Pd-catalyzed oxidative coupling with organometallic reagents via C–H activation
Chang-Liang
Sun
,
Bi-Jie
Li
and
Zhang-Jie
Shi
*
Beijing National Laboratory of Molecular Sciences (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, Peking University, Beijing, China. E-mail: zshi@pku.edu.cn; Fax: 86-010-62760890; Tel: 86-010-62760890
First published on 5th January 2010
Abstract
Direct selective palladium catalyzed C–H functionalization has become a highly attractive strategy in organic synthesis and represents a highly desirable goal. Compared with cross-coupling reactions of C–H bonds with aryl or alkyl halides/pseudohalides, the strategy of cross-coupling reactions of C–H bonds with organometallic reagents is of great significance and obvious advantages. This feature article provides a comprehensive summary of recent advances and an intensive analysis on Pd-catalyzed C–H activation and oxidative coupling with various organometallic reagents.
 Chang-Liang Sun | Chang-Liang Sun was born in Jilin, China, in 1983. He received his BSc in chemistry from Jilin University in 2006. In September 2006, he joined the Shi group at Peking University in the College of Chemistry and Molecular Engineering. His current research interests include Ni-catalyzed activation of C–O bonds and transition metal catalyzed activation of carbon dioxide. |
 Bi-Jie Li | Bi-Jie Li was born in Hubei, China, in 1985. He received his BSc from Peking University in 2007. He is currently a second-year graduate student in the Shi group at Peking University in the College of Chemistry and Molecular Engineering. His current research interests focus on transition metal catalyzed sp3 C–H bond activation. |
 Zhang-Jie Shi | Zhang-Jie Shi was born in Anhui, China, in 1974. He obtained his BSc at East China Normal University in 1996, and PhD in the Shanghai Institute of Organic Chemistry (SIOC), CAS in 2001. After his postdoctoral studies at Harvard University and the University of Chicago, he joined the chemistry faculty of Peking University in 2004, where he was promoted to a full Professor in 2008. |
Introduction
Green and sustainable chemistry has attracted much attention and chemists have developed many novel synthetic methods to avoid the use of toxic chemicals and shorten synthetic routes. The C–H bond is the most common chemical bond in organic compounds and can be transformed into other functional groups through direct or indirect processes.1 Undoubtedly, direct and selective C–H functionalization has become a highly attractive strategy to approach green, clean and efficient transformations.2In the past several decades, transition metal catalyzed cross-coupling reactions have been well developed and widely applied in organic synthesis, which provides useful methods to construct complicated scaffolds.3 For example, Suzuki–Miyaura coupling,4 Stille coupling,5 Kumada coupling,6 Hiyama coupling,7 and Negishi coupling,8 have been well studied as powerful methods in the toolbox of organic chemists. It is well known that traditional cross-coupling involves two kinds of fully functionalized starting materials, including organic halides and organometallic reagents. Taking the place of either C–X or C–M or both with C–H is an ideal design to make cross-coupling more efficient and cleaner (Scheme 1).
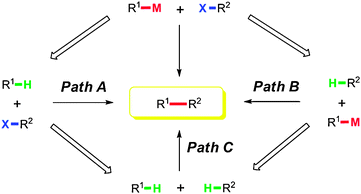 | ||
| Scheme 1 Introduction of C–H bond activation into cross-couplings. | ||
In the past two decades, many research groups have reported the intermolecular and intramolecular cross-coupling reactions involving direct C–H activation with organic halides/pseudohalides as coupling partners.9 Mechanistically, these processes were initiated by oxidative addition of organic halides/pseudohalides to Pd(0) species, followed by the electrophilic attack of the generated Pd(II) species to alkyl/aryl groups. Finally, deprotonation and reductive elimination took place and Pd(0) species were regenerated to complete the catalytic cycle (Scheme 2). Different parameters, such as bases, phosphine ligands, high-polarity solvents and high temperature were necessary to facilitate these transformations. More recently, cross-couplings directly starting from two C–H bonds have also been reported,10 which showed the potential to approach the most efficient way to construct C–C bonds starting from simple arenes, although the selectivity and conditions might not reach practical levels as of now.
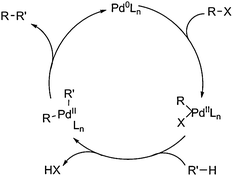 | ||
| Scheme 2 General mechanism of cross-couplings of C–H bonds with organic halides/pseudohalides. | ||
Compared with the remarkable processes of Pd(0)-catalyzed cross-coupling reactions of C–H bonds with aryl or alkyl halides/pseudohalides and their equivalents, the strategy of Pd(II)-catalyzed cross-coupling reactions of C–H bonds with organometallic reagents has only recently received more attention. Undoubtedly, such transformations involve different pathways and remain a challenge. Direct C–H functionalizations with different organometallic reagents via Pd catalysis has recently been developed extensively by our group amongst others.
In the proposed catalytic cycle, the electrophilic substitution toward aliphatic or aromatic systems with/without directing groups by Pd(II) species, initiates the transformation and generates aryl Pd(II) species. Subsequently, the alkyl or aryl Pd species are generated by transmetalation between organometallic reagents and the aryl Pd(II) precursor. Followed by reductive elimination to produce the desired product, the Pd(0) species are generated, which are reoxidized to Pd(II) species to complete the catalytic cycle (Scheme 3). Therefore, appropriate oxidants are necessary and acidic conditions would promote these processes. Thus, such a process is defined as an oxidative cross-coupling.
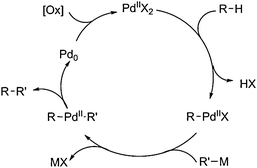 | ||
| Scheme 3 General mechanism of cross-couplings of C–H bonds with organometallic reagents. | ||
Mechanistically, neither ligands nor bases are needed, which makes the resulting system less toxic and pollutive. However, there are still several challenges: (1) the homo-coupling of organometallic reagents caused by Pd(II) species or oxidants in the system; (2) the incompatibility of reaction conditions between C–H activation and transmetalation; (3) the stability of organometallic reagents under the electrophilic C–H activation. Among the typical organometallic reagents, Grignard reagents (organomagnesium reagents), organolithium, organoaluminium and organozinc reagents are of high reactivity and low stability. As a result, they can hardly survive in the C–H activation process and thus are rarely introduced into such kinds of cross-couplings. Considering their intrinsic nature and wide applications in organic synthesis, organoboron, organosilicon and organotin reagents are ideal choices as coupling partners in these transformations.
Organoboron reagents as the coupling partners
Organoboron reagents, especially boronic acids, have been widely applied in organic synthesis as a particularly attractive class of reagents due to their availability, stability, nontoxicity and ease of handling. Suzuki–Miyaura coupling has become a powerful tool to construct complicated C–C scaffolds. Other analogs of boronic acids, such as cyclic boroxines, corresponding boronic esters and trifluoroborate salts, also showed great reactivities in many transformations (Scheme 4). Traditionally, organoboron reagents could be easily prepared by hydroboration or exchange with organometallic reagents. With the recent development of direct borylation of aryl and alkyl C–H bonds, organoboron reagents could be obtained in a more efficient and atom-economic way.11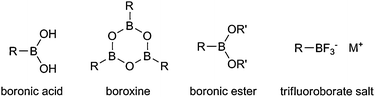 | ||
| Scheme 4 Various forms of organoboron reagents. | ||
C–C bonds can be constructed by direct C–H activation using various methods.12 However, processes to form C–C bonds starting from C–H bonds by Suzuki–Miyaura type coupling are rarely reported. Murai and Sames originally reported direct arylation of sp3 and sp2 C–H bonds directed by a heterocyclic group via Ru catalysis with aromatic boronic esters.13 Our goal is to introduce the simplest and most common boronic acids into such direct arylations to develop more useful and efficient methodologies. In this field, Yu’s group has been prominent, reporting Pd-catalyzed alkylation and arylation of sp2 and sp3 C–H bonds with boronic esters and boroxines with different directing groups.14 Up to now, the application of directing groups is still the best means to tune the reactivity and control the regioselectivity.
Ortho-arylation of acetanilides
As our design, we first tried direct arylation of acetanilides, which showed high reactivity in various transformations.15 The palladacycle obtained by electrophilic substitution (Scheme 5, path a) was proposed as a key intermediate, which is identical to that generated from oxidative addition with the corresponding halide and Pd(0) species (Scheme 5, path b). Such an intermediate may undergo transmetalation and subsequent reductive elimination to form the C–C bond. However, significant challenges remain: (1) after the catalytic cycles, Pd(0) species different from the initiated Pd(II) are obtained; (2) acidic conditions enhance electrophilicity of Pd(II) species, while most Suzuki–Miyaura couplings proceed under basic conditions. Thus, the proposed key point for this design is the choice of an appropriate base and oxidant.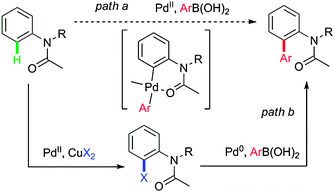 | ||
| Scheme 5 Design of a Suzuki–Miyaura coupling via C–H activation. | ||
The substrate 1a was chosen as a model substrate due to its high reactivity towards direct arylation with phenylboronic acid. After screening, we finally obtained the optimized reaction conditions (Table 1) and then different aromatic boronic acids were subjected. We observed that different substituents on the aromatic rings of the boronic acids were compatible with such arylation though electron-withdrawing groups decreased the yields (3ae, 3af). The position of the substituents showed significant influence on yields and meta or para substituents provided the best yields (3ac, 3ad). When ortho-substituted boronic acids (for example, o-tolylboronic acid) were used, the coupling reactions did not work well due to steric hindrance (3ab).
| a All the reactions were carried out with 1 (0.2 mmol) and arylboronic and 2 (0.4 mmol). |
|---|
|
Various N-alkyl acetanilide derivatives were investigated (Table 1). N-Acetyl-2,3-dihydroindole and its derivatives were also good substrates for this transformation (3ca, 3da). Notably, the relatively stable chloride group can survive for further functionalization (3da).
As already mentioned, this coupling is initiated by electrophilic attack of a Pd(II) center to the aromatic ring with the assistance of the acetamino group, followed by transmetalation and reductive elimination to produce the desired product. The other possibility is transmetalation of the boronic acid with Pd(II) salts to form initially an arylated Pd(II) species,16 which may attack the aromatic ring in an electrophilic manner to form a diaryl palladium species, which then would undergo reductive elimination to give the final product.
Ortho-arylation of O-methyl oximes and their subsequential transformations
Due to the synthetic variety of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X (X = O, N) groups,17 investigation of the ortho arylation of aryl aldehydes and their derivatives using arylboronic acids is of great importance. Since the O-methyl oximyl group has been applied for ortho acetoxylation and amination of sp2 C–H as well as sp3 C–H bonds, it was further considered as an efficient directing group for direct arylation. The challenge for this arylation is to avoid the direct addition of arylboronic acids to the CX group in the presence of Pd(II) species, which has been well studied by Lu and others.18
X (X = O, N) groups,17 investigation of the ortho arylation of aryl aldehydes and their derivatives using arylboronic acids is of great importance. Since the O-methyl oximyl group has been applied for ortho acetoxylation and amination of sp2 C–H as well as sp3 C–H bonds, it was further considered as an efficient directing group for direct arylation. The challenge for this arylation is to avoid the direct addition of arylboronic acids to the CX group in the presence of Pd(II) species, which has been well studied by Lu and others.18
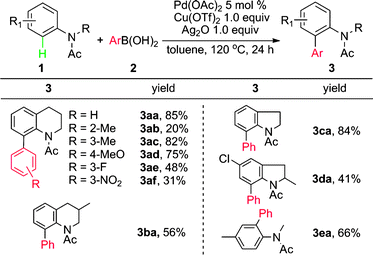
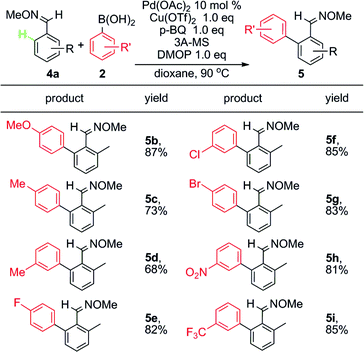
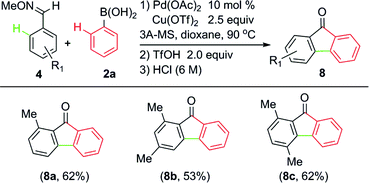
In fact, when the ortho-arylation of O-methyl (E)-2-methylbenzaldoxime 4a with phenylboronic acid 2a was tested in the presence of Pd(OAc)2 and Cu(OTf)2, annulated product 9H-fluoren-9-one and other byproducts were also observed in a low efficiency process. However, the addition to CX (X = O or N) was suppressed under these conditions and the in situ generated acid was proposed to make the system complicated. Thus, further efforts was made to control the reaction at different stages by addition of bases or acids.19 After many trials we found that 2,6-dimethoxypyridine (DMOP) is the most efficient base to improve the yield of direct arylation (eqn (1)), which may arise from its appropriate steric hindrance and basicity.
 | (1) |
Various boronic acids are suitable for this ortho arylation. (Table 2). The electronic properties of aromatic substituents did not significantly affect this transformation. Arylboronic acids bearing both electron-donating groups, such as MeO and Me (5b, 5c, 5d), and electron-withdrawing groups, such as NO2 and CF3, worked well as aryl sources (5h, 5i). Notably, C–Br and C–Cl can be presented as a functional group on boronic acid for further functionalization (5f, 5g).
Unfortunately, such conditions of direct ortho arylation of aldoximes can not be directly applied into the similar arylation of aryl ketoximes, which resulted in an incomplete conversion, arising from its relatively high steric hindrance (eqn (2)). Thus, the additional base was removed to promote the efficiency. As designed, the ortho arylated product 7a was obtained in 81% yield in the absence of DMOP (eqn (2)).
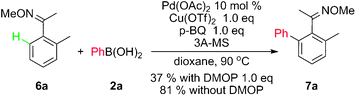 | (2) |
9H-fluoren-9-one and its derivatives are common scaffolds existing in many natural products and synthetic drugs20 as well as intermediates in materials chemistry,21 and much attention has been directed to this unique scaffold. After further screening, 8a was isolated in 62% yield by sequentially adding TfOH and HCl in one pot (Table 3). Through this process, polysubstituted 9H-fluoren-9-ones 8a–8c were obtained in one pot in moderate to good yields. Although the yields are not high, such a one-pot transformation provided a straightforward method to construct such scaffolds. It is important to note that, almost at the same time, Cheng and Daugulis reported similar strategies to achieve this goal by using aryl halides.22
Ortho-arylation directed by pyridinyl group
Yu and co-workers first reported Pd-catalyzed direct methylation of sp2 and sp3 C–H bonds with methylboroxine by using pyridine (Py) as a directing group.14a Screening of coupling partners and reaction conditions indicated that the combination of Pd(OAc)2, methylboroxine, Cu(OAc)2 and benzoquinone provided an excellent efficiency and the desired product 10a was obtained in 72% yield (Scheme 6). Functional groups attached to aryl rings, such as MeO, vinyl, CHO and CF3, were tolerated (10b–10e), though electron-withdrawing groups obviously decreased the yields (10d, 10e).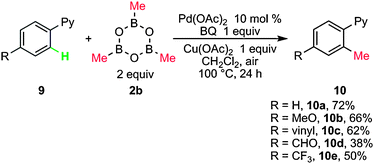 | ||
| Scheme 6 Methylation of sp2 C–H bonds with methylboroxine.a | ||
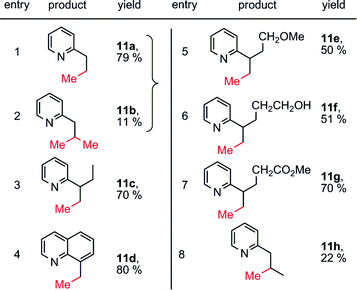
Importantly, this strategy can be applied in the direct functionalization of sp3 C–H bonds. With methylboroxine as the methyl source, methylation was also achieved with the direction of the pyridinyl group, by running the reaction in acetic acid/O2 (1 atm) rather than CH2Cl2/air (Table 4). Ether, alcohol and ester groups (entries 5–7) are compatible with this reaction. The methylation of the secondary sp3 C–H bond was also possible (entry 8), albeit in lower yield.
| a 10 mol% Pd(OAc)2, 2 equiv. of BQ, 2 equiv. of Cu(OAc)2, 2 equiv. of 2b and 100 °C, 24 h, HOAc, O2. |
|---|
|
Ortho alkylation and arylation of carboxylic acids
Very recently, Yu and co-workers further reported the first example of Pd-catalyzed β-C–H activation of simple aromatic acids. A promising protocol was established for direct alkylation and arylation of sp2 of carboxylic acids by using boronic acids and their derivatives (Scheme 7).14b Use of a suitable base highly improved the efficiency, according to the mechanism of the Suzuki–Miyaura coupling reaction. In their cases, K2HPO4 increased the yields of 13b and 13c to 75 and 63%, respectively (Scheme 7). Since the presence of K2HPO4 led to the in situ formation of potassium carboxylate, benzoic acids could be used instead of sodium carboxylates as substrates to facilitate this transformation.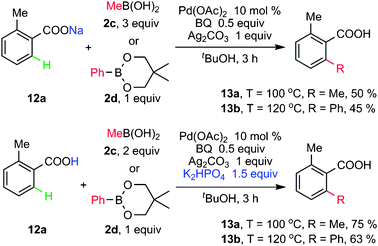 | ||
| Scheme 7 Yu's protocol of C–H methylation and phenylation using boronic acids or esters. | ||
The couplings of β-C–H bonds in aliphatic acids with 2d were also attempted. The potassium carboxylate of 14 generated in situ using K2HPO4 afforded 13a in 38% isolated yield (Scheme 8). This is the first example of arylation of an sp3 C–H bond in the Suzuki–Miyaura cross-coupling manner.
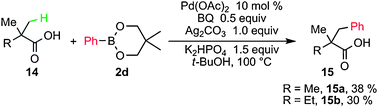 | ||
| Scheme 8 β-Arylation of aliphatic acids using PhB(OR)2. | ||
Moreover, Yu and co-workers disclosed a new catalytic system for C–H activation/C–C coupling. In this system, the scope of substrates was greatly extended by using aryltrifluoroborate salts as the coupling partners.23 The use of 1 atm of O2 or air led to the desired products in 60–70% yields after 72 h. However, the use of 20 atm of air or O2 could shorten the reaction time and improve the yields (Scheme 9). This system could be expanded to the coupling of phenyl acetic acid with PhBF3K, giving the diarylated product 18a in 69% yield.
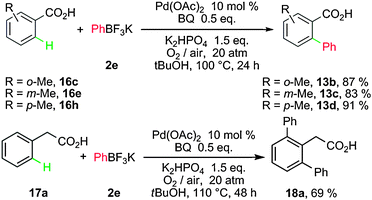 | ||
| Scheme 9 Coupling of arylacetic acids with potassium aryltrifluoroborates. | ||
β-Arylation and alkylation of O-methyl hydroxamic acids
Very recently, Yu and co-workers further developed the first protocol for the coupling of sp3 C–H bonds of O-methyl hydroxamic acids with both sp2 and sp3 boronic acids.24 Since the CONHOMe group can be readily converted to esters and amides, or reduced to alkane fragments, this reaction is likely to find broad synthetic utility. It is noteworthy that the use of 2,2,5,5-tetramethyltetrahydrofuran as the solvent allows the coupling of sp3 C–H bonds with alkylboronic acids. This solvent might serve as a sterically bulky ligand to prevent homo-coupling and β-hydride elimination from the alkyl fragments of the sp3 boronic acids. Some typical products are shown in Table 5.
a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Conditions A: 19a (0.5 mmol), arylboronic acid (0.8 mmol), Pd(OAc)2 (0.05 mmol, 10 mol%), Ag2O (1 mmol), BQ (0.25 mmol), K2CO3 (1 mmol), t-BuOH (3 mL), 70 °C, 18 h. bConditions B: 19a (0.5 mmol), alkylboronic acid (0.8 mmol), Pd(OAc)2 (0.05 mmol, 10 mol%), Ag2O (1 mmol), K2CO3 (1 mmol), BQ (0.25 mmol), 2,2,5,5-tetramethylTHF (3 mL, inhibitor free, anhydrous), 70 °C, 18 h. Conditions A: 19a (0.5 mmol), arylboronic acid (0.8 mmol), Pd(OAc)2 (0.05 mmol, 10 mol%), Ag2O (1 mmol), BQ (0.25 mmol), K2CO3 (1 mmol), t-BuOH (3 mL), 70 °C, 18 h. bConditions B: 19a (0.5 mmol), alkylboronic acid (0.8 mmol), Pd(OAc)2 (0.05 mmol, 10 mol%), Ag2O (1 mmol), K2CO3 (1 mmol), BQ (0.25 mmol), 2,2,5,5-tetramethylTHF (3 mL, inhibitor free, anhydrous), 70 °C, 18 h. |
|---|
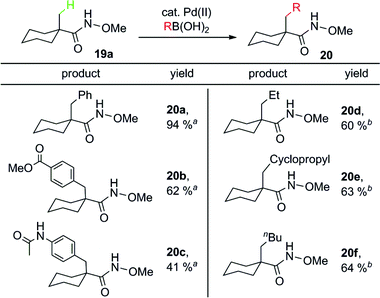
|
Direct arylation of electron-rich arenes and heterocycles
Although the cross-coupling between C–H bonds and organoboronic acids and their derivatives has advanced Suzuki–Miyaura coupling, the relatively low yields, the requirement of a directing group, and the complicated reaction conditions made them less attractive for real applications. Our efforts were made to develop new methods of Pd(II)-catalyzed cross-couplings between general C–H bonds and arylboronic acids.25We tested the direct arylation of mesitylene (21a) with phenylboronic acid (2a) under various conditions. Finally we found that such transformation could progress under relatively strong acidic conditions in the presence of Cu(OAc)2 as a co-catalyst and O2 as a terminal oxidant. However, the substrate scope of the arenes is quite narrow. In general, electron-rich arenes bearing methyl substitutents showed good reactivities and the corresponding phenylated products 22 were obtained in good yields (Table 6). Electron-rich polyarenes and methoxy substituent were also beneficial for this transformation.
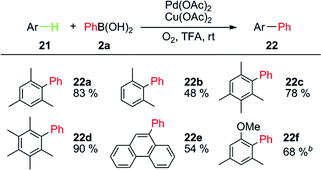
This system is perfectly suitable for direct arylation of electron-rich heteroaromatic systems, even under simpler conditions (Table 7). 2,3-Benzothiophene (21g) and 2,3-benzofuran (21h) were suitable substrates and the corresponding phenylated products at the 2-position were obtained in excellent regioselectivities. Moreover, N-heterocycles, such as pyrroles 21i, 21j and indoles 21k–21r, were only mono-phenylated. No protection was required for the arylation of pyrrole 21j and indoles 21k–21m. The relatively low yields arose from the instability of substrates and products under acidic conditions. More functionalized indole derivatives (21n–21q) could be arylated at the 2-position with high efficiency, excellent selectivity, and good yields. Substrates 21m and 21o showed that C–Cl bonds were tolerated. N-Acetyl protection (21r) significantly diminished the yield, due to the decrease of electron density.
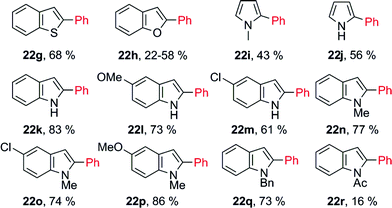
Later on, Zhang and co-workers also developed mild conditions to achieve Pd(OAc)2-catalyzed regioselective cross-coupling between indoles and potassium aryltrifluoroborates in the presence of a catalytic amount of Cu(OAc)2 in acetic acid at room temperature (Scheme 10).26
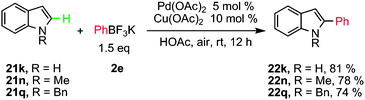 | ||
| Scheme 10 Pd-catalyzed direct arylation of indoles with potassium phenyltrifluoroborate. | ||
In summary, employing aryl/alkyl boronic acids and their derivatives as reagents, novel methods to construct C–C bonds via Pd(II)-catalyzed Suzuki–Miyaura type reactions have been developed starting from aryl C–H bonds, as well as aliphatic C–H bonds, with/without directing groups. Acetanilides, O-methyl oximes, pyridines, carboxylic acids, O-methyl hydroxamic acids and (hetero)arenes were successfully subjected to such transformations. Our studies also resulted in the direct arylation of common electron-rich arenes. The milder reaction conditions enabled these transformations to tolerate different functional groups very well. Further extension of substrate scope and application of such methods are still pictured in near future.
Organosilicon reagents as the coupling partners
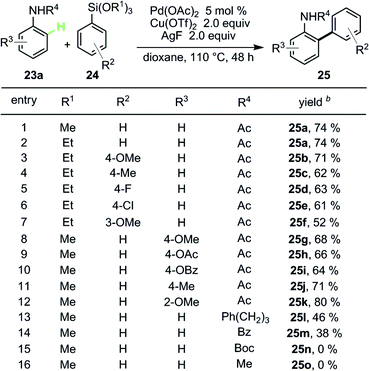
Other than organoboron reagents, organosilicon reagents are another class of important organometallic reagents and are widely applied in organic synthesis. The Pd-catalyzed Hiyama coupling reaction is one of the most important examples using organosilicon reagents as coupling partners. Compared with other organometallic reagents, organosilicon reagents possess the lowest reactivity and a better tolerance of functional groups. The C–Si bonds are of only slight polarity due to the slightly higher electronegativity of carbon over silicon. In previous studies, fluorides were frequently used as additives to activate C–Si bonds by formation of the five- or six-coordinated silicates and facilitate the transmetalation of organosilicon reagents. Those facts made the cross-coupling between general arenes and organosilicon reagents more challenging. Our studies resulted in a unique case up to the present.27In our research, Hiyama type coupling between acetanilide 38a and trimethoxy(phenyl)silane 39a could be promoted by AgF, which might play dual roles as both co-oxidant and fluoride source. In fact, the best oxidant for this transformation is Cu(OTf)2 and the desired product was afforded in excellent yield (Table 8, entry 1).
Different trialkyloxy(aryl)silanes exhibited excellent reactivities in this transformation, regardless of electron-withdrawing groups or electron-donating groups on the phenyl rings of the phenylsilanes (Table 8, entries 2–7). C–Cl on phenylsilane survived, providing a great chance for further functionalization (entry 6). Under this condition, different acetanilides were also studied, with broad functional group compatibility (40g–40k). The effect of different groups on the N atom were also studied; Ph(CH2)3 and Bz groups led to relatively lower yields (40l and 40m) while Boc and methyl groups were not effective at all in this process (40n and 40o).
Organotin reagents as the coupling partners
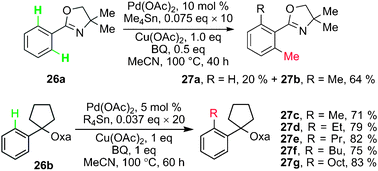
Stille coupling using organotin reagents have been widely investigated, though usually only one of the four groups on tin was transferred, leaving the other three as spectators.28 Due to their relatively high tolerance of functional groups, moderate reactivities and mild reaction conditions, organotin compounds were frequently used in total synthesis. However, the toxicity of organotin reagents is a considerable drawback which limits their applications.Stille type cross-coupling starting from C–H activation was earlier studied than other cross-couplings. In 2005, Yu and co-workers discovered the first protocol for Pd(II)-catalyzed alkylations of aryl C–H bonds with a variety of primary-alkyl tin regents using a combination of directed C–H activation and batchwise addition of the organotin reagents. They found that catalytic alkylation of 26a can be achieved by using 1 equiv. of Cu(OAc)2 and 0.5 equiv. of benzoquinone in MeCN under air to afford mainly the dialkylated product 27b directed by the oxazolinyl (Oxa) group (Scheme 11). A variety of primary alkyl tin reagents were tested under these new conditions, and the alkylated products were obtained in good yields (Scheme 11).29 The addition of tin reagents in batches was necessary to avoid homo-coupling of tin reagents, but resulted in longer reaction time.
 | ||
| Scheme 11 Catalytic methylation of aryl C–H bonds using organotin reagents. | ||
Very recently, Inoue and co-workers reported a direct Pd-catalyzed C–H bond arylation of simple arenes with aryltin reagents in the presence of CuCl2, which was proved to be an activator for the Pd catalyst as well as an oxidant (Scheme 12). The use of aryltin trichloride not only diminished the toxicity of reagents, but also improved the atom-economy of carbon atoms. Moreover, the absence of the directing group highly expanded the substrate scope.30
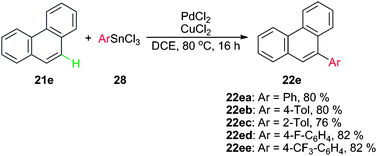 | ||
| Scheme 12 Arylation of phenanthrene using aryltin trichlorides. | ||
Conclusions
In this feature article, we have summarized recent advances on Pd-catalyzed direct C–H functionalization via oxidative-couplings with organometallic reagents. Concerning the substrates, acetanilides, O-alkyl oximes, aryl and alkyl carboxylic acids, 2-aryl or alkyl pyridines, 2-aryl or alkyl oxazolines, O-methyl hydroxamic acids, and even common electron-rich (hetero)arenes without directing groups have been successfully introduced into these transformations. Some of the cross-coupling products could be further transformed into other useful scaffolds and compounds by condition-controlled cascade reactions. Concerning the oxidants, Cu(OAc)2 was the most efficient in the system of palladium catalysis and Ag(I) salts, p-BQ were also used frequently. The use of oxidants containing heavy metals depreciated these methods to some extent. In some cases, even O2 or air could react as terminal oxidants, which provided a potential opportunity of developing more efficient and greener transformations.In another aspect, the organometallic reagents involved were mainly organoboron, organosilicon and organotin reagents due to the consonance of their reactivities and stabilities. Moreover, the reactions using other organometallic reagents with higher reactivities, such as Grignard reagents and organozinc reagents, remain as a substantial challenge in this field. Importantly, recent developments via Fe catalysis by Nakamura and co-workers showed great potential in this area.31
Summarizing, Pd-catalyzed C–H activation and cross-coupling with organometallic reagents presents a new strategy in the field of cross-coupling reactions. Undoubtedly, these reactions have great prospects of applications in organic syntheses and industrial processes. In spite of their great significance and obvious advantages, there are still many problems to be solved, such as the relatively narrow substrate scope and the harsh and complicated reaction conditions. The clear understanding of the catalytic pathway is still elusive. We are striving in further developments to make these transformations more promising and practical.
Acknowledgements
Support of this work by grants from NSFC (No. 20672006, 20821062 and 20925207) and the “973” program from MOST of China (2009CB825300) is gratefully acknowledged.References
- G. Dyker, Handbook of C–H Transformations. Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) W. Jones and F. Fehe, Acc. Chem. Res., 1989, 22, 91 CrossRef CAS; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS.
- (a) A. F. Browning and N. Greeves, Palladium-Catalyzed Carbon–Carbon Bond Formation, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, New York, 1997, pp. 35–64 Search PubMed; (b) Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, New York, 1998 Search PubMed; (c) J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS; (d) A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2004 Search PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) P. Lloyd-Williams and E. Giralt, Chem. Soc. Rev., 2001, 30, 145 RSC.
- J. K. Stille and D. Milstein, J. Am. Chem. Soc., 1978, 100, 3636 CrossRef.
- (a) R. J. P. Corriu and J. P. Masse, J. Chem. Soc., Chem. Commun., 1972, 144 Search PubMed; (b) K. Tamao, Y. Kiso, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 9268 CrossRef CAS.
- (a) K. Takahashi, T. Minami, Y. Ohara and T. Hiyama, Tetrahedron Lett., 1993, 34, 8263 CrossRef CAS; (b) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 835 CrossRef CAS.
- (a) S. Baba and E. Negishi, J. Am. Chem. Soc., 1976, 98, 6729 CrossRef CAS; (b) E. Negishi and S. Baba, J. Chem. Soc., Chem. Commun., 1976, 596 Search PubMed; (c) E. Negishi, Acc. Chem. Res., 1982, 15, 340 CrossRef CAS.
- (a) G. Dyker, Angew. Chem., Int. Ed. Engl., 1992, 31, 1023 CrossRef; (b) T. Harayama, T. Akiyama and Y. Nakano, Chem. Pharm. Bull., 1997, 45, 1723 CAS; (c) T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed. Engl., 1997, 36, 1740 CrossRef CAS; (d) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 1473 CrossRef CAS; (e) Y. Kawamura, T. Satoh, M. Miura and M. Nomura, Chem. Lett., 1999, 961 CrossRef CAS; (f) J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360 CrossRef CAS; (g) Y. Terao, H. Wakui, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2001, 123, 10407 CrossRef CAS; (h) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (i) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112 CAS; (j) K.-I. Fujita, M. Nonogawa and R. Yamaguchi, Chem. Commun., 2004, 1926 RSC; (k) Q. Huang, A. Fazio, G. Dai, M. A. Campo and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 7460 CrossRef; (l) H. Wakui, S. Kawasaki, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2004, 126, 8658 CrossRef CAS; (m) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS; (n) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS; (o) L. Ackermann, A. Althammer and R. Born, Angew. Chem., Int. Ed., 2006, 45, 2619 CrossRef CAS; (p) L.-C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581 CrossRef CAS; (q) R. Ferraccioli, D. Carenzi, E. Motti and M. Catellani, J. Am. Chem. Soc., 2006, 128, 722 CrossRef CAS; (r) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS.
- (a) T. Itahara, J. Org. Chem., 1985, 50, 5272 CrossRef CAS; (b) K. L. Hull, E. L. Lanni and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 14047 CrossRef CAS; (c) R. Li, L. Jiang and W. Lu, Organometallics, 2006, 25, 5973 CrossRef CAS; (d) Y. Rong, R. Li and W. Lu, Organometallics, 2007, 26, 4376 CrossRef CAS; (e) J.-B. Xia and S.-L. You, Organometallics, 2007, 26, 4869 CrossRef CAS; (f) D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172 CrossRef CAS; (g) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904 CrossRef CAS; (h) D. R. Stuart, E. Villemure and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 12072 CrossRef CAS; (i) T. A. Dwight, N. R. Rue, D. Charyk, R. Josselyn and B. DeBoef, Org. Lett., 2007, 9, 3137 CrossRef CAS; (j) B. Li, S. Tian, Z. Fang and Z. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115 CrossRef CAS; (k) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 9651 CrossRef CAS.
- D. G. Hall, Boronic Acids, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005 Search PubMed.
- (a) S. J. Tremont and H. U. Rahman, J. Am. Chem. Soc., 1984, 106, 5759 CrossRef CAS; (b) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS; (c) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS; (d) M. A. Campo, Q. Huang, T. Yao, Q. Tian and R. C. Larock, J. Am. Chem. Soc., 2003, 125, 11506 CrossRef CAS; (e) E. J. Hennessy and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 12084 CrossRef CAS; (f) V. G. Zaitsev and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 4156 CrossRef CAS; (g) X. Wang, B. S. Lane and D. Sames, J. Am. Chem. Soc., 2005, 127, 4996 CrossRef CAS; (h) C. Bressy, D. Alberico and M. Lautens, J. Am. Chem. Soc., 2005, 127, 13148 CrossRef CAS.
- (a) N. Chatani, T. Asaumi, T. Ikeda, S. Yorimitsu, Y. Ishii, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2000, 122, 12882 CrossRef CAS; (b) N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935 CrossRef CAS; (c) S. J. Pastine, D. V. Gribkov and D. Sames, J. Am. Chem. Soc., 2006, 128, 14220 CrossRef CAS.
- (a) X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634 CrossRef CAS; (b) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS.
- X. Wan, Z. Ma, B. Li, K. Zhang, S. Cao, S. Zhang and Z. Shi, J. Am. Chem. Soc., 2006, 128, 7416 CrossRef CAS.
- (a) J. P. Parrish, Y. C. Jung, S. I. Shin and K. W. Jung, J. Org. Chem., 2002, 67, 7127 CrossRef CAS; (b) C. H. Yoon, K. S. Yoo, S. W. Yi, R. K. Mishra and K. W. Jung, Org. Lett., 2004, 6, 4037 CrossRef CAS; (c) J. H. Delcamp and C. M. White, J. Am. Chem. Soc., 2006, 128, 15076 CrossRef CAS; (d) Z. Shi, B. Li, X. Wan, J. Cheng, Z. Fang, B. Cao, C. Qin and Y. Wang, Angew. Chem., Int. Ed., 2007, 46, 5554 CrossRef CAS.
- (a) M. A. Sprung, Chem. Rev., 1940, 26, 297 CrossRef CAS; (b) C. K. Bradsher, Chem. Rev., 1987, 87, 1277 CrossRef CAS; (c) L. G. Wade, Organic Chemistry, Prentice-Hall, Englewood, Cliffs, New Jersey, 1995, pp. 48–78 Search PubMed; (d) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS; (e) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS.
- (a) C. Zhou and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 2302 CrossRef CAS; (b) T. Yamamoto, T. Ohta and Y. Ito, Org. Lett., 2005, 7, 4153 CrossRef CAS; (c) G. Liu and X. Lu, J. Am. Chem. Soc., 2006, 128, 16504 CrossRef CAS; (d) C. Zhou and R. C. Larock, J. Org. Chem., 2006, 71, 3551 CrossRef.
- C. Sun, N. Liu, B. Li, D. Yu, Y. Wang and Z. Shi, Org. Lett. DOI:10.1021/ol902552v.
- (a) M. L. Greenlee, J. B. Laub, G. P. Rouen, F. DiNinno, M. L. Hammond, J. L. Huber, J. G. Sundelof and G. G. Hammond, Bioorg. Med. Chem. Lett., 1999, 9, 3225 CrossRef CAS; (b) P. J. Perry, M. A. Read, R. T. Davies, S. M. Gowan, A. P. Reszka, A. A. Wood, L. R. Kelland and S. Neidle, J. Med. Chem., 1999, 42, 2679 CrossRef CAS.
- M. Gross, D. C. Muller, H.-G. Nothofer, U. Scherf, D. Neher, C. Brauchle and K. Meerholz, Nature, 2000, 405, 661 CrossRef CAS.
- (a) V. S. Thirunavukkarasu, K. Parthasarathy and C. Cheng, Angew. Chem., Int. Ed., 2008, 47, 9462 CrossRef CAS; (b) D. Shabashov, J. R. M. Maldonado and O. Daugulis, J. Org. Chem., 2008, 73, 7818 CrossRef CAS.
- D.-H. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676 CrossRef CAS.
- D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS.
- S. Yang, C. Sun, Z. Fang, B. Li, Y. Li and Z. Shi, Angew. Chem., Int. Ed., 2008, 47, 1473 CrossRef CAS.
- J. Zhao, Y. Zhang and K. Cheng, J. Org. Chem., 2008, 73, 7428 CrossRef CAS.
- S. Yang, B. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 6066 CrossRef CAS.
- A. G. Davies, Organotin Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2003 Search PubMed.
- X. Chen, J.-J. Li, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 78 CrossRef CAS.
- H. Kawai, Y. Kobayashi, S. Oi and Y. Inoue, Chem. Commun., 2008, 1464 RSC.
- J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5858 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |