Synthesis and modification of octafluoro[2.2]paracyclophane (parylene AF4)
Craig
Hicks
,
Brendan
Duffy
and
Gráinne C.
Hargaden
*
Centre for Research in Engineering Surface Technology (CREST), Focas Institute and School of Chemical and Pharmaceutical Sciences, Dublin Institute of Technology, Kevin Street, Dublin 8, Ireland. E-mail: grainne.hargaden@dit.ie
First published on 7th May 2014
Abstract
Parylenes are a class of organic compounds which have gained significant attention due to their application as coatings in areas such as medical devices and electronics. This review highlights the development of synthetic pathways to Octafluoro[2.2]Paracyclophane, commonly referred to as parylene AF4. Methods which have been used to functionalise AF4 are also presented.
 Craig Hicks | Craig Hicks received his BSc in Chemical Science and Medicinal Chemistry from the Dublin Institute of Technology in 2011. In the same year he began PhD studies in the Centre for Research in Engineering Surface Technology (CREST) in the area of materials science. His current research interests are the applications of organic synthesis in developing novel surface coating materials. |
 Brendan Duffy | Dr Brendan Duffy joined Centre for Research in Engineering Surface Technology (CREST) in the Dublin Institute of Technology in 2004. He has been the Centre Manager since 2012 and his main interests include protective coatings on engineering surfaces and materials analysis. He is involved in European and nationally funded research projects and has patented and licensed several technologies to Irish and multinational based companies. |
 Gráinne C. Hargaden | Dr Gráinne Hargaden received her BSc and PhD from University College Dublin. Following three years as a postdoctoral research associate at the University of Oxford, she was appointed assistant lecturer in organic chemistry at Dublin Institute of Technology in 2010 and progressed to lecturer in 2013. She is a principal investigator in the Centre for Research in Engineering Surface Technology (CREST), and her research interests lie at the interface of synthetic organic chemistry and materials science. |
Introduction
Parylene is a commonly used industrial term to describe a number of vapour deposited poly(p-xylylene) polymers. Since their development in the late 1940s1 these materials have garnered attention as protective coatings due to their attractive physical and chemical properties and are widely used by the electronic and medical device industries. The conformal coating can be applied at ambient temperatures with tailored thickness, and specific properties of the coating can be altered by adjusting the chemistry of the precursors by the addition of various chemical substituents. The most commonly used precursors to produce these films are [2.2]paracyclophanes of which a variety have been produced (Scheme 1).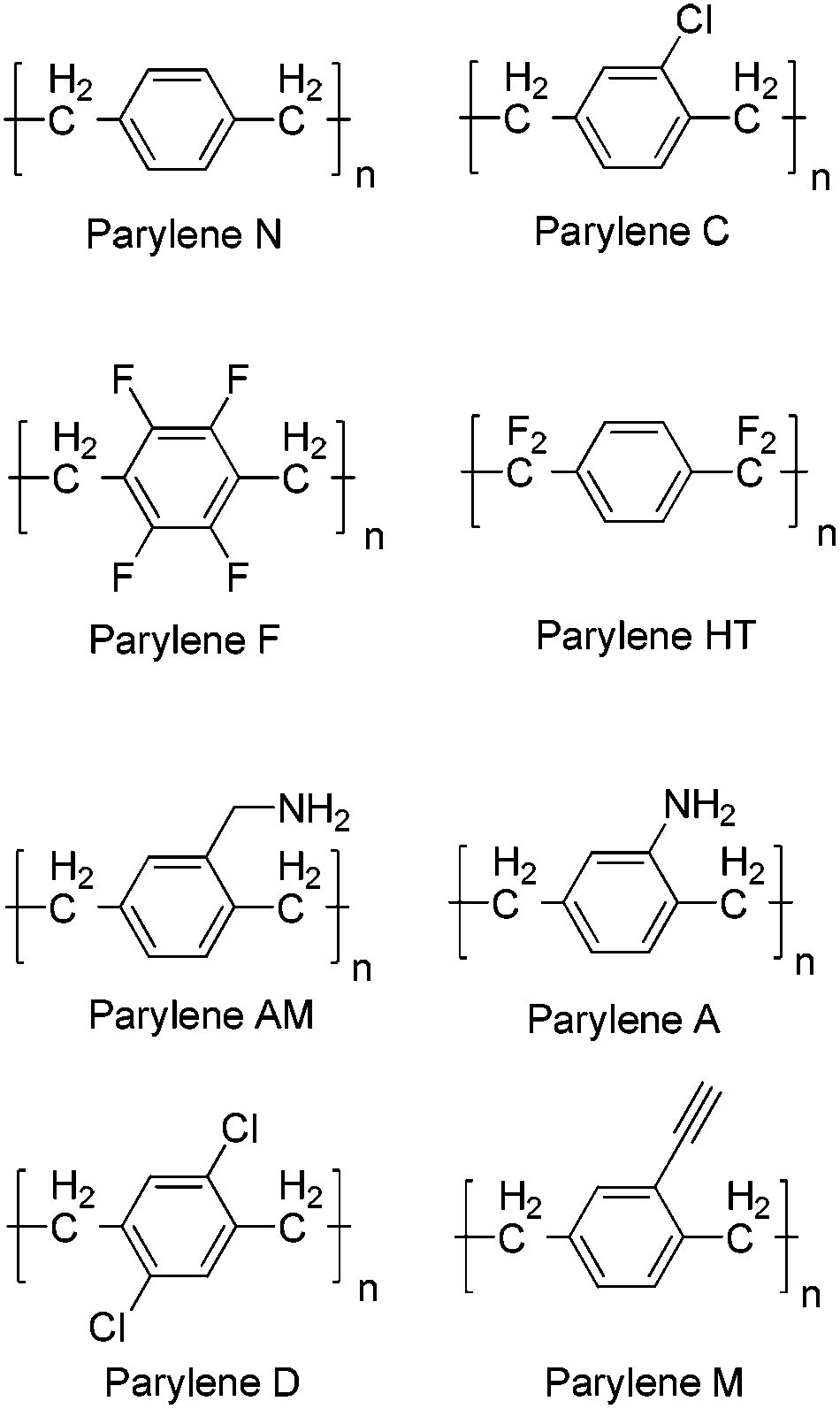 | ||
| Scheme 1 Examples of parylene coatings. | ||
The development by Gorham2 of a deposition process in 1966 ultimately led to the widespread industrial use of these coatings. The cyclophanes are pyrolysed at temperatures of approximately 550 °C in a vacuum chamber resulting in the formation of the reactive p-xylylene monomers which are deposited and begin chain growth polymerisation on the surface; typical chain lengths are of the order of 2000–4000 units (Scheme 2). This is considered to be a “green process” as no initiators or terminators are used and the process produces no by-products. As a result, the coatings are free of contaminants and chemically pure.
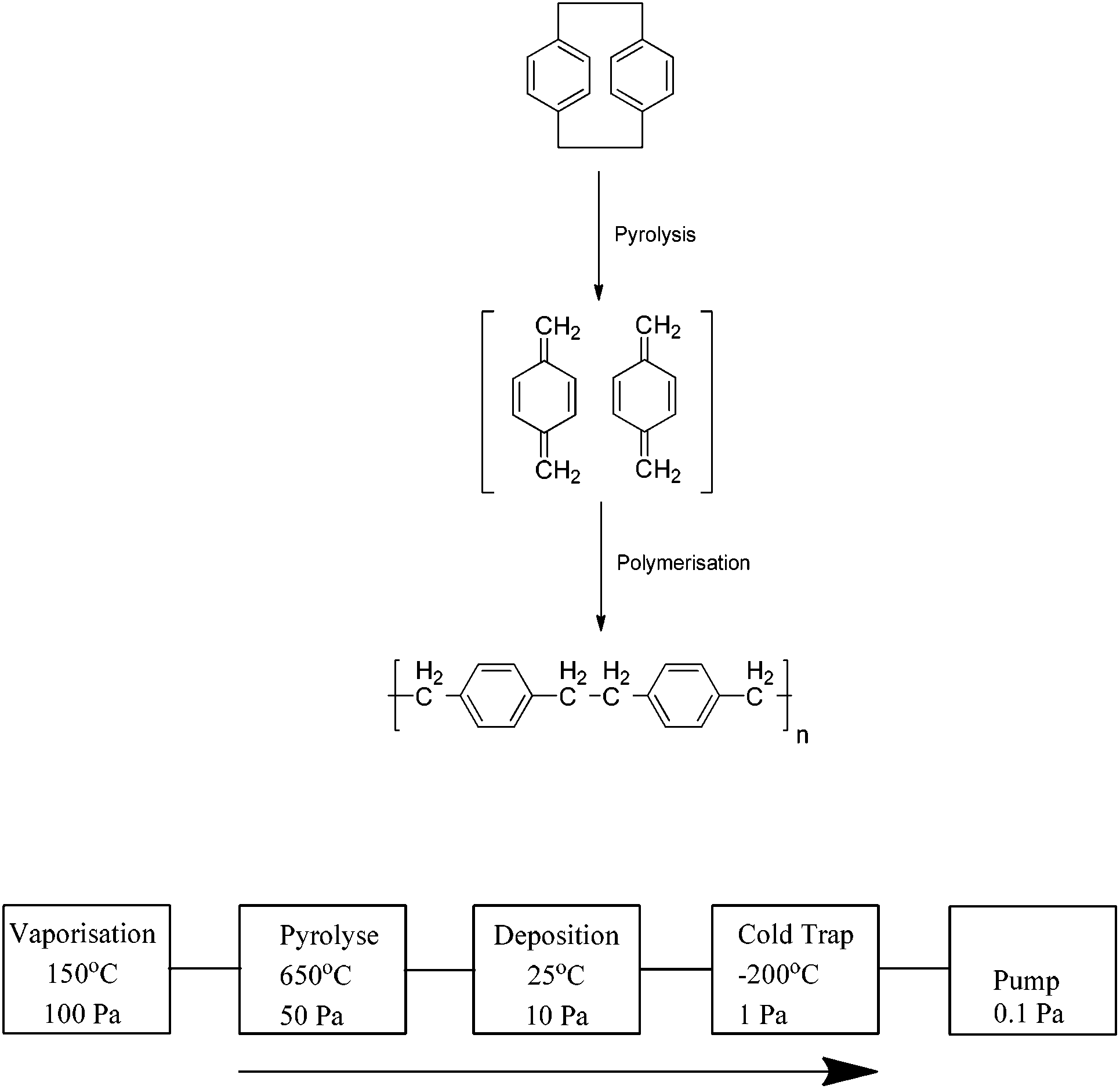 | ||
| Scheme 2 The polymerisation of [2.2]paracyclophane into parylene polymer and a schematic representation of the parylene fabrication process. | ||
Of particular commercial interest is the parylene HT coating which is produced from the precursor octafluoro[2.2]paracyclophane 1, commonly referred to as AF4. This compound is structurally similar to the parent [2.2]paracyclophane but the alkyl hydrogen atoms are replaced by fluorine atoms. This chemical modification results in a coating that displays significantly superior thermal and UV stability in addition to a low dielectric constant and excellent barrier properties.3,4 However a facile and high yielding synthesis of 1 has proven difficult and herein is discussed the advances to date in its preparation and modification.
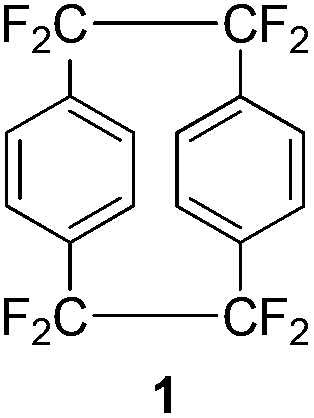
The preparation of octafluoro[2.2]paracyclophane
The initial procedures for the preparation of 1 were developed in the early 1960s when a number of p-bis-alkylsulfonyls 2 were pyrolysed at high temperatures (600–800 °C) to yield 1.5 Around the same time, a similar high temperature procedure in the presence of copper was developed using p-bis-alkylhalo analogues 3 and 4 (Scheme 3).6 These methods did not exclusively yield 1 and a variety of cyclic and linear oligomers were also formed contributing to a yield of approximately 30% by either method. Subsequent research has focused on pathways which would lead exclusively to 1.7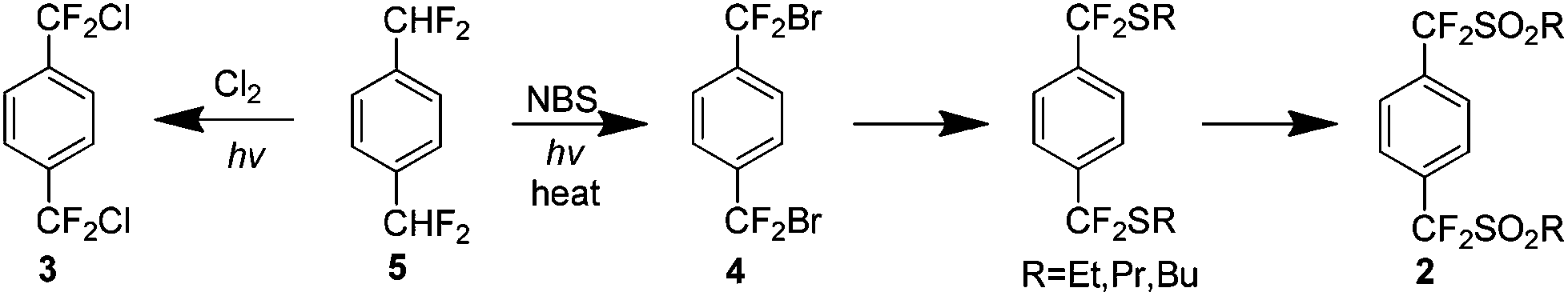 | ||
| Scheme 3 The preparation of p-bis-alkylsulfonyl and p-bis-alkylhalo precursors for the high temperature synthesis of AF4. | ||
The high temperature synthesis optimised by Wheelwright et al.7 remained the only route to produce 1 for almost 30 years. It was not until the early 1990s that a new method was presented. Dolbier et al. attempted to extend the Hoffman elimination of an amino tetrafluoroborate salt 6 to yield 1, based on a previous synthesis of the parent hydrocarbon paracyclophane8 (Scheme 4). However this proved unsuccessful as did an attempt to use carboxylic acid salt 7. It was found that treatment of compound 4 with a combination of titanium tetrachloride and lithium aluminium hydride afforded 1 in a 32% yield.9
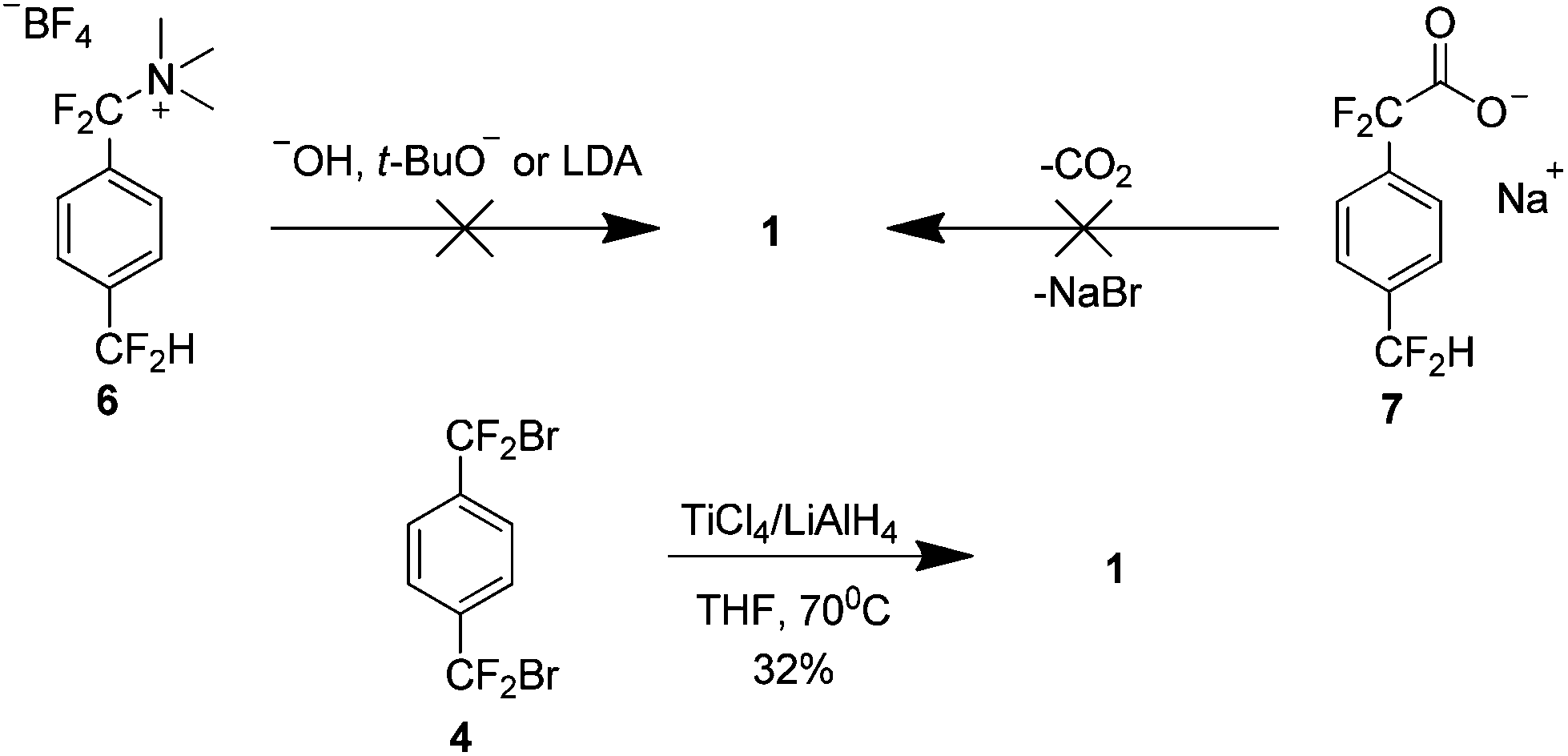 | ||
| Scheme 4 Development of a low temperature preparation of AF4. | ||
Although this work has removed the necessity for high temperatures, it presented a new issue. An appreciable yield was only obtained when the reaction was carried out under highly dilute conditions, 1.5 L of solvent yielded 1.7 g of 1 (32%) and extremely slow addition of reactants was necessary. This limitation is explained on consideration of the reaction mechanism which proceeds via formation of the reactive tetrafluoro-p-xylylene 8, as evidenced by a small amount of the trimer 9 in the reaction mixture (Scheme 5). There is an increase in the concentration of 8 as the dilution factor is decreased so it must be maintained to limit the collisions with the di-radical intermediate 10. This dilution also provides the optimal kinetic environment to minimise the bimolecular oligomerisation of the extended conformer 11.
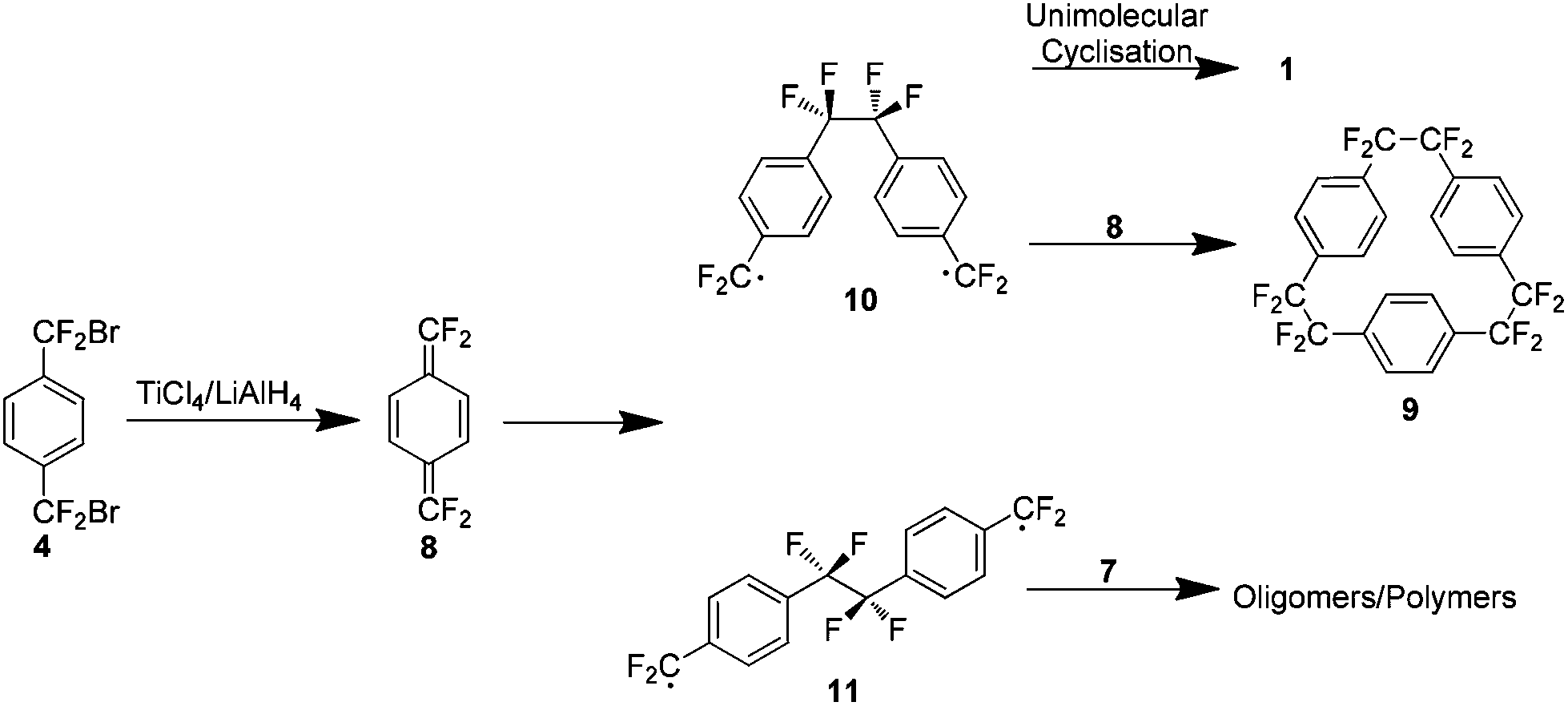 | ||
| Scheme 5 The possible reaction pathways of the intermediate tetrafluoro-p-xylylene. | ||
Dolbier reported an improved method several years later which obviated the need for high dilution. Based on a study into Diels–Alder reactions by Mori et al.,10,11 it was found that a combination of trimethylsilyl tributyltin and caesium fluoride could affect the required debromination of 4 to yield 1 in 40% yield (Scheme 6). It was found that a solvent system of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 tetrahydrofuran and dimethyl sulfoxide was crucial for this to proceed. A mechanistic reasoning explains the need for this and the formation of 8. Caesium fluoride has limited solubility in this solvent system so it impedes the rate of formation of the necessary intermediate such that at any one time its concentration is low. This essentially presents the same favourable kinetic environment that high dilution facilitates but without the increased solvent volume.12
:1 tetrahydrofuran and dimethyl sulfoxide was crucial for this to proceed. A mechanistic reasoning explains the need for this and the formation of 8. Caesium fluoride has limited solubility in this solvent system so it impedes the rate of formation of the necessary intermediate such that at any one time its concentration is low. This essentially presents the same favourable kinetic environment that high dilution facilitates but without the increased solvent volume.12
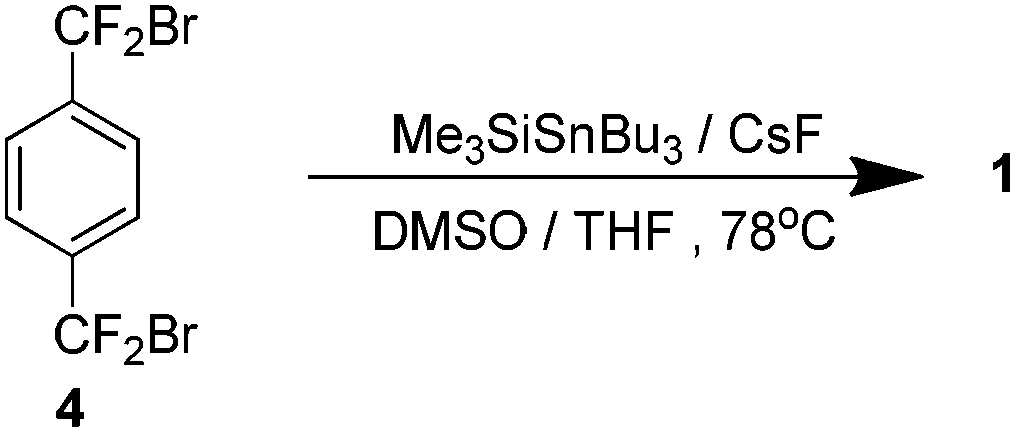 | ||
| Scheme 6 A tin-catalysed debromination to yield AF4. | ||
There was a synthesis available for the production of 1 that did not require high dilution technology and it was industrially feasible to produce the material on a kilogram scale but some issues remained. The reaction was still low yielding at 40%, there is significant cost associated with the reagents, and the use of stoichiometric quantities of an organostannane is undesirable.
In a report which followed and may be considered amongst the best to date, again by Dolbier, it was shown that 1 could be produced in a 60% yield without the use of stannanes. Treating the chloro analogue 3 with zinc dust in dimethylacetamide yielded the product without needing high dilution conditions (Scheme 7). The mechanism is not fully understood but it is believed that the coupling occurs on the metal surface. Not requiring high dilution would imply that the pathway does not involve generation of free intermediate 8.13
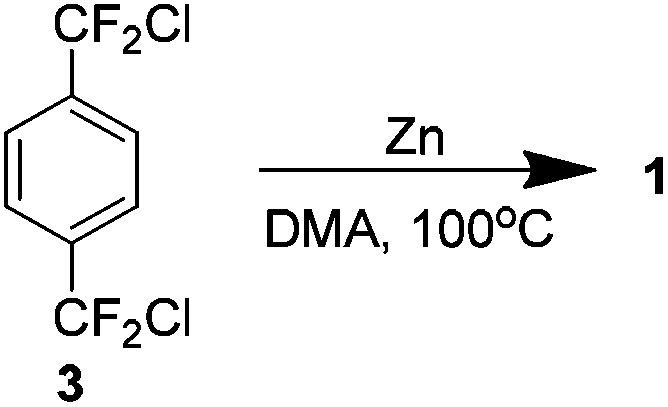 | ||
| Scheme 7 The zinc-catalysed coupling of p-bis-chloro difluoromethyl benzene to yield AF4. | ||
A year following this report Uneyama et al. presented a new carbon–fluorine bond cleavage route to 1 from the inexpensive starting material p-bis-trifluoromethyl benzene 12 (Scheme 8). A magnesium-promoted defluorinative silylation of one of the trifluoromethyl groups was used to yield the corresponding trimethylsilyl compound 13 which was then heated at reflux in anisole in the presence of catalytic amounts of caesium fluoride and tris(dibenzylideneacetone)dipalladium(0) providing 1 in an isolated yield of 53%.14
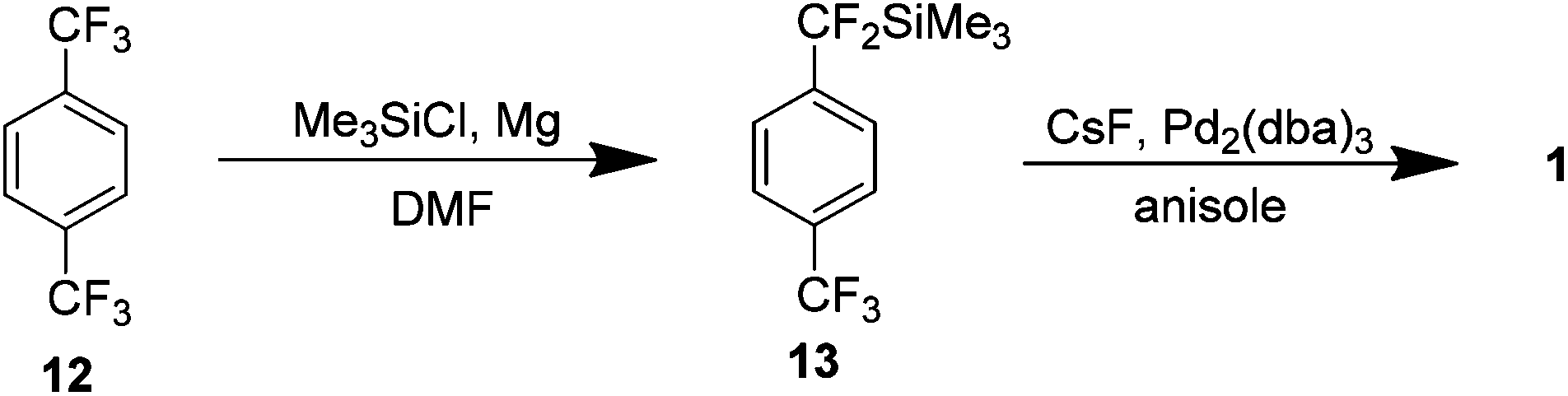 | ||
| Scheme 8 The preparation of AF4 via the formation of an organosilane and a subsequent Pd/CsF catalysed coupling. | ||
This procedure, along with the previously mentioned zinc-catalysed method, represent the current leading technology for the synthesis of 1. The yields of both procedures are comparable so when deciding which route is best it would be prudent to consider the precursor compounds required. While 12 is inexpensive and readily available in bulk commercially, 3 is not and its preparation from an inexpensive precursor could prove laborious.
There are two pathways presented in the literature to this end and these will now be considered. The first is described in a patent from the mid 1990s15 and is summarised in Scheme 9. Terephthalaldehyde 14 is fluorinated using sulphur tetrafluoride to yield p-bis-difluoromethyl benzene 516 and this is subsequently chlorinated (or brominated if desired) by a photo halogenation process to yield the desired precursor 3. Sulphur tetrafluoride is a gas and a highly corrosive species which liberates hydrogen fluoride on contact with moisture. Such a material is undesirable industrially. However, the use of this material can be circumvented in two steps by chlorinating 14 with boron trichloride to yield p-bis-dichloromethyl benzene 1517 and a subsequent solid state halogen exchange process with caesium fluoride to give 518 or the use of phase transfer agents.19 Thionyl chloride may be utilised in place of boron trichloride to yield p-bis-dichloromethylbenzene 15.20 The second approach to this precursor was published in 2007 by Dolbier and describes its production from p-bis-trichloromethyl benzene 16 (readily produced itself via photochlorination of p-xylene) using anhydrous hydrogen fluoride with reported yields of 79% for 3.18
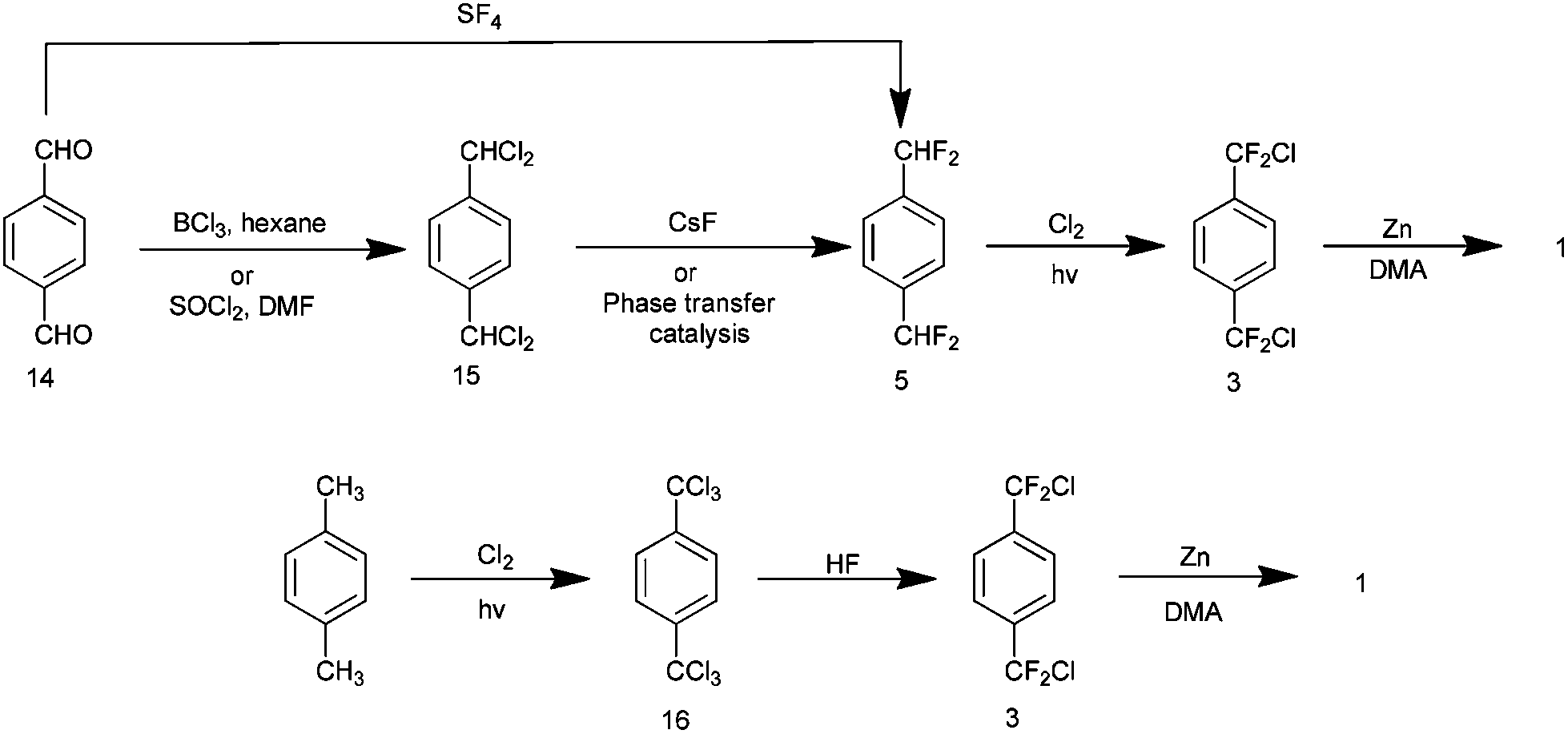 | ||
| Scheme 9 Two chemical pathways to the AF4 precursor p-bis-chlorodifluoro benzene. | ||
The preparation of substituted octafluoro[2.2]paracyclophane analogues
There have been numerous reports to date examining analogues of 1 and they range from the preparation of commercially useful substituted analogues to studies of chiral di-paracyclophanes. Paracyclophane systems have elicited much academic interest due to their unique chemical behaviours such as unusual reactivities, transannular communication and spectroscopic abnormalities.3 It is worth noting that the reports discussed in the following were all carried out by Dolbier's group. Work published in 1999 details procedures for the electrophilic aromatic substitution of 1. Methods were developed to introduce an impressive variety of functionalities, beginning with nitration using nitronium tetrafluoroborate NO2BF4 in sulfolane (Scheme 10).21 This work is particularly relevant because these additional groups can act as linkers for the attachment of other chemical entities to the precursors or the subsequent coating they generate.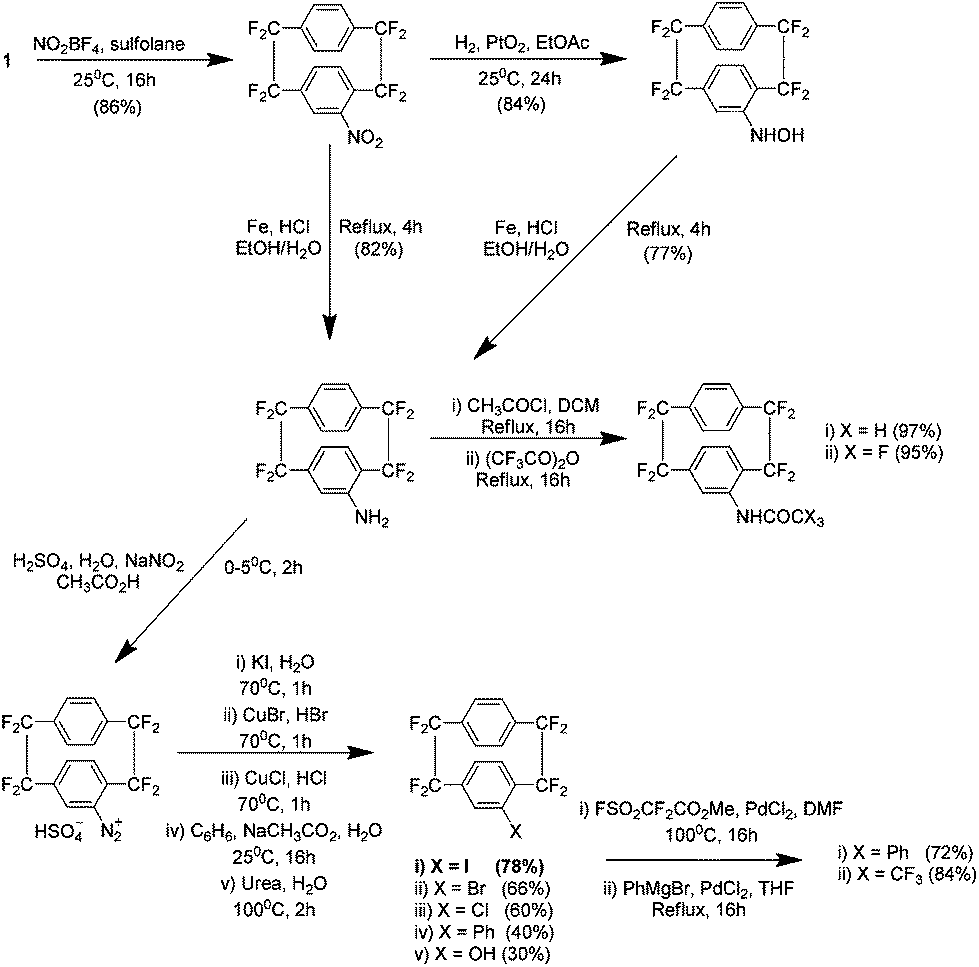 | ||
| Scheme 10 The nitration of AF4 and subsequent modifications to yield a variety of substituted analogues. | ||
Subsequently it was shown that utilising harsher nitrating conditions could yield a di-nitro analogue of 1 in 81% yield, again using nitronium tetrafluoroborate in sulfolane but at a higher temperature. However the product was shown to be an equivalent mixture of three heteroannularly substituted isomers which may be considered pseudo-ortho17, pseudo-meta18 and pseudo-para19 (Scheme 11). No pseudo-geminal or homoannular substitution was detected. Interestingly the pseudo-ortho isomer could be readily separated by column chromatography (along with any mono nitrated or starting material). These isomers could all be subsequently reduced to the corresponding amines followed by substitution to bromo or iodo derivatives. Attempts at the reduction of a single nitro group were achieved but were found to be low yielding at 11%.22
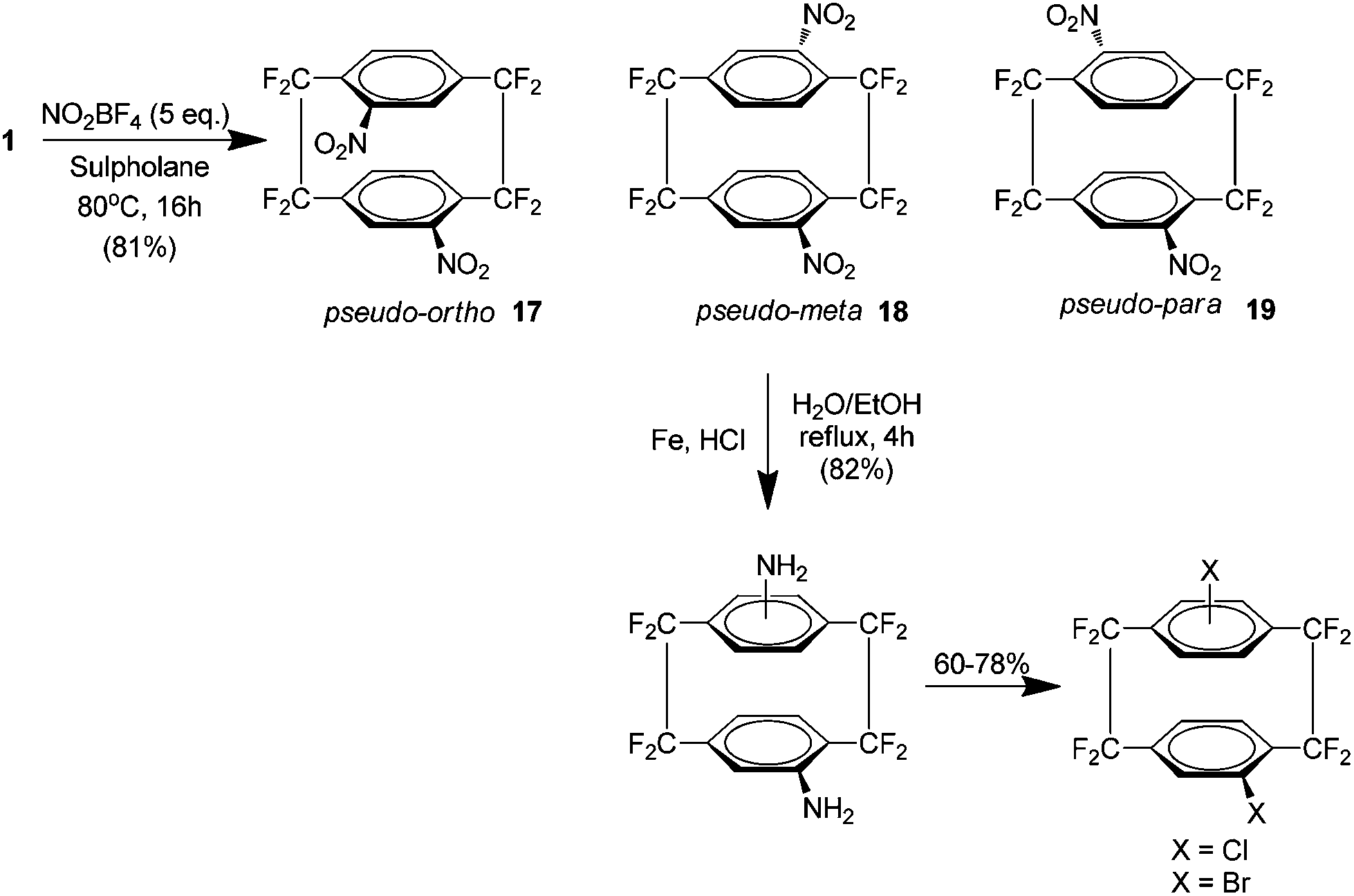 | ||
| Scheme 11 The preparation of di-nitro derivatives of AF4 and their subsequent reduction and halogenation. | ||
Direct bromination was also carried out on 1 using N-bromosuccinimide and sulfuric acid and gave an unexpected result. The reaction proceeded in 55% yield and the product was exclusively a homoannular p-dibromide which is unusual given the deactivating nature of halo substituents.22 This provides a method for the synthesis of novel homoannularly and heteroannularly di-substituted analogues of 1. Such compounds may prove superior to their hydrocarbon counterparts as ligands for asymmetric synthesis because they show a significantly higher resistance to thermal isomerisation.22,23
As shown previously, the trifluoromethyl derivative of 1 could be produced in 4 steps (Scheme 10). Attempts were made to extend Sawada's free radical trifluoromethylation procedure to 1 which utilises decomposition of trifluoroacetyl peroxide in dichloromethane.24 Although the trifluoromethyl radical added to one of the rings, no rearomatisation was observed. Instead the resulting cyclohexadienyl radical 20 proved stable enough to dimerise to one of the isomers 21 or 22 (Scheme 12). The value of these isomers is in the generation of trifluoromethyl radicals.25 Usually these are generated using trifluoroacetyl peroxide which is thermally unstable even at room temperature or by the use of Scherer's radical which is considered an excellent clean source.26 The isomers shown here are stable crystalline compounds which produce trifluoromethyl radicals at temperatures above 150 °C with the only by product being the essentially chemically inert 1.
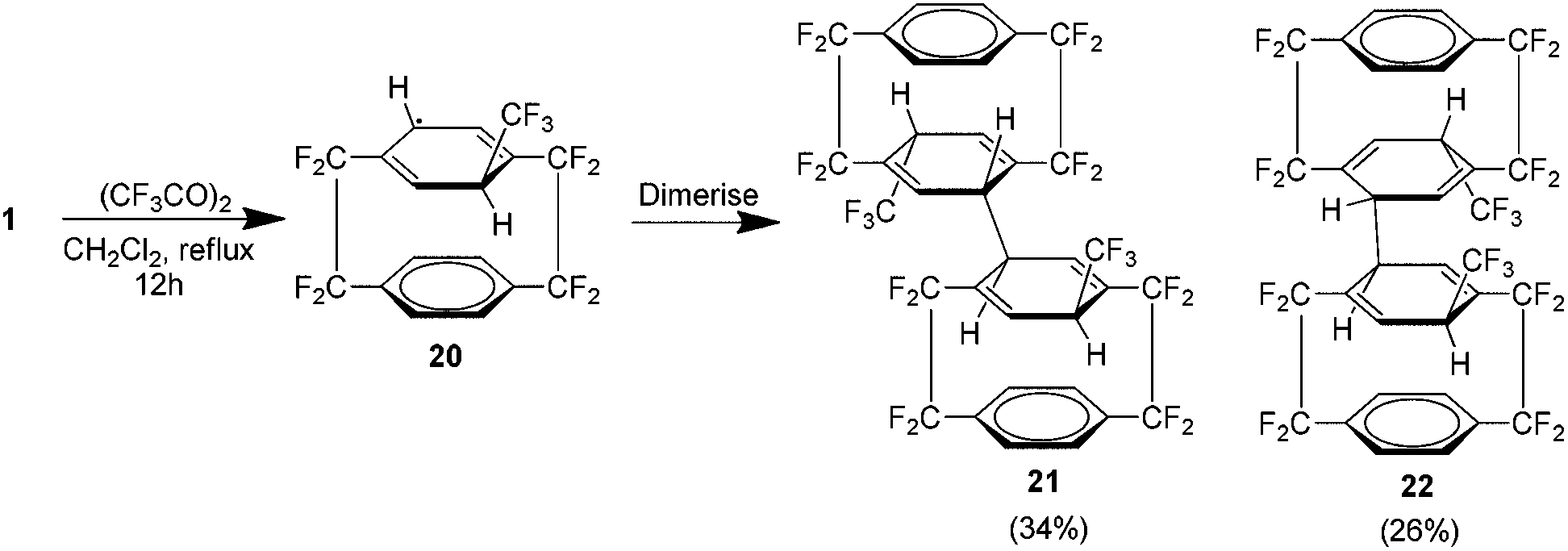 | ||
| Scheme 12 The preparation of trifluoromethyl substituted bis AF4 analogues. | ||
Following this was the first report of a di-octafluoro[2.2]paracyclophane produced via the palladium catalysed reductive homocoupling of the cyclophane analogue 23 (Scheme 13). The result was again an isomeric mixture of 24 and 25. Low temperature 19F NMR studies showed that the meso compound 25 is actually a rotamer. The introduction of a trifluoromethyl group as in 26 yielded only the meso compound 27 by a similar procedure.27
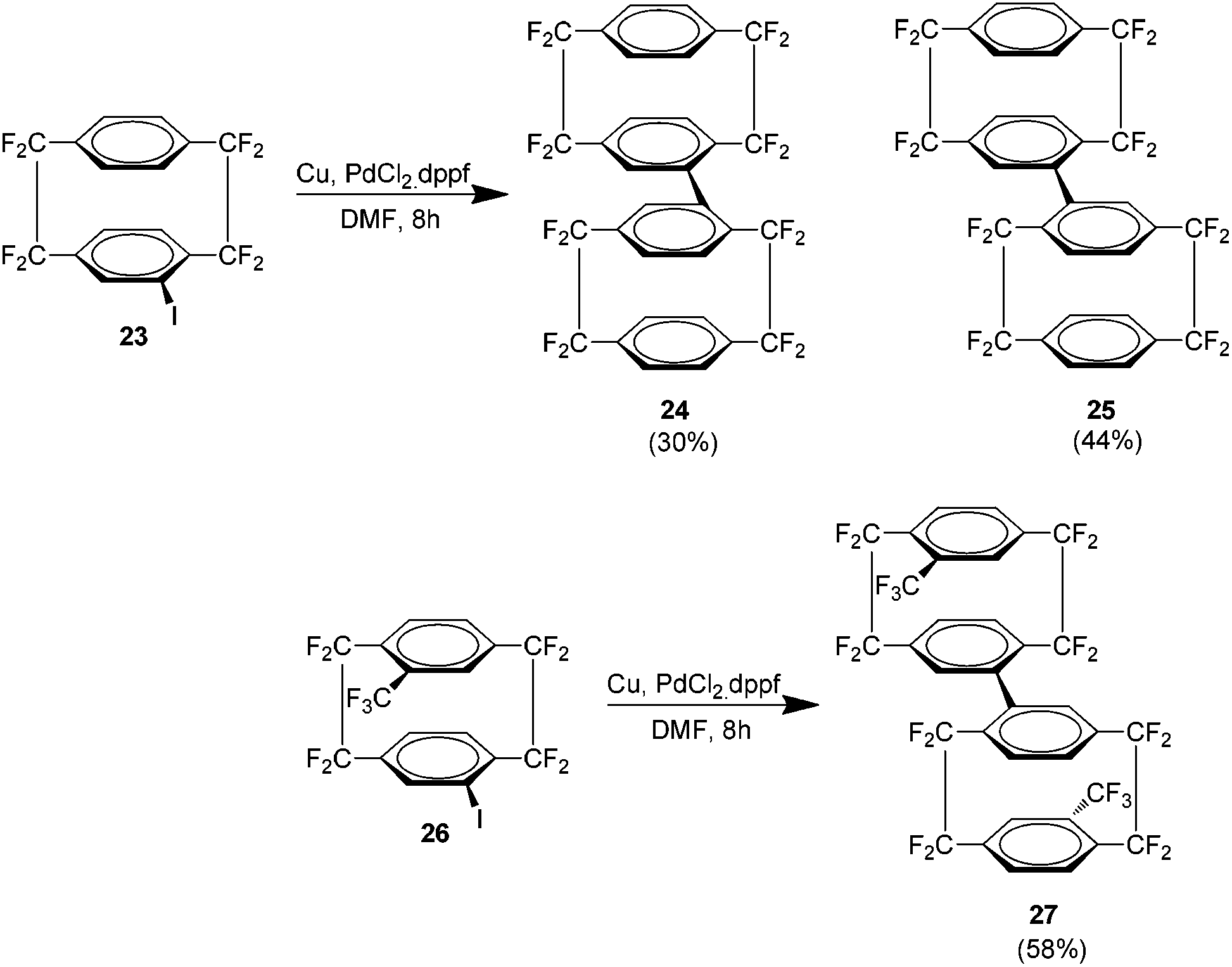 | ||
| Scheme 13 The preparation of di-octafluoro[2.2]paracyclophanes and trifluoromethyl derivatives. | ||
Facile access to halo analogue 23 also allowed an investigation into the use of Suzuki cross couplings with these fluorinated cyclophanes (Scheme 14). The reactions proved very effective when using non-hindered boronic acids and pinacol esters. A method was developed for the production of a boronic acid derivative of the fluorinated cyclophane 28. This novel material showed comparable yields under the same conditions when coupled with non-hindered aryl iodides.28
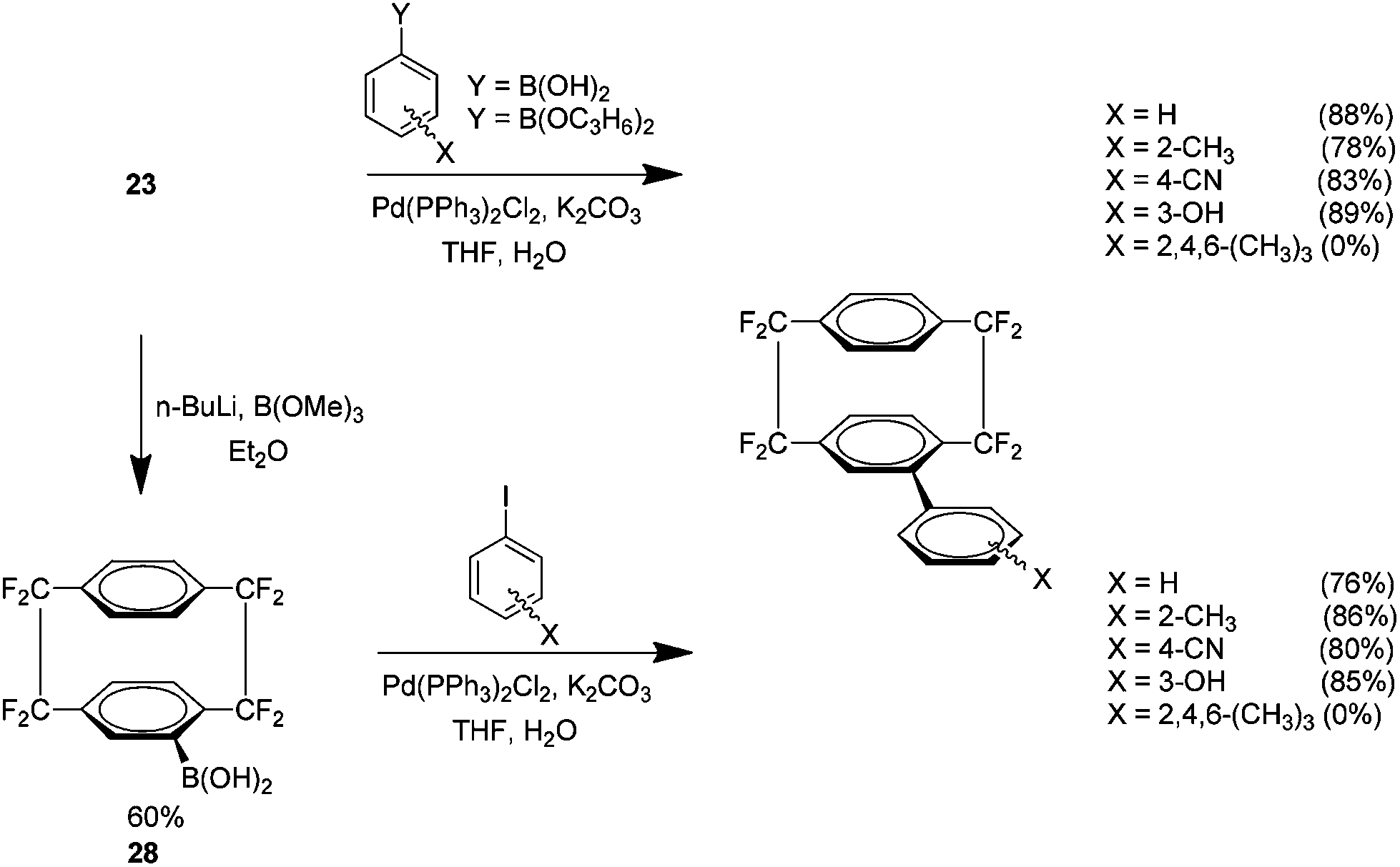 | ||
| Scheme 14 Suzuki cross couplings of octafluoro[2.2]paracyclophanes derivatives. | ||
It was found that phenyl spacer groups could be introduced between the cyclophane units under appropriate conditions to yield novel compounds 29 and 30 (Scheme 15).
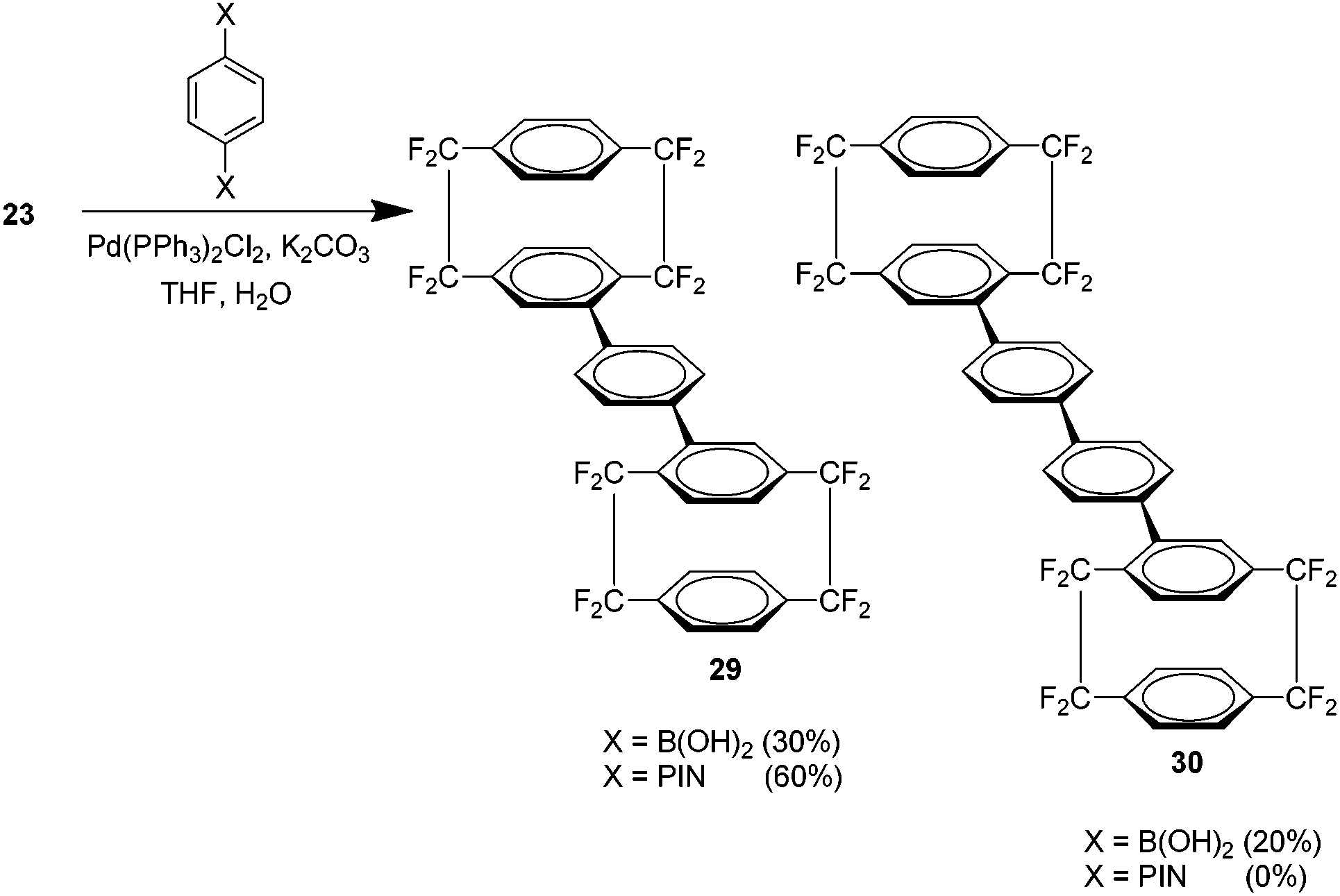 | ||
| Scheme 15 The preparation of di-cyclophane derivatives using Suzuki cross couplings. | ||
An examination of the SRN1 reactivity of 23 showed that it reacts with certain aryl thiolates and stabilised enolates (Scheme 16). This result was promising because it indicated that many novel fluorinated cyclophane derivatives may be produced from SRN1 nucleophiles. These reactions did however require photochemical initiation and did not proceed via thermal methods alone.29
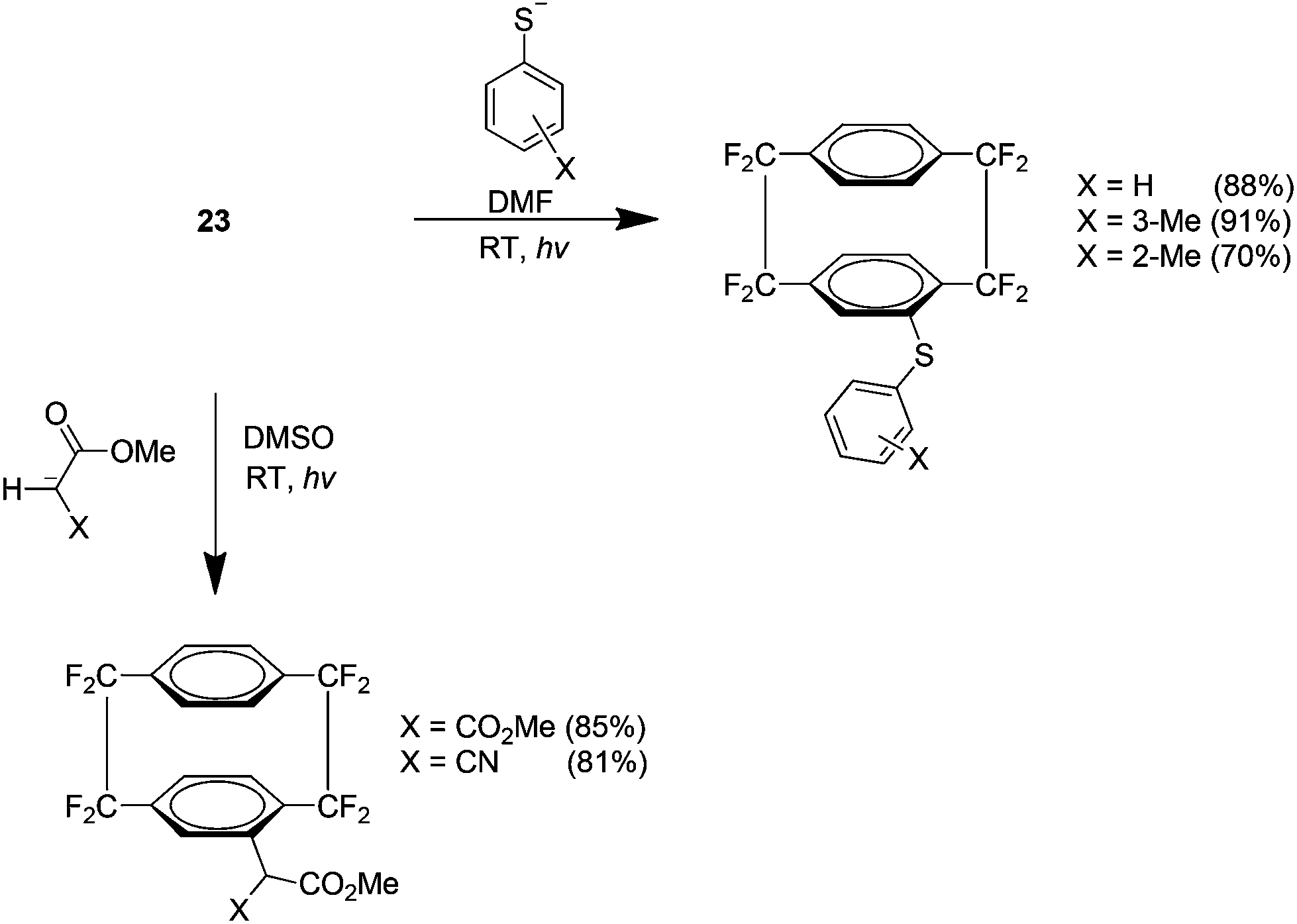 | ||
| Scheme 16 The preparation of substituted AF4 analogues by an SRN1 pathway. | ||
An unusual reaction product of nucleophilic attack of the nitro analogue 31 led to the discovery of a novel ring cleaving reaction of fluorinated cyclophanes (Scheme 17).30 Although there were reports of opening the cyclophane system with the parent hydrocarbon,31,32 this occurred via the alkyl bridge carbons whereas in this case the cleavage occurs at the alkyl–aryl bond. It is suggested that this occurs via an SNAR pathway.
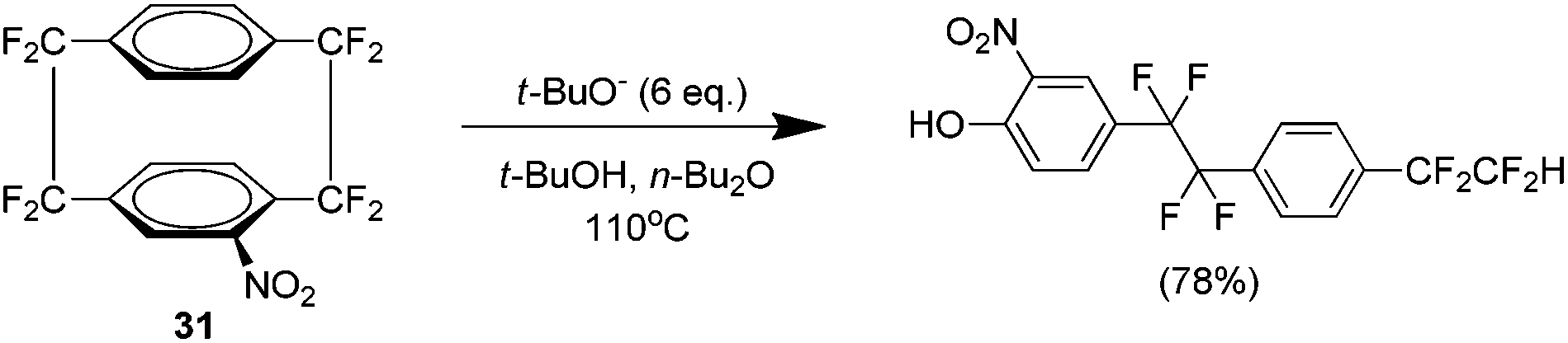 | ||
| Scheme 17 The ring cleaving reaction of nitro AF4 with a nucleophile. | ||
The preparation of substituted octafluoro[2.2]paracyclophane analogues by Diels–Alder reactions
Arynes (or benzynes) are a highly reactive intermediary species that are known to readily take part in Diels–Alder reactions. It was shown in a report from 2002 that benzynes 32 and 33 can be produced from mono and di-iodo substituted analogues of 1 (Scheme 18).33,34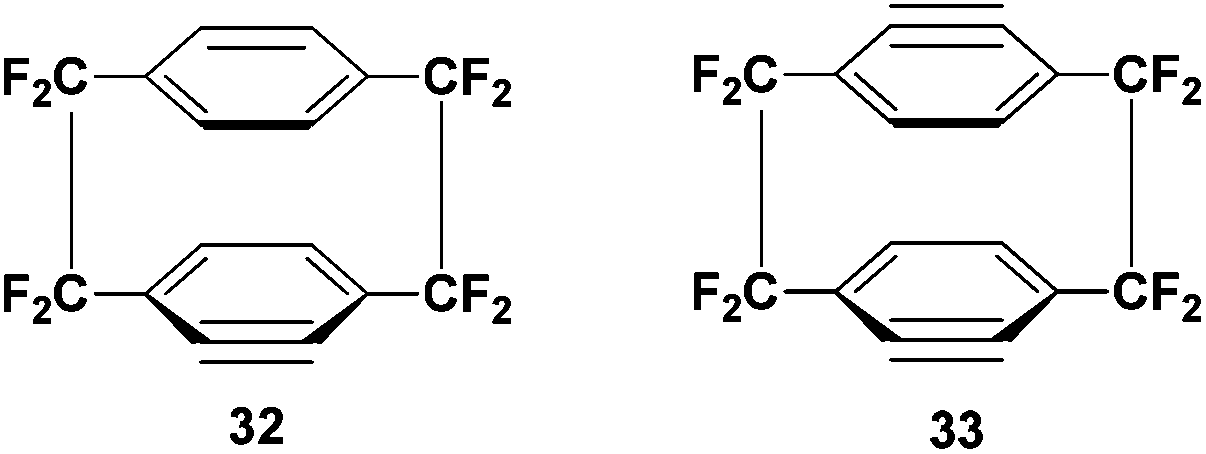 | ||
| Scheme 18 The structure of 4,5 dehydrooctafluoro[2.2]paracyclophane and 4,5,15,16-bis-dehydrooctafluoro[2.2]paracyclophane, intermediates in the Diels–Alder cycloaddition. | ||
Prior to this report in 1969, Longone et al. had used the parent hydrocarbon of 1 in the presence of anthracene to produce the Diels–Alder adduct however the yield was only 15%.35 In the same year Cram et al. also reported a bis Diels–Alder product via the dehalogenation of 4,5,15,16 tetrabromo[2.2]paracyclophane.36 Longone generated the necessary aryne using potassium t-butoxide a method normally avoided since its original reporting.37 This is because arynes typically react with the nucleophilic t-butoxide ion leading to significant amounts of by-product. In the case of arynes generated from 1 however this method is ideal giving excellent yields with virtually none of this by-product detected. As an example, Scheme 19 shows the Diels Alder products produced from iodo analogues of 1 (which occur via the formation of the corresponding arynes 32 and 33) with anthracene as a substrate which gave excellent yields. Other substrates employed were benzene, naphthalene, t-butylbenzene, furan and [2.2]paracyclophane, each of which demonstrated comparable yields with anthracene.
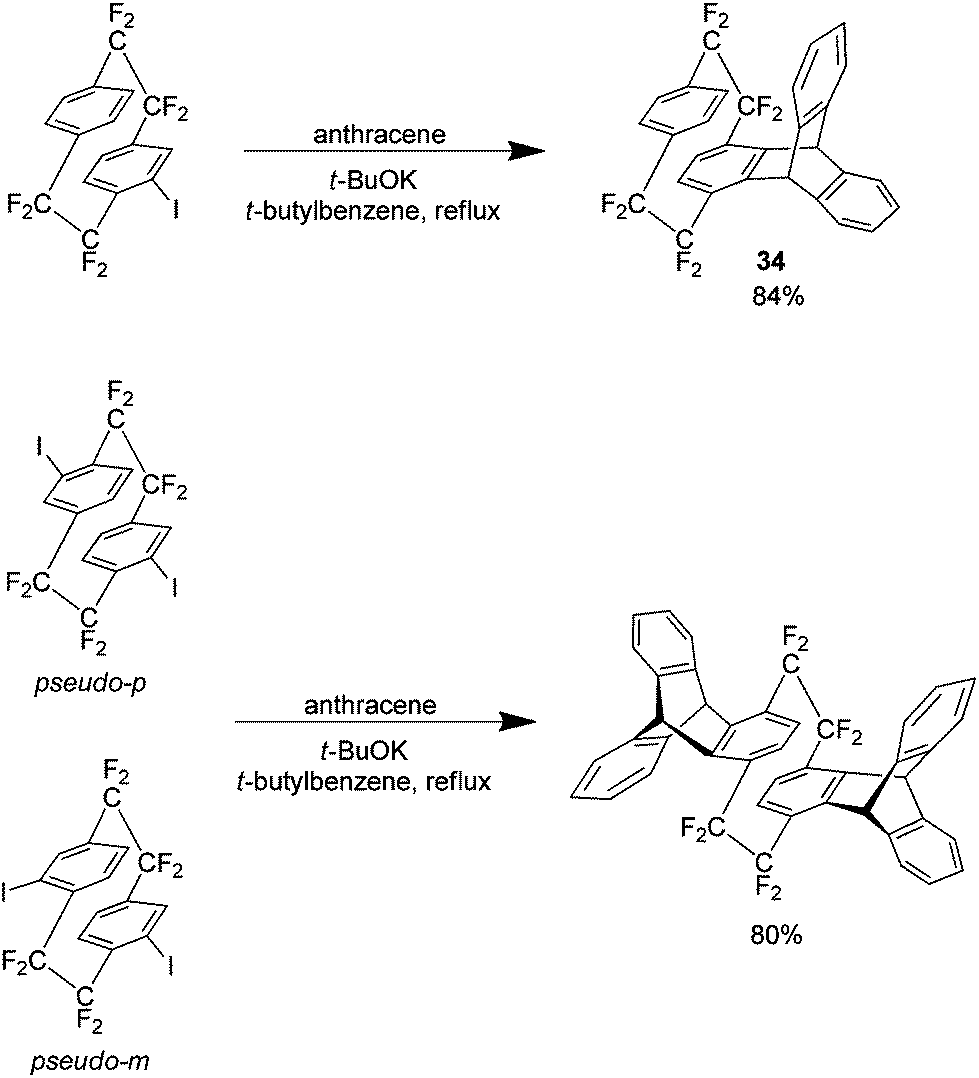 | ||
| Scheme 19 Diels–Alder cycloaddition reactions yielding substituted AF4 derivatives. | ||
As a continuation of this synthesis it was found that the Diels–Alder adduct 34 (amongst others) could be used to prepare novel cyclophanes such as the anthraceno analogue 35 (Scheme 20). This compound is of note because it was found to form a dimer when exposed to UV light. The structure is unusual in that the anthracene ring directly involved in the cyclophane system breaks aromaticity to form a cyclobutane ring.38
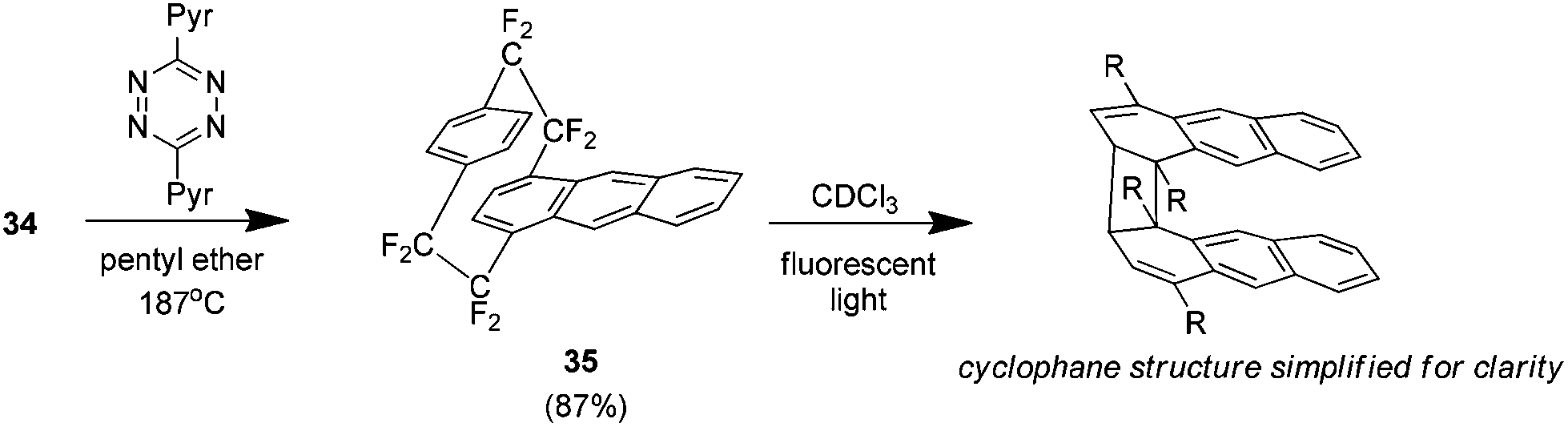 | ||
| Scheme 20 The preparation of (1,4)anthraceno-octafluoro[2.2]paracyclophane and its dimerisation. | ||
The approach used thus far in this area was the Cram methodology but Diels Alder adducts may also be produced using the Cadogan method.39 This method generates the aryne by reaction of an N-nitroacetanilide, produced in situ by the reaction of acetanilide and 4-chlorobenzoyl nitrite, which proceeds to react with the desired arynophile. This method is typically low yielding with the reaction of acetanilide and anthracene for example, yielding only 16% of the Diels–Alder adduct. When extended to the acetanilide derivative 36 however the yield is 90% (Scheme 21). The increased efficiency of the reaction is ascribed to the bridge fluorine atoms because they increase the ring hydrogen acidity and produce a more electron deficient aryne.
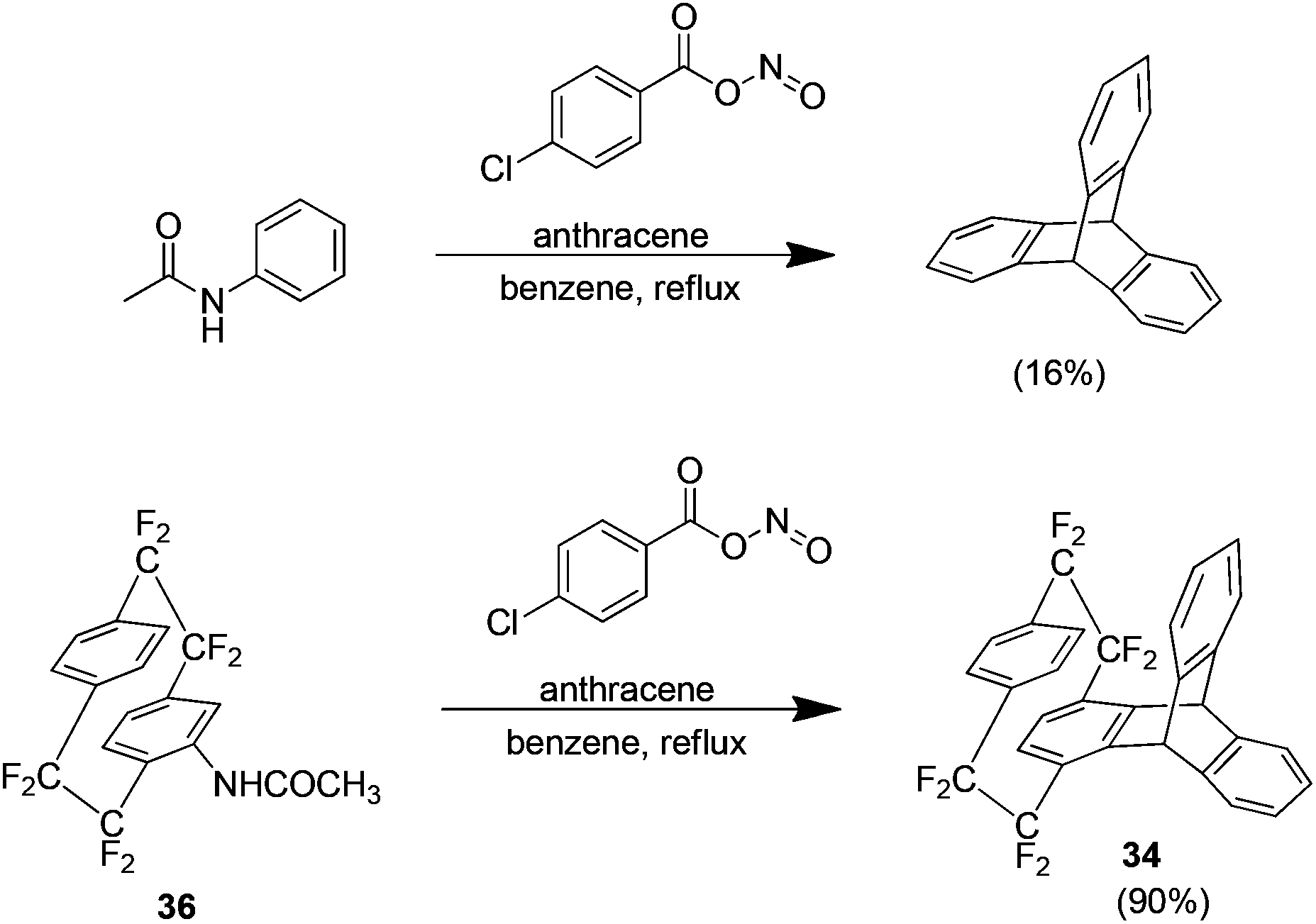 | ||
| Scheme 21 The preparation of the anthracene Diels–Alder adduct of octafluoro[2.2]paracyclophane. | ||
The Cadogan methodology was also found to be very effective for reactions with alkenes. For example the reaction with oct-1-ene yielded 91% of the ene product (Scheme 22). Interestingly the same reaction under Cram conditions yielded no product, only reduction of the aryl iodide. No alkene utilised was shown to produce any ene product under Cram conditions.
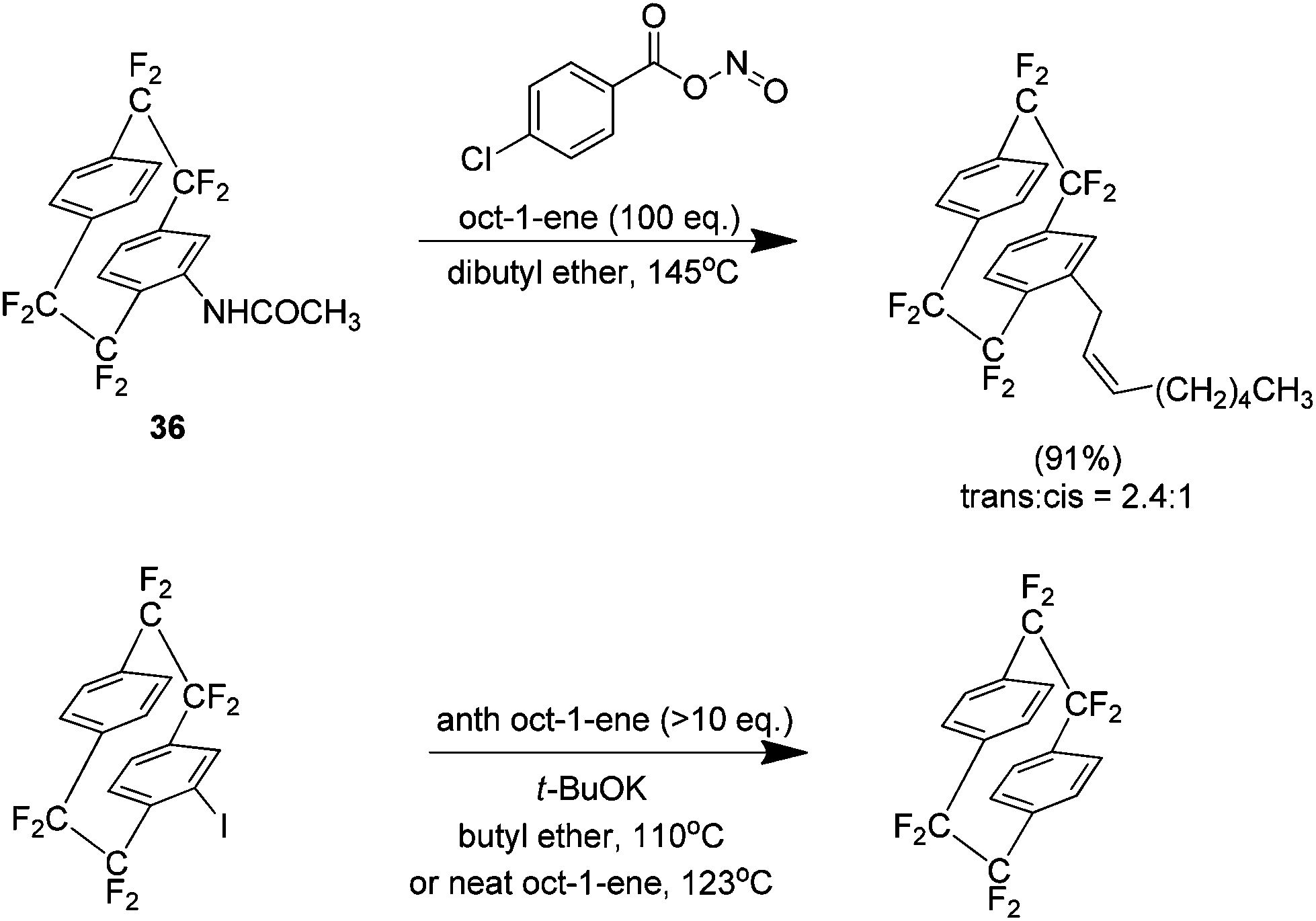 | ||
| Scheme 22 Ene reactivity of octafluoro[2.2]paracyclophane derivatives under Cadogan and Cram conditions. | ||
The final report relating to this area details a sequential ene reaction followed by a Diels–Alder reaction with cyclohepta-1,3,5-triene. The ene adduct produced via the Cadogan method reacts further with an additional aryne to yield a Diels–Alder adduct (Scheme 23). The product was synthesised as a diastereomeric 2:1 mixture in a yield of 25%. It was possible to fully characterise the mixture solely by NMR.40
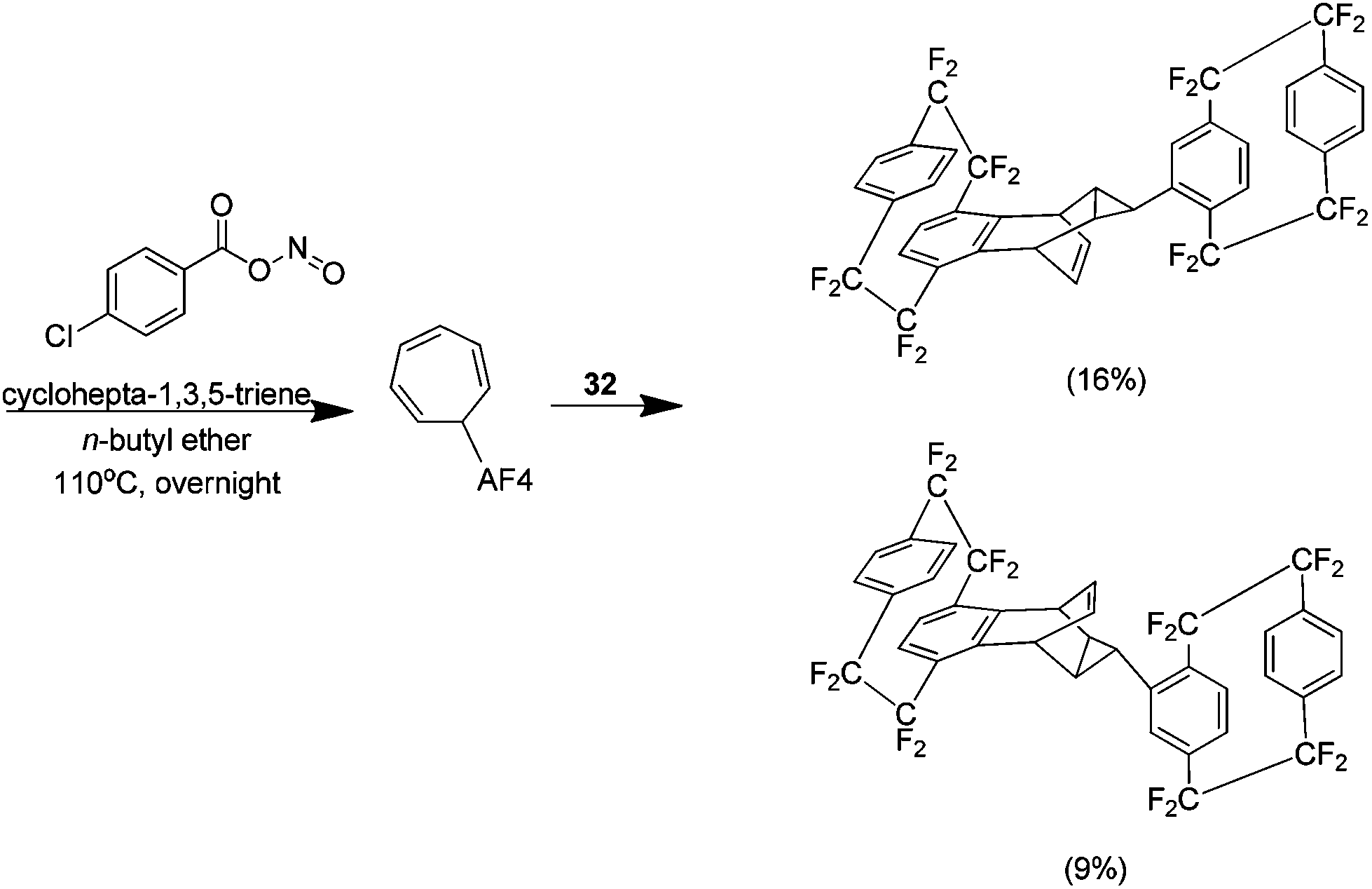 | ||
| Scheme 23 Sequential ene and Diels–Alder reaction of AF4-acetamide and cyclohepta-1,3,5-triene. | ||
The preparation, modification and analysis of perfluoro[2.2]paracyclophane
A logical progression from the preparation of 1 was the synthesis of perfluoro[2.2]paracyclophanes 37 (Scheme 24). The parylene coating produced from such a material was predicted to have an even lower dielectric constant and friction coefficient than parylene HT produced from 1. The initial preparation of this compound detailed two synthetic routes, a five step procedure beginning with 2,3,5,6-tetrachloroterphthalonitrile and a second refined pathway beginning with 1,2,4,5-tetrachlorobenzene (Scheme 24). Although a zinc-catalysed homocoupling is again utilised to produce the paracyclophane the presence of aromatic fluorine instead of hydrogen meant a different approach was required to make the necessary chlorodifluoro precursor. It was determined that ultimately 37 may be produced in a 39% yield when the coupling is done in acetonitrile at 100 °C for 38 hours.41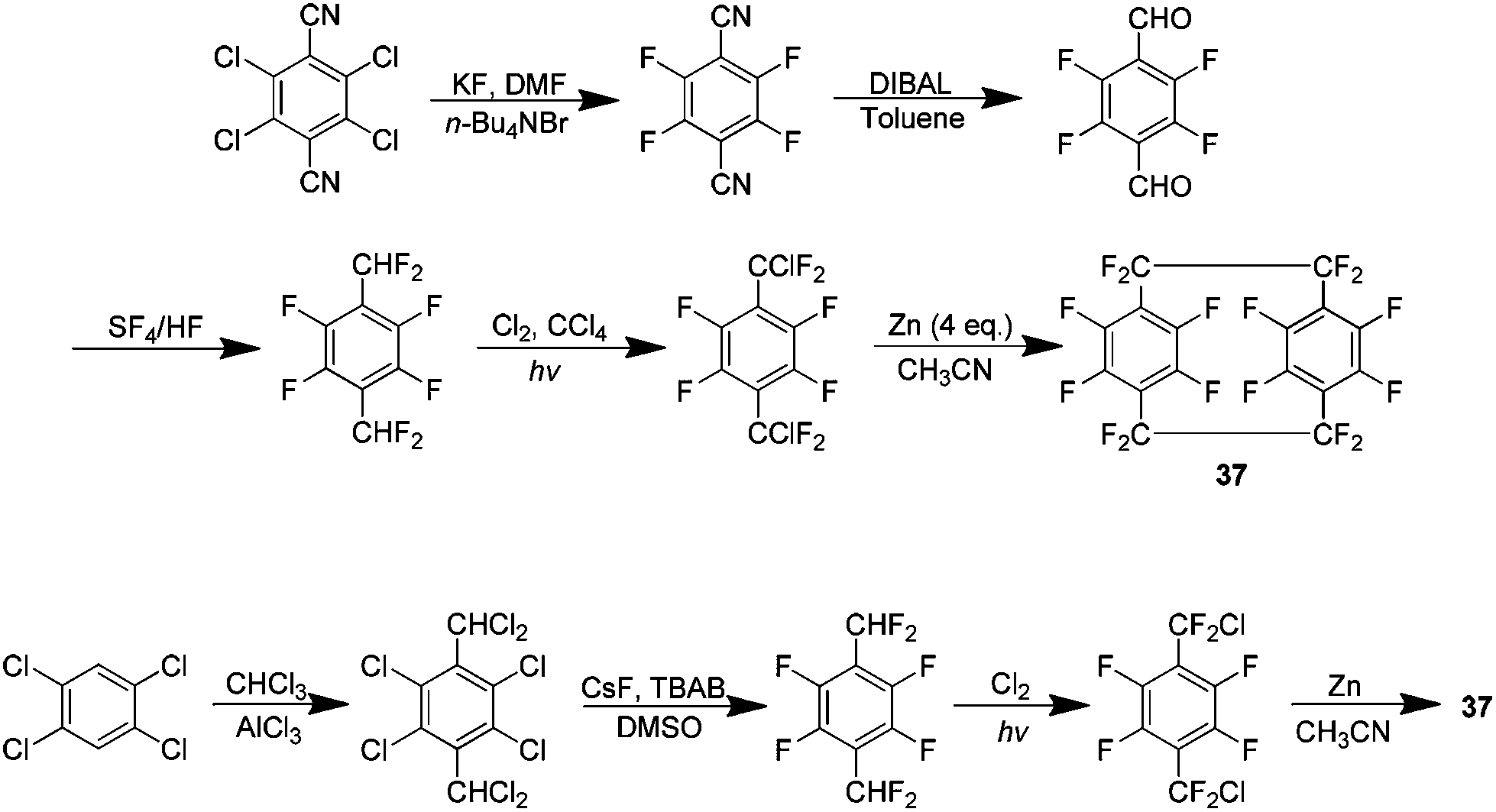 | ||
| Scheme 24 Two synthetic pathways for the production of perfluoro[2.2]paracyclophanes. | ||
Following the preparation of 37, an investigation was carried out to examine the reactivity of this compound to nucleophilic aromatic substitution.42 It was found that 37 could indeed be modified in this manner and several examples were reported using both mono and bidentate nucleophiles. The presence of such significant amounts of fluorine produces an extremely electron deficient aromatic system which is then quite reactive to nucleophiles (Scheme 25). No disubstitution is observed in the phenol derivative and this is likely due to the strongly electron donating ability of the phenoxide. It is noteworthy that this effect is transmitted to the other ring of the system. This then explains why the more weakly donating methoxy derivative shows some disubstitution exclusively in the form of the pseudo-para isomer. The reaction of 37 with sodium thiophenoxide yielded no monosubstituted product but gave a para isomer, indicating that this group had an activating effect. This is in agreement with a previous study of the reaction of sodium thiophenoxide and 2,3,5,6-tetrafluorobenzene.43 The most successful bidentate ligand class were found to be catechols followed closely by bis-amines. In a reaction with ethylene glycol no cyclisation product was observed. Competition experiments showed that 37 was in fact much more reactive than hexafluorobenzene under these conditions. It was determined that the most likely mechanistic pathway for these reactions is an SNAr mechanism.
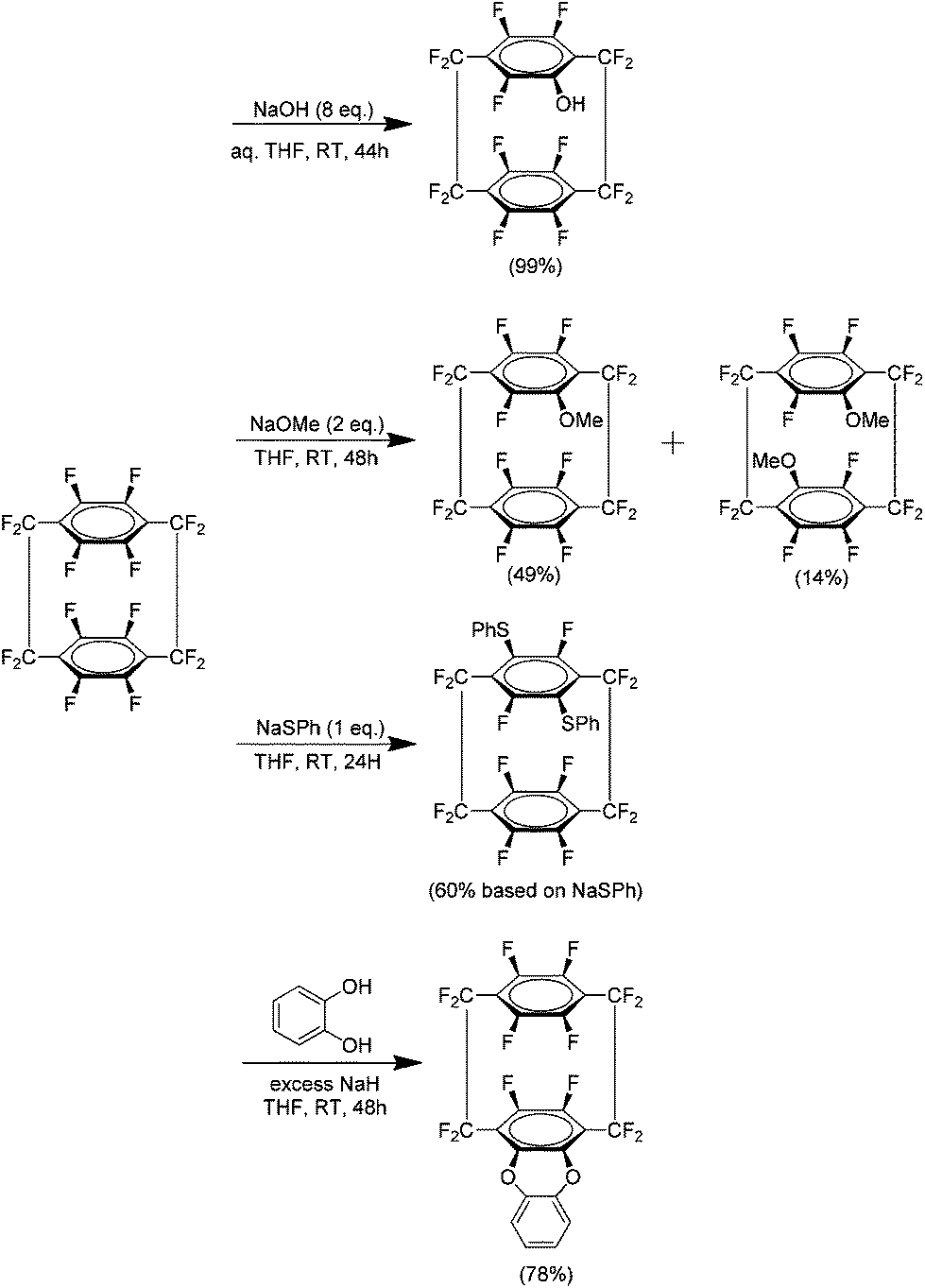 | ||
| Scheme 25 Nucleophilic substitution reactions of perfluoro[2.2]paracyclophanes. | ||
Monosubstituted analogues of 37 constitute a break in the symmetry and this allowed a detailed study of the spectral and conformational properties of these new analogues. Examination of the 19F coupling constants, particularly long range four and five bond couplings, allowed a detailed characterisation of the system and any disubstituted products could be differentiated. It was found that substitution skews the geometry of the system such that the unsubstituted ring may be drawn toward or away from the new substituent (Scheme 26). The results showed that this was not just due to the size of the substituent but is likely to be heavily dependent on the electronic environment of the aromatic rings. The experimental data was found to be in good agreement with computational models.44
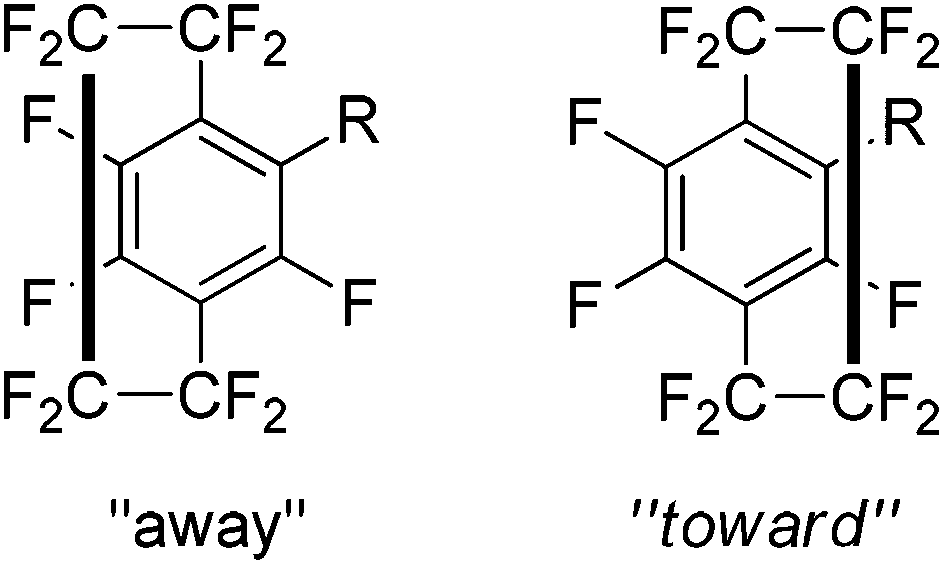 | ||
| Scheme 26 Possible conformations of the paracyclophanes system with respect to new substituent. | ||
The most recent report concerning 37 is the atropoisomerism of the acetoacetic ester produced by the reaction of 37 and ethyl acetoacetate.45 Atropisomers are stereoisomers resulting from restricted rotation about a single bond where the barrier of rotation is sufficiently high that both isomers can be isolated. In this case the isomers were produced as a mixture (Scheme 27) in the form of a crystalline solid and it was readily determined that the compound exists purely in the enol form with no keto tautomer detected. In solution the energy barrier to rotation was found to be 23.5 Kcal mol−1 and it is suggested this is due to interference from the close fluorinated methylene bridge group. The ester was then cyclised under basic conditions to a furan product which was confirmed by NMR. Finally, computational models were used to support mechanistic theories proposed and the conformations suggested by the NMR results.
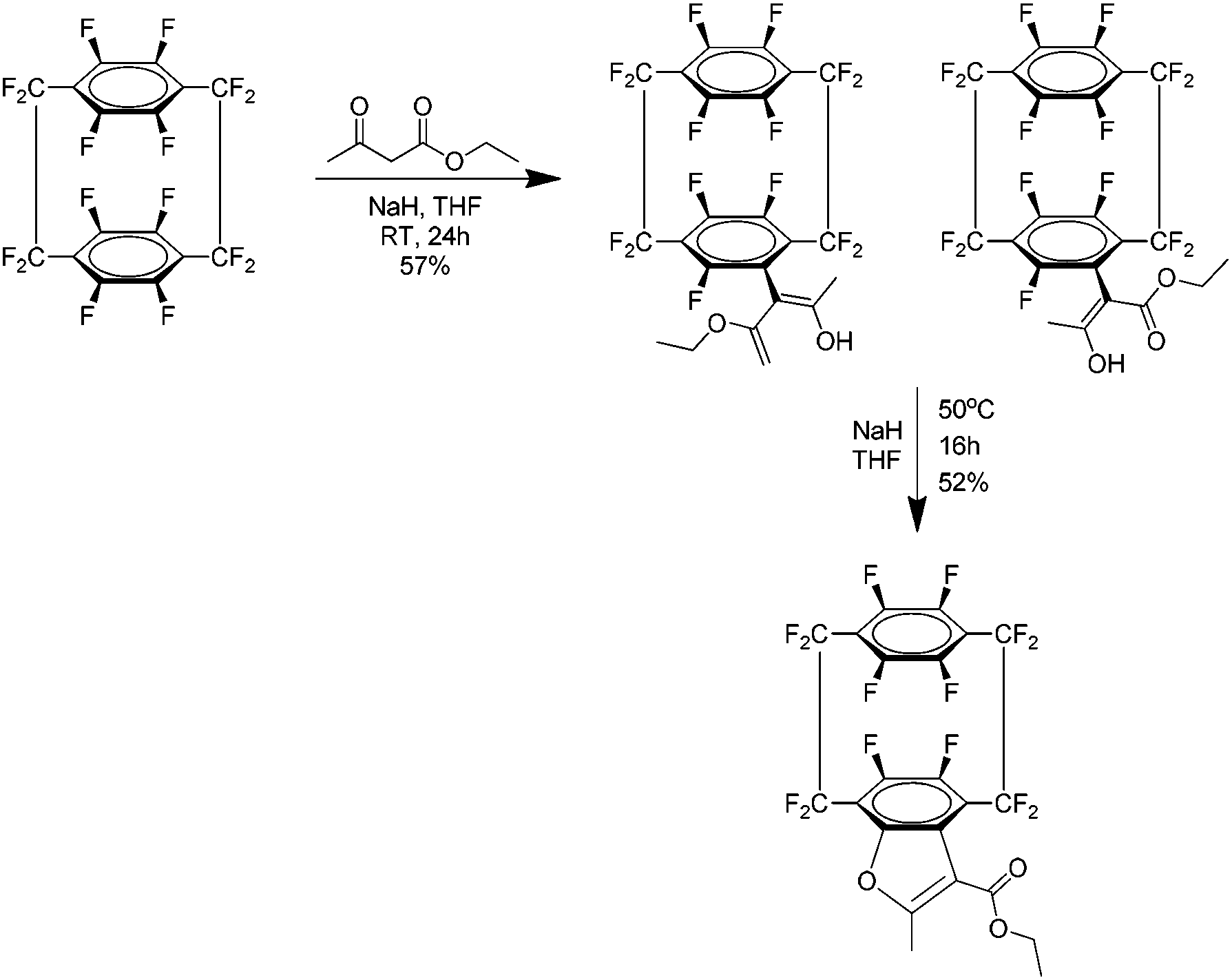 | ||
| Scheme 27 The preparation of the acetoacetic ester derivative of perfluoro[2.2]paracyclophane and its subsequent cyclisation under basic conditions. | ||
Conclusions
This review has discussed the history and development of octafluoro[2.2]paracyclophane and subsequent investigations into its chemistry. The development of facile synthetic pathways to the compound and the subsequent functionalisation of its structure have demonstrated that it is amenable to a broad variety of chemistry. Its unusual electronic properties, ascribed to the highly fluorinated structure, have led to it being surprisingly efficacious under a range of conditions. At present it is a highly desirable material in industry for hydrophobic protective coatings. Research to date has demonstrated that it would be an ideal precursor to a new generation of multifunctional coatings as it can be readily modified. The chirality of the substituted paracyclophanes framework, along with its unique electronic properties, means that it could have applications as a scaffold in asymmetric synthesis and catalyst design, areas which have not yet been fully explored.Acknowledgements
The authors would like to acknowledge the support of the Metal Improvement Company and Enterprise Ireland for funding this work through the Innovation Partnership program.Notes and references
- C. J. Brown and A. C. Farthing, Nature, 1949, 164, 915 CrossRef CAS.
- W. F. Gorham, J. Polym. Sci., 1966, 4, 3027 CrossRef CAS.
- H. Hopf and R. Gleiter, Modern Cyclophane Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2004 Search PubMed.
- W. R. Dolbier and W. F. Beach, J. Fluorine Chem., 2003, 122, 97 CrossRef CAS.
- D. F. Pollart, Pet. Chem. Repr. 150th Natl. Meet. Am. Chem. Soc., Atl. City, NJ, 1965, 175.
- W. R. Hertler, J. Org. Chem., 1963, 28, 2877 CrossRef CAS.
- S. W. Chow, L. A. Pilato and W. L. Wheelwright, J. Org. Chem., 1969, 35, 20 CrossRef.
- H. E. Winberg, F. S. Fawcett, W. E. Mochel and C. W. Theobald, J. Am. Chem. Soc., 1960, 82, 1428 CrossRef CAS.
- W. R. Dolbier, M. A. Asghar, H. Pan and L. Celewicz, J. Org. Chem., 1993, 58, 1827 CrossRef CAS.
- H. Sato, N. Isono, K. Okamura, T. Date and M. Mori, Tetrahedron Lett., 1994, 35, 2035 CrossRef CAS.
- H. Sato, N. Isono, I. Miyoshi and M. Mori, Tetrahedron, 1996, 52, 8143 CrossRef CAS.
- W. F. Beach, W. R. Dolbier, X. X. Rong and Y. Xu, J. Org. Chem., 1997, 62, 7500 CrossRef.
- W. R. Dolbier, J. Duan and A. J. Roche, Org. Lett., 2000, 2, 1867 CrossRef CAS PubMed.
- H. Amii, Y. Hatamoto, M. Seo and K. Uneyama, J. Org. Chem., 2001, 66, 7216 CrossRef CAS.
- W. R. Dolbier, Jr., M. A. Asghar and H.-Q. Pan, EU Pat, 0549980A1, 1993 Search PubMed.
- W. R. Hasek, W. C. Smith and V. A. Engelhardt, J. Am. Chem. Soc., 1960, 82, 543 CrossRef CAS.
- G. W. Kabalka and Z. Wu, Tetrahedron Lett., 2000, 41, 579 CrossRef CAS.
- W. R. Dolbier, J.-X. Duan and X. X. Rong, J. Fluorine Chem., 2007, 128, 1091 CrossRef CAS PubMed.
- C.-Y. Ho, T.-F. Lin, C.-H. Lin and S.-J. Wang, US Pat, 7173159B2, 2007 Search PubMed.
- S.-J. Jong, C.-H. Lin, C.-C. Tu and C.-C. Chang, US Pat, 7060862B2, 2006 Search PubMed.
- A. J. Roche and W. R. Dolbier, J. Org. Chem., 1999, 64, 9137 CrossRef CAS.
- A. J. Roche and W. R. Dolbier, J. Org. Chem., 2000, 65, 5282 CrossRef CAS.
- H. J. Reich and D. J. Cram, J. Am. Chem. Soc., 1969, 91, 3517 CrossRef CAS.
- H. Sawada and M. Nakayama, J. Fluorine Chem., 1990, 46, 423 CrossRef CAS.
- W. R. Dolbier, J. Duan, K. Abboud and B. Ameduri, J. Am. Chem. Soc., 2000, 122, 12083 CrossRef CAS.
- K. V. Scherer, T. Ono, K. Yamanouchi, R. Fernandez and P. Henderson, J. Am. Chem. Soc., 1985, 107, 718 CrossRef CAS.
- A. J. Roche, W. R. Dolbier and K. A. Abboud, J. Org. Chem., 2001, 66, 7055 CrossRef CAS PubMed.
- A. J. Roche and B. Canturk, J. Fluorine Chem., 2005, 126, 481 CrossRef PubMed.
- K. Wu, W. R. Dolbier, M. A. Battiste and Y. Zhai, Mendeleev Commun., 2006, 16, 146 CrossRef PubMed.
- W. R. Dolbier, Y. Zhai, W. Xu, W. Wheelus, F. Dulong, E. Goldberg, I. Ghiviriga and M. a. Battiste, J. Fluorine Chem., 2008, 129, 1133 CrossRef CAS PubMed.
- W. Adam, A. M. Miranda, F. Mojarrad and H. Sheikh, Chem. Ber., 1994, 127, 875 CrossRef CAS.
- S. Sankararaman, H. Hopf, I. Dix and P. G. Jones, Eur. J. Org. Chem., 2000, 2711 CrossRef CAS.
- M. A. Battiste, J. Duan, Y. Zhai, I. Ghiviriga, K. A. Abboud and W. R. Dolbier, J. Org. Chem., 2003, 68, 3078 CrossRef CAS PubMed.
- M. A. Battiste, J. Duan, Y. Zhai, I. Ghiviriga, K. A. Abboud, A. Roitberg, G. R. Shelton and W. R. Dolbier, Tetrahedron Lett., 2002, 43, 7047 CrossRef CAS.
- D. T. Longone and G. R. Chipman, J. Chem. Soc. D, 1969, 1358 RSC.
- H. J. Reich and D. J. Cram, J. Am. Chem. Soc., 1969, 91, 3527 CrossRef CAS.
- J. I. G. Cadogan, J. K. A. Hall and J. T. Sharp, J. Chem. Soc., 1967, 1860 CAS.
- W. R. Dolbier, Y. Zhai, M. A. Battiste, K. A. Abboud and I. Ghiviriga, ARKIVOC, 2006, 2006, 97 CrossRef.
- W. R. Dolbier, Y.-A. Zhai, W. Wheelus, M. A. Battiste, I. Ghiviriga and M. D. Bartberger, J. Org. Chem., 2007, 72, 550 CrossRef CAS PubMed.
- W. R. Dolbier, Y. Zhai, W. Wheelus, M. a. Battiste and I. Ghiviriga, J. Fluorine. Chem., 2008, 129, 28 CrossRef CAS PubMed.
- W. R. Dolbier, P. Xie, L. Zhang, W. Xu, Y. Chang and K. A. Abboud, J. Org. Chem., 2008, 73, 2469 CrossRef CAS PubMed.
- L. Zhang, K. Ogawa, I. Ghiviriga and W. R. D. Jr., J. Org. Chem., 2009, 74, 6831 CrossRef CAS PubMed.
- K. R. Langille and M. E. Peach, J. Fluorine Chem., 1971, 4, 407 Search PubMed.
- I. Ghiviriga, L. Zhang, H. Martinez, R. H. Contreras, C. F. Tormena, L. Nodin and W. R. Dolbier, Magn. Reson. Chem., 2011, 49, 93 CrossRef CAS PubMed.
- I. Ghiviriga, H. Martinez, C. Kuhn, L. Zhang and W. R. Dolbier, Org. Biomol. Chem., 2012, 10, 882 CAS.
| This journal is © the Partner Organisations 2014 |