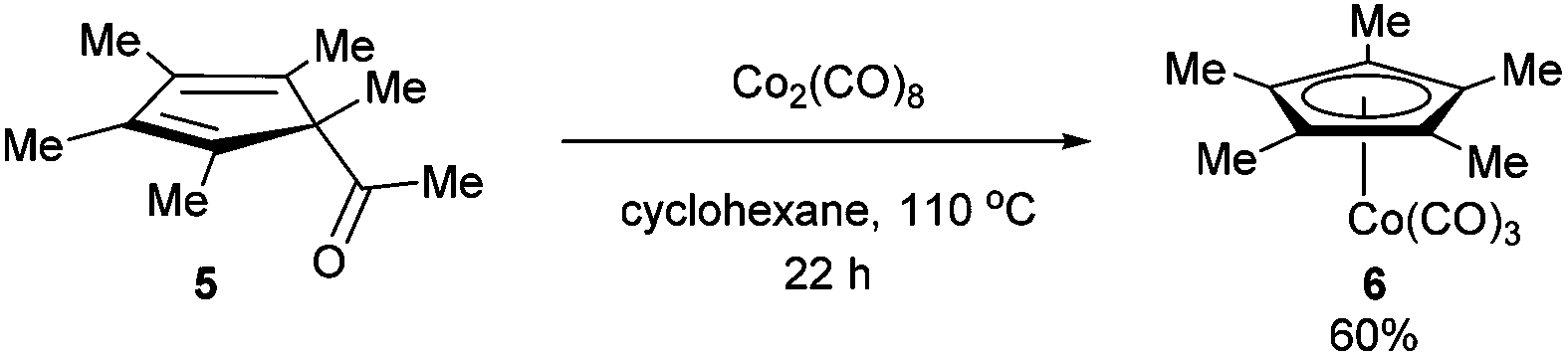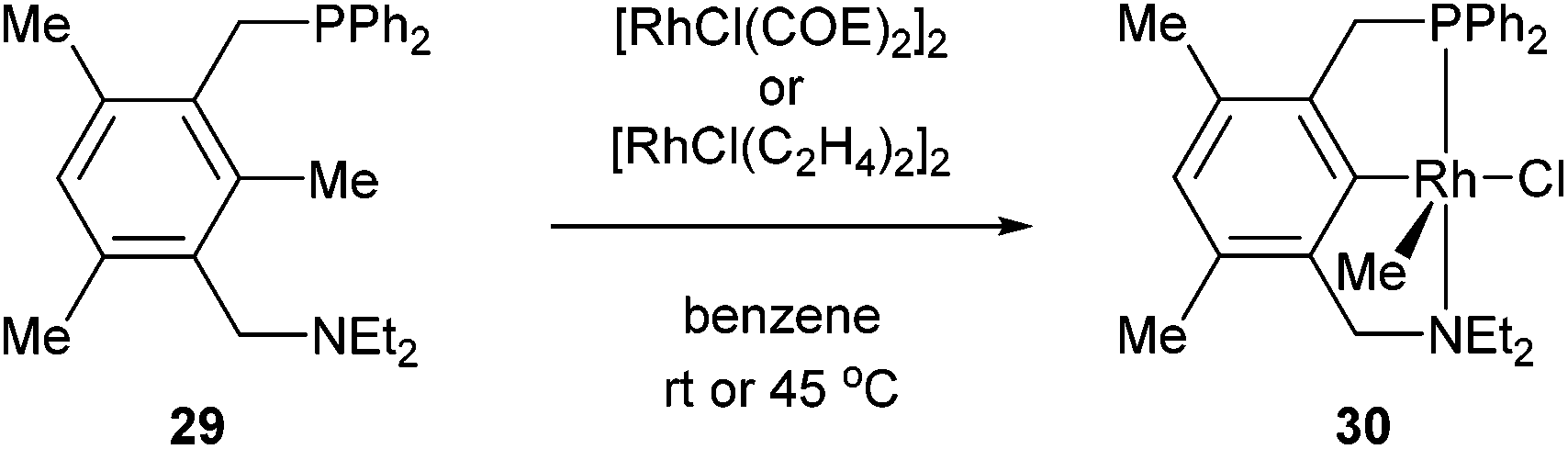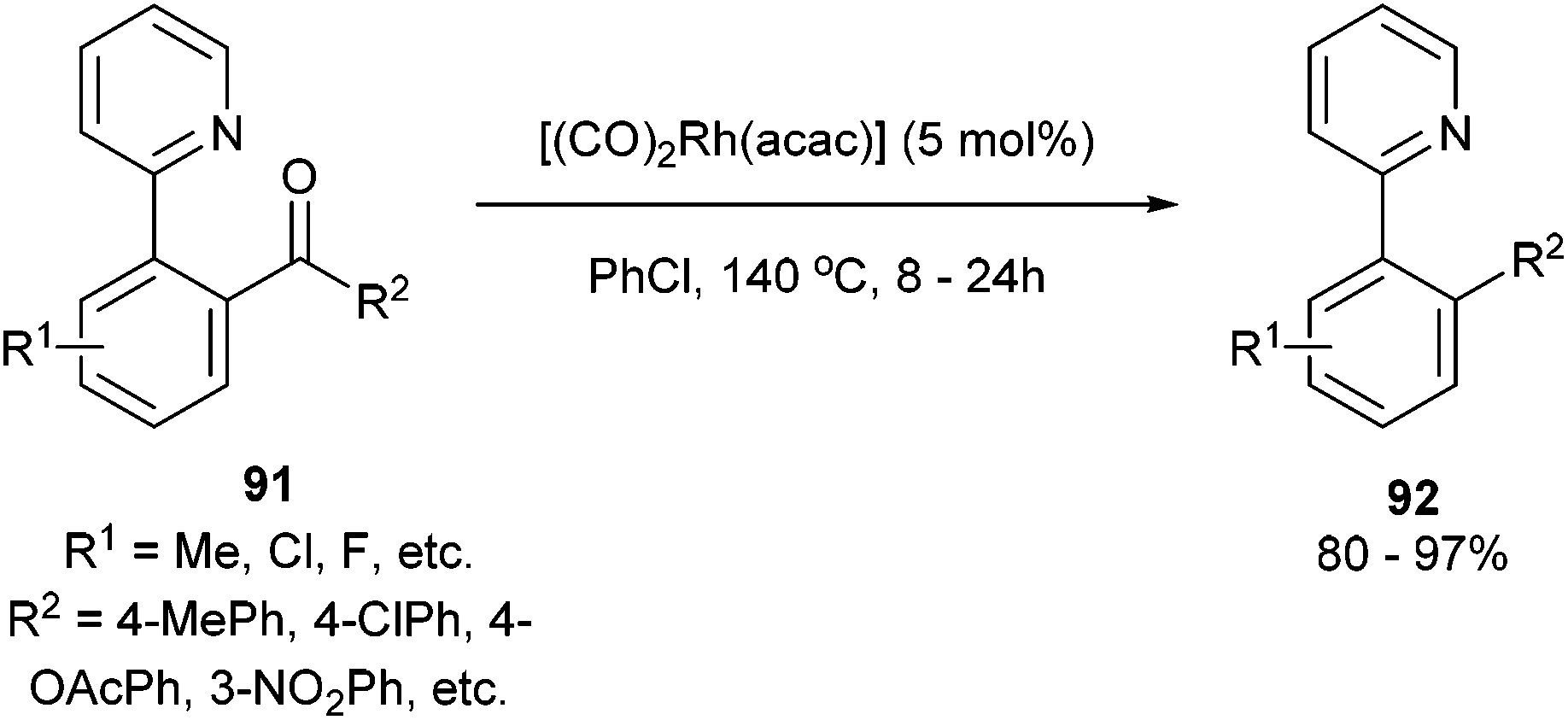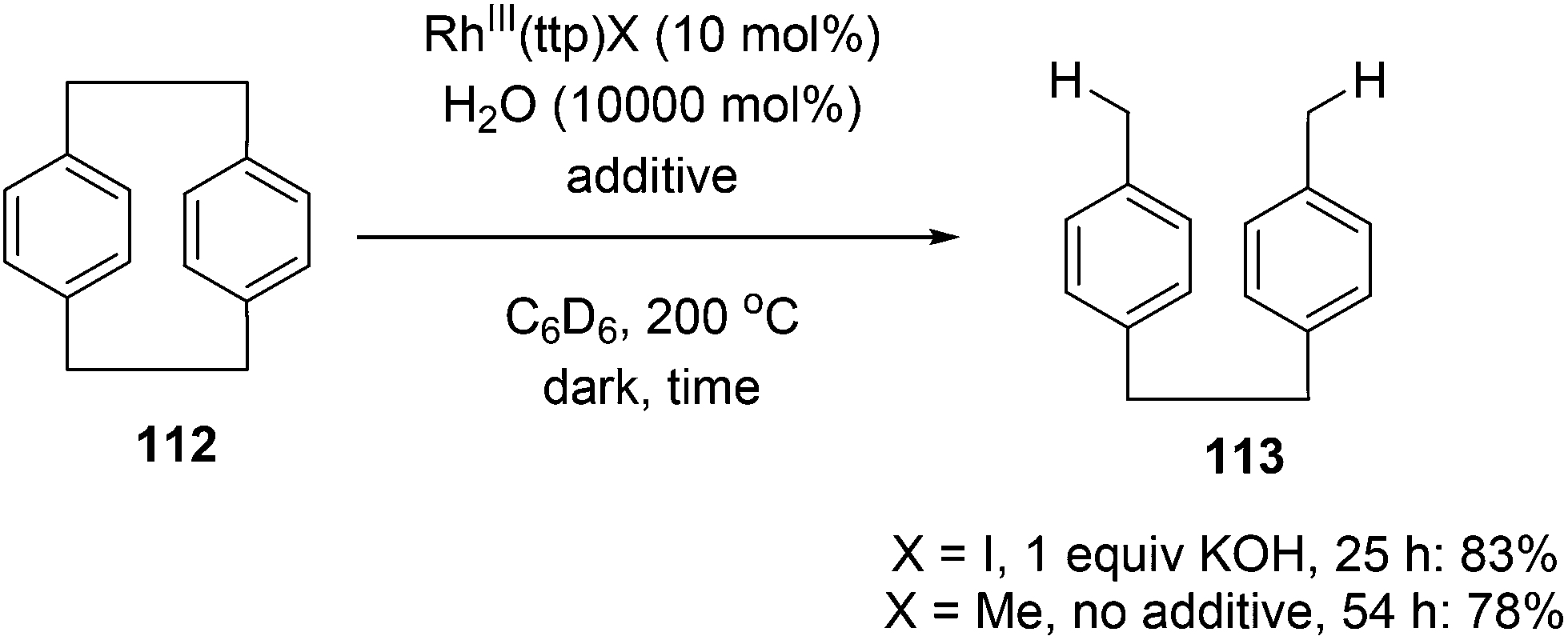DOI:
10.1039/C4QO00053F
(Review Article)
Org. Chem. Front., 2014,
1, 567-581
Direct activation of relatively unstrained carbon–carbon bonds in homogeneous systems
Received
25th February 2014
, Accepted 27th March 2014
First published on 27th March 2014
Abstract
New modes of chemical reactivity are of high value to synthetic organic chemistry. In this vein, carbon–carbon (C–C) activation is an emerging field that offers new possibilities for synthesizing valuable complex molecules. This review discusses the pioneering stoichiometric discoveries in this field up to the most recent synthetic applications that apply catalytic transformations. Specifically, the review focuses on C–C activation in relatively unstrained systems, including stoichiometric reactions, chelation-directed and chelation-free catalytic reactions. While the field of C–C activation of relatively unstrained systems is underdeveloped, we expect that this review will provide insight into new developments and pave the path for robust, practical applications.
 Alpay Dermenci | Alpay Dermenci was born in Huntington Beach, CA (USA) in 1984. He received his B.S. from University of California, Irvine and carried out his doctoral studies under the guidance of Prof. Scott J. Miller at Yale University. His research focused on the application and development of amino acids and peptides as catalysts for the synthesis of natural products. Following his Ph.D., he conducted post-doctoral studies with Prof. Guangbin Dong at UT Austin focusing on the development of C–C activation methodology. Alpay is currently a research scientist at Pfizer, Inc. in Groton, CT. |
 Jotham W. Coe | Jotham W. Coe, a Research Fellow at Pfizer, is a medicinal chemist in BioTherapeutics working towards treatments for inflammatory conditions. Coe spent 15 years in the Neuroscience Department (CNS) pursuing medicines for depression, schizophrenia, ADHD and addiction, where he was directly involved in the discovery of Chantix (Champix), an aid to smoking cessation. Coe earned his A.B. degree in chemistry from Harvard College in 1981, worked 2 years at SISA Pharmaceutical Laboratories, then entered MIT to study with Professor William R. Roush. He received his Ph.D. degree in 1988 and joined Pfizer Central Research. Coe has published extensively in the area of organic and medicinal chemistry with a focus on reaction mechanism and synthesis and has received numerous patents in the areas of oncology, neuroscience and inflammation. |
 Guangbin Dong | Guangbin Dong received his B.S. degree from Peking University and completed his Ph.D. degree in chemistry from Stanford University with Professor Barry M. Trost, where he was a Larry Yung Stanford Graduate fellow. In 2009, He began to research with Prof. Robert H. Grubbs at California Institute of Technology, as a Camille and Henry Dreyfus Environmental Chemistry Fellow. In 2011, he joined the department of chemistry and biochemistry at the University of Texas at Austin as an assistant professor and a CPRIT Scholar. His research interests lie at the development of powerful chemical tools for addressing questions of biological importance. |
New modes of catalytic chemical reactivity can provide powerful transformations for synthetic chemists to attain higher efficiency and more direct routes to desired molecules. Two areas of research that fit the criteria for novel mechanisms and transformations include carbon–hydrogen (C–H) and carbon–carbon (C–C) activation. While catalytic C–H activation has garnered significant recent attention by the synthetic community, reports of C–C activation methods are less prominent in the literature. C–C Activation is often viewed as a destructive mode of reactivity, counter to the primary focus on developing methods to form C–C bonds rather than to break them. This review will show how selective functionalization of the metal center in conjunction with C–C activation can lead to unique methods for constructive bond formation.
When compared to C–H activation, C–C activation is kinetically challenging. C–H bonds are typically more abundant and C–C bonds are hindered. Therefore, C–H bonds more readily display proper orbital overlap trajectories with the metal center than C–C bonds. Thermodynamics play less of a role as C–C oxidative addition can in fact be exothermic, although cleavage of C–C bonds can also be thermodynamically uphill. Methods for transition-metal-mediated C–C activation employ strategies mainly involving strain-release, aromatization, and chelation-assistance (proximity effect) to overcome these barriers. The latter two have proven useful for the activation of unstrained C–C bonds, which remain particularly challenging in the context of C–C activation of unstrained C–C bonds.
This review primarily focuses on the direct activation of C–C bonds in relatively unstrained systems, i.e. non-three or four-membered ring systems. While not intended to comprehensively cover all literature references, it rather provides a perspective on the scope of reactivity through selected examples to highlight representative reactions types. Chronological order is roughly followed to loosely track the progress of research for the activation of unstrained C–C bonds. Three main sections will be discussed: (1) stoichiometric C–C activation reactions, (2) chelation-directed catalytic C–C activation reactions and (3) chelation-free catalytic C–C activation reactions. Olefin metathesis, retro-aldol reactions, β-carbon elimination reactions, C–CN activation,1 decarboxylation,2 deallylation,3 solid-state photochemical4 reactions, diazoalkane-carbonyl homologation,1 Baeyer–Villiger oxidation and other oxidative C–C cleavage reactions, while all involving C–C bond breaking or rearrangement, will not be discussed here as these topics are either beyond the scope of the review or have been thoroughly reviewed elsewhere.5
Stoichiometric reactions
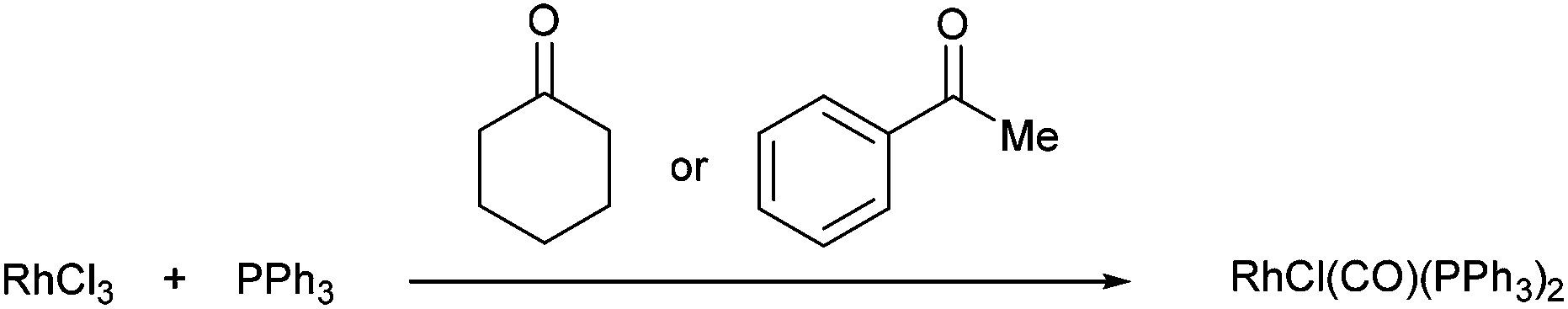
One of the first examples of C–C activation in an unstrained system was reported by Rusina and co-workers in 1965.6 They observed that when RhCl3 and PPh3 were heated in a number of different alcoholic solvents, they obtained golden-yellow crystals of RhICl(CO)(PPh3)2. The same complex was isolated when conducted in cyclohexanone and acetophenone at reflux. Although no mechanism was proposed, it is likely that RhICl(CO)(PPh3)2 formation is the result of a C–C activation event. Furthermore, they suggest that elevated temperatures were required as no complex was formed when the reaction was conducted in acetone (b.p. 57 °C).| | 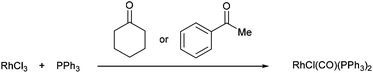 | (1) |
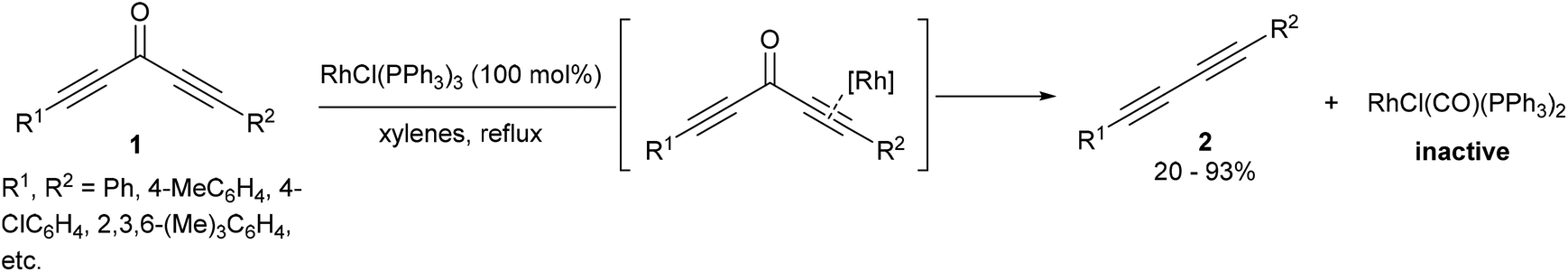
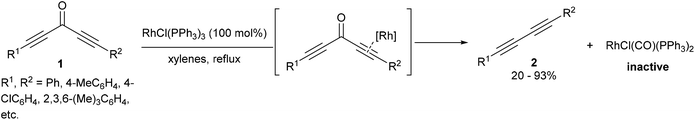
A few years after the work of Rusina, Müller and co-workers7,8 showed that Wilkinson's complex RhCl(PPh3)3 can cleave the sp–sp2 bonds of diynones (1), resulting in decarbonylation and reductive elimination to produce diynes (2) and RhCl(CO)(PPh3)2 (eqn (2)). A stoichiometric amount of RhCl(PPh3)3 was required for full conversion as the RhCl(CO)(PPh3)2 carbonyl byproduct does not catalyze the reaction. This method was demonstrated to work on a number of different symmetrical and unsymmetrical diynones with varying electronic and sterics properties to produce the corresponding diynes (20–93%). In one example, a monoynone produced a disubstituted alkyne in 8% yield. A mechanistic discussion in the catalytic version of this reaction will be presented in a later section of this review.
| |  | (2) |
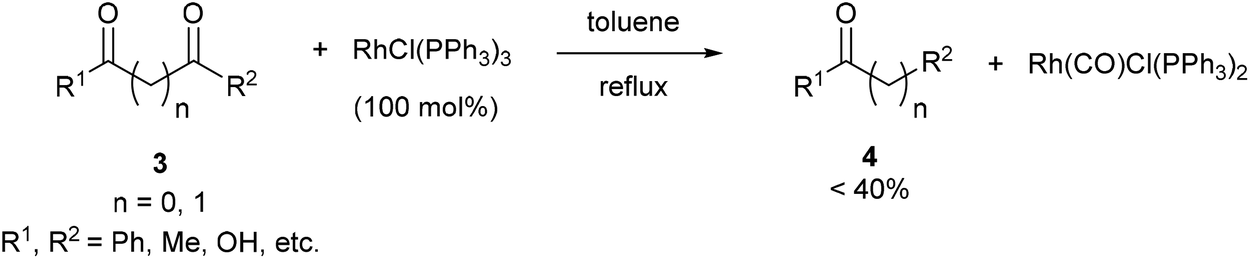
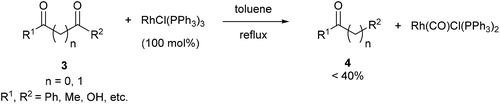
Wilkinson's complex effectively decarbonylates unstrained 1,2- and 1,3-diketones (3), as shown by Teranishi and co-workers9 who observed monoketone products (4) under refluxing toluene (eqn (3)). They reported the isolation of an acetylacetonato complex, which proved inactive as a catalyst. However, the Rh-carbonyl complex isolated from the reaction was found to be active in the decarbonylation of acetylacetone to provide methyl ethyl ketone. Their studies highlighted specificity of Rh in the decarbonylation of 1,2- and 1,3 diketones as other catalysts, such as RhCl3–3H2O, IrCl3–3H2O, PdCl2, and CoCl2–6H2O, were screened but failed to promote decarbonylation chemistry.
| |  | (3) |
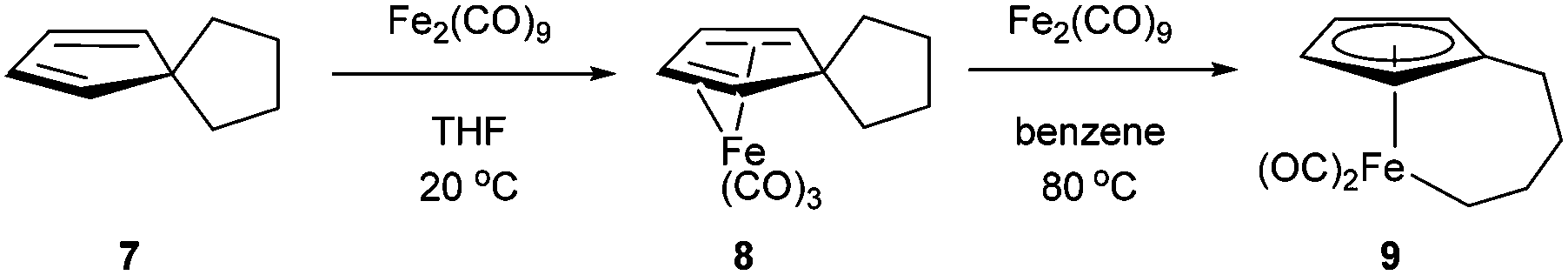
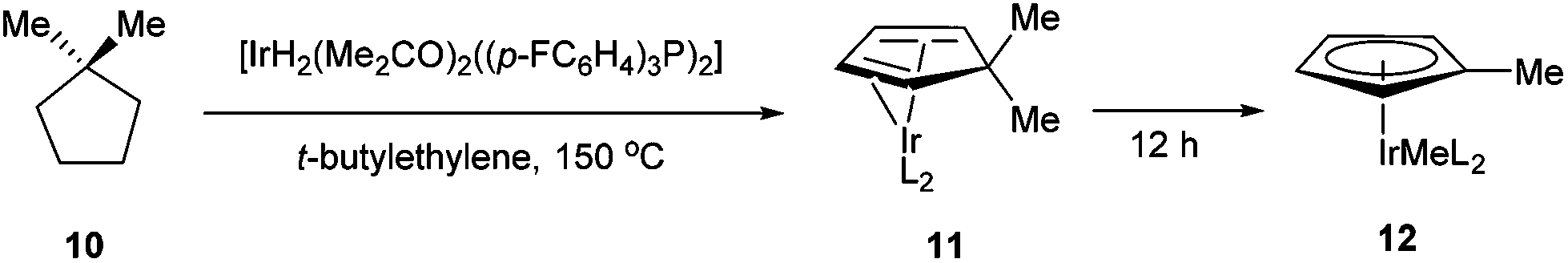
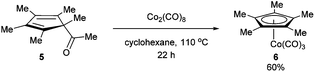
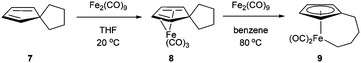
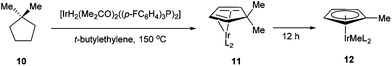
In 1971, King and co-workers showed that 5-acetyl-1,2,3,4,5-pentamethylcyclopentadiene (5) undergoes C–C bond cleavage when treated with Co2(CO)8 in cyclohexane at 110 °C for 22 h to form stable complex 6 (eqn (4)). Eilbracht and Dahler10 later demonstrated that alkyl substituents can also participate in C–C activation via aromatization. Diene 7 reacts with Fe2(CO)9 to form a stable complex (8), which when heated with additional Fe2(CO)9 in benzene at 80 °C provides a cyclic Fe species 9. Furthermore, Crabtree and co-workers11 also showed that iridium complexes can form Cp-complex via C–C bond cleavage in various gem-dialkyldienes (eqn (6)). Initially, [IrH2(Me2CO)2((p-FC6H4)3P)2] underwent dehydrogenation with compound 10 to form diene complex 11, and demethylation via C–C activation provided complex 12. While not all examples of aromatization strategies for C–C activation are covered here,12 these examples demonstrate that aromatization can drive C–C activation with a variety of different metals and substrates.
| |  | (4) |
| |  | (5) |
| |  | (6) |
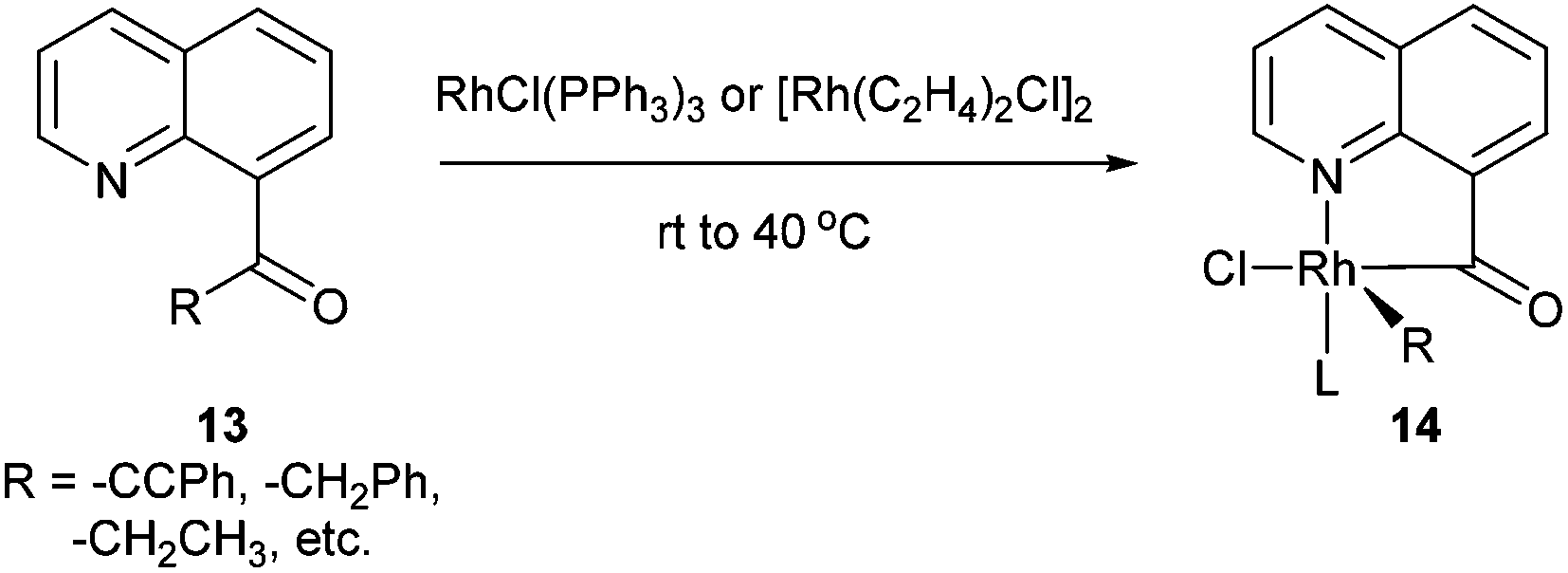
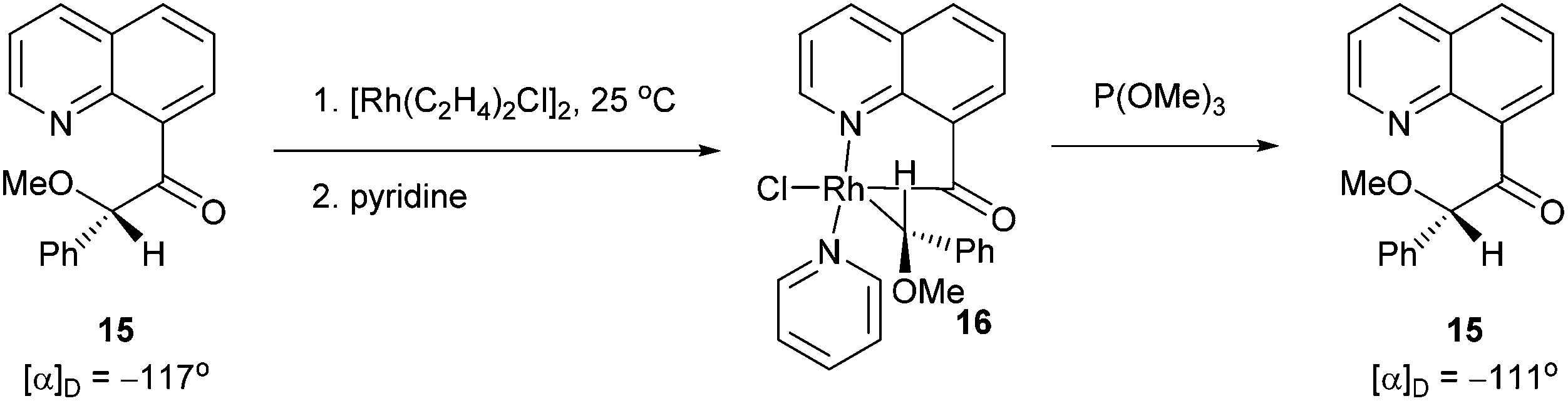
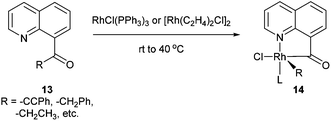
Chelation-assisted activation of α C–C bonds to a carbonyl group was first demonstrated by Suggs and co-workers13 through insertion of Rh into a series of quinoline-derived substrates under mild conditions (rt to 40 °C) (13, eqn (7)). This strategy was found applicable for the activation of alkynyl and alkyl ketones; however, styryl ketones did not react even at high temperature. Deuterium labeling studies showed that in this case C–H activation of the alkyl substituents did not occur prior to C–C activation. Furthermore, this approach was used to synthesize chiral rhodium alkyl species through the incorporation of an α chiral substituent (15, Scheme 1). When the chiral metal-complex 16 was treated with P(OMe)3, reductive elimination returned the starting material with no loss of optical purity. Given that reductive elimination typically occurs with retention of configuration14 C–C activation must also occur through retention of configuration. The authors proposed that the reaction followed a mechanism similar to the Baeyer–Villiger reaction through a tetrahedral intermediate.
| |  | (7) |
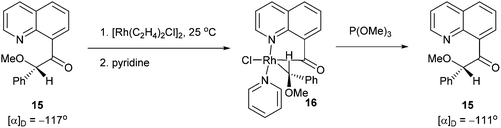 |
| | Scheme 1 Rh-promoted C–C activation of a chiral quinoline derived substrate. | |
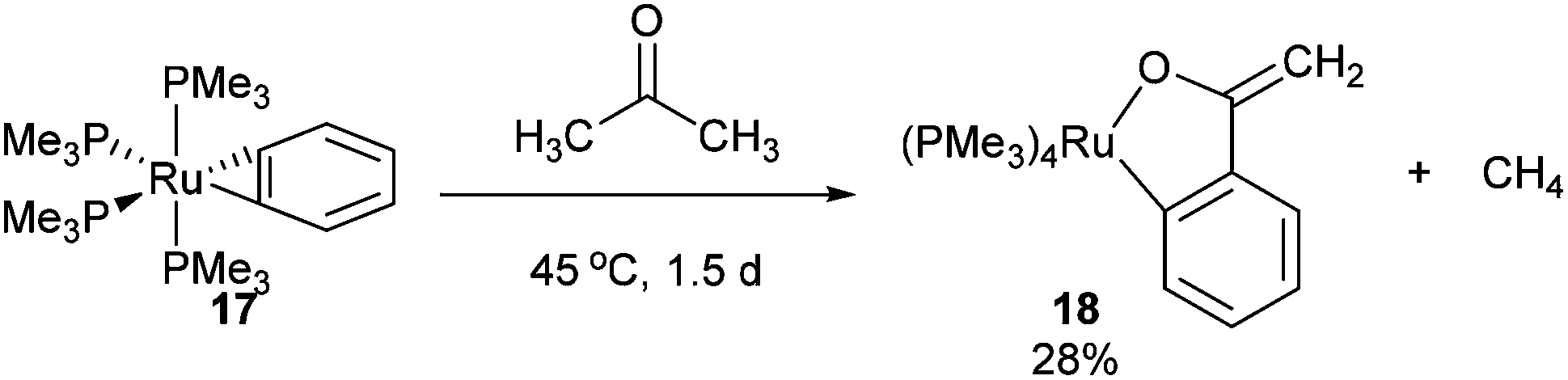
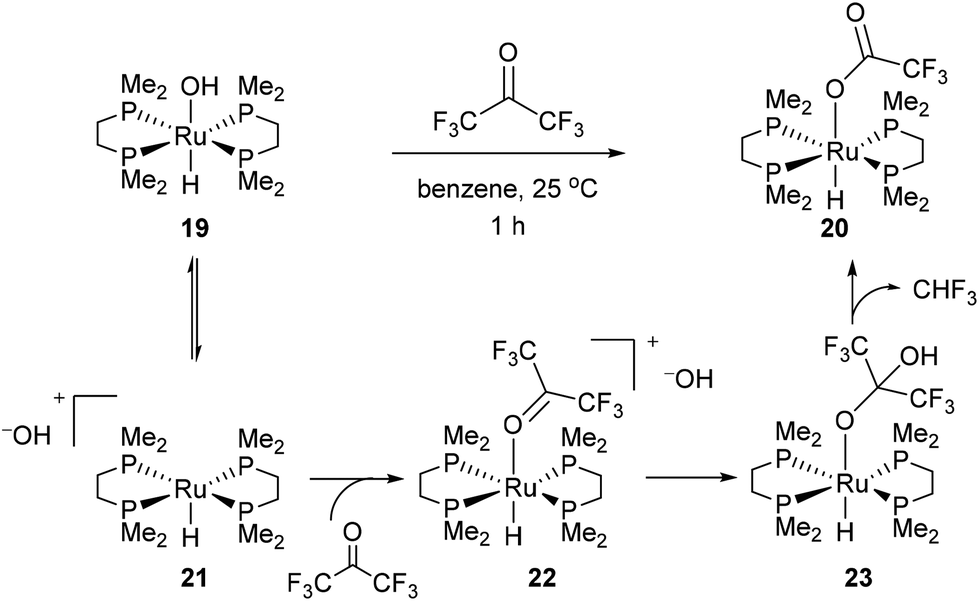
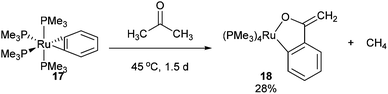
In 1989 Bergman and co-workers15 reported the synthesis of a highly reactive Ru benzyne complex 17 that was able to active C–H, C–N, N–H, O–H and C–C bonds under relatively mild conditions.16 When the catalyst is heated with acetone at 45 °C for 1.5 days, Ru complex 18 was obtained in 28% isolated yield and 1H NMR analysis identified methane as a byproduct (eqn (8)). Bergman later identified a hydroxyruthenium complex 19, also capable of promoting C–C activation under mild conditions. When hexafluoroacetone was treated with 19 in benzene at room temperature for 1 h, complex 20 was formed along with an equivalent of fluoroform. By the mechanism shown in Scheme 2, dissociation of hydroxide permits reversible formation of cationic complex 21, allowing the substrate to bind giving intermediate 22. Hydroxide ion attack on the carbonyl forms species 23, which undergoes C–C cleavage to produce fluoroform and complex 20.
| |  | (8) |
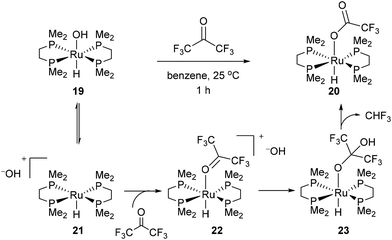 |
| | Scheme 2 Ru-promoted C–C activation of hexafluoroketone. | |
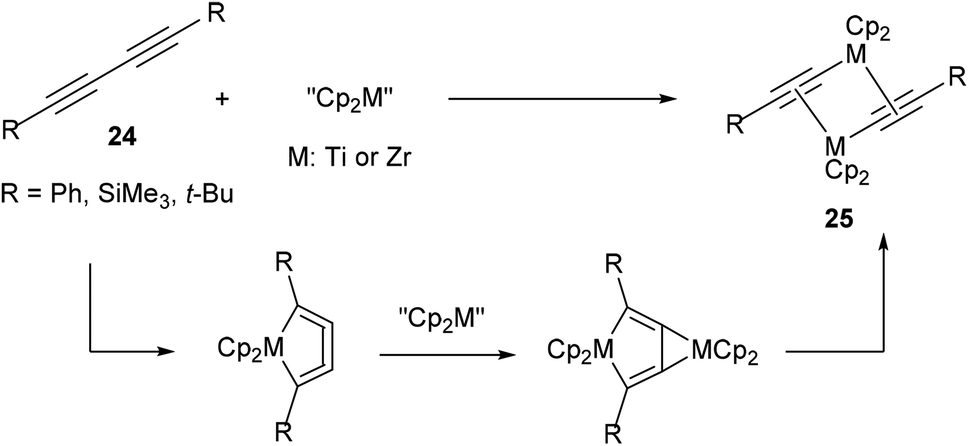
In 199217 and subsequent papers18 in following years, Rosenthal and co-workers reported that titanium and zirconium could be used to activate the internal C–C bond of diynes (24, Scheme 3). A general scheme is provided to illustrate the basic mechanism of this reaction. Most characteristic to the mechanism is that the reaction goes through a metallocyclocumulene to give the observed dimeric products (25). Crystallographic data was obtained to support the proposal of this mechanism. This work demonstrates that early transition metals are also capable of C–C bond activation.
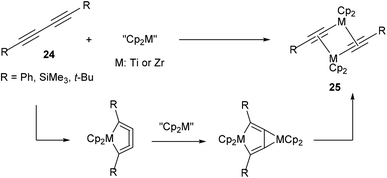 |
| | Scheme 3 Zr- and Ti-mediated C–C activation of diynes. | |
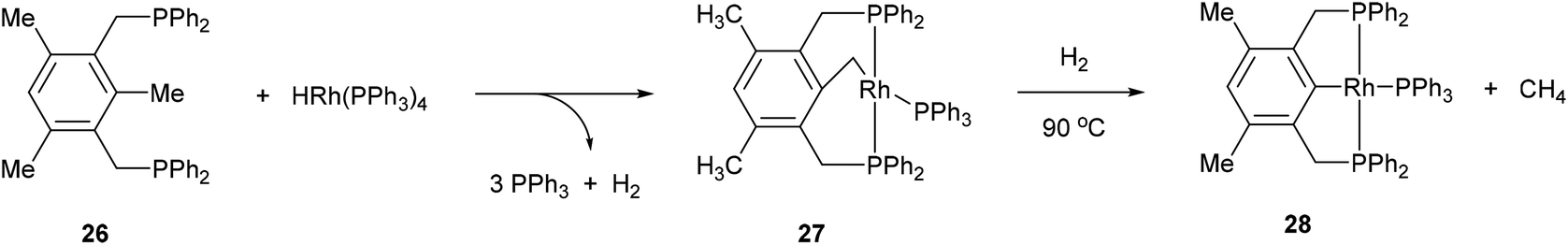
The activation of strong unstrained C–C bonds was demonstrated by Milstein and co-workers19 with pincer complexes. When bisphosphine 26 was reacted with HRh(PPh3)3 at room temperature in THF, a thermally stable C–H activation product was obtained in quantitative yield (27, Scheme 4). When this complex was heated at 90 °C in the presence of H2, the C–C activation product (28) was formed quantitatively. The mechanism of C–C activation likely involves reversible C–H activation followed by subsequent C–C activation. These results suggest that the C–C activation and formation of a strong Rh–Ar bond is thermodynamically more favorable than C–H activation. Milstein and co-workers20,21 indicated that the C–C activation event proceeds through a three-centered mechanism rather than an η2-arene complex suggesting that the metal center is perfectly positioned for direct C–C activation. Milstein and co-workers22 also studied an unsymmetrical pincer ligand bearing a phosphine and amine chelate (29). C–C Activation occurs exclusively (eqn (9)) under mild conditions (rt to 45 °C) to provide complex 30 with no evidence for C–H activation or initial complexation of Rh to the ligand. This observation may be explained by the less sterically demanding amine ligand with its hard electronic influence versus a soft phosphine promoting rapid reversible C–H activation. These experiments demonstrate the feasibility of activating unstrained C–C bonds, and support the notion that the cleavage can be favored from both kinetic and thermodynamic respects under the correct circumstances.
| | 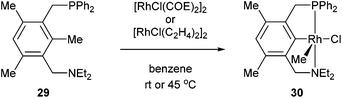 | (9) |
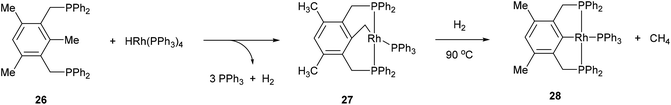 |
| | Scheme 4 Activation of strong C–C bonds in pincer ligands reported by Milstein and co-workers. | |
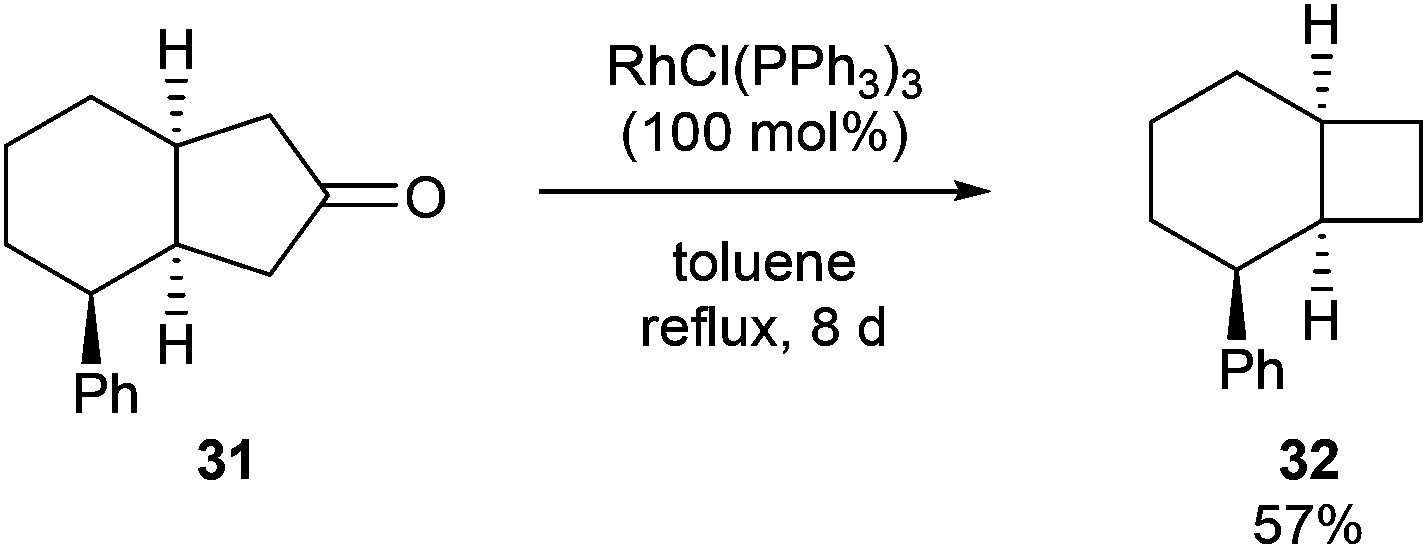
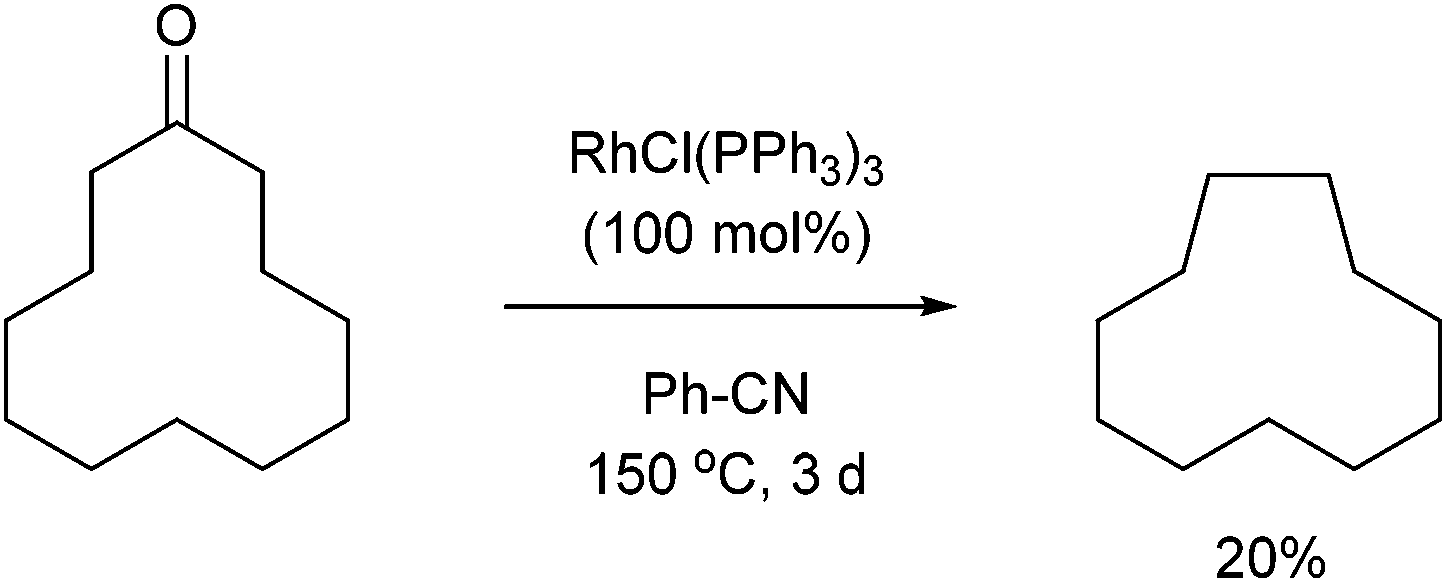
In 1994, Ito and co-workers showed that Rh-catalyzed C–C activation can take place with strain-free cycloalkanones to provide strained ring-contracted products.23 Cyclopentanone derivative 31 was decarbonylated using an equimolar amount of Wilkinson's catalyst to provide a strained cyclobutane (32) in 57% yield after refluxing for 8 days (eqn (10)). Larger rings, such as cyclododecanone, were decarbonylated after heating at higher temperature (150 °C) for 3 d to provide an eleven-membered carbocycle, albeit in much lower yield (20%, eqn (11)).
| | 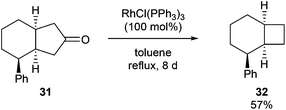 | (10) |
| | 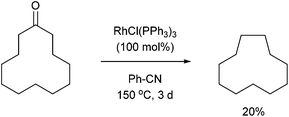 | (11) |
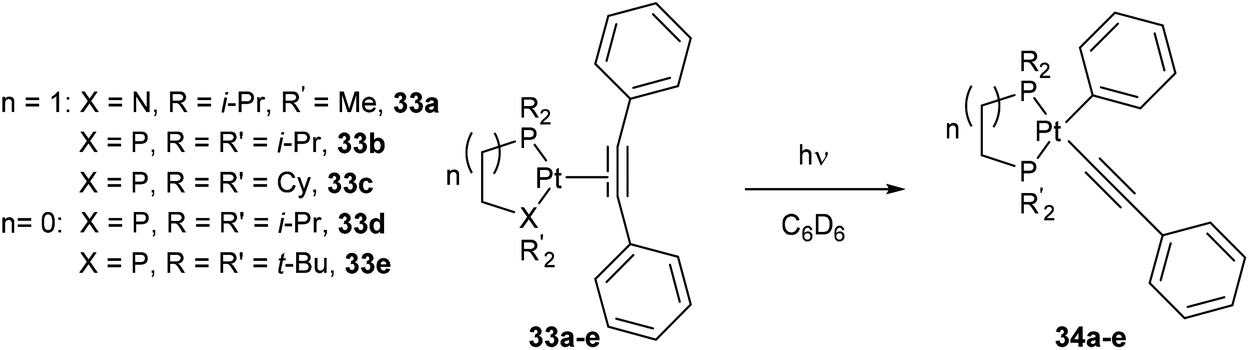
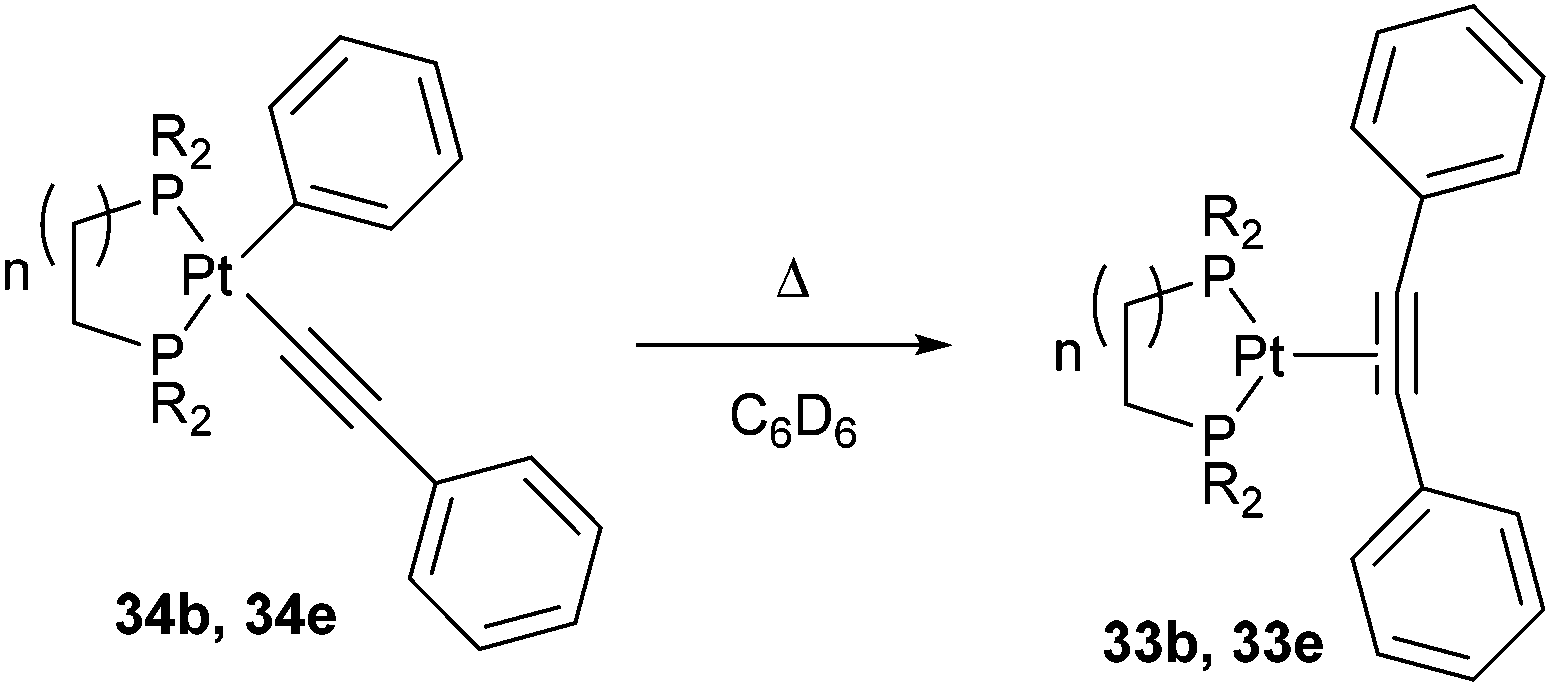
Jones and co-workers24 presented a unique case where Pt can cleave the sp2–sp C–C bonds of diphenyl acetylene under photochemical conditions.25 Shedding UV light onto a series of air- and moisture-sensitive Pt-η2-alkyne-complexes (33a–e) afforded unsymmetrical PtII complexes 34a–e with a phenyl group σ-bonded to the metal center (eqn (12)). The structures were confirmed by single crystal X-ray diffraction, and 1H NMR analysis shows platinum satellites for a single set of ortho-phenyl protons indicating that the phenyl group is indeed σ-bonded to the Pt center. Interestingly, when specific complexes 34b and 34e were thermally activated, a reductive elimination took place to reform the Pt-η2-alkyne complexes (33b and 33e, eqn (13)). The reversible nature of the reaction under thermal conditions suggests that oxidative cleavage of sp2–sp C–C bonds in these particular complexes is thermodynamically uphill.26
| | 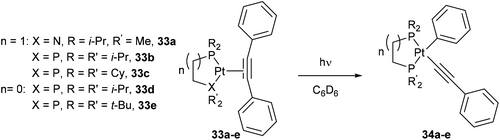 | (12) |
| | 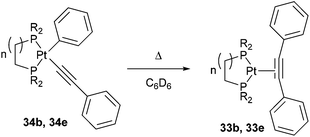 | (13) |
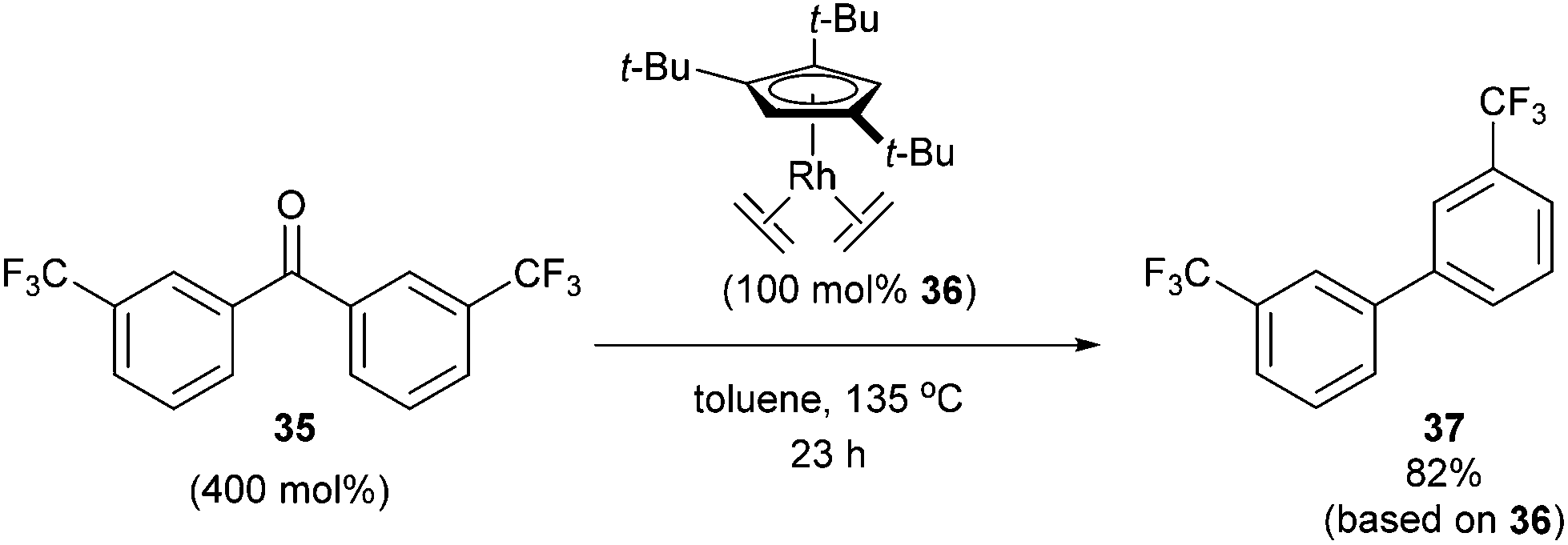
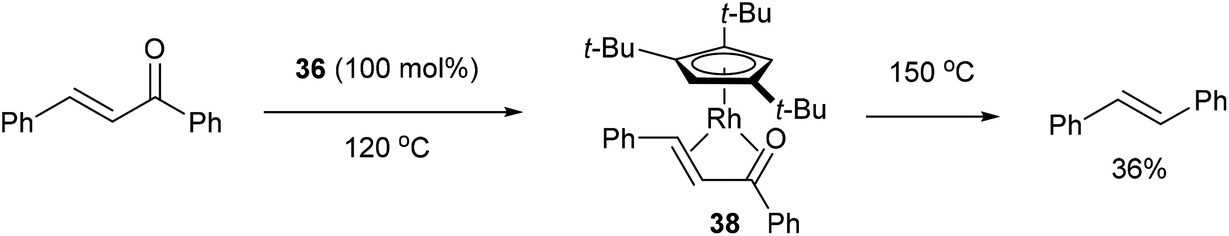
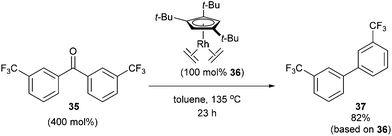
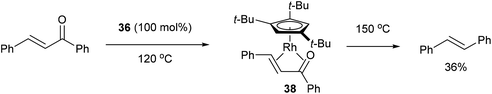
In 2004, Daugulis and Brookhart27 reported the use of a Rh complex capable of decarbonylation. For example, 3,3′-bis(trifluoromethyl) benzophenone (35) was decarbonylated with Cp*Rh(C2H4)2 (36) in refluxing toluene to give biaryl product 37 in 82% yield (based on catalyst, eqn (14)). Catalyst 36 was also found to successfully decarbonylate chalcone. Heating chalcone with Cp*Rh(C2H4)2 at 120 °C forms complex 38, however upon further heating at elevated temperatures (150 °C) decarbonylation occurs to form stilbene in 36% yield (Scheme 5). A number of other benzophenones and acetophenones were decarbonylated and while electronic factors did not influence the rate of decarbonylation with benzophenones, more electron-withdrawing substituents (e.g. CF3) on acetophenones gave substantially faster reaction rates.
| |  | (14) |
 |
| | Scheme 5 Decarbonylation of chalcone by unique Rh complex 36. | |
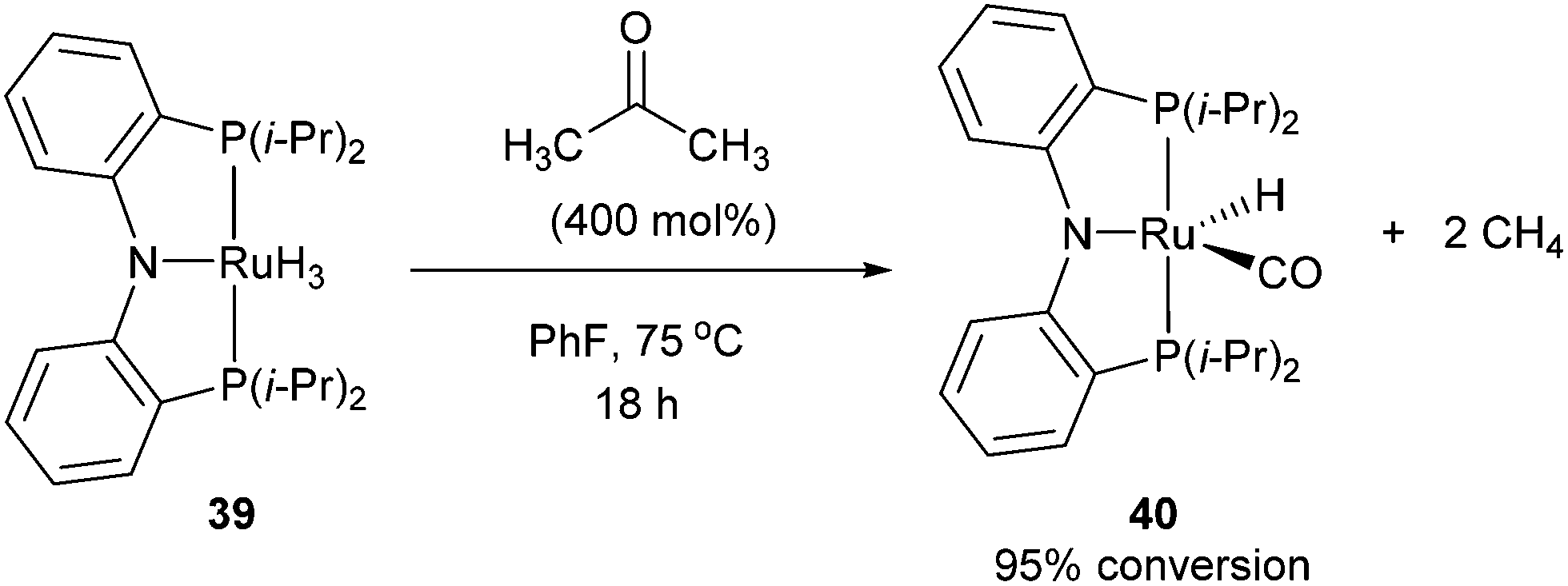
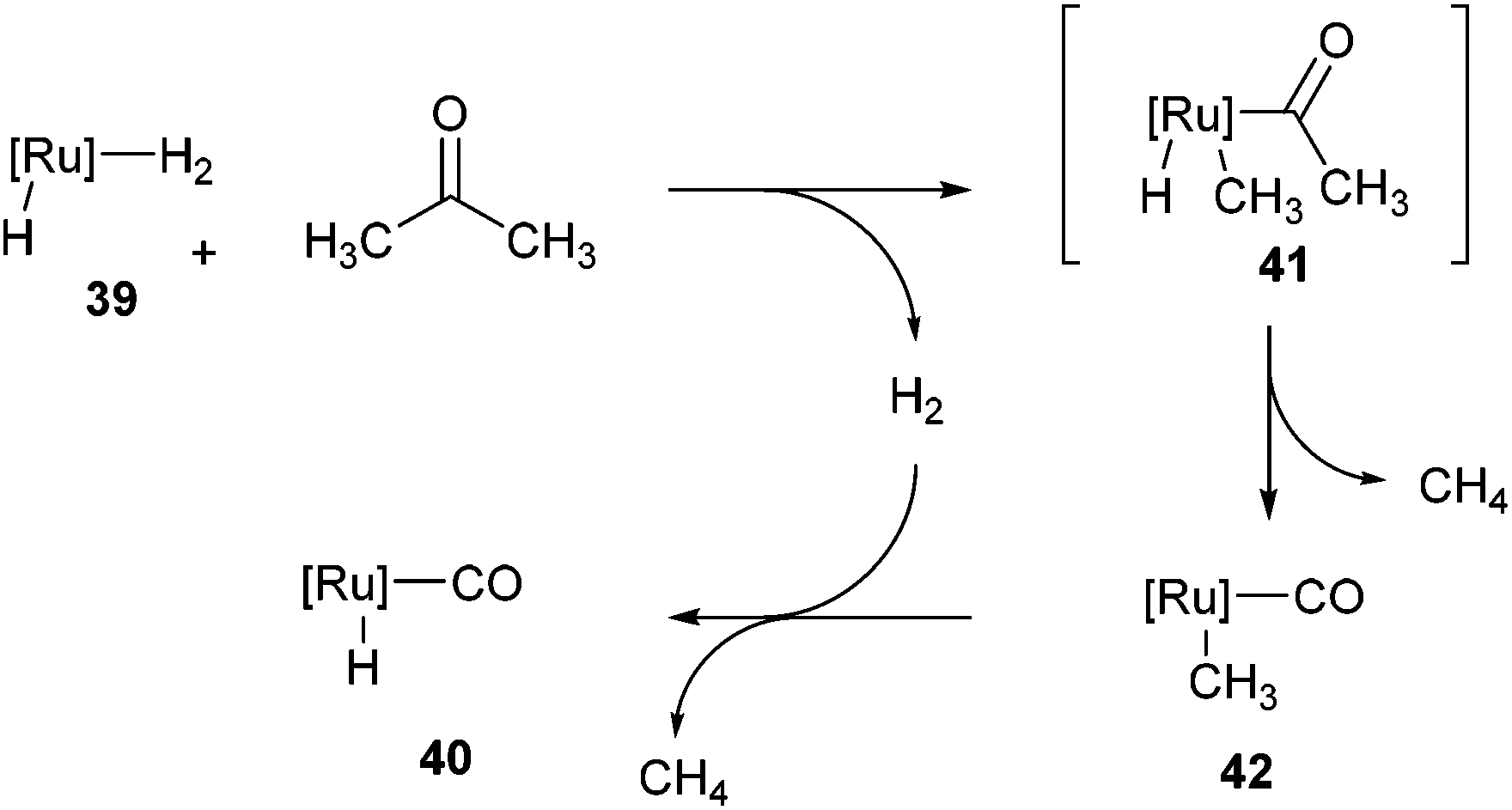
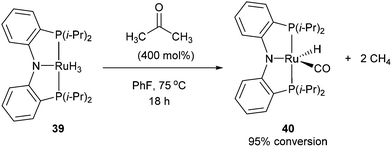
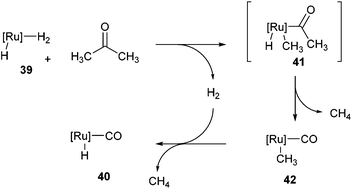
Following the work of Daugulis and Brookhart, Ozerov and co-workers28 presented an electron-rich pincer-ligated Ru complex (PNP)(Ru)H3, 39) that can successfully decarbonylate acetone. Heating of acetone (4 equiv.) with 39 in fluorobenzene at 75 °C for 18 h resulted in the decarbonylation of acetone to form 40 in 95% conversion (eqn (15)). The proposed mechanism is shown in Scheme 6, where 39 cleaves a single α C–C bond of acetone to provide ruthenium-acyl complex 41. Reductive elimination produces an equivalent of methane (observed by 1H NMR) and complex 42, which can react with hydrogen to extrude another equivalent of methane and result in the formation of complex 40. Ozerov and co-workers conducted DFT studies, in addition to 1H NMR experiments, to support the mechanistic rationale and the exothermic nature of the reaction (−51.5 kcal mol−1). They proposed two main thermodynamic driving forces for the reaction. The first involves the favorable binding of CO over H2 by 38.9 kcal mol−1 to the metal center. The second is preferable formation of C–H bonds over C–C bonds by 19.9 kcal mol−1, supporting the production of two equivalents of CH4 rather than one equivalent of C2H6.
| |  | (15) |
 |
| | Scheme 6 Demethylation of acetone via C–C activation. | |
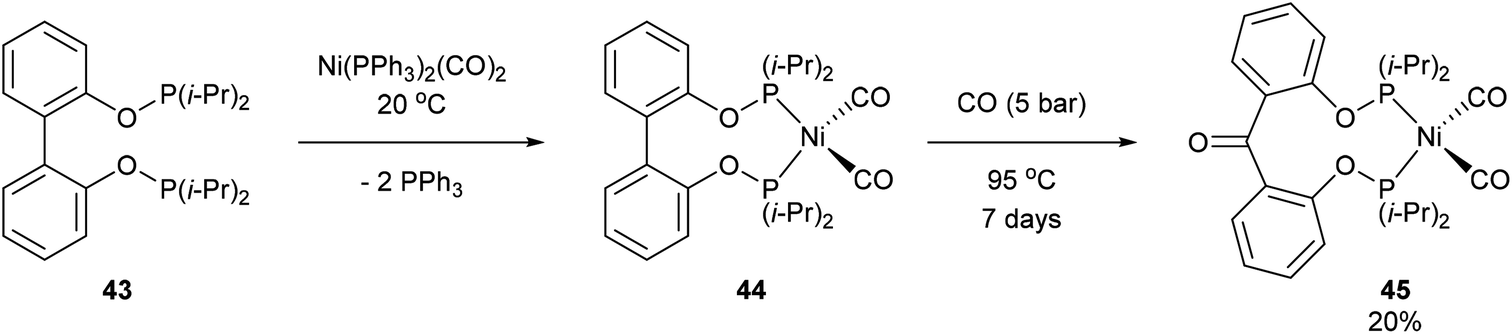
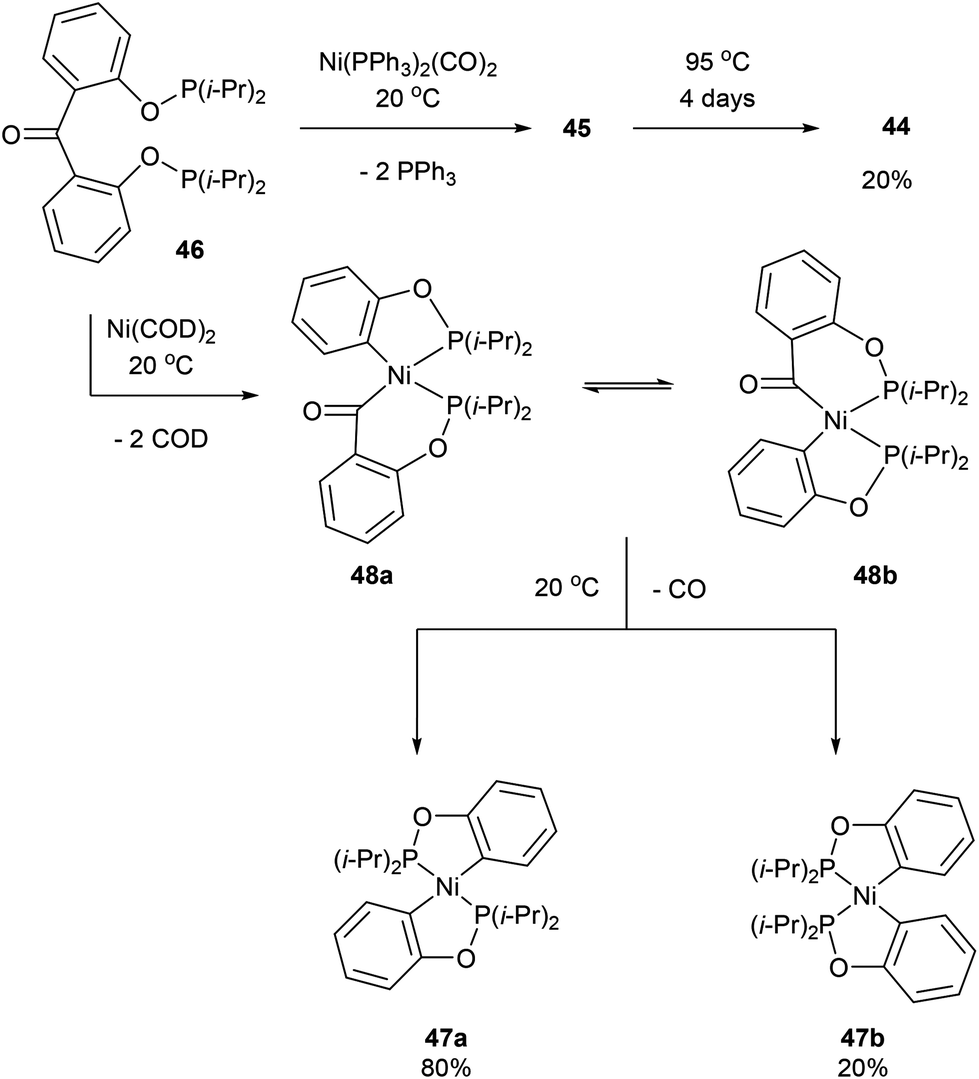
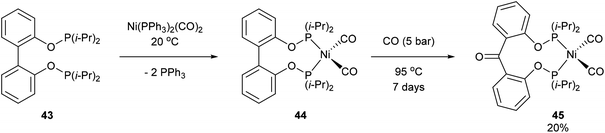
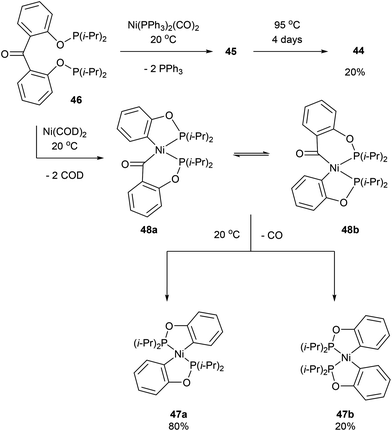
Another form of chelate-assisted C–C activation was reported by Ruhland and co-workers showing that sp2–sp2 C–C bonds in biaryls and benzophenones can be successfully cleaved with phosphonite directing groups.29 For instance, when biaryl compound 43 was treated with Ni(PPh3)2(CO)2 at room temperature, the two phosphinite groups coordinated to the metal center and extruded two equivalents of PPh3 to provide complex 44 (Scheme 7). Upon heating of 44 under 5 bar CO, benzophenone complex 45 was afforded via C–C activation and CO insertion The reverse reaction was also shown to be possible in the absence of CO, where ligand 46 was treated with Ni(PPh3)2(CO)2 to afford complex 45 and upon heating (95 °C) decarbonylation occurred to give complex 44 (20%, 4 days). Decarbonylation was also observed when 46 was treated with Ni(COD)2, to give a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of complexes 47a and 47b, respectively (Scheme 8). The formation of 47a and 47b arises from two degenerate intermediates 48a and 48b, which are in equilibrium as observed by NMR analysis. Extensive experimentation and further NMR analysis provided a mechanistic understanding of the carbonylation and decarbonylation reactions. In the case of carbonylation reaction, they proposed that CO dissociation occurred prior to C–C activation, whereas in the decarbonylation pathway CO deinsertion occurred after electron-rich Ni(0) oxidatively inserted into the C–C bond. Similar C–C activation of benzophenone derivatives was observed by Obenhuber and Ruhland with other transition metals such as Ir (e.g. Vaska complex)30 and Rh (e.g. [Rh(COE)2Cl]2).31
:1 mixture of complexes 47a and 47b, respectively (Scheme 8). The formation of 47a and 47b arises from two degenerate intermediates 48a and 48b, which are in equilibrium as observed by NMR analysis. Extensive experimentation and further NMR analysis provided a mechanistic understanding of the carbonylation and decarbonylation reactions. In the case of carbonylation reaction, they proposed that CO dissociation occurred prior to C–C activation, whereas in the decarbonylation pathway CO deinsertion occurred after electron-rich Ni(0) oxidatively inserted into the C–C bond. Similar C–C activation of benzophenone derivatives was observed by Obenhuber and Ruhland with other transition metals such as Ir (e.g. Vaska complex)30 and Rh (e.g. [Rh(COE)2Cl]2).31
 |
| | Scheme 7 Phosphonite-directed C–C activation and carbonylation with Ni. | |
 |
| | Scheme 8 C–C activation and decarbonylation of benzophenone with Ni as the metal center. | |
Catalytic chelation-directed C–C activation of unstrained substrates
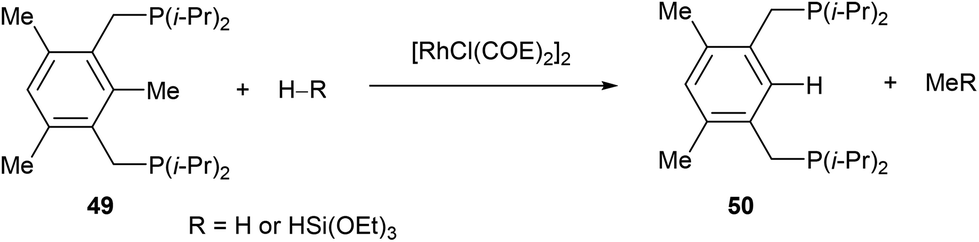
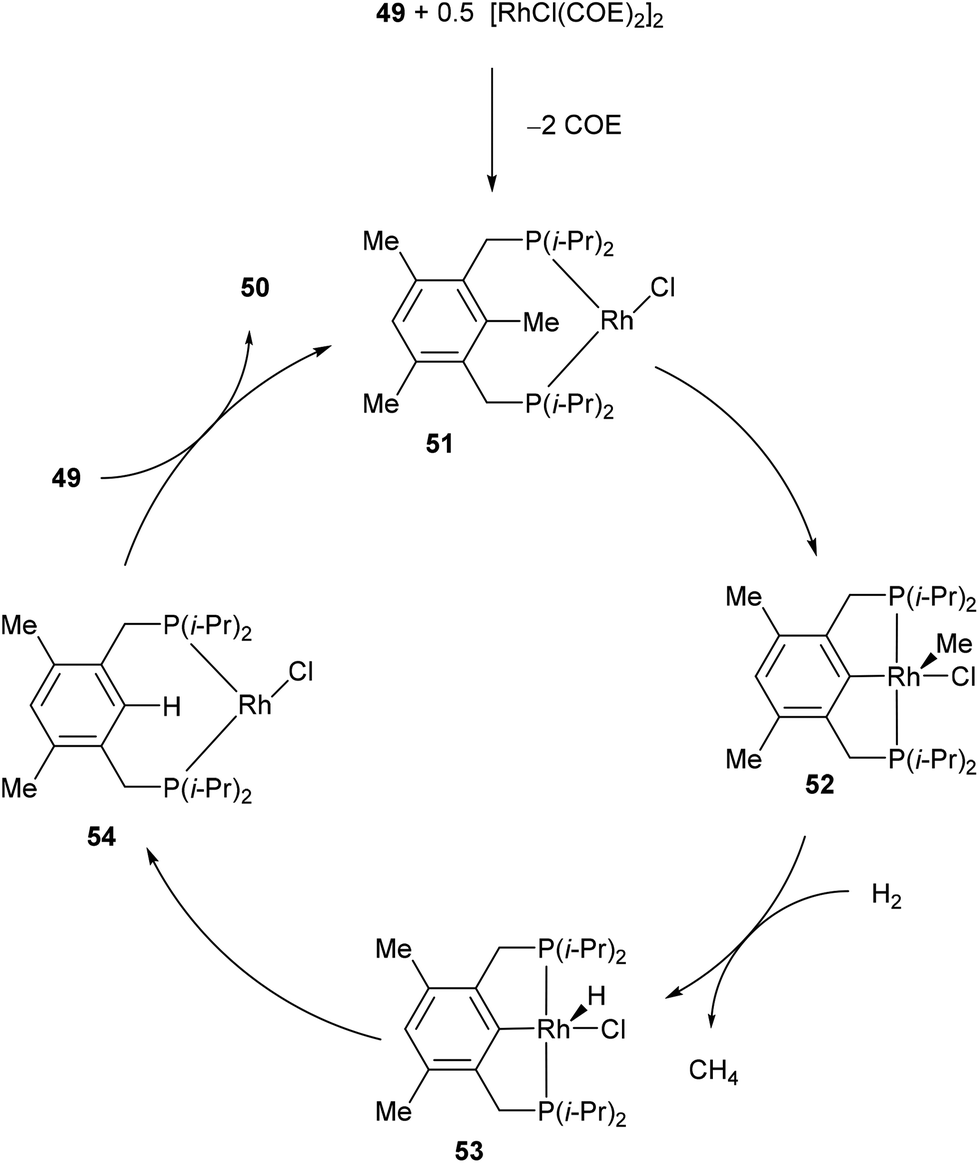
In 1998, Milstein and co-workers32 described a catalytic version of the C–C activation with their bidentate phosphine substrates in converting 49 to 50 under H2 pressure or with excess HSi(OEt)3 (eqn (16)). Although not optimized at the time, [RhCl(COE)2]2 showed 100 turnovers with H2. The proposed catalytic cycle is shown in Scheme 9. The first step involves hydrogenation of the COE ligand and complexation to the bidentate substrate to form 51. At this stage C–C activation can ensue to achieve RhIII intermediate 52, at which point hydrogenolysis of the Rh–methyl bond produces methane and complex 53. Reductive elimination results in RhI54, which can undergo exchange with the substrate to produce 50 and allow RhI to reenter the catalytic cycle.| | 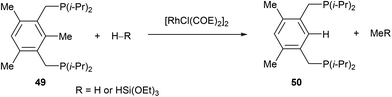 | (16) |
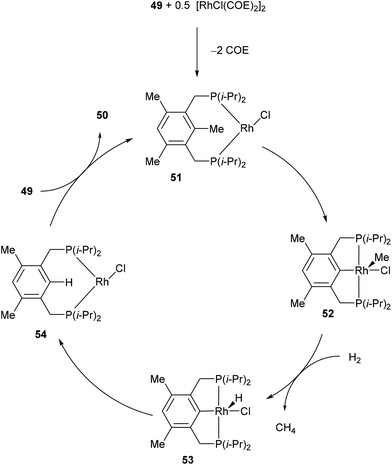 |
| | Scheme 9 Proposed catalytic cycle for C–C activation of PCP ligand. | |
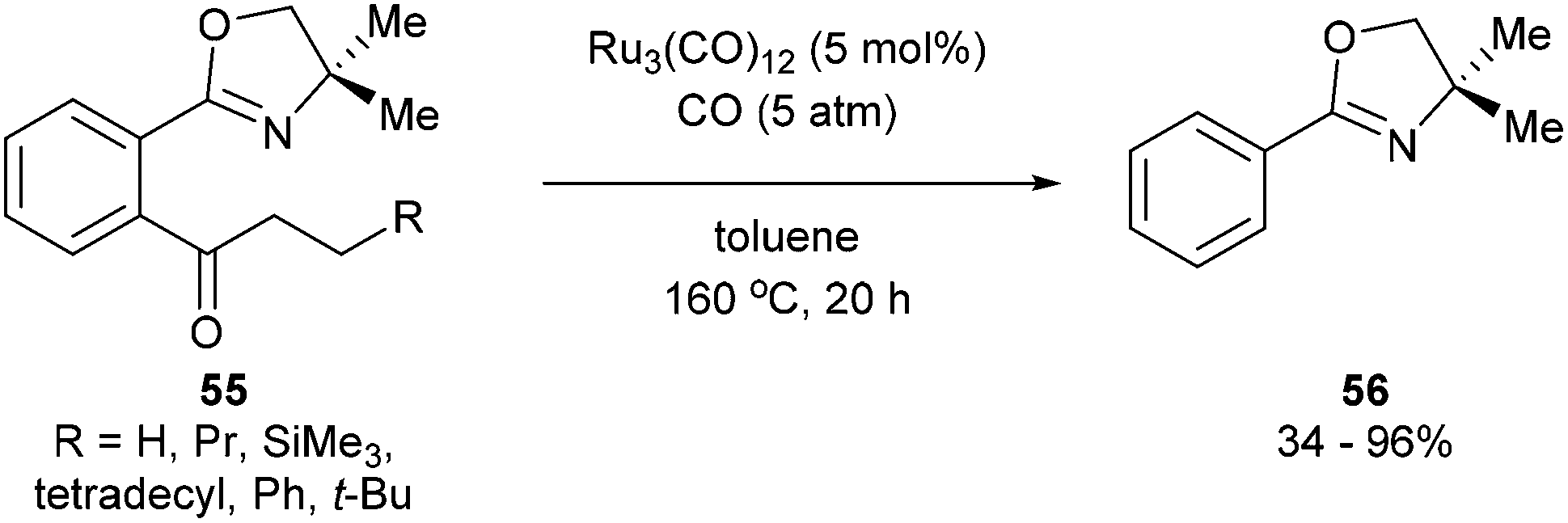
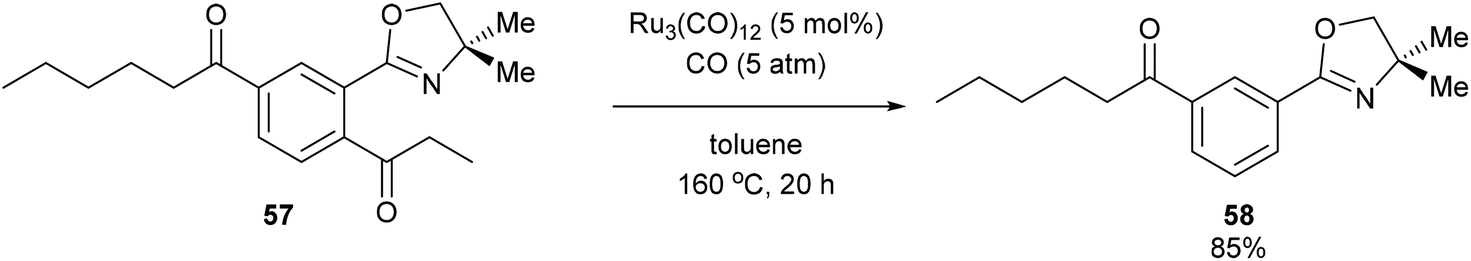
A year after Milstein's report on catalytic C–C activation, Murai and co-workers reported a chelation-assisted decarbonylation reaction via activation of unstrained C–C bonds.33 They reported the use of Ru3(CO)12 (5 mol%) under 5 atm CO in toluene at 160 °C to undergo oxazoline-directed decarbonylative C–C cleavage of alkyl phenyl ketones (55) to the corresponding products in 34–96% yield (56, eqn (17)). They also demonstrated that the directing group is necessary, as in substrate 57 decarbonylation only occurred ortho to the directing group to furnish 58 in 85% yield (eqn (18)).
| | 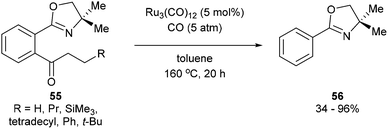 | (17) |
| | 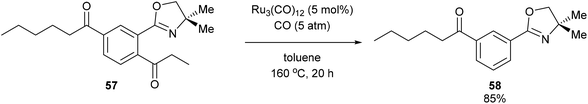 | (18) |
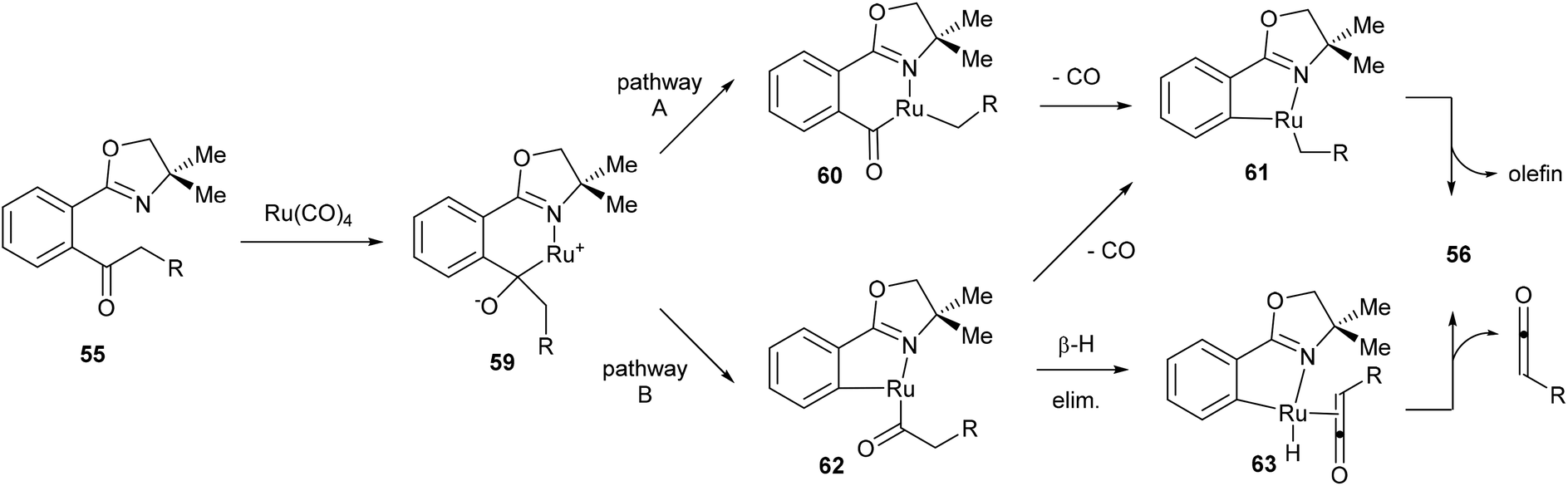
Murai and co-workers propose two possible mechanistic pathways that lead to the observed product (Scheme 10). Coordination of Ru to the starting material (55) followed by nucleophilic attack at the carbonyl provides complex 59. At this point there are two possible C–C bonds that can be cleaved, the Ar–CO C–C bond or the CO–alkyl C–C bond. Cleavage through pathway A would lead to six-membered rhodacycle 60, which after decarbonylation (to give 61) and β-hydride elimination would provide the desired product. Alternatively, pathway B would provide the five-membered rhodacycle 62, which can undergo decarbonylation to provide the same intermediate (61) or it can first undergo β-hydride elimination to give complex 63, which would release a molecule of ketene and reductively eliminate to give the desired product. Murai and co-workers suggest that pathway B is more plausible. Benzyl ketone derived substrate (R = Ph) was reacted in methanol to give 73% yield of the desired product (56) isolated along with 34% of methyl phenylacetate through methanolysis of intermediate 62 or trapping of phenyl ketene.
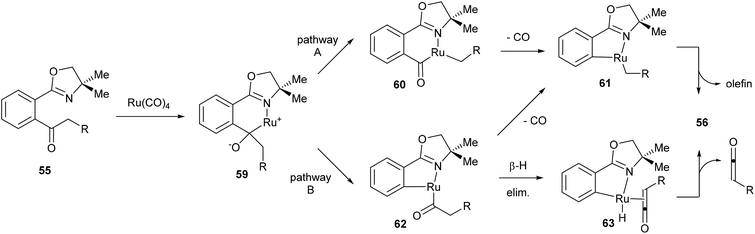 |
| | Scheme 10 Proposed mechanism for oxazoline-directed C–C activation. | |
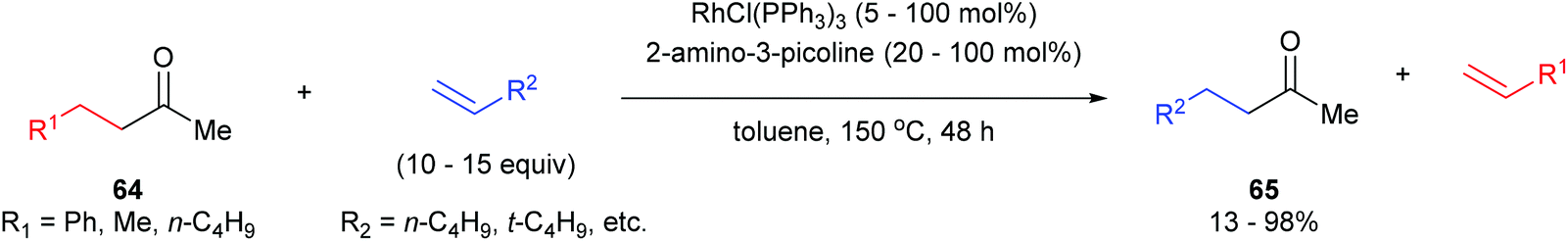
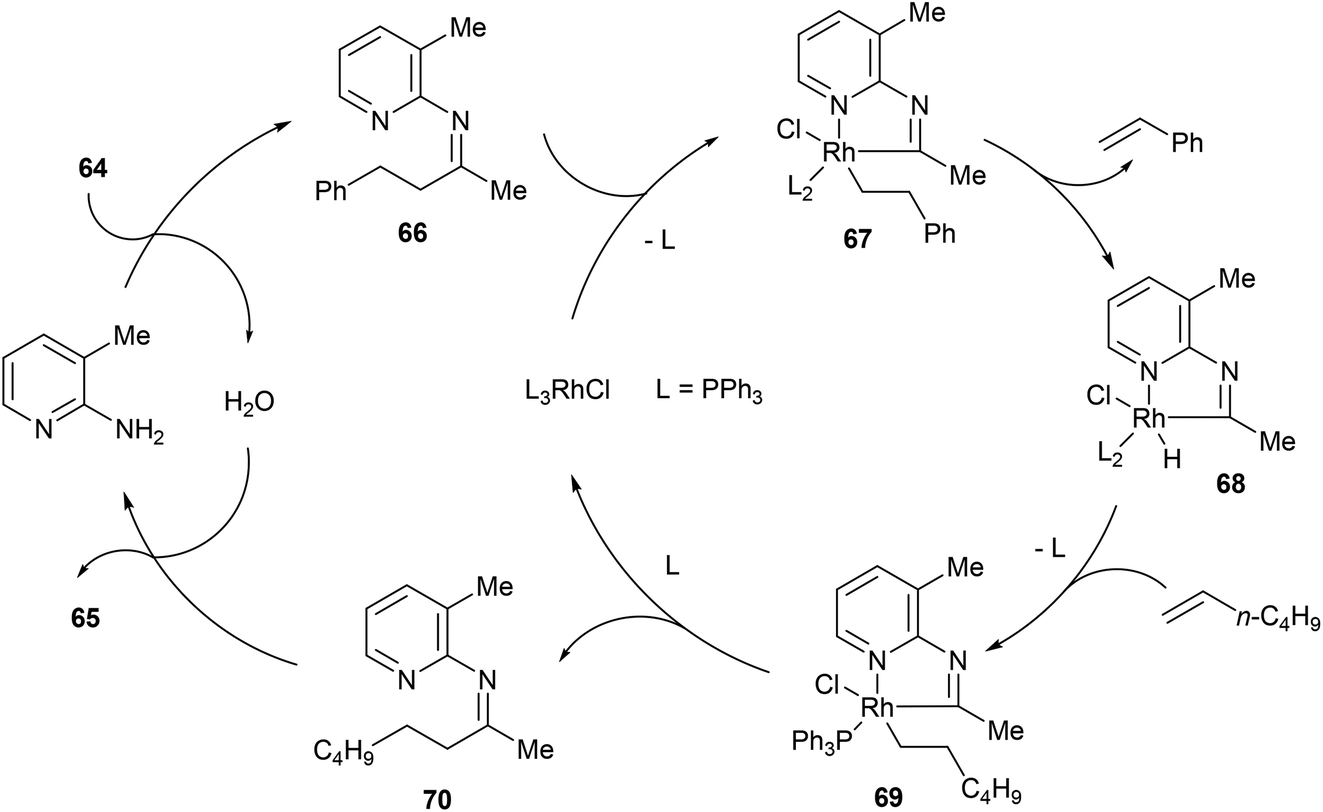
In the same year, Jun and co-workers34 reported a catalytic C–C activation of unstrained ketones using 2-amino-3-picoline as a cofactor.35 As shown in eqn (19), Wilkinson's catalyst and 2-amino-3-picoline promote C–C activation and alkyl group transfer in the presence of ketones (64) and terminal olefins with good yields overall (42–98%). Internal olefins typically did not work as well giving lower yields. The proposed mechanism of the reaction explains the preferred C–C cleavage on the homobenzyl side of the ketone (Scheme 11). The first step of the reaction involves condensation of 2-amino-3-picoline to provide imine 66 and H2O. Coordination of Rh(I) to the substrate followed by C–C activation gives rhodacycle 67, which can undergo β-hydride elimination, possible only on the homobenzylic side, to form an equivalent of styrene and complex 68. Migratory insertion into 1-hexene affords intermediate 69, which after reductive elimination provides ketimine 70 and regenerates the Rh(I) catalyst. Hydrolysis of ketimine 70 provided product 65 and 2-amino-3-picoline which can reenter the catalytic cycle. This process was also demonstrated with the C–C activation of secondary alcohols, where alkyl-group-exchange takes place after initial oxidation of the secondary alcohol to the ketone via transfer hydrogenation.36
| |  | (19) |
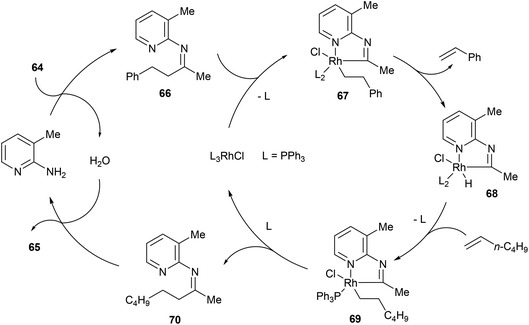 |
| | Scheme 11 Mechanism of alkyl-exchange via C–C bond activation. | |
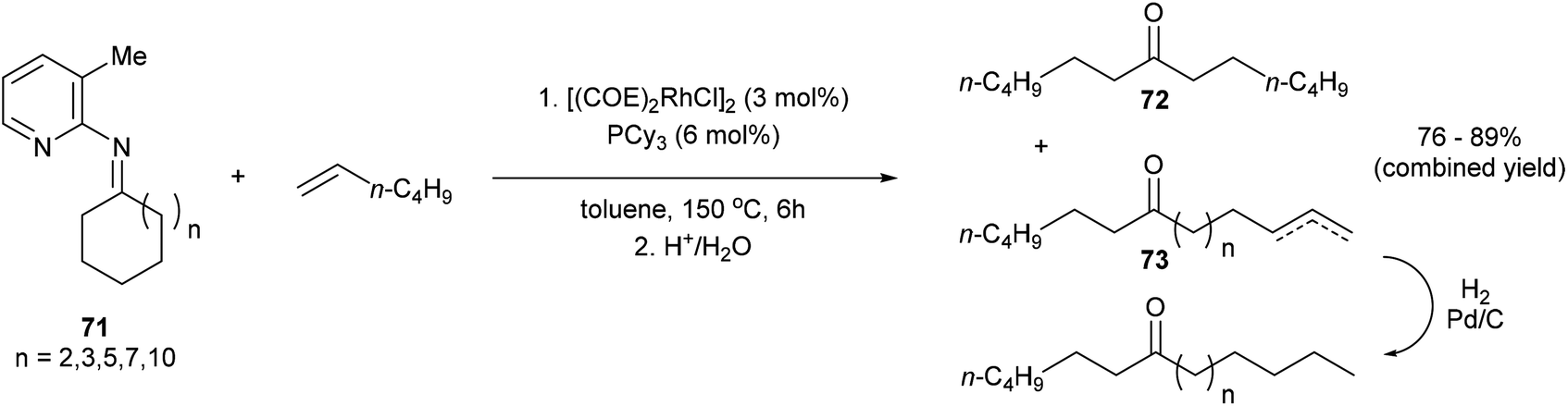
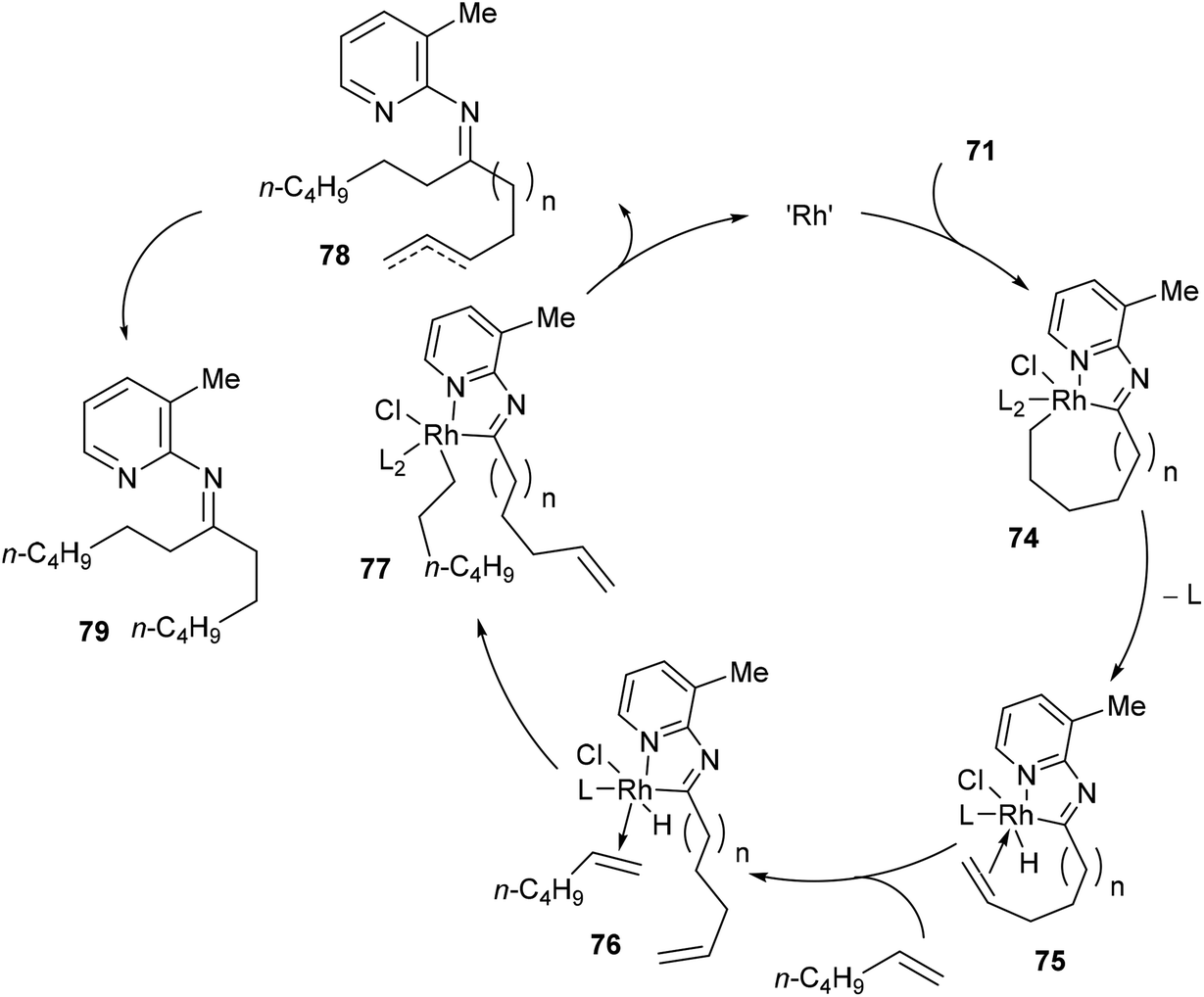
Jun and co-workers further developed a picoline-directed C–C activation strategy for ring-opening and skeletal rearrangement of several different cycloalkanone imines.37 For example, when cycloalkanoketimine 71 is subjected to [(COE)2RhCl]2 (3 mol%) and PCy3 (6 mol%) in toluene at 150 °C for 6 h a mixture of symmetrical and unsymmetrical ring-opened products (72 and 73, respectively) were obtained in good yields (76–89%, eqn (20)). Smaller ring systems such as cyclopentanoketimine and cyclohexanoketimine reacted poorly and gave yields of less than 10%. The mechanism of this reaction (Scheme 12) is similar to that of the previously discussed reaction, where 2-amino-3-picoline directs Rh(I) to cleave the α C–C bond of the ketimine providing intermediate 74. β-Hydride elimination gives Rh–H 75, which can then add across an equivalent of 1-hexene to provide complex 78. Hydrolysis of 78 gives product 73; alternatively, 78 can undergo another cycle of alkyl-group transfer to give the symmetrical ketimine 79, ultimately leading to ketone 72.
| | 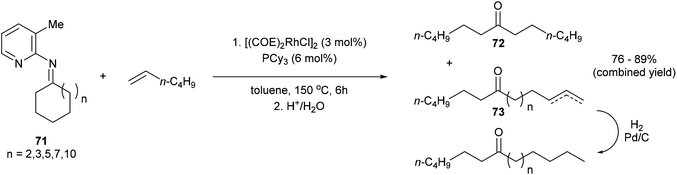 | (20) |
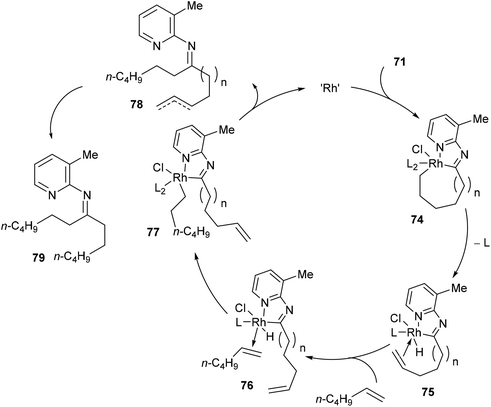 |
| | Scheme 12 Proposed mechanism for Rh-catalyzed ring-opening of cycloalkanoketimines. | |
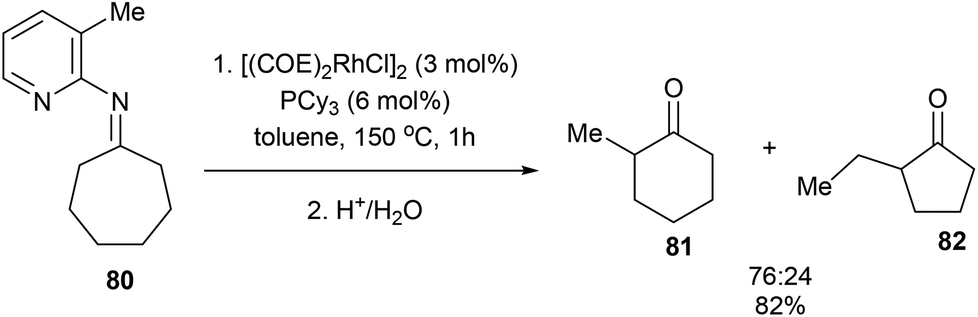
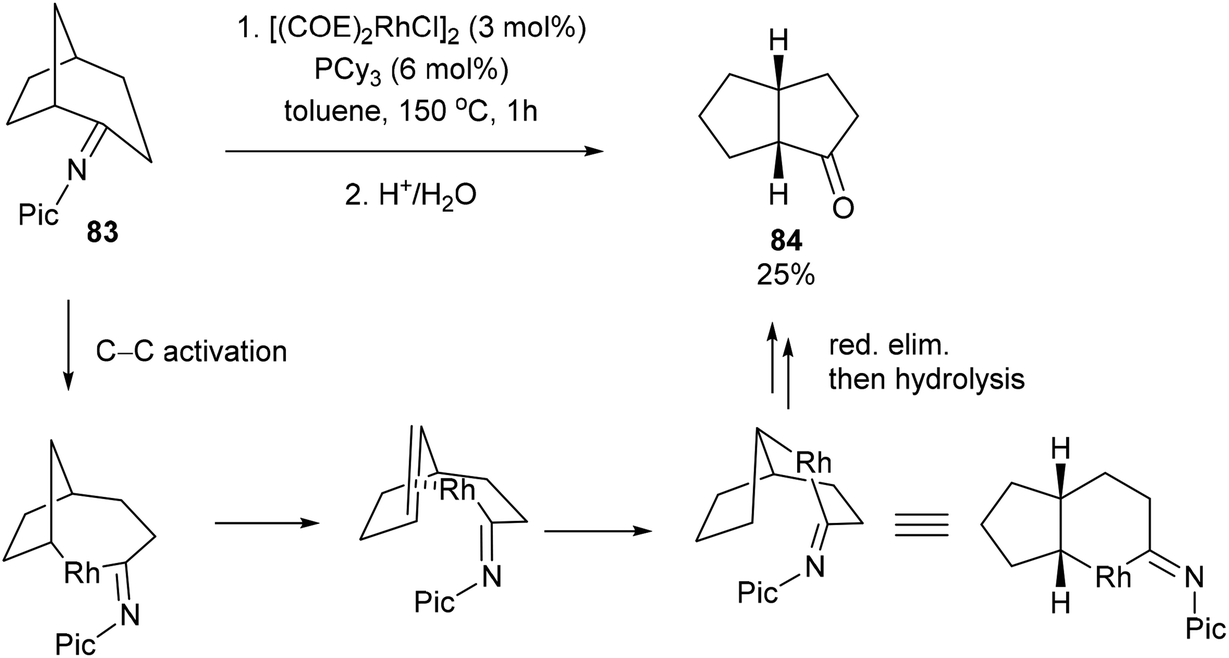
When the reaction with seven-membered ketimine 80 was run without 1-hexene, a rearrangement occurred followed by hydrolysis to produce cyclohexanone 81 and cyclopentanone 82 in 82% yield with 76:24 ratio, respectively. Other rings sizes gave lower yields (0–21%). The mechanism of this rearrangement follows a similar sequence as that in Scheme 12. For example, rather than addition to 1-hexene, the RhIII intermediate 75 reinserts into the appended olefin in a 6-exo fashion to give an intermediate that can reductively eliminate to provide cyclohexanone product 81. On the other hand, for product 82, after reinsertion of the appending olefin, a second β-hydride elimination (alkene chain-walk) followed by another addition across the olefin and reductive elimination would result in cyclopentanone 82. Jun and co-workers also showed that when bicyclic systems such as bicycle[3.2.1]octan-2-one 83 were subjected to the reaction conditions, a 5,5-fused bicyclic ketone 84 is obtained in 25% yield following the mechanism proposed below (Scheme 13).
| | 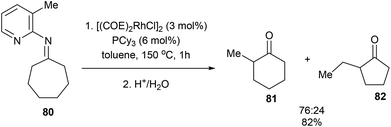 | (21) |
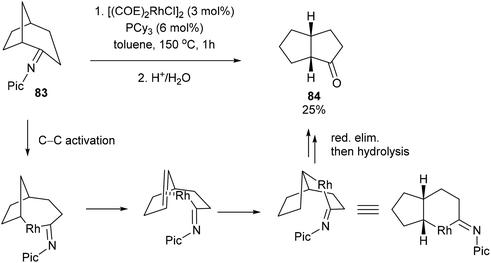 |
| | Scheme 13 Rh-catalyzed skeletal rearrangement of bicycle[3.2.1]octan-2-one. | |
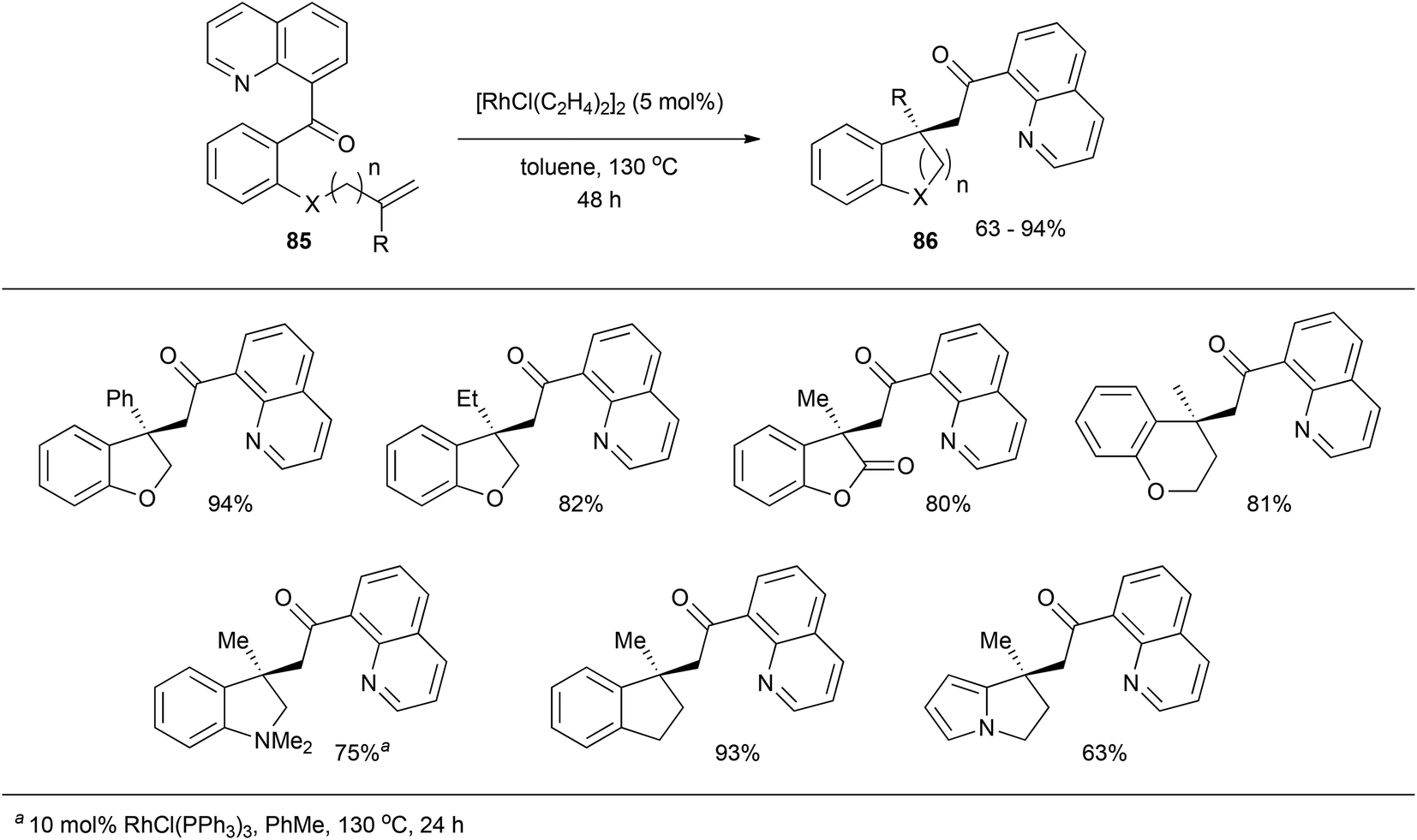
In 2009, Douglas and Dreis38 reported an intramolecular carboacylation reaction. Quinolines were utilized as directing groups for catalytic C–C σ bond activation with Rh. Refluxing substrate 85 with 5 mol% [RhCl(C2H4)2]2 in toluene effectively results in all-carbon quaternary stereocenter-containing compound 86 in moderate to excellent yields (63–94%, eqn (22)). A number of different substrates with various linkers participate. An intermolecular version of this transformation was reported later that year by the same group on insertion of norbornene-type alkenes.39,40
| | 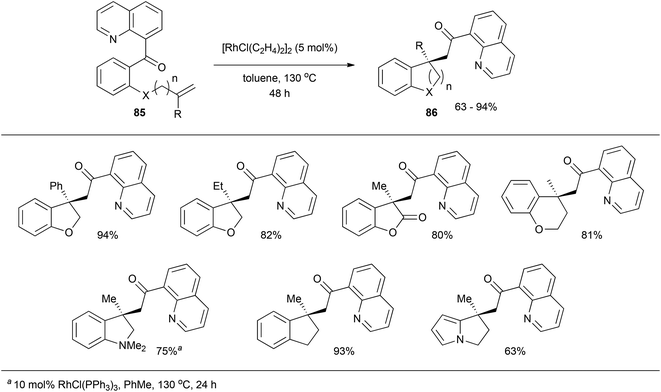 | (22) |
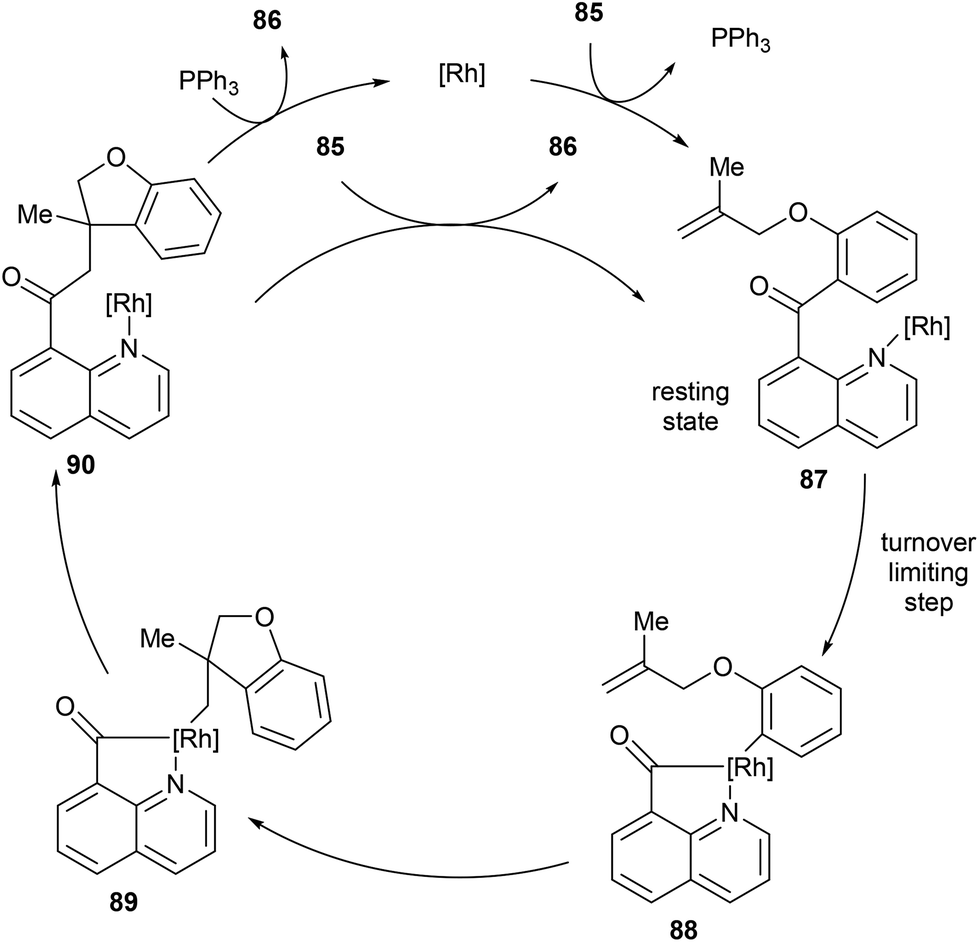
Johnson and co-workers published mechanistic studies regarding the catalytic cycle for these transformations (Scheme 14).41 They found slightly different mechanistic pathways by investigating the use of RhCl(PPh3)3 and [RhCl(C2H4)2]2 as catalysts, respectively. With RhCl(PPh3)3 the proposed catalytic cycle is shown in Scheme 13, wherein the reaction was found to be zero-order in substrate and first-order in catalyst, and the C–C activation step was determined to be rate-limiting. The resting state of the reaction is when catalyst is bound to the substrate in complex 87. Next, C–C activation and acylation of the metal center provides intermediate 88, which rearranges to the quaternary center in complex 89. Reductive elimination would then produce 90, where the product can be exchanged with another equivalent of substrate allowing the catalyst to reenter the catalytic cycle. PPh3 may also intercept 90 to free the catalyst. On the other hand, when the reaction was studied for [RhCl(C2H4)2]2 a slightly different catalytic cycle was proposed. The reaction is second-order overall, first-order in both substrate and catalyst, suggesting that the resting state of the catalyst is when it is not bound to either substrate or product. The rate-limiting step was determined to be C–C activation with 1,1′-disubstituted alkenes or substrates with longer linkers influencing the reaction rate. The main intermediates of catalytic cycle for [RhCl(C2H4)2]2 are similar to RhCl(PPh3)3, with the exception of the resting state of the catalyst.
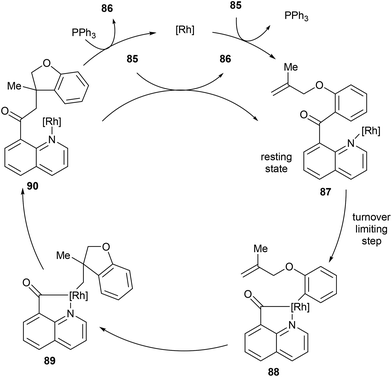 |
| | Scheme 14 Mechanism for the synthesis of all carbon quaternary centers. | |
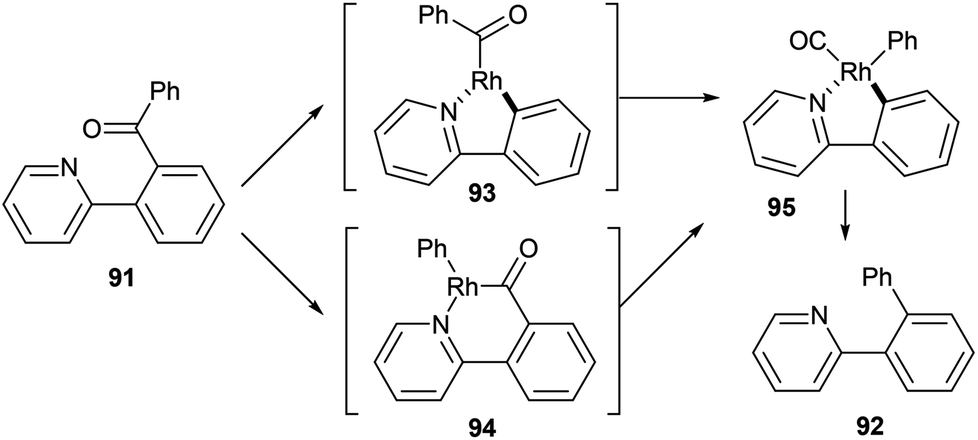
In 2012, Shi and co-workers42,43 published a pyridine-directed C–C activation strategy for the decarbonylation of biaryl ketones and alkyl/alkenyl aryl ketones (91). The optimized conditions employ 5 mol% [(CO)2Rh(acac)] in refluxing chlorobenzene to provide the decarbonylated products (92) in 80–97% yield (eqn (23)). A number of different aryl, alkyl, and alkenyl groups participate under the reaction conditions and both electron-donating and electron-withdrawing groups afforded good yields. Nitrogen-based directing groups other than pyridine (e.g. pyrazolyl and oxazolyl) provided decarbonylation products in lower yields (52 and 44%, respectively). The proposed mechanism for the decarbonylation is shown in Scheme 15. The reaction is initiated by coordination and subsequent oxidative addition to form either intermediate 93 or 94, and decarbonylation forms 95 followed by reductive elimination giving the observed product. Shi and co-workers propose the five-membered rhodacycle (93) as the favored pathway, although the other pathway cannot be ruled out. The reaction was not promoted by photoirradiation (as the reaction takes place in the dark).
| | 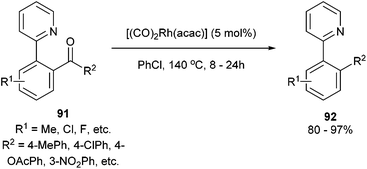 | (23) |
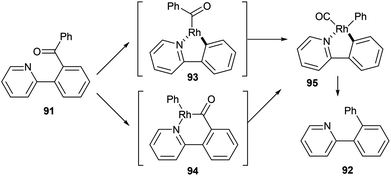 |
| | Scheme 15 Proposed catalytic cycle for the C–C activation and decarbonylation of biaryl ketones. | |
Catalytic C–C activation without chelation
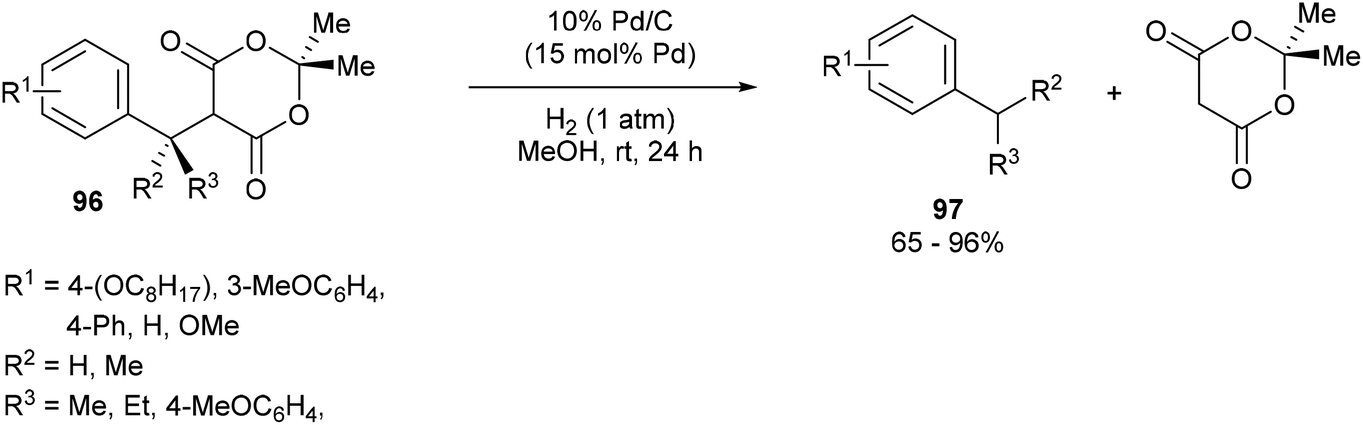
Fillion and co-workers44 showed that unstrained C–C σ bonds in benzylic Meldrum's acid derivatives (96) could be hydrogenolyzed to produce various benzylic products 97 (eqn (24)). When subjected to 15 mol% of 10% Pd/C under 1 atm of H2 for 24 h in MeOH at room temperature, benzylic products were obtained in good to excellent yields (65–96%). Electronic effects were observed on the phenyl moiety. meta-Substituted substrates (OC8H17) gave no product whereas the ortho- and para-substituted analogs gave facile hydrogenolysis and high yields. At the benzylic position, sterics played an important factor such that incorporation of an i-Pr group hampered the reactivity giving only modest conversion; whereas methyl substitution gave near full conversion and high yields. When enantioenriched substrates were hydrogenolyzed, an inversion of stereochemistry is observed with only slight erosion of the enantiomeric ratio. Furthermore, deuterium labeling studies showed that full incorporation of deuterium was observed only when both the solvent and gas were labeled and only partial incorporation was observed when run separately. Therefore, it is likely that the reaction follows an SN2 type mechanism that involves nucleophilic attack by palladium to obtain a benzylic organopalladium intermediate, which undergoes protonation by methanol.| | 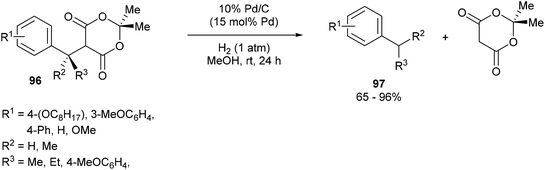 | (24) |
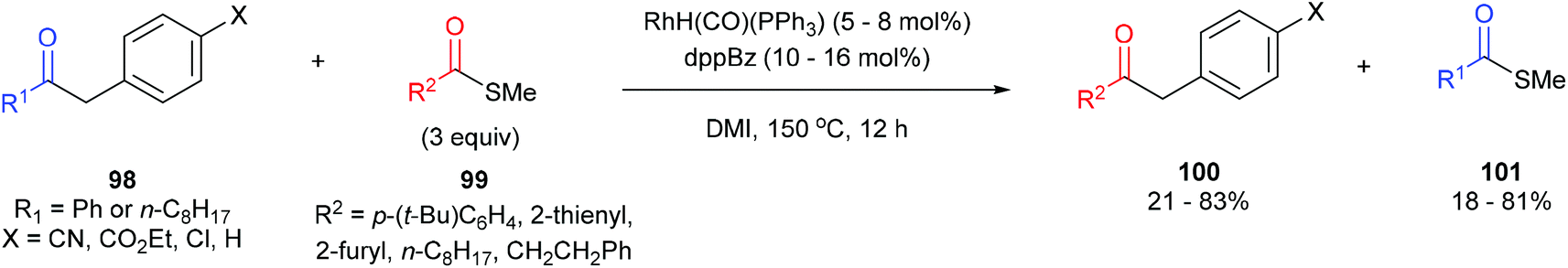
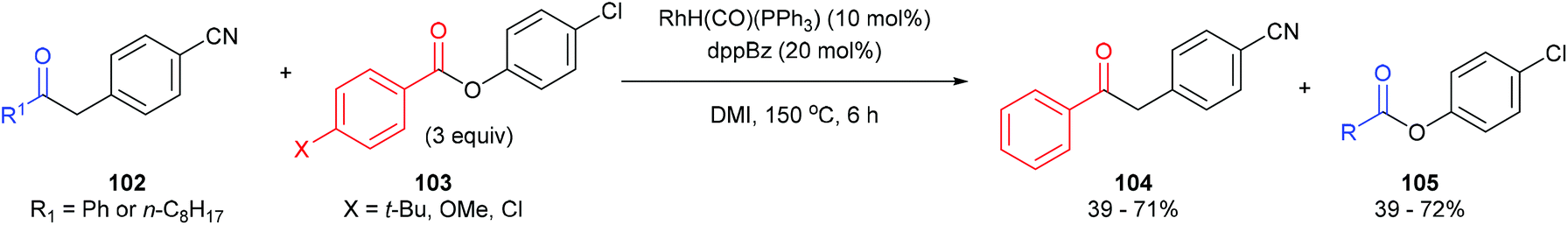
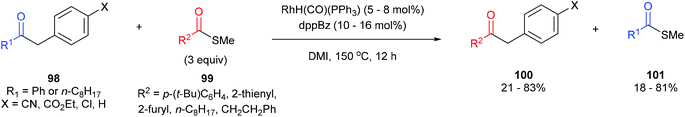
In 2012, Arisawa, Yamaguchi and co-workers45 reported a method for preparing unsymmetrical ketones through a Rh-catalyzed acyl-transfer reaction that requires no chelation assistance. Under optimized conditions RhH(CO)(PPh3)3 with 1,2-bis(diphenylphosphino)benzene (dppBz) in N,N′-dimethylimidazolidinone (DMI) at 150 °C for 12 h results in the transfer of acyl groups between a variety of benzyl ketones (98) and thioesters/aryl esters (99) to provide the respective products 100 and 101. In general, a combination of different substrates gave good yields with the exception of 1,2-diphenylethanone, which gave 21% yield of unsymmetrical benzyl ketone and 18% yield of the thioester. The reaction was also applied to the acyl transfer of benzyl ketones (102) with aryl esters (103) and gave modest to good yields (39–71%) of the benzyl ketone and thioester. Multiple equivalents of the starting thioester favor the forward direction in this equilibrium driven reaction. While the exact mechanism is still unclear, the authors propose that the reaction first undergoes CO–benzyl bond cleavage by a low-valent Rh, followed by exchange with the thioester and reductive elimination to give the product.
| |  | (25) |
| |  | (26) |
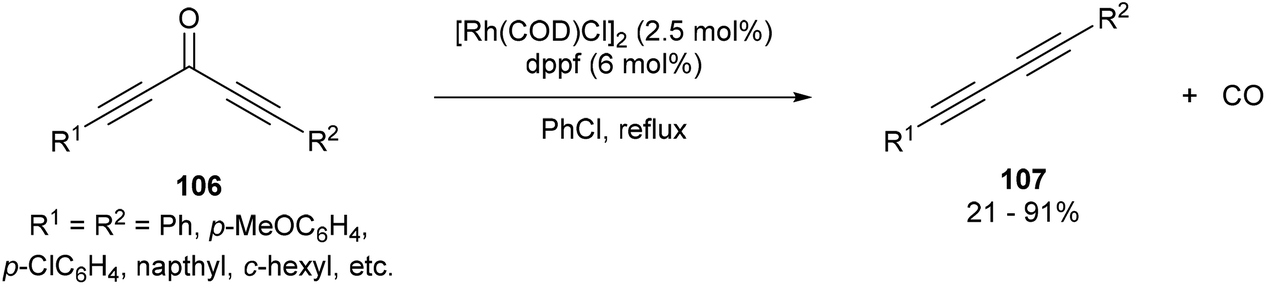
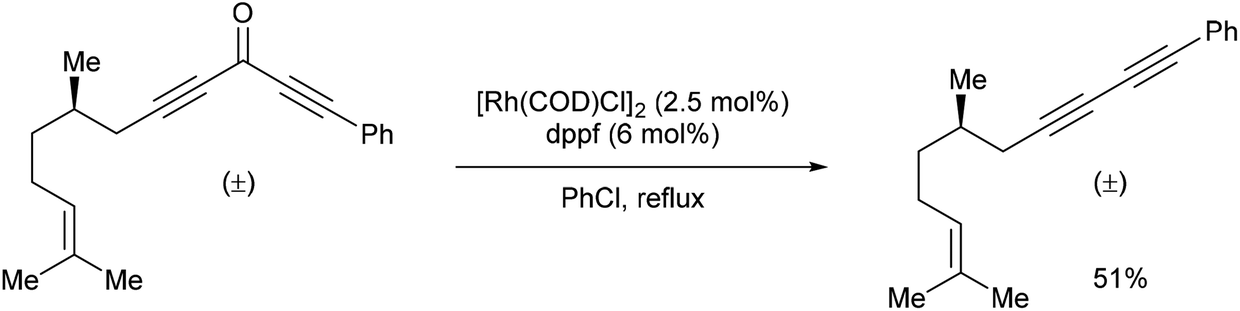
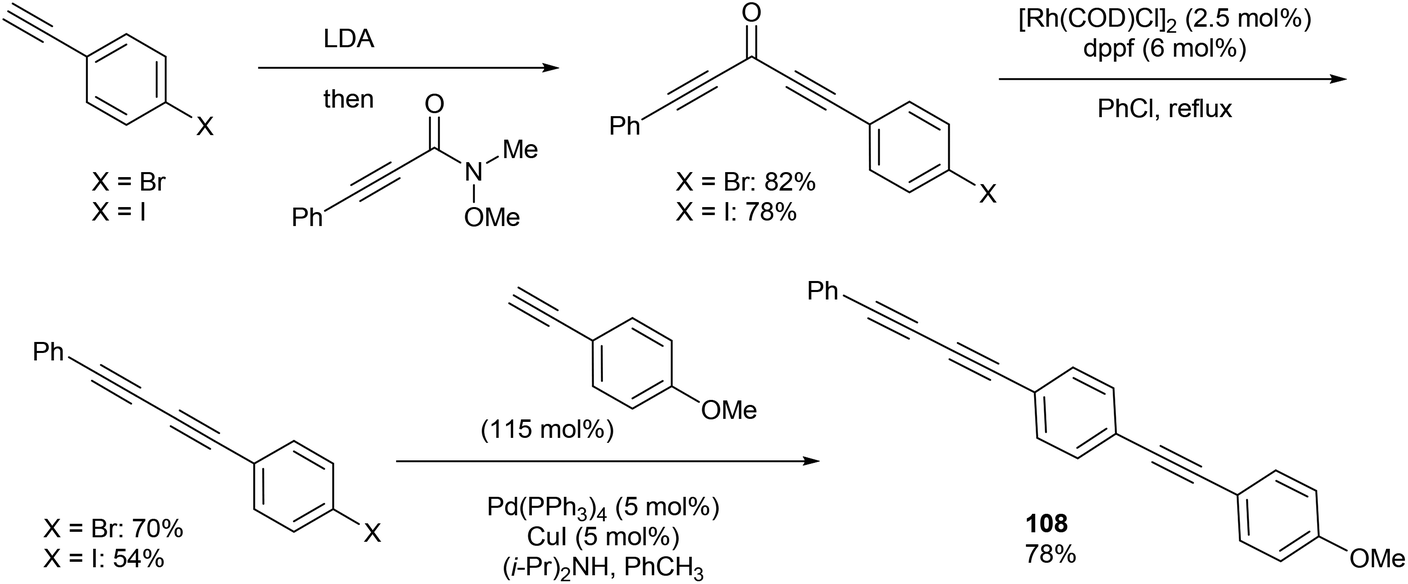
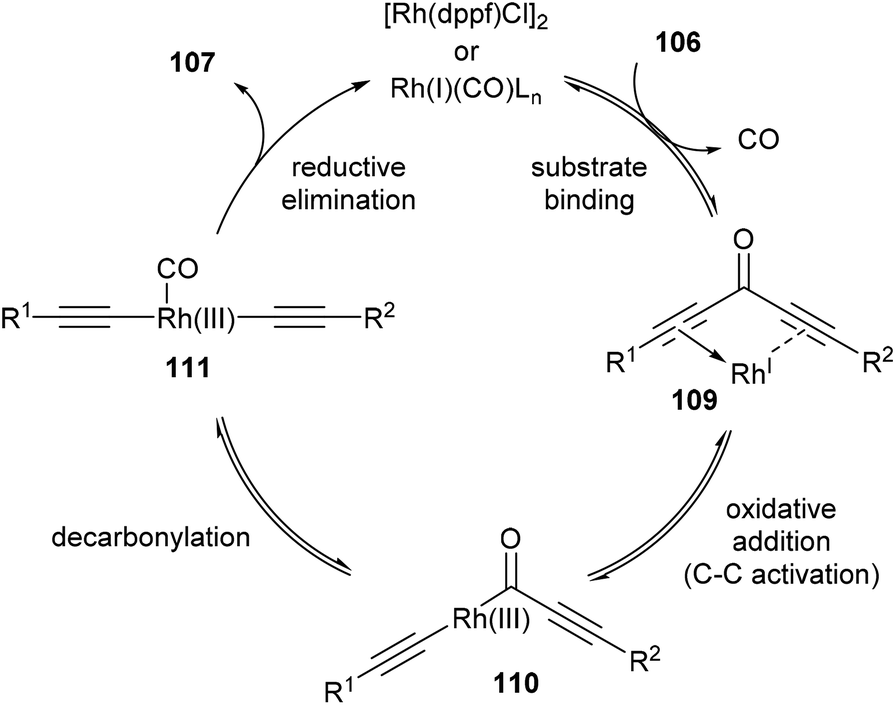
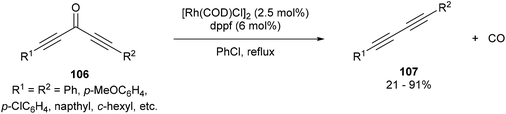
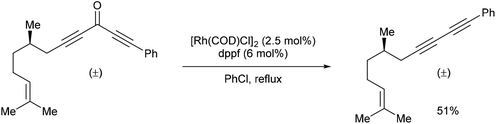
In 2013, Dong and co-workers46 reported a catalytic version of the Rh-mediated decarbonylation of diynones initially reported by Muller. The key factor for promoting this catalytic process was a bidentate phosphine ligand, which is believed to assist in CO elimination from the metal center allowing regeneration of the active catalyst. The reaction works well for a number of symmetrical and unsymmetrical diynones 106 using 2.5 mol% [Rh(COD)Cl]2 and 6 mol% 1,1′-bis(diphenylphosphino)ferrocene (dppf) in refluxing chlorobenzene to obtain the diyne products 107 (21–91%). The reaction is amenable to substrates that present both electronic and steric variations. This method has been used for natural product modification (eqn (28)). It was also applied to the synthesis of a highly conjugated ynediyne 108 through a decarbonylation followed by Sonagashira coupling, in which the orthogonality of this process to other Pd and Cu-catalyzed methods (i.e. tolerance of aryl halides) was demonstrated, making it a complimentary strategy (Scheme 16).
| |  | (27) |
| |  | (28) |
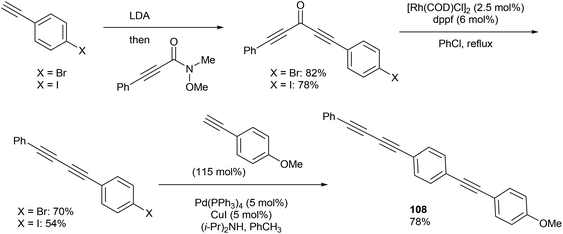 |
| | Scheme 16 Utilization of the Rh-catalyzed decarbonylation to synthesize highly conjugated rod-like structures. | |
The proposed mechanism for the decarbonylation is illustrated in Scheme 17. The first step involves coordination of the substrate to the metal center (109), bringing the metal into close proximity for C–C bond cleavage. Oxidative addition of RhI gives RhIII-complex 110, which undergoes decarbonylation and CO migration to form complex 111. Rapid reductive elimination provides the product and Rh carbonyl complex, which is regenerated into its active form through ligand-assisted release of CO upon substrate binding. The first three steps are in principle all reversible. Also, in the case of unsymmetrical diynones, only unsymmetrical diynes are obtained, suggesting that the reaction is strictly intramolecular without observed intermolecular exchange of acetylene units.
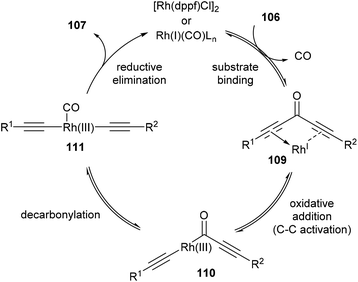 |
| | Scheme 17 Proposed mechanism for the Rh-catalyzed decarbonylation of diynones. | |
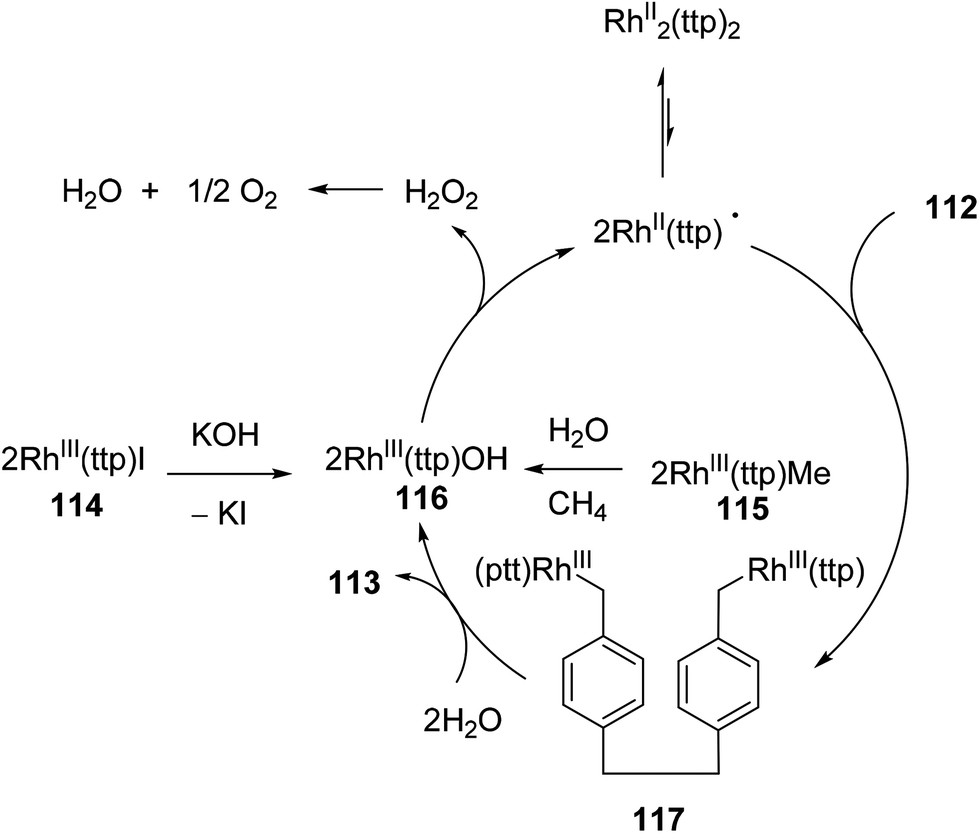
Chan and co-workers have developed a rhodium phorphyrin for the catalytic C–C activation of aliphatic [2.2]paracyclophane (112, PCP).47 Similar chemistry was developed by the group previously in the context of C–C activation using stoichiometric rhodium phorphyrin complexes.48,49 In this transformation, PCP is catalytically converted to 4,4′-dimethylbibenzyl 113 under two possible conditions (eqn (29)). RhIII(ttp)I (ttp = tetratolylporphyrinato dianion) in the presence of KOH (1 equiv.) provided the cleaved product 113 in 83% yield in 25 h; with RhIII(ttp)Me, the product was obtained in 78% after 54 h. Independent deuterium labeling experiments showed that water is the source of hydrogen and that the C–C activation does not involve C–H activation intermediates. The proposed catalytic cycle, which is supported by experimental data, is shown in Scheme 18. Hydrolysis of either precatalyst 114 or 115 provides RhIII(ttp)OH (116), which can decompose to RhII(ttp) radical and exist in an equilibrium with RhII(ttp)2. Two equivalents of RhII(ttp) react with PCP to form complex 117; kinetic data supports an overall third-order reaction, first order in PCP and second order in RhII(ttp). Finally, reaction with H2O provides 113 and regenerates the initial catalyst 116.
| | 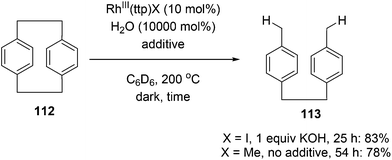 | (29) |
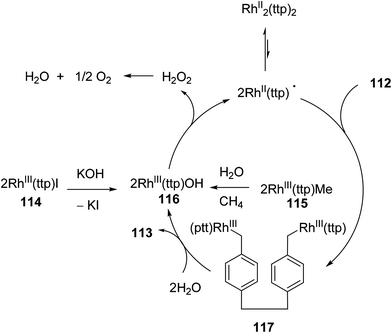 |
| | Scheme 18 The proposed mechanism for Rh-catalyzed cleavage of PCP. | |
Conclusion
In summary, a variety of stoichiometric and catalytic C–C activation reactions have been presented with a discussion of the differing mechanistic pathways that are involved. Although many examples may appear to be special cases, they demonstrate the potential to develop generalized methodologies and strategies. With improvements in this field, it should become possible to construct compounds non-traditionally using novel bond disconnections. In fact, as the field has moved forward from stoichiometric reactions to the development of new catalytic C–C activation transformations, the synthetic utility of this approach has become more apparent. Future improvements in this field would involve moving away from specific examples and developing more general methods for catalytic C–C activation of more common substrates. This will likely require the evolution of new types of catalysts or strategies. We expect, in the coming years, C–C activation will emerge to have a profound impact, in both academia and industry, on accessing synthetically useful molecules.
Abbreviations
| Acac | Acetylacetone, |
| C–C | Carbon–carbon, |
| C–H | Carbon–hydrogen, |
| C2H4 | Ethylene, |
| COD | Cyclooctadiene, |
| COE | Cyclooctene, |
| DMI |
N,N′-Dimethylimidazolidinone, |
| dppBz | 1,2-Bis(diphenylphosphino)benzene, |
| dppf | 1,1′-Bis(diphenylphosphino)ferrocene, |
| NMR | Nuclear magnetic resonance, |
| PCy3 | Tricyclohexylphosphine, |
| PCP | [2.2]paracyclophane, |
| P(OMe)3 | Trimethylphosphite, |
| PPh3 | Triphenylphosphine, |
| THF | Tetrahydrofuran, |
| ttp | Tetratolylporphyrinato dianion. |
Acknowledgements
We thank UT Austin and CPRIT for a start up fund, NIGMS (R01GM109054-01) and the Welch Foundation (F 1781) for research grants. G. D. is a Searle Scholar. We also thank Johnson Matthey for a generous donation of Rh salts.
References
-
(a)
Y. Nakao, Catalytic C–CN Bond Activation, ed. G. Dong, C–C Activation; Topics in Current Chemistry, Springer-Verlag, Berlin, Heidelberg, 2014, in press Search PubMed;
(b)
D. C. Moebius, V. L. Rendina and J. S. Kingsbury, Catalysis of Diazoalkane–Carbonyl Homologation. How New Developments in Hydrazone Oxidation Enable the Carbon Insertion Strategy for Synthesis, ed. G. Dong, C–C Activation; Topics in Current Chemistry, Springer-Verlag, Berlin, Heidelberg, 2014, in press Search PubMed.
- N. Rodríguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- For key references, see: D. Necas and M. Kotora, Org. Lett., 2008, 10, 5261–5263 CrossRef CAS PubMed; D. Necas, M. Tursky and M. Kotora, J. Am. Chem. Soc., 2004, 126, 10222–10223 CrossRef PubMed and references therein.
- For a lead reference, see: G. Kuzmanich, A. Natarajan, Y. Shi, B. O. Patrick, J. R. Scheffer and M. A. Garcia-Garibay, Photochem. Photobiol. Sci., 2011, 10, 1731–1734 CAS and references therein.
- For reviews on C−C activation, see:
(a) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870–883 CrossRef;
(b) C.-H. Jun and J. H. Lee, Pure Appl. Chem., 2004, 76, 577–587 CrossRef CAS;
(c) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC;
(d) T. Kondo and T.-a. Mitsudo, Chem. Lett., 2005, 34, 1462–1467 CrossRef CAS;
(e) T. Kondo, Synlett, 2008, 629–644 CrossRef CAS PubMed;
(f) K. Ruhland, Eur. J. Org. Chem., 2012, 2683–2706 CrossRef CAS;
(g) J. E. M. N. Klein and B. Pliekter, Org. Biomol. Chem., 2013, 11, 1271–1279 RSC;
(h) D. Necas and M. Kotora, Curr. Org. Chem., 2007, 11, 1566–1591 CrossRef CAS;
(i) A. Korotvicka, D. Necas and M. Kotora, Curr. Org. Chem., 2012, 16, 1170–1214 CrossRef CAS.
- A. Rusina and A. A. Vlček, Nature, 1965, 206, 295–296 CrossRef CAS.
- E. Muller, A. Stegnitz and E. Langer, Tetrahedron Lett., 1969, 14, 1129–1132 CrossRef.
- B. Müller and A. Segnitz, Liebigs Ann. Chem., 1973, 1583–1591 CrossRef.
- K. Kaneda, H. Azuma, M. Wayaku and S. Teranishi, Chem. Lett., 1974, 215–216 CrossRef CAS.
- P. Eilbracht and P. Dahler, J. Organomet. Chem., 1977, 135, C23–C25 CrossRef CAS.
- R. H. Crabtree, R. P. Dion, D. J. Gibboni, D. V. McGrath and E. M. Holt, J. Am. Chem. Soc., 1986, 108, 7222–7227 CrossRef CAS.
- M. A. Halcrow, F. Urbanos and B. Chaudret, Organometallics, 1993, 12, 955–957 CrossRef CAS.
-
(a) J. W. Suggs and S. D. Cox, J. Organomet. Chem., 1981, 221, 199–201 CrossRef CAS;
(b) J. W. Suggs and C.-H. Jun, J. Am. Chem. Soc., 1984, 106, 3054–3056 CrossRef CAS;
(c) J. W. Suggs and C.-H. Jun, J. Am. Chem. Soc., 1986, 108, 4679–4681 CrossRef CAS.
- T. C. Flood, Top. Stereochem., 1981, 12, 37 CAS.
- J. F. Hartwig, R. A. Anderson and R. G. Bergman, J. Am. Chem. Soc., 1989, 111, 2717–2719 CrossRef CAS.
- For similar reactions following a β-aryl or -alkyl elimination, see: P. Zhao and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 11618–11619 CrossRef CAS PubMed.
- U. Rosenthal and H. Gorls, J. Organomet. Chem., 1992, 439, C36–C41 CrossRef CAS.
-
(a) U. Rosenthal, A. Ohff, W. Baumann, R. Kempe, A. Tillack and V. V. Burlakov, Organometalics, 1994, 13, 2903–2906 CrossRef CAS;
(b) U. Rosenthal, A. Ohff, W. Baumann, R. Kempe, A. Tillack and V. V. Burlakov, Angew. Chem., Int. Ed. Engl., 1994, 33, 1605–1607 CrossRef;
(c) U. Rosenthal, S. Pulst, P. Arndt, A. Ohff, A. Tillack, W. Baumann, R. Kempe and V. V. Burlakov, Organometallics, 1995, 14, 2961–2968 CrossRef CAS;
(d)
U. Rosenthal, P. Arndt, W. Baumann, V. V. Burlakov and A. Spannenberg, J. Organomet. Chem., 2003, 670, 84–96 Search PubMed.
- M. Gozin, A. Welsman, Y. Ben-David and D. Milstein, Nature, 1993, 364, 699–701 CrossRef CAS.
- S.-Y. Liou, M. Gozin and D. Milstein, J. Am. Chem. Soc., 1995, 117, 9774–9775 CrossRef CAS.
- B. Rybtchinski, A. Vigalok, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 1996, 118, 12406–12415 CrossRef CAS.
- M. Gandelman, A. Vigalok, L. J. W. Shimon and D. Milstein, Organometallics, 1997, 16, 3981–3986 CrossRef CAS.
- M. Murakami, M. H. Amii and Y. Ito, Nature, 1994, 370, 540–541 CrossRef CAS.
- C. Muller, C. N. Iverson, R. J. Lachiocotte and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 9718–9719 CrossRef CAS.
- For a report on the activation of diynes, see: U. Rosenthal, S. Pulst, P. Arndt, A. Ohff, A. Tillack, W. Baumann, R. Kempe and V. V. Burlakov, Organometallics, 1995, 14, 2961–2968 CrossRef CAS.
- For further analysis of this system, see:
(a) A. Gunay and W. D. Jones, J. Am. Chem. Soc., 2007, 129, 8729–8735 CrossRef CAS PubMed;
(b) A. Gunay, C. Müller, R. J. Lachicotte, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 6524–6530 CrossRef CAS.
- O. Daugulis and M. Brookhart, Organometallics, 2004, 23, 527–534 CrossRef CAS.
- R. Celenligil, L. A. Watson, C. Guo, B. M. Foxman and O. V. Ozerov, Organometallics, 2005, 24, 186–189 CrossRef.
- A. Obenhuber and K. Ruhland, Organometallics, 2008, 27, 3482–3495 CrossRef.
- A. Obenhuber and K. Ruhland, Organometallics, 2011, 30, 171–186 CrossRef CAS.
- A. Obenhuber and K. Ruhland, Organometallics, 2011, 30, 4039–4051 CrossRef CAS.
- S.-Y. Liou, M. E. van der Boom and D. Milstein, Chem. Commun., 1998, 687–688 RSC.
- N. Chatani, Y. Ie, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 1999, 121, 8645–8646 CrossRef CAS.
- C.-H. Jun and H. Lee, J. Am. Chem. Soc., 1999, 121, 880–881 CrossRef CAS.
- C.-H. Jun, C. W. Moon, H. Lee and D.-Y. Lee, J. Mol. Catal. A: Chem., 2002, 189, 145–156 CrossRef CAS.
- C.-H. Jun, D.-Y. Lee, Y.-H. Kim and H. Lee, J. Am. Chem. Soc., 2001, 20, 2928–2931 CAS.
- C.-H. Jun, H. Lee and S.-G. Lim, J. Am. Chem. Soc., 2001, 123, 751–752 CrossRef CAS.
- A. M. Dreis and C. J. Douglas, J. Am. Chem. Soc., 2009, 131, 412–413 CrossRef CAS PubMed.
- For a separate report on related intermolecular C–C and C–H bond activation, see: M. T. Wentzel, V. J. Reddy, T. K. Hyster and C. J. Douglas, Angew. Chem., Int. Ed., 2009, 48, 6121–6123 CrossRef CAS PubMed.
- For a related oxidative C–C cleavage/C–C formation with 8-acylquinolines, see: J. Wang, W. Chen, S. Zuo, L. Liu, X. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 12334–12338 CrossRef CAS PubMed.
-
(a) C. M. Rathbun and J. B. Johnson, J. Am. Chem. Soc., 2011, 133, 2031–2033 CrossRef CAS PubMed;
(b) J. P. Lutz, C. M. Rathbun, S. M. Stevenson, B. M. Powell, T. S. Boman, C. E. Baxter, J. M. Zona and J. B. Johnson, J. Am. Chem. Soc., 2012, 134, 715–722 CrossRef CAS PubMed.
- Z.-Q. Lei, H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 2690–2694 CrossRef CAS PubMed.
- For similar examples of pyridine-directed C–C activation of benzylic alcohols, see:
(a) H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2011, 133, 15244–15247 CrossRef CAS PubMed;
(b) K. Chen, H. Li, Y. Li, X.-S. Zhang, Z.-Q. Lei and Z.-J. Shi, Chem. Sci., 2012, 3, 1645–1649 RSC.
- A. Wilsily, Y. Nguyen and E. Fillion, J. Am. Chem. Soc., 2009, 131, 15606–15607 CrossRef CAS PubMed.
- M. Arisawa, M. Kuwajima, F. Toriyama, G. Li and M. Yamaguchi, Org. Lett., 2012, 14, 3804–3807 CrossRef CAS PubMed.
- A. Dermenci, R. Whittaker and G. Dong, Org. Lett., 2013, 15, 2242–2245 CrossRef CAS PubMed.
- C. T. To, K. S. Choi and K. S. Chan, J. Am. Chem. Soc., 2012, 134, 11388–11391 CrossRef CAS PubMed.
- Y. W. Chan and K. S. Chan, J. Am. Chem. Soc., 2010, 132, 6920–6922 CrossRef CAS PubMed.
- K. S. Chan, X. Z. Li, W. I. Dzik and B. de Bruin, J. Am. Chem. Soc., 2008, 130, 2051–2061 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.