Redox-responsive polymers for drug delivery: from molecular design to applications
Meng
Huo
ab,
Jinying
Yuan
*a,
Lei
Tao
b and
Yen
Wei
*b
aKey Lab of Organic Optoelectronics and Molecular Engineering of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing, 100084, P. R. China. E-mail: yuanjy@mail.tsinghua.edu.cn; Web: http://www.yuanjinyinggroup.com Tel: +86-1062783668
bKey Lab of Bioorganic Phosphorus Chemistry & Chemical Biology of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing, 100084, P. R. China. E-mail: weiyen@mail.tsinghua.edu.cn; Tel: +86-1062772674
First published on 29th October 2013
Abstract
Glutathione has been regarded as a significant signal for distinguishing between tumor and normal tissue. Recently, reactive oxygen species have attracted much attention for their close connection with many diseases. Taking advantage of the physiological signals, redox-responsive polymeric drug carriers constitute a significant research area in the various stimuli-responsive polymers for biomedical applications. During the rapid development of redox-responsive polymers, molecular design and related synthetic methodology plays a crucial role. In this review, we discuss the reduction- and oxidation-responsive polymeric drug carriers from the view of functional groups, as well as their applications in controlled release.
 Meng Huo | Mr Meng Huo received his B.Sc. from the College of Chemistry, Sichuan University in 2013. He is now studying for his Ph.D degree under the supervision of Prof. Yen Wei and Jinying Yuan at the Department of Chemistry, Tsinghua University. His research interests are focused on developing new stimuli for biomedical applications and exploring biomimetic applications of stimuli-responsive polymers and supramolecular systems. |
 Jinying Yuan | Dr Jinying Yuan received her B.Sc from the Department of Applied Chemistry, University of Science and Technology of China (USTC) in 1987, and received her Ph.D. in the Department of Polymer Science and Engineering, USTC in 2000. After two years of postdoctoral research at the College of Chemistry and Molecular Engineering, Peking University, she joined the Department of Chemistry, Tsinghua University in 1987, where she became a full professor in 2011. Her research interests are focused on the methodology of polymer synthesis and functional polymer materials, especially on controlled polymerization and stimuli-responsive polymers. |
 Lei Tao | Dr Lei Tao graduated from the University of Science and Technology of China, receiving his Bachelor and Master degrees in 1999 and 2002, respectively. After his PhD study in Warwick University (2003–2006), he moved to the University of California, Los Angels (UCLA, USA) in 2006, and then the University of New South Wales (UNSW, Australia) in 2008 as a postdoctoral research assistant. In 2010, he joined the Department of Chemistry, Tsinghua University. His research interests are focused on the synthesis of well-defined polymers through controllable polymerization methods and the application of those polymers in biological areas. |
 Yen Wei | Dr Yen Wei received his M.S. from Peking University in 1981. He then earned his Ph.D. from City University of New York in 1986. After postdoctoral work at MIT, he joined Drexel University in 1987, where he became a Herman B. Wagner Professor in 2004. He is now a Chair Professor of Chemistry and Director of the Center for Frontier Polymer Research at Tsinghua University in Beijing. He has contributed to various fields of polymer, materials, and biological chemistry, and coauthored 589 scientific articles with over 13 |
1. Introduction
Drug delivery systems are regarded as one promising approach to alter the pharmacokinetics and/or bio-distribution of drugs.1 By means of the enhanced permeability and retention (EPR) effect discovered by Maeda in 1989, or active targeting molecules which are decorated on the surface of the system for specific recognition of the target area, to some extent, drug delivery systems have alleviated the main problems of many drugs, such as poor solubility and bio-distribution, inappropriate pharmacokinetics, and severe side effects.2–6 As in the exhaustive research papers in the past decades, “2P” should be taken into account when designing drug delivery systems. The first “P” refers to “protect”, that is, the drug carriers are required to protect the drugs from metabolism in blood circulation.7,8 Besides particle size control, PEGylation was also considered as an ideal tool for reducing the chance of both immunological and kidney clearance.9–11 The other “P” is “preserve”, which means that the carriers should preserve the drugs for a suitable or even programmable time interval before entering malignant tissues or cancer cells.12 To realize this function, new generations of drug delivery systems have been endowed with stimuli-responsiveness.13,14 After navigation along the blood stream, drug carrier systems with pre-designed stimuli-responsiveness accumulated around or even entered the cancerous cells and disintegrated under the carcinoma physiological environment or external stimuli.15Most of the drugs would be pumped out of the tumor cell though the multi-drug resistance mechanism. As a result, they could not take effect even though they have entered the cells.16 One goal of stimuli-responsive polymeric drug carriers was to enable the drugs to instantaneously release after entering the cell, so as to increase the drug concentration to the threshold to kill the cancer cells.17 To better control the drug carriers, they have up to now been endowed with pH-, redox-, light-, magnet-, thermal-, gas- and ultrasonic-responsiveness.18–33 In this review article, we focus on the redox-responsive polymeric drug delivery systems. After a brief introduction to the redox-responsive polymeric drug delivery systems, we will further discuss reduction-responsive and oxidation-responsive systems.
2. Redox-responsive polymeric drug delivery systems
The basic principle of redox-responsive polymeric drug delivery systems is to utilize the distinct differences in redox potentials between tumors and normal tissues. It has been demonstrated in many papers that GSH/glutathione disulfide (GSSG) is the most abundant redox couple in animal cells. In the cytosol and nuclei, the concentration of GSH reaches 10 mM under the reduction of NADPH and glutathione reductase, while outside the cell the concentration drops to about 2–20 μM.34 Moreover, in vivo research has demonstrated that the tumor tissues showed at least 4-fold higher GSH concentrations than that of normal tissue in mice.35 On the other hand, reactive oxygen species (ROS) are believed to be implicated with some serious diseases like arteriosclerosis, heart injury and cancer.36 All of these pathological signals can be exploited as guidance for redox-responsive drug delivery systems.2.1 Reduction-responsive polymeric drug delivery systems
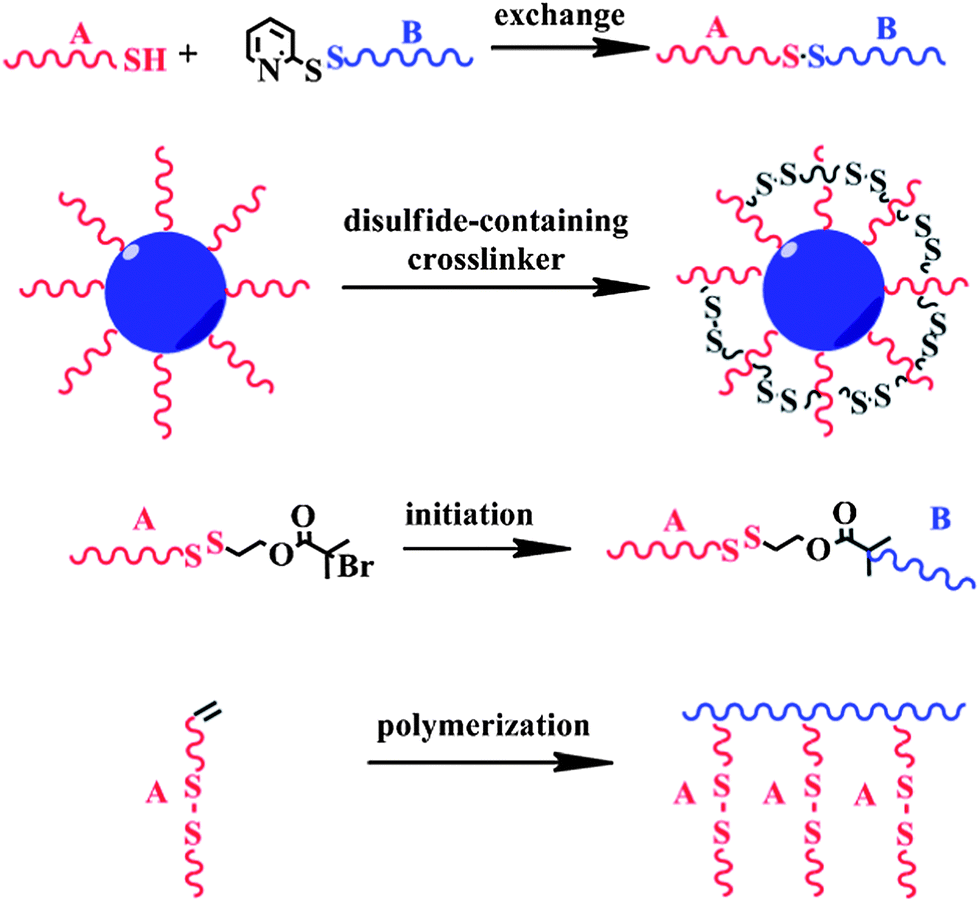 | ||
| Scheme 1 Schematic illustration of the synthetic methods for incorporating the disulfide linkage. | ||
In biological areas, the thiol–disulfide exchange reaction has been found to be closely related to signal transduction, thiol protection and switching between the different conformational and functional states of enzymes.50–53 In biomedical applications, it has been exploited extensively for constructing redox-responsive prodrugs, and gene and drug carriers.34,54–58 For example, Meng and Zhong's group designed a facile way to prepare the disulfide-linked dextran-b-poly(ε-caprolactone) amphiphilic block copolymer using the thiol–disulfide exchange reaction under mild conditions (Scheme 2).59 With an average size of 60 nm in phosphate buffer solution (PBS), the micelles released their cargo in a zero-order manner and almost all of the doxorubicin (DOX) anti-cancer drugs could be released in 10 h at 10 mM dithiothreitol (DTT). Confocal laser scanning microscopy (CLSM) images showed that DOX-loaded micelles showed effective inhibition to RAW 264.7 cells, while the left empty micelles showed non-toxicity. Using modular design, Thayumanavan and his group prepared a disulfide bond-containing ATRP initiator 1 (Scheme 3) and used it to initiate N-isopropylacrylamide and tetrahydropyran protected 2-hydroxyethyl methacrylate, respectively.60 Disulfide bonds featured in the resulting di-block copolymer enabled the successful integration of three stimuli-responsive patterns, which not only showed synergistic effects, but also provided multiple-mode control of the resulting micelle system. One possible problem of the thiol–disulfide exchange reaction may lie in the oxygen sensitivity of thiol-containing compounds, which may increase the complexity of the pretreatment procedure.
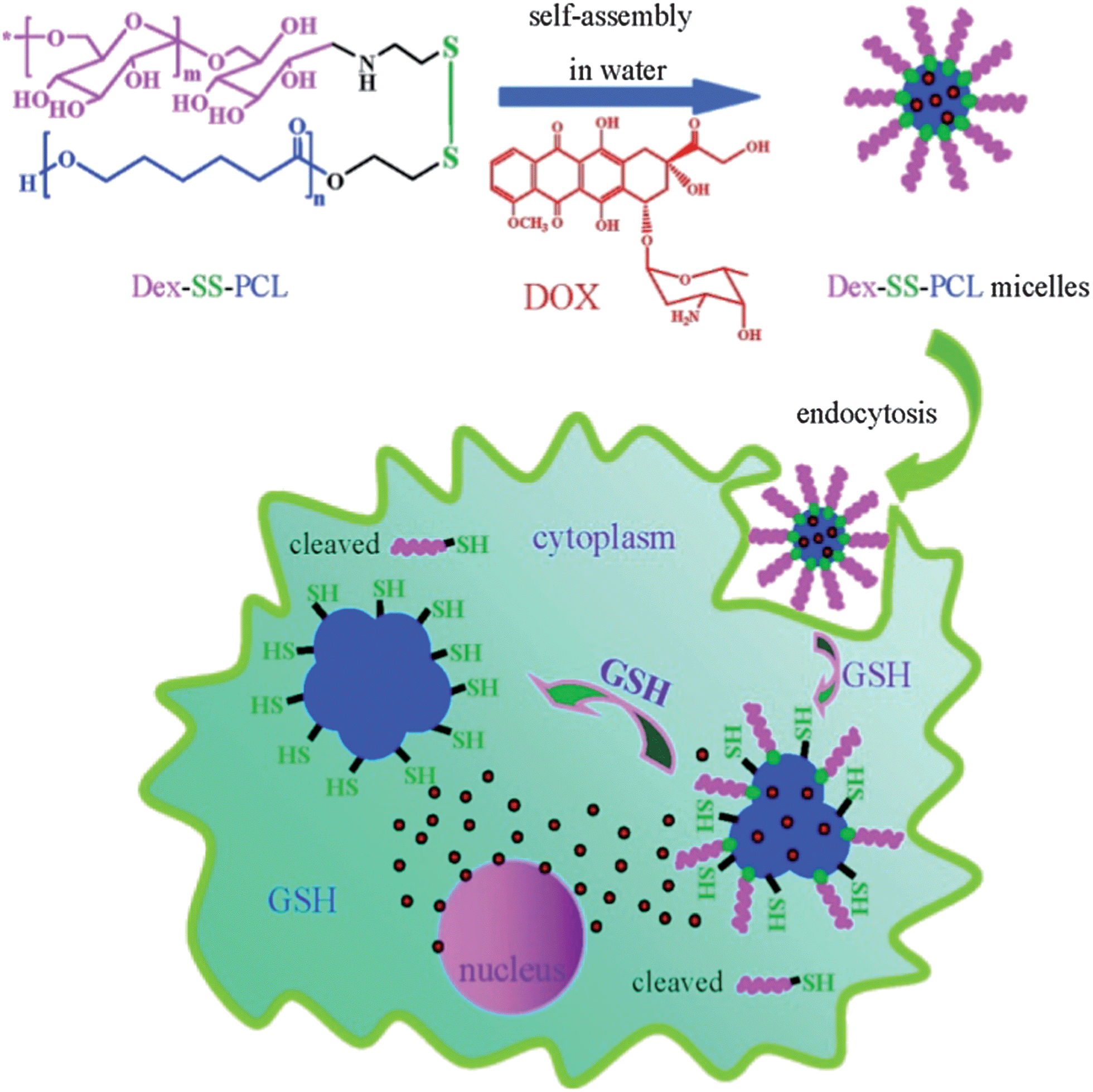 | ||
| Scheme 2 Schematic presentation of redox-responsive Dex–SS–PCL micelles and the intracellular release of DOX.59 | ||
 | ||
| Scheme 3 Synthetic route of a multi-sensitive block copolymer.60 | ||
Apart from the direct disulfide linkage formation reaction, one of the most common strategies was crosslinking through disulfide-containing cross-linkers.61 Up to now, there have been numerous structures built on the basis of cross-linking reactions.62–65 Dithiodipropionic acid, bis(2,2′-hydroxyethyl)disulfide, cystamine and their derivatives were the most useful small molecules for disulfide functionality.56 The resulting cross-linked micelle was observed to efficiently prevent drug leakage in circulation before reaching the target. After core-cross-linked micelles, Wooley's group raised the concept of “shell-cross-linked knedel-like” (SCK) particles, and latter Armes' group and McCormick's group applied this method to stimuli-responsive polymers.66–68 However, cross-linking may slow down the response speed. Recently, Zhang and Zhao reported a surface-cross-linked micelle with extremely rapid release of the encapsulated pyrene (Scheme 4).69 The reason for the fast release profile was associated with the electrostatic repulsion among the headgroups. Once the covalent crosslinking shell was broken by the stimuli, the electrostatic repulsion would be predominant and the micelle exploded rapidly. This may provide inspiration for cross-linked micelles.
 | ||
| Scheme 4 Schematic representation of the surface cross-linked micelle.69 | ||
During recent decades, disulfide-containing ring-opening initiators, ATRP initiators and RAFT agents have been increasingly exploited as a convenient approach to disulfide-containing polymers with defined-structures.60,70–72 For instance, Huang and Yan's group have recently synthesized a functional monomer with a disulfide bond, which was further polymerized into hyperbranched polyphosphate by self-condensing ring-opening polymerization.71 With a hydrophobic disulfide domain and hydrophilic phosphate, these homopolymers could self-assemble into multi-core–shell micelles with potential applications in drug release. Kataoka and coworkers synthesized a series of block copolymers (PEG–SS–P(Asp), PEG–SS–P[Asp(DET)] and PEG–P[Asp(DET)]) by ring-opening polymerization, and the electrostatic attractions between the polycations and polyanions blocks drove them to self-assemble into polyion complex micelles (PICmicelles) (Scheme 5).70
 | ||
| Scheme 5 Self-assembly of PEG–SS–P(Asp) and PEG–SS–P[Asp(DET) and morphology transition under DTT reduction.70 | ||
Interestingly, these micelles re-assemble into hollow nanocapsules under the reduction of the DTT, thus providing a “self-templating” approach towards hollow nanocapsules. Apart from introducing a disulfide bond into the initiator, nucleophilic attack of the thiocarbonyl group was a simple way to convert the end-group of RAFT polymers to thiols, which can not only react with the disulfide but extend the reaction to thiol chemistry.72
Olefin-based polymers with disulfide bonds hanging on the side chain could be obtained by the polymerization of disulfide-containing monomers. This strategy satisfied a tunable balance between the hydrophobic and hydrophilic blocks, and once the balance was broken by cleaving the disulfide bond, the assemblies would collapse to release the drugs. Moreover, according to the amount of DTT added, there could be a micelle-to-nanogel transition that provided a more complex release profile.73 This micelle-to-nanogel transition afforded a facile technique to solve the drug leakage problem of uncross-linked polymeric carriers in clinical applications. Thayumanavan et al. synthesized self-cross-linked nanogels based on RAFT polymerization of oligoethyleneglycol methacrylate (OEGMA) and pyridyl disulfide ethyl methacrylate (PDSMA).74 Nanogels with controlled size and guest release rate formed after adding a certain amount of DTT into the precursor solution. As presented in Scheme 6, fluorescence resonance energy transfer (FRET) was used to study the stability of the nanogels and their encapsulation ability: no green fluorescence emission indicated that the FRET occurred among the dye molecules in the network, while after adding 20 mM DTT, the breakage of the nanogels enabled the dyes to migrate to the hydrophobic domain of the dioleoyl phosphatidylcholine bilayer vesicles, which caused the decrement of the FRET ratio.
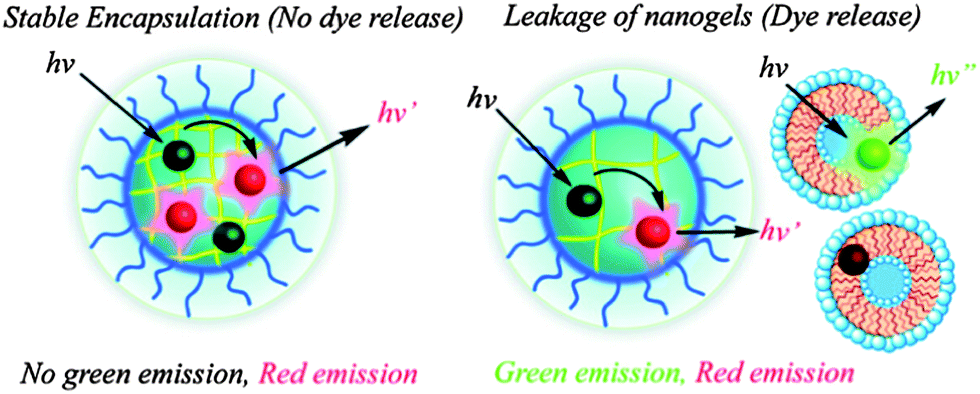 | ||
| Scheme 6 Mechanism of the FRET experiment in comparing the stability of nanogels with or without a reducing agent.74 | ||
In 2009, Xu and Zhang's group reported a diselenide-containing polyurethane triblock copolymer, PEG–PUSeSe–PEG, using toluene diisocyanate as the chain extension agent (Scheme 7).76 They investigated both the reduction- and oxidation-responsive behaviors of these assemblies, and the results indicated that the diselenide was much more sensitive: even under 0.01 mg mL−1 GSH the encapsulated Rhodamine B could still be released almost entirely in only 5 h, while they were stable without redox stimuli. To confirm its use in drug delivery and tumor inhibition, Pang, Huang and Yan et al. introduced the diselenium bond into a hyperbranched structure and found that the hyperbranched polydiselenide could not only be used as a biocompatible drug carrier but itself had the ability to inhibit tumor proliferation.77
 | ||
| Scheme 7 Redox-responsive diselenide-containing PEG–PUSeSe–PEG block copolymers.76 | ||
Until now, diselenide-containing polymers were still limited by the difficulty in the incorporation of the diselenide bond. To some extent, Xu and Zhang's work has depicted their fascinating future in controlled release and enzyme mimics. Recently, Zhu et al. reported the development of a new RAFT mediator based on diselenocarbonyl compounds.78 After optimization, they obtained a comparatively universal selenium-containing RAFT agent which may be of significance for preparing well-defined selenium-containing polymers. With more attempts in this field, diselenide-containing polymers will definitely grow into an important class of biomaterials.
 | ||
| Scheme 8 Synthetic scheme of the platinum(IV)-coordinate polymers.79 | ||
 | ||
| Scheme 9 Reduction of a cisplatin derivative in tumor cells.79 | ||
Trimethyl-locked benzoquinone (TMBQ) and the corresponding hydrocoumarin have been studied extensively, from the transformation mechanism and kinetics to their applications in prodrug design, solid-phase synthesis, probes and biological switches.80 McCarley and his group used it as a trigger of responsive liposomes for the first time.81 However, little attention has been paid to exploit it as switch for redox-responsive polymers until in 2012 Jo et al. applied the TMBQ redox-responsive chemistry for the design of polymeric drug carriers (Scheme 10).82 Under the reduction of sodium dithionite, the TMBQ was shed from the polymer backbone, resulting in the disassembly of the nanoparticles. In vitro drug release experiments showed that this new nano-vehicle could release 52% of the drugs within 3 h in the presence of sodium dithionite, while only 13% of the drugs were released over 12 h without the reducing agent. Yet, the reducing concentration used in the experiment was relatively high compared with that in the cell, and there was no experimental data about the in vitro cytotoxity.
 | ||
| Scheme 10 TMBQ shed from the polymer backbone upon reaction with sodium dithionate.82 | ||
Another redox-responsive system was based on 4-N-amino-2,2,6,6-tetramethylpiperidin-1-oxyl-4-yl (TEMPO), which has vast applications in selective catalysis and battery materials.83,84 Recently, it was used to tune the lower critical solution temperature of poly(N-isopropylacrylamide) (PNIPAM).85 Further applications may be exploited in the field of redox-responsive drug delivery.
2.2 Oxidation-responsive polymeric drug delivery systems
Oxidation-responsive drug carriers rely on reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) and hydroxyl radicals, whose origin is byproducts from aerobic metabolism. Like glutathione, ROS can be found in nearly every corner of the body, the concentration of which would increase significantly during surgery or when suffering from many diseases, resulting in the damaging of cells.36,86 As a result, the ROS can be used as signal molecules of arteriosclerosis, some nerve diseases and heart injury.87 Taking advantage of ROS, oxidation-responsive polymeric drug delivery systems have enlarged the limits of redox-responsive drug carriers.88As sulfur is relative stable, sulfide-containing oxidation-responsive polymers have not been reported extensively. In fact, the architecture of sulfide-containing polymers is almost, if not all, linear. More synthetic methods were needed for polymers with various architectures to function in different applications.
Compared with the conventional covalent bond, supramolecular tools have the intrinsic advantages to overcome the complex synthetic routes required to incorporate the selenium. For example, Xu and Zhang's group developed approaches to selenium-containing oxidation-responsive systems with a selenium-containing surfactant and poly(ethylene glycol)-b-acrylic acid.96 Hydrophobic interactions and electrostatic attractions between the surfactant and poly(acrylic acid) anion served as the driving force for the assemblies, which disassociated under the incubation of 0.1% H2O2 solution. The model molecule fluorescein sodium was loaded and released in a controllable manner for further exploring the potential applications. However, the toxicity of the surfactant used should be considered in clinical applications.
Vancso's group reported an oxidation-responsive polymeric hybrid capsule fabricated by LBL assembly on colloidal microparticles followed by core removing, which is depicted with more detail in Scheme 11.108 Oxidation-responsive poly(ferrocenylsilane)-containing polycations and polyanions was chosen as the main building blocks, while the outer layers of the capsules were composed of poly(styrene sulfonate) and poly(allylamine hydrochloride) (PAH) in order to suppress the excess swell in the oxidation state. Interestingly, both the size and permeation could be well-tuned via the redox state change, which can be very meaningful in biomedical applications and biomimetic research.
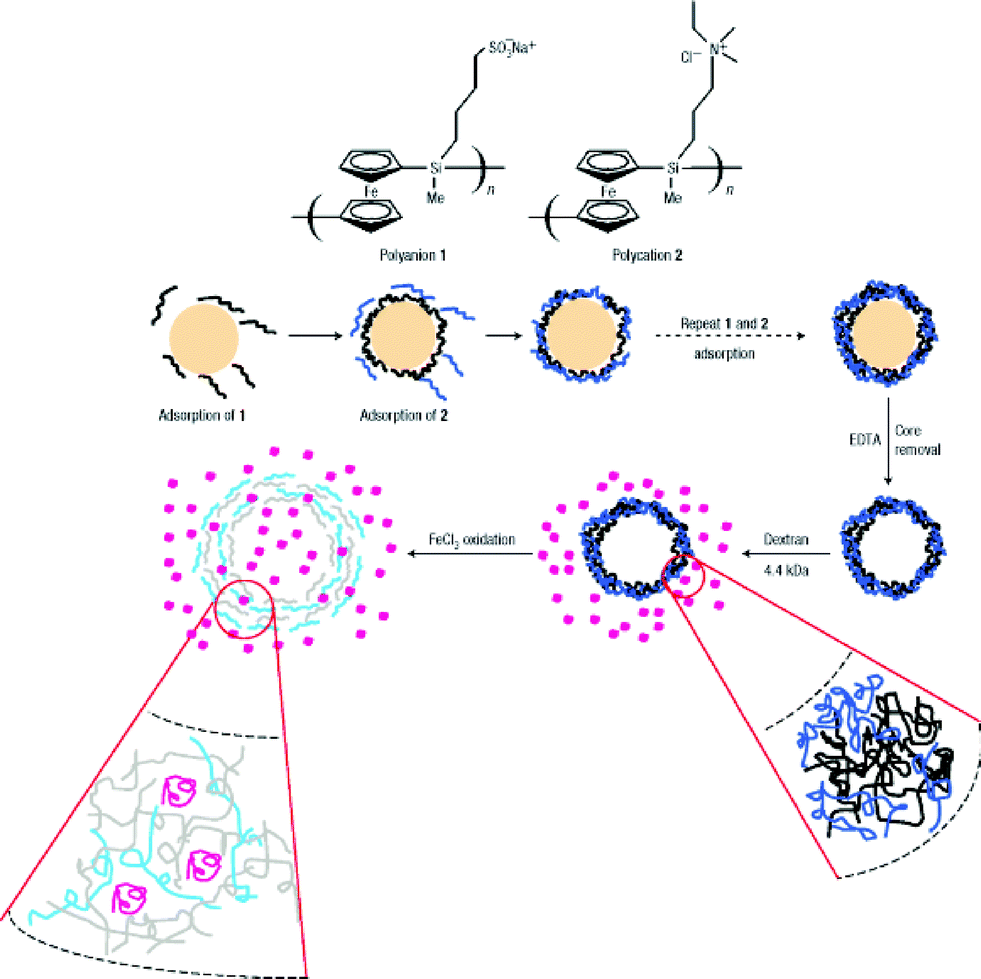 | ||
| Scheme 11 LBL process of the polymeric hybrid capsule and its redox-responsive “breath”.108 | ||
Gallei and Crespy's group successfully fabricated poly(vinylferrocene)-block-poly(methyl methacrylate) (PVFc-b-PMMA) nanocapsules with ferrocene on the side chain.109 Microphase separation in the nanoparticles led to a patchy structure, with PVFc patches surrounded by the PMMA phase. Furthermore, these patches could be oxidized into hydrophilic ferrocenium, forming many leaky tunnels for the encapsulated liquid. Gao et al. developed another facile way to synthesize ferrocene-containing polymers.110 Instead of direct polymerization, they prepared the single-component microcapsules by the in situ reaction of PAH with ferrocenecarboxaldehyde in CaCO3 nanoparticles. In contrast to the typical electrostatic attractions, the microcapsules were stabilized by the hydrophobic interactions of the ferrocene group and the protection of hydrophilic PAH after the core removal.
Polymers decorated with ferrocene as the terminal group are usually used to construct more complex systems, utilizing ferrocene-related host–guest chemistry. Depending on the redox state of ferrocene, the inclusion complexation between ferrocene and cyclodextrin has been verified to associate and disassociate reversibly. For example, Chen and Jiang have exploited the ferrocene-terminated diblock copolymer poly(N,N′-dimethylacrylamide)-b-poly-(N-isopropylacrylamide) and cyclodextrin-decorated CdS quantum dots to obtain a redox-responsive hybrid hydrogel at elevated temperatures.111 Our group have prepared a pseudo-block copolymer via orthogonal assembly between cyclodextrin-modified poly(styrene) and ferrocene end-functionalized poly(ethylene oxide) in aqueous solution (Scheme 12).112 These supramolecular block copolymers could further self-assemble into polymeric vesicles with voltage-responsiveness. The association–disassociation balance could be changed by electro-stimuli: upon a +1.5 V voltage stimulus, the vesicles disassembled into small pieces in less than 5 h, while under a −1.5 V voltage, the fragments could reassemble into vesicles. Further controlled release experiments elucidated that the release rate can be well-controlled by slightly tuning the voltage strength, exhibiting great potential in electrochemical therapeutic applications.113 To further explore the application of this host–guest chemistry based electrical-responsive pattern, we decorated oligo(ethylene glycol) with ferrocene and β-cyclodextrin to obtain Fc–OEG–Fc and β-CD–OEG-β-CD, respectively.114 These two monomers would form a supramolecular polymer with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio as a result of host–guest inclusion, and the supramolecular polymers further hierarchically self-assemble into fibres with voltage-responsive degradation and self-healing properties.
:1 ratio as a result of host–guest inclusion, and the supramolecular polymers further hierarchically self-assemble into fibres with voltage-responsive degradation and self-healing properties.
 | ||
| Scheme 12 Orthogonal assembly of PS-β-CD and PEO–Fc and their voltage responsiveness.112 | ||
Ferrocene-containing polymers have found applications in switches, probes and electric devices. In biomedical applications, they may serve as long-term drug delivery pumps that release drugs upon exerting electricity, and thus may be helpful to ensure maximal therapeutic effects.
 | ||
| Scheme 13 The degradation of self-immolative nanoparticles caused by H2O2 in inflamed tissue.115 | ||
Aside from boronic esters, some other redox-responsive groups such as tetrathiafulvalene, mesoporous silicon, polythioketal and oligoproline have also been used in bio-related polymer synthesis.36,118–120 For example, Cooke and Woisel have found that tetrathiafulvalene (TTF) end-modified PNIPAM homopolymers would self-assemble into micelles in aqueous solution (Scheme 14).118 The main reason for the stability of this micelle is presumably the integration of hydrophobic interactions of TTF groups, S⋯S interactions and π–π stacking. These micelles could be broken by oxidation and host–guest inclusion of the TTF group and tetracationic macrocycle cyclobis(paraquat-p-phenylene) (CBPQT4+). Further, the amphiphilicity of this polymer could be manipulated by adding CBPQT4+ or the randomly methylated β-cyclodextrin.121 Because of TTF's diverse redox and inclusion chemistry, this polymer may be used in drug carrier, biological probe, redox-responsive switch, and molecular machine.
 | ||
| Scheme 14 Formation of the micelles and their oxidation- and host-responsive disassembly.118 | ||
3. Conclusion and perspective
Taking advantage of the specific micro-environment of cancer tissues, redox-responsive carriers satisfy the trend of non-invasive therapy. The incorporation of various redox-responsive functional groups onto the polymer rendered the polymer with “intelligence”, and thus the assembled drug carriers would be able to transport to the required region. In fact, the physicochemical discrepancy between cancerous and normal tissues is rather small for conventional chemical stimuli, so the development of redox-responsive polymeric drug carriers moves towards a more sensitive trigger. To realize the goal, more attention should be focused on the relative concepts in organic chemistry. Moreover, the hetereogeniety of tumor cells will inevitably increase the difficulty of drug carriers to take effect in vivo. Shen and Gu's group have pointed out recently that polymeric nanoparticles responsive to both GSH and ROS may provide an approach to the hetereogeneity of tumor micro-environment.122 In fact, there were indeed some redox-responsive functional groups, for example, sulfide, disulfide, selenium, diselenium and ferrocene, that have been studied for both reduction- and oxidation-responsive drug delivery. However, few comprehensive in vivo experiments have been performed, and more attention should be paid to this field. Additionally, designing drug carriers with specific structures (core–shell, compartmental nanoparticles and hybrid nanoparticles) while lowering the cost is still a dilemma. Furthermore, integrating multiple functionalities into one drug carrier is still a challenge. For instance, to visualize the drug release, combining imaging and treatment would be attractive, which would aid the drug distribution studies as well as the controlled release.Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (21374053, 51073090), the National Basic Research Program of China (2011CB935700) and the Specialized Research Fund for the Doctoral Program of High Education of China (20120002110015).Notes and references
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- H. Maeda and Y. Matsumura, Crit. Rev. Ther. Drug Carrier Syst., 1989, 6, 193–210 CAS.
- S. Dufort, L. Sancey and J.-L. Coll, Adv. Drug Delivery Rev., 2012, 64, 179–189 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- J. A. Barreto, W. O'Malley, M. Kubeil, B. Graham, H. Stephan and L. Spiccia, Adv. Mater., 2011, 23, H18–H40 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- S. Mitragotri and J. Lahann, Adv. Mater., 2012, 24, 3717–3723 CrossRef CAS PubMed.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- S. Nie, Nanomedicine, 2010, 5, 523–528 CrossRef PubMed.
- J. A. Hubbell and A. Chilkoti, Science, 2012, 337, 303–305 CrossRef PubMed.
- Q. Sun, M. Radosz and Y. Shen, J. Controlled Release, 2012, 164, 156–169 CrossRef CAS PubMed.
- A. Chan, R. P. Orme, R. A. Fricker and P. Roach, Adv. Drug Delivery Rev., 2013, 65, 497–514 CrossRef CAS PubMed.
- Z. L. Tyrrell, Y. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128–1143 CrossRef CAS.
- G. B. Sukhorukov and H. Möhwald, Trends Biotechnol., 2007, 25, 93–98 CrossRef CAS PubMed.
- Q. Yin, J. Shen, Z. Zhang, H. Yu and Y. Li, Adv. Drug Delivery Rev., 2013, 65, 1699–1715 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10–16 CrossRef CAS PubMed.
- C. d. l. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- J. M. Spruell and C. J. Hawker, Chem. Sci., 2011, 2, 18–26 RSC.
- H. Wei, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- G. Pasparakis and M. Vamvakaki, Polym. Chem., 2011, 2, 1234–1248 RSC.
- C. P. McCoy, C. Brady, J. F. Cowley, S. M. McGlinchey, N. McGoldrick, D. J. Kinnear, G. P. Andrews and D. S. Jones, Expert Opin. Drug Delivery, 2010, 7, 605–616 CrossRef CAS PubMed.
- A. E. Felber, M.-H. Dufresne and J.-C. Leroux, Adv. Drug Delivery Rev., 2012, 64, 979–992 CrossRef CAS PubMed.
- Y. Xin and J. Yuan, Polym. Chem., 2012, 3, 3045–3055 RSC.
- I. Tomatsu, K. Peng and A. Kros, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef CAS PubMed.
- J. Qin, I. Asempah, S. Laurent, A. Fornara, R. N. Muller and M. Muhammed, Adv. Mater., 2009, 21, 1354–1357 CrossRef CAS.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2011, 50, 4923–4927 CrossRef CAS PubMed.
- Q. Yan, J. Wang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2013, 52, 5070–5073 CrossRef CAS PubMed.
- J. Xuan, O. Boissière, Y. Zhao, B. Yan, L. Tremblay, S. Lacelle, H. Xia and Y. Zhao, Langmuir, 2012, 28, 16463–16468 CrossRef CAS PubMed.
- H.-J. Zhang, Y. Xin, Q. Yan, L.-L. Zhou, L. Peng and J.-Y. Yuan, Macromol. Rapid Commun., 2012, 33, 1952–1957 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- P. Kuppusamy, H. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307–312 CAS.
- S. H. Lee, M. K. Gupta, J. B. Bang, H. Bae and H.-J. Sung, Adv. Healthcare Mater., 2013, 2, 908–915 CrossRef CAS PubMed.
- S. Aleksanian, B. Khorsand, R. Schmidt and J. K. Oh, Polym. Chem., 2012, 3, 2138–2147 RSC.
- Y. Yan, Y. Wang, J. K. Heath, E. C. Nice and F. Caruso, Adv. Mater., 2011, 23, 3916–3921 CrossRef CAS PubMed.
- Z.-Q. Yu, J.-T. Sun, C.-Y. Pan and C.-Y. Hong, Chem. Commun., 2012, 48, 5623–5625 RSC.
- D. Han, X. Tong and Y. Zhao, Langmuir, 2012, 28, 2327–2331 CrossRef CAS PubMed.
- R. L. McCarley, Annu. Rev. Anal. Chem., 2012, 5, 391–411 CrossRef CAS PubMed.
- T. Thambi, V. G. Deepagan, H. Ko, D. S. Lee and J. H. Park, J. Mater. Chem., 2012, 22, 22028–22036 RSC.
- W. Yuan, H. Zou, W. Guo, T. Shen and J. Ren, Polym. Chem., 2013, 4, 2658–2661 RSC.
- J. Zhang, F. Yang, H. Shen and D. Wu, ACS Macro Lett., 2012, 1, 1295–1299 CrossRef CAS.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. B. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- H. F. Gilbert, J. Biol. Chem., 1982, 257, 12086–12091 CAS.
- K. S. Jensen, R. E. Hansen and J. R. Winther, Antioxid. Redox Signaling, 2009, 11, 1047–1058 CrossRef CAS PubMed.
- Y. M. Go and D. P. Jones, Free Radical Biol. Med., 2011, 50, 495–509 CrossRef CAS PubMed.
- W. Wang, Y. Huang, S. Zhao, T. Shao and Y. Cheng, Chem. Commun., 2013, 49, 2234–2236 RSC.
- R. Ondarza, Biosci. Rep., 1989, 9, 593–604 CrossRef CAS PubMed.
- A. J. van der Vlies, U. Hasegawa and J. A. Hubbell, Mol. Pharmaceutics, 2012, 9, 2812–2818 CrossRef CAS PubMed.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- S. Son, R. Namgung, J. Kim, K. Singha and W. J. Kim, Acc. Chem. Res., 2011, 45, 1100–1112 CrossRef PubMed.
- G. T. Zugates, D. G. Anderson, S. R. Little, I. E. Lawhorn and R. Langer, J. Am. Chem. Soc., 2006, 128, 12726–12734 CrossRef CAS PubMed.
- H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS PubMed.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- Y. Li, R. Tong, H. Xia, H. Zhang and J. Xuan, Chem. Commun., 2010, 46, 7739–7741 RSC.
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS PubMed.
- X.-Q. Li, H.-Y. Wen, H.-Q. Dong, W.-M. Xue, G. M. Pauletti, X.-J. Cai, W.-J. Xia, D. Shi and Y.-Y. Li, Chem. Commun., 2011, 47, 8647–8649 RSC.
- Z. Zhou, X. Ma, E. Jin, J. Tang, M. Sui, Y. Shen, E. A. Van Kirk, W. J. Murdoch and M. Radosz, Biomaterials, 2013, 34, 5722–5735 CrossRef CAS PubMed.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1996, 118, 7239–7240 CrossRef CAS.
- S. Liu and S. P. Armes, J. Am. Chem. Soc., 2001, 123, 9910–9911 CrossRef CAS PubMed.
- Y. Li, B. S. Lokitz and C. L. McCormick, Macromolecules, 2005, 39, 81–89 CrossRef.
- S. Zhang and Y. Zhao, J. Am. Chem. Soc., 2010, 132, 10642–10644 CrossRef CAS PubMed.
- W.-F. Dong, A. Kishimura, Y. Anraku, S. Chuanoi and K. Kataoka, J. Am. Chem. Soc., 2009, 131, 3804–3805 CrossRef CAS PubMed.
- J. Liu, W. Huang, Y. Pang, P. Huang, X. Zhu, Y. Zhou and D. Yan, Angew. Chem., 2011, 123, 9328–9332 CrossRef.
- A. P. Vogt and B. S. Sumerlin, Soft Matter, 2009, 5, 2347–2351 RSC.
- Q. Zhang, S. Aleksanian, S. M. Noh and J. K. Oh, Polym. Chem., 2013, 4, 351–359 RSC.
- J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS PubMed.
- N. Ma, Y. Li, H. Xu, Z. Wang and X. Zhang, J. Am. Chem. Soc., 2009, 132, 442–443 CrossRef PubMed.
- J. Liu, Y. Pang, J. Chen, P. Huang, W. Huang, X. Zhu and D. Yan, Biomaterials, 2012, 33, 7765–7774 CrossRef CAS PubMed.
- J. Zeng, J. Zhu, X. Pan, Z. Zhang, N. Zhou, Z. Cheng, W. Zhang and X. Zhu, Polym. Chem., 2013, 4, 3453–3457 RSC.
- J. Yang, W. Liu, M. Sui, J. Tang and Y. Shen, Biomaterials, 2011, 32, 9136–9143 CrossRef CAS PubMed.
- M. N. Levine and R. T. Raines, Chem. Sci., 2012, 3, 2412–2420 RSC.
- W. Ong, Y. Yang, A. C. Cruciano and R. L. McCarley, J. Am. Chem. Soc., 2008, 130, 14739–14744 CrossRef CAS PubMed.
- H. Cho, J. Bae, V. K. Garripelli, J. M. Anderson, H.-W. Jun and S. Jo, Chem. Commun., 2012, 48, 6043–6045 RSC.
- P. L. Bragd, H. van Bekkum and A. C. Besemer, Top. Catal., 2004, 27, 49–66 CrossRef CAS.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS PubMed.
- H. Fu, D. M. Policarpio, J. D. Batteas and D. E. Bergbreiter, Polym. Chem., 2010, 1, 631–633 RSC.
- Y. L. Colson and M. W. Grinstaff, Adv. Mater., 2012, 24, 3878–3886 CrossRef CAS PubMed.
- B. D'Autreaux and M. B. Toledano, Nat. Rev. Mol. Cell Biol., 2007, 8, 813–824 CrossRef PubMed.
- E. Lallana and N. Tirelli, Macromol. Chem. Phys., 2013, 214, 143–158 CrossRef CAS.
- A. Napoli, M. Valentini, N. Tirelli, M. Muller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- D. Velluto, D. Demurtas and J. A. Hubbell, Mol. Pharmaceutics, 2008, 5, 632–642 CrossRef CAS PubMed.
- P. Hu and N. Tirelli, Bioconjugate Chem., 2012, 23, 438–449 CrossRef CAS PubMed.
- E. A. Mahmoud, J. Sankaranarayanan, J. M. Morachis, G. Kim and A. Almutairi, Bioconjugate Chem., 2011, 22, 1416–1421 CrossRef CAS PubMed.
- N. Ma, Y. Li, H. Ren, H. Xu, Z. Li and X. Zhang, Polym. Chem., 2010, 1, 1609–1614 RSC.
- H. Ren, Y. Wu, N. Ma, H. Xu and X. Zhang, Soft Matter, 2012, 8, 1460–1466 RSC.
- J. Liu, Y. Pang, Z. Zhu, D. Wang, C. Li, W. Huang, X. Zhu and D. Yan, Biomacromolecules, 2013, 14, 1627–1636 CrossRef CAS PubMed.
- P. Han, N. Ma, H. Ren, H. Xu, Z. Li, Z. Wang and X. Zhang, Langmuir, 2010, 26, 14414–14418 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- M. Nakahata, Y. Takashima, A. Hashidzume and A. Harada, Angew. Chem., Int. Ed., 2013, 52, 5731–5735 CrossRef CAS PubMed.
- K. Kulbaba and I. Manners, Macromol. Rapid Commun., 2001, 22, 711–724 CrossRef CAS.
- K. Kulbaba, A. Cheng, A. Bartole, S. Greenberg, R. Resendes, N. Coombs, A. Safa-Sefat, J. E. Greedan, H. D. H. Stover, G. A. Ozin and I. Manners, J. Am. Chem. Soc., 2002, 124, 12522–12534 CrossRef CAS PubMed.
- H. Wang, X. Wang, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2008, 130, 12921–12930 CrossRef CAS PubMed.
- X. Sui, X. Feng, M. A. Hempenius and G. J. Vancso, J. Mater. Chem. B, 2013, 1, 1658–1672 RSC.
- M. F. R. Fouda, M. M. Abd-Elzaher, R. A. Abdelsamaia and A. A. Labib, Appl. Organomet. Chem., 2007, 21, 613–625 CrossRef CAS.
- B. Lal, A. Badshah, A. A. Altaf, N. Khan and S. Ullah, Appl. Organomet. Chem., 2011, 25, 843–855 CrossRef CAS.
- O. N. Kadkin and G. G. Yu, Russ. Chem. Rev., 2012, 81, 675 CrossRef CAS.
- R. Gracia and D. Mecerreyes, Polym. Chem., 2013, 4, 2206–2214 RSC.
- X. Sui, X. Feng, A. Di Luca, C. A. van Blitterswijk, L. Moroni, M. A. Hempenius and G. J. Vancso, Polym. Chem., 2013, 4, 337–342 RSC.
- Y. Ma, W.-F. Dong, M. A. Hempenius, H. Mohwald and G. Julius Vancso, Nat. Mater., 2006, 5, 724–729 CrossRef CAS PubMed.
- R. H. Staff, M. Gallei, M. Mazurowski, M. Rehahn, R. Berger, K. Landfester and D. Crespy, ACS Nano, 2012, 6, 9042–9049 CrossRef CAS PubMed.
- Z. Wang, H. Möhwald and C. Gao, Langmuir, 2010, 27, 1286–1291 CrossRef PubMed.
- P. Du, J. Liu, G. Chen and M. Jiang, Langmuir, 2011, 27, 9602–9608 CrossRef CAS PubMed.
- Q. Yan, J. Yuan, Z. Cai, Y. Xin, Y. Kang and Y. Yin, J. Am. Chem. Soc., 2010, 132, 9268–9270 CrossRef CAS PubMed.
- L. Peng, A. Feng, H. Zhang, H. Wang, C. Jian, B. Liu, W. Gao and J. Yuan, Polym. Chem. 10.1039/c3py01204b.
- Q. Yan, A. Feng, H. Zhang, Y. Yin and J. Yuan, Polym. Chem., 2013, 4, 1216–1220 RSC.
- C.-C. Song, R. Ji, F.-S. Du, D.-H. Liang and Z.-C. Li, ACS Macro Lett., 2013, 2, 273–277 CrossRef CAS.
- C. de Gracia Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758–15764 CrossRef CAS PubMed.
- K. E. Broaders, S. Grandhe and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 133, 756–758 CrossRef PubMed.
- J. Bigot, B. Charleux, G. Cooke, F. Delattre, D. Fournier, J. Lyskawa, L. Sambe, F. Stoffelbach and P. Woisel, J. Am. Chem. Soc., 2010, 132, 10796–10801 CrossRef CAS PubMed.
- E. C. Wu, J.-H. Park, J. Park, E. Segal, F. d. r. Cunin and M. J. Sailor, ACS Nano, 2008, 2, 2401–2409 CrossRef CAS PubMed.
- S. S. Yu, R. L. Koblin, A. L. Zachman, D. S. Perrien, L. H. Hofmeister, T. D. Giorgio and H.-J. Sung, Biomacromolecules, 2011, 12, 4357–4366 CrossRef CAS PubMed.
- L. n. Sambe, F. o. Stoffelbach, J. Lyskawa, F. o. Delattre, D. Fournier, L. Bouteiller, B. Charleux, G. Cooke and P. Woisel, Macromolecules, 2011, 44, 6532–6538 CrossRef CAS.
- J. Wang, X. Sun, W. Mao, W. Sun, J. Tang, M. Sui, Y. Shen and Z. Gu, Adv. Mater., 2013, 25, 3670–3676 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |