DOI:
10.1039/D6MA00202A
(Paper)
Mater. Adv., 2026, Advance Article
Hydrophobic block-structured PDMS/Pt mesoporous silica catalysts for sustainable water–hydrogen isotope exchange
Received
12th February 2026
, Accepted 15th May 2026
First published on 1st June 2026
Abstract
Efficient separation of hydrogen isotopes is essential for sustainable hydrogen management and environmental protection in nuclear and other industrial systems. Conventional hydrogen isotope separation technologies, including cryogenic distillation, the Girdler sulfide process, and ammonia–hydrogen exchange, consume massive energy and involve corrosive or toxic reagents, leading to significant environmental and operational challenges. In contrast, the liquid-phase catalytic exchange (LPCE) process, based on water–hydrogen gas isotope exchange, provides an efficient and energy-conserving alternative consistent with sustainable catalytic processes. In this study, a platinum-decorated mesoporous silica-polydimethylsiloxane composite (Pt@SiO2-PDMS) was developed and applied as a catalyst for the LPCE process. Physico-chemical characterizations verified the decoration of Pt into mesoporous silica without destruction of the mesoporous structure. The Pt@SiO2 particles were homogeneously distributed in the PDMS matrix, and the resulting composite maintained an intrinsic hydrophobic surface with a water contact angle above 108°. The catalyst composite exhibited effective hydrogen isotope exchange performance up to a column efficiency of 67.7% at 80 °C when using Pt@SiO2-PDMS randomly packed with Dixon rings in a volume ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the LPCE process. Furthermore, stable operation was maintained continuously for 14 days at 80 °C, demonstrating the durability of the catalyst composite and its application under actual processing conditions. The selective hydrogen isotope exchange between liquid water and hydrogen gas using the Pt@SiO2-PDMS catalyst is attributed to the effective porous structure of SiO2, the Pt loading in Pt@SiO2 and the hydrophobic nature of the PDMS matrix.
:1 in the LPCE process. Furthermore, stable operation was maintained continuously for 14 days at 80 °C, demonstrating the durability of the catalyst composite and its application under actual processing conditions. The selective hydrogen isotope exchange between liquid water and hydrogen gas using the Pt@SiO2-PDMS catalyst is attributed to the effective porous structure of SiO2, the Pt loading in Pt@SiO2 and the hydrophobic nature of the PDMS matrix.
Introduction
Hydrogen isotope separation has gained increasing attention due to its growing demand, ranging from nuclear technology to advanced manufacturing of electronic devices.1,2 The expanded use of deuterium (D) in industrial processes and the continuous generation of tritium (T) in nuclear systems have further highlighted the need for reliable and efficient isotope separation.1 In nuclear power systems, heavy-water reactors utilize deuterium oxide (D2O) as a moderator and coolant, providing outstanding neutron economy and efficient moderation.3 Beyond nuclear applications, D2O, as a valuable resource, is utilized in advanced industrial and electronic fields, such as manufacturing of organic light-emitting diodes.4 On the other hand, T, a radioactive hydrogen isotope, is produced inside nuclear reactors through neutron-capture and nuclear transmutation reactions, and its quantity is significant in heavy-water reactors and fusion research facilities.5 Since T exhibits physicochemical properties nearly identical to those of protium (H) and D, separation of hydrogen isotopes is extremely hard to achieve via conventional chemical separation processes.6 Once T is released into the environment, it easily spreads into the atmosphere, hydrosphere and biosphere, thereby causing harmful effects on living organisms. Thus, proper long-term management plans, including separation technology for hydrogen isotopes, are required.7 Furthermore, the recent release of T-contaminated water from the Fukushima Daiichi Nuclear Power Plant has intensified the need for developing efficient and cost-effective hydrogen isotope separation technologies.8
Various hydrogen isotope separation techniques have been developed, including water distillation,9 cryogenic distillation10 and chemical isotope exchange.11 Water distillation, which relies on the vapor pressure difference between water isotopologues, has a low separation factor (αH/T = 1.04–1.07), leading to high energy consumption (∼80 GJ kg−1 D2O).12,13 Cryogenic distillation, which separates hydrogen gas isotopologues at liquefaction temperatures, is limited by a low separation factor (αD/H ≈ 1.5 at −249 °C),14 high operational costs (∼22 GJ kg−1 D2O),15 and difficulties in system maintenance.16 On the other hand, chemical isotope exchange, driven by equilibrium reactions between hydrogen gas and hydrogen-containing molecules (such as H2S, NH3 or H2O), is considered commercially valuable due to its high separation factors and the feasibility of operation under technically viable temperature conditions.17 For example, chemical isotope exchange methods, such as the Girdler sulfide process and ammonia–hydrogen exchange, were primarily developed and commercialized for D enrichment in D2O production.18 Although these techniques possess high isotope separation efficiency, they involve the use of toxic or corrosive reagents and require a large amount of energy (∼30 GJ kg−1 D2O for the Girdler sulfide process and ∼11 GJ kg−1 D2O for ammonia–hydrogen exchange),15 and exhibit a relatively low separation factor of approximately 2,19 which limits their environmental and economic sustainability.20 Meanwhile, the chemical isotope exchange between hydrogen gas and liquid water via the liquid-phase catalytic exchange (LPCE) process is considered the most promising approach for environmentally benign isotope separation and detritiation.14 This process allows T to be either concentrated in or extracted from water, depending on the equilibrium reaction conditions. Also, LPCE offers advantages such as low energy consumption, as it can be operated under ambient pressure and relatively mild temperatures ranging from 60 to 80 °C, without requiring extreme energy,21 and offers a high separation factor (αH/D ≈ 5 at 70 °C) and environmental sustainability.22,23 In South Korea, the Wolsong Nuclear Power Plant operates a T-removal facility, where the LPCE process is industrially applied as the primary step to extract T from tritiated D2O, followed by cryogenic distillation to further enrich tritium as T2 (Fig. S1 of SI).11
The performance of the LPCE process is highly dependent on the catalyst that facilitates the isotope exchange reaction between hydrogen isotopes. The activity and stability of the catalyst are critical factors that determine the reaction efficiency, and the size of the column is directly correlated to the cost-effectiveness,24 because the functional and structural properties of the catalyst surface critically influence the catalytic performance.25–27 Currently, platinum (Pt)-based catalysts are most widely used in LPCE due to their high reaction rate; however, their catalytic activity may gradually decline over long-term operation due to surface poisoning and structural deformation.28 Even worse, the active sites can be blocked due to the penetration of liquid water into the catalyst. Since the LPCE process is a multi-phase reaction involving liquid water, water vapor and hydrogen gas, excessive exposure and physical as well as chemical adsorption of liquid water and water vapor on the catalyst surface block the active sites, thereby reducing reaction efficiency.29 Therefore, catalysts for LPCE should possess hydrophobicity and a large active surface area to enhance catalytic activity, while facilitating the diffusion of water vapor. To address this issue, catalyst designs that rely on the combination of porous and hydrophobic frameworks have been studied. Metal–organic frameworks (MOFs),30 zeolites,31 carbon-based materials32 and mesoporous silica33 serve as catalyst supports, and they provide a large number of active sites due to their high specific surface area,21 where controlled nanostructure and dispersion can significantly influence catalytic behavior.34 However, MOFs reported in previous studies exhibited poor stability to water and are prone to structural changes, which may restrict their practical application.35 Similarly, zeolites readily take up water due to their hydrophilic nature, then the adsorbed water promotes hydrolysis of the Si–O(H)–Al bonds and induces structural changes under aqueous or humid conditions.36 Carbon-based materials also strongly interact with water and the adsorbed moisture, inducing oxidation or changes in the pore structure under humid environments.37 To improve their water stability, diverse surface modifications of support materials have been studied to tailor the hydrophobicity. In MOFs, post-synthetic treatments, such as alkyl-group incorporation and silylation, have been employed, but these approaches often lead to reduced specific surface area and pore size.38 Zeolites have also been modified by surface grafting of organosilanes onto silanol groups, which is accompanied by a reduction in pore volume and specific surface area.39 Activated carbon has similarly been silylated to provide hydrophobicity, although such modifications can induce moisture-related pore structure changes and diminish adsorption performance.40 To overcome the side effects of surface modifications, applying a hydrophobic polymer framework to form a composite catalyst has been proposed recently, as polymer matrices can provide structural stability and chemical functionality in hybrid systems.41 Pt/styrene–divinylbenzene copolymer42 and Pt/carbon black/polytetrafluoroethylene43 significantly improve structural stability. However, these composite catalysts still exhibit non-uniform distribution and surface aggregation of Pt.44 Collectively, these limitations highlight a fundamental challenge in existing hydrophobic LPCE catalysts: achieving a balanced integration of water stability, efficient mass transport, and uniform dispersion of active Pt sites without compromising structural integrity or porosity. Therefore, novel catalyst designs are required to enhance hydrophobicity while optimizing the performance of the hydrogen isotope exchange reaction. Polydimethylsiloxane (PDMS) was selected as the hydrophobic framework material due to its unique combination of high hydrophobicity, exceptional gas permeability, and chemical stability under LPCE conditions.45 Unlike conventional hydrophobic polymers, such as fluoropolymers (e.g., Teflon), which can limit mass transfer due to their dense and rigid structure, PDMS exhibits high hydrogen permeability, facilitating efficient transport of H2 to the active Pt sites.46 In addition, its elastomeric nature allows a conformal coating on the catalyst surface, preserving active site accessibility while providing a stable hydrophobic environment.47 These properties make PDMS particularly well-suited for maintaining an effective gas–liquid–solid interface and enhancing the catalytic performance in LPCE systems.
In this study, a composite based on Pt-decorated mesoporous silica and PDMS is fabricated and applied to the LPCE process as a hydrophobic catalyst for the durable and efficient separation of hydrogen isotopes. Mesoporous silica has a high specific surface area and abundant surface hydroxyl groups that provide available space for uniform modification and dispersion of Pt.48 In addition, the relatively wide mesopores avoid severe diffusion limitation, thereby offering active transport that is aligned with the operating conditions of this study.49 Hydrophobicity and catalyst stability are achieved by employing PDMS as a framework to prevent liquid water infiltration, while ensuring sufficient exposure of Pt active sites, thereby ensuring stable catalytic activity in the LPCE process. Ultimately, this offers improved hydrogen isotope separation techniques by enabling efficient and stable catalytic performance under diverse liquid-phase exchange conditions.
Experimental
Materials
The following chemicals and gases were used as received without further purification. Tetraethyl orthosilicate (TEOS, 98%), cetyltrimethylammonium bromide (CTAB, >98%), ethanol (EtOH, 99.99%) and ammonium hydroxide solution (NH4OH, 28.0–30.0 wt%) were purchased from Sigma-Aldrich. Tetraammineplatinum(II) nitrate (Pt(NH3)4(NO3)2) was purchased from Alfa Aesar. PDMS (Sylgard® 184), including a silicone elastomer base and its curing agent, was purchased from Dow Corning. Deionized (DI) water was supplied by a Direct-Q® 3 water purification system with a resistivity of 18.2 MΩ cm at 25 °C. The stainless steel Dixon ring (KT-DR) of 3 mm diameter and height for hydrophilic column packing was purchased from Beihai Kaite Chemical Packing Co., Ltd. H2 (99.999%) and nitrogen gas (N2, 99.999%) were purchased from Daesung Industrial Gases Co., Ltd. Air (78% of N2, 21% of O2 and around 0.9% of trace amounts of other gases) was purchased from Joongang Industrial Gases Co., Ltd and argon gas (Ar, 99.999%) was obtained from Sejong Industrial Gases Co., Ltd.
Preparation of mesoporous silica powder
Mesoporous silica was prepared according to a previously reported method.50 CTAB (2.4 g) was dissolved in an equivolume mixture of DI water (50 mL) and ethanol (50 mL). After 10 min of vigorous stirring, NH4OH (12 mL) was added (Fig. 1a). Subsequently, TEOS (3.4 g) was dispersed dropwise into the solution mixture (Fig. 1b). The mixed solution was additionally stirred for another 2 h until it formed a gel (Fig. 1c). The molar composition of the final gel is TEOS: 0.43 CTAB: 12.5 NH3: 54.3 EtOH: 416 H2O. The resulting gel was washed 5 times with DI water, and the solid/liquid was separated by centrifugation at each cycle. The resulting solid was then dried overnight in an oven at 65 °C. The CTAB template, a structure-directing agent, was removed by calcination at 550 °C with a heating rate of 1 °C min−1 for 6 h under stagnant air conditions in a furnace (Fig. 1d). The prepared mesoporous silica powder is denoted as SiO2.
 |
| | Fig. 1 Schematic of the fabrication process for Pt@SiO2-PDMS. (a) Formation of micelles with CTAB as a structure-directing agent in a solution containing DI water, EtOH and NH4OH; (b) addition of TEOS as the silica source into the solution mixture; (c) formation of a silica-template composite structure through TEOS-induced hydrolysis and condensation; (d) template removal by calcination activating uniform, well-ordered mesopores; (e) deposition of Pt onto the mesoporous silica framework through H2 reduction; (f) homogeneously mixed PDMS and Pt@SiO2 mixture in the mold for curing; (g) final catalyst composite, Pt@SiO2-PDMS. | |
Decoration of Pt on mesoporous silica powder
To impregnate Pt onto the silica framework, 0.52 g of Pt(NH3)4(NO3)2 was thoroughly mixed with 5.0 g of prepared mesoporous silica, resulting in a 5 wt% Pt loading. Subsequently, 9.6 mL of DI water, corresponding to 1.2 times the mesopore volume (measured by N2 physisorption), was slowly added dropwise to the mixture while continuously spatula mixing at room temperature to ensure homogeneous dispersion. Then, the Pt(NH3)4(NO3)2 infiltrated into the surface and pores of the mesoporous silica during drying. The dried sample was calcined under a flow of air at 400 °C with a heating rate of 2 °C min−1 for 6 h. This decomposed the Pt precursor ligands and formed Pt oxide, which was stably incorporated onto the surface and pores of the mesoporous silica. Following calcination, the sample was purged with Ar gas for 30 min to remove residual air and volatile impurities. Afterward, it was reduced with H2 under the same heating rate and temperature conditions, which converted the Pt oxide into metallic Pt and integrated it uniformly into the mesoporous silica (Fig. 1e). The final catalyst powder was denoted as Pt@SiO2.
Fabrication of the catalytic composite based on Pt@SiO2 and PDMS
The silicone elastomer base and its curing agent were mixed with a weight ratio of 5:2, corresponding to 2.9 g of elastomer base and 1.1 g of its curing agent, until a uniform mixture was obtained. Then, 1.0 g of Pt@SiO2 was added and manually mixed for 10 min to ensure even distribution. The mixture was degassed under 1.3 kPa at room temperature for 10 min to eliminate air bubbles that could cause potential discontinuities in the final catalyst. After degassing, the mixture was poured into a lattice mold consisting of multiple trapezoidal prisms with dimensions of 2.1 mm top base, 3.6 mm bottom base, and 1.2 mm height, without overflow (Fig. 1f). After curing at 60 °C for 4 h, the composite was carefully recovered from the mold (Fig. 1g). The resulting composite, denoted as Pt@SiO2-PDMS, had 1 wt% Pt loading. Additionally, pristine PDMS and mesoporous silica-incorporated PDMS were fabricated without the Pt decoration procedure; the products were denoted as PDMS and SiO2-PDMS, respectively.
Characterizations
X-ray diffraction (XRD) patterns of the powder samples were measured using a D2 PHASER (Bruker) from 0 to 8° (low angle) and 10 to 80° (high angle) of 2θ at a rate of 0.002° per step, with a counting time of 0.5 s per step using a Cu-Kα radiation source. The scanning electron microscopy (SEM) images were obtained with an SU-8020 (Hitachi) at a landing energy of 15 kV. The powder and composite samples were prepared on top of carbon tape and coated with gold to avoid surface discharge. Transmission electron microscopy (TEM) coupled with energy-dispersive X-ray spectroscopy (EDS) analyses was performed with a JEM-2100F (JEOL) operated at an accelerating voltage of 200 kV. The powder samples were prepared by dispersing in ethanol and drop-casting the suspension onto a carbon-coated Cu grid, followed by natural drying. The N2 physisorption isotherms were measured at −196 °C using an Autosorb iQ-XR Viton (Quantachrome). Before measurement, the powder samples were degassed overnight at 150 °C under vacuum to remove the pre-adsorbed gases, moisture and other impurities. The N2 physisorption isotherms were analyzed to specify the pore volume, specific surface area and pore-size distribution (PSD). The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method, and the PSD was determined by the Broekhoff–de Boer/Frenkel–Halsey–Hill (BdB-FHH) method. The Pt content in Pt@SiO2 was measured using inductively coupled plasma optical emission spectrometry (ICP-OES) with an Avio 500 (PerkinElmer). The Pt@SiO2 was digested by microwave-assisted acid digestion using aqua regia mixed with hydrofluoric acid, followed by treatment at 230 °C for 30 min in an ultraWAVE system (Milestone). The morphology of the fabricated PDMS, SiO2-PDMS, Pt@SiO2-PDMS and Dixon rings was observed using a stereomicroscope (M205 C, Leica) equipped with an MC170 HD camera. The Fourier-transform infrared/attenuated total reflectance (FT-IR/ATR) spectra of the samples were measured using a Nicolet iS5 (Thermo Fisher Scientific). The spectra were recorded in the range of 500–4000 cm−1 at a resolution of 4 cm−1. The wettability of the fabricated catalytic composites was evaluated by measuring the water contact angle (WCA) with a Cam 200 (KV Instruments). H2 chemisorption measurements were carried out using an AutoChem II 2920 (Micromeritics). Prior to the measurement, the samples were treated under a 10% H2/argon (Ar) flow at 180 °C for 30 min with a heating rate of 10 °C min−1. The samples were then cooled to 40 °C under flowing Ar, followed by an Ar purge for 10 min. Subsequently, H2 pulse chemisorption was conducted at 40 °C, and the amount of chemisorbed H2 was used to determine the metal dispersion of Pt. Thermogravimetric analysis (TGA) was performed using a SETSYS Evolution TGA (SETARAM Instrumentation). 3 mg of Pt@SiO2-PDMS samples were heated from room temperature to 800 °C at a heating rate of 10 °C min−1 under Ar with a flow rate of 50 mL min−1.
Liquid-phase catalytic exchange performance
The catalytic reactor, as shown in Fig. S2 of the SI, was designed as a glass column with an inner diameter of 15 mm and a length of 175 mm. The exterior of the column was insulated to maintain reaction temperature by circulating water coming from a water bath, which was also controlled at the reaction temperature. The top of the column was connected to a peristaltic pump, allowing the liquid water (DI water) to flow at a constant rate. The liquid water was placed in a 2 L beaker on a hot plate and the connecting tubing was pre-heated to the reaction temperature by wrapping it with heating tape. At the bottom of the column, a gas inlet was connected to supply N2 or H2 gas, with flow rates controlled by a mass flow controller. The liquid outlet of the column was a narrow glass tube, where the packing structure prevented the loss of the catalyst composites and Dixon rings, allowing only the liquid to discharge. The top of the column was designed with a gas outlet to vent the reacted gas. Before and after running, the column interior was purged with N2 gas at a flow rate of 2 standard liters per minute (SLPM) for 10 min. The LPCE performance was evaluated with a H2 (g) flow rate of 0.365 SLPM and a DI water flow rate of 0.3 mL min−1. This is a 1:1 mole ratio of hydrogen in gas and liquid. The catalyst bed consisted of randomly packed catalyst and Dixon rings with a fixed bed length of 155 mm, and the catalytic performance was tested under various volume ratios of 1:4, 1:1 and 2:1 at temperatures ranging from 30 to 80 °C. The liquid samples were collected every 30 min in the liquid outlet for 180 min. The long-term performance test was conducted with a packing volume ratio of 2:1 using a mixture of catalyst and Dixon rings at 80 °C for 14 days. The liquid samples were collected every 24 h. After each cycle, the column containing the catalyst and Dixon rings was stored in a 65 °C oven overnight for regeneration before the recyclability test. Column efficiency (η) was evaluated using eqn (1):| |
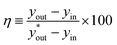 | (1) |
here, yin represents the atomic concentration of D at the gas inlet. The isotopic compositions xin and xout, corresponding to the atomic concentration of D at the liquid inlet and outlet, respectively, were experimentally measured. The gas-phase outlet composition yout was determined from a mass-balance calculation using xin and xout. The theoretical outlet composition 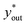 was calculated using the temperature-dependent water–hydrogen equilibrium constant.24 The D concentrations in the H2 gas and DI water were 135 µmol mol−1 (D/(D + H)) and 147 µmol mol−1 (D/(D + H)), respectively. For the H2 gas, the D concentration was measured with a quadrupole mass spectrometer using the PrismaPro® QMG 250 M1 (Pfeiff Vacuum GmbH). The D concentration in the liquid samples was determined with a cavity ring-down spectroscopy with a L2140-i (PICARRO).
was calculated using the temperature-dependent water–hydrogen equilibrium constant.24 The D concentrations in the H2 gas and DI water were 135 µmol mol−1 (D/(D + H)) and 147 µmol mol−1 (D/(D + H)), respectively. For the H2 gas, the D concentration was measured with a quadrupole mass spectrometer using the PrismaPro® QMG 250 M1 (Pfeiff Vacuum GmbH). The D concentration in the liquid samples was determined with a cavity ring-down spectroscopy with a L2140-i (PICARRO).
Results and discussion
Characterization of Pt@SiO2
Fig. 2 shows the XRD patterns for SiO2 and Pt@SiO2. As presented in Fig. 2a, both display a low-angle diffraction peak at 2θ = 3.3°, indicating the presence of an ordered mesoporous structure.51 The preservation of this peak even after Pt decoration suggests that the mesopore structure of the silica remains without collapse. In Fig. 2b, a broad peak at 2θ = 21°–23° is observed, indicating the amorphous nature of the mesoporous silica. For Pt@SiO2, additional peaks appear at 2θ = 39.8°, 46.3° and 67.8°, which are attributed to metallic Pt0. Overall, Pt was successfully decorated without any significant change in the mesoporous structure of the support.52
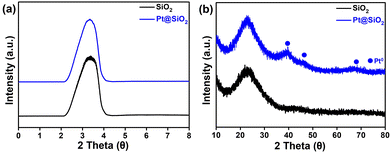 |
| | Fig. 2 The (a) low- and (b) high-angle XRD patterns of SiO2 and Pt@SiO2. | |
Fig. 3a and b display SEM images of SiO2 and Pt@SiO2, respectively. They exhibit the spherical morphology commonly observed in a novel synthesis of mesoporous silica.50 The particle sizes range from approximately 200 to 900 nm, without any noticeable difference after Pt decoration. Fig. 3c presents a TEM image of Pt@SiO2, in which homogeneously dispersed Pt is observed within the mesoporous silica framework. Correspondingly, the EDS elemental mappings of Si, O and Pt (Fig. 3d–f) show uniform Si and O originating from mesoporous silica, and a homogeneous distribution of Pt. Thus, the presence and dispersion of Pt followed by Pt decoration is visually as well as elementally validated, which is consistent with the crystallography results shown in Fig. 2.
 |
| | Fig. 3 SEM images of (a) SiO2 and (b) Pt@SiO2. TEM image of (c) Pt@SiO2 and corresponding EDS elemental mappings of (d) O, (e) Si, and (f) Pt. | |
Fig. 4 and Table 1 present the N2 physisorption isotherms and textural properties of SiO2 and Pt@SiO2. As shown in Fig. 4a, they exhibit Type IV isotherms in IUPAC classification, which stands for the typical mesoporous structure. The adsorption capacity decreased from 612 to 342 mmol g−1 after Pt decoration on SiO2. This reduction indicates partial blockage or narrowing of the pore structure. In Fig. 4b, SiO2 shows a PSD centered at 3.8 nm. After Pt loading, the main peak is slightly shifted to 3.5 nm, and its intensity decreases significantly. This coincided well with the reduction in adsorption capacity. According to Table 1, the BET surface area and pore volume also decrease from 1800 to 710 m2 g−1 and 0.80 to 0.53 cm3 g−1 after Pt decoration, respectively. Since the pore size, ranging from 3.0 to 3.9 nm, still remains within the mesopore framework, the mesoporous silica framework is preserved after Pt decoration. Based on the ICP-OES results, the Pt content in Pt@SiO2 was determined to be 5.12 wt%, which closely matches the intended loading of 5.00 wt%. Overall, Pt is successfully incorporated into the mesoporous silica framework.
 |
| | Fig. 4 (a) N2 physisorption isotherms for SiO2 and Pt@SiO2 at −196 °C (filled square: adsorption; open square: desorption) and (b) pore size distributions for SiO2 and Pt@SiO2 calculated by the BdB-FHH method. | |
Table 1 Textural properties of SiO2 and Pt@SiO2
| |
BET surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore size (nm) |
| SiO2 |
1800 |
0.83 |
3.3–4.3 |
| Pt@SiO2 |
710 |
0.53 |
3.0–3.9 |
Characterization of Pt@SiO2-PDMS
Fig. 5 presents stereomicroscopy images of PDMS, SiO2-PDMS, Pt@SiO2-PDMS, and a Dixon ring. The Dixon ring is used as a hydrophilic packing material in our LPCE process. The composite morphology and packing structure are clearly distinguishable and can be visually identified. Pristine PDMS (Fig. 5a) exhibits a transparent and smooth surface with a uniform appearance, reflecting its homogeneous molecular arrangement and flexibility.53 SiO2-PDMS (Fig. 5b) appears white and opaque due to the uniform dispersion of mesoporous silica particles within the PDMS polymer matrix. Fig. 5c shows Pt@SiO2-PDMS with a dark-brown color that is close to black. This is because of the presence of metallic Pt supported on silica and suggests its uniform distribution within the PDMS matrix as demonstrated in Fig. 3. Moreover, the cross-sectional SEM image (Fig. S3 of the SI) indicates that Pt@SiO2 is well incorporated within the PDMS matrix. Fig. 5d presents the Dixon ring, which is composed of a regular stainless-steel mesh. It promotes vapor–liquid equilibrium, here the equilibrium between H2O(v) and H2O(l). Further, this enhances interfacial contact, which reduces mass transfer resistance during the isotope exchange process.54
 |
| | Fig. 5 Stereomicroscopy images of (a) PDMS, (b) SiO2-PDMS, (c) Pt@SiO2-PDMS and (d) Dixon ring (scale bar is 1 mm). | |
Fig. 6 presents the FT-IR/ATR spectra of SiO2, Pt@SiO2, PDMS, SiO2-PDMS and Pt@SiO2-PDMS, which were obtained to investigate the surface and near-surface chemical structures. In SiO2, a strong asymmetric Si–O–Si stretching band is observed in the range of 1100–1150 cm−1, corresponding to the primary bonding network within the silica framework.55 For Pt@SiO2, the same band appears with increased intensity and a slight shift toward lower wavenumbers, which indicates structural modification induced by Pt decoration.56 The spectrum of PDMS shows a C–H stretching band in the range of 2900–2950 cm−1, representing the presence of methyl groups in the polymer structure. This peak is also observed in SiO2-PDMS, suggesting that the organic structure of PDMS remains stable after silica incorporation.57 In Pt@SiO2-PDMS, both the Si–O–Si and C–H stretching bands are clearly retained, indicating the coexistence of organic and inorganic hybrid components. These results indicate that the structural features of PDMS and SiO2 are preserved near the surface. Also, the Pt decoration and composite formation do not disrupt the integrity of each component.
 |
| | Fig. 6 FT-IR/ATR spectra of SiO2, Pt@SiO2, PDMS, SiO2-PDMS and Pt@SiO2-PDMS. | |
Fig. 7 presents the WCA of PDMS, SiO2-PDMS and Pt@SiO2-PDMS, which were measured to evaluate their surface wettability. Surface wettability is a critical parameter that influences the interaction between the catalyst and liquid reactants. In the LPCE process, an excessively hydrophilic surface may lead to the accumulation and coverage of water due to reactions on the catalyst composite surface, potentially causing catalyst poisoning.28 Therefore, appropriate hydrophobicity is necessary for maintaining catalyst activity and operational stability. As shown in Fig. 7a, the pristine PDMS exhibits a high WCA of 118.3°, which is attributed to abundant methyl groups.58 SiO2-PDMS (Fig. 7b) has a slightly reduced WCA of 108.3°, which is associated with the incorporation of hydrophilic SiO2 particles within the PDMS matrix. Pt@SiO2-PDMS (Fig. 7c) has a similar WCA of 108.5°, indicating that the decoration of Pt does not affect the intrinsic surface wettability of SiO2-PDMS. The surface wettability suggests that the PDMS-based composites maintain an appropriate hydrophobic surface that is suitable for efficient isotope exchange under liquid-phase conditions.
 |
| | Fig. 7 The WCA of (a) PDMS, (b) SiO2-PDMS, (c) Pt@SiO2-PDMS. | |
As shown in Table 2, H2 chemisorption analysis revealed a metal dispersion of 3.5%, corresponding to a metallic surface area of 8.6 m2 g−1 metal and an average particle size of 32.4 nm. It should be noted that the H2 chemisorption measurements primarily reflect the fraction of Pt sites accessible to H2 under the measurement conditions (40 °C, pulse chemisorption). In our Pt@SiO2-PDMS system, Pt species partially embedded within the PDMS layer may not be fully probed, potentially leading to an underestimation of the actual dispersion. To further evaluate batch-to-batch reproducibility of Pt@SiO2-PDMS, TGA was performed as shown in Fig. S4 of the SI. Two different Pt@SiO2-PDMS samples from different batches show 20.5% and 20.7% weight loss, which are marginally identical. This confirms the reproducibility of Pt@SiO2-PDMS.
Table 2 H2 chemisorption results for Pt@SiO2-PDMS
| Metal dispersion (%) |
Metallic surface area (m2 g−1 metal) |
Particle size (nm) |
| 3.5 |
8.6 |
32.4 |
Liquid-phase catalytic exchange performance
Fig. 8 demonstrates the column efficiency (%) of the PDMS, SiO2-PDMS and Pt@SiO2-PDMS randomly packed with Dixon rings in a volume ratio of 1:4, measured at 30 min intervals over 180 min at 60 °C. The corresponding percentage values are summarized in Table S1 of the SI. Both PDMS and SiO2-PDMS exhibited negligible column efficiency over the 180 min reaction period. In contrast, Pt@SiO2-PDMS clearly demonstrated catalytic activity. It reached a maximum column efficiency of 23.0% at 90 min reaction time and maintained values of around 20% steadily up to 180 min. This indicates that the newly developed Pt@SiO2-PDMS effectively promotes the hydrogen isotope exchange reaction, facilitating the transfer of D from the liquid to the gas phase.
 |
| | Fig. 8 Column efficiency (%) of PDMS, SiO2-PDMS and Pt@SiO2-PDMS randomly packed with Dixon rings in a volume ratio of 1:4 measured at 60 °C. | |
Fig. 9 displays the column efficiency of Pt@SiO2-PDMS randomly packed with Dixon rings in a volume ratio of 1:4, 1:1 and 2:1, measured at 30 min intervals over 180 min at 60 °C. The corresponding percentage values are summarized in Table S2 of the SI. When the catalyst amount was increased from 1:4 to 1:1 ratio, the overall column efficiency increased by around 14.8%, and the maximum value was 37.7% at 60 min. Further increasing the ratio of catalyst amount from 1:1 to 2:1, the overall column efficiency increases further by around 10.3%, with the highest efficiency of 47.4% at 120 min. This trend indicates that increasing the catalyst amount enhances the catalytic efficiency of the LPCE column. In conventional LPCE processes, the gas–liquid interfacial area is largely fixed by the packing configuration, thereby increasing the catalyst fraction alone does not guarantee a proportional improvement in column efficiency.21 In the present LPCE process, the performance enhancement observed is attributed to the greater availability of active Pt and the expanded interfacial contact between the catalyst and reactants, H2O(l) and H2(g), thereby reducing mass transfer resistance and promoting the hydrogen isotope exchange reaction.21 Across every volume ratio, the column efficiency remained stable over the 180 min reaction period within a standard deviation of ±0.4%–1.2%. This demonstrates that Pt@SiO2-PDMS maintained both activity and durability under the tested conditions.
 |
| | Fig. 9 Column efficiency (%) of Pt@SiO2-PDMS randomly packed with Dixon rings in a volume ratio of 1:4, 1:1 and 2:1 measured at 30 min intervals over 180 min at 60 °C. | |
With the volume ratio of 2:1 having the highest column efficiency, Fig. 10 presents the column efficiency of Pt@SiO2-PDMS measured at 180 min at temperatures ranging from 30 to 80 °C. Detailed time-dependent profiles are provided in Fig. S5 of the SI, and the corresponding percentage values are summarized in Table S3 of the SI. Between 30 and 70 °C, the column efficiency was enhanced with increasing temperature while maintaining stable profiles over 180 min. The maximum efficiencies in each temperature were 28.7% (30 °C, 60 min), 33.6% (40 °C, 150 min), 46.0% (50 °C, 60 min), 48.8% (60 °C, 30 min) and 54.2% (70 °C, 60 min). Throughout the entire reaction time, the column efficiencies remained relatively stable, exhibiting only minor fluctuations within a standard deviation range of ±0.2–1.8% at temperatures ranging from 30 to 70 °C. On the other hand, the catalyst exhibited a much better performance at 80 °C. The efficiency increased with the reaction time, reaching 67.7% at 180 min. Overall, it exhibited minor fluctuations within a standard deviation of ±2.4%, indicating a stable and steady catalytic exchange reaction. This temperature-dependent trend suggests that the hydrogen isotope exchange reaction is governed by reaction kinetics, where increasing temperature promotes more frequent and energetic molecular collisions, thereby accelerating the D–H exchange.59 The catalyst retained its activity under the tested temperature conditions, and excellent performance was observed at 80 °C. Thus, this condition was selected for the subsequent long-term durability evaluation to evaluate its feasibility for industrial application.
 |
| | Fig. 10 Column efficiency (%) of Pt@SiO2-PDMS randomly packed with Dixon rings in a volume ratio of 2:1 measured at 180 min under temperatures ranging from 30 to 80 °C. | |
With the optimized volume ratio and temperature, the long-term operational stability of Pt@SiO2-PDMS was evaluated under continuous LPCE operation for 14 days at 80 °C (Fig. 11). The corresponding column efficiency values are summarized in Table S4 of the SI. Throughout the entire test period, the column efficiency fluctuated slightly around an average value of 64.9% ± 2.3%, indicating stable catalytic performance without noticeable deactivation. Minor variations were observed around day 3 and 12, during which the efficiency temporarily increased to 69.3% and subsequently returned to the average level. This stable efficiency profile demonstrates that the Pt active sites were effectively protected by the PDMS matrix during prolonged liquid-phase operation. After 14 days of LPCE operation and subsequent regeneration, recyclability was evaluated under the same conditions as those shown in Fig. 9 to assess the stability of the catalytic performance (Fig. S6 of the SI). The corresponding percentage values are summarized in Table S5 of the SI, providing an average column efficiency of 45.6 ± 1.1% over 180 min. This is consistent with the values obtained in Fig. 9, thereby demonstrating the stable and consistent catalytic performance under repeated operation. In addition, the hydrophobic characteristic of the catalyst was maintained, with a WCA of 102.8° measured after the LPCE test (Fig. S7 of the SI). This value is comparable to that of the fresh catalyst (108.5°), indicating that the hydrophobicity of Pt@SiO2-PDMS is well preserved after long-term operation, with the surface functional groups of the PDMS framework remaining stable under LPCE conditions. H2 chemisorption results of the used catalyst after long-term operation (Table S6 of the SI) show a metal dispersion of 3.1%, a metallic surface area of 7.6 m2 g−1 metal, and a Pt particle size of 36.7 nm. These metrics are comparable to those of the fresh catalyst, which were 3.5%, 8.6 m2 g−1 metal, and 32.4 nm, respectively. Thus, the result demonstrates that the catalytic structure is well preserved.
 |
| | Fig. 11 Column efficiency (%) of Pt@SiO2-PDMS randomly packed with Dixon rings at a volume ratio of 2:1 measured over 14 days at 80 °C. | |
In Table 3, the Pt@SiO2-PDMS catalyst is compared with previously reported Pt-based catalysts for hydrogen isotope exchange in the LPCE process. The Pt@SiO2-PDMS catalyst shows column efficiencies of 49%–68% at 60–80 °C, while Pt/200-S-NH2-GR exhibits 75%–90% at 40–60 °C,60 Pt@MIL-101/PVDF shows 75% at 60 °C,61 and Pt0.5Ru0.5/C exhibits 72%–88% at 50 °C.62 Pt@SBA-15-Tetramethyldisilazane and Pt/s-C3N4-7 also show efficiencies of 63–73% and 64%–85% over 30–70 °C and 40–80 °C, respectively.33,44 The Pt@SiO2-PDMS catalyst exhibits lower column efficiency under similar operating conditions, such as column diameter, height, and particularly temperature. This difference is attributed to the limited availability of exposed Pt active sites, as evidenced by the large Pt particle size. The H2 chemisorption of Pt@SiO2-PDMS shows a low metal dispersion (3.1%) and relatively large Pt particle size (∼36.7 nm), as shown in Table 2. Significant work remains to be done to optimize mesoporous silica materials and Pt decoration techniques to allow better control of metal dispersion and Pt particle size, thereby increasing the number of accessible active sites. Despite the lower column efficiency, the catalyst can be prepared through a simple fabrication procedure using mesoporous silica and PDMS with a laboratory-scale LPCE column, offering advantages in terms of cost and scalability. The LPCE process itself is successfully demonstrated under the present conditions, and further studies should focus on scaling up the processes and optimizing catalyst fabrication.
Table 3 Summary of previously reported Pt-based catalysts for hydrogen isotope exchange in the LPCE process
| Catalyst |
Column diameter (mm) |
Column height (mm) |
Pt particle size (nm) |
Operating temperature (°C) |
Column efficiency (%) |
Ref. |
| Pt/200 µm trimethoxyoctylsilane dual modified 2-dimensional aminated graphene. Pt@MIL-101/polyvinylidene fluoride. Pt0.5Ruthenium0.5/carbon. Pt@Santa Barbara amorphous-15-tetramethyldisilazane. Pt/PDMS modified carbon nitride-7 (where 7 indicates the theoretical mass percentage of Pt in the catalyst). |
| Pt@SiO2-PDMS |
15 |
155 |
32.4 |
30–80 |
29–68 |
This work |
| Pt/200-S-NH2-GRa |
38 |
32.2 |
1.85 ± 0.35 |
40–60 |
75–90 |
60 |
| Pt@MIL-101/PVDFb |
25 |
450 |
2.9 |
60 |
75 |
61 |
| Pt0.5Ru0.5/Cc |
16 |
450 |
1.9 ± 0.5 |
50 |
72–88 |
62 |
| Pt@SBA-15-tetramethyldisilazaned |
15 |
350 |
2.03 |
30–70 |
63–73 |
33 |
| Pt/s-C3N4-7e |
0.65 (cross-section) |
630 |
0.82 ± 0.08 |
40–80 |
64–85 |
44 |
Conclusions
Pt@SiO2-PDMS was successfully fabricated by decorating Pt on a mesoporous silica framework, followed by embedding within a PDMS matrix. The catalyst demonstrated high column efficiency and stability for the hydrogen isotope exchange reaction in the LPCE process. The highest performance was achieved when Pt@SiO2-PDMS was randomly packed with Dixon rings in a volume ratio of 2:1 and measured at 80 °C, resulting in a column efficiency of 67.7% with a catalyst bed length of 155 mm. The stable column efficiency observed in up to 14 days of continuous operation indicated that Pt@SiO2-PDMS retained its hydrophobicity and structural integrity without noticeable deactivation or poisoning. The enhanced performance is attributed to the effective exposure of Pt active sites and the hydrophobic surface provided by the PDMS matrix, which minimized liquid water penetration and preserved catalytic activity. These results demonstrate that Pt@SiO2-PDMS serves as a durable and efficient catalyst for the LPCE process, enabling long-term hydrogen isotope exchange from liquid to gas phase. This study provides a promising platform for designing hydrophobic composite catalysts applicable to the sustainable and scalable hydrogen isotope exchange process. Further, hydrophobic composite catalysts can be extended to catalytic systems that require stable operation under moisture-rich conditions, including hydrogen purification and gas-phase reaction processes.
Author contributions
Daeuk Kang: data curation, investigation, methodology, writing – original draft, writing – review & editing. Huiryung Heo: investigation, methodology. Euna Jeong: investigation. Hee-Man Yang: conceptualization. Dong-Yeun Koh: funding acquisition. Chan Woo Park: conceptualization, funding acquisition, project administration, supervision, writing – review & editing. Hyung-Ju Kim: conceptualization, investigation, methodology, resources, supervision, writing – original draft, writing – review & editing.
Conflicts of interest
The authors declare no conflict of interest.
Data availability
The data supporting the findings of this study are available within the article and its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6ma00202a.
Acknowledgements
The authors are grateful for the financial support provided by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. RS-2022-00155422) and by the Korea Evaluation Institute of Industrial Technology, funded by the Korean government, Ministry of Trade, Industry and Energy (Grant No. 20022479). This research was also financially supported by the Institute of Civil Military Technology cooperation funded by the Defense Acquisition Program Administration and the Ministry of Trade, Industry and Energy of the Korean government under grant No. 22-CM-BR-14.
References
- B. Wang, X. Xiao, C. Chang, F. Huang, T. Y. K. Qin, H. He and J. Xu, J. Mater. Chem. A, 2025, 13, 18081–18114 Search PubMed.
- M. Chen, K. Zhao, J. Li, G. Lin, D. Banham, L. Du and M. Chen, J. Mater. Chem. A, 2025, 13, 7687–7691 RSC.
- R. Cameron, in Nuclear Fission Reactors, ed I. R. Cameron, Springer, Boston, MA, 1982, pp. 269–282 Search PubMed.
- S. Jung, W.-L. Cheung, S. Li, M. Wang, W. Li, C. Wang, X. Song, G. Wei, Q. Song, S. S. Chen, W. Cai, M. Ng, W. K. Tang and M.-C. Tang, Nat. Commun., 2023, 14, 6481 CrossRef CAS PubMed.
- X. Hou, J. Nucl. Fuel Cycle Waste Technol., 2018, 16, 11–39 CrossRef.
- T. Tanabe, J. Nucl. Mater., 2011, 417, 545–550 CrossRef CAS.
- N. Momoshima, Radiat. Prot. Dosimetry, 2022, 198, 896–903 Search PubMed.
- A. Cauquoin, M. Gusyev, H. Bong, A. Okazaki and K. Yoshimura, Environ. Sci. Pollut. Res., 2025, 32, 3649–3663 Search PubMed.
- C. Chen, J. Hou, J. Li, X. Chen, C. Xiao, Q. Wang, Y. Gong, L. Yue, L. Zhao, G. Ran, X. Fu, X. Xia and H. Wang, Fusion Eng. Des., 2020, 153, 111460 Search PubMed.
- M. Enoeda, T. Yamanishi, H. Yoshida, Y. Naruse, H. Fukui and K. Muta, Fusion Eng. Des., 1989, 10, 319–323 Search PubMed.
- K. M. Song, S. H. Sohn, D. W. Kang, S. W. Paek and D. H. Ahn, Fusion Eng. Des., 2007, 82, 2264–2268 CrossRef CAS.
- E. P. Magomedbekov, I. L. Rastunova and N. N. Kulov, Theor. Found. Chem. Eng., 2021, 55, 1–11 Search PubMed.
- M. Lozada-Hidalgo, S. Zhang, S. Hu, A. Esfandiar, I. V. Grigorieva and A. K. Geim, Nat. Commun., 2017, 8, 15215 Search PubMed.
- H. K. Rae, Separation of Hydrogen Isotopes, American Chemical Society, 1978, vol. 68, pp. 1–26 Search PubMed.
- M. Rethinasabapathy, S. M. Ghoreishian, S.-K. Hwang, Y.-K. Han, C. Roh and Y. S. Huh, Adv. Mater., 2023, 35, 2301589 Search PubMed.
- G. Ana, I. Cristescu, M. Draghia, C. Bucur, O. Balteanu, M. Vijulie, G. Popescu, C. Costeanu, N. Sofilca, I. Stefan, R. Daramus, A. Niculescu, A. Oubraham, I. Spiridon, F. Vasut, C. Moraru, S. Brad and G. Pasca, Fusion Eng. Des., 2016, 106, 51–55 Search PubMed.
- J. Hou, J. Li, C. Xiao, H. Wang and S. Peng, Separations, 2024, 11, 219 CrossRef CAS.
- J. L. Aprea, Int. J. Hydrogen Energy, 2002, 27, 741–752 CrossRef CAS.
- D.-W. Lim, J. Ha, Y. Oruganti and H. R. Moon, Mater. Chem. Front., 2021, 5, 4022–4041 Search PubMed.
- K. D. Kok, Nuclear
Engineering Handbook, CRC Press, Boca Raton, FL, 2nd edn, 2016 Search PubMed.
- S. M. Mhd Yusof, S. S. M. Lock, N. N. Abdul Talib and L. C. Seng, Sustainability, 2024, 16, 4796 Search PubMed.
- H. Yamai, S. Konishi, M. Hara, K. Okuno and I. Yamamoto, Fusion Technol., 1995, 28, 1591–1596 Search PubMed.
- A. Fedorchenko, I. A. Alekseev, S. D. Bondarenko and T. V. Vasyanina, Fusion Sci. Technol., 2017, 71, 432–437 CrossRef.
- J. Hou, S. Hu, L. Xiong, K. Weng, T. Yang and Y. Luo, Processes, 2024, 12, 2549 Search PubMed.
- B. Siddique, M. A. Shaheen, A. Abbas, Y. Zaman, H. M. A. Amin, M. M. Alam, N. K. Alharbi, F. Alshehri, A. Shami, F. A. Al-Joufi and A. Ali, Int. J. Environ. Anal. Chem., 2025, 105, 9414–9436 CrossRef.
- A. Jabbar, A. Abbas, N. Assad, M. Naeem-ul-Hassan, H. A. Alhazmi, A. Najmi, K. Zoghebi, M. Al Bratty, A. Hanbashi and H. M. A. Amin, RSC Adv., 2023, 13, 28666–28675 RSC.
- J. Tang, L. Shen, K. Zhao, J. Peng, Q. Chen, C. Yu, Y. Li, A. Abbas, S. Wang and Z. Liu, Appl. Surf. Sci., 2024, 643, 158574 CrossRef CAS.
- G. Ionita, C. Bucur, I. Spiridon and I. Stefanescu, J. Radioanal. Nucl. Chem., 2015, 305, 117–126 CrossRef CAS.
- S. H. Sohn and K. J. Lee, J. Nucl. Sci. Technol., 2006, 43, 874–883 CrossRef CAS.
- H. Heo, J. Jang, E. Jeong, H.-J. Kim, Y. J. Kim, C. W. Park, J. So and D.-Y. Koh, Energy Environ. Mater., 2025, 8, e12815 CrossRef CAS.
- L. He, H. Ruan, Y. Liu, Q. Jia, D. Wu, L. Huang, X. Wang and Y. Yin, Appl. Catal. Open, 2025, 201, 207034 Search PubMed.
- S. Hu, J. Hou, L. Xiong, K. Weng, T. Yang and Y. Luo, Sep. Purif. Technol., 2011, 77, 214–219 CrossRef CAS.
- Z. Lu, X. Fu, J. Li, J. Hou, G. Ran, C. Xiao and X. Wang, Int. J. Hydrogen Energy, 2023, 48, 1979–1987 Search PubMed.
- L. Shen, X. Chen, Y. Chen, J. Peng, A. Abbas, J. Wei, C. Yu, J. Li and Y. Li, J. Electron. Mater., 2022, 51, 3092–3100 Search PubMed.
- C. Xiao, J. Tian, Q. Chen and M. Hong, Chem. Sci., 2024, 15, 1570–1610 RSC.
- S. Prodinger and M. A. Derewinski, Pet. Chem., 2020, 60, 420–436 CrossRef CAS.
- F. He, J. Zhang, A. Liu and G. Zhu, E3S Web Conf., 2023, 416, 01001 Search PubMed.
- D. Meili and J. Hai-Long, CCS Chem, 2020, 2, 2740–2748 Search PubMed.
- H.-T. Vu, F. M. Harth and N. Wilde, Front. Chem., 2018, 6, 1–9 Search PubMed.
- F. Cosnier, A. Celzard, G. Furdin, D. Bégin, J. F. Marêché and O. Barrès, Carbon, 2005, 43, 2554–2563 CrossRef CAS.
- A. Ali, M. A. Hussain, M. T. Haseeb, M. U. Ashraf, M. Farid-ul-Haq, T. Tabassum, G. Muhammad and A. Abbas, J. Braz. Chem. Soc., 2023, 34, 906–917 CAS.
- X. Li, C. Liu, K. Gou, H. Yang, X. Ren and B. Peng, RSC Adv., 2015, 5, 45420–45425 Search PubMed.
- F. Huang and C. Meng, Int. J. Hydrogen Energy, 2010, 35, 6108–6112 Search PubMed.
- Q. Fu, F. Xin, X. Yin, Y. Song and Y. Xu, Int. J. Hydrogen Energy, 2021, 46, 22446–22453 CrossRef CAS.
- P. Bian, Y. Wang and T. J. McCarthy, Macromol. Rapid Commun., 2021, 42, 2000682 Search PubMed.
- B. Sebők, M. Schülke, F. Réti and G. Kiss, Polym. Test., 2016, 49, 66–72 CrossRef.
- J. Liu, L. Ye, S. Wooh, M. Kappl, W. Steffen and H.-J. Butt, ACS Appl. Mater. Interfaces, 2019, 11, 27422–27425 CrossRef CAS PubMed.
- X. Yu and C. T. Williams, Catal. Sci. Technol., 2022, 12, 5765–5794 RSC.
- C. Martínez Casillas, M. P. Longinotti, M. M. Bruno, F. Vaca Chávez, R. H. Acosta and H. R. Corti, J. Phys. Chem. C, 2018, 122, 3638–3647 CrossRef.
- K. Schumacher, M. Grün and K. K. Unger, Microporous Mesoporous Mater., 1999, 27, 201–206 CrossRef CAS.
- T. Kimura and K. Kuroda, in Comprehensive Inorganic Chemistry II, ed J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2013, pp. 133–150 Search PubMed.
- D. Kiadehi and M. Taghizadeh, Int. J. Hydrogen Energy, 2018, 43, 4826–4838 CrossRef.
- A. Gökaltun, Y. B. Kang, M. L. Yarmush, O. B. Usta and A. Asatekin, Sci. Rep., 2019, 9, 7377 CrossRef PubMed.
- Y. Zhang and R. Wang, J. Membr. Sci., 2013, 443, 170–180 CrossRef CAS.
- R. Ellerbrock, M. Stein and J. Schaller, Sci. Rep., 2022, 12, 11708 CrossRef CAS PubMed.
- Y. Borodko, J. W. Ager, G. E. Marti, H. Song, K. Niesz and G. A. Somorjai, J. Phys. Chem. B, 2005, 109, 17386–17390 Search PubMed.
- A. Gavran, M. V. Pergal, T. Vicentic, M. Rasljic Rafajilovic, I. A. Pasti, M. V. Boskovic and M. Spasenovic, Sensors, 2025, 25, 5238 CrossRef CAS PubMed.
- T. S. Vo, T. S. Nguyen, S.-H. Lee, D. C. T. Vo, D. Kim and K. Kim, J. Sci. Adv. Mater. Devices, 2023, 8, 100554 CrossRef.
- N. J. Pester, M. E. Conrad, K. G. Knauss and D. J. DePaolo, Geochim. Cosmochim. Acta, 2018, 242, 191–212 Search PubMed.
- Y. Xu, F. Xin, Y. Jiang and X. Yin, ACS Appl. Mater. Interfaces, 2021, 13, 31660–31667 CrossRef CAS PubMed.
- X. Fu, J. Hou, C. Chen, J. Li, L. Yue, X. Chen, L. Zhao, G. Ran, X. Xia, Y. Gong, W. Ding, C. Xiao and H. Wang, J. Hazard. Mater., 2019, 380, 120904 Search PubMed.
- S. Hu, L. Xiong, J. Hou, K. Weng, Y. Luo and T. Yang, Int. J. Hydrogen Energy, 2010, 35, 10118–10126 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Chan Woo Park
c,
Chan Woo Park

 was calculated using the temperature-dependent water–hydrogen equilibrium constant.24 The D concentrations in the H2 gas and DI water were 135 µmol mol−1 (D/(D + H)) and 147 µmol mol−1 (D/(D + H)), respectively. For the H2 gas, the D concentration was measured with a quadrupole mass spectrometer using the PrismaPro® QMG 250 M1 (Pfeiff Vacuum GmbH). The D concentration in the liquid samples was determined with a cavity ring-down spectroscopy with a L2140-i (PICARRO).
was calculated using the temperature-dependent water–hydrogen equilibrium constant.24 The D concentrations in the H2 gas and DI water were 135 µmol mol−1 (D/(D + H)) and 147 µmol mol−1 (D/(D + H)), respectively. For the H2 gas, the D concentration was measured with a quadrupole mass spectrometer using the PrismaPro® QMG 250 M1 (Pfeiff Vacuum GmbH). The D concentration in the liquid samples was determined with a cavity ring-down spectroscopy with a L2140-i (PICARRO).









