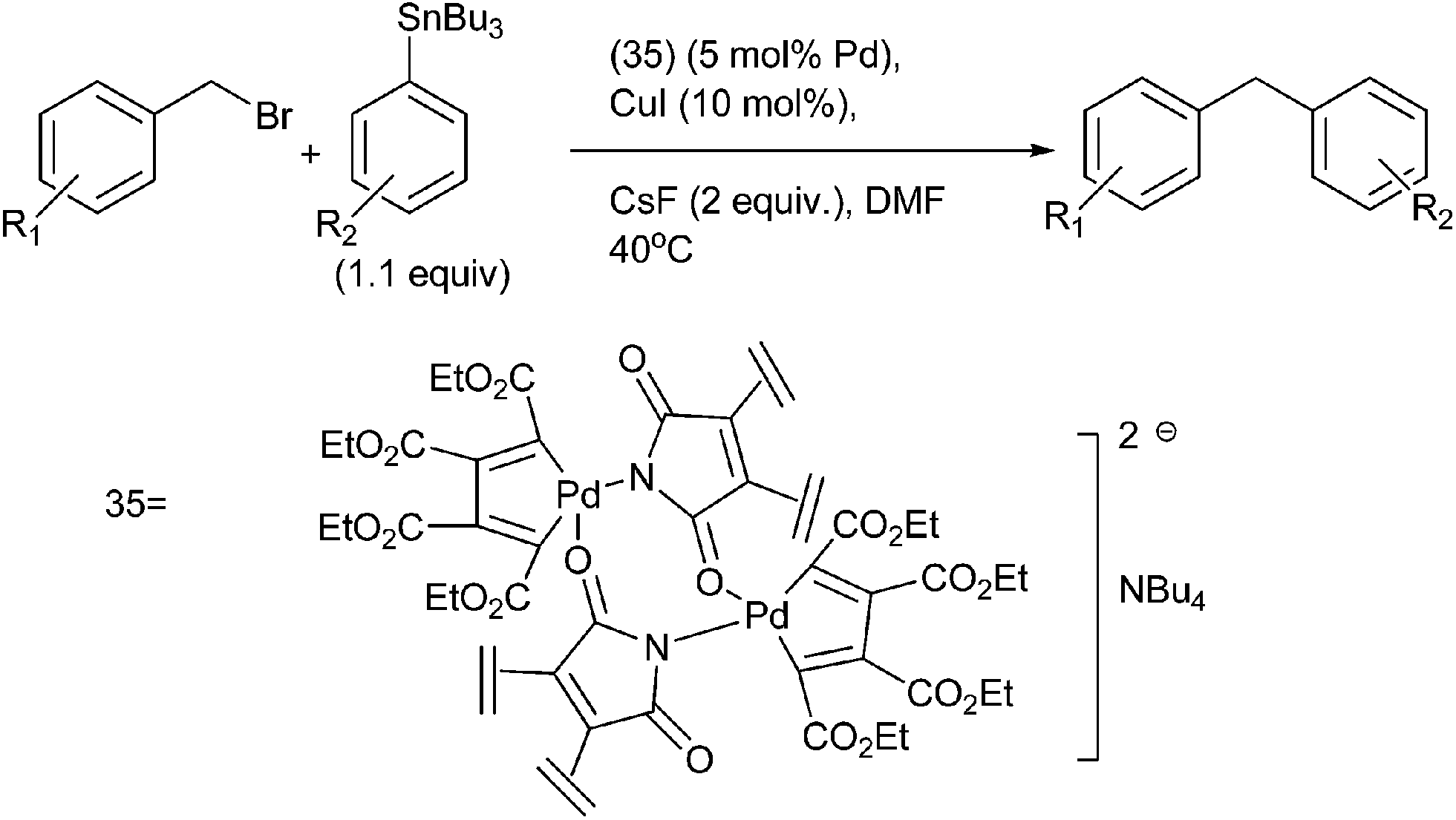 | ||
| Scheme 44 | ||
G. A. Molander et al. reported Suzuki coupling of benzyl halides with potassium aryltrifluoroborates for the preparation of diarylmethanes (Scheme 45).68 The pre-catalyst used was PdCl2(dppf)·CH2Cl2. This cross coupling procedure was high yielding and was compatible with various functional groups. The authors found that increasing the catalyst loading from the optimal amount increased the percentage of the homocoupled product instead of the cross coupled product. Also the authors found that in the ethereal solvents the percentage of the homocoupled product was minimum and use of higher boiling cyclopentyl methyl ether instead of tetrahydrofuran caused an increase in the reaction rate. However interestingly, for the cross coupling with potassium (4-carboxyphenyl) trifluoroborate only cyclopentyl methyl ether had to be used as in tetrahydrofuran the reaction only yielded the homocoupled product.
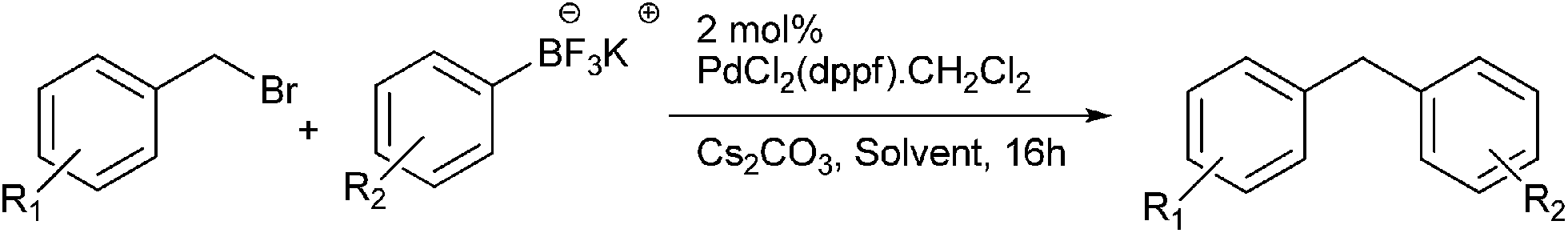 | ||
| Scheme 45 | ||
R. J. K. Taylor et al. in 2005 reported the preparation of PdBr(N-Succ)(PPh3)2 (36) and utilized the complex for the Stille coupling with vinyl and aryl stannanes.69,70 In 2007, they reported trans-36 as an effective precatalyst for the Suzuki–Miyaura coupling of the benzylic halides with aryl as well as heteroaryl boronic acids (Scheme 46).
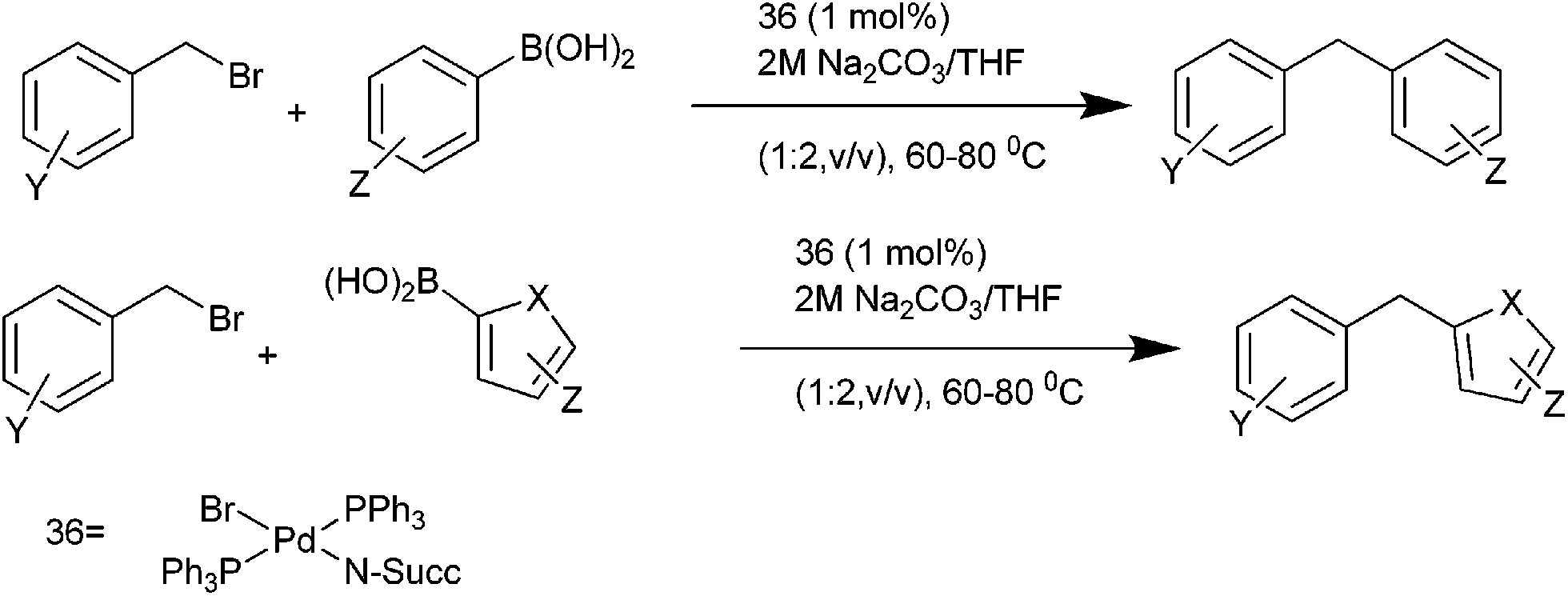 | ||
| Scheme 46 | ||
A. Sarkar and coworkers in 2008 reported benzaldimines (37) as efficient ligands for the Suzuki–Miyaura Coupling of aryl and heteroaryl halides with boronic acids (Scheme 47).71 The authors could extend this methodology for the efficient cross coupling of the benzyl halides with aryl boronic acids to obtain the diarylmethanes in high yield and short reaction time.
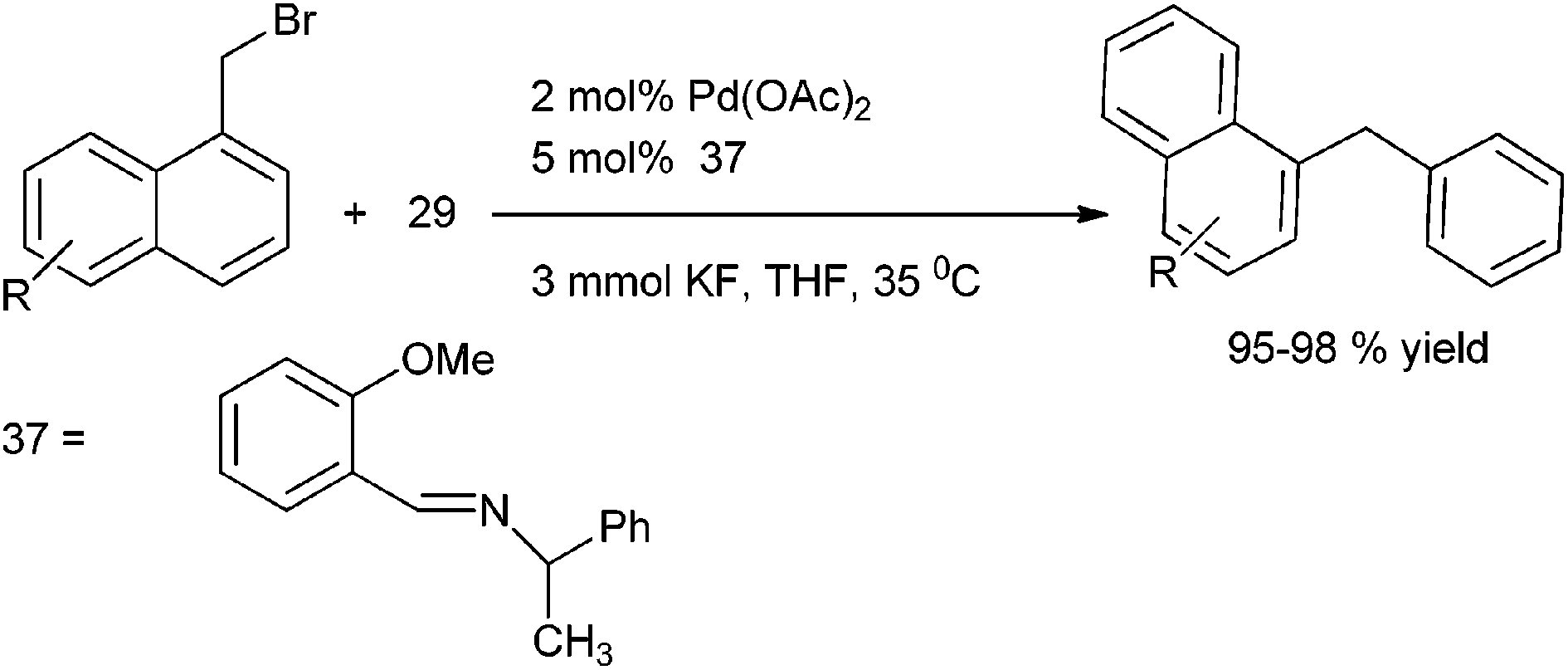 | ||
| Scheme 47 | ||
Later on in 2013 M. T. Tudge et al. reported palladium catalyzed cross coupling of nitrogen bearing heterocyclic chloromethyl compounds with aryl and heteroaryl boronic acids for the preparation of nitrogen containing diarylmethanes.72
R. S. Martin et al. first reported pincer complexes of palladium (38) for the Suzuki–Miyaura coupling of benzylic halides with aryl boronic acids for the preparation of diarylmethanes (Scheme 48).73 The authors observed that this reaction methodology was inert towards the electronic effects in the boronic acid as well as the benzyl halides but were vulnerable to the steric effects in the boronic acid.
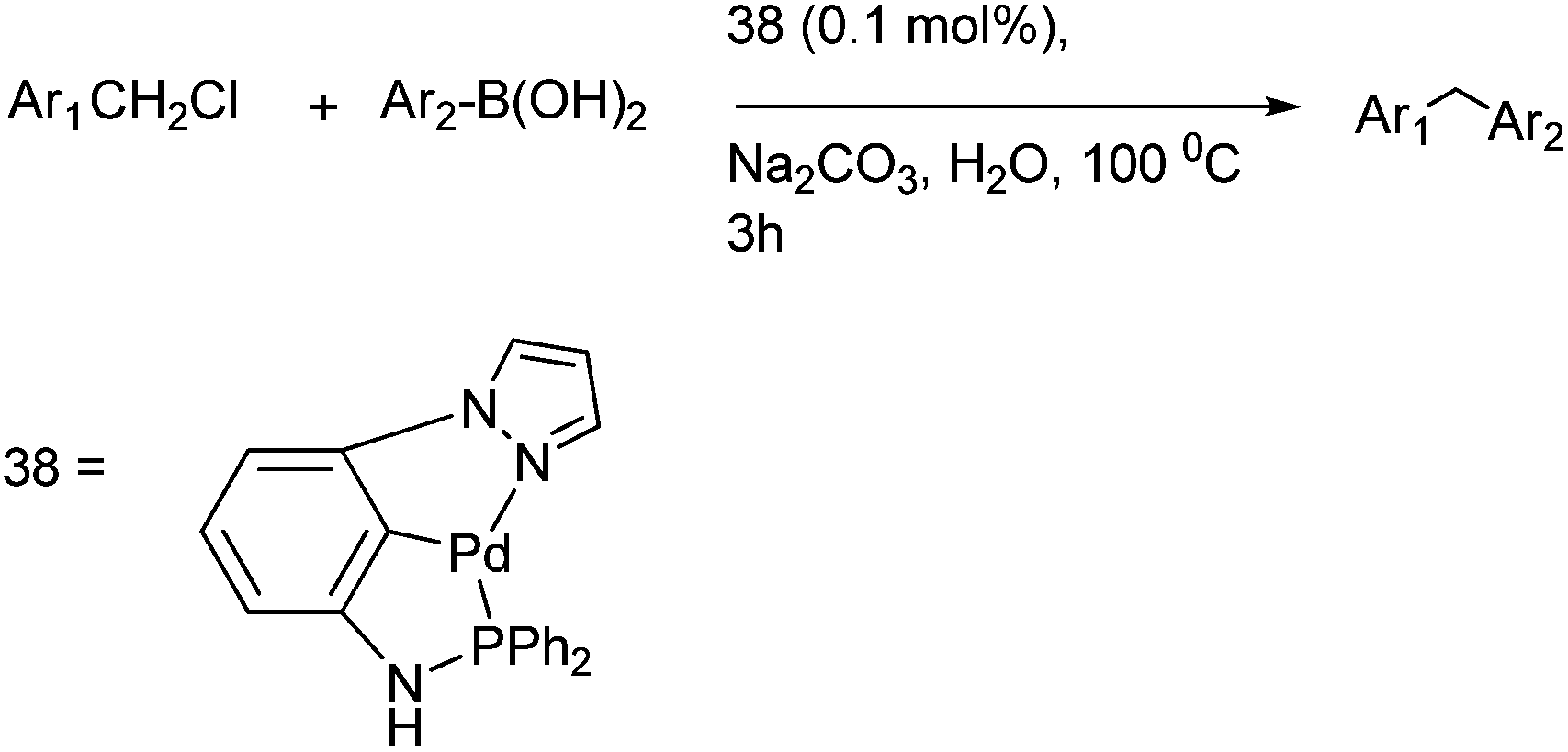 | ||
| Scheme 48 | ||
8.3 Stille coupling
A. L. Monteiro et al. reported Stille coupling of benzylic halides with aryl tributyl tin for the preparation of diarylmethanes (Scheme 49)74 with palladium acetate as the palladium source, diphenylphosphinyl ferrocene (dppf) as the ligand and potassium fluoride as a base in dioxane as solvent. However, when the reaction was carried out in the absence of a base, triphenyl phosphine was found to be a better ligand.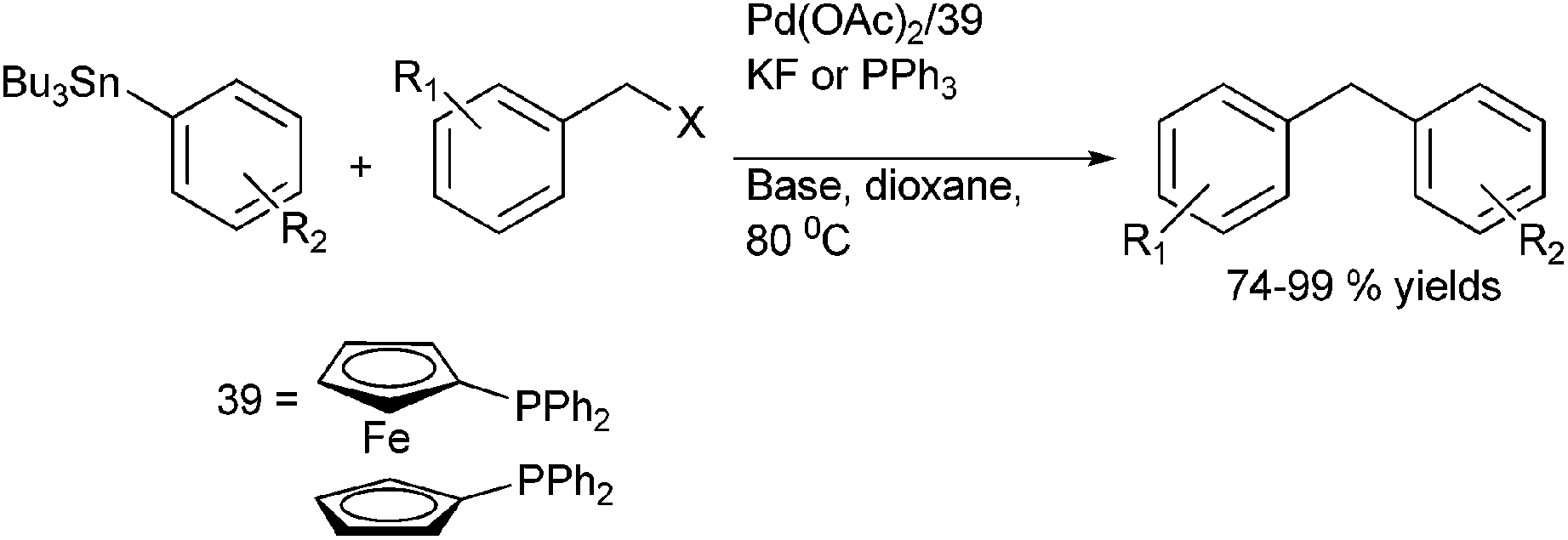 | ||
| Scheme 49 | ||
8.4 Kumada coupling
A. Sarkar and coworkers in 2010 developed a novel air stable bidentate ligand (40) having an indole moiety and N–P donor atom for the Nickel catalyzed Kumada coupling of aryl and benzyl chlorides with aryl magnesium halides (Scheme 50).75 The Kumada coupling reactions of the benzyl chlorides with the aryl magnesium halides were highly reactive yielding the corresponding diarylmethanes in high yields (upto 97% yield).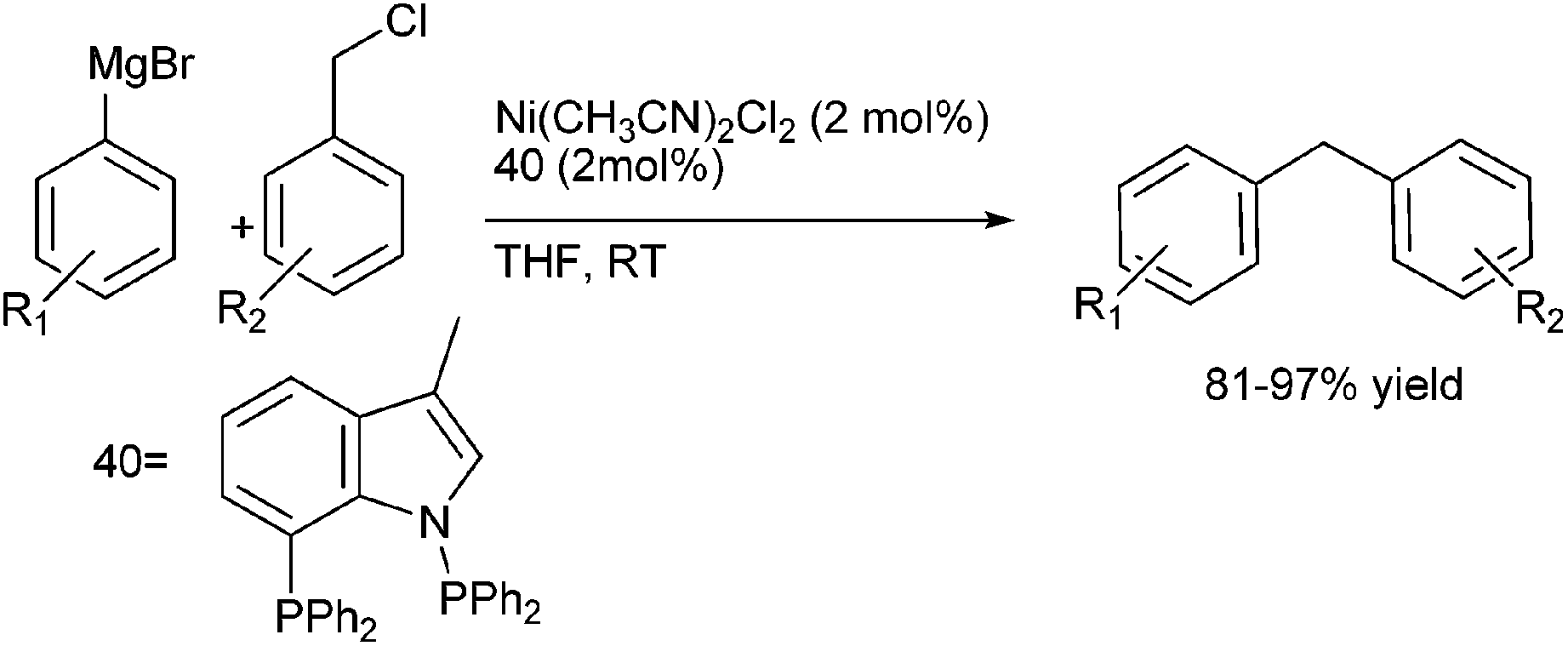 | ||
| Scheme 50 | ||
8.5 Hiyama coupling
A. Sarkar and coworkers in 2010 reported palladium nanoparticles as catalyst for the Hiyama coupling of benzylic halides with aryl trialkoxy silanes (Scheme 51).76 Interestingly the authors found that on decreasing the concentration of the palladium nanoparticles from 4 mol% to 2 mol% decreased the rate of the reaction as well as the product yield. The authors further demonstrated the importance of this methodology by synthesizing the natural product 2,4-bis(4-hydroxy benzyl) phenol in only 3 steps starting from the readily available anisole (Scheme 52).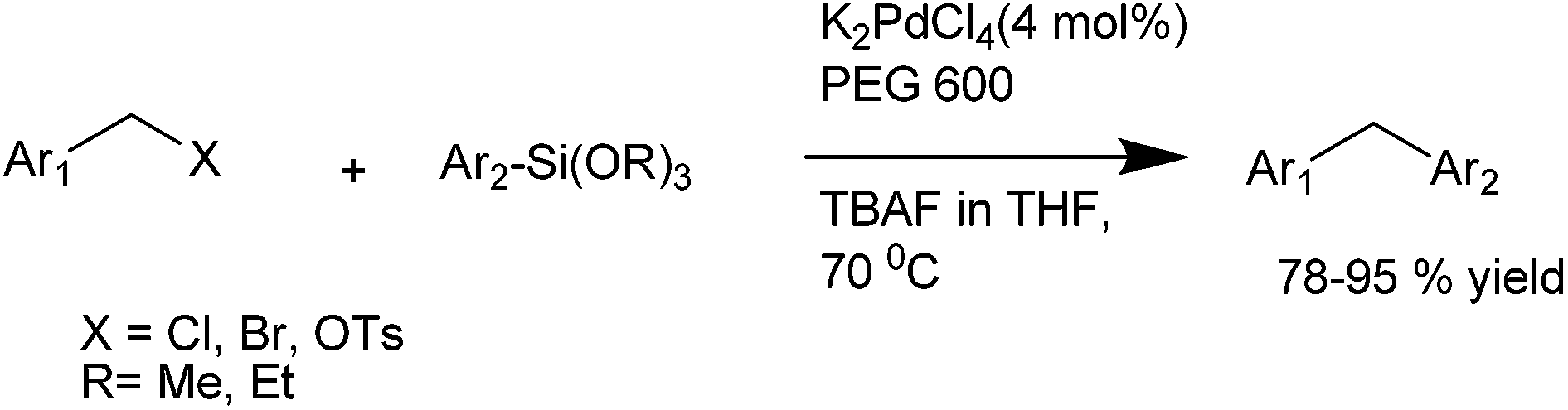 | ||
| Scheme 51 | ||
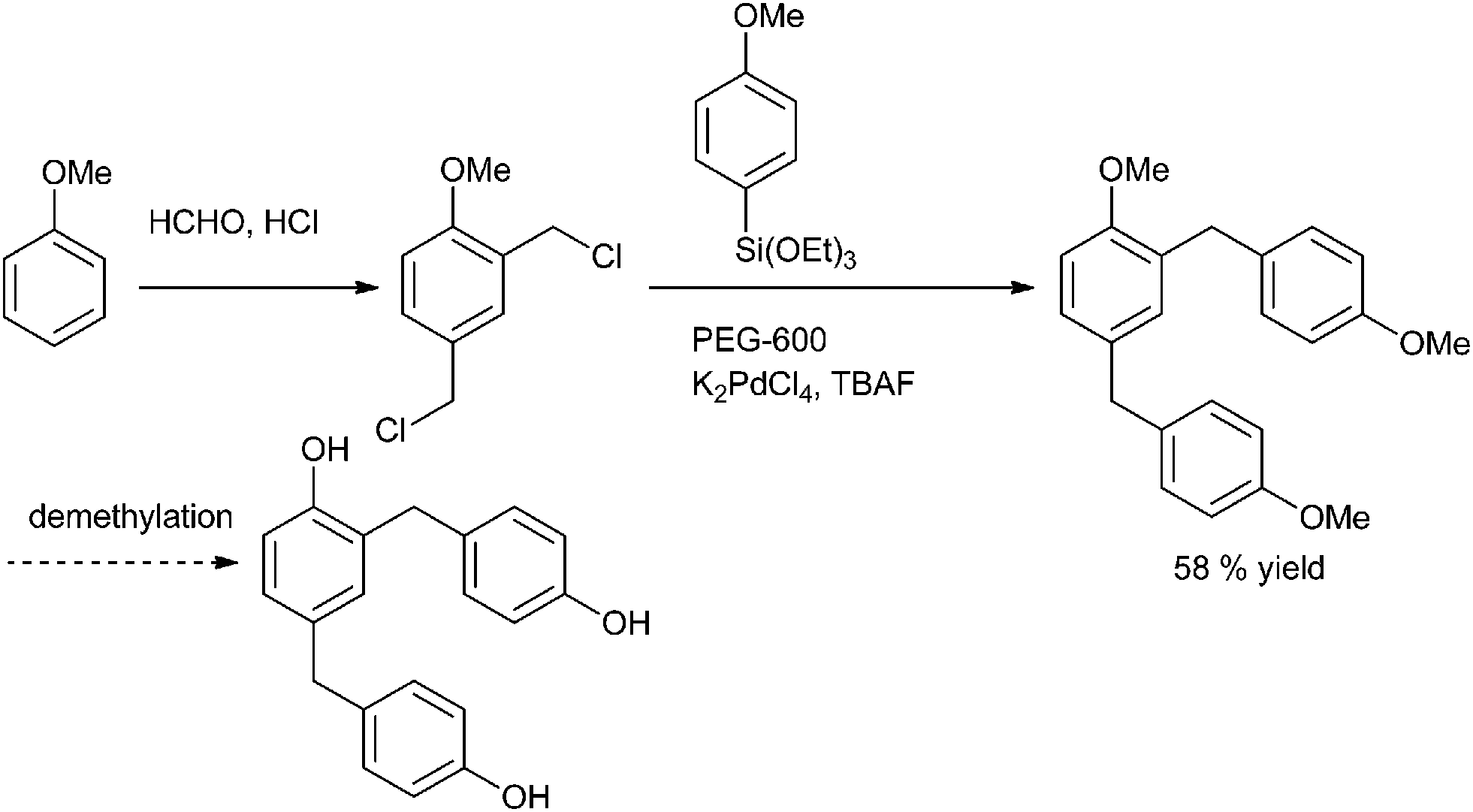 | ||
| Scheme 52 | ||
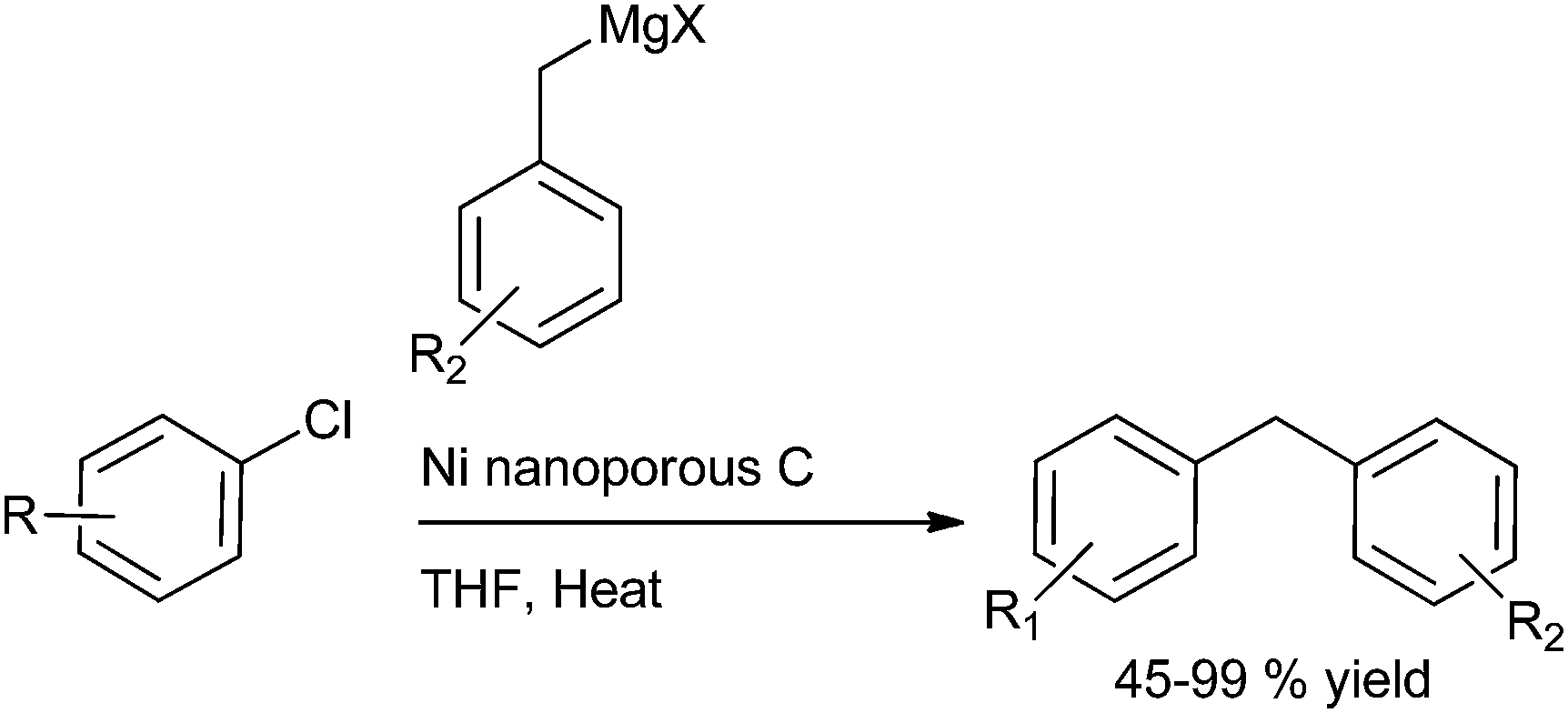
I. M. Lee et al. in 2005 reported nickel(II) nanoporous carbon catalyzed Kumada cross coupling for the preparation of diarylmethanes (Scheme 53).77 When electron donating groups were used in the aromatic ring the cross coupling reaction was very clean while homocoupling as well as cross coupling products were obtained when electron withdrawing groups were used.
 | ||
| Scheme 53 | ||
P. Knoechel et al. in 2006 reported copper(I) catalyzed cross coupling of benzylic phosphates and aryl Grignard reagents for the preparation of diarylmethanes (Scheme 54).78 The authors observed that tetra butyl ammonium iodide had to be added as an additive and for the stabilization of the aryl copper reagent triethyl phosphite had to be used. The authors observed that this protocol was highly general and heteroaryl magnesium intermediate could be coupled with heterocyclic phosphates. The authors further demonstrated the utility of their protocol by synthesizing the antibiotic agent Trimethoprim in four steps in overall yield of 52% (Scheme 55).
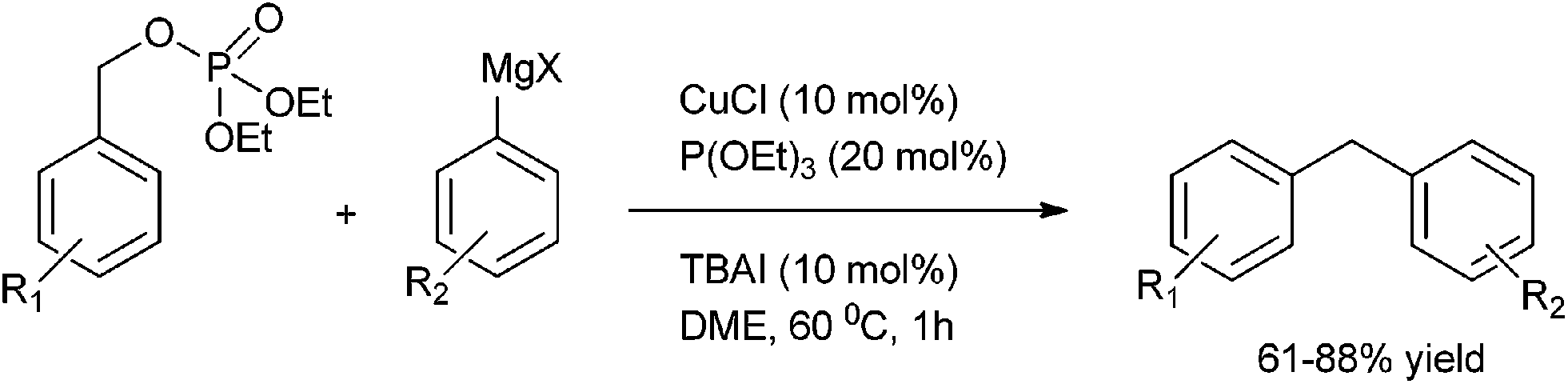 | ||
| Scheme 54 | ||
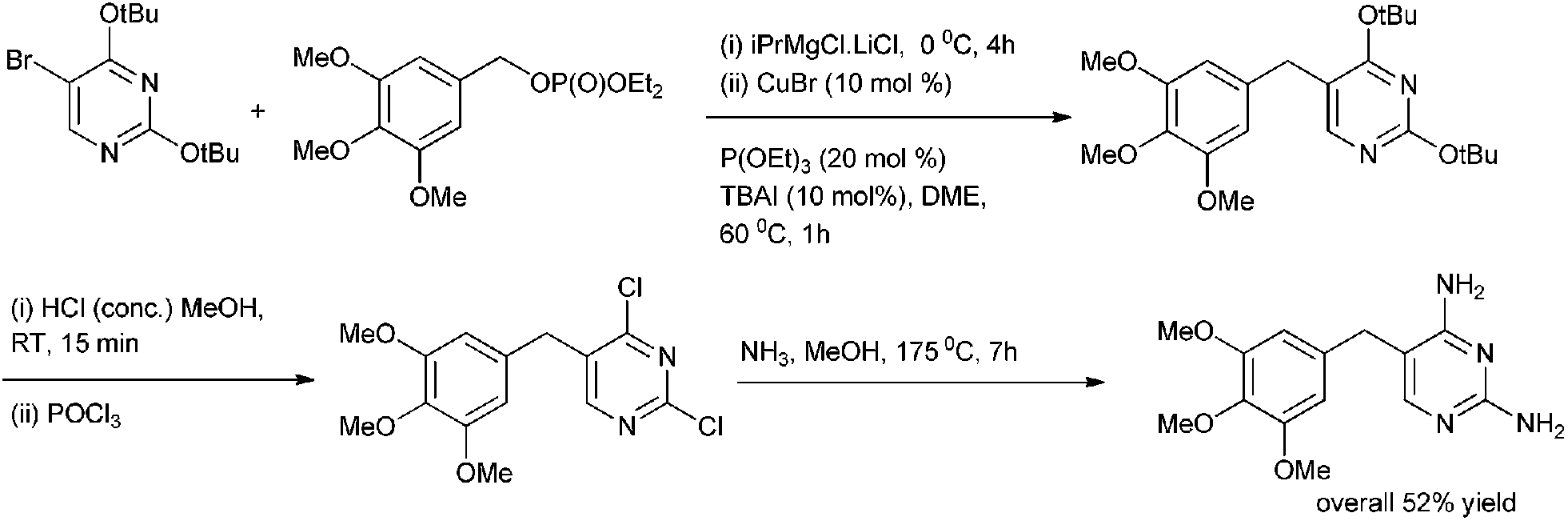 | ||
| Scheme 55 | ||
C. Gosmini et al. first reported cobalt catalyzed reductive cross coupling between the benzyl halides and the aryl halides to form the diarylmethanes in high yields (Scheme 56).79 The authors found that the homocoupled product was formed in low amount. In order to eliminate the formation of homocoupled product, the authors carried out the reaction under Barbier reaction condition. However instead of reducing the homocoupled products, it led to formation of more side products. The authors proposed this was due to the fact that both the halides competitively formed the aryl zinc reagents leading to the homocoupled product. The authors were able to overcome this problem by adding the more reactive benzyl halides in excess.
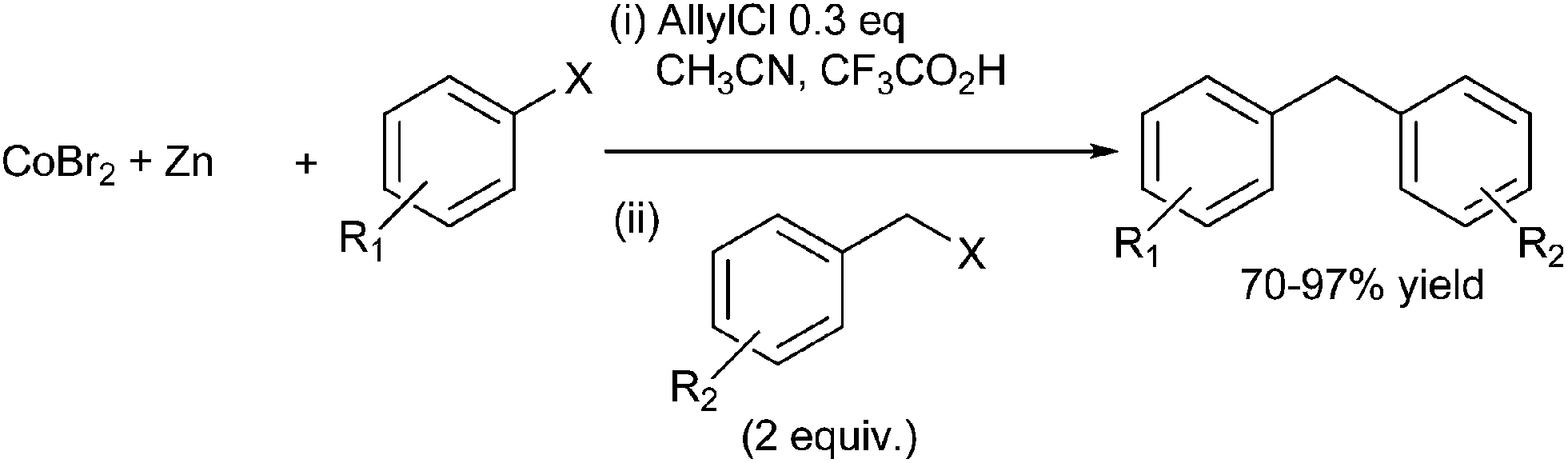 | ||
| Scheme 56 | ||
8.6 Negishi coupling
R. B. Bedford et al. reported iron catalyzed Negishi coupling of benzyl halides and phosphates with aryl zinc reagents (Scheme 57)80 with 1,3-diphenylphosphino propane (41) and 1,2-diphenylphosphino benzene (42) as ligands. Without the ligands, the authors observed this cross coupling reaction was unsatisfactory yielding various side products along with a small amount of the cross coupled product. The authors observed diaryl zinc was better than aryl zinc halide and further addition of a non-transferable dummy ligand like trimethylsilyl methyl group or N,N-dimethylbenzyl amine increases the practicality of the reaction when costlier aryl zinc reagents have to be transferred. Interestingly the authors found that the aryl zinc reagents formed from the corresponding Grignard reagents were far more active than that formed from the aryl lithium reagents. The authors proposed that the magnesium halide formed as by product during the formation of the aryl zinc reagents from the corresponding Grignard reagents helped in the formation of the corresponding aryl zincates through Schlenk equilibrium. The Zincate complexes were more nucleophilic and increased the transmetallation reaction.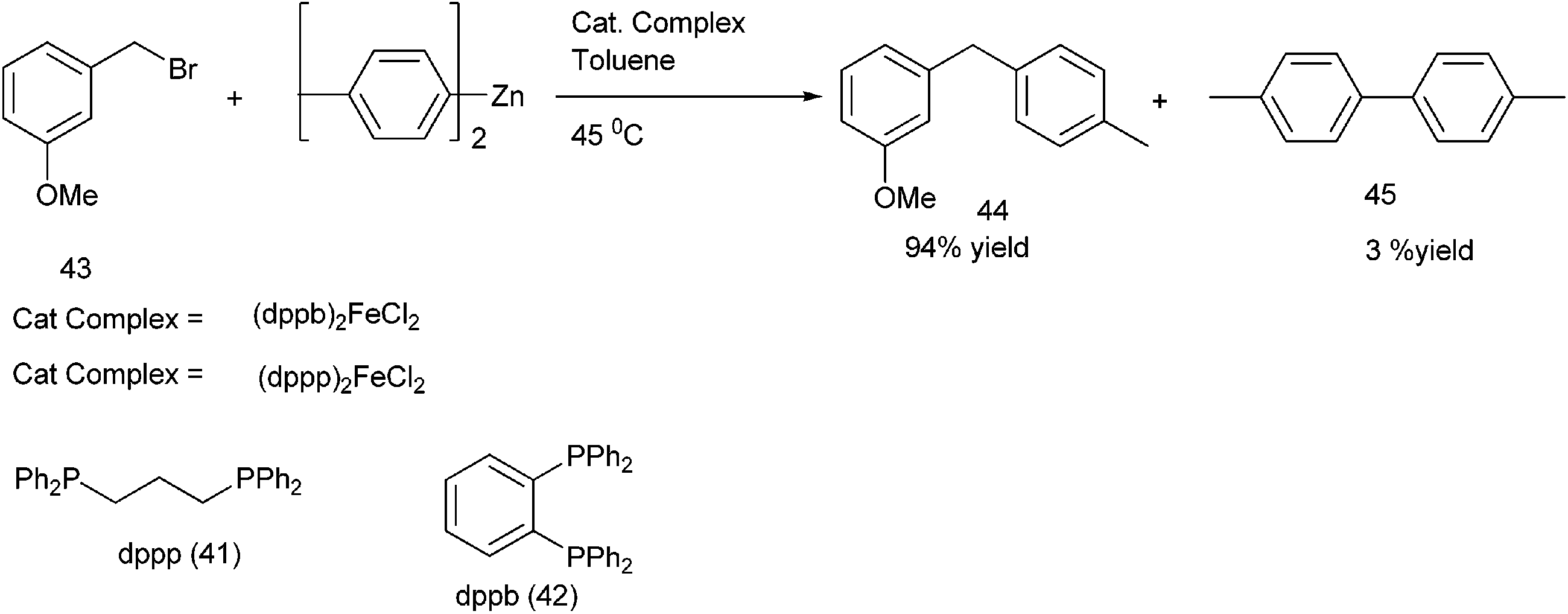 | ||
| Scheme 57 | ||
The authors proposed the iron diphenylphosphino benzene complex gets arylated with the aryl zincate complex to form the neutral species (42A) (Scheme 58) which losses a halide ion to form a five coordinate species (42B). The species (42B) undergoes by either a bimolecular pathway or by the second arylation followed by a reductive elimination. The authors also observed that the zinc atom helps in the stabilization of the intermediate (42B) through the intermediate bimetallic intermediates (42C) and (42D).
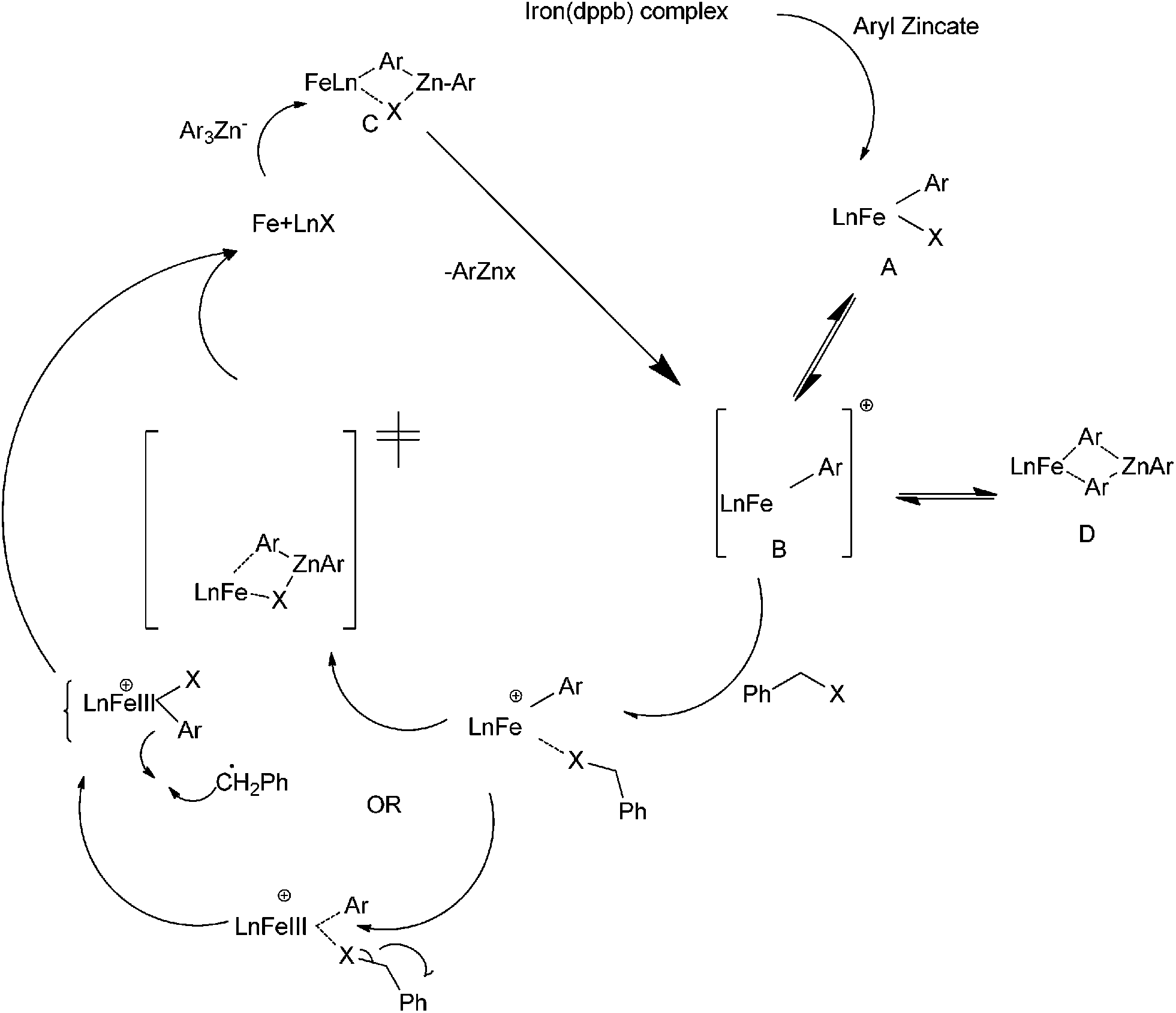 | ||
| Scheme 58 | ||
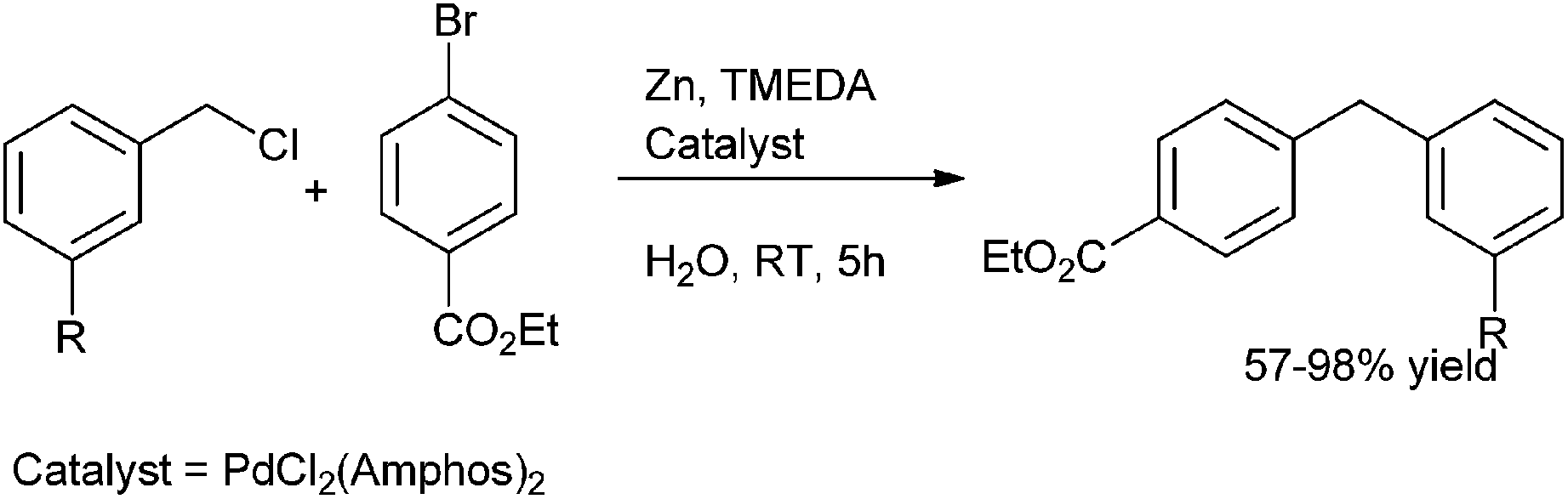
B. H. Lipshutz et al. in 2010 reported in situ organozinc formation followed by palladium catalyzed cross coupling between benzylic halides and aryl halides on water for the preparation of diarylmethanes (Scheme 59)81 with PdCl2 (Amphos)2 as the palladium catalyst and TMEDA as an additive. The authors found that only 25 mol% of TMEDA was sufficient for the reaction and the use of excess TMEDA caused uncontrollable rate of zinc insertion followed by protonation of the organozinc intermediate. Interestingly the authors found that the use of the surfactant for micellar catalysis neither increased the reaction rate nor the yield of the cross coupled product.
 | ||
| Scheme 59 | ||
P. A. Smith et al. in 2011 reported Pd(DPEPhos)Cl2 catalyzed Negishi cross coupling of aryl and benzyl halides with 2,4,6-trimethoxy phenyl zinc chloride for the synthesis of functionalized biaryls and diarylmethanes containing phloroglucinol motif.82 The authors observed both electron donating as well as electron withdrawing groups was tolerated in this cross coupling methodology.
8.7 Cross coupling using benzyl metals and aryl halides
P. Knoechel et al. in 1997 reported palladium catalyzed cross coupling of benzyl metals with organic halides for the preparation of diarylmethanes (Scheme 60).83 Sequential palladium catalyzed cross coupling of benzyl zinc halides with aryl halides followed by cross coupling of the triflates (previously incorporated in the benzyl zinc halides) with aryl halides led to the formation of polyfunctional aromatic compounds.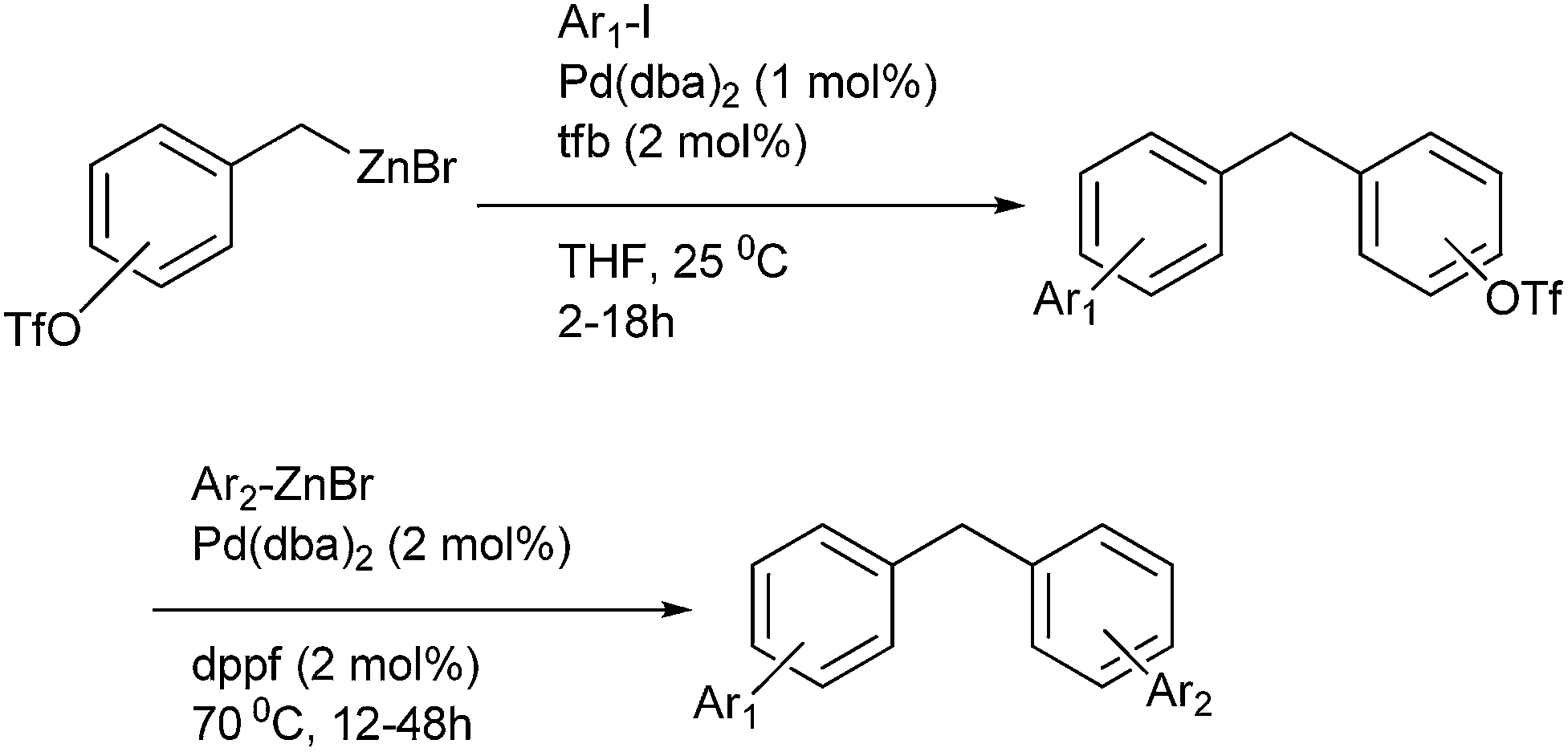 | ||
| Scheme 60 | ||
P. A. Worthington et al. in 2000 reported cross coupling reactions of benzylic zinc halides with 2-(6-bromo-2-pyridyl) pyrimidines for the preparation of agrochemicals (fungicides) based on the pyridyl pyrimidines core84 with 31 as the catalyst.
A. G. Martínez et al. in 2000 reported Stille cross coupling between a hypervalent tin reagent (tetrabutylammonium difluorotribenzylstannate) (46) with aryl triflates for the preparation of unsymmetrical diarylmethanes (Scheme 61).85 Small amount (less than 10%) of the homocoupling products (biaryls and diaryl ethanes) was formed. The authors observed the completion of the reaction varied (within 1 min to 2 h) depending on the nature of the substrates.
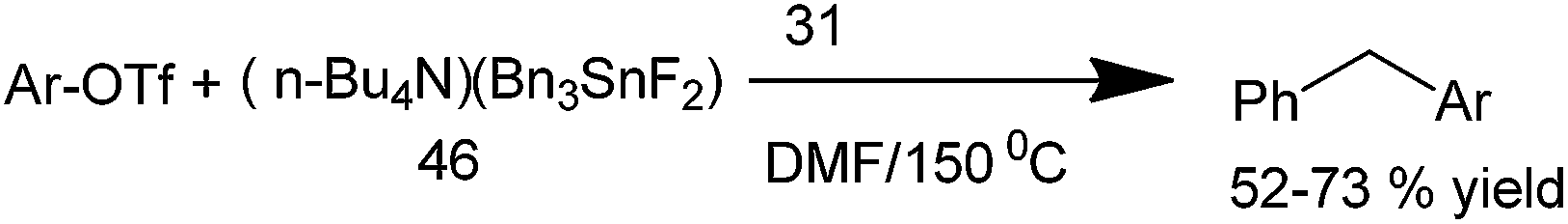 | ||
| Scheme 61 | ||
R. D. Rieke et al. in 2000 reported the facile preparation of benzyl manganese from benzyl halides, phosphates and sulfonates and utilized it in various cross coupling reactions (Scheme 62).86 Thus (31) catalyzed cross coupling with aryl iodides yielded diarylmethanes.
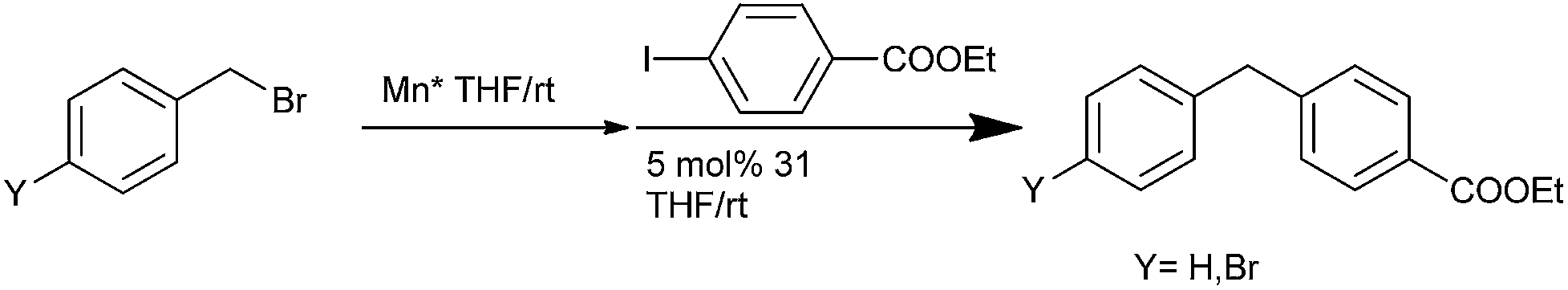 | ||
| Scheme 62 | ||
C. Mioskowski et al. in 2000 reported the solid phase synthesis of the diarylmethanes (Scheme 63).87 Alkyl thiol resins were first alkylated with 4-cyano benzyl bromide. Further alkylation of the resin with triethyl oxonium tetrafluoroborate through palladium catalyzed cross coupling with aryl boronic acids released the ethylated alkyl resin as well as the diaryl methane. The authors observed the homocoupled biaryls as side products. The authors also observed that both electrons donating as well as the electron withdrawing group was tolerated in the thiol resin. Interestingly, the authors observed that acetal, ester, amide and nitrile groups in the benzyl bromide which were assumed to be a threat to the activation step were well tolerated in the alkylation step as well as the cross coupling step. To illustrate the versatility of this protocol, the 4-bromo benzyl bromide was attached to the alkyl thiol resin followed by a Heck coupling, activation of the resin and then a palladium catalyzed cross coupling with aryl boronic acids to yield the diaryl methyl cinnamates.
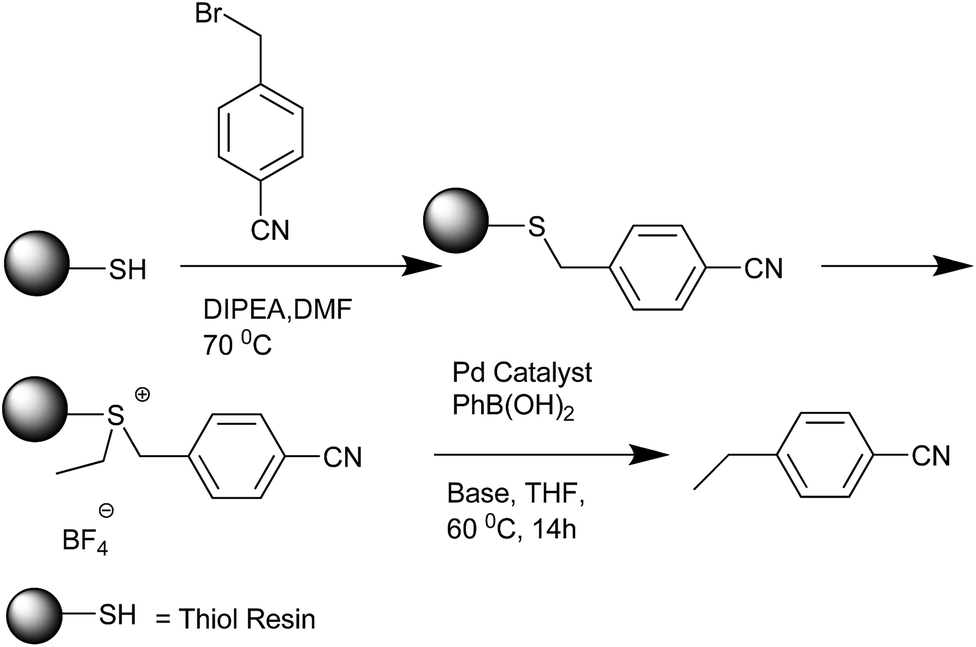 | ||
| Scheme 63 | ||
K. Itami et al. in 2002 reported diversity oriented synthesis of diarylmethanes through sequential cross coupling reactions of a mixed gem-dimetalmethanes using 2-PyMe2SiCH2–SnBu3 as the cross coupling agent (Scheme 64).88 The authors envisioned that a palladium catalyzed cross coupling with aryl iodide would give a benzyl metallic species which on further palladium catalyzed cross coupling with another aryl iodide (Hiyama coupling) would generate the unsymmetrical diarylmethanes. However, interestingly the authors noticed that the Hiyama coupling was unsuccessful without the use of additives. With additives like potassium fluoride, TBAF and TASF, protodesilylation was the only product and the reaction was unsuccessful with silver(I) sulphide, silver fluoride, silver tetrafluoro borate, cupric oxide, cuprous oxide and auric oxide. However the reaction was smooth when silver oxide or silver acetate was used as an additive with silver oxide being higher yielding. The authors deciphered that strong silver oxygen bond was a requisite factor for the reaction. When the pyridyl group was changed to phenyl group or methoxy group, the reaction did not proceed and also with the protonated pyridyl group, the reaction failed. Thus the authors proposed that the pyridyl group coordinates the silver atom through a proximity effect while the Lewis base activation through oxygen and silicon helps the Hiyama coupling to proceed.
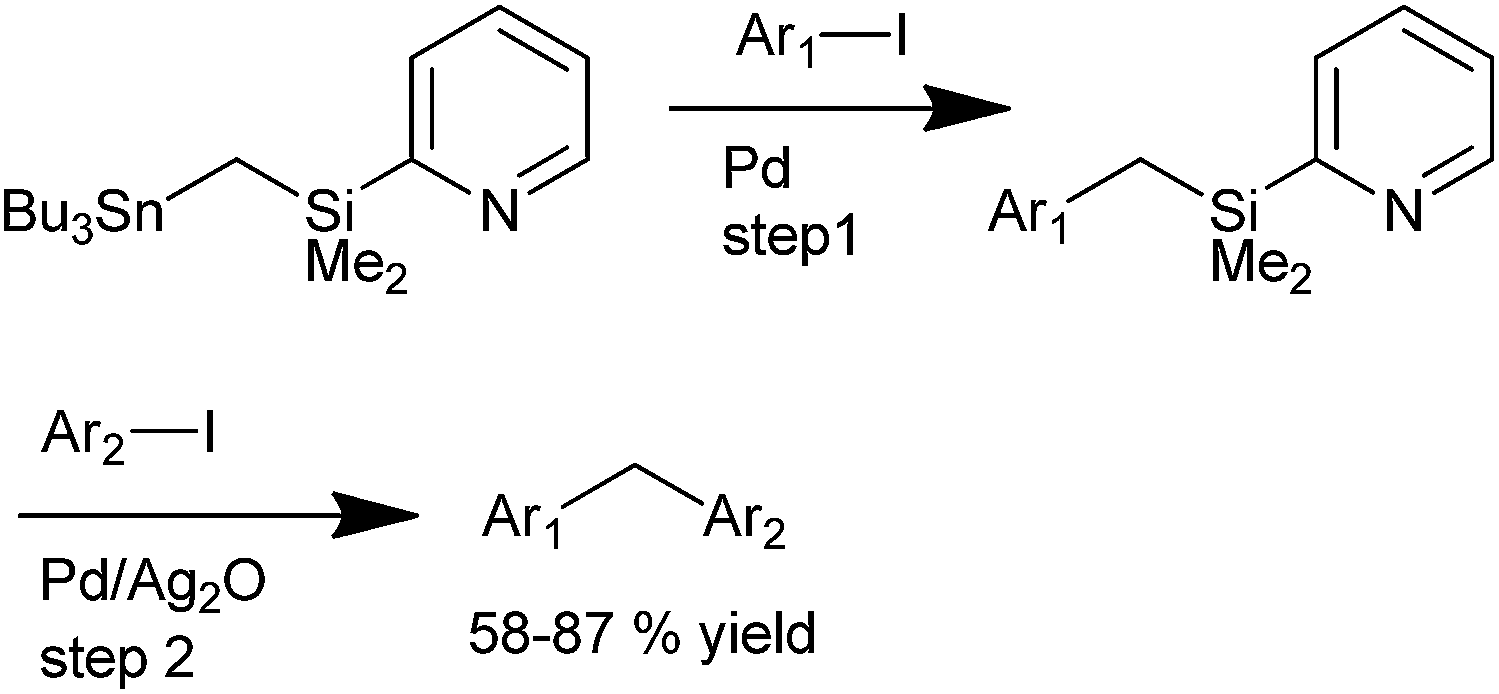 | ||
| Scheme 64 | ||
A. Flaherty et al. reported in 2005 palladium catalyzed Suzuki–Miyaura of B-benzyl-9-borabicyclo [3.3.1] nonane with aryl halides and aryl triflates for the synthesis of diarylmethanes (Scheme 65).89 Probably due to proper dissolving of the base, water was found to be an accelerating solvent and the coupling was complete within one hour. For most of the electron deficient aryl halides, (31) was found to be an efficient catalyst. However 4-chlorobenzonitrile failed to respond to this cross coupling reaction. A further investigation revealed that S-phos ligated palladium acetate was a better catalyst than Pd(PPh3)4.
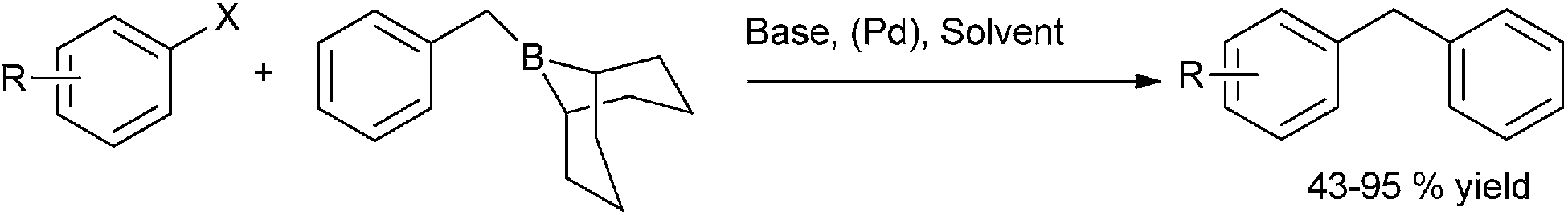 | ||
| Scheme 65 | ||
Later on in 2012, T. Shibata et al. reported the Suzuki–Miyaura cross coupling of diborylmethanes with aryl halides for the preparation of symmetrical as well as unsymmetrical diarylmethanes in one pot (Scheme 66).90 Interestingly the authors found that the reaction course could be controlled by adjusting the temperature as well as the amount of the added base. The synthesis of the unsymmetrical diarylmethanes demanded the use of 2 equivalent diboryl methanes in the first cross coupling sequence and excess base in the second step of the sequence was to negate the deleterious effect of the excess diborylmethane.
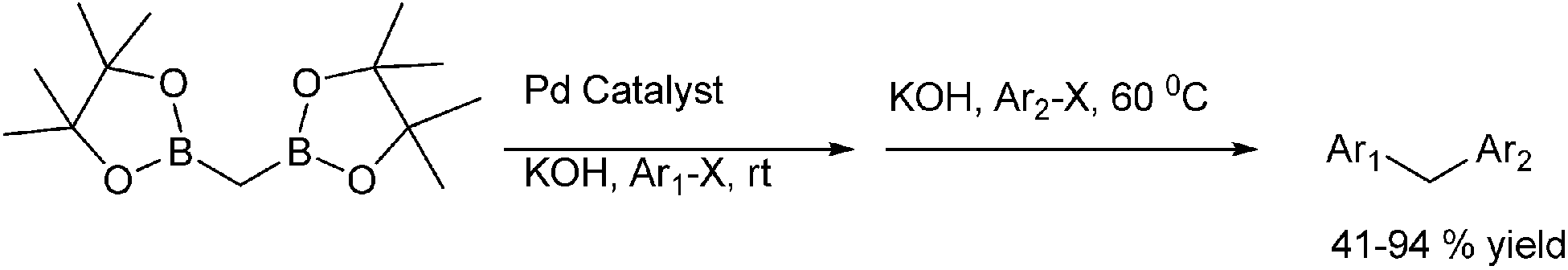 | ||
| Scheme 66 | ||
P. Knoechel et al. in 2008 reported nickel catalyzed cross coupling of benzyl zinc reagents with aryl halides for the preparation of the diarylmethanes (Scheme 67).91 The benzyl zincs were generated by the insertion of metallic zinc to benzylic chlorides in presence of lithium chlorides. This method was highly chemoselective tolerating various electrophilic functional groups like ester, carbonyl and the cyano functional groups.
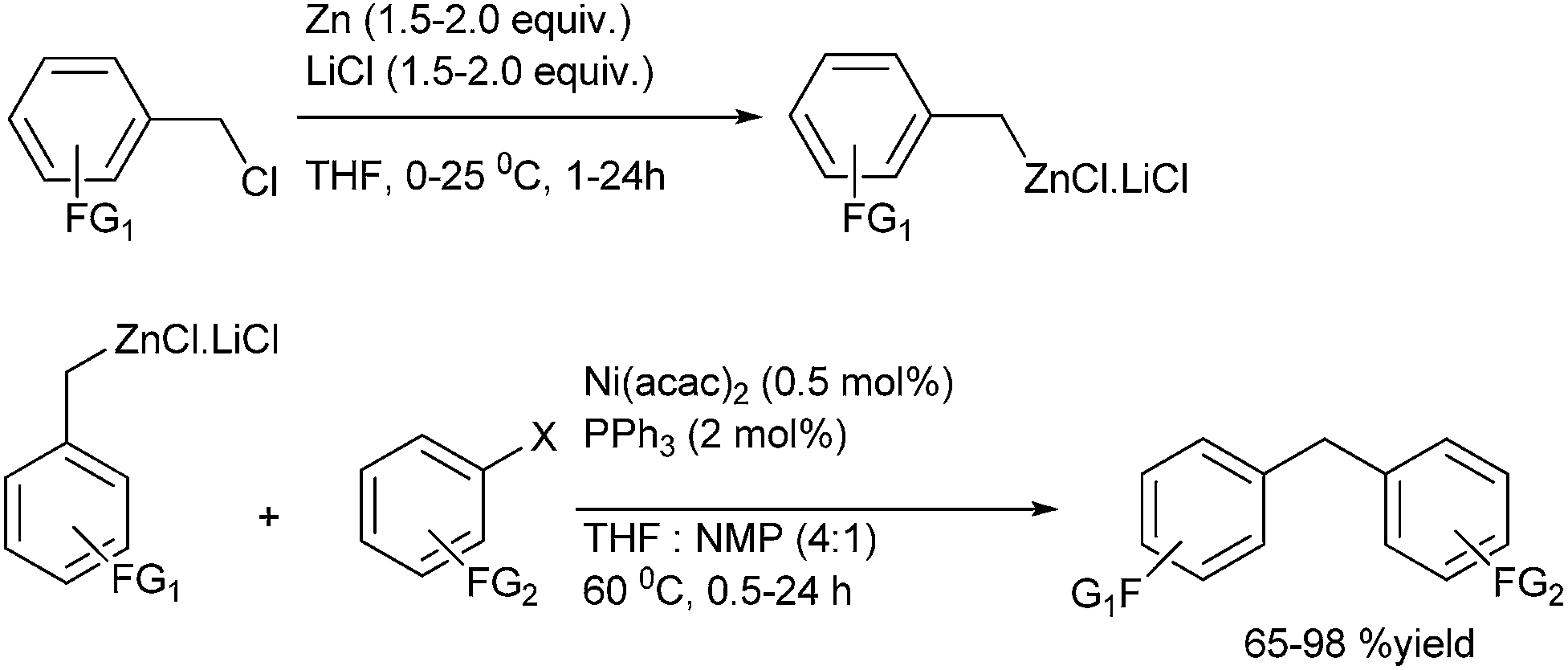 | ||
| Scheme 67 | ||
L. S. Chupak et al. in 2009 first reported Pd(PPh3)4 (31) catalyzed cross coupling of benzyl indium reagents with aryl halides for the preparation of diarylmethanes (Scheme 68).92 The benzyl indium reagents were generated in situ by the addition of indium powder to benzyl halides in DMF. Though the reaction was tolerant to the electron withdrawing effects in the electrophilic halides as well as the aryl halides, electron rich gave modest yields. Particularly noteworthy was the fact that ortho and meta methoxy benzyl bromide as well as para methoxy benzyl chloride failed to give any cross coupled product. However, interestingly the para-trifluoromethoxy benzyl bromide, having attenuated electron rich ability of the oxygen atom gave high yield of the cross coupled product. Also interesting was the notion that the para substituted benzyl bromides gave consistent low yield of the cross coupled product compared to the ortho and the meta isomer. Also, interestingly the authors found that the reaction was inert towards steric effects in the electrophilic coupling partner. However α-methyl benzyl bromide, though formed the organometallic intermediate was inert to this coupling reaction.
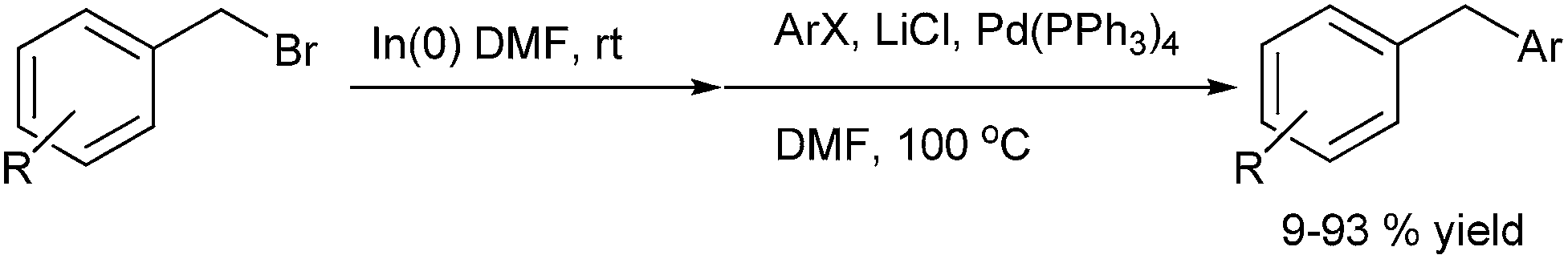 | ||
| Scheme 68 | ||
Later in 2010, X. Zhang et al. reported triphenyl phosphine ligated palladium acetate to be an efficient catalyst for the cross coupling of the benzyl halides with perfluoroarenes.93 The authors observed the reaction to be dependent on the nature of the solvent, reaction temperature and the nature of the base used. Also they observed the reaction to be depended on the electronic nature of the benzyl chlorides with electron withdrawing benzyl halides giving unsatisfactory yield of the cross coupled product while the electron rich benzyl halides giving excellent yield of the cross coupled product.
L. Liu et al. in 2011 reported palladium catalyzed decarboxylative coupling of potassium nitro phenyl acetates with aryl halides for the preparation of diarylmethanes (Scheme 69).94 [Pd(allyl)Cl]2 (47) was used as a palladium catalyst and among the phosphine ligands tested, X-Phos was the best yielding the diarylmethanes in high yields. The use of nitro functionality allowed the authors to prepare a diverse set of diarylmethanes prepared through the functional group interconversion of the nitro functionality after the decarboxylative coupling with aryl halides. They also found that the decarboxylative protonation was a major side reaction yielding nitrotoluenes as the side products.
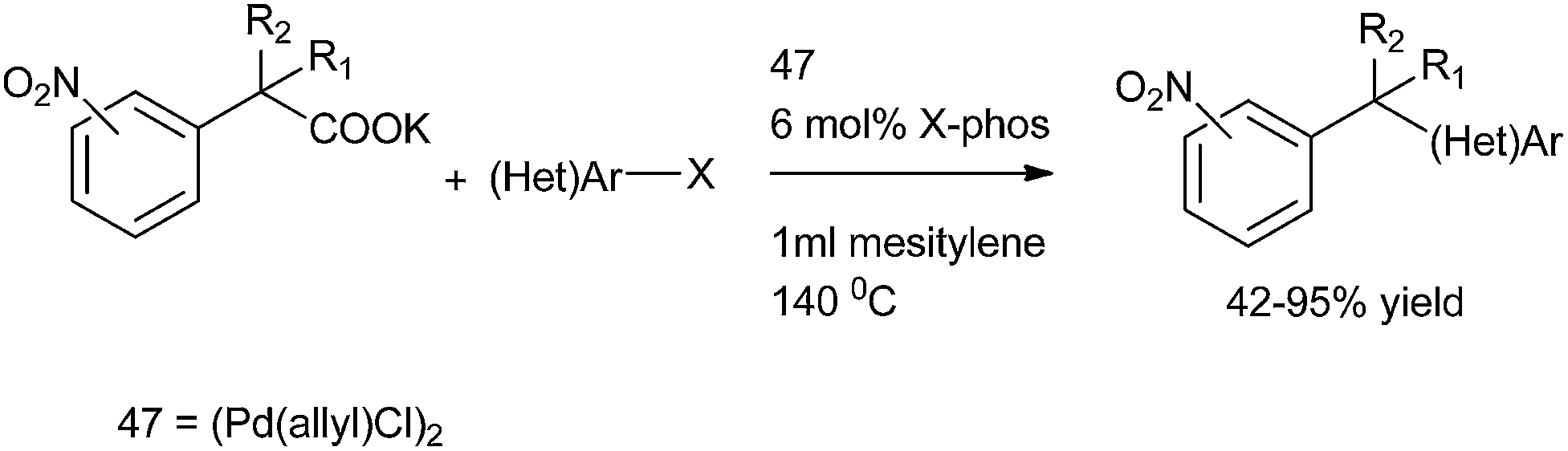 | ||
| Scheme 69 | ||
N. E. Leadbeater et al. in 2009 reported ligand free palladium catalyzed α-arylation of benzyl ketones for the synthesis of diarylmethanes in water (Scheme 70).95
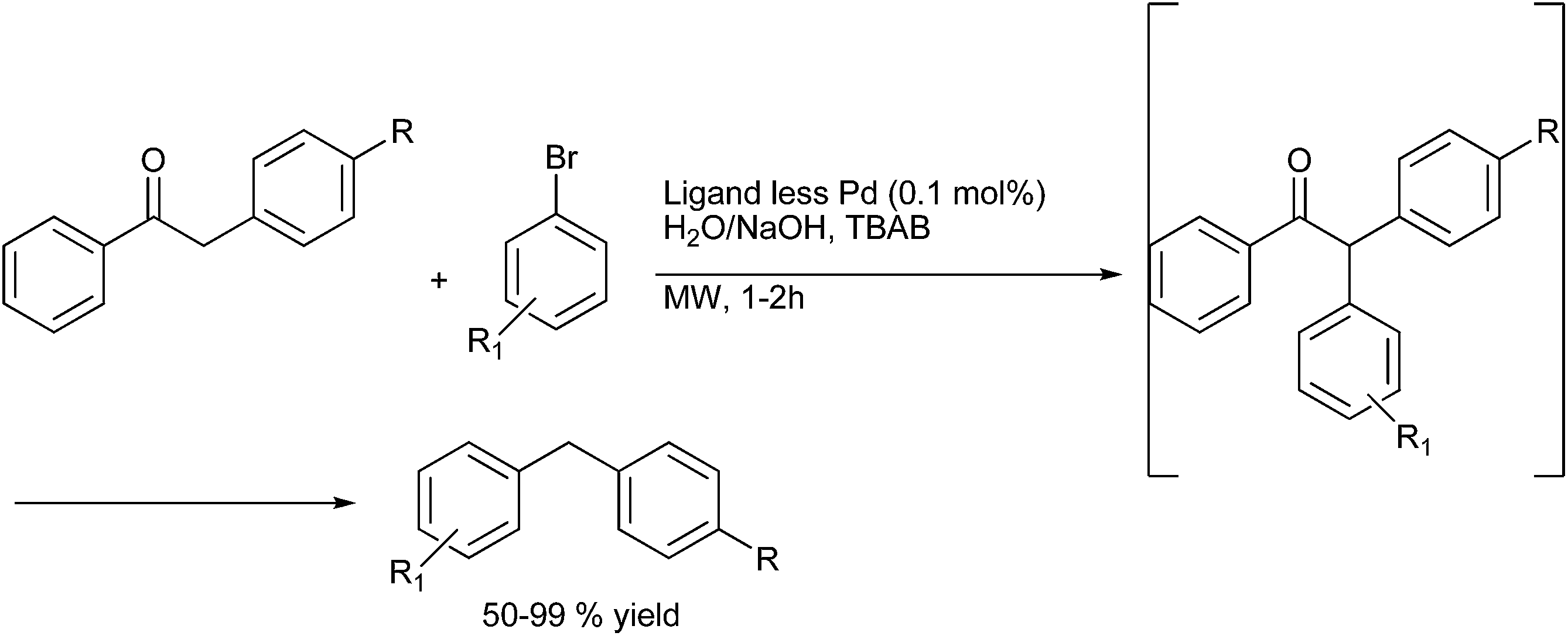 | ||
| Scheme 70 | ||
G. J. Deng et al. in 2013 reported palladium catalyzed desulfitative cross coupling of sodium sulfinates with benzyl halides for the synthesis of diarylmethanes (Scheme 71).96 At first oxidative addition of the benzyl halide with the palladium catalyst forms an benzyl palladium intermediate (A) (Scheme 72). The replacement of the chloride with aryl sulfinite yields the intermediate (B) which loses a molecule of sulfur trioxide to form intermediate (C). Reductive elimination generates the diarylmethanes.
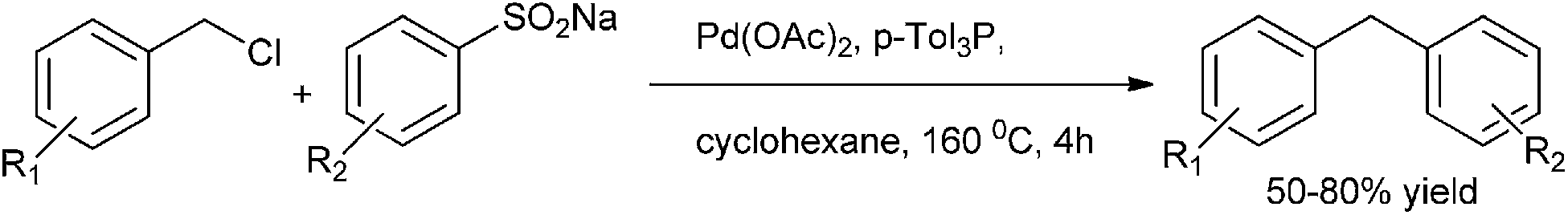 | ||
| Scheme 71 | ||
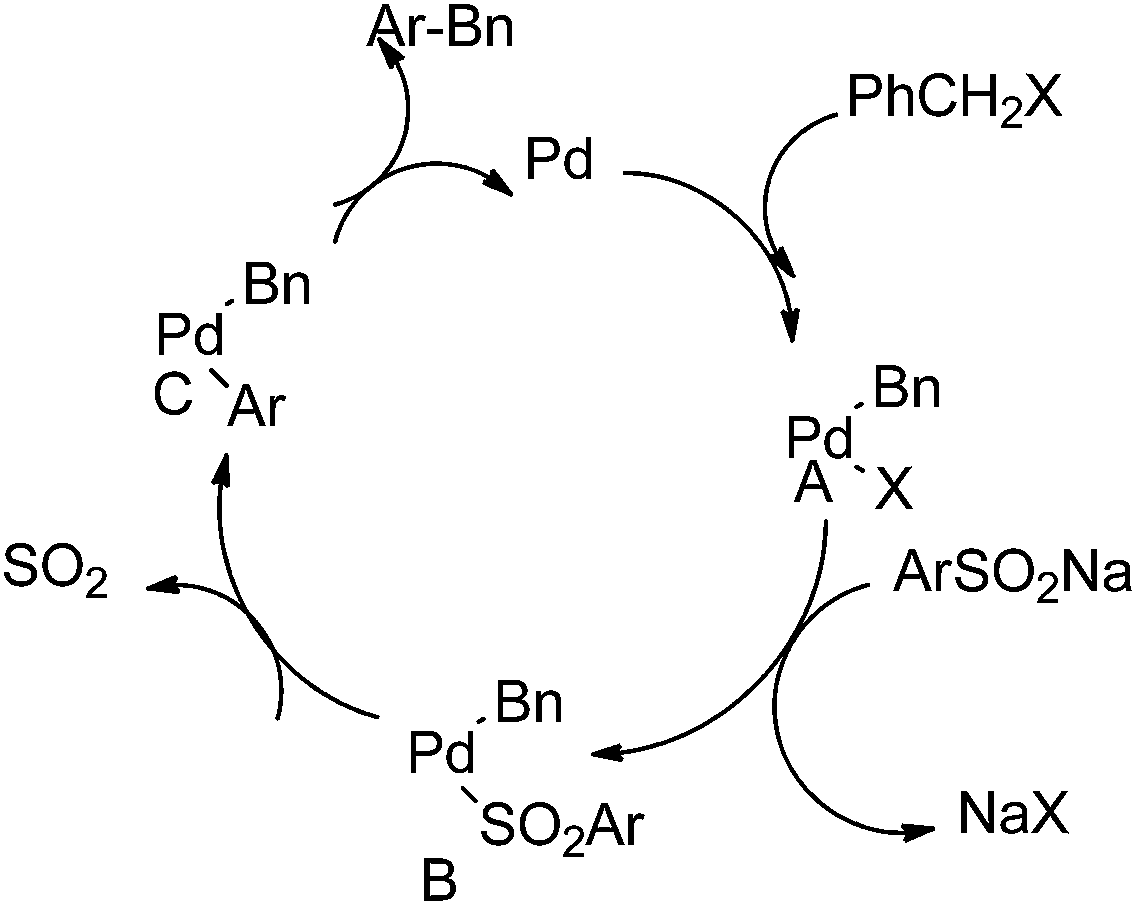 | ||
| Scheme 72 | ||
9. Miscellaneous routes for the preparation of diarylmethanes
9.1 Preparation of diarylmethanes through direct reduction of diaryl ketones
B. Hatano et al. in 2003 reported the direct reduction of diaryl ketones to diarylmethanes using supercritical isopropanol (Scheme 73).97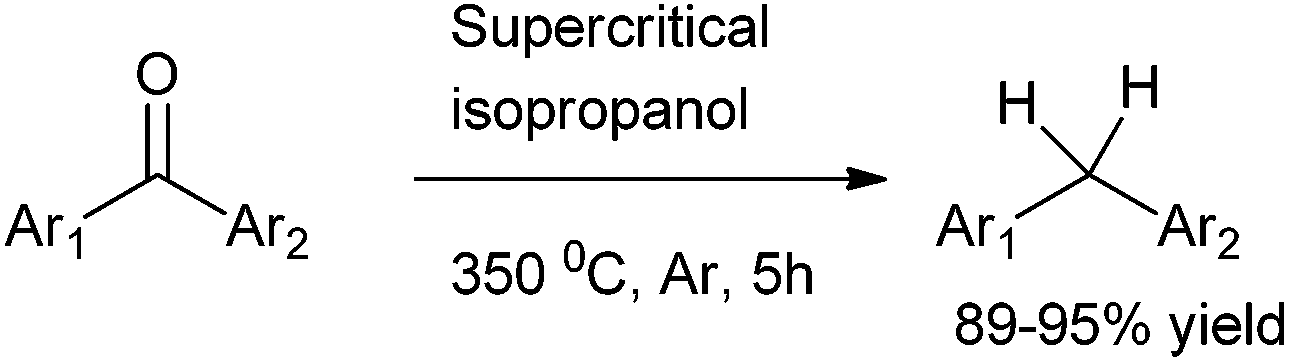 | ||
| Scheme 73 | ||
9.2 Preparation of diarylmethanes through activation of C–O bond
 | ||
| Scheme 74 | ||
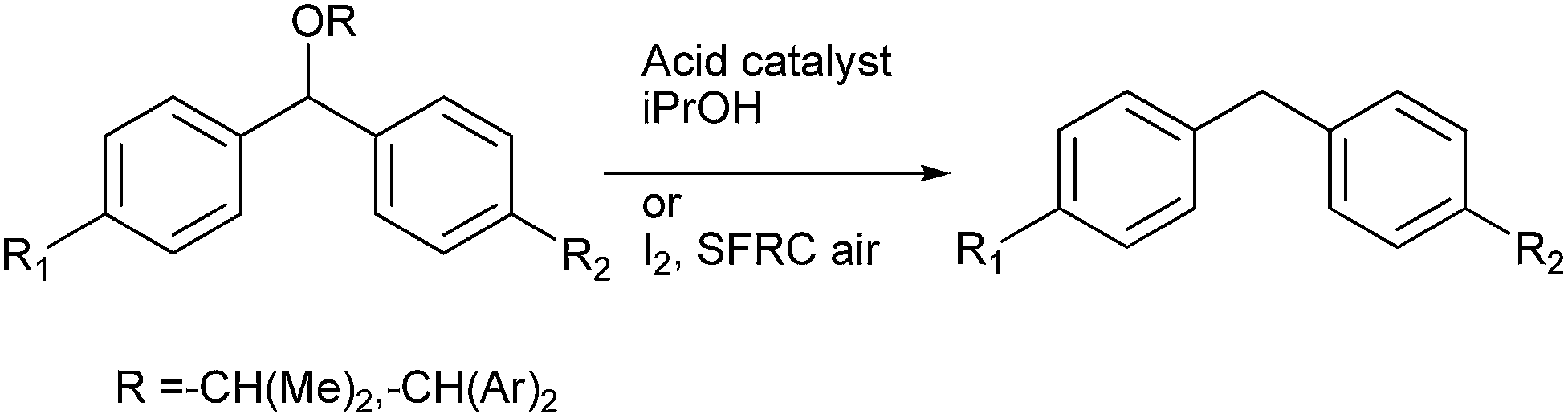
Later in 2013 M. Jereb et al. demonstrated molecular iodine to be an efficient, selective catalyst for the dispropotionation of the aryl ethers to the corresponding diarylmethanes and the diaryl ketones under solvent free reaction condition (SFRC) (Scheme 74).99 The authors observed that the reaction could be carried out under solvent free conditions and that the crucial hydride transfer for the dispropotionation took place from the more electronically poorer side yielding the more stable carbocation.
9.3 Lewis acid mediated sp3 C–O bond activation
Z. J. Shi et al. in 2008 reported FeCl3 mediated sp3 C–O activation of benzyl ethers for the Friedel–Crafts alkylation of benzyl ethers with arenes (Scheme 75).100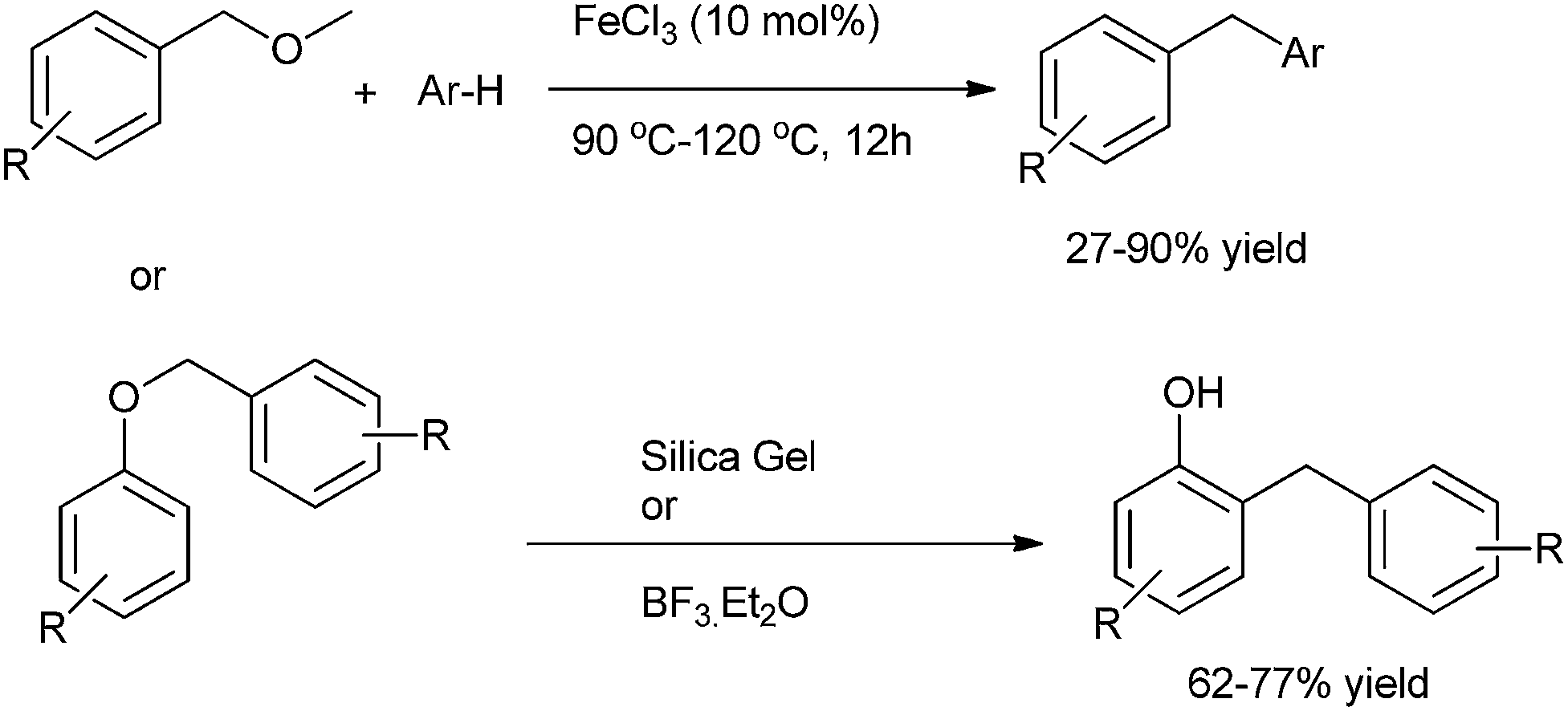 | ||
| Scheme 75 | ||
G. A. Kraus et al. in 2012 reported Lewis acid catalyzed rearrangement of benzylic ethers for the facile preparation of diarylmethanes (Scheme 75).101
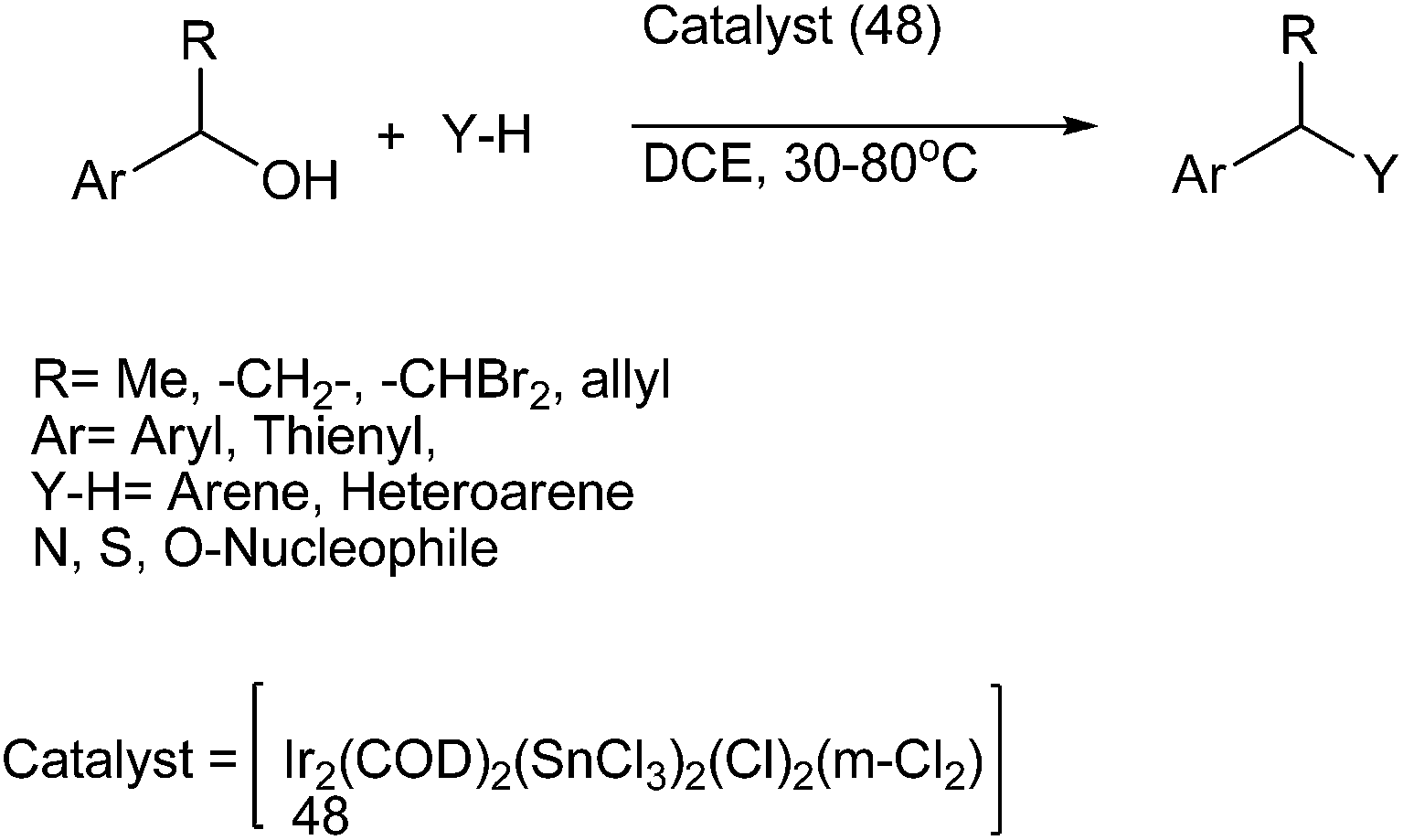
S. Roy et al. in 2007 reported hetero bimetallic Ir–Sn complex (48) for the benzylations of the benzylic alcohols having β-hydrogens for the preparation of diarylmethanes (Scheme 76).102,103 The hard main group metal center of the complex activated the hard donor atom while the soft transition metal center activated the soft donor center such as the π-electron cloud. The authors observed that at room temperature the reaction was sluggish and much of the starting material remains unreacted. Also the reaction yielded dibenzyl ether instead of the diaryl methane. Interestingly, they noted that on increasing the temperature to 50 °C, the reaction yielded the dibenzyl ether as well as the alkane with the ether being the major product while further increasing the temperature to 80 °C yielded the diaryl methane as the sole product. The authors noted that this methodology could be extended towards the preparation of diheteroaryl as well as aryl heteroaryl alkane.
 | ||
| Scheme 76 | ||
M. Lautens et al. in 2008 reported benzyl chloride as an aprotic surrogate of benzyl alcohol for the reductive benzylation (Scheme 77).104 The authors observed benzyl chloride was the hydride transfer agent.
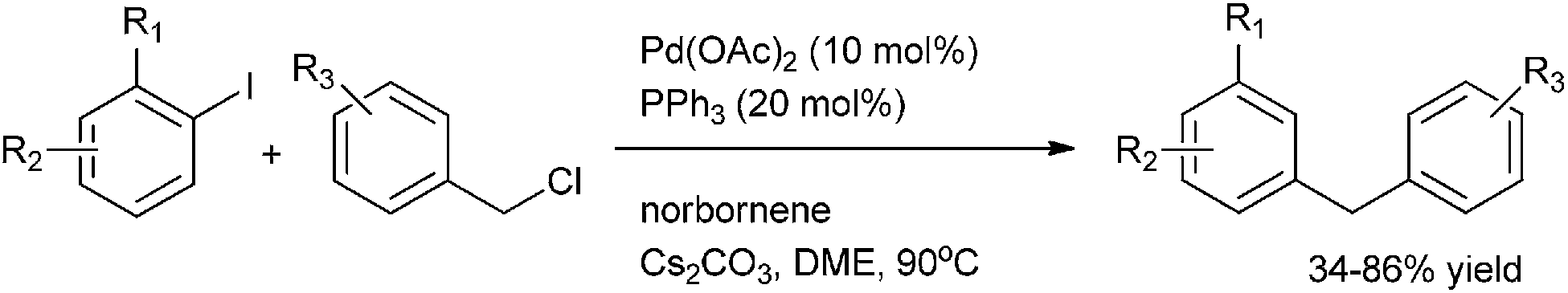 | ||
| Scheme 77 | ||
9.4 Other miscellaneous approaches
A. Kumar et al. in 2009 reported organocatalytic multi component coupling procedure for the synthesis of diarylmethanes (Scheme 78).105 The authors observed that with ordinary acid catalysts, the reaction yielded the diaryl methane containing the two amine groups in major amounts while with proline the diarylmethane containing the β-naphthol unit was formed as major product (Scheme 79).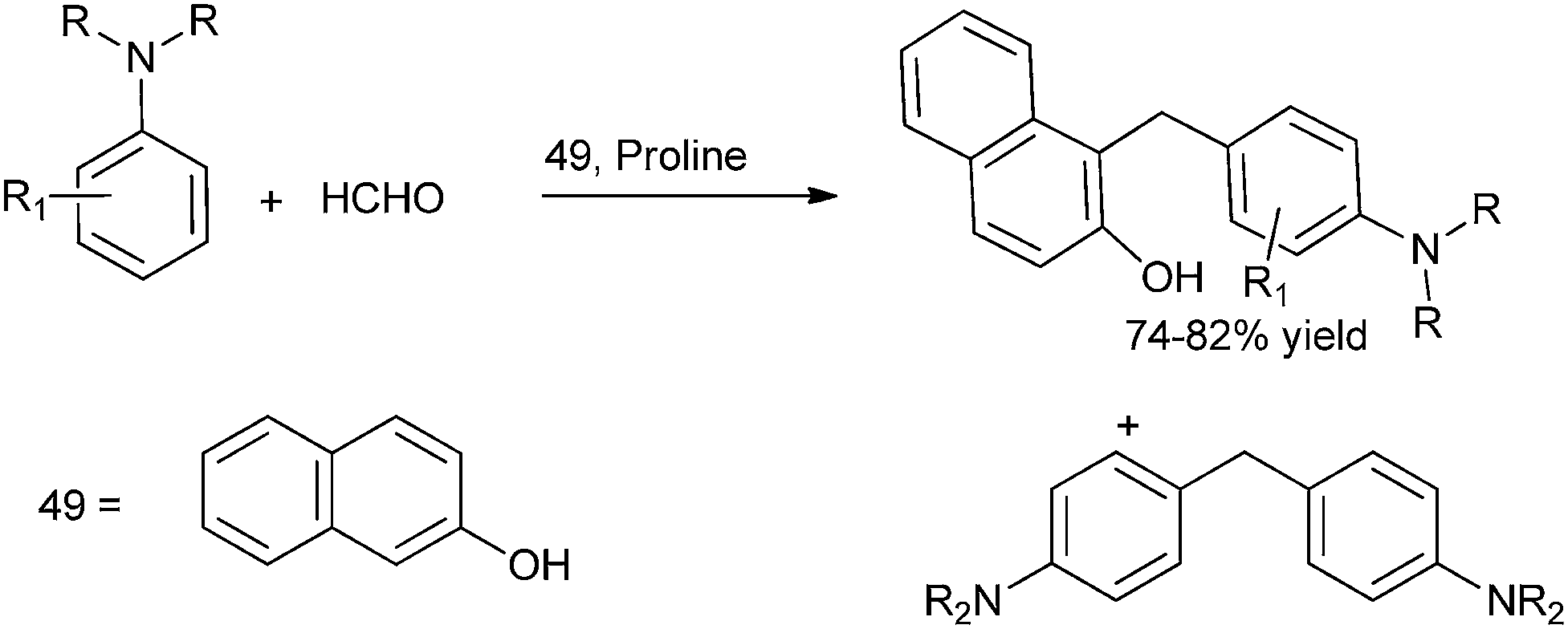 | ||
| Scheme 78 | ||
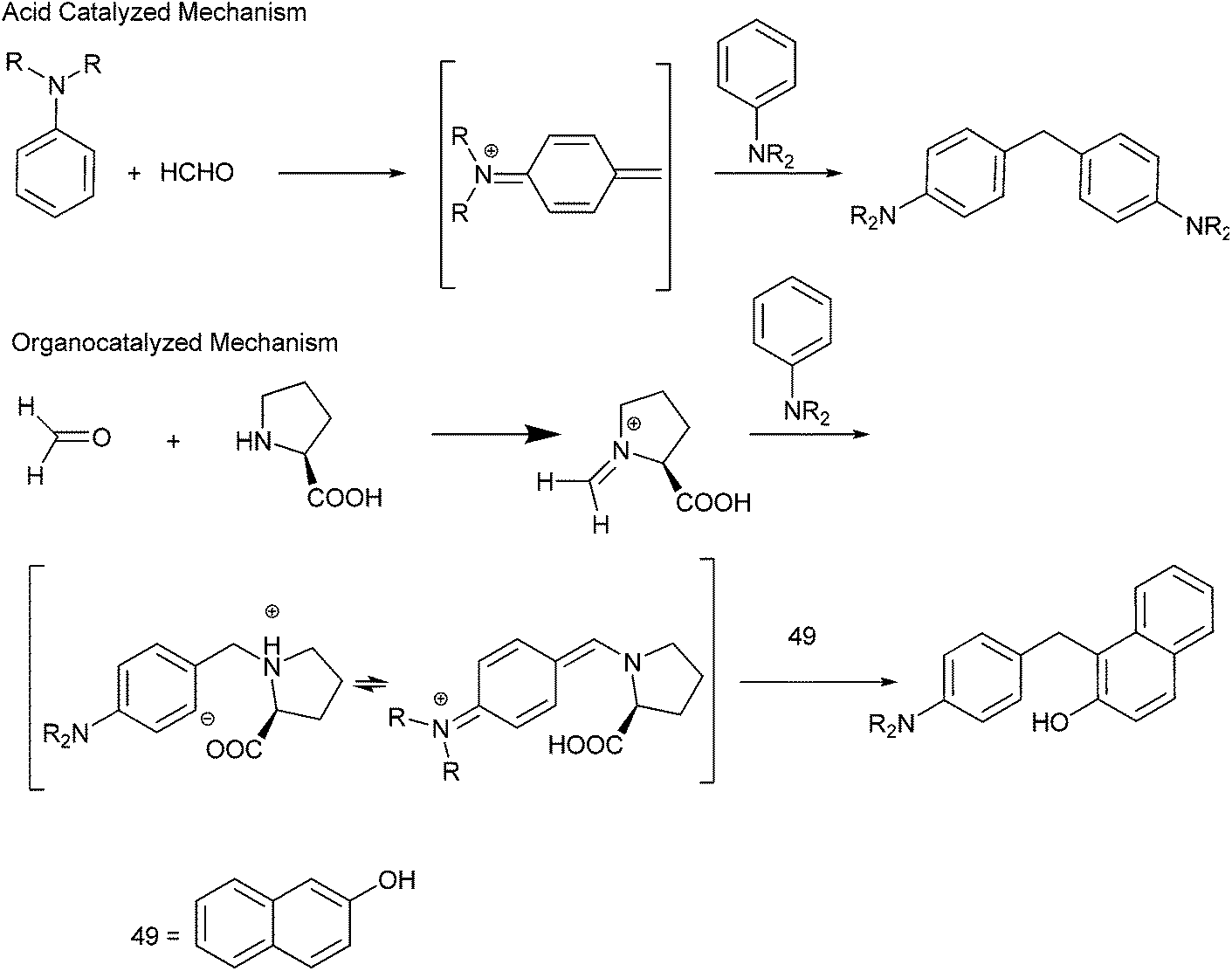 | ||
| Scheme 79 | ||
M. S. Sigman et al. in 2012 reported acid catalyzed hydroarylation of vinyl indoles to form the bis-indolyl ethanes and aryl indolyl ethanes.106 The authors found that though the reaction proceeded in dichloromethane, dichloroethane or in toluene, the yield was very poor as the indoles tend to polymerise in the acid medium. Addition of a Lewis basic solvent (dimethyl acetamide) as a co-solvent increased the reaction yield and in the Lewis basic solvent alone the reaction yield jumped up to about 89%.
N. Sakai et al. in 2011 reported InBr3–PhSiH3 mediated direct reduction of aromatic carboxylic acid to diarylmethanes (Scheme 80).107
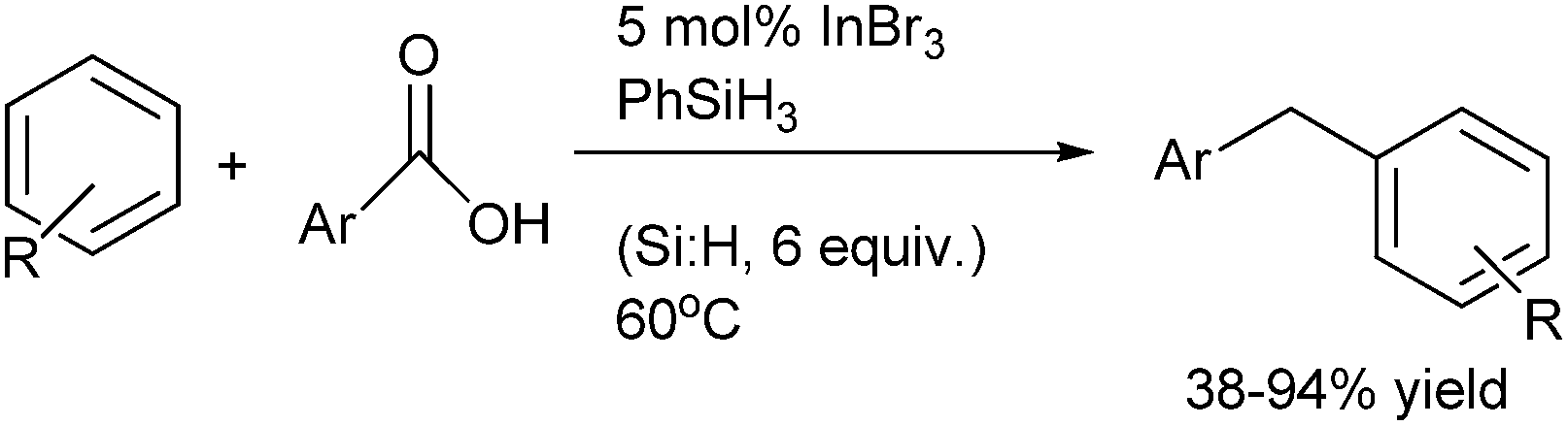 | ||
| Scheme 80 | ||
B. L. Ashfeld et al. reported in 2012 a multicomponent titanocene dichloride catalyzed coupling of aldehydes, iodoalkynes and arenes for the preparation of diaryl ethynyl methanes.108 Use of indoles as the arenes led to the formation of functionalized indoles, a common structural motif in many pharmaceutically relevant molecules and natural products.
10. Triarylmethanes-overview
The importance of triarylmethanes in the dye,109a–d bioorganic chemistry,109e–g material science109c,h–j as well as in organic synthesis is well established and documented.109k Triarylmethanes like malachite green, sunset orange, crystal violet, pararosanilin, victoria blue are very important in dye industry. Moreover, due to the steric hindrance among the ortho protons, the triarylmethanes exhibit chiral helical conformation and are subject of many theoretical and conformational studies.109l Caged molecules bicapped with trithienylmethanes have also been prepared and these types of caged molecules have shown that all the sulphur atoms of these caged molecules are directed inwards. These trithienylmethanes can thereby act as electron donors from the sulphur atom and π-electron donor from the hetero aromatic rings.109m Tri-(2-alkoxy-5-ureido-phenyl) methanes also represent a novel self complementary motif which forms hydrogen bonded homo and hetero dimers in non polar aprotic solvents. Moreover due to the stability of the carbocations generated from the acid treatment or photo irradiation of the trityl protected alcohols, the deprotectection of the trityl protected ethers are relatively easy. This property makes the trityl group a well celebrated protecting group in carbohydrate chemistry, peptide chemistry, and also in nucleoside chemistry.109k Trityl tags are also used in encoding in combinatorial synthesis, in labeling oligonucleotides with biotin, to purify the oligonucleotides by immobilizing it on solid support after synthesis. Also the modified trityl groups are used for creating internucleotide bond in phosphotriester approach for the solid phase synthesis of oligonucleotides. Chiral triarylmethyl carbocations generated from triarylmethyl chlorides or from triarylmethanols are used as chiral catalysts in various organic transformations like Mukaiyama aldol reactions,109n enantioselective hydride abstraction,109o allylations of the aldehydes with allyl tributyl stannane109p etc.Though there are a number of reviews on this topic, they mostly covered the photochemical and photophysical properties of the triarylmethanes, applications of triarylmethanes in the dye industry, mass spectrometric application, application in bioconjugation, cross linking, fluorescence and in optics.110 However to the best of our knowledge, the synthesis and bioactivities of the triarylmethanes have not been reviewed. We would cover the synthesis of the triarylmethanes and the application of triarylmethanes as active pharmaceutical agents from (1995–2013) leaving behind the materials that are covered in the previous reviews.
11. Synthetic approaches towards symmetrical and unsymmetrical triarylmethanes
The synthetic approaches followed in the preparation of symmetrical and unsymmetrical triarylmethanes broadly fall under the following categories:(i) Electrophilic aromatic substitution-Lewis acid catalyzed and protic acid catalyzed Friedel–Crafts alkylation and hydroxyarylation reactions of aldehydes.
(ii) Metal catalyzed cross coupling reactions of activated diarylmethanols and aryl boronic acids, C–H bond activations and decarboxylative coupling.
(iii) Miscellaneous approaches.
12. Electrophilic aromatic substitution
12.1 Lewis & protic acids catalyzed hydroxyarylation of aldehydes & Friedel–Crafts arylation reaction
J. C. Carretero et al. in 2006 reported the copper catalyzed aza-Friedel–Crafts reaction of imines for the preparation of unsymmetrical diarylmethylamines and triarylmethanes (Scheme 81).111 The authors found treatment of N-aryl sulfonyl aldimines with Cu(I) triflate and an aromatic hydrocarbon yielded the N-sulfonated diaryl methyl amines whereas the treatment with Cu(II) triflate yielded the triarylmethanes. However N-(2-pyridyl sulfonyl) aldimines did not give the expected triarylmethanes even on treatment with Cu(II) triflate. The authors could convert them to the respective triarylmethanes on treating N-(2-pyridyl) sulfonated diaryl methyl amines with scandium triflate in acetonitrile solvent at 60 °C.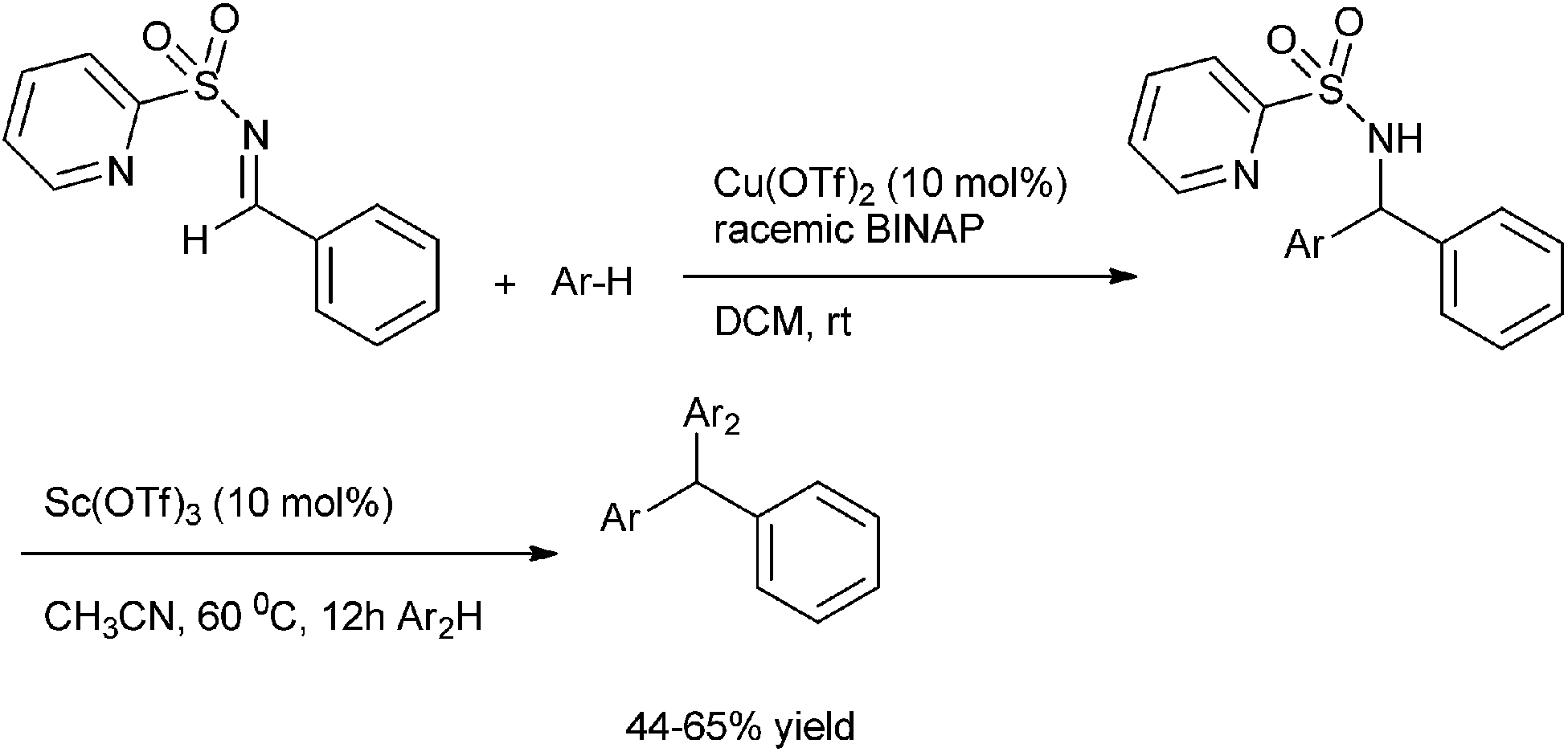 | ||
| Scheme 81 | ||
Later in 2008, the authors observed that with aromatic sulfonyl substituted aldimines and copper(II) triflate and on reducing the reaction temperature, the N-aryl sulfonated diaryl methyl amines could be isolated albeit in low yields along with the triarylmethanes and on increasing the reaction temperature, there was a steady decrement in the percentage yields of the N-sulfonated diarylmethyl amines. At room temperature, only the triarylmethanes could be isolated. However for the N-(2-pyridyl) sulfonated imines even at room temperature, N-(2-pyridyl) sulfonated diarylmethyl amines were obtained. Heating the reaction mixture to 45 °C with the aromatic hydrocarbon however converted the amines to the triarylmethanes.
A DFT study confirmed that in case of the N-aryl sulfonated imines the generated N-aryl sulfonated diarylmethylamines coordinated the metal ion through the sulphonamide nitrogen thereby activating the requisite C–N bond for the aza-Friedel–Crafts reaction. However in case of 2-pyridyl substituted sulphonamides, the pyridyl nitrogen coordinated with the metal ion thereby hindering the requisite activation of the C–N bond for aza-Friedel–Crafts reaction.
Later on in 2008 J. Wu et al. reported FeCl3 catalyzed aza-Friedel–Crafts reaction of aziridines and imines with electron rich arenes.111c When aryl imines were used, triarylmethanes were formed.
S. Kobayashi et al. in 2006, reported carboxylic acid catalyzed multi component reaction between an aldehyde, primary amine and an indole to yield triarylmethanes (Scheme 82).112 Interestingly, the authors found that the reaction was sluggish in organic solvents like THF and DCM but the reaction was comparatively faster in water. The authors found that among the carboxylic acid used, the decanoic acid was the most suitable.
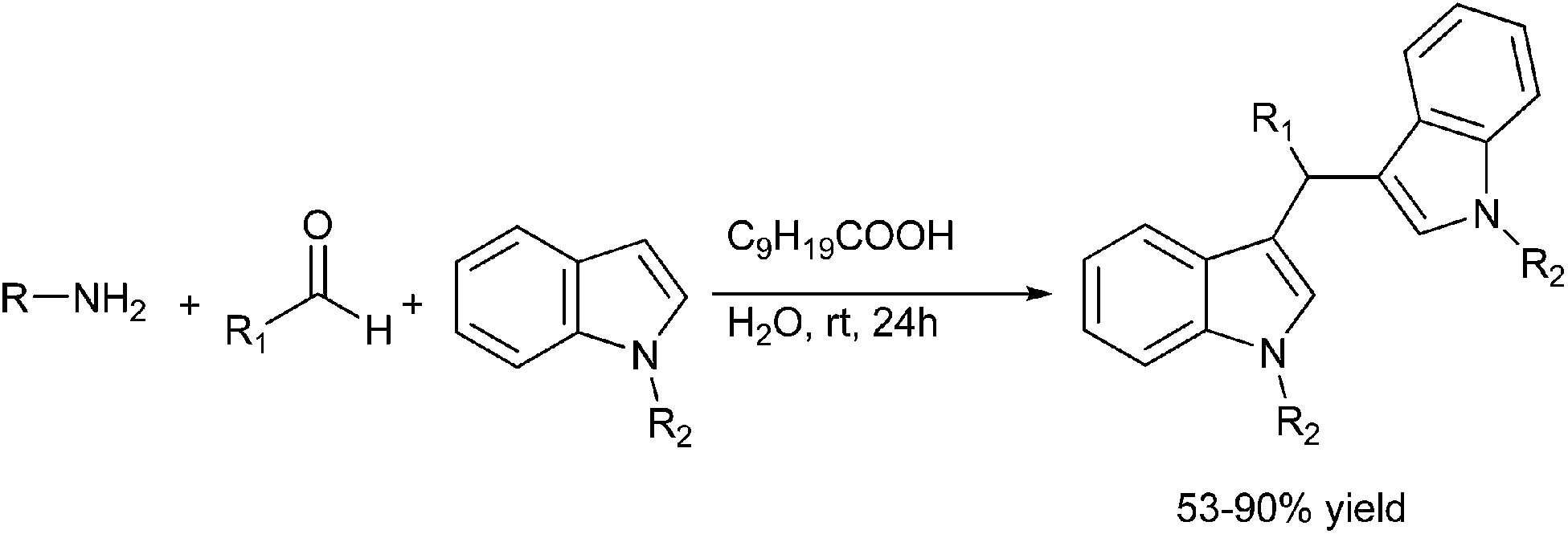 | ||
| Scheme 82 | ||
Later on in 2007, when S. L. You et al. carried out chiral phosphoric acid catalyzed Friedel–Crafts reaction of indoles with imines, they observed that a small amount of triarylmethane was always formed.113 Inspired by this side reaction in 2009, the same group reported Bronsted acid (50) catalyzed aza-Friedel–Crafts reactions of indoles to synthesize the unsymmetrical indole containing triarylmethanes (Scheme 83).
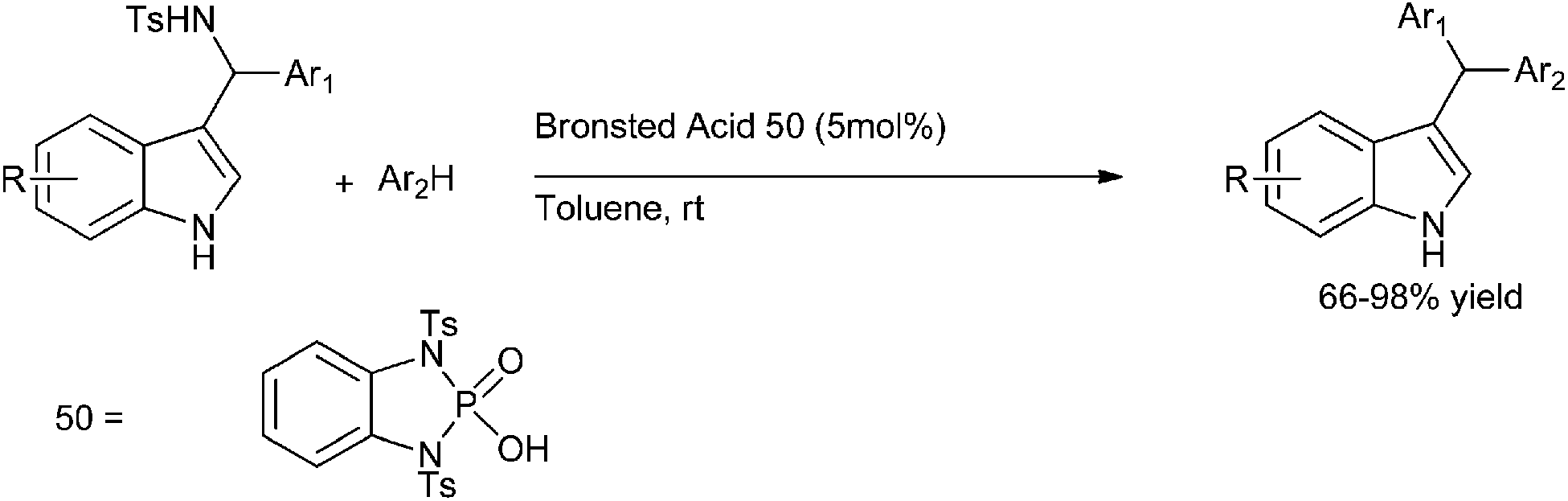 | ||
| Scheme 83 | ||
H. U. Reissig et al. reported in 2013 trifluoromethane sulfonic acid catalyzed Friedel–Crafts alkylation of electron rich arene 1,2,4-trimethoxy benzene with benzyl alcohols or benzaldehydes for the preparation of triarylmethanes (Scheme 84).114
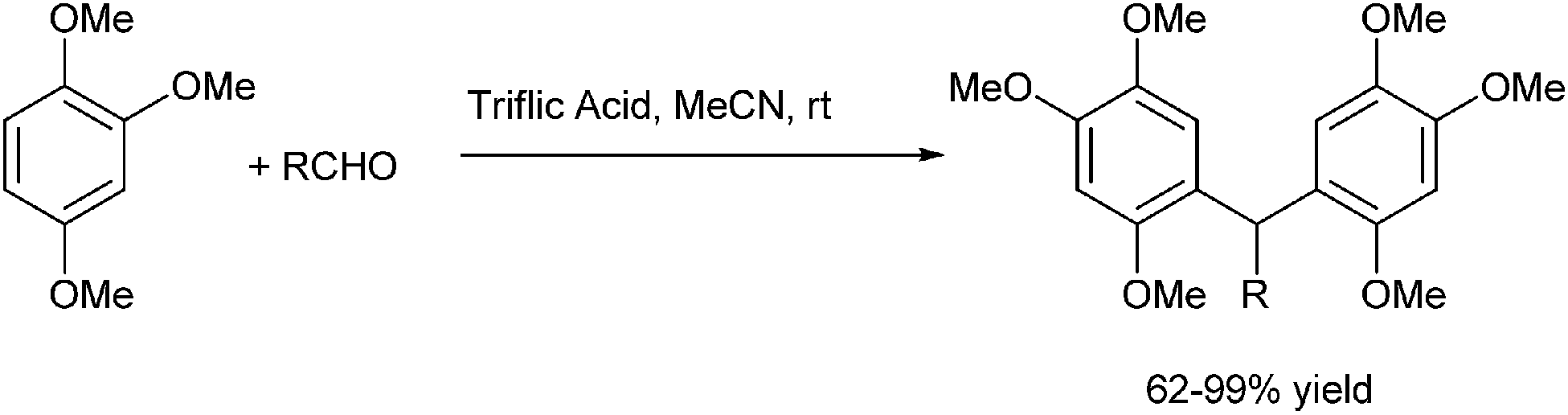 | ||
| Scheme 84 | ||
S. Kumar et al. reported the synthesis of di(uracilyl) arylmethanes from 1-benzyl uracil (Scheme 85).115 Heating of 1-benzyl uracil with aromatic aldehydes in presence of HBr–acetic acid yielded the respective di(uracilyl) aryl methanes in high yields. However the authors observed that heating 1-benzyl uracil with para methoxy benzaldehyde yielded the (4-hydroxy phenyl) bis uracil indicating either demethylation of the aldehyde followed by the condensation with uracil takes place or first the condensation of the aldehydes with uracil followed by demethylation takes place. On lowering the temperature to 60 °C yielded the (4-methoxy phenyl) bis uracil product.
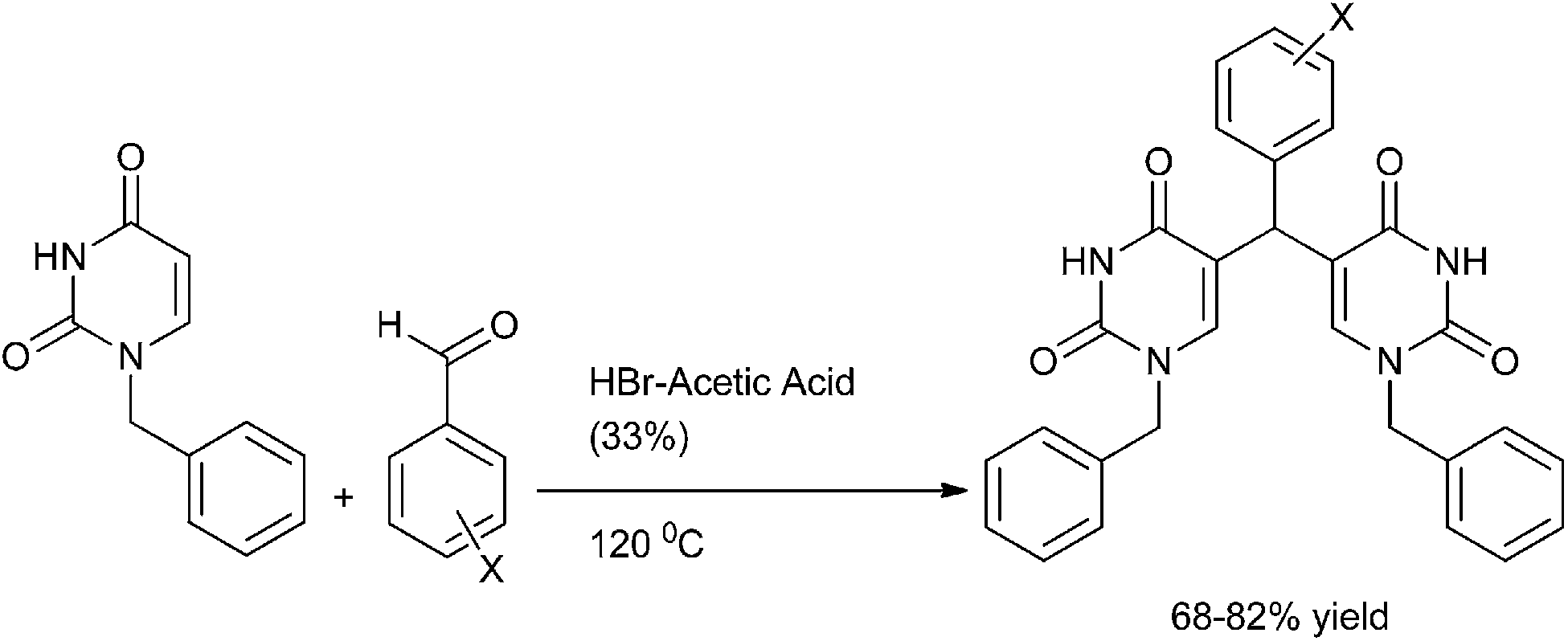 | ||
| Scheme 85 | ||
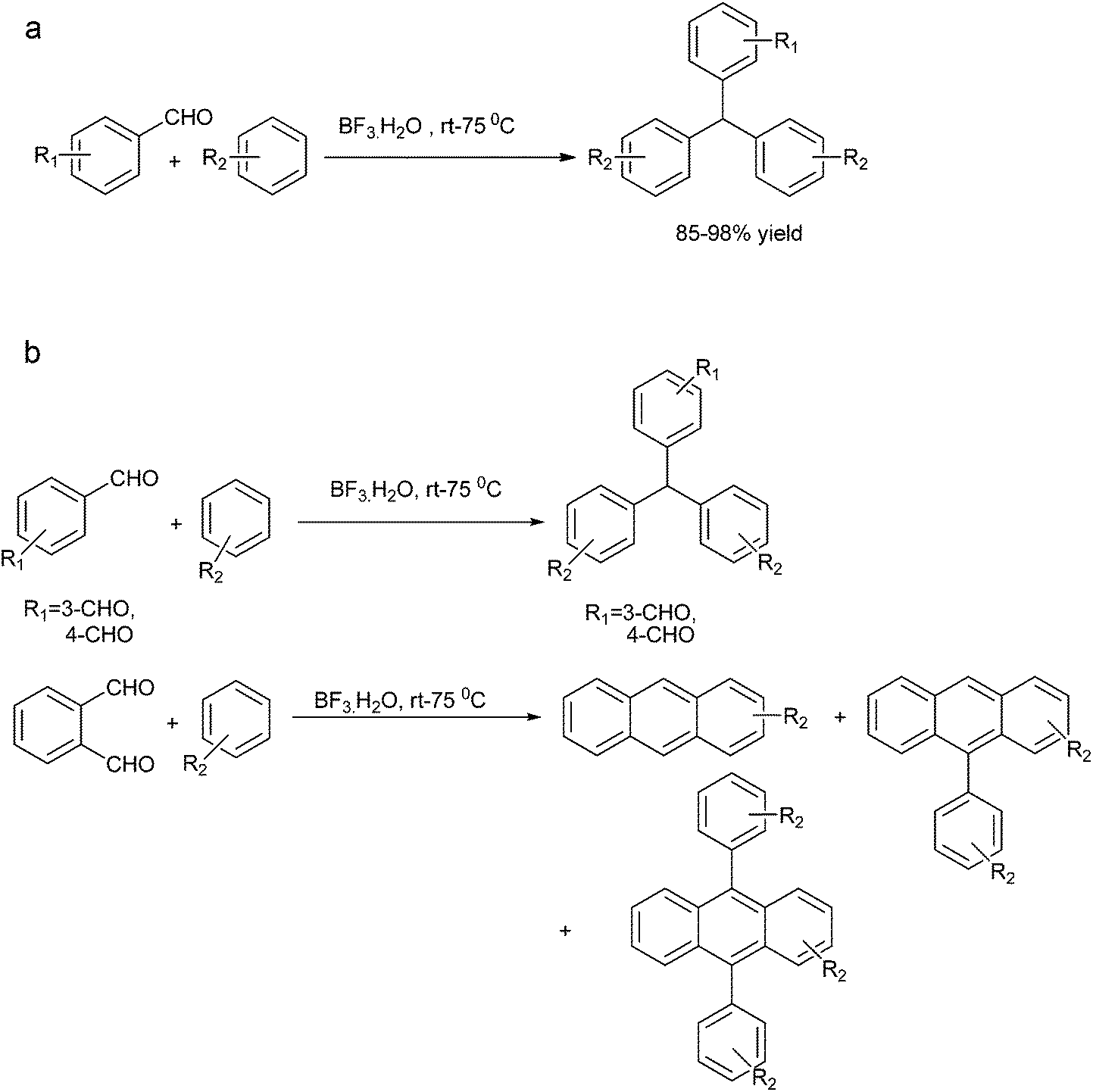
J. Xu et al. reported solvent free anhydrous aluminium chloride catalyzed tandem Friedel–Crafts arylation of arenes and aldehydes to form the triarylmethanes (Scheme 86a).116
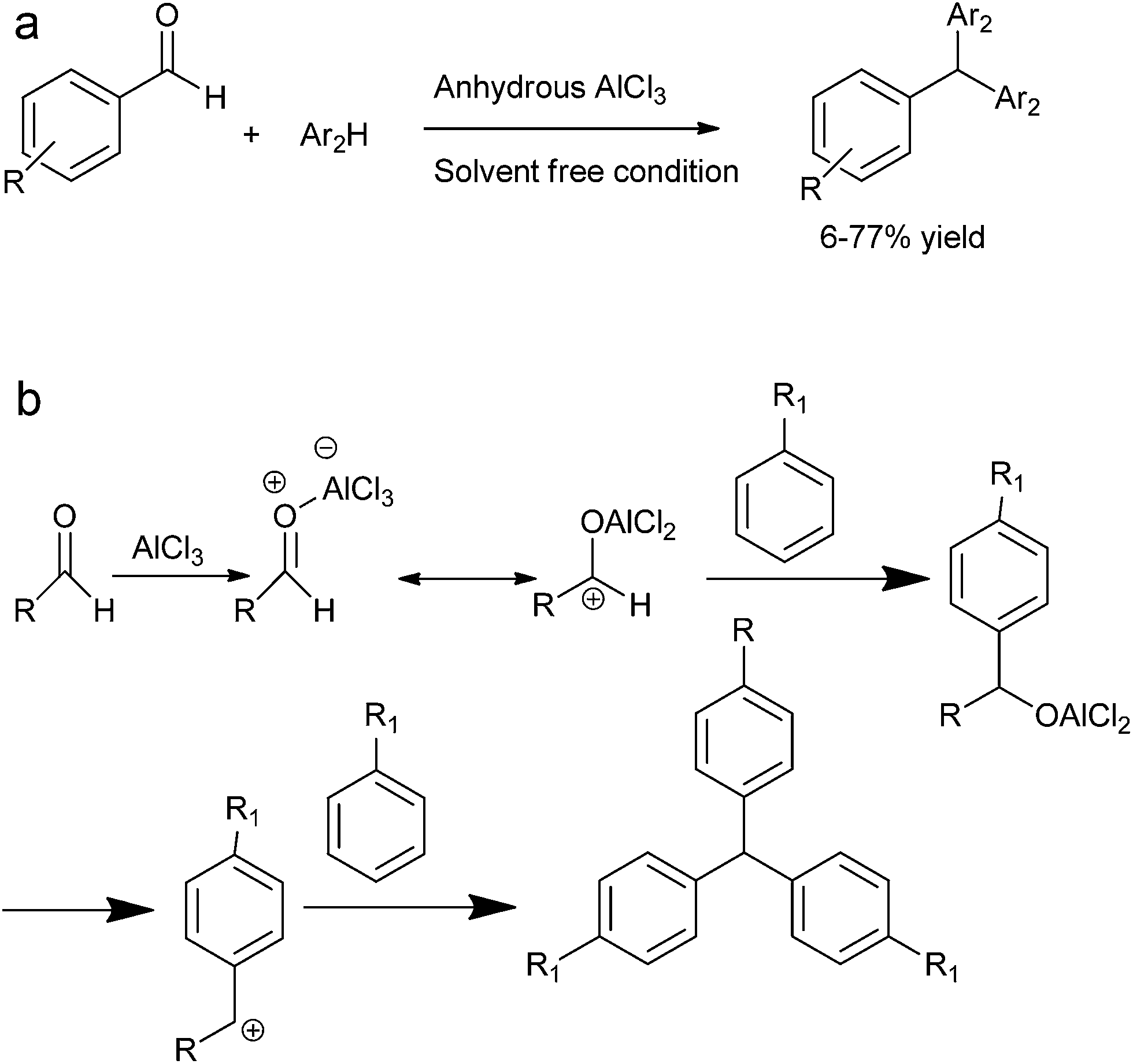 | ||
| Scheme 86 | ||
The author explained the mechanism of the reaction by proposing that at first the Lewis acid coordinates with the carbonyl oxygen followed by Friedel–Crafts reaction to form the aluminium alkoxide which further generates the benzylic carbocation followed by Friedel–Crafts arylation to form the observed triarylmethane (Scheme 86b).
S. Roy et al. in 2007 reported [Ir(COD)Cl]2SnCl4 (51a) system for the hydroxy arylation of aromatic aldehydes for the preparation of triarylmethanes in high yields (Scheme 87a).117 The authors found that the Ir–Sn domain played a dual-reagent catalyst role. Also the authors found that only triarylmethanes were formed.
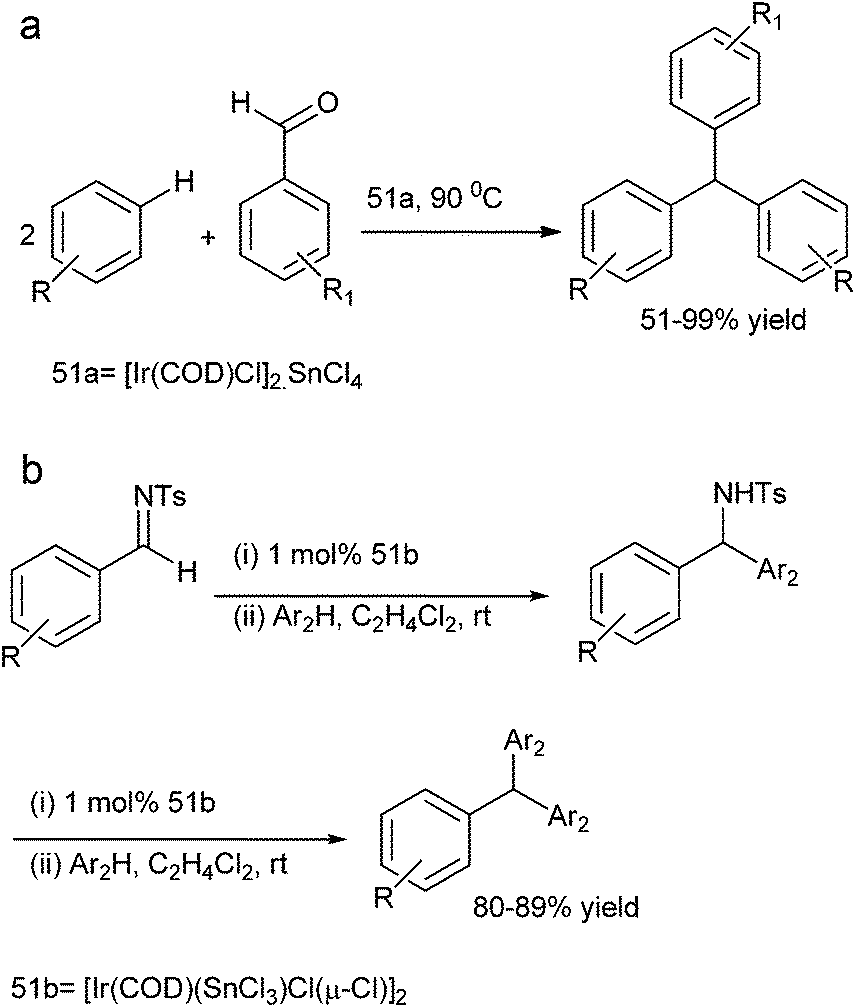 | ||
| Scheme 87 | ||
Later on in 2013, they reported [Ir(COD)(SnCl3)Cl(μ-Cl)]2 (51b) catalyzed AFCR of N-sulfonyl aromatic aldimines with electron rich arenes and heteroarenes for the synthesis of symmetrical and unsymmetrical triarylmethanes (Scheme 87b).118
M. Periasamy et al. in 2007 reported a simple and efficient titanium tetrachloride mediated electrophilic substitution of benzaldehyde dimethyl acetal to produce triarylmethanes in good yields (Scheme 88).119
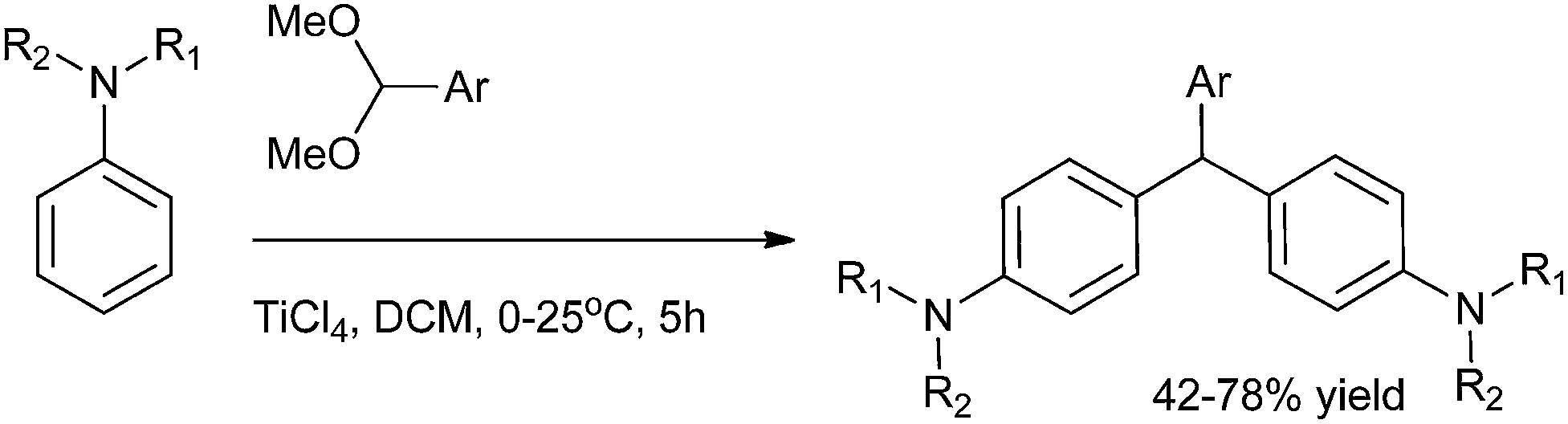 | ||
| Scheme 88 | ||
J. S. Yadav et al. in 2007 reported niobium pentachloride as an efficient catalyst for the nucleophilic substitution of diaryl carbinols with C, O, N, and S based nucleophiles.120 When the ambidented (49) was used, the authors found exclusive C-alkylation. The authors extended this concept for the preparation of the triarylmethanes.
M. Kodomari et al. in 2008 reported Friedel–Crafts alkylation of aromatic aldehydes using acetyl bromide and silica gel supported Zinc bromide in benzene solvent (Scheme 89).121 The authors found that the ratio of arene to aromatic aldehyde was a crucial factor controlling the reaction product. When the ratio of the arene to aldehyde was 4:1, the triarylmethane was the only product whereas the only product 9,10-diaryl anthracene was isolated, when the ratio of the arene to aldehyde was 1:3. The author proposed that the aldehyde at first reacts with the acetyl bromide in presence of zinc bromide to yield the α-bromo ester which further reacts with the arene to give the triarylmethane.
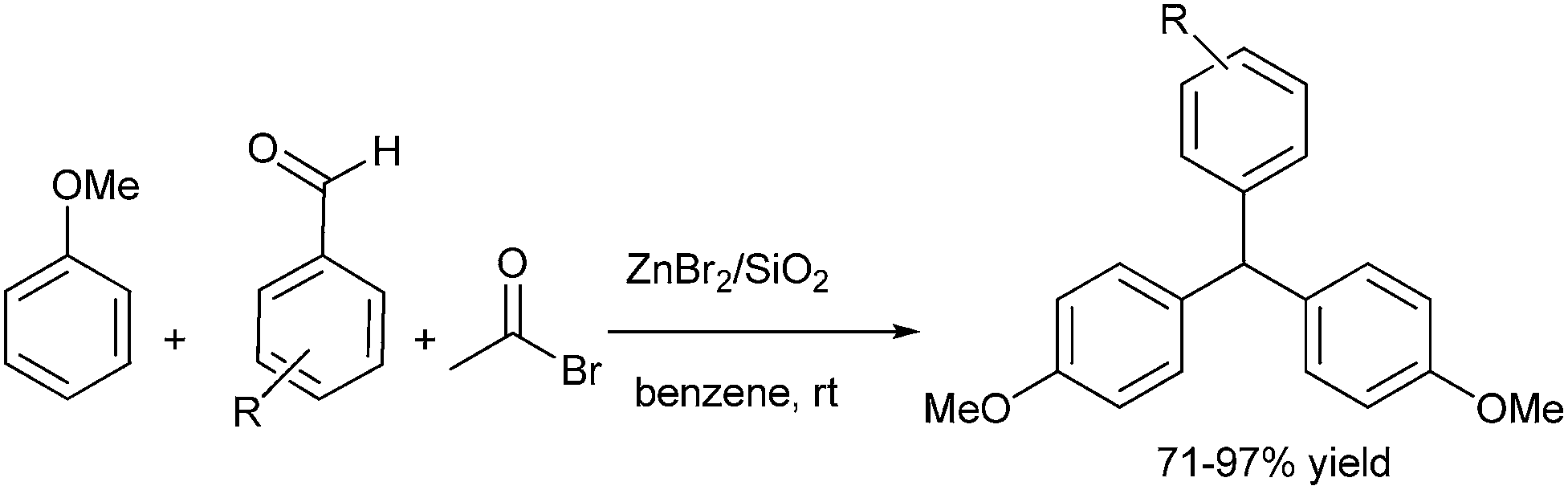 | ||
| Scheme 89 | ||
S. K. Tian et al. reported the bismuth sulphate trimethylsilyl chloride catalyzed double Friedel–Crafts arylation of imines with phenols, anisoles and thioanisoles to prepare triarylmethanes in high yields (Scheme 90).122 With aliphatic imines, the diarylamines were observed.
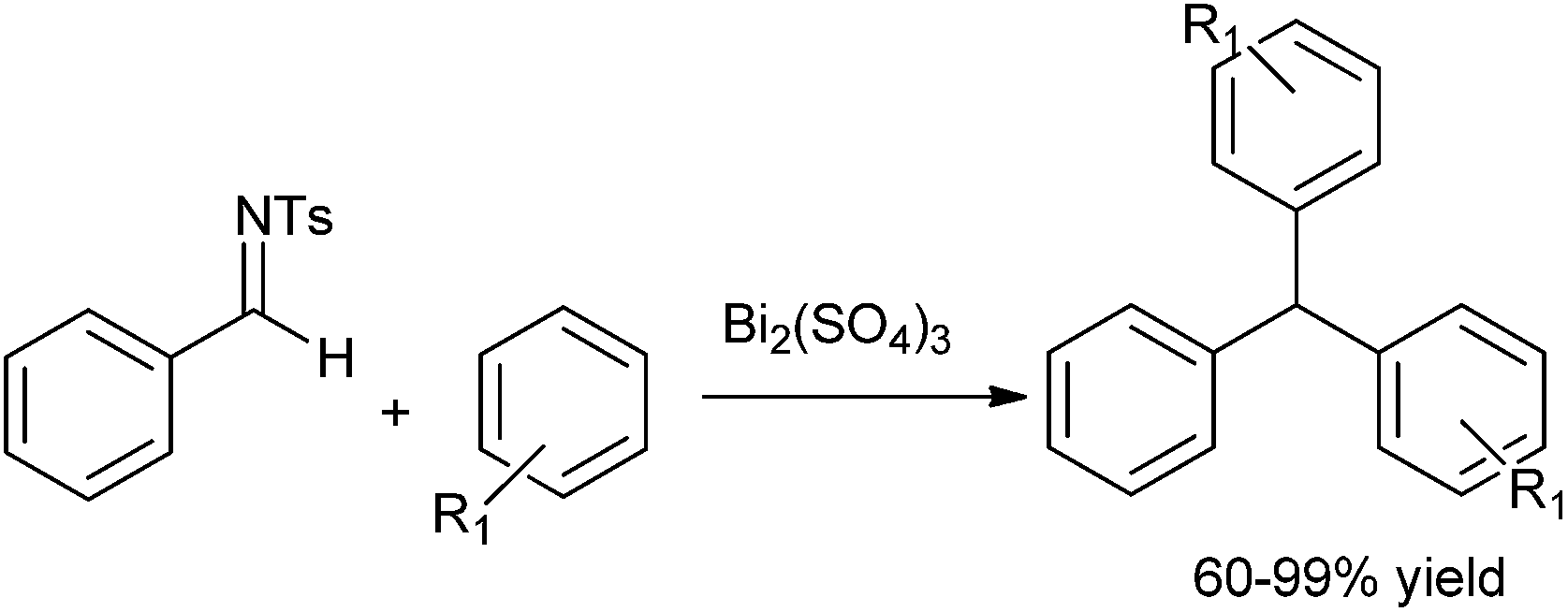 | ||
| Scheme 90 | ||
S. Genovese et al. in 2009 reported the use of lanthanide salt Ytterbium triflate as a catalyst for the bis-arylation of aromatic aldehydes under neat condition to form the triarylmethanes in high yields.123 Precipitation of the Lewis acid catalyst and further regeneration of it allowed for the recycling of the catalyst system without significant loss in the reaction yield. The reaction was highly regioselective as reflected from the fact that when anisole was used as the arene component, only the para substituted product was observed. When 2-methylfuran was treated with (6) under the optimized condition the corresponding triarylmethane was obtained in very good yield (Scheme 91).
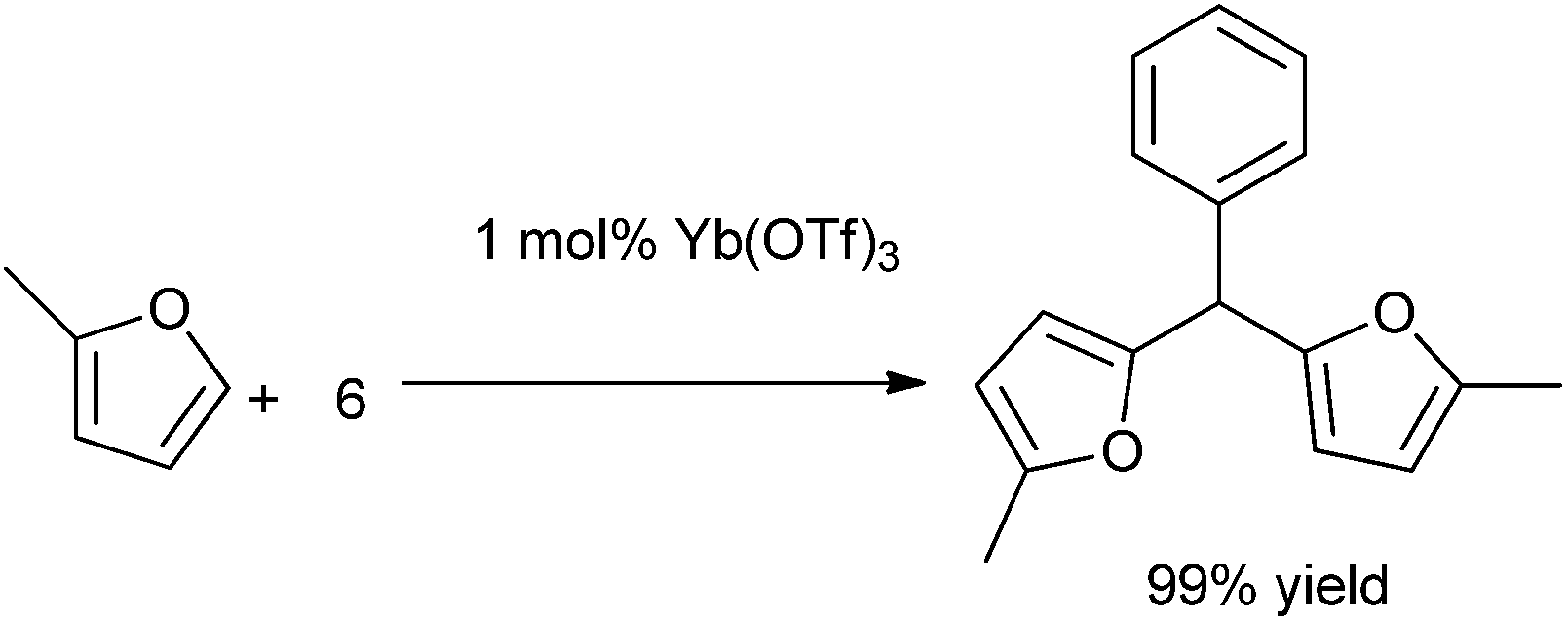 | ||
| Scheme 91 | ||
The authors proposed that high oxophilicity of the Lewis acid catalyst caused coordination of the Lewis acid to the carbonyl oxygen thereby increasing its electrophilicity and facilitating the nucleophilic attack by the π-electron rich arene.
Later in 2009 Ch. S. Reddy et al. generalized this concept and reported zirconium oxy chloride catalyzed condensation of aryl aldehydes with N,N-dimethyl anilines to yield 4,4′-(arylmethylene)bis(N,N-dimethylaniline) (Scheme 92).124,125
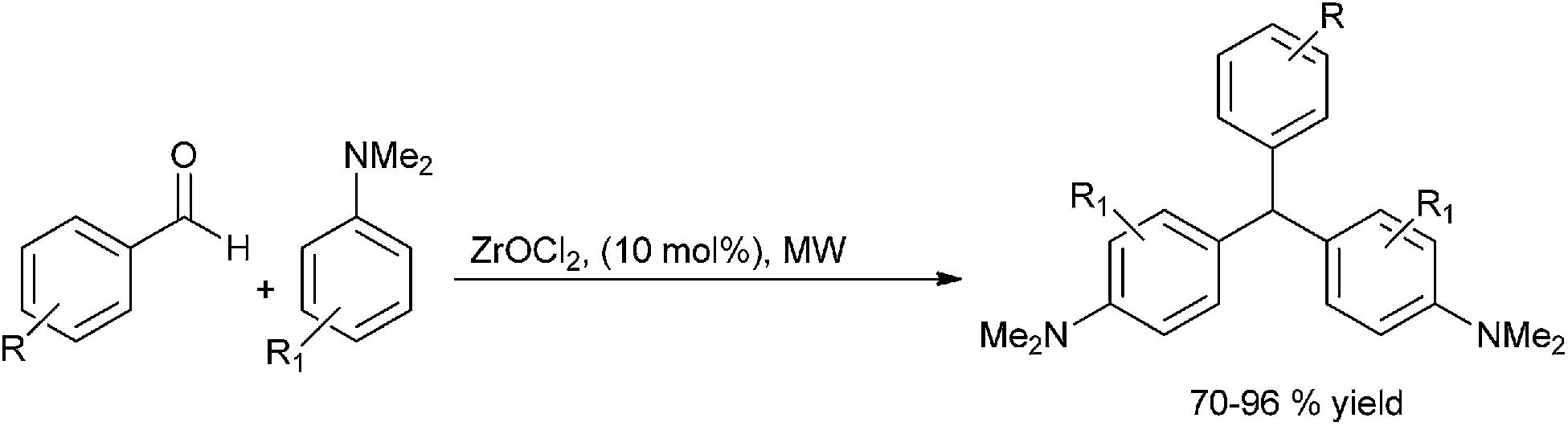 | ||
| Scheme 92 | ||
S. S. Kim et al. in 2009 reported indium bromide catalyzed Friedel–Craft reactions of α-amido sulfones, N-sulfonyl aldimines and sulphonamide sulfones to yield the triarylmethanes in good yield (Scheme 93).126,127 Under low catalyst loading at room temperature and mild reaction condition, the authors could achieve high yield of the triarylmethanes. Later in 2010, they improved the protocol by using Ferric Chloride as the Lewis Acid.
 | ||
| Scheme 93 | ||
G. Panda and coworkers in 2006 utilized the HSAB principle for the Friedel–Crafts alkylation of anthracen-9-yl (4-methoxy phenyl) carbinols with phenols, thiols and anisoles for the synthesis of anthracene based triarylmethanes (Scheme 94).128,129 Interestingly the authors observed that in protic acids the Friedel–Craft alkylation of the carbinols with phenols as well as anisoles gave the diarylmethanes in major amounts along with the anthracene based triaryl methanes in minor amounts. However in presence of Lewis acids the amount of the diaryl methane product decreased while that of the triarylmethanes increased. In presence of anhydrous zinc chloride the triarylmethanes were obtained in the major amounts.
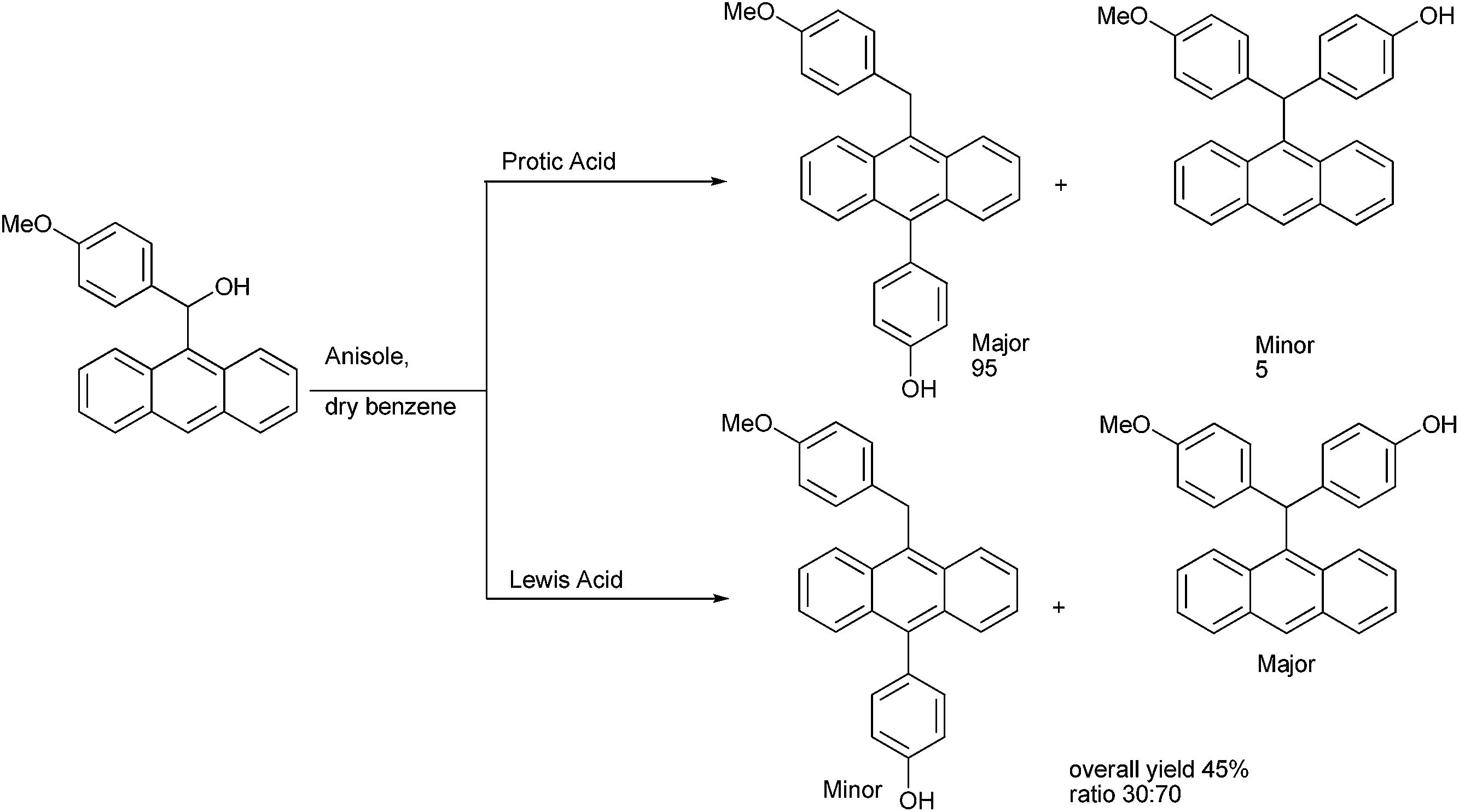 | ||
| Scheme 94 | ||
The authors carried out a DFT study to understand the mechanism of the reaction. In 2013 they extended this concept for the preparation of trisubstituted methanes containing aryl and heteroaryl rings.
G. A. Olah et al. in 2009 reported hydrated boron triflouride catalyzed hydroxy arylation of aromatic aldehydes to form triarylmethanes (Scheme 95a).130 The authors found that hydrated boron triflouride acted not only as the catalyst but also an efficient protosolvating medium generating efficiently the super electrophiles. The authors observed that treatment of electron rich aromatic aldehydes like p-tolualdehyde, p-anisaldehyde and 2-naphthaldehyde respectively with benzene yielded mostly triphenylmethane for p-tolualdehyde, triphenyl methane and phenol for p-anisaldehyde and triphenylmethane, naphthalene and (49) for 2-naphthaldehyde. They suggested transformylation reaction occurs before the arylation of the aldehyde. Also with electron rich arenes, the reaction was very fast while for electron poor system, the reaction was sluggish.
 | ||
| Scheme 95 | ||
When the reaction was extended for dialdehydes for 1,3 and 1,4 systems, only one aldehyde group responded to the reaction and diarylmethyl benzaldehydes were obtained. However for phthaldehyde, 9-arylanthracenes along with anthracene and 9,10-diarylanthracene was obtained (Scheme 95b).
T. Murai et al. on 2009 reported sequential, iodine and pyridine promoted cyclization and condensation reactions of N-thioacyl-1-(2-pyridyl)-1,2-amino alcohols derived from secondary thioamides and aromatic aldehydes for the preparation of nitrogen containing triarylmethanes with 10π electrons (Scheme 96).131
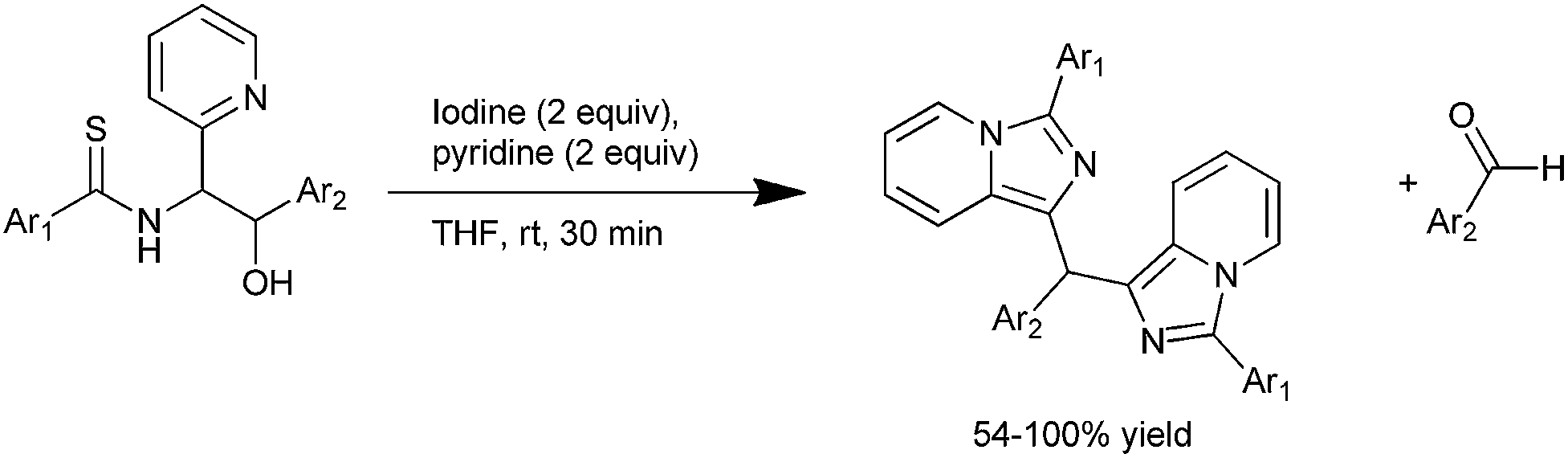 | ||
| Scheme 96 | ||
The authors observed that when the thioamide was treated with iodine in presence of electron rich imidazo pyridine, unsymmetrical triarylmethanes were observed. The authors explained the mechanism of the reaction by stating that at first the thioamides react with iodine to form the carbinol intermediate which further takes part in an iodine mediated Friedel–Crafts reaction to give the triarylmethanes.
Later on in the same year J. Jaratjaroonphong et al. reported iodine as an efficient and mild Lewis acid for the Friedel–Crafts alkylation of aldehydes with arenes for the preparation of diarylalkanes and triarylmethanes (Scheme 97).131b Iodine catalyzed Friedel–Crafts alkylation of aliphatic aldehydes with arenes yielded diaryl alkanes while iodine catalyzed Friedel–Crafts alkylation of aromatic aldehydes yielded triarylmethanes. The authors observed a remarkable solvent effect for this reaction with dichloromethane and toluene proved to the best. The authors observed for propanal in dichloromethane the corresponding diarylalkane was obtained in low yield while in toluene the yield was very high.
 | ||
| Scheme 97 | ||
S. S. Kim et al. reported an environmentally benign and mild condition for the preparation of triarylmethanes (Scheme 98).132 Thus using hydrated ferric perchlorate as the catalyst and another electron rich arene, the authors could arylate the benzhydrols to the corresponding symmetrical triarylmethanes in good yields. The author noticed that temperature played an important role in the reaction as at low temperature, the diarylether from the diarylmethanol was the major product whereas on increasing the temperature the triarylmethanes were formed exclusively.
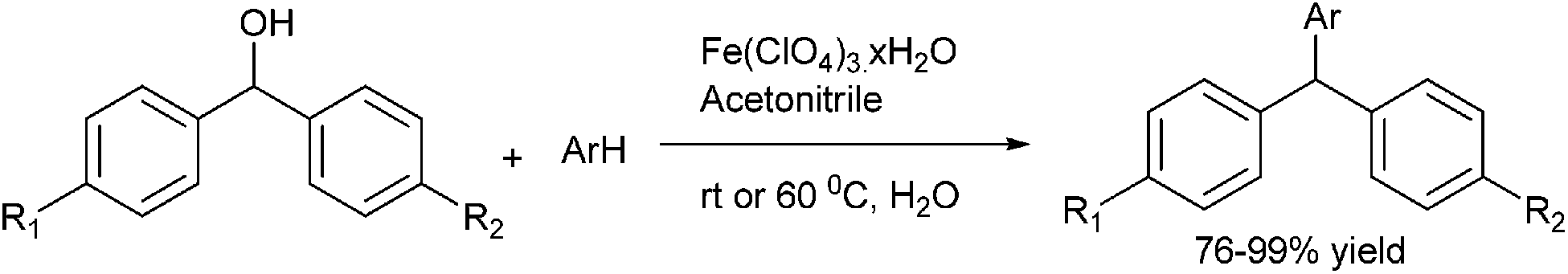 | ||
| Scheme 98 | ||
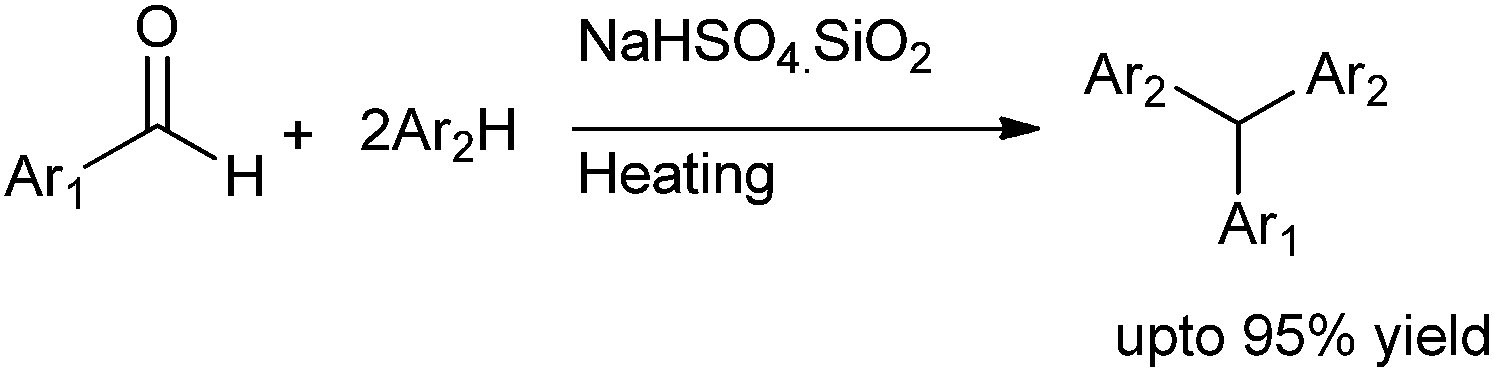
W. Duan et al. in 2010 reported bis-arylation of aryl aldehydes using silica-gel supported sodium hydrogen sulphate as the catalyst (Scheme 99).133 The use of thiophene as the aromatic hydrocarbon allowed the reaction to be carried out in neat condition. The authors noticed that bis arylation of electronically poor aldehydes were faster and yielded the triarylmethanes in higher yields than the electron rich aldehydes.
 | ||
| Scheme 99 | ||
G. Panda and coworkers in 2005 reported conc. sulfuric acid catalyzed Friedel–Crafts alkylation of phenols with aryl heteroaryl carbinols for the facile preparation of heteroaryl diarylmethanes.134
Later in 2010, G. A. Olah et al. reported the use of Nafion-H catalyzed hydroxy arylation of benzaldehydes to form triarylmethanes in high yield.135 They reported that for mono substituted aromatics, microwave heating caused the formation of more o, o′ product whereas conventional heating caused the formation of p, p′ product. Along with the triarylmethane, diarylmethane was also formed.
K. A. Jørgensen et al. in 2001 reported Friedel–Crafts hydroxyalkylation of pyridine carboxyaldehydes with N,N-dimethyl aniline. Aluminium complexes acted as the Lewis acids.136 Though pydidine-3 and -4-carboxyaldehydes gave the desired triarylmethanes pyridine-2-carboxyaldehydes did not gave the triarylmethane, instead it gave the diaryl methanol. Also benzaldehydes failed to give the triarylmethane product.
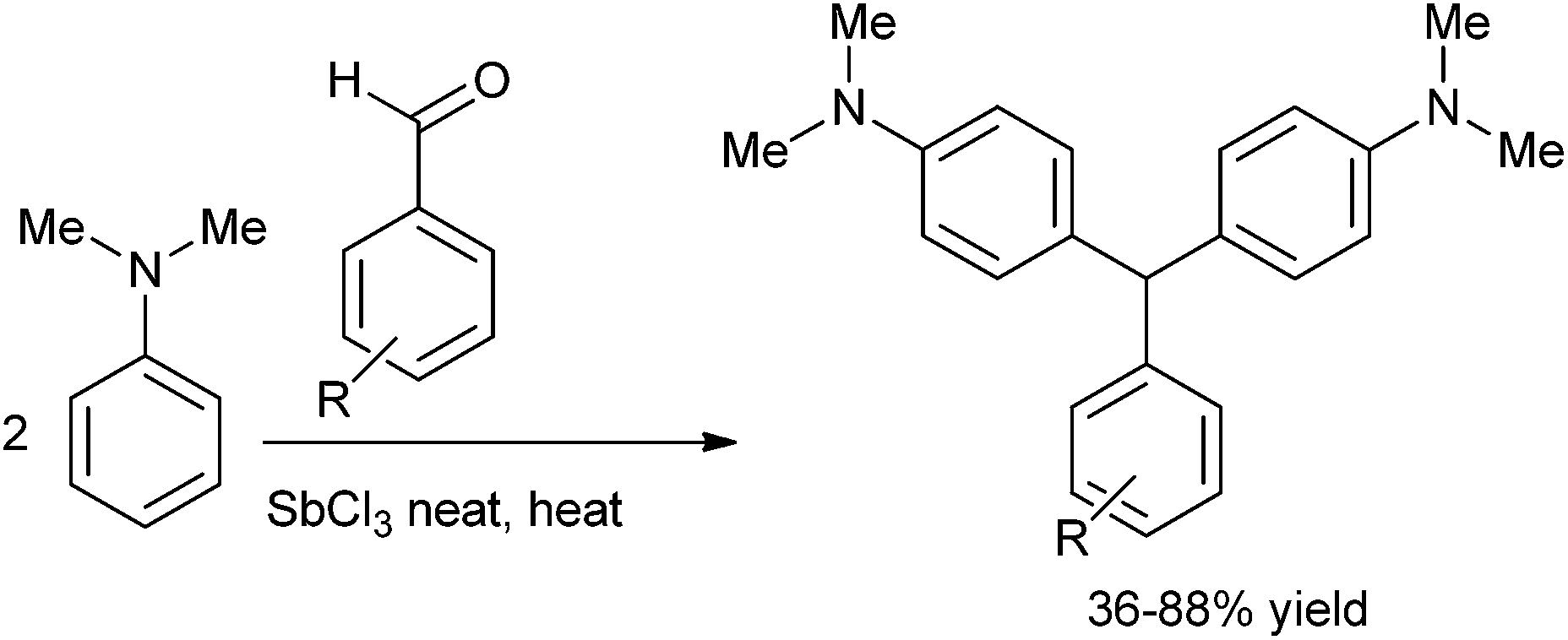
Later on in 2011 G. R. Bardajee et al. reported antimony(III) chloride catalyzed solvent free Friedel–Crafts hydroxyalkylation of aldehydes with N,N-dimethyl anilines in a single pot to achieve the synthesis of triarylmethanes (Scheme 100).137 This procedure was operationally simple, high yielding and was widely applicable. The authors proposed that the Lewis acid antimony(III) chloride forms a complex with the oxygen atom of the carbonyl group thereby increasing the electrophilicity of the carbonyl group. Friedel–Crafts arylation with the arene generated the triarylmethanes in high yield.
 | ||
| Scheme 100 | ||
I. M. Baltork et al. reported phosphotungstic acid as the catalyst for the bis arylation of aromatic aldehydes for the preparation of triarylmethanes and difurylaryl methanes in one pot under neat conditions with conventional or microwave heating.138 The authors observed that electron withdrawing groups in the aldehyde gave the corresponding triarylmethanes in very high yields however with aldehydes having electron donating groups the reaction failed. However treatment of veratole with aromatic aldehydes under conventional heating or under microwave conditions yielded the triarylmethanes in high yields (Scheme 101). Further increasing the amount of the arene gave diveratryl triarylmethanes. Further treatment of the diveratryl triarylmethanes with the aromatic aldehyde in presence of acetic acid and acetic anhydride gave the 9,10-diarylanthracene.
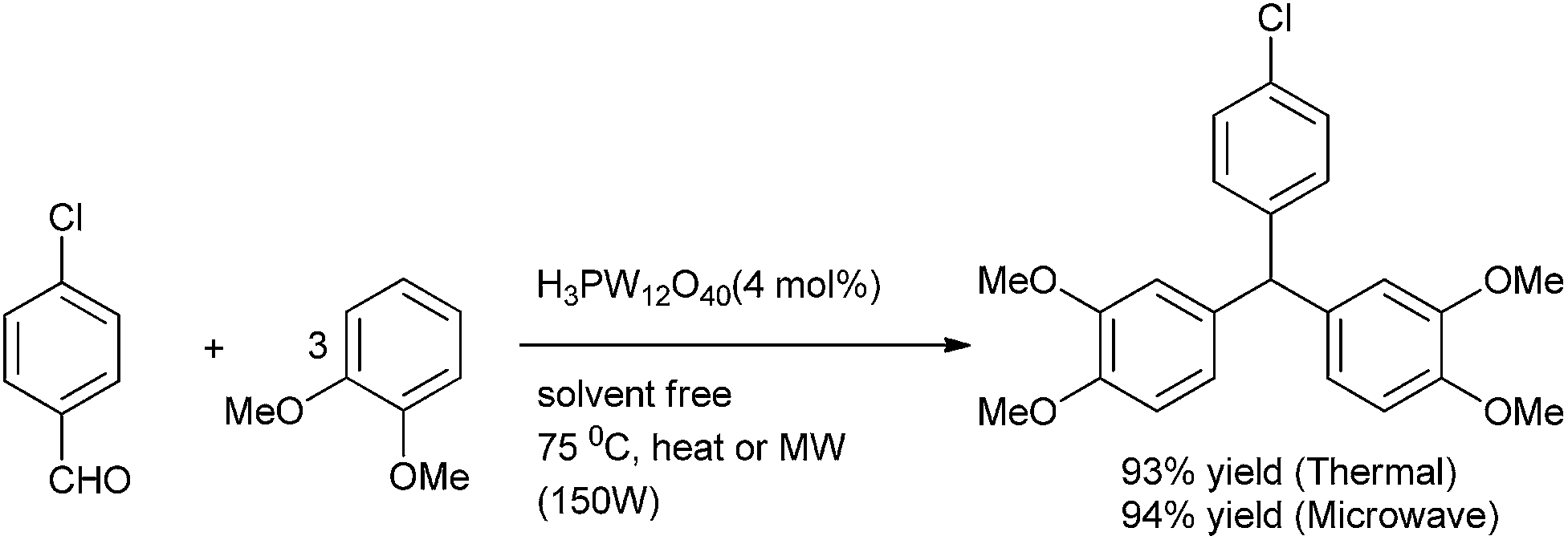 | ||
| Scheme 101 | ||
H. Hikawa et al. in 2013 reported Au(III) chloride catalyzed chemoselective benzylation of unprotected anthranilic acids. When electron rich diarylmethanols were used, instead of N-benzylation, C-benzylation (Friedel–Crafts alkylation) was observed to form the triarylmethanes (Scheme 102).139
 | ||
| Scheme 102 | ||
13. Metal catalyzed C (sp3)–C (sp2) coupling
H. Yorimitsu et al. in 2007 reported palladium catalyzed coupling of aryl chlorides with aryl (aza-aryl) methanes through benzylic (sp3) C–H bond activation for the preparation of triarylmethanes (Scheme 103).140 Interestingly, the author observed for electron poor 4-(tertiarybutoxycarbonyl) chlorobenzene the reaction was low yielding, did not reach to completion and significant amount of ester bond cleaved product was obtained. For such substrates the ligand had to be changed to di(tert-butyl)(2-phenylphenyl) phosphine for high yields.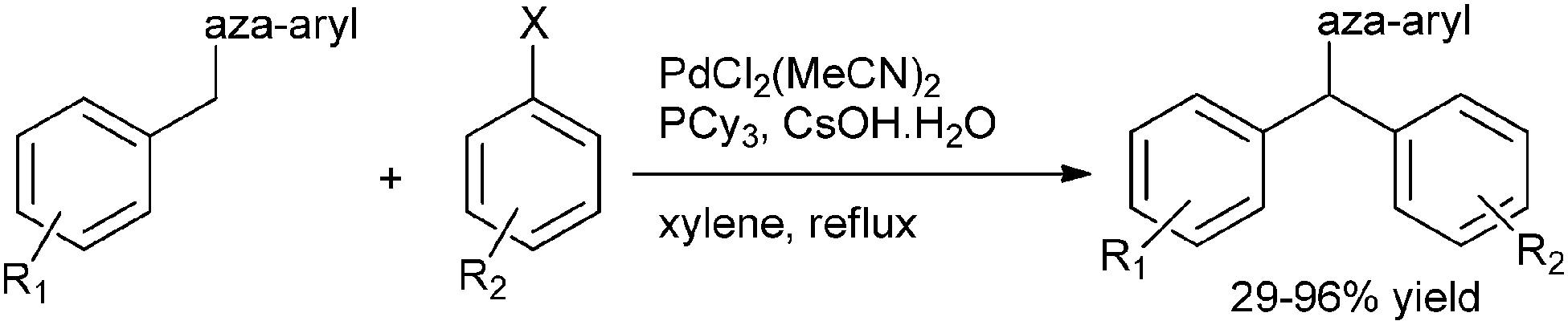 | ||
| Scheme 103 | ||
The authors explained this observation by proposing that the reductive elimination from the intermediate [4-(tert-butoxycarbonyl) phenyl][phenyl(pyrimidyl)methyl]palladium was hindered by the electron withdrawing nature of the carbonyl group.
R. Kuwano et al. in 2008 reported palladium acetate catalyzed Suzuki–Miyaura Coupling of diaryl methyl carbonates with aryl boronic acids to form triarylmethanes in high yields (Scheme 104).141 The N-heterocyclic carbene (33) acted as the ligand while dimeric η3allyl palladium(II) chloride was used as a catalyst. When monophosphine ligands were used, the yields were low. Among the solvents screened tertiary amyl alcohol was found to be the best solvent. The authors proposed that at first a molecule of carbon dioxide is lost to form the diaryl palladium intermediate. Subsequently the methoxy group attached to palladium undergoes the transmetallation with boronic acid to form the intermediate (B) which undergoes reductive elimination to give the triarylmethane product.
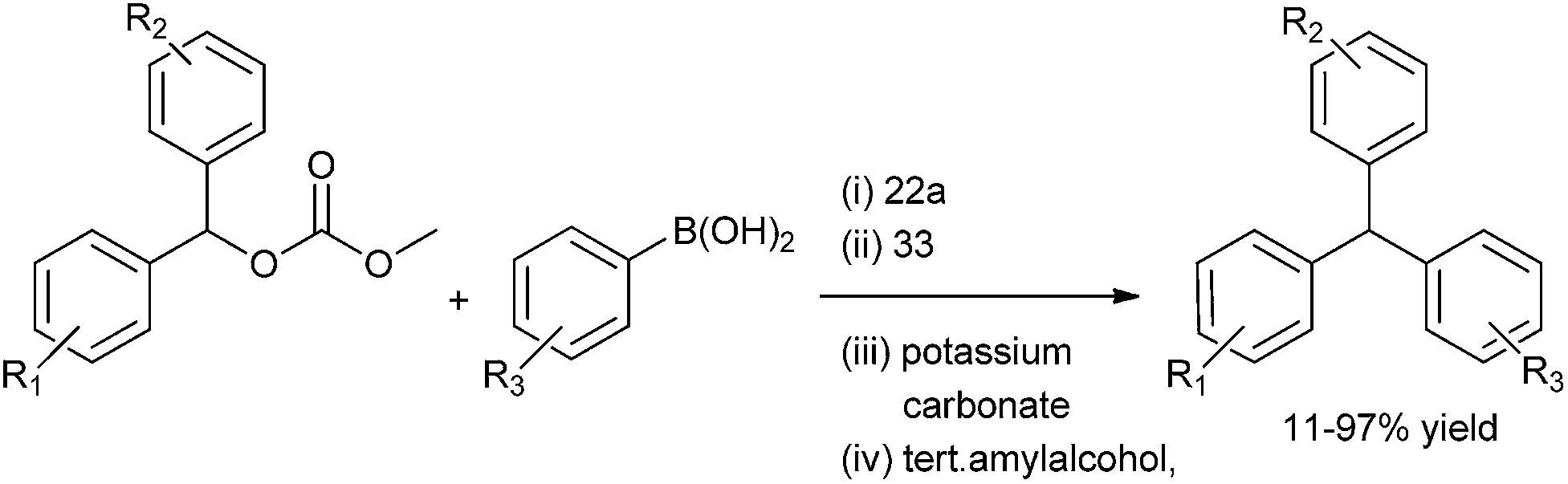 | ||
| Scheme 104 | ||
P. J. Walsh et al. reported C–H bond activation of unactivated and non acidic benzylic sp3 C–H for the preparation of the triarylmethanes (Scheme 105).142,143 η6-Tricarbonyl chromium was used to increase the acidity of the C–H bonds. From diarylmethanes, triarylmethanes could easily be formed. However the corresponding formation of triarylmethanes from toluene required heating at 60° C. This was probably due to decrement in the acidity of the benzylic proton. Also the reaction was sensitive to steric encumbrance in the aryl halide as the 2,6-disubstituted aryl bromide gave the mono coupled product. Interestingly, with 4-bromoacetophenone, no aldol product was observed and the triarylmethanes were formed. Later in 2013, the authors investigated the role of additives in this deprotonative cross coupling procedure.144 The authors carried out high throughput experimentation (HTE) for the identification of the optimal additive.
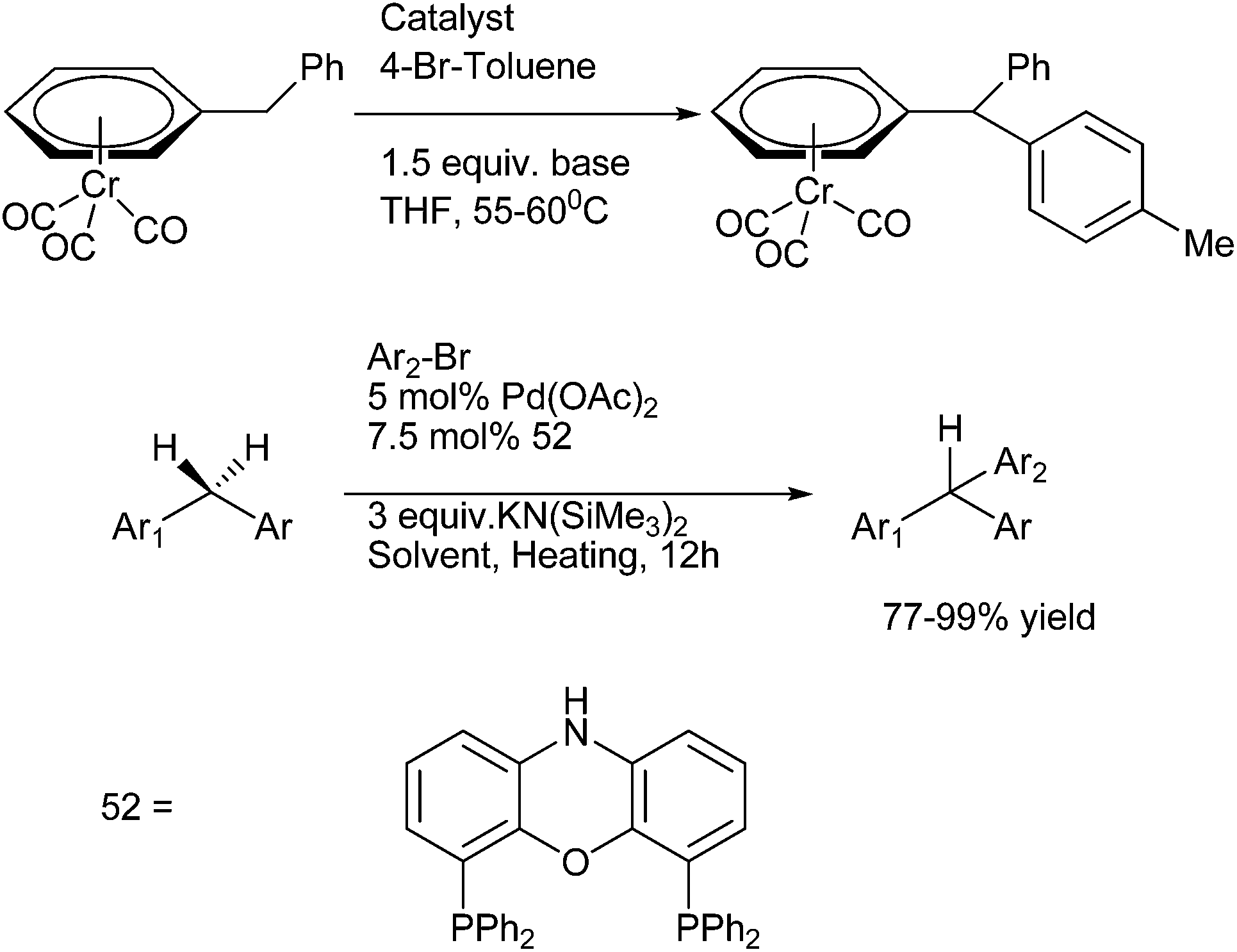 | ||
| Scheme 105 | ||
X. Li et al. in 2011 reported palladium catalyzed diarylation of sp3 C–H bond of aza aryl aryl methanes by aromatic halides to form triarylmethanes (Scheme 106).145 The authors found that chelating phosphine ligands were inefficient for the reaction. Though the authors found that both diarylmethanes and triarylmethanes were formed, the yield of the triarylmethanes was increased when 3 equivalent of aryl bromide was used. The use of mesityl bromide led to the formation of the mono arylated product in major amounts. Interestingly, the diarylated product obtained in minor amount was not a triarylmethane but a product formed from the activation of the sp3 C–H bond of the methyl group of the mestiyl group. This was probably due to the steric hindrance. In order to explain the reaction mechanism, the competition reactions were carried out. Reaction between 1,2-dimethylbenzimidazole and N-methyl benzoxazole led to the formation of triarylmethanes through the sp3 C–H bond activation of the benzoxazole group. Similar endeavor with 2-methyl picoline and 2-methyl quinoline led to the formation of triarylmethane through C–H bond activation of 2-methyl quinoline. This disproved the formation of palladacyclic intermediates as 1,2-dimethylbenzimidazole and 2-picoline would show greater chelation assistance. DFT studies were also carried out to explain the reaction mechanism.
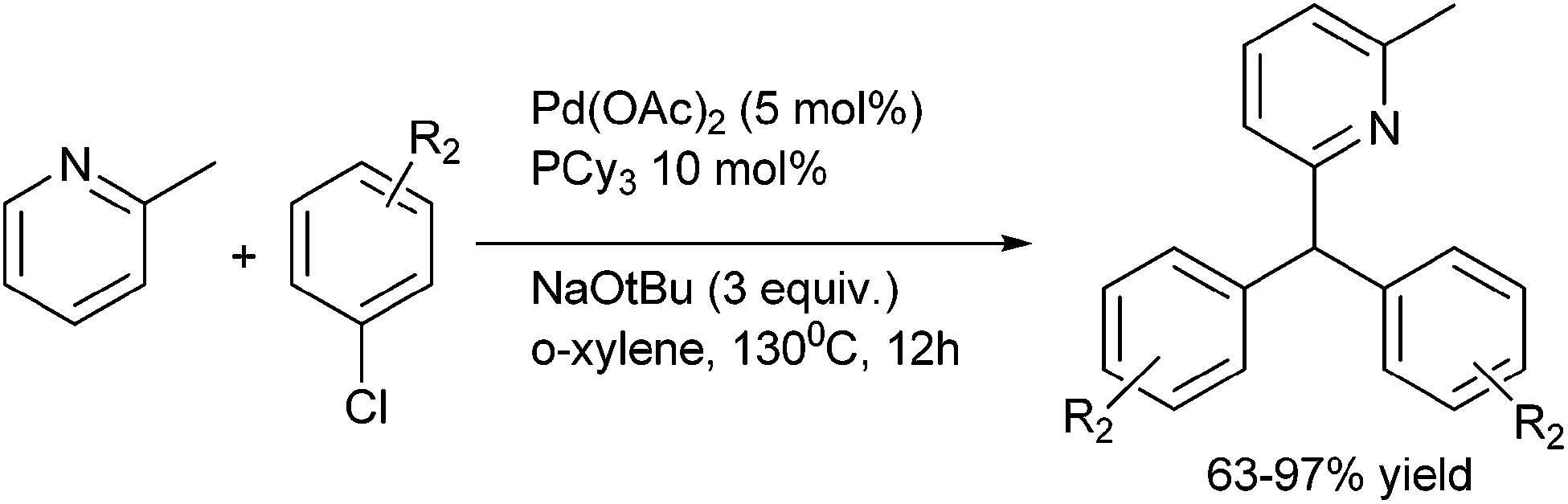 | ||
| Scheme 106 | ||
Z. J. Shi et al. in 2012 reported the use of direct benzyl alcohols (or their magnesium salts) as electrophiles for the metal catalyzed cross coupling with various Grignard reagents through sp3 C–O activation.146 When aryl Grignard reagents were used, diarylmethanes were formed in good yields. Interestingly the authors observed an unprecedented formation of benzyl Grignard reagents when the benzyl alcohols were treated with n-hexylmagnesium chloride under nickel, cobalt or iron catalysis. When the authors used α-arylated benzyl alcohols, the triarylmethanes were formed albeit in low yields.
Y. Zhang et al. in 2013 reported palladium catalyzed coupling of aryl bromides and N-tosyl hydrazones for the preparation of triarylmethanes (Scheme 107).147 Among the tested ligands, 1,1′-2-biphenyl-2-yl diphenyl phosphine (53) were found to be the best. Cesium carbonate and ammonium formate were used as a base and a reducing agent respectively. The authors observed the reduction of aryl bromide to the corresponding arene was a common side reaction. Not only ortho or para-substituted triarylmethanes, but also all meta-substituted triarylmethanes could be produced which were inaccessible through traditional methods.
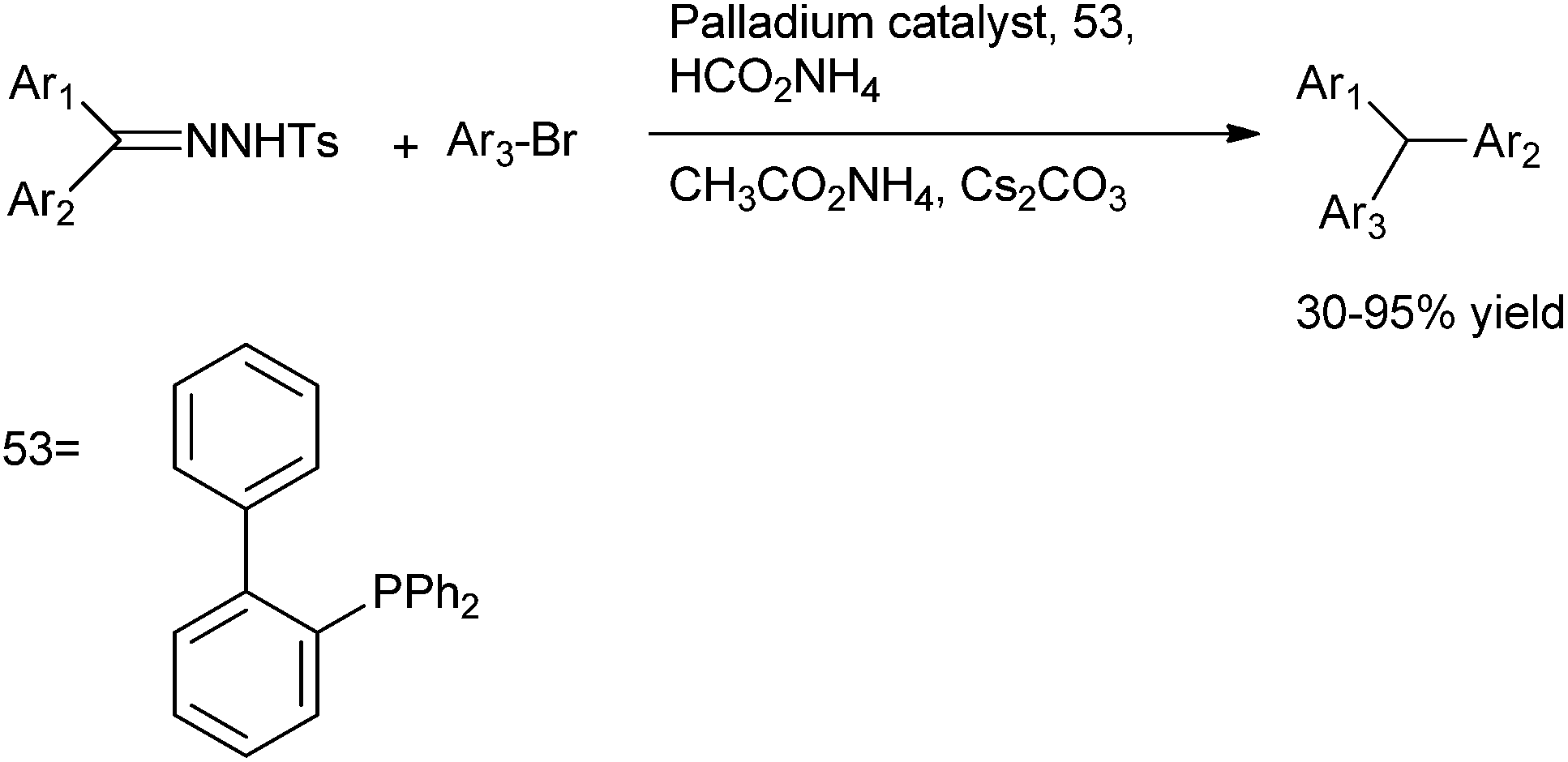 | ||
| Scheme 107 | ||
The authors proposed that at first the aryl halide undergoes an oxidative addition with the palladium complex (53A) to form the intermediate (53B) which forms the metalocarbene complex (53C) generated by treating the N-tosyl hydrazone with a base in presence of the palladium complex. A migratory insertion into the metal carbene bond generates the intermediate (53D). Reduction of this intermediate with the ammonium formate followed by reductive elimination forms the triarylmethane (Scheme 108). The side product was obtained from the reduction of the intermediate (53A) with ammonium formate.
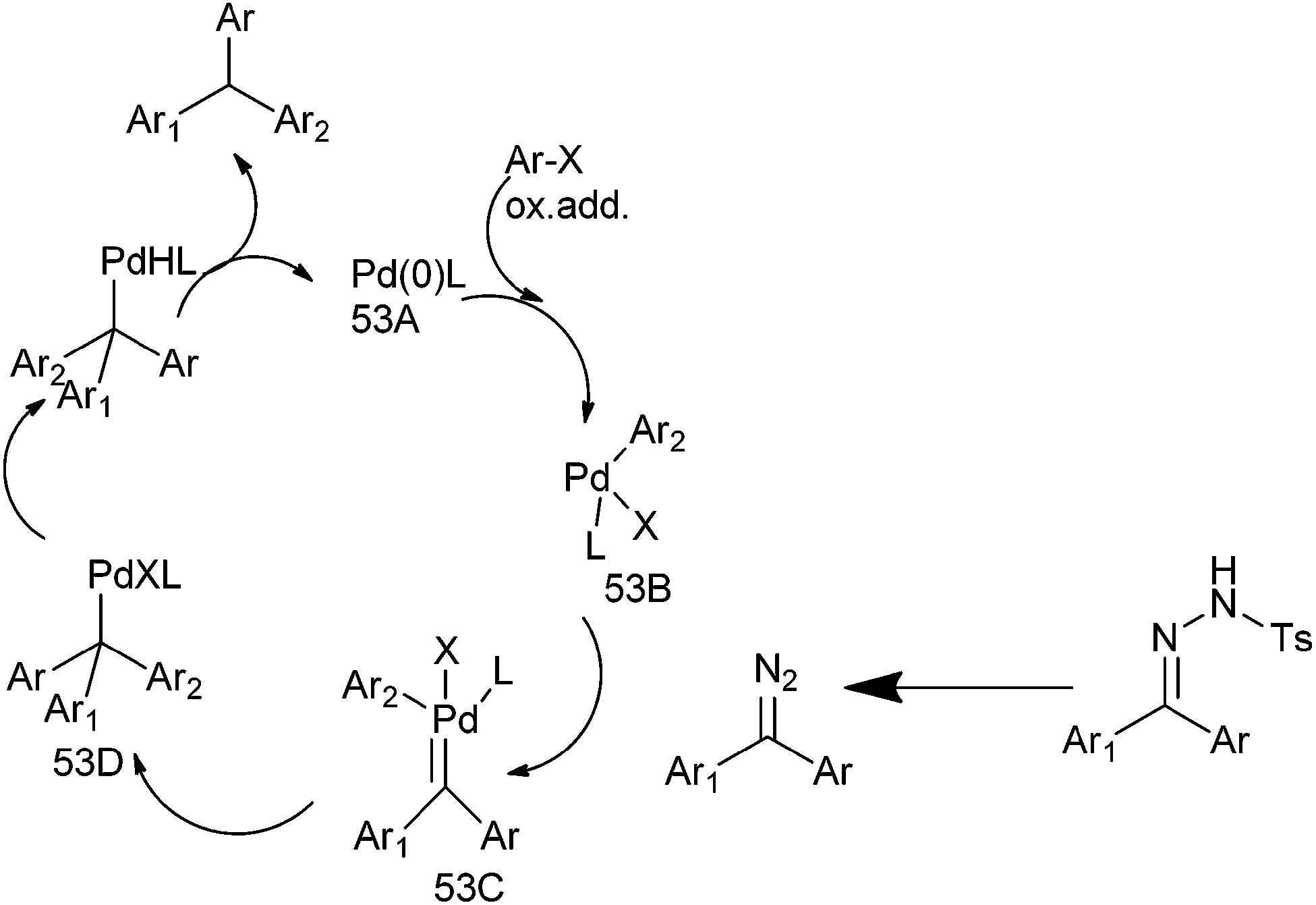 | ||
| Scheme 108 | ||
In the same year L. A. Lopez et al. reported the activation of O–H and N–H bonds of the alcohols or azoles with enynones for the preparation of furans and triarylmethanes (Scheme 109).148 They performed a computational study to establish the mechanism of the reaction.
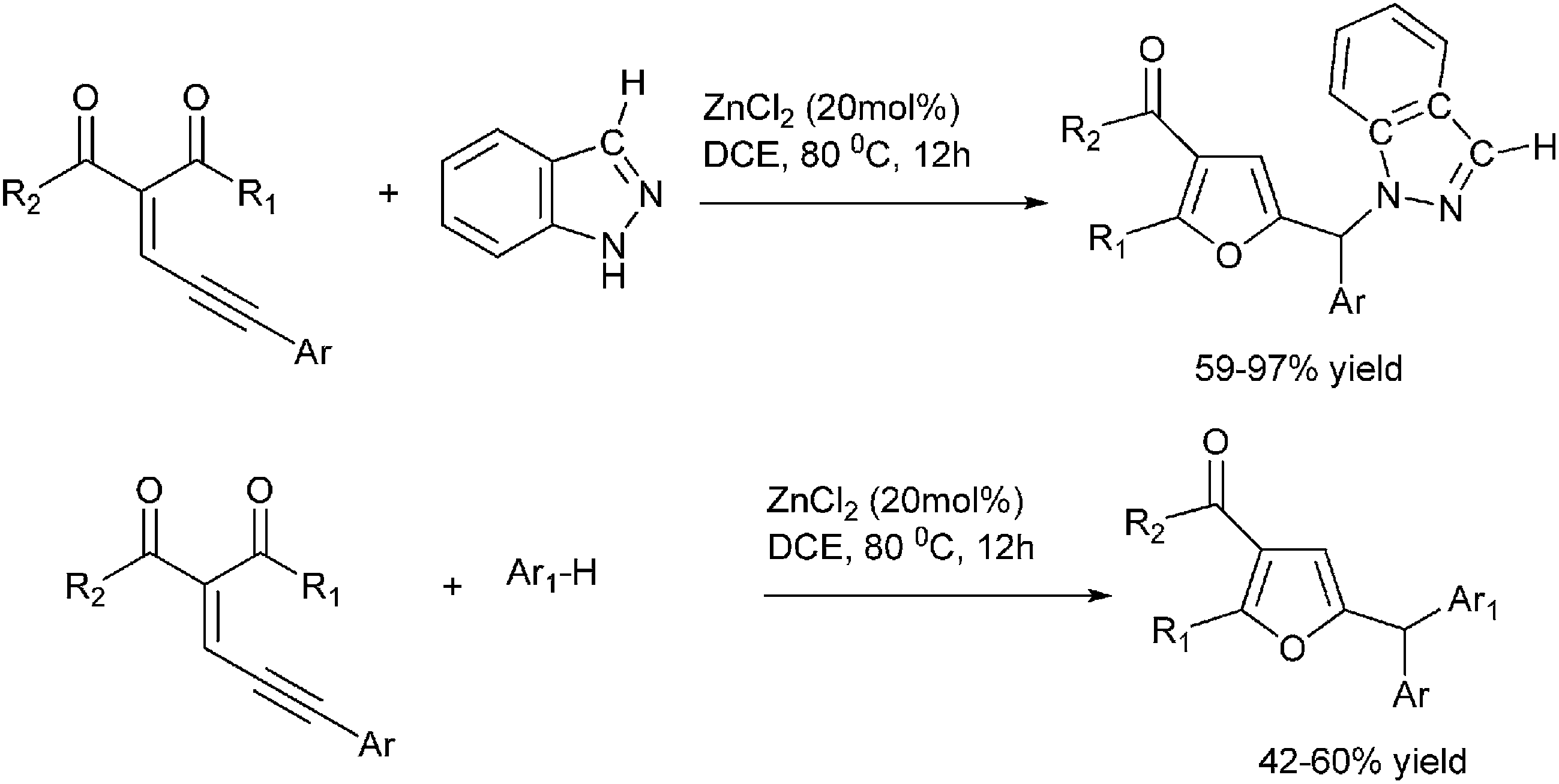 | ||
| Scheme 109 | ||
H. Hikawa et al. in 2013 reported palladium catalyzed domino reactions of indoles with benzyl alcohols for the synthesis of bis-indolyl aryl methanes. The authors used palladium acetate as the catalyst and sodium diphenylphosphinobenzene-3-sulfonate (TPPMS) as the ligand in water for this transformation (Scheme 110).149,150 The authors established the mechanism of the reaction.
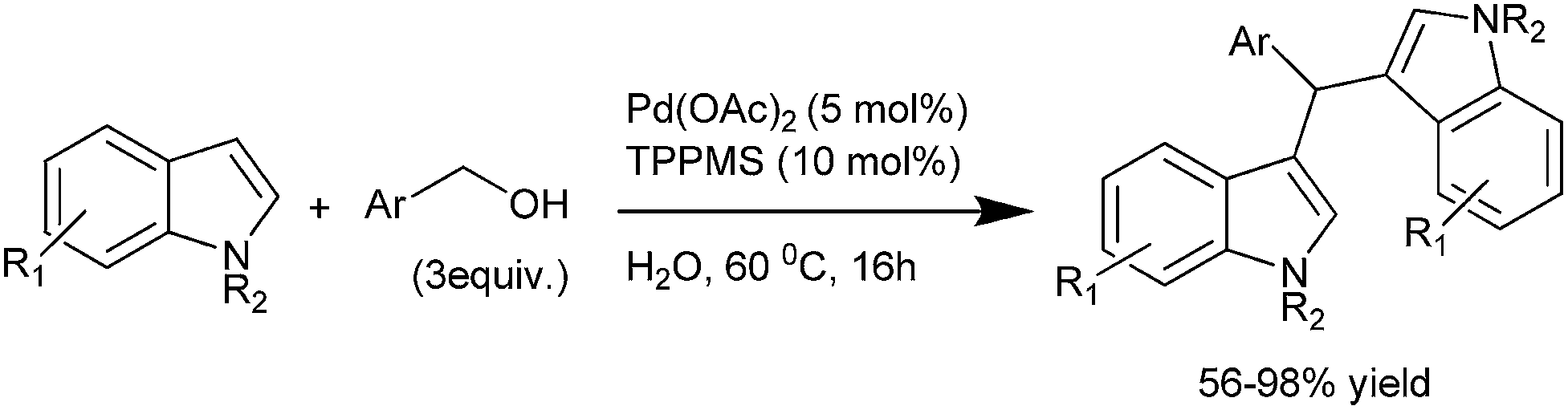 | ||
| Scheme 110 | ||
14. Miscellaneous approaches towards synthesis of triarylmethanes
14.1 Use of benzotriazolylalkylation for the synthesis of triarylmethanes
A. R. Katritzky et al. reported the use of benzenesulphonyl benzotriazole for the preparation of symmetrical and usymmetrical triarylmethanes (Scheme 111).151 They observed coupling of benzenesulphonyl benzotriazole with aromatic aldehydes followed by arylation with electron rich aromatic hydrocarbons gave the benzotriazolyl arylated compounds which on lewis acid mediated displacement with other aromatic hydrocarbons yielded the triaylmethanes. | ||
| Scheme 111 | ||
14.2 Preparation of triarylmethanes through reduction of triarylmethanols
R. Engel et al. in 1997 reported the preparation of triarylmethanes by hypo phosphorus acid mediated reduction of triaryl methanols in acetonitrile solvent (Scheme 112).152 The use of unsubstituted triarylmethanols only led to longer reaction time under harsher reaction conditions proving that the reaction proceeds via trityl ion formation.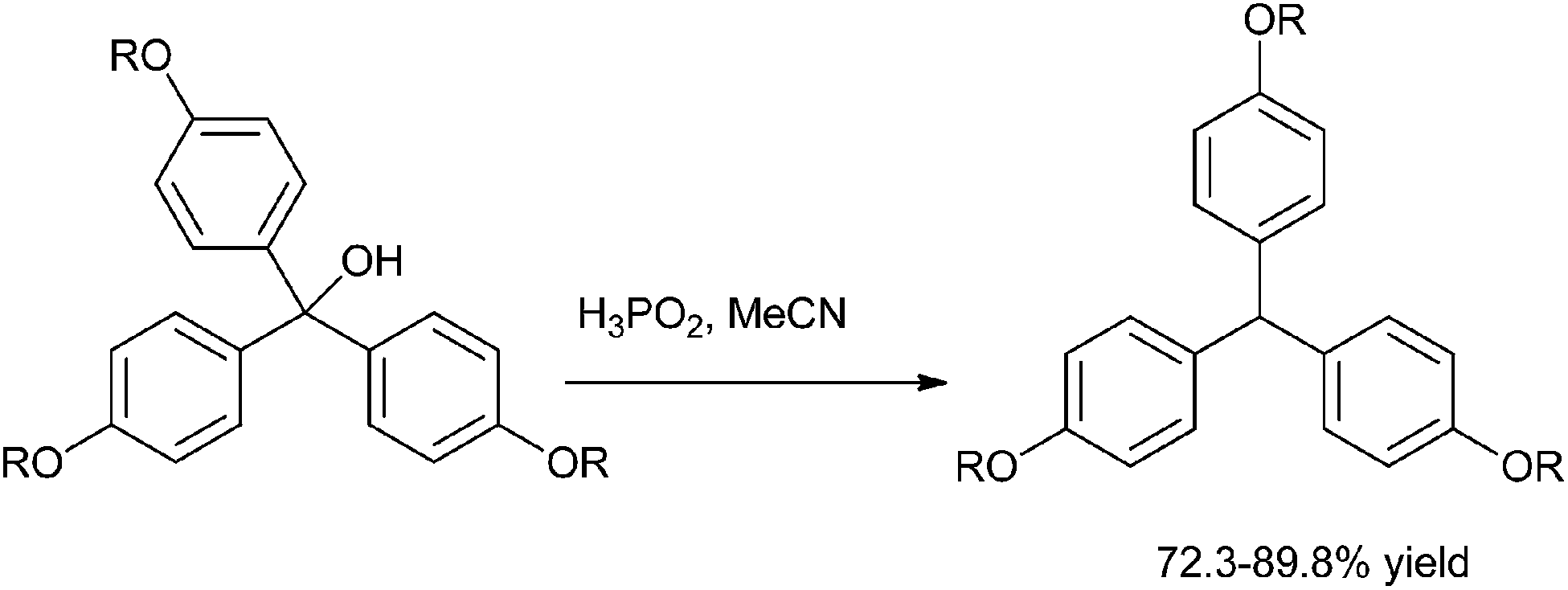 | ||
| Scheme 112 | ||
14.3 Vicarious nucleophilic substitution (VNS)
A. R. Katritzky et al. in 1997 reported the synthesis of (p-nitroaryl) diarylmethanes from diarylmethanols using the concept of vicarious nucleophilic substitution of hydrogen (Scheme 113).153 Para toluene sulfonic acid treatment of the diarylmethanols in presence of (54) yielded the benzotriazoyl diarylmethane which when treated with 5 equivalent of potassium tertiary butoxide followed by treatment with the nitro arenes yielded the corresponding triarylmethane in good yield along with the oxidative nucleophilic substitution product.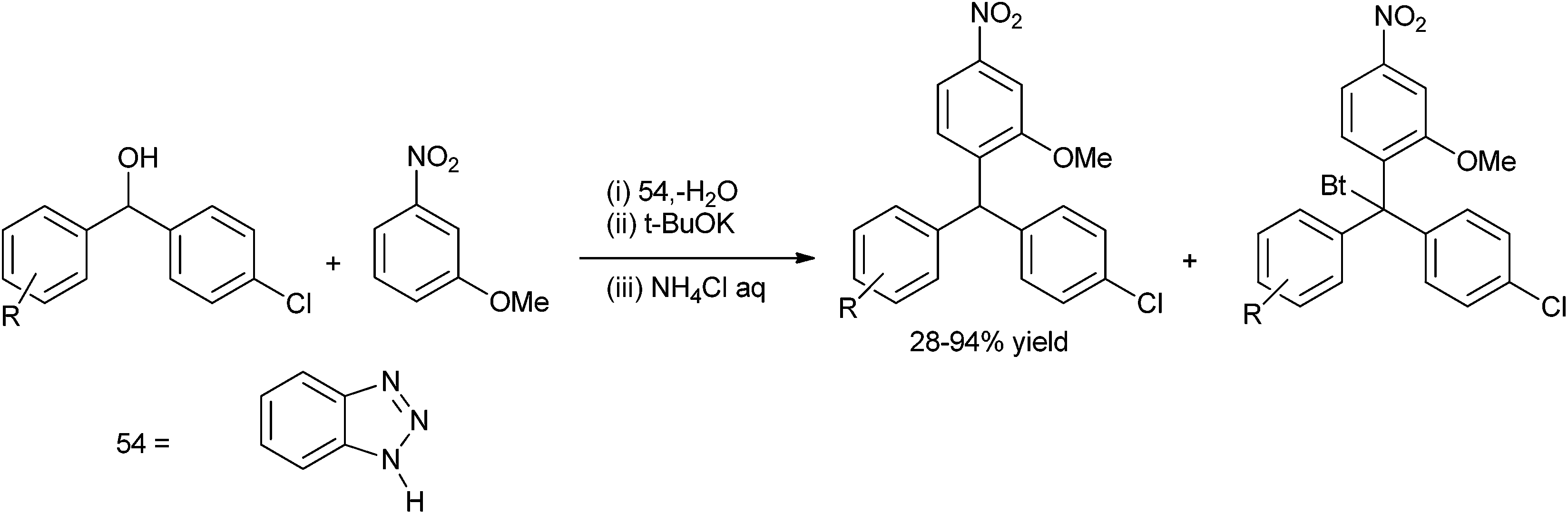 | ||
| Scheme 113 | ||
14.4 Preparation of 2,2′-dihydroxy containing triarylmethanes through condensation of metal phenolates with aryl aldehydes
K. Sumoto et al. in 2000 reported condensation of oxophilic metal phenolates having electron rich substituents attached to the phenolic ring with aromatic aldehydes for the preparation of 2,2′-dihydroxytriphenyl methanes (Scheme 114).154 The reaction was regiospecific giving rise to only the ortho substituted product.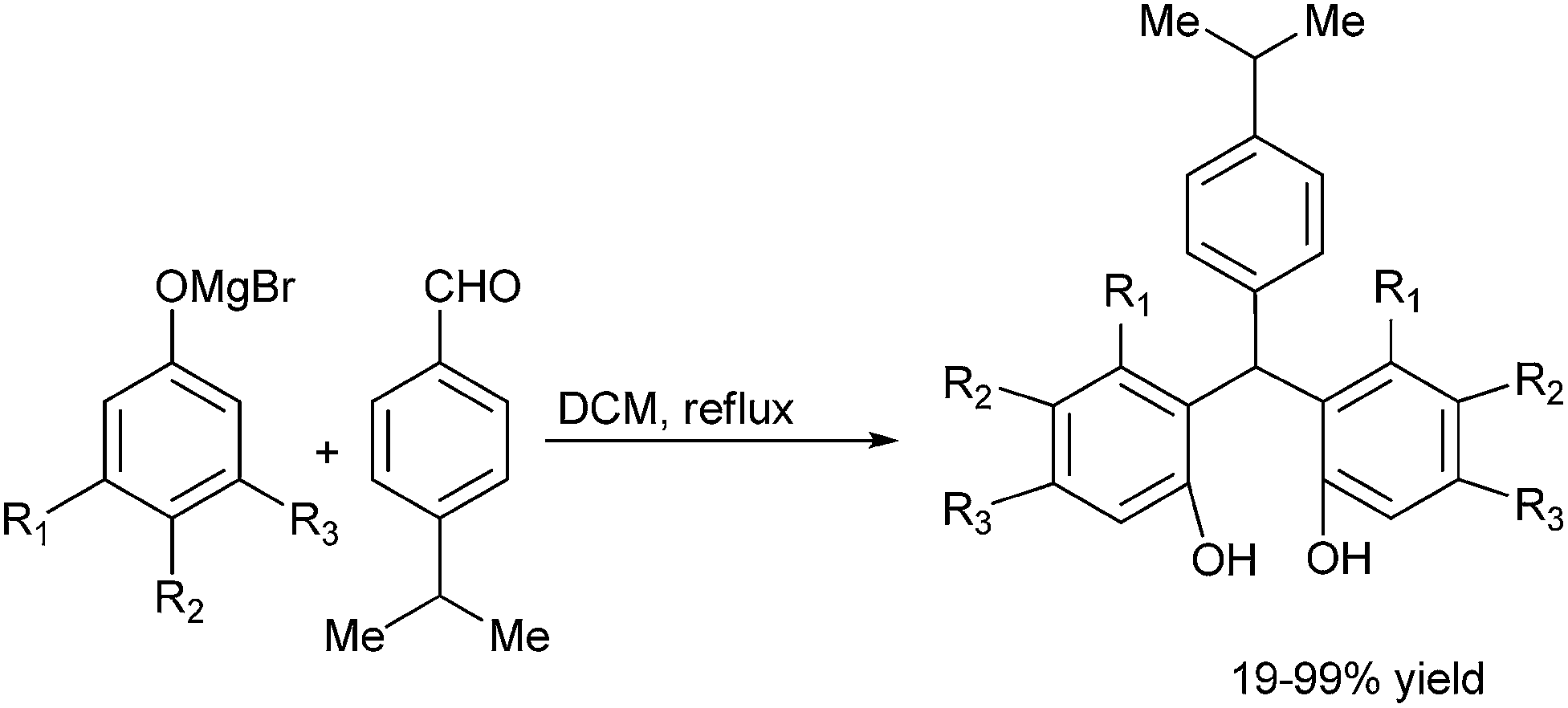 | ||
| Scheme 114 | ||
14.5 Cationic palladium complex catalyzed addition of aryl boronic acids to aldehydes followed by electrophilic aromatic substitution
X. Lu et al. in 2007 reported two step reaction sequences for the preparation of unsymmetrical triarylmethanes from aryl aldehydes (Scheme 115).155 At first a cationic palladium complex catalyzed arylation of aromatic aldehydes with aryl boronic acids gave diarylmethanols which, after completion (after about half an hour) of the reaction, were treated with the electron rich arenes. The authors proposed a plausible mechanism for the reaction. The authors proposed that at first a transmetallation between the boronic acids and the cationic palladium complex occurs to form the active aryl palladium species. A coordination of the aryl aldehyde with this palladium species followed by addition to the carbonyl carbon yields the intermediate II which undergoes a transmetallation with the aryl boronic acids followed by protonolysis to form the diarylmethanol. An activation of the hydroxy group of the methanol with the cationic palladium complex followed by electrophilic aromatic substitution with the electron rich arene generates the unsymmetrical triarylmethanes.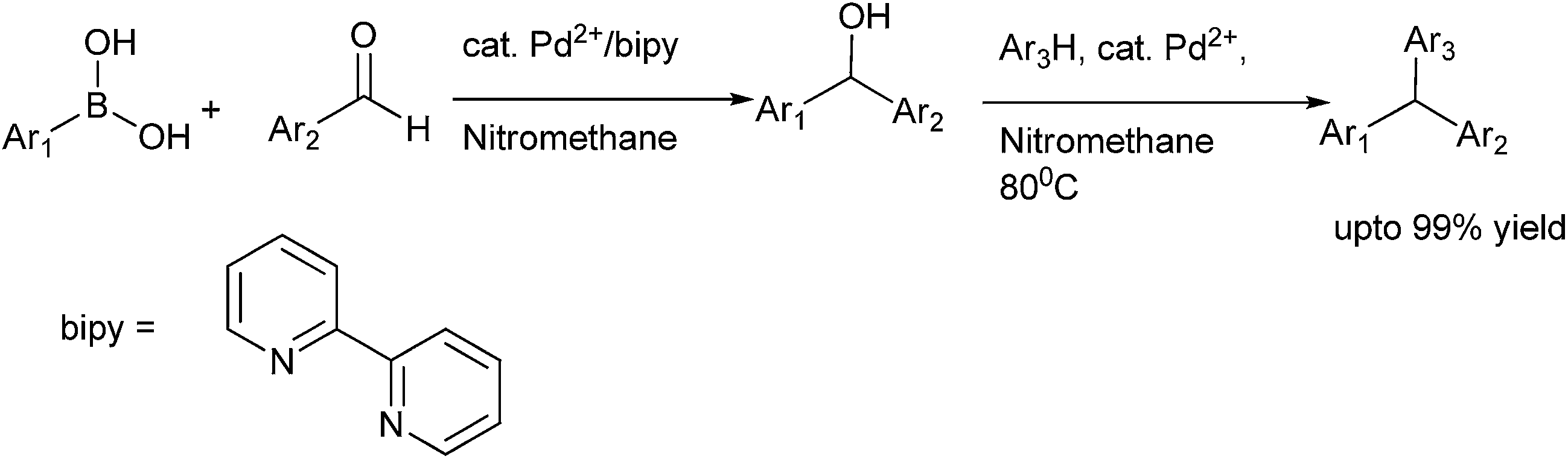 | ||
| Scheme 115 | ||
14.6 Use of silica sulfuric acid for the preparation of triarylmethanes
M. Shiri et al. in 2008 reported silica supported sulphuric acid for the preparation of triindolyl methanes from aromatic dialdehydes.156 Later on in 2011 A. Khazei et al. reported P2O5–SiO2 and/or P2O5–Al2O3 as efficient system for the arylation of benzyl alcohols for the synthesis of di and triarylmethanes in high yields.157I. M. Baltork et al. in 2011 reported a highly eco-friendly, efficient and environmentally benign process for the preparation of triarylmethanes (Scheme 116).158 They used silica sulphuric acid under ultra sound irradiation in green solvent cyclohexane–ethyl acetate system for the hydroarylation of the aromatic aldehydes for the preparation of triarylmethanes.
 | ||
| Scheme 116 | ||
T. Aoyama et al. in 2011 reported silica sulphuric acid promoted deacylation of α-bromo-β-diketones for the preparation of α-bromo ketones. When the reaction was carried out in benzene with 3-(secalkyl)-2,4-pentane-diones, the triarylmethane was formed via Friedel–Crafts alkylation.159
14.7 3 + 3 Cyclocondensation for the preparation of triarylmethanes
P. Langer et al. in 2009 reported ferric chloride catalyzed benzylations with diarylmethanols to prepare 3-(diaryl methyl) pentane 2,4-diones followed by formal [3 + 3] cyclizations with 1,3-bis(1,3-trimethylsilyloxy)-1,3-dienes to prepare highly functionalized triarylmethanes that are inaccessible by conventional synthetic approaches (Scheme 117a).160,161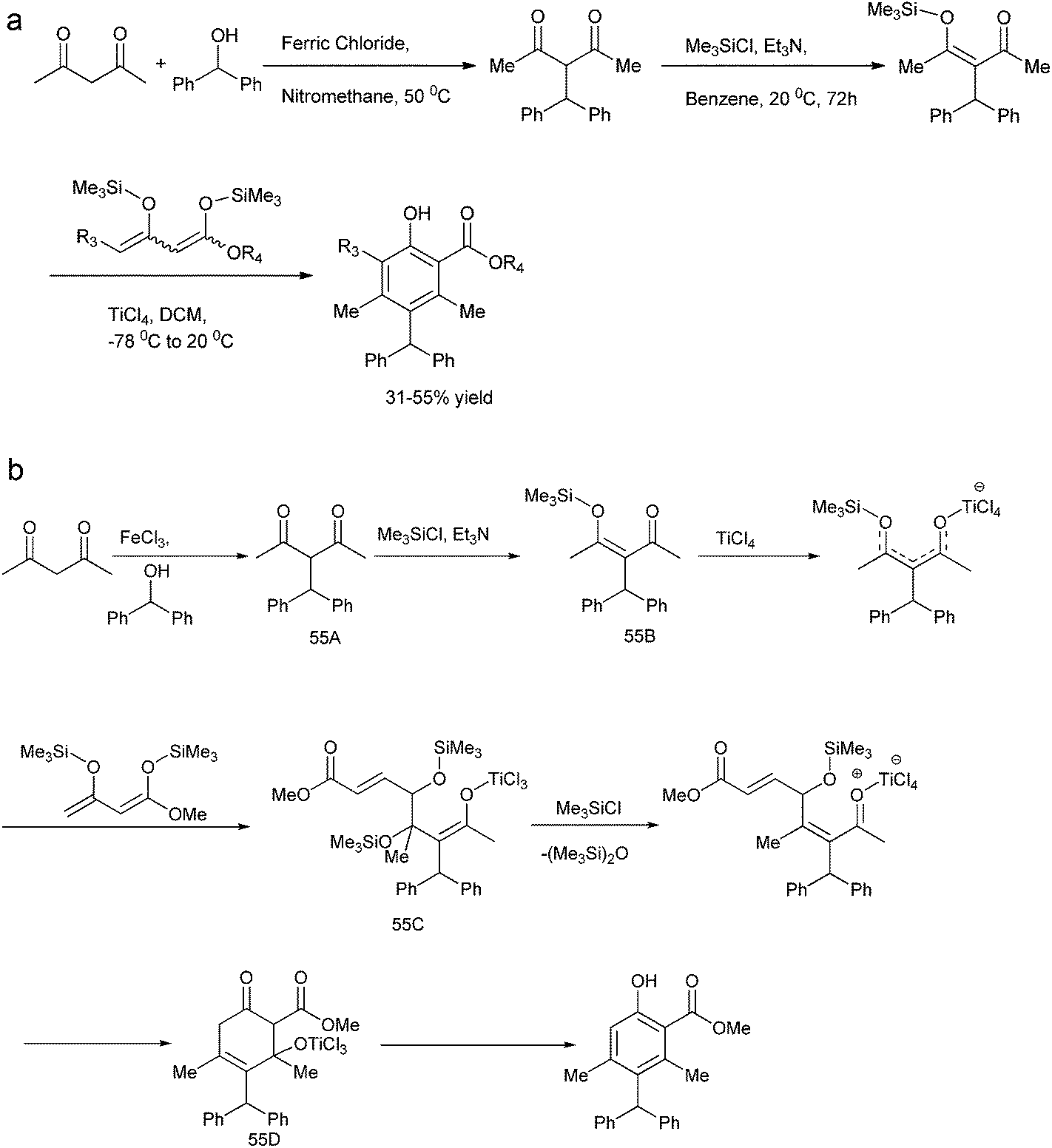 | ||
| Scheme 117 | ||
In order to explain the reaction pathway, the author proposed the following mechanism (Scheme 117b). At first the pentane diones react with diarylmethanols in presence of ferric chloride to form the 3-(diaryl methyl) pentane-2,4-diones (55A). This on treatment with trimethylsilyl chloride and trimethyl amine gives (55B). Treatment of (55B) with titanium tetrachloride followed by the bis trimethylsilyloxy diene led to the formation of the intermediate (55C). Intermediate (55C) is formed by the attack of the bis trimethylsilyloxy diene with (55B) from the terminal end of the diene. Elimination of the trimethylsiloxane followed by cyclization gives compound (55D). Loss of titanium hydroxide followed by aromatization of the intermediate (55D) gives the product.
14.8 Ferric chloride And scandium triflate catalyzed arylation-cyclization cascade of aldehydes
Y. Li et al. in 2009 reported ferric chloride catalyzed arylation of the 2-aryloxy benzaldehydes for the preparation of the bis-indolyl triarylmethanes.162 However within 3 h the bis-indolyl triarylmethanes undergoes a further C–C bond cleavage resulting in the formation of the 9-aryl xanthenes in high yield (Scheme 118).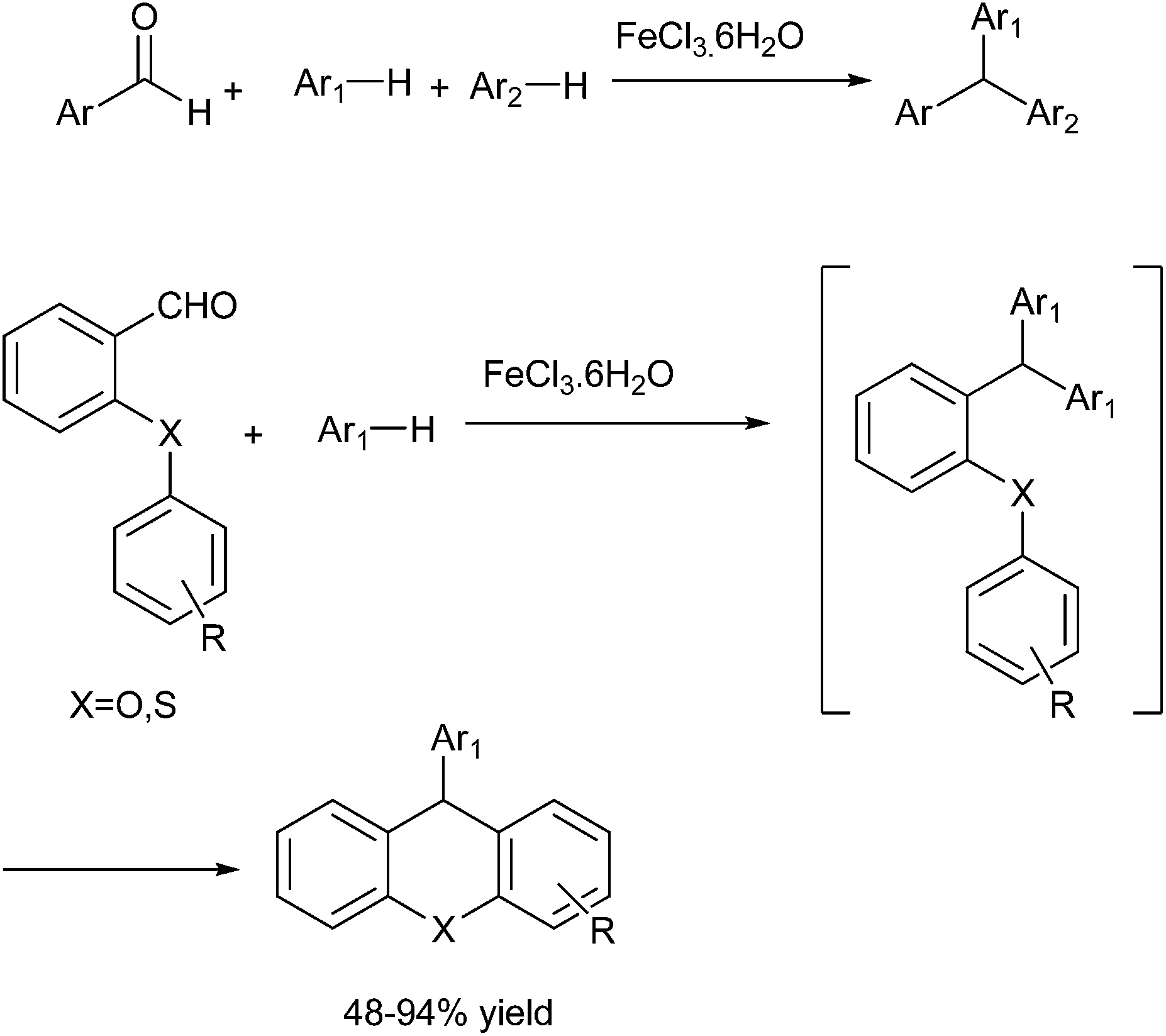 | ||
| Scheme 118 | ||
The authors explained the reaction pathway (Scheme 119). At first a bis-arylation of the aldehyde takes place to form the bis-indolyl methanes (56A) followed by electrophilic substitution of the indolyl ring at C-3 position by the Ferric chloride. A C–C bond cleavage generates a diaryl methyl carbocation (56B) which undergoes a further intramolecular Friedel–Crafts alkylation reaction to form the 9-aryl xanthenes.
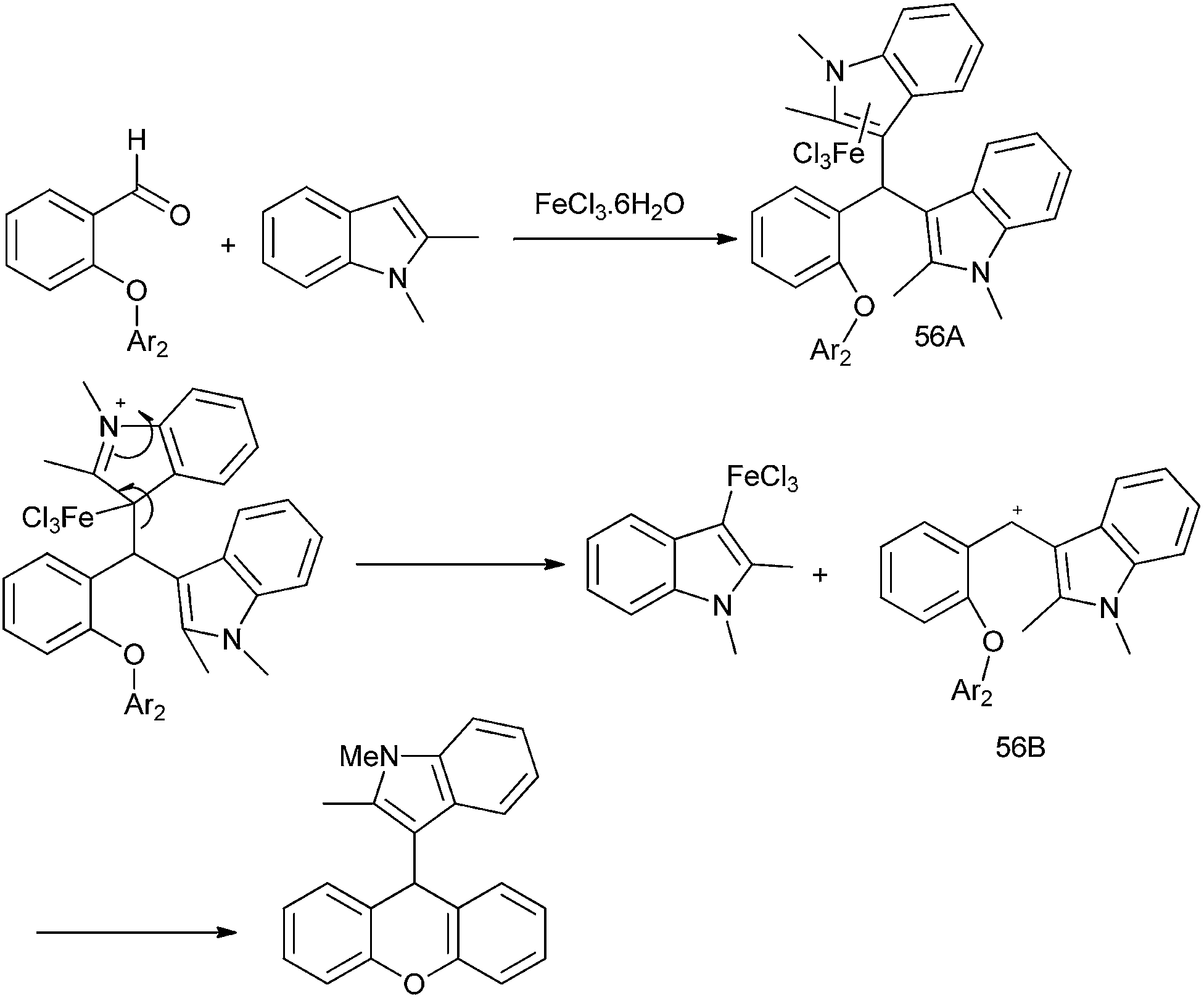 | ||
| Scheme 119 | ||
Later on in 2010, G. Panda and coworkers reported mild Lewis acid Scandium triflate catalyzed bis arylation of 2-naphthoxy benzaldehydes for the preparation of the 9-Arylxanthenes (Scheme 120).163
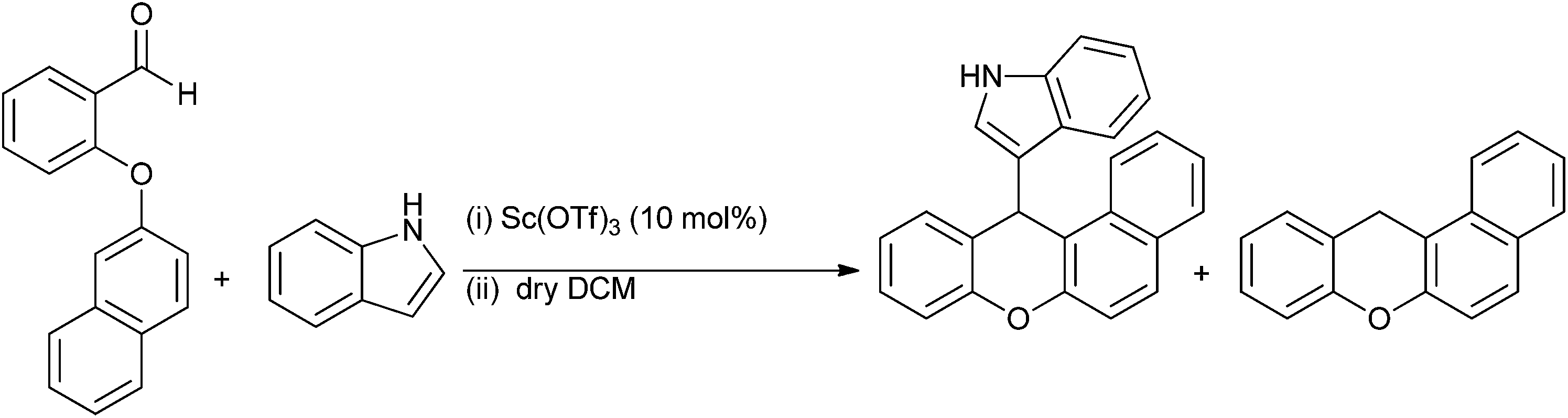 | ||
| Scheme 120 | ||
14.9 Catalyst free green synthesis of triarylmethanes
Y. Gu et al. in 2009 developed a green procedure for the electrophilic activation of the aromatic aldehydes (Scheme 121).164 They reported the use of glycerol as a medium for the electrophilic activation of the aldehydes. Thus they could avoid use of toxic metal catalysts for the hydroxyalkylation of aldehydes and thus prepare the triarylmethanes in high yield. The authors observed that both the steric effect as well as the electronic effect plays an important role in the reaction rate as well as in the successful achievement of the reaction.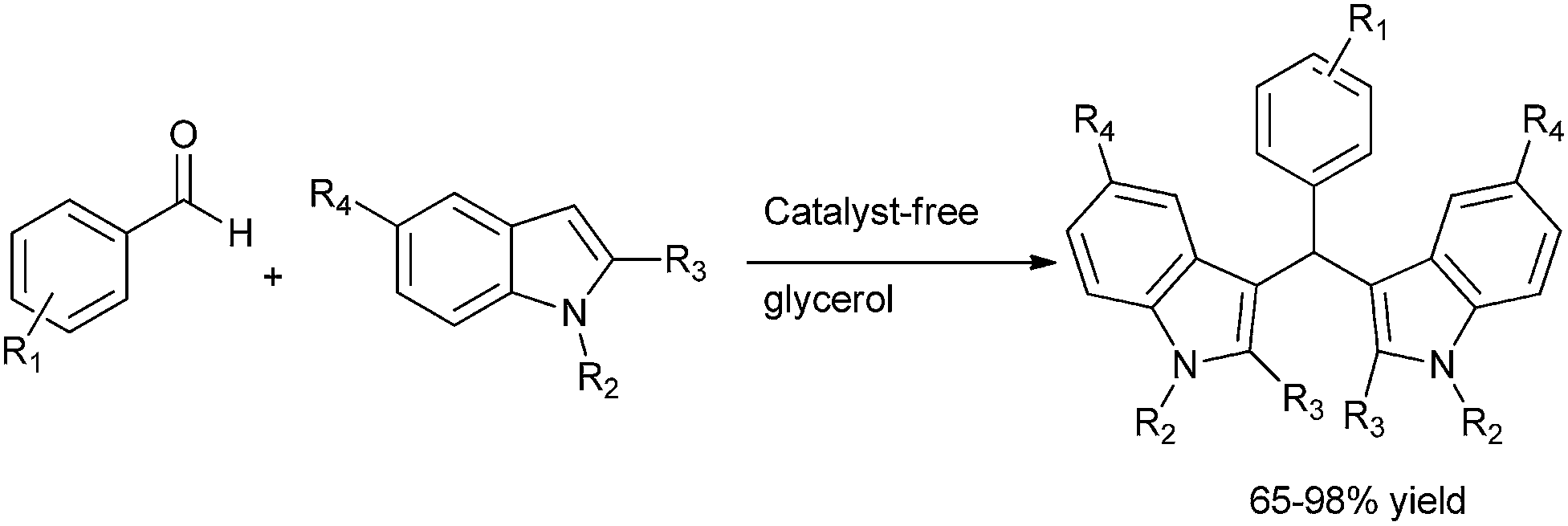 | ||
| Scheme 121 | ||
15. Organocatalytic approach for the synthesis of triarylmethanes
15.1 Organocatalytic Friedel–Crafts reaction
J. A. McCubbin et al. in 2010 reported boronic acid catalyzed Friedel–Craft reaction of diarylmethanols to prepare triarylmethanes in high yields (Scheme 122).165 The author observed that the stability of the carbocations derived from the diaryl methanol was crucial for the success of the reaction as neither phenyl ethyl alcohol nor diaryl alcohols possessing electron withdrawing substituents in one of the rings responds to the reaction.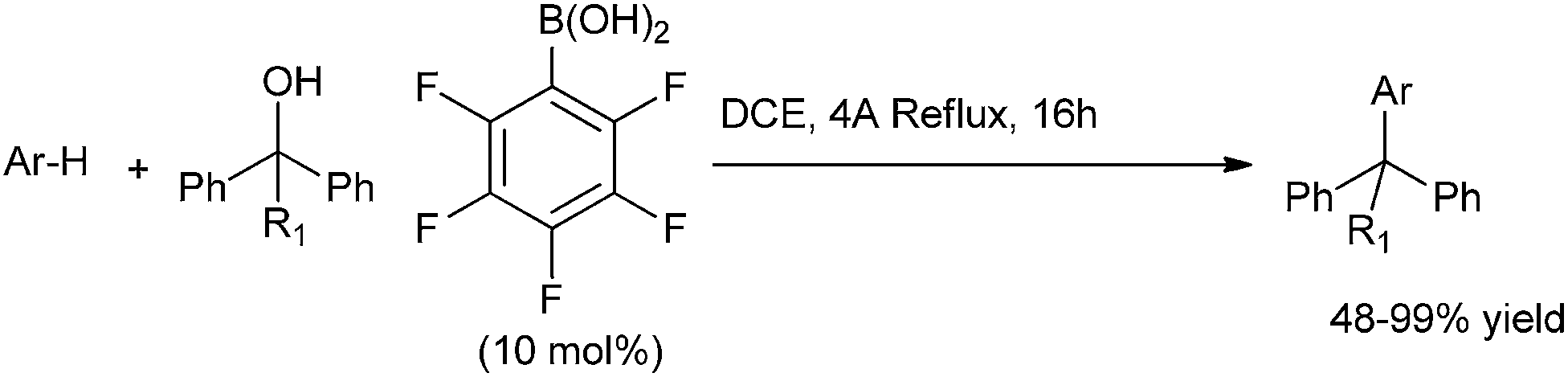 | ||
| Scheme 122 | ||
15.2 Organocatalytic hydroxyalkylation reaction
M. Barbero et al. in 2011, reported o-benzenedisulfonimide 57 catalyzed hydroxyarylation reaction of aromatic aldehyds to obtain the triarylmethanes in good yield (Scheme 123).166 The authors observed that the temperature played a crucial role in the reaction as at room temperature the reaction did not proceed while at 100 °C the product was obtained in very small amount. The authors also observed that regioisomeric para, para product was always the major product whereas the ortho, para isomer was obtained in very small amount but the ortho, ortho isomer was not observed at all.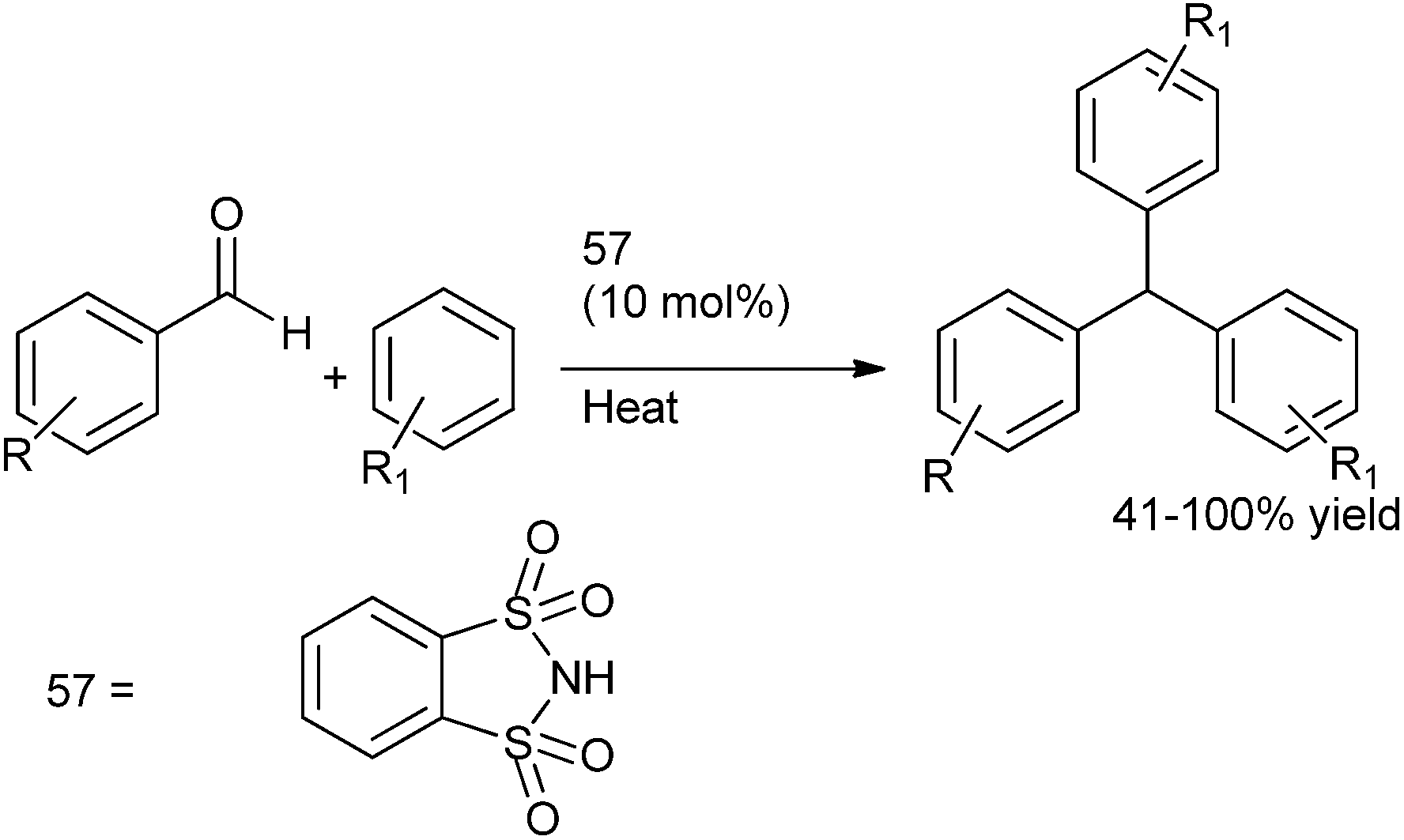 | ||
| Scheme 123 | ||
16. Biomedical application of diaryl & triarylmethanes
In the last fifteen years there has been an explosion in the utilization of diaryl and triarylmethane derivatives in the biomedical arena. The triarylmethane derivatives have shown various diverse bioactivities like as anticancer, antitubercular, antimalarial, and antiviral and anti HIV agents. Moreover due to photo toxicity of various triaryl methyl dyes towards tumor cells, many triarylmethane derivatives are used in photodynamic therapy. Though such compounds have such a wide domain of applicability, to the best of our knowledge there is no review covering this topic. We will be covering the biomedical applications of diaryl as well as triarylmethane derivatives that were reported during 1995–2013.16.1 Diaryl and triarylmethane derivatives as antiproliferative agents
C. Brugnara et al. in 1997 reported triaryl methane and their derivatives thereof wherein one or more aryl moieties have be replaced by heteroaryl, cycloalkyl or heterocycloalkyl &/or wherein the tertiary carbon has been replaced by Si, Ge, N or P167 as active agents against sickle cell disease and diseases characterized by unwanted or abnormal cell proliferation.S. Safe et al. in 1998 reported diindolylmethanes to have anti estrogenic and anti tumourigenic properties (Fig. 4).168–174 The authors reported that the diindolyl methanes bound to the aryl hydrocarbon receptor. Later in 2000 they extended this concept and reported symmetrical dihalo substituted bis-indoles acted as inhibitors of carcinogen induced rat mammary tumour growth and estrogen depended responses. In 2004 they reported 1,1-bis (3′-indolyl)-1-p-(trifluoromethylphenyl) methanes and various p-substituted 1,1-bis (3′-indolyl) methanes as new class of PPARγ inhibitors. In the same year they reported 1,1-bis (3′-indolyl)-1-p-substituted phenyl methanes containing p-trifluoromethyl, p-tertiary butyl and p-phenyl groups as PPARγ mediated transactivators in HT-29,HCT-15,RKO and SW480 colon cancer cell lines. In 2005 they investigated the antileukemic activity of 1,1-bis[(3′-(5-methoxyindolyl)]-1-(p-t-butyl phenyl) methane (58). In 2006 they reported 3,3′-diindolyl methanes and 1,1-bis (3′-indolyl)-1-(p-substituted phenyl) methanes inhibiting the growth of Panc-1 and Panc-28 pancreatic cancer cells. The authors found that the 3,3′-diindolyl methanes activated aryl hydrocarbon receptor and the 1,1-bis(3′-indolyl)-1-(p-4-t-Bu phenyl) methanes activated the peroxisome proliferator-activated receptor γ.
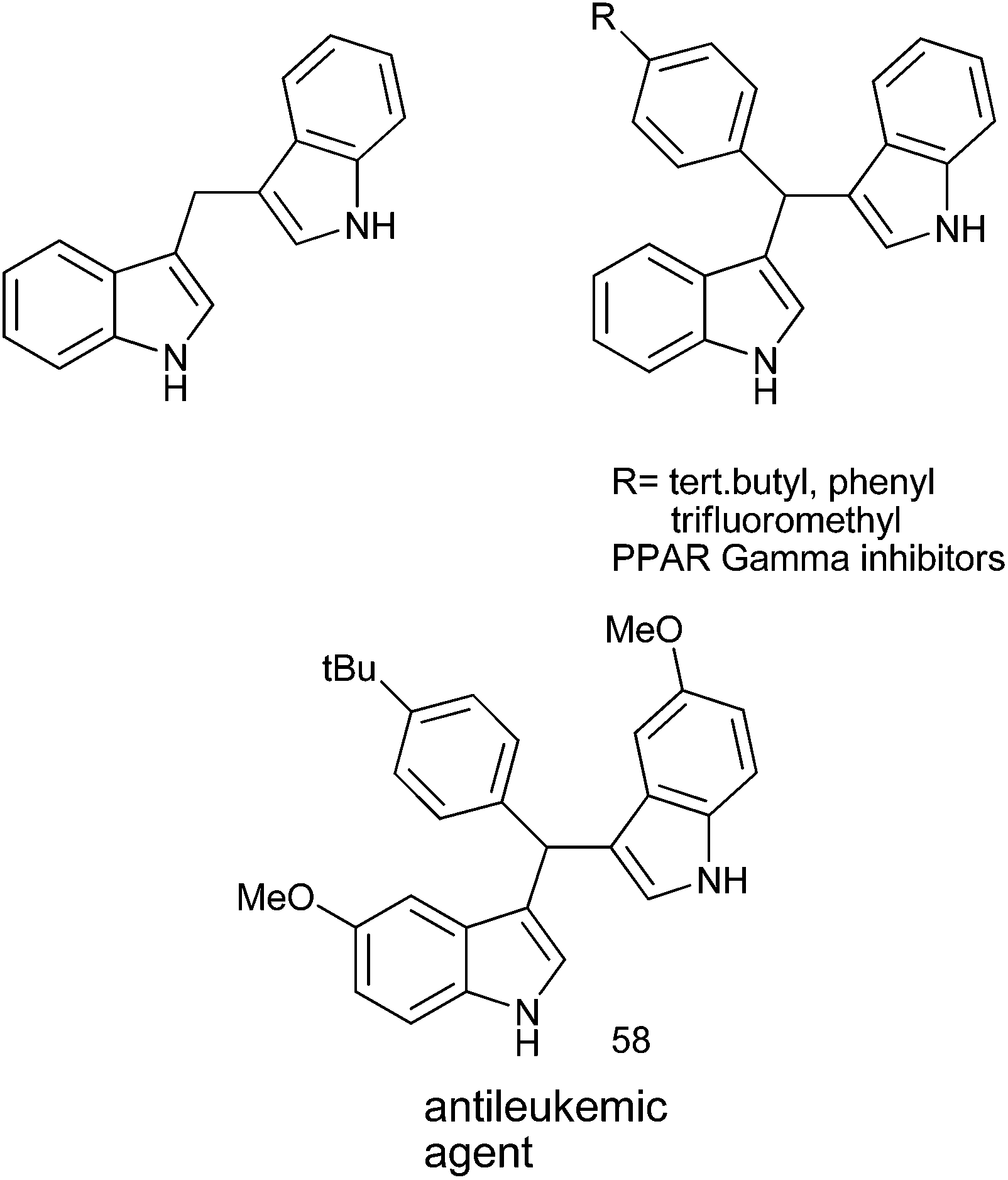 | ||
| Fig. 4 Diindolyl methanes as anti-breast cancer & antileukemic agents. | ||
In order to investigate the physiochemical change on the photodynamic activity of the dyes based on the crystal violet structure, M. Wainwright et al. in 1999 prepared a series of compounds based on the Victoria blue BO core structure.175 The compounds were compared to VBBO with respect to dark toxicity and photodynamic activity.
G. L. Indig et al. reported crystal violet exhibited pronounced phototoxicity towards L1210 leukemia cells but small toxic effects towards hematopoietic cells.176 The authors established a structure activity relationship between the phototoxicity and the molecular structure.
D. A. Skoufias et al. in 2004 reported S-trityl L-cysteines as potent anticancer agents (Fig. 5).177,178 The reported compounds inhibited the human kinesin Eg5 that specifically arrested the mitosis of the cells.
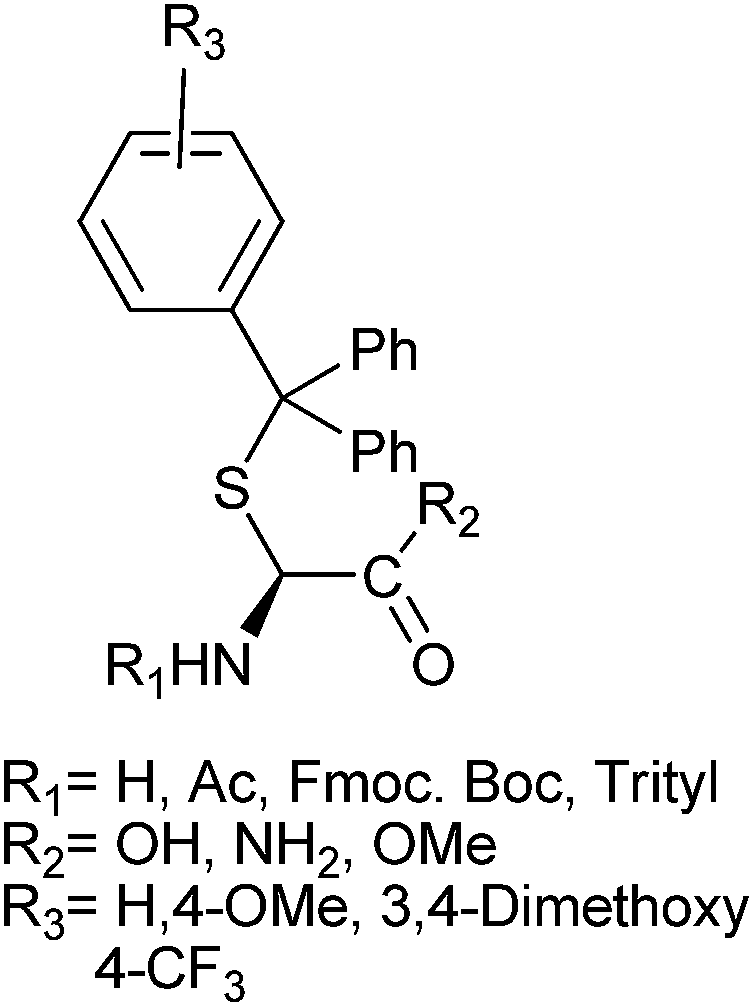 | ||
| Fig. 5 S-Trityl L-cysteine derivatives as anti cancer agents. | ||
Later on in 2007 Akira Asai et al. performed a SAR study on the S-trityl L-cysteine derivatives. They found that protection of the amino group resulted in the loss of the activity while conversion to the ester or the amide caused little loss in activity.179
Towards the development of Clotrimazole derivatives with no hepatoxicity yet retaining the efficacy of Clotrimazole, R. A. Al-Qawasmeh et al. in 2004 reported compounds (59a–v) (Fig. 6).180 The compounds had IC50 values in the range 0.2–3.8 μM. Interestingly the compounds 59c, 59g, 59i, 59k, 59s, 59t, 59u, 59v were more potent than Clotrimazole and at 10 μM concentration inhibited 99% of the cell cycle.
 | ||
| Fig. 6 Triarylmethane based antiproliferative agents. | ||
J. A. Halperin et al. in 2004 reported substituted 3,3-diphenyl-1,3-dihydro-indol-2-ones (60a–g) as potent cell growth inhibitors (Fig. 7).181 The reported compounds depleted the intracellular calcium levels thereby phosphorylating the eIF2α cells and inhibiting the translation process. A SAR study revealed m-tert-butyl and o-hydroxy substituted diphenyl oxindole as the lead compound. Later on they reported arylsulfoanilide-oxindole hybrid as anticancer agents.182 The reported compounds showed partial depletion of intracellular calcium ion stores and inhibited growth of Lung Cancer cells (A549) with the most potent compound having GI50 value of 0.8 μM.
 | ||
| Fig. 7 3,3-Diaryl-1,3-dihydro-indol-2-ones as cell growth inhibitors. | ||
G. L. Indig et al. in 2005 reported four bromo derivatives of Rhodamine-123 a xanthenes derivative as potential phototoxic dyes towards human uterine sarcoma cells and towards green monkey kidney (CV-1) cells.183
P. J. Hergenrother et al. in 2005 reported triphenyl methyl amides as active anticancer agents (Fig. 8).184 The compounds arrested the growth of melanoma cells in the G1 phase of the cell cycle. Interestingly the authors found that these compounds also reduced the levels of active nuclear factor κ-B (NF κ-B) which is actively expressed in the melanoma cells.
 | ||
| Fig. 8 Triaryl methyl amides as anti cancer agents. | ||
In 2008 they reported triphenylmethane motif containing phosphonates and phosphonochloridates as potential anticancer agents (Fig. 9).185 The compounds induced apoptosis of the melanoma and other cancer cell lines and arrests cellular growths in the G1phase or the M phase of the cell cycle.
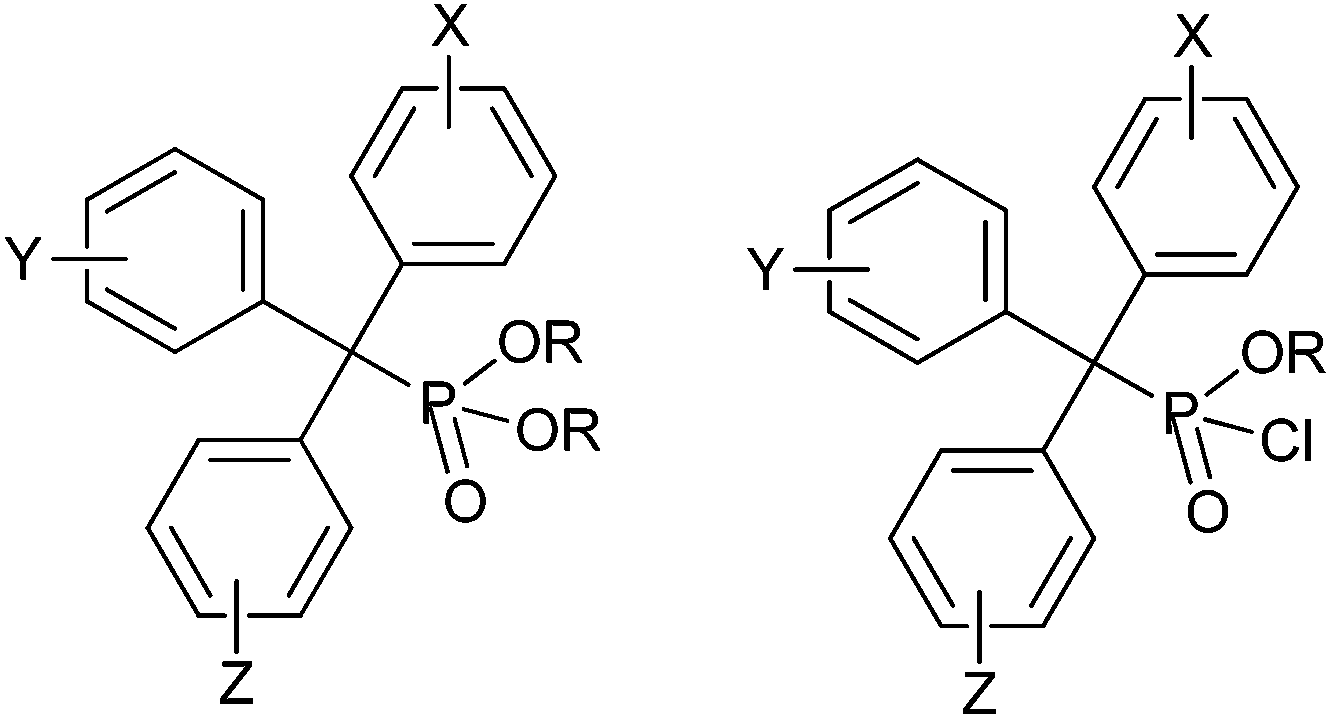 | ||
| Fig. 9 Triaryl methyl phosphonates and phosphonidates as anticancer agents. | ||
J. L. Arbiser et al. in 2010 reported triaryl methane analogues with steroids and steroid precursors like cholesterol, progesterone, testosterone or estrogens, dyes such as indigo and chrysin, benzophenones, nucleosides such as adenine, thymidine, cytosine, guanine and uracil, and aromatic amino acids like phenyl alanine and folic acid as inhibitors of tNOX expression, and hence probable anticancer agents.186 The compounds were also active against inflammatory diseases, degenerative and vascular diseases including various ocular and parasitic infections.
16.2 Triarylmethane derivatives as anti-breast cancer agents
C. Simons et al. in 2006 reported 1-[(benzofuran-2-yl)phenylmethyl]-pyridines, imidazoles and triazoles as potent aromatase (P450AROM,CYP19) inhibitors and hence possible antibreast cancer agents.187 The authors found that the 6-methoxy (61) (i.e. R = 6-methoxy) and the 6-hydroxy compounds (62) (i.e. R = 6-hydroxy) were the more active than the unsubstituted ones (63) (i.e. R = H) (Fig. 10).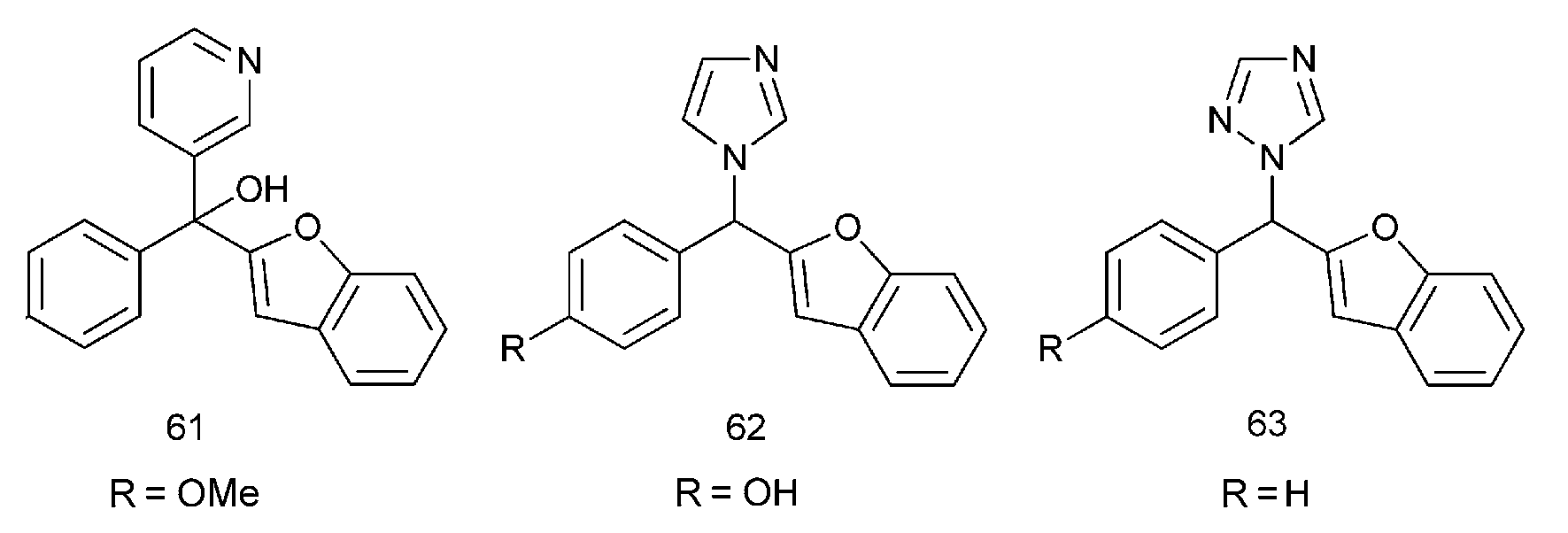 | ||
| Fig. 10 Benzofuran phenyl methyl-pyridines, imidazoles & triazoles as aromatase inhibitors. | ||
G. Panda and co-workers in 2006 reported aryl aryl phenanthrenes based triarylmethanes with basic amino side chains as the probable anti breast cancer agents (Fig. 11).188 The authors found that the ortho methoxy substituted compounds were inactive whereas corresponding meta isomer and para isomer were active. However, interestingly the compounds with meta methoxy group and N,N-dimethyl amine chain or meta methoxy group and N,N-diethyl amine chain were both inactive.
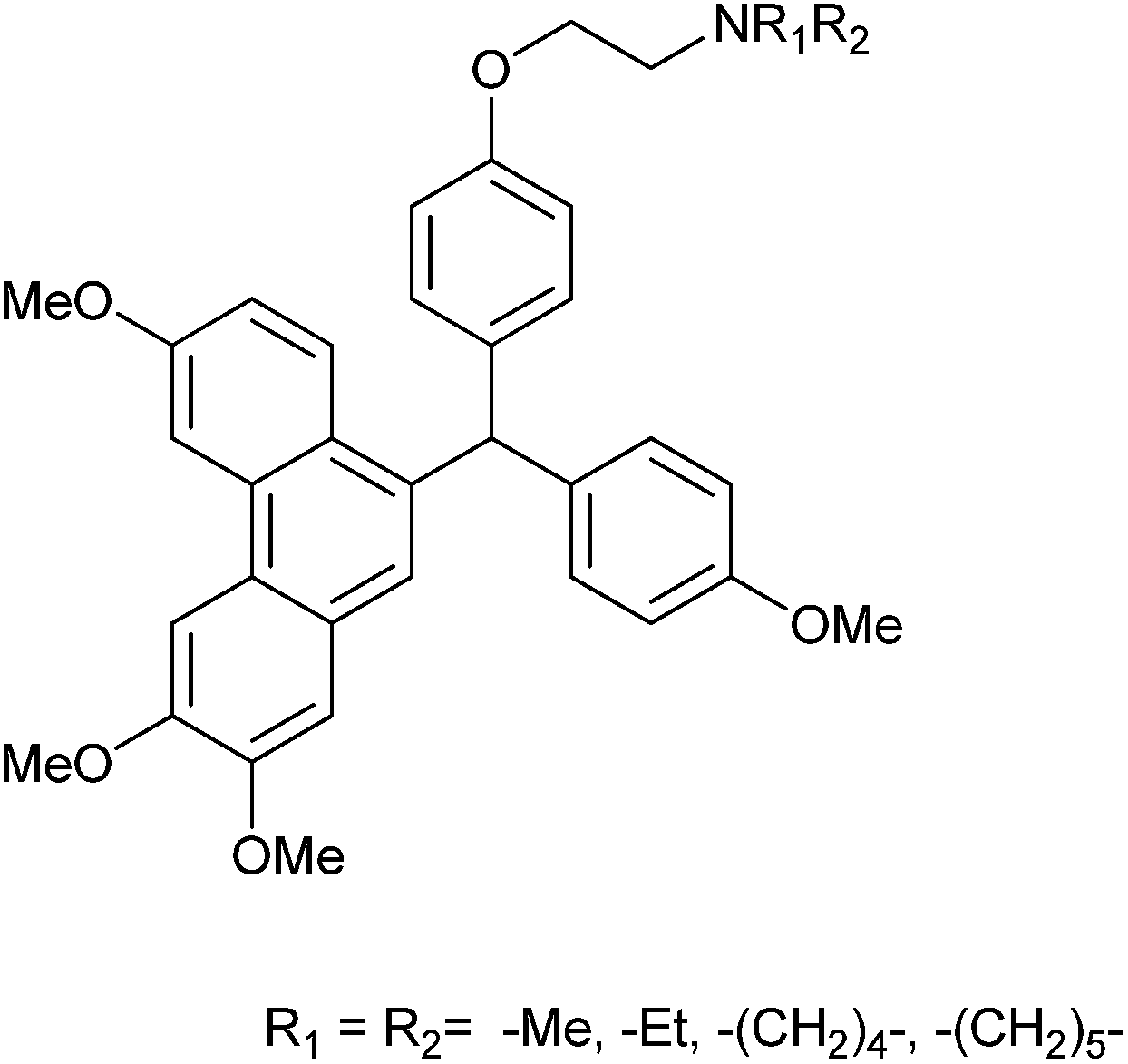 | ||
| Fig. 11 Aryl aryl phenanthrene based triarylmethanes as antibreast cancer agents. | ||
S. Gobbi et al. in 2001 designed various non-steroidal aromatase inhibitors using results from the previously deduced COMFA model. Later on in 2005 in order to identify enantioselective nonsteroidal aromatase inhibitors they performed a multidisplinary medicinal chemistry approach.189–192 Later on in 2007 they reported imidazolylmethylbenzophenones as highly potent aromatase inhibitors. The authors observed the compounds were highly selective to 17α-hydroxylase/17, 20-lyase. Later on in 2010 the same group further modified their previously reported scaffold and performed a SAR study on it. They observed that substitution of the oxygen atom of the xanthone ring with the sulfur atom always caused an increase in the potency of the compounds; the nitro group in their previous molecule was also not a compulsion as it could be changed to carbonyl group however removal of both was detrimental to the inhibitory activity.
B. V. L. Potter et al. in 2005 reported letrozole based dual aromatase-sulphatase inhibitors. They incorporated the pharmacophore of the sulfatase inhibitors to the Letrozole core.193 In 2007 they reported the synthesis and bioevaluation of novel diarylmethane based dual aromatase-sulfatase inhibitors based on the anastrozole template (Fig. 12a).194 In 2008 the same group further improved on the bioactivity of the dual aromatase sulfatase inhibitors by reporting enantiopure non-steroidal inhibitors (Fig. 12b).195 The authors found that (64) was the most potent aromatase inhibitor. Interestingly they reported that the (R) isomer was a potent aromatase inhibitor while the (S) isomer was a potent sulfatase inhibitor. Later in the year 2010 they reported single compound dual inhibitors of the aromatase and steroid sulfatase enzymes.196
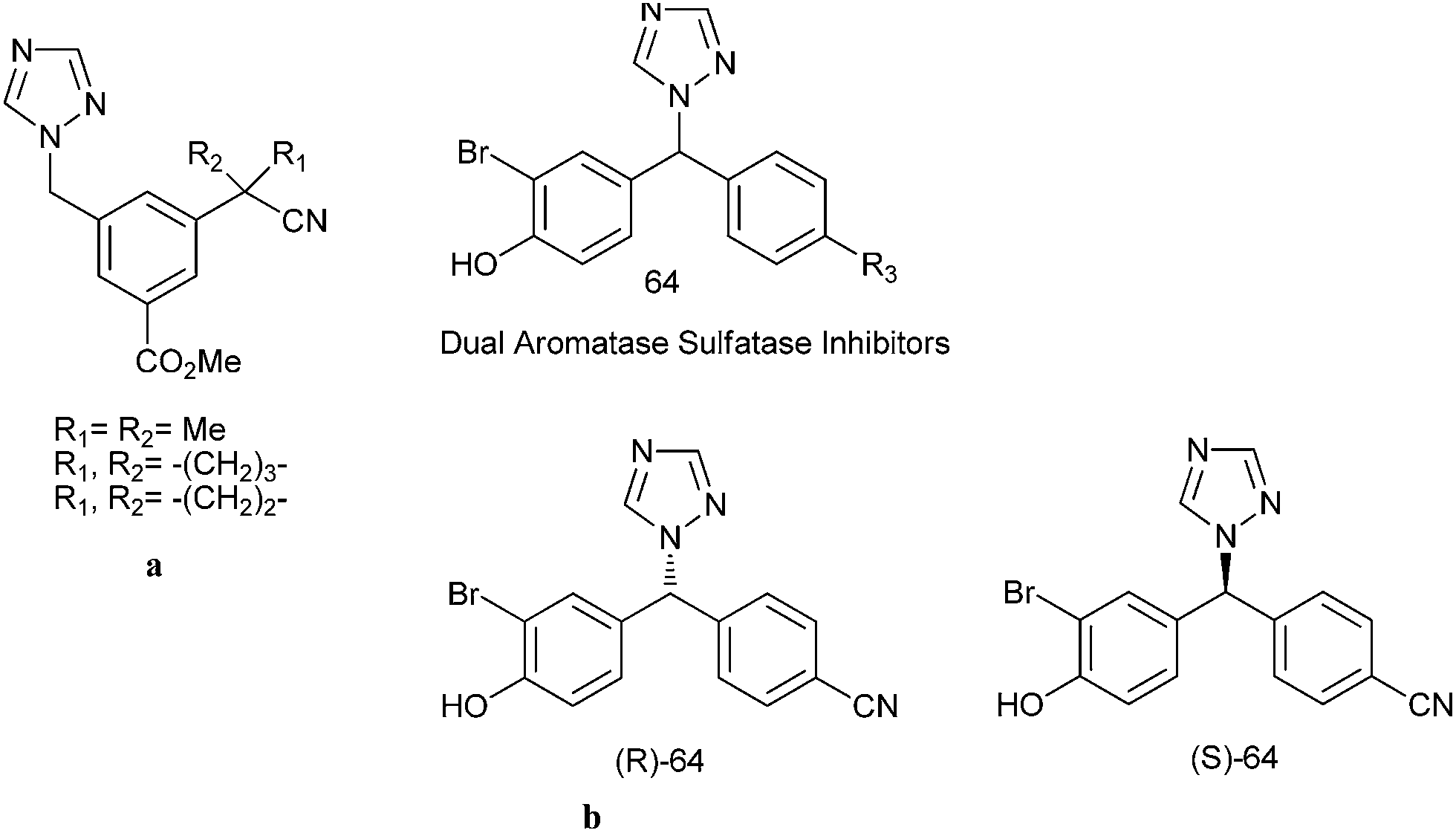 | ||
| Fig. 12 (a) Diarylmethane based dual aromatase sulfatase inhibitors and (b) chiral non-stroidal dual aromatase–sulfatase inhibitors based on the letrozole template. | ||
A. Carotti et al. in 2011 reported coumarin imidazolyl derivatives as aromatase inhibitors which had high selectivity over 17-α-hydroxylase/C17-20 Lyase.197 The authors noted that most of the compounds had IC50 value in the nanomolar range.
R. W. Hartmann et al. in 2013 reported pyridyl methyl substituted 1,2,5,6-tetrahydro pyrrolo [3,2,1-ij] quinolin-4-ones as dual inhibitors of aromatase/aldosterone synthase as anti breast cancer agents (Fig. 13).198 Interestingly the authors found that increase in the inhibition of one enzyme leads to the decrease in the inhibition of the other enzyme. However the optimal compound was 1,2,5,6-tetrahydro (3-toluyl) pyrrolo [3,2,1-ij] quinolin-4-one (65) with an IC50 values of 32 and 41 nm respectively for CYP19 and CYP11B2 enzymes.
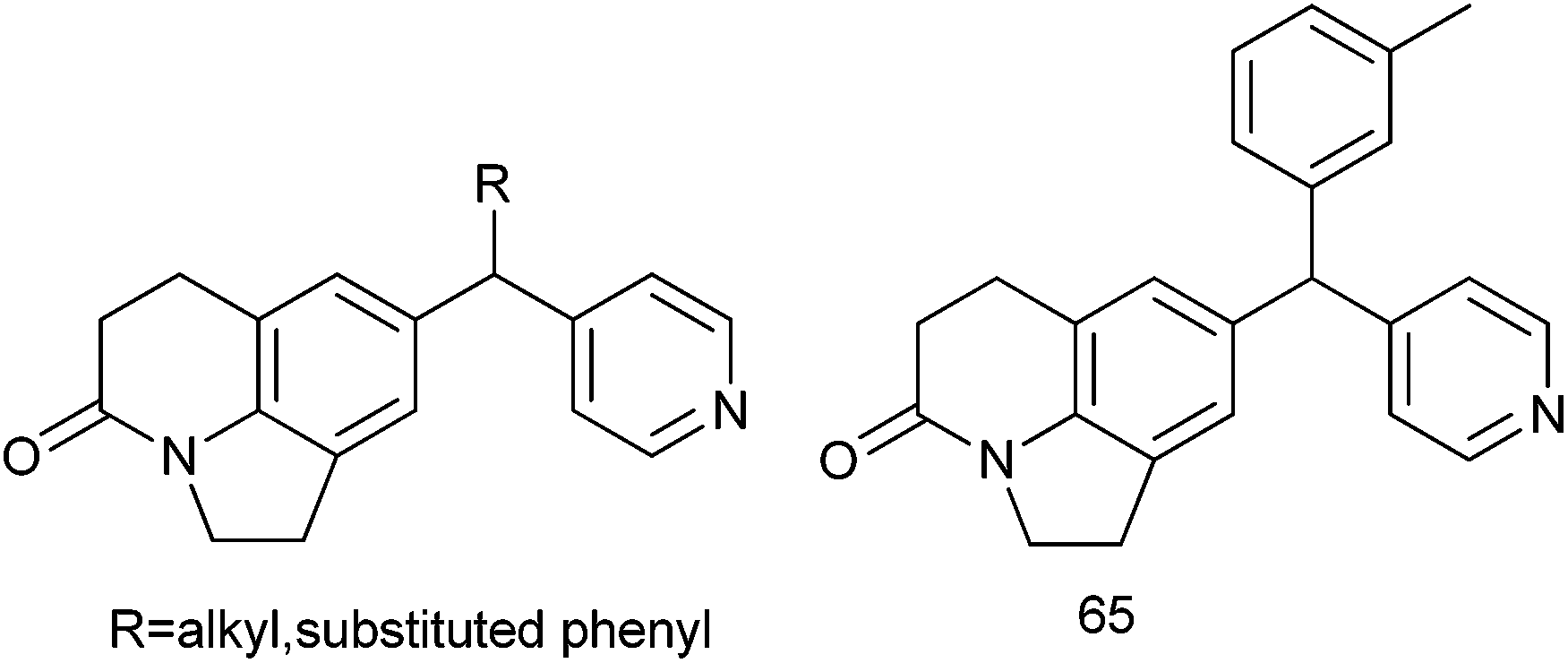 | ||
| Fig. 13 Tetrahydropyrroloquinolinones as dual aromatase/aldosterone synthase inhibitors. | ||
16.3 Diaryl and triarylmethane derivatives as antimalarials
K. Kirk et al. in 1998 reported clotrimazole inhibited the growth of the chloroquinine resistant Plasmodium falciparum in vitro.199 The IC50 value was 1 μM and for full inhibition less than 2.5 μM of Clotrimazole was required. In 2000 Virgio L. Lew et al. reported clotrimazole to inhibit five different strains of the Plasmodium falciparum species.200 The IC50 value for the compound for different stains were in the range 0.2–1.1 μM. U. Bandyopadhyay et al. in 2005 proposed the mechanism behind the antimalarial action of Clotrimazole.201a The authors proposed that Clotrimazole inhibited hemoperoxidase of Plasmodium falciparum and thereby induced oxidative stress in the parasite.G. Campiani et al. in 2007 reported triarylmethanes containing the imidazole ring based on the Clotrimazole scaffold as chloroquinine resistant antimalarial agents (Fig. 14).201b The compounds were selective for heme and did not show inhibitory activity against P 450 cyctochrome. The authors found that the protonatable lateral chain was requisite for maintaining the pharmacokinetic properties, the aromatic or heteroaromatic group was required to stabilize the trityl radical while the imidazole ring was required for binding to the heme molecule.
 | ||
| Fig. 14 Imidazole based clotrimazole analogs as antimalarials. | ||
Later in the year 2009 the authors hybridized 4-amino quinoline and clotrimazole to develop potent antimalarial agents. The molecules were extremely potent and were highly active towards chloroquinine resistant Plasmodium falciparum.
G. Panda et al. & U. Bandyopadhyay et al. in 2008 reported antiplasmodial activity of [(aryl) aryl sulfanylmethyl] pyridines (66) (Fig. 15).202,203 The authors found that the compounds inhibited hemozoin formation and formed complexes with free heme at a pH close to the pH of the food vacuole of the plasmodium parasite.
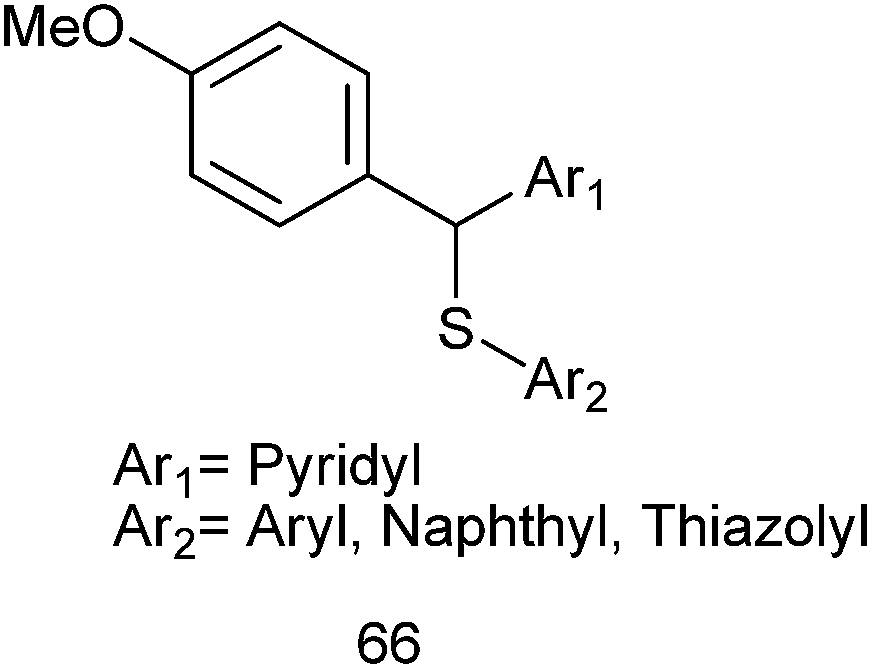 | ||
| Fig. 15 [(Aryl) aryl sulfanylmethyl] pyridines as antimalarial agents. | ||
Furthermore the authors observed that inhibition of the hemozoin formation led to the accumulation of free heme thereby creating an oxidative stress in the parasite, generating free hydroxyl radical and hence showing antimalarial activity.
G. Panda and coworkers in 2012 reported the synthesis and antimalarial activities of aryl aryl thioarenes.203 The aryl aryl thioarenes were found to interact with free heme and inhibit hemozoin formation.
16.4 Diarylmethanes & triarylmethanes as selective inhibitors of calcium activated potassium channel
H. Wulff et al. in 2000 designed Clotrimazole analogs as the selective inhibitors of calcium activated potassium channel IKCa1 (Fig. 16).204 The compounds selectively inhibited the IKCa1 channel without inhibiting the P450 cytochrome enzymes. The authors found that the active pharmacophore was 2-chlorophenyl diphenyl methane moiety substituted by unsubstituted polar π electron rich heterocycle pyrazole or tetrazole while for the inhibition of the P450 cytochrome enzymes a imidazole ring is a must. | ||
| Fig. 16 Clotrimazole analogs as selective calcium activated potassium channel inhibitors. | ||
16.5 Diarylmethanes and triarylmethanes as antiviral agents
J. S. Wai in 2000 reported diaryl methane as antiviral agents. The reported compounds acted as inhibitors of HIV-Integrase and inhibited viral cell replication.205 K. Sumoto et al. in 2002 reported new triarylmethane based 2,2′-dihydroxy bis phenols to be potent antiviral agents.206 The authors found the 4,4′-dihydroxy bis phenols to have higher potency than the 2,2′ compounds. Later on in 2003, the same group reported that the 4,4′-dihydroxy triphenyl methanes synthesized by using Bronstead or Lewis acid catalyst showed very high activity against the Herpes simplex virus type-1(Fig. 17a).207 The most potent compound was found to be 4,4′-dihydroxy triphenyl methane with an EC50 value of 0.79 μg ml−1. In 2010, they prepared heteroaryl substituted triarylmethanes as probable antiviral agents (Fig. 17b).207,208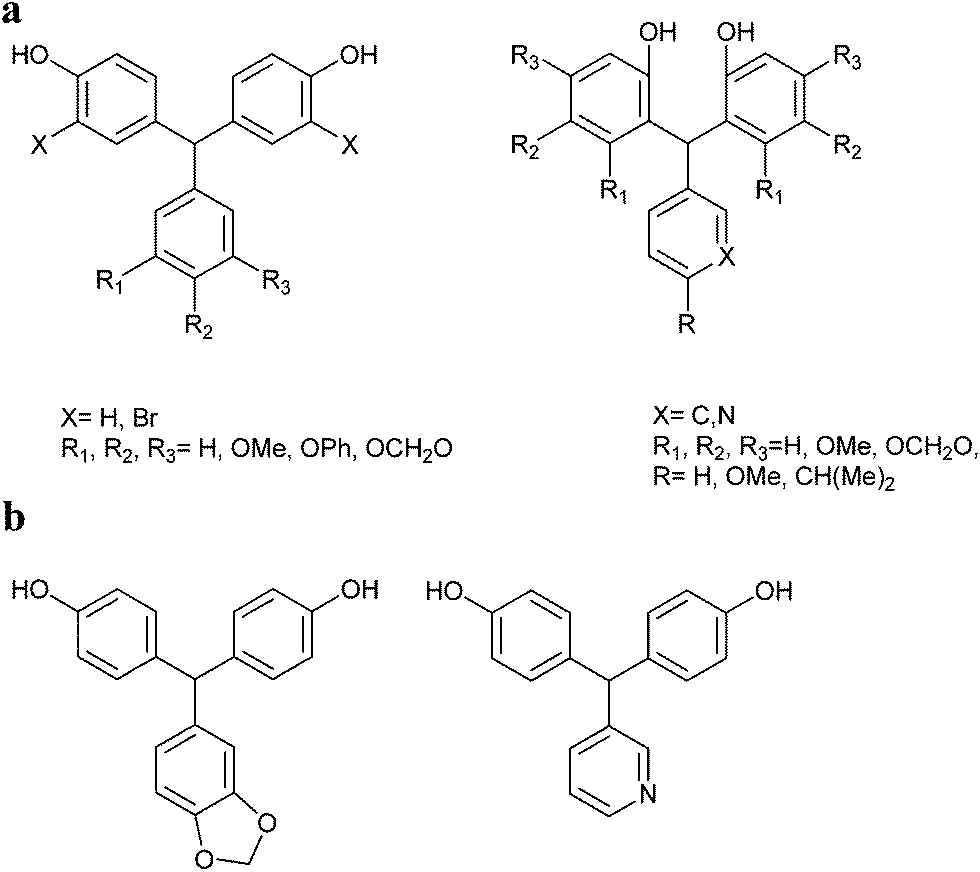 | ||
| Fig. 17 (a) Triarylmethanes as antiviral agents and (b) triarylmethanes and triheteroaryl methanes as antiviral agents. | ||
16.6 Diaryl and triarylmethanes as antitubercular agents
G. Panda and coworkers in 2004 reported diaryloxy methano phenanthrenes as novel antitubercular agents (Fig. 18a).209,210 The IC50 values were in the range 6.25–25 μg ml−1. In order to improve upon the bioactivities, in 2005 they reported 2-hydroxy aminoalkyl derivatives of diaryloxy methano phenanthrenes. The authors performed a 3D QSAR study to design more potent anti tubercular compounds.211 Later in 2007 the authors reported amino alkyl derivatives of diarylmethanes to be antitubercular agents. The compounds had the IC50 value in the range 6.25–25 μg ml−1. In 2007 the authors reported the antitubercular activity of diaryl methyl naphtholoxy ethylamines.212 The compounds were tested on the Mycobacterium Tuberculosis H37Rv. The MIC value was in the range 3.25–25 μg ml−1. Later, in 2008 the authors explored the antitubercular activity of the thiophene containing triarylmethanes (Fig. 18b).213 In order to further investigate new pharmacophores having antitubercular activity, the authors reported the synthesis and bioevaluation of aryl and heteroaryl groups with alkyl aminoethyl chain on aryl-chromenyl carbinol, bis and tris alkyl aminoethyl chains on aryl and triarylmethanes.214 However the authors observed that the compounds with bis and tris alkyl amino ethyl chains on the triaryl methane were proved to be inactive. Moreover among the heteroaryl containing alkyl aminoethyl chain containing triarylmethanes, the thiophene containing the triarylmethanes showed good activity. In 2012 R. Srivastava et al. induced simultaneously acute and persistent infection of M. fortium and used this model to screen a library of thiophene containing Triarylmethanes.215 The authors further identified two compounds that were bactericidal against M. fortium, non tubercular Mycobacterium tuberculosis.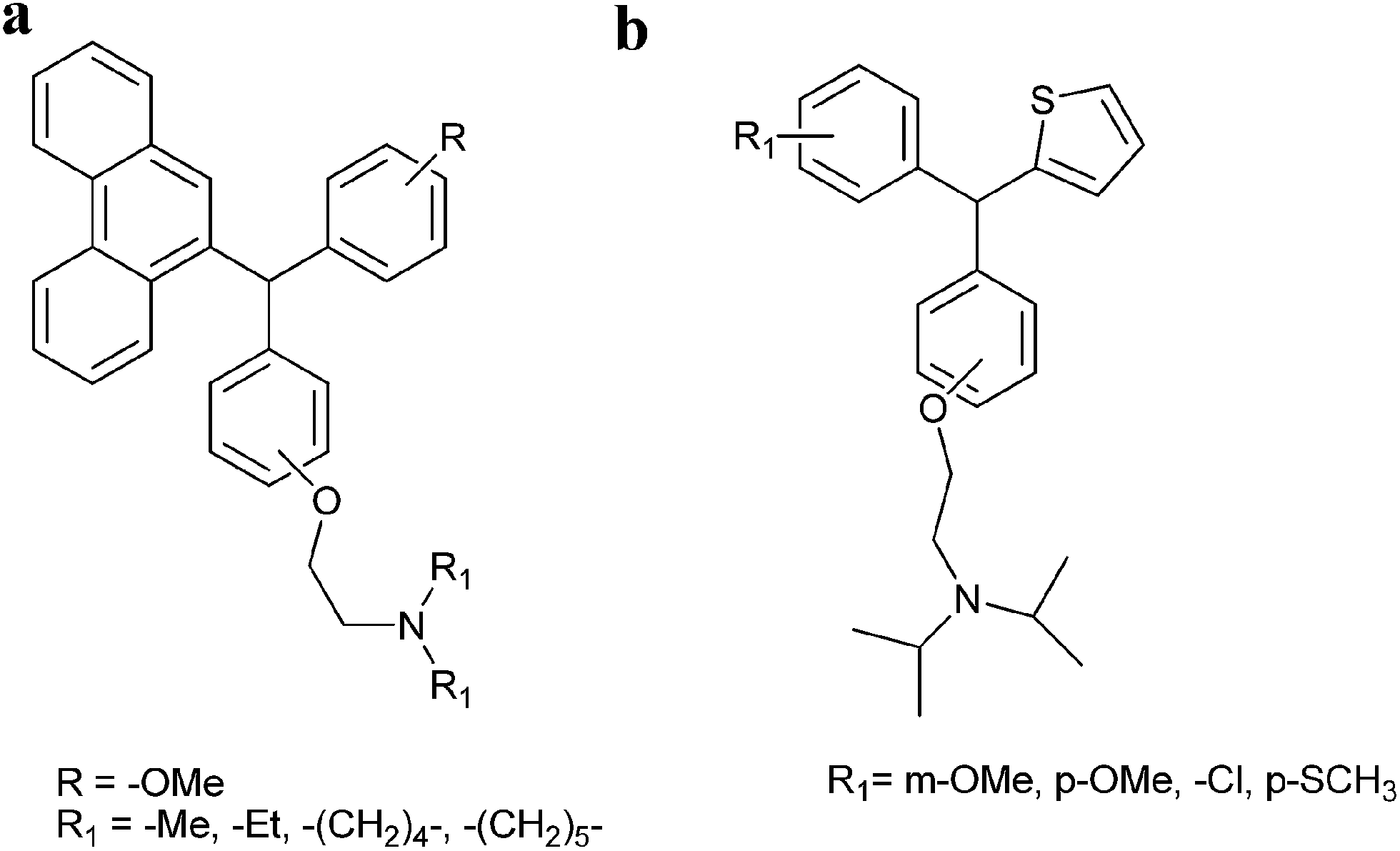 | ||
| Fig. 18 (a) Diaryloxy methano phenanthrene as antitubercular agents and (b) thiophene containing triarylmethanes as antitubercular agents. | ||
J. Chattopadhyaya et al. in 2009 reported the antitubercular activity of derivatives of 3-benzyl 6-bromo-2-methoxy-quinolines (67) and amides of 2-[(6-bromo-2-methoxy-quinolin-3-yl)-phenyl-methyl]malonic acid monomethyl ester (68) (Fig. 19).216 The authors observed that the 2-[(6-bromo-2-methoxy-quinolin-3-yl)-phenyl methyl] malonic acids were less potent than the 3-benzyl 6-bromo-2-methoxy-quinolines and among the 3-benzyl 6-bromo-2-methoxy-quinolines, the triarylmethanes containing the imidazolyl group, pyrazolyl group, 6-amino-chromen-2-one and the 1-(3-trifluoromethyl-phenyl)-piperazinyl group were most active (only most active compounds are drawn).
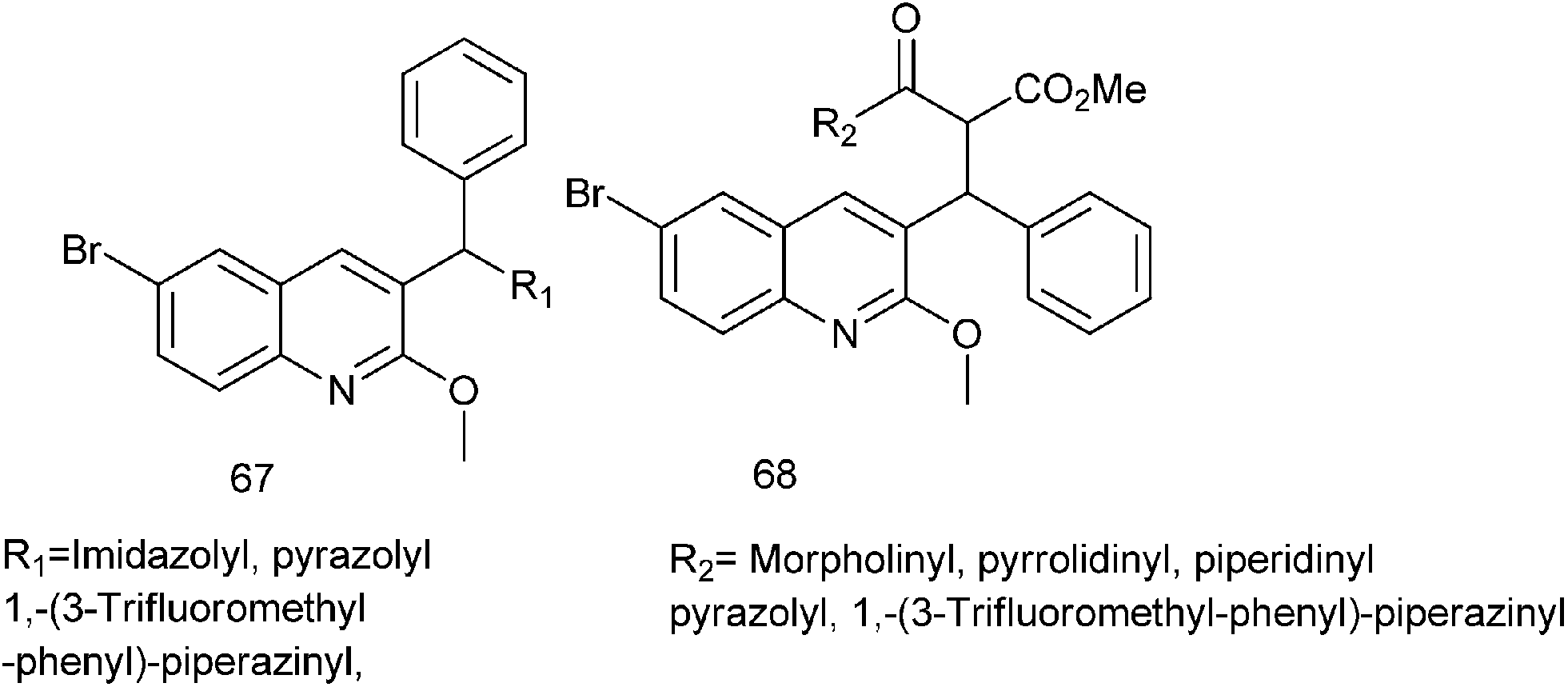 | ||
| Fig. 19 Quinoline derivatives as anti tubercular compounds. | ||
16.7 Miscellaneous bioactivities of compounds containing diaryl and triaryl methane core
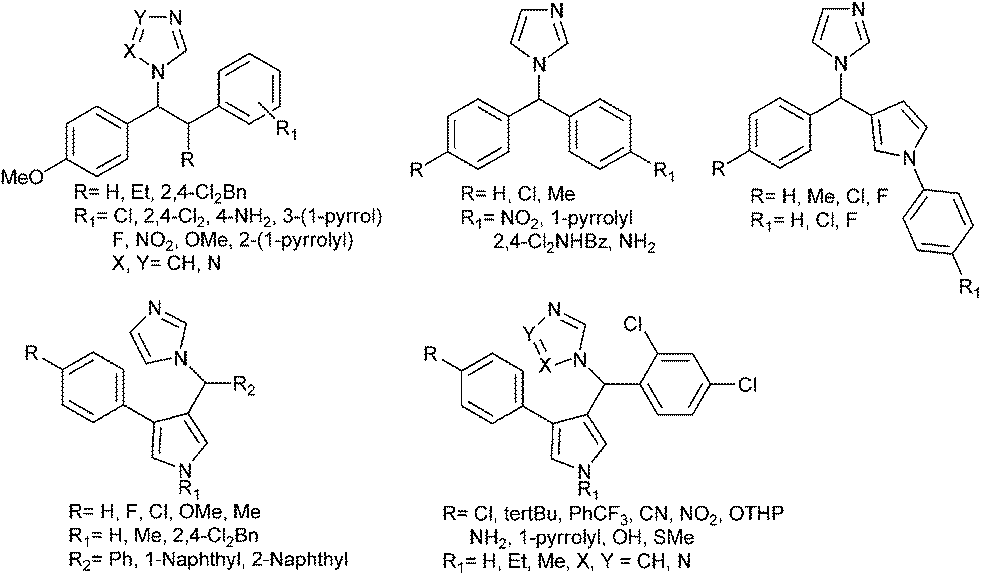 | ||
| Fig. 20 Imidazole based triarylmethanes as antifungal agents | ||
J. W. Stocker et al. in 2008 reported the synthesis and bioevaluation of triaryl methane derivatives as inhibitors of the Gardos channel in the sickle cell disease.218
R. W. Hartmann et al. in 2012 reported Imidazol-1-ylmethyl substituted tetrahydropyrrolo quinolin-4-ones as potent CYP11B1 inhibitors for the cushings syndrome (Fig. 21).219 Among the compounds tested the cyclopropyl compound was the most potent with IC50 value 2.2 nm.
 | ||
| Fig. 21 Imidazoyl-1-yl methyl tetrahydropyrrolo quinolin-4-ones as inhibitors of cushings syndrome. | ||
17. Summary & outlook
There is an increase in the number of pharmaceutically important molecules as well as molecules important from the material science perspective containing the diaryl or triaryl methane motif. Also various triarylmethane based architectures that are important in the supramolecular field are being reported. Steric hindrance between the ortho protons of the triarylmethanes leads to helical conformation of these classes of molecules.221 Light of suitable wavelength causes an interconversion between the helical conformations and this concept has been successfully applied in photochromatic devices. Diphenyl pyridyl ethylamines, a class of diarylmethanes, were also found to be potent inhibitors of CETP.222 Moreover due to antitumour activities of various triaryl methane derivatives; they are extensively used in the photodynamic therapy. Also trisubstituted methanes having imidazolyl ring and thiophene ring are reported to be potent analgesic agents.223 The present review discusses the recent developments in the chemistry and biology of the triarylmethane derivatives that came up in the last fifteen years (1995–2013) and hence can be an impetus for the further expansion of these family of molecules towards both material science as well as small molecules as therapeutic agents. The great utility of this class of molecules has created a high demand for the synthetic strategies for the synthesis of this class of compounds and has thus led to various developments in synthetic organic chemistry.Acknowledgements
Sankalan Mondal highly acknowledges Council of Scientific and Industrial Research for Senior Research Fellowship. We would like to thank the referees for their valuable comments which helped us a great deal to improve the quality of the paper. This is CDRI communication no 8712. We thank CSIR network project BSC 0104 for funding.References
- (a) F. Schmidt, R. T. Stemmler, J. Rudolph and C. Bolm, Chem. Soc. Rev., 2006, 35, 454 CAS; (b) M. W. Paixao, A. L. Braga and D. S. Ludtke, J. Braz. Chem. Soc., 2008, 19(5), 813 CrossRef CAS PubMed.
- (a) M. Braun, Angew. Chem., Int. Ed., 1996, 35(5), 519 CrossRef CAS; (b) G. P. Cozzi, F. Berfatti and L. Zoli, Angew. Chem., Int. Ed., 2009, 48, 1313 CrossRef PubMed; (c) L. Zhang, L. Cui, X. Li, J. Li, S. Luo and P. J. Chen, Chem.–Eur. J., 2010, 16, 2045 CrossRef CAS PubMed; (d) G. Bergonzini, S. Vera and P. Melchiorre, Angew. Chem., Int. Ed., 2010, 49, 9685 CrossRef CAS PubMed; (e) E. Emer, R. Sinisi, G. M. Capdevila, D. Petruzziello, F. Vincentiis and G. P. Cozzi, Eur. J. Org. Chem., 2011, 647 CrossRef CAS; (f) M. Shibata, M. Ikeda, K. Motoyama, Y. Miyake and Y. Nishibayashi, Chem. Commun., 2012, 48, 9528 RSC; (g) J. Xiao, K. Zhao and P. T. Loh, Chem. Commun., 2012, 48, 3548 RSC; (h) L. Song, X. Q. Guo and C. X. Li, Angew. Chem., Int. Ed., 2012, 51, 1899 CrossRef CAS PubMed; (i) M. Trifonidou and G. C. Kokotos, Eur. J. Org. Chem., 2012, 1563 CrossRef CAS; (j) B. Xu, L. Z. Guo, Y. W. Jin, P. Z. Wang, G. Y. Peng and X. Q. Guo, Angew. Chem., Int. Ed., 2012, 51, 1059 CrossRef CAS PubMed.
- (a) K. Nakata, Y. Onda, K. Ono and I. Shiina, Tetrahedron Lett., 2010, 51, 5666 CrossRef CAS PubMed; (b) I. Shiina, K. Nakata, K. Ono, Y. Onda and M. Itagaki, J. Am. Chem. Soc., 2010, 132(33), 11629 CrossRef CAS PubMed.
- (a) S. K. Manna, M. K. Parai and G. Panda, Tetrahedron Lett., 2011, 52, 5951 CrossRef CAS PubMed; (b) S. K. Das, R. Singh and G. Panda, Eur. J. Org. Chem., 2009, 4757 CrossRef CAS.
- L. Boymond, M. Rottlander, G. Cahiez and P. Knochel, Angew. Chem., Int. Ed., 1998, 37, 1701 CrossRef CAS.
- I. Sapountzis and P. Knochel, Angew. Chem., Int. Ed., 2002, 41(9), 1610 CrossRef CAS.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS PubMed.
- F. Kopp, A. Krasovskiy and P. Knochel, Chem. Commun., 2004, 2288 RSC.
- A. Inoue, K. Kitagawa, H. Shinokubo and K. Oshima, J. Org. Chem., 2001, 66(12), 4333 CrossRef CAS PubMed.
- M. Durandetti, J. Perichon and J. Y. Nedelec, Tetrahedron Lett., 1999, 40, 9009 CrossRef CAS.
- M. Durandetti, J. Y. Nedelec and J. Perichon, Org. Lett., 2001, 3(13), 2073 CrossRef CAS PubMed.
- K. K. Majumdar and H. C. Cheng, Org. Lett., 2000, 2(15), 2295 CrossRef CAS PubMed.
- G. Takahashi, E. Shirakawa, T. Tsuchimoto and Y. Kawakami, Chem. Commun., 2005, 1459 RSC.
- L. Zhou, X. Du, R. He, Z. Ci and M. Bao, Tetrahedron Lett., 2009, 50, 406 CrossRef CAS PubMed.
- J. Bouffard and K. Itami, Org. Lett., 2009, 11(19), 4410 CrossRef CAS PubMed.
- C. H. Xing and Q. S. Hu, Tetrahedron Lett., 2010, 51, 924 CrossRef CAS PubMed.
- C. J. Li, J. Am. Chem. Soc., 2000, 122, 9538 CrossRef CAS.
- S. U. K. Son, S. B. Kim, J. A. Reingold, G. B. Carpenter and D. A. Sweigart, J. Am. Chem. Soc., 2005, 127, 12238 CrossRef CAS PubMed.
- J. R. White, G. J. Price, P. K. Plucinski and C. G. Frost, Tetrahedron Lett., 2009, 50, 7365 CrossRef CAS PubMed.
- T. Yamamoto, T. Ohta and T. Ito, Org. Lett., 2005, 7(19), 4153 CrossRef CAS PubMed.
- K. Suzuki, T. Arao, S. Ishii, Y. Maeda, K. Kondo and T. Aoyama, Tetrahedron Lett., 2006, 47, 5789 CrossRef CAS PubMed.
- P. He, Y. Lu, C. G. Dong and Q. S. Hu, Org. Lett., 2007, 9(2), 343 CrossRef CAS PubMed.
- P. He, Y. Lu and Q. S. Hu, Tetrahedron Lett., 2007, 48, 5283 CrossRef CAS PubMed.
- C. Qin, H. Wu, J. Cheng, X. Chen, M. Liu, W. Zhang, W. Su and J. Ding, J. Org. Chem., 2007, 72, 4102 CrossRef CAS PubMed.
- A. Yu, B. Cheng, Y. Wu, J. Li and K. Wei, Tetrahedron Lett., 2008, 49, 5405 CrossRef CAS PubMed.
- M. Kuriyama, R. Shimazawa and R. Shirai, J. Org. Chem., 2008, 73, 1597 CrossRef CAS PubMed.
- M. Kuriyama, R. Shimazawa, T. Enomoto and R. Shirai, J. Org. Chem., 2008, 73, 6939 CrossRef CAS PubMed.
- M. Kuriyama, N. Ishiyama, R. Shimazawa and O. Onomura, Tetrahedron, 2010, 66, 6814 CrossRef CAS PubMed.
- Y. Luo and J. Wu, Chem. Commun., 2010, 46, 3785 RSC.
- Y. X. Liao, H. C. Xing, P. He and Q. S. Hu, Org. Lett., 2008, 10(12), 2509 CrossRef CAS PubMed.
- H. Zheng, Q. Zhang, J. Chen, M. Liu, S. Cheng, J. Ding, H. Wu and W. Su, J. Org. Chem., 2009, 74, 943 CrossRef CAS PubMed.
- Y. X. Liao and Q. S. Hu, J. Org. Chem., 2011, 76, 7602 CrossRef CAS PubMed.
- T. Zou, S. S. Pi and J. H. Li, Org. Lett., 2009, 11(2), 453 CrossRef CAS PubMed.
- X. Jia, L. Fang, A. Lin, Y. Pan and C. Zhu, Synlett, 2009, 495 CAS.
- A. Jasat and J. C. Sherman, Chem. Rev., 1999, 99(4), 931 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS PubMed.
- (a) M. M. Conn and J. Rebek, Jr, Chem. Rev., 1997, 97, 1647 CrossRef CAS PubMed; (b) M. Alami, S. Messaoudi, A. Hamze, O. Provot, J. D. Brion, J. M. Liu, J. Bignon and J. Bakala, Internation Patent, Patent Number WO/2009/147217A1, December 10 2009; (c) A. V. Chetsov, M. Aoyagi, A. Aleshin, E. C. W.Yu, T. Gilliland, D. Zhai, A. A. Bobkov, J. C. Reed, R. C. Liddington and R. Abagya, J. Med. Chem., 2010, 53, 3899 CrossRef PubMed; (d) Q. Z. Hu, L. N. Yin, C. Jagusch, U. E. Hille and R. W. Hartmann, J. Med. Chem., 2010, 53, 5049 CrossRef CAS PubMed; (e) S. Messaoudi, A. Hamze, O. Provot, B. Treguier, J. R. De Losada, J. Bignon, J. M. Liu, J. WdzieczakBakala, S. Thoret, J. Dubois, J. D. Brion and M. Alami, ChemMedChem, 2011, 6, 488 CrossRef CAS PubMed; (f) T. P. Pathak, K. M. Gligorich, B. E. Welm and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 7870 CrossRef CAS PubMed; (g) C. Canel, R. M. Moraes, F. E. Dyan and D. Ferreira, Phytochemistry, 2000, 54, 115 CrossRef CAS; (h) C. Tsutsui, Y. Yamada, M. Ando, D. Toyama, J. L. Wu, L. Wang, S. Taketani and T. Kataoka, Bioorg. Med. Chem. Lett., 2009, 19, 4084 CrossRef CAS PubMed; (i) P. V. Kerrebroeck, K. Kreder, U. Jonas, N. Zinner and A. Wein, Urology, 2001, 57(3), 414 CrossRef; (j) L. Gennari, D. Merlotti, G. Martini and R. Nuti, Expert Opin. Invest. Drugs, 2006, 15, 1091 CrossRef CAS PubMed.
- P. E. Georghiou, J. Org. Chem., 1995, 60, 7284 CrossRef CAS.
- S. Fukuzawa, T. Tsuchimoto and T. Hiyama, J. Org. Chem., 1997, 62, 151 CrossRef CAS PubMed.
- T. Tsuchimoto, K. Tobita, T. Hiyama and S. Fukuzawa, J. Org. Chem., 1997, 62, 6997 CrossRef CAS.
- H. B. Sun, R. Hua and Y. Yin, Tetrahedron Lett., 2006, 47, 2291 CrossRef CAS PubMed.
- H. B. Sun, B. Li, S. Chen, J. Li and R. Hua, Tetrahedron, 2007, 63, 10185 CrossRef CAS PubMed.
- V. Y. Sosnovskikh and R. A. Irgashev, Tetrahedron Lett., 2007, 48, 7436 CrossRef CAS PubMed.
- M. M. Rudolf and B. Myrboh, Indian Chem. Eng., Sect. B, 2009, 48, 146 Search PubMed.
- O. Mendoza, G. Rossey and L. Ghosez, Tetrahedron Lett., 2011, 52, 2235 CrossRef CAS PubMed.
- J. Gao, J. Q. Wang, Q. W. Song and L. N. He, Green Chem., 2011, 13, 1182 RSC.
- K. Tangdenpaisal, W. Phakhodee, S. Ruchirawat and P. Ploypradith, Tetrahedron, 2013, 69, 933 CrossRef CAS PubMed.
- J. Barluenga, M. T. Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494 CrossRef CAS PubMed.
- M. M. Khodaei and E. Nazari, Tetrahedron Lett., 2012, 53, 5131 CrossRef CAS PubMed.
- Y. Y. Ku, R. R. Patel and D. P. Sawick, Tetrahedron Lett., 1996, 37(12), 1949 CrossRef CAS.
- J. Srogl, G. D. Allred and L. S. Liebeskind, J. Am. Chem. Soc., 1997, 119, 12376 CrossRef CAS.
- S. Zhang, D. Marshall and L. S. Liebeskind, J. Org. Chem., 1999, 64, 2796 CrossRef CAS PubMed.
- S. Chowdhury and P. E. Georghiou, Tetrahedron Lett., 1999, 40, 7599 CrossRef CAS.
- I. Pérez, J. P. Sestelo and L. A. Sarandeses, J. Am. Chem. Soc., 2001, 123, 4155 CrossRef PubMed.
- L. Botella and C. Nájera, Angew. Chem., Int. Ed., 2002, 41, 179 CrossRef CAS.
- S. Langle, M. Abarbri and A. Duchêne, Tetrahedron Lett., 2003, 44, 9255 CrossRef CAS PubMed.
- S. M. Nobre, S. I. Wolke, R. G. da Rosa and A. L. Monteiro, Tetrahedron Lett., 2004, 45, 6527 CrossRef CAS PubMed.
- B. P. Bandgar, S. V. Bettigeri and J. Phopase, Tetrahedron Lett., 2004, 45, 6959 CrossRef CAS PubMed.
- S. M. Nobre and A. L. Monteiro, Tetrahedron Lett., 2004, 45, 8225 CrossRef CAS PubMed.
- R. Kuwano and M. Yokogi, Org. Lett., 2005, 7(5), 945 CrossRef CAS PubMed.
- R. Kuwano and M. Yokogi, Chem. Commun., 2005, 5899 RSC.
- M. McLaughlin, Org. Lett., 2005, 7(22), 4875 CrossRef CAS PubMed.
- R. Singh, M. S. Viciu, N. Kramareva, O. Navarro and S. P. Nolan, Org. Lett., 2005, 7(9), 1829 CrossRef CAS PubMed.
- C. Song, Y. Ma, Q. Chai, C. Ma, W. Jiang and M. B. Andrus, Tetrahedron, 2005, 61, 7438 CrossRef CAS PubMed.
- Y. Zhang, M. T. Feng and J. M. Lu, Org. Biomol. Chem., 2013, 11, 2266 CAS.
- A. Cwik, Z. Hell and F. Figueras, Org. Biomol. Chem., 2005, 3, 4307 CAS.
- C. M. Crawforth, I. J. S. Fairlamb, A. R. Kapdi, J. L. Serrano, R. J. K. Taylor and G. Sanchez, Adv. Synth. Catal., 2006, 348, 405 CrossRef CAS.
- G. A. Molander and M. D. Elia, J. Org. Chem., 2006, 71, 9198 CrossRef CAS PubMed.
- C. M. Crawforth, S. Burling, I. J. S. Fairlamb, A. R. Kapdi, R. J. K. Taylor and A. C. Whitwood, Tetrahedron, 2005, 61, 9736 CrossRef CAS PubMed.
- J. M. Burns, I. J. S. Fairlamb, R. A. Kapdi, P. Sehnal and R. J. K. Taylor, Org. Lett., 2007, 9(26), 5397 CrossRef PubMed.
- D. Srimani and A. Sarkar, Tetrahedron Lett., 2008, 49, 6304 CrossRef CAS PubMed.
- J. R. Schmink and M. T. Tudge, Tetrahedron Lett., 2013, 54, 15 CrossRef CAS PubMed.
- B. Inés, I. Moreno, R. S. Martin and E. Domínguez, J. Org. Chem., 2008, 73, 8448 CrossRef PubMed.
- T. Z. Nichele and A. L. Monteiro, Tetrahedron Lett., 2007, 48, 7472 CrossRef CAS PubMed.
- R. Ghosh and A. Sarkar, J. Org. Chem., 2010, 75, 8283 CrossRef CAS PubMed.
- D. Srimani, A. Bej and A. Sarkar, J. Org. Chem., 2010, 75, 4296 CrossRef CAS PubMed.
- S. Y. Park, M. Kang, J. E. Yie, J. M. Kim and I. M. Lee, Tetrahedron Lett., 2005, 46, 2849 CrossRef CAS PubMed.
- C. C. Kofink and P. Knochel, Org. Lett., 2006, 8(18), 4121 CrossRef CAS PubMed.
- M. Amatore and C. Gosmini, Chem. Commun., 2008, 5019 RSC.
- R. B. Bedford, M. Huwe and M. C. Wilkinson, Chem. Commun., 2009, 600 RSC.
- C. Duplais, A. Krasovskiy, A. Wattenberg and B. H. Lipshutz, Chem. Commun., 2010, 46, 562 RSC.
- E. G. Dennis, D. W. Jeffery, M. V. Perkins and P. A. Smith, Tetrahedron, 2011, 67, 2125 CrossRef CAS PubMed.
- M. Rottländer and P. Knochel, Tetrahedron Lett., 1997, 38(10), 1749 CrossRef.
- S. L. Hargreaves, B. L. Pilkington, S. E. Russell and P. A. Worthington, Tetrahedron Lett., 2000, 41, 1653 CrossRef CAS.
- A. G. Martínez, J. O. Barcina, M. d. R. C. Heras and A. d. F. Cerezo, Org. Lett., 2000, 2(10), 1377 CrossRef PubMed.
- S. H. Kim and R. D. Rieke, J. Org. Chem., 2000, 65, 2322 CrossRef CAS.
- C. Vanier, F. Lorgé, A. Wagner and C. Mioskowski, Angew. Chem., Int. Ed., 2000, 39, 1679 CrossRef CAS.
- K. Itami, M. Mineno, T. Kamei and J. Yoshida, Org. Lett., 2002, 4(21), 3635 CrossRef CAS PubMed.
- A. Flaherty, A. Trunkfield and W. Barton, Org. Lett., 2005, 7(22), 4975 CrossRef CAS PubMed.
- K. Endo, T. Ishioka, T. Ohkubo and T. Shibata, J. Org. Chem., 2012, 77, 7223 CrossRef CAS PubMed.
- M. A. Schade, A. Metzger, S. Hug and P. Knochel, Chem. Commun., 2008, 3046 RSC.
- L. S. Chupak, J. P. Wolkowski and Y. A. Chantigny, J. Org. Chem., 2009, 74, 1388 CrossRef CAS PubMed.
- S. Fan, C. Y. He and X. Zhang, Chem. Commun., 2010, 46, 4926 RSC.
- R. Shang, Z. Huang, L. Chu, Y. Fu and L. Liu, Org. Lett., 2011, 13(16), 4240 CrossRef CAS PubMed.
- J. R. Schmink and N. E. Leadbeater, Org. Lett., 2009, 11(12), 2575 CrossRef CAS PubMed.
- F. Zhao, Q. Tan, F. Xiao, S. Zhang and G. J. Deng, Org. Lett., 2013, 15(7), 1520 CrossRef CAS PubMed.
- B. Hatano and H. Tagaya, Tetrahedron Lett., 2003, 44, 6331 CrossRef CAS.
- L. Hermite, G. A. Nathalie, O. Provot, J. F. Peyrat, M. Alami and J. D. Brion, Tetrahedron, 2006, 62, 11994 CrossRef PubMed.
- M. Jereb and D. Vražič, Org. Biomol. Chem., 2013, 11, 1978 CAS.
- B. Q. Wang, S. K. Xiang, Z. P. Sun, B. T. Guan, P. Hu, K. Q. Zhao and Z. J. Shi, Tetrahedron Lett., 2008, 49, 4310 CrossRef CAS PubMed.
- G. A. Kraus and D. Chaudhary, Tetrahedron Lett., 2012, 53, 7072 CrossRef CAS PubMed.
- S. Podder, J. Choudhury and S. Roy, J. Org. Chem., 2007, 72, 3129 CrossRef CAS PubMed.
- S. Roy, S. Podder and J. Choudhury, J. Chem. Sci., 2008, 120(5), 429 CrossRef CAS PubMed.
- A. Martins and M. Lautens, Org. Lett., 2008, 10(21), 5095 CrossRef CAS PubMed.
- A. Kumar, M. Kumar and M. K. Gupta, Tetrahedron Lett., 2009, 50, 7024 CrossRef CAS PubMed.
- T. P. Pathak, J. G. Osiak, R. M. Vaden, B. E. Welm and M. S. Sigman, Tetrahedron, 2012, 68, 5203 CrossRef CAS PubMed.
- N. Sakai, K. Kawana, R. Ikeda, Y. Nakaike and T. Konakahara, Eur. J. Org. Chem., 2011, 3178 CrossRef CAS.
- J. B. Gianino and B. L. Ashfeld, J. Am. Chem. Soc., 2012, 134, 18217 CrossRef CAS PubMed.
- (a) C. D. Mason and F. F. Nord, J. Org. Chem., 1951, 16, 722 CrossRef CAS; (b) C. D. Mason and F. F. Nord, J. Org. Chem., 1952, 17, 778 CrossRef CAS; (c) C. A. Gindre, C. G. Screttas, C. Fiorini, C. Schmidt and J. M. Nunzi, Tetrahedron Lett., 1999, 40, 7413 CrossRef; (d) S. Sengupta and P. Purkayastha, Org. Biomol. Chem., 2003, 1, 436 RSC; (e) D. A. Stetsenko, E. N. Lubyako, V. K. Potapov, T. L. Azhikina and E. D. Sverdiov, Tetrahedron Lett., 1996, 37, 3571 CrossRef CAS; (f) M. Baptista and G. Indig, Chem. Commun., 1997, 1791 RSC; (g) R. Schirrmaker, W. Hamkens, M. Piel, U. Schmitt, H. Luddens, C. Hiemke and F. Rosch, J. Labelled Compd. Radiopharm., 2001, 44, 627 CrossRef; (h) M. Ota, S. Otani and K. Kobayashi, Chem. Lett., 1989, 1175 CrossRef CAS; (i) L. Strekowski, H. Lee, S. Y. Lin, A. Czarny and D. V. Deerveer, J. Org. Chem., 2000, 65, 7703 CrossRef CAS; (j) A. Noack, A. Schroder and H. Hartmann, Angew. Chem., Int. Ed., 2001, 40, 3008 CrossRef CAS; (k) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 3rd edn, 1999 CrossRef; (l) M. J. Sabacky, S. M. Johnson, J. C. Martin and I. C. Paul, J. Am. Chem. Soc., 1969, 91(26), 7542 CrossRef CAS; (m) H. Kurata, H. Nakaminami, K. Matsumoto, K. Takeshi and M. Oda, Chem. Commun., 2001, 529 RSC; (n) S. E. Denmark and C. T. Chen, Tetrahedron Lett., 1994, 35, 4327 CrossRef CAS; (o) D. Magdziak, L. H. Pettus and T. R. R. Pettus, Org. Lett., 2001, 3(4), 557 CrossRef CAS PubMed; (p) C. T. Chen and S. D. Dao, J. Org. Chem., 1999, 64, 1090 CrossRef CAS.
- (a) D. F. Duxbury, Chem. Rev., 1993, 93, 381 CrossRef CAS; (b) M. S. Shchepinov and V. A. Korshun, Chem. Soc. Rev., 2003, 32, 170 RSC; (c) V. Nair, S. Thomas, S. C. Mathew and K. G. Abhilash, Tetrahedron, 2006, 62, 6731 CrossRef CAS PubMed.
- (a) J. Esquivias, R. G. Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 629 CrossRef CAS PubMed; (b) I. Alonso, J. Esquivias, R. G. Arrayás and J. C. Carretero, J. Org. Chem., 2008, 73, 6401 CrossRef CAS PubMed; (c) Z. Wang, X. Sun and J. Wu, Tetrahedron, 2008, 64, 5013 CrossRef CAS PubMed.
- S. Shirakawa and S. Kobayashi, Org. Lett., 2006, 8(21), 4939 CrossRef CAS PubMed.
- Q. L. He, F. L. Sun, X. J. Zheng and S. L. You, Synlett, 2009, 7, 1111 Search PubMed.
- M. Wilsdorf, D. Leichnitz and H. U. Reissig, Org. Lett., 2013, 15(10), 2494 CrossRef CAS PubMed.
- S. Kumar, V. Malik, N. Kaur and K. Kaur, Tetrahedron Lett., 2006, 47, 8483 CrossRef CAS PubMed.
- X. Wang, Y. Wang, D. M. Du and J. Xu, J. Mol. Catal. A: Chem., 2006, 255, 31 CrossRef CAS PubMed.
- S. Podder, J. Choudhury, U. K. Roy and S. Roy, J. Org. Chem., 2007, 72, 3100 CrossRef CAS PubMed.
- P. N. Chatterjee, A. K. Maity, S. S. Mohapatra and S. Roy, Tetrahedron, 2013, 69, 2816 CrossRef CAS PubMed.
- M. Periasamy, N. Kishorebabu and K. N. Jayakumar, Tetrahedron Lett., 2007, 48, 1955 CrossRef CAS PubMed.
- J. S. Yadav, D. C. Bhunia, K. V. Krishna and P. Srihar, Tetrahedron Lett., 2007, 48, 8306 CrossRef CAS PubMed.
- M. Kodomari, M. Nagamatsu, M. Akaike and T. Aoyama, Tetrahedron Lett., 2008, 49, 2537 CrossRef CAS PubMed.
- C. R. Liu, M. B. Li, C. F. Yang and S. K. Tian, Chem. Commun., 2008, 1249 RSC.
- S. Genovese, F. Epifano, C. Pelucchini and M. Curini, Eur. J. Org. Chem., 2009, 1132 CrossRef CAS.
- C. S. Reddy, A. Nagaraj, A. Srinivas and G. P. Reddy, Indian Chem. Eng., Sect. B, 2009, 48, 248 Search PubMed.
- M. A. Pasha and S. Nagashree, Int. J. Res. Chem. Envior., 2013, 3(2), 54 CAS.
- P. Thirupathi and S. S. Kim, J. Org. Chem., 2009, 74, 7755 CrossRef CAS PubMed.
- P. Thirupathi and S. S. Kim, J. Org. Chem., 2010, 75, 5240 CrossRef CAS PubMed.
- G. Panda, J. K. Mishra, Shagufta, T. C. Dinadayalane, G. N. Sastry and D. S. Negi, Indian Chem. Eng., Sect. B, 2006, 45, 276 Search PubMed.
- P. Singh, S. K. Dinda, Shagufta and G. Panda, RSC Adv., 2013, 3, 12100 RSC.
- G. K. Surya Prakash, C. Panja, A. Shakhmin, E. Shah, T. Mathew and G. A. Olah, J. Org. Chem., 2009, 74, 8659 CrossRef PubMed.
- (a) T. Shinsuke, F. Shibahara, T. Maruyama and T. Murai, Chem. Commun., 2009, 7009 Search PubMed; (b) J. Jaratjaroonphong, S. Sathalalai, P. Techasauvapak and V. Reutrakul, Tetrahedron Lett., 2009, 50, 6012 CrossRef CAS PubMed.
- P. Thirupathi and S. S. Kim, Tetrahedron, 2010, 66, 2995 CrossRef CAS PubMed.
- Y. Leng, F. Chen, L. Zuo and W. Duan, Tetrahedron Lett., 2010, 51, 2370 CrossRef CAS PubMed.
- S. K. Das, Shagufta and G. Panda, Tetrahedron Lett., 2005, 46, 3097 CrossRef CAS PubMed.
- G. K. SuryaPrakash, G. Fogassy and G. A. Olah, Catal. Lett., 2010, 138, 155 CrossRef.
- A. S. Gothelf, T. Hansen and K. A. Jørgensen, J. Chem. Soc., Perkin Trans. 1, 2001, 854 RSC.
- G. R. Bardajee, Beilstein J. Org. Chem., 2011, 7, 135 CrossRef PubMed.
- I. M. Baltork, M. Moghadam, S. Tangestaninejad, V. Mirkhani, M. K. Abbasabadi and H. R. Khavasi, Eur. J. Org. Chem., 2011, 1357 CrossRef.
- H. Hikawa, H. Suzuki, Y. Yokoyama and I. Azumaya, J. Org. Chem., 2013, 78, 6714 CrossRef CAS PubMed.
- T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9(12), 2373 CrossRef CAS PubMed.
- J. Y. Yu and R. Kuwano, Org. Lett., 2008, 10(5), 973 CrossRef CAS PubMed.
- G. I. McGrew, J. Temaismithi, P. J. Carroll and P. J. Walsh, Angew. Chem., Int. Ed., 2010, 49, 5541 CrossRef CAS PubMed.
- J. Zhang, A. Bellomo, A. D. Creamer, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2012, 134, 13765 CrossRef CAS PubMed.
- A. Bellomo, J. Zhang, N. Trongsiriwat and P. J. Walsh, Chem. Sci., 2013, 4, 849 RSC.
- G. Song, Y. Su, X. Gong, K. Han and X. Li, Org. Lett., 2011, 13(8), 1968 CrossRef CAS PubMed.
- D. G. Yu, X. Wang, R. Y. Zhu, S. Luo, X. B. Zhang, B. Q. Wang, L. Wang and Z. J. Shi, J. Am. Chem. Soc., 2012, 134, 14638 CrossRef CAS PubMed.
- Y. Xia, F. Hu, Z. Liu, P. Qu, R. Ge, C. Ma, Y. Zhang and J. Wang, Org. Lett., 2013, 15(7), 1784 CrossRef CAS PubMed.
- J. González, J. González, C. P. Calleja, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2013, 52, 5853 CrossRef PubMed.
- H. Hikawa and Y. Yokoyama, RSC Adv., 2013, 3, 1061 RSC.
- H. Hikawa, H. Suzuki, Y. Yokoyama and I. Azumaya, Catalysts, 2013, 3, 486 CrossRef CAS PubMed.
- A. R. Katritzky, V. Gupta, C. Garot, C. V. Stevens and M. F. Gordeev, Heterocycles, 1994, 38(2), 345 CrossRef CAS.
- V. Shevchenko and R. Engel, Heteroat. Chem., 1997, 8(6), 501 CrossRef CAS.
- A. R. Katritzky and D. Toader, J. Org. Chem., 1997, 62, 4137 CrossRef CAS.
- N. Mibu and K. Sumoto, Chem. Pharm. Bull., 2000, 48(11), 1810 CrossRef CAS.
- S. Lin and X. Lu, J. Org. Chem., 2007, 72, 9757 CrossRef CAS PubMed.
- M. A. Zolfigol, P. Salehi, M. Shiri, A. Sayadi, A. Abdoli, H. Keypour, M. N. Rezaeivala and K. E. Kolvari, Mol. Diversity, 2008, 12, 203 CrossRef CAS PubMed.
- A. Khazaei, M. N. S. Rad, K. M. Moradian, M. K. Borazjani and M. H. Zebardian, S. Afr. J. Chem., 2011, 64, 120 CAS.
- M. I. Baltork, M. Moghadam, S. Tangestaninejad, V. Mirkhani, M. K. Abbasabdi and M. A. Zolfigol, J. Iran. Chem. Soc., 2011, 8(3), 840 CrossRef.
- T. Aoyama, S. Kubota, T. Takido and M. Kodomari, Chem. Lett., 2011, 40, 484 CrossRef CAS.
- R. Ahmad, A. Riahi and P. Langer, Tetrahedron Lett., 2009, 50, 1490 CrossRef CAS PubMed.
- R. A. Khera, I. Ullah, R. Ahmad, A. Riahi, N. T. Hung, M. Sher, A. Villinger, C. Fischer and P. Langer, Tetrahedron, 2010, 66, 1643 CrossRef CAS PubMed.
- H. Li, J. Yang, Y. Liu and Y. Li, J. Org. Chem., 2009, 74, 6797 CrossRef CAS PubMed.
- R. Singh and G. Panda, Org. Biomol. Chem., 2010, 8, 1097 CAS.
- F. He, P. Li, Y. Gu and G. Li, Green Chem., 2009, 11, 1767 RSC.
- J. A. McCubbin and O. V. Krokhin, Tetrahedron Lett., 2010, 51, 2447 CrossRef CAS PubMed.
- M. Barbero, S. Cadamuro, S. Dughera, C. Magistris and P. Venturello, Org. Biomol. Chem., 2011, 9, 8393 CAS.
- C. Brugnara, J. Halperin, E. M. Bellot, M. Froimowitz, R. J. Lombardy and J. J. Clifford, International Patent, Patent NumberWO 97/34589, 25 September 1997.
- I. Chen, A. McDougal, F. Wang and S. Safe, Carcinogenesis, 1998, 19(9), 1631 CrossRef CAS PubMed.
- A. McDougal, S. M. Gupta, K. Ramamoorthy, G. Sun and S. H. Safe, Cancer Lett., 2000, 151(2), 169 CrossRef CAS.
- A. McDougal, S. M. Gupta, D. Morrow, K. Ramamoorthy, J. E. Lee and S. H. Safe, Breast Cancer Res. Treat., 2001, 66, 147 CrossRef CAS.
- C. Qin, D. Morrow, J. Stewart, K. Spencer, W. Porter, R. Smith, T. Phillips, M. Abdelrahim, I. Samudio and S. Safe, Mol. Cancer Ther., 2004, 3, 247 CAS.
- S. Chintharlapalli, R. Smith, I. Samudio, W. Zhang and S. Safe, Cancer Res., 2004, 64, 5994 CrossRef CAS PubMed.
- R. Contractor, I. Samudio, J. Estrov, Z. D. Harris, J. A. McCubrey, S. H. Safe, M. Andreeff and M. Konopleva, Cancer Res., 2005, 65, 2890 CrossRef CAS PubMed.
- M. Abdelrahim, K. Newman, K. Vanderlaag, I. Samudio and S. Safe, Carcinogenesis, 2006, 27(4), 717 CrossRef CAS PubMed.
- M. Wainwright, S. M. Burrow, S. G. R. Guinot, D. A. Phoenix and J. Waring, Cytotechnology, 1999, 29, 35 CrossRef CAS.
- G. L. Indig, G. S. Anderson, M. G. Nichols, J. A. Bartlett, W. S. Mellon and F. Sieber, J. Pharmaceut. Sci., 2000, 89(1), 88 CrossRef CAS.
- S. DeBonis, D. A. Skoufias, L. Lebeau, R. Lopez, G. Robin, R. L. Margolis, R. H. Wade and F. Kozielski, Mol. Cancer Ther., 2004, 3, 1079 CAS.
- D. A. Skoufias, S. DeBonis, Y. Saoudi, L. Lebeau, I. Crevel, R. Cross, R. H. Wade, D. Hackney and F. Kozielski, J. Biol. Chem., 2006, 281(26), 17559 CrossRef CAS PubMed.
- N. Ogo, S. Oishi, K. Matsuno, J. I. Sawada, F. Nobutaka and A. Asai, Bioorg. Med. Chem. Lett., 2007, 17, 3921 CrossRef CAS PubMed.
- R. A. Al-Qawasmeh, Y. Lee, M. Y. Cao, X. Gu, A. Vassilakos, A. J. Wright and A. Young, Bioorg. Med. Chem. Lett., 2004, 14, 347 CrossRef CAS PubMed.
- A. Natarajan, Y. H. Fan, H. Chen, Y. Guo, J. Iyasere, F. Harbinski, W. J. Christ, H. Aktas and J. A. Halperin, J. Med. Chem., 2004, 47, 1882 CrossRef CAS PubMed.
- A. Natarajan, Y. Guo, F. Harbinski, Y. H. Fan, H. Chen, L. Luus, J. Diercks, H. Aktas, M. Chorev and J. A. Halperin, J. Med. Chem., 2004, 47, 4979 CrossRef CAS PubMed.
- S. H. D. Lacerda, B. Abraham, T. C. Stringfellow and G. L. Indig, Photochem. Photobiol., 2005, 81, 1430 CrossRef CAS PubMed.
- R. S. Dothager, K. S. Putt, B. J. Allen, B. J. Leslie, V. Nesterenko and P. J. Hergenrother, J. Am. Chem. Soc., 2005, 127, 8686 CrossRef CAS PubMed.
- R. Palchaudhuri, V. Nesterenko and P. J. Hergenrother, J. Am. Chem. Soc., 2008, 130(31), 10274 CrossRef CAS PubMed.
- J. L. Arbiser, International Patent, Patent Number US 2010/0189762 A1, 29 July 2010.
- M. R. Saberi, T. K. Vinh, S. W. Yee, N. B. J. Griffiths, P. J. Evans and C. Simons, J. Med. Chem., 2006, 49, 1016 CrossRef CAS PubMed.
- Shagufta, A. K. Srivastava, R. Sharma, R. Mishra, A. K. Balapure, P. S. R. Murthy and G. Panda, Bioorg. Med. Chem., 2006, 14, 1497 CrossRef CAS PubMed.
- M. Recanatini, A. Bisi, A. Cavalli, F. Belluti, S. Gobbi, A. Rampa, P. Valenti, M. Palzer, A. Palusczak and R. W. Hartmann, J. Med. Chem., 2001, 44, 672 CrossRef CAS PubMed.
- B. C. A. Alessandra, B. Carlo, R. Carlo, E. G. P. A. S. Giorgio, A. Rampa, F. Belluti, L. Piazzi, P. Valenti, R. W. Hartmann and M. Recanatini, J. Med. Chem., 2005, 48, 7282 CrossRef PubMed.
- S. Gobbi, A. Cavalli, M. Negri, K. E. Schewe, F. Belluti, L. Piazzi, R. W. Hartmann, M. Recanatini and A. Bisi, J. Med. Chem., 2007, 50, 3420 CrossRef CAS PubMed.
- S. Gobbi, C. Zimmer, F. Belluti, A. Rampa, R. W. Hartmann, R. Maurizio and A. Bisi, J. Med. Chem., 2010, 53, 5347 CrossRef CAS PubMed.
- P. M. Wood, L. W. L. Woo, A. Humphreys, S. K. Chander, A. Purohit, M. J. Reed and V. L. B. Potter, J. Steroid Biochem. Mol. Biol., 2005, 94, 123 CrossRef CAS PubMed.
- T. Jackson, L. W. L. Woo, M. N. Trusselle, S. K. Chander, A. Purohit, M. J. Reed and B. V. L. Potter, Org. Biomol. Chem., 2007, 5, 2940 CAS.
- P. M. Wood, L. W. L. Woo, J. R. Labrosse, M. N. Trusselle, S. Abbate, G. Longhi, E. Castiglioni, F. Lebon, A. Purohit, M. J. Reed and B. V. L. Potter, J. Med. Chem., 2008, 51, 4226 CrossRef CAS PubMed.
- L. W. L. Woo, T. Jackson, A. Putey, G. Cozier, P. Leonard, K. R. Acharya, S. K. Chander, A. Purohit, M. J. Reed and B. V. L. Potter, J. Med. Chem., 2010, 53, 2155 CrossRef CAS PubMed.
- A. Stefanachi, A. D. Favia, O. Nicolotti, F. Leonetti, L. Pisani, M. Catto, C. Zimmer, R. W. Hartmann and A. Carotti, J. Med. Chem., 2011, 54, 1613 CrossRef CAS PubMed.
- L. Yin, Q. Hu and R. W. Hartmann, J. Med. Chem., 2013, 56, 460 CrossRef CAS PubMed.
- K. J. Saliba and K. Kirk, Trans. R. Soc. Trop. Med. Hyg., 1998, 92, 666 CrossRef CAS.
- T. Tiffert, H. Ginsburg, M. Krugliak, B. C. Elford and V. L. Lew, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(1), 331 CrossRef CAS.
- (a) V. Trivedi, P. Chand, K. Srivastava, S. K. Puri, P. R. Maulik and U. Bandyopadhyay, J. Biol. Chem., 2005, 280, 41129 CrossRef CAS PubMed; (b) S. Gemma, G. Campiani, S. Butini, G. Kukreja, B. P. Joshi, M. Persico, B. Catalanotti, E. Novellino, E. Fattorusso, V. Nacci, L. Savini, D. Taramelli, N. Basilico, G. Morace, V. Yardley and C. Fattorusso, J. Med. Chem., 2007, 50, 595 CrossRef CAS PubMed.
- S. Kumar, S. K. Das, S. Dey, P. Maity, M. Guha, V. Choubey, G. Panda and U. Bandyopadhyay, Antimicrob. Agents Chemother., 2008, 52(2), 705 CrossRef CAS PubMed.
- M. Goyal, P. Singh, A. Alam, S. K. Das, M. S. Iqbal, S. Dey, S. Bindu, C. Pal, S. K. Das, G. Panda and U. Bandyopadhyay, Free Radicals Biol. Med., 2012, 53, 129 CrossRef CAS PubMed.
- H. Wulff, M. Miller, W. Hänsel, S. Grissmer, M. D. Cahalan and K. G. Chandy, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(14), 8151 CrossRef CAS.
- S. J. Wai, M. S. Egbertson, L. S. Payne, T. E. Fisher, M. W. Embrey, L. O. Tran, J. Y. Melamed, H. M. Langford, J. P. Guare Jr, L. Zhuang, V. E. Grey, J. P. Vacca, M. K. Holloway, A. M. Naylor Olsen, D. J. Hazuda, P. J. Felock, A. L. Wolfe, K. A. Stillmock, W. A. Schleif, L. J. Gabryelski and S. D. Young, J. Med. Chem., 2000, 43(26), 4923 CrossRef CAS; K. Sumoto, N. Mibu, K. Yokomizo and M. Uyeda, Chem. Pharm. Bull., 2002, 50(2), 298 CrossRef.
- N. Mibu, K. Yokomizo, M. Uyeda and K. Sumoto, Chem. Pharm. Bull., 2003, 51(11), 1325 CrossRef CAS.
- N. Mibu, K. Yokomizo, M. Uyeda and K. Sumoto, Chem. Pharm. Bull., 2005, 53(9), 1171 CrossRef CAS.
- N. Mibu, K. Yokomizo, T. Miyata and K. Sumoto, J. Heterocycl. Chem., 2010, 47, 1434 CrossRef CAS.
- G. Panda, Shagufta, J. K. Mishra, V. Chaturvedi, A. K. Srivastava, R. Srivastava and B. S. Srivastava, Bioorg. Med. Chem., 2004, 12, 5269 CrossRef CAS PubMed.
- G. Panda, Shagufta, A. K. Srivastava and S. Sinha, Bioorg. Med. Chem. Lett., 2005, 15, 5222 CrossRef CAS PubMed.
- Shagufta, A. Kumar, G. Panda and M. I. Siddiqi, J. Mol. Model., 2007, 13, 99 CrossRef CAS PubMed.
- G. Panda, M. K. Parai, S. K. Das, Shagufta, M. Sinha, V. Chaturvedi, A. K. Srivastava, Y. S. Manju, A. N. Gaikwad and S. Sinha, Eur. J. Med. Chem., 2007, 42, 410 CrossRef CAS PubMed.
- G. Panda, M. K. Parai, A. K. Srivastava, V. Chaturvedi, Y. K. Manju and S. Sinha, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2009, 48, 1121 Search PubMed.
- M. K. Parai, G. Panda, V. Chaturvedi, Y. K. Manju and S. Sinha, Bioorg. Med. Chem. Lett., 2008, 18, 289 CrossRef CAS PubMed.
- V. K. Kashyap, R. K. Gupta, R. Shrivastava, B. S. Srivastava, R. Srivastava, M. K. Parai, P. Singh, S. Bera and G. Panda, J. Antimicrob. Chemother., 2012, 67(5), 1188 CrossRef CAS PubMed.
- R. S. Upadhayaya, J. K. Vandavasi, N. R. Vasireddy, V. Sharma, S. S. Dixit and J. Chattopadhyaya, Bioorg. Med. Chem., 2009, 17, 2830 CrossRef CAS PubMed.
- A. Tafi, R. Costi, M. Botta, R. D. Santo, F. Corelli, S. Massa, A. Ciacci, F. Manetti and M. Artico, J. Med. Chem., 2002, 45, 2720 CrossRef CAS PubMed.
- G. A. McNaughtonSmith, J. F. Burns, J. W. Stocker, G. C. Rigdon, C. Creech, S. Arrington, T. Shelton and L. Franceschi de, J. Med. Chem., 2008, 51, 976 CrossRef CAS PubMed.
- L. Yin, S. Lucas, F. Maurer, U. Kazmaier, Q. Hu and R. W. Hartmann, J. Med. Chem., 2012, 55(13), 6629 CrossRef CAS PubMed.
- S. B. Bodendiek, C. Rubinos, M. P. Trelles, N. Coleman, D. P. Jenkins, H. Wulff and M. Srinivas, Front. Pharmacol., 2012, 3, 106 CAS.
- M. J. Sabacky, S. M. Johnson, J. C. Martin and I. C. Paul, J. Am. Chem. Soc., 1969, 91, 26 CrossRef.
- L. S. Harikrishnan, H. J. Finlay, J. X. Qiao, M. G. Kamau, J. Jiang, T. C. Wang, J. Li, C. B. Cooper, M. A. Poss, L. P. Adam, D. S. Taylor, A. Y. A. Chen, X. Yin, P. G. Sleph, R. Z. Yang, D. F. Sitkoff, M. A. Galella, D. S. Nirschl, K. V. Kirk, A. V. Miller, C. S. Huang, M. Chang, X. Q. Chen, E. M. Salvati, R. R. Wexler and R. M. Lawrence, J. Med. Chem., 2012, 55(13), 6162 CrossRef CAS PubMed.
- R. Boyd, E. C. R. Rasmussen, J. B. Press, R. B. Raffa, E. E. Codd, C. D. Connelly, Q. S. Li, R. P. Martinez, M. A. Lewis, H. R. Almond and A. B. Reitz, J. Med. Chem., 2001, 44, 863 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |