
Isoxerophilins A and B, two diterpene heterodimers from Isodon xerophilus: structural elucidation and semisynthesis of isoxerophilin analogues†
Jia-Meng
Dai‡
ab,
Bing-Chao
Yan‡
a,
Ling-Mei
Kong
a,
Kun
Hu
a,
Xing-Ren
Li
a,
Xiao-Nian
Li
a,
Han-Dong
Sun
 a,
Yan
Li
a and
Pema-Tenzin
Puno
*a
a,
Yan
Li
a and
Pema-Tenzin
Puno
*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, and Yunnan Key Laboratory of Natural Medicinal Chemistry, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China. E-mail: punopematenzin@mail.kib.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 7th November 2023
Abstract
Two types of novel diterpene heterodimers, isoxerophilins A (1, ent-kaurane–sempervirane) and B (2, ent-kaurane–abietane), were isolated from the roots of Isodon xerophilus. Their structures and absolute configurations were determined by spectroscopic data and single crystal X-ray diffraction analysis. A bioinspired semisynthesis of ten isoxerophilin analogues, featuring a tandem oxidative dearomatization/intermolecular Diels–Alder reaction, has been achieved, and the transformation of ferruginol into a sempervirane scaffold was accomplished by the SIBX-mediated tandem reaction. Compound 12 promotes MICA/B expression on SMMC-7721 cells.
Introduction
The great structural diversity of natural products serves as an important inspiration source for innovation in drug discovery.1 Naturally occurring diterpene dimers, over 300 members to date, have emerged as an essential and unique class of natural products, arousing increasing interest from the chemistry community.2 Most diterpene dimers are homodimers while heterodimers from two different skeleton units are rare, such as hispidanins A–D3 (labdanetotarane), taiwaniadducts B–J4 and taiwanoids A–D5 (abietanelabdane). And they display a broad spectrum of biological effects, such as cytotoxic,6 anti-inflammatory,7 antimicrobial,8 and antimalarial9 activities. A few diterpene dimers from the genus Isodon, such as bisrubescensin A,10 hispidanin B,3 and bistenuifolin B,11 were reported to have cytotoxic activity. Intriguingly, enanderinanin J is a potential antiviral agent that inhibits autophagosome–lysosome fusion.12Isodon xerophilus H. Hara, a perennial shrub, has long been used in Yunnan Province, China, for its anti-tumor, anti-inflammatory, and anti-bacterial properties. Previous studies on the aerial parts of I. xerophilus have disclosed the presence of some ent-kaurane diterpenoids, meroditerpenoids, and homodimers.13 To further explore the structural and biological diversity of diterpene dimers, we investigated the secondary metabolites in one-year-old roots of I. xerophilus for the first time, which was cultivated in Kunming Botanical Garden. Isoxerophilins A (1) and B (2) (Fig. 1) represented two classes of novel diterpene heterodimers formed from ent-kaurane and sempervirane units, and ent-kaurane and abietane units, respectively. Furthermore, a divergent bioinspired semisynthesis of ten isoxerophilin analogues was accomplished starting from natural dehydroabietic acid (3), which facilitated the discovery of a new type of dimer exhibiting tumor immunotherapy activity. Herein, we describe the structural elucidation and plausible biogenetic pathways of 1 and 2. Moreover, we present a bioinspired semisynthesis and biological evaluation of isoxerophilin analogues.
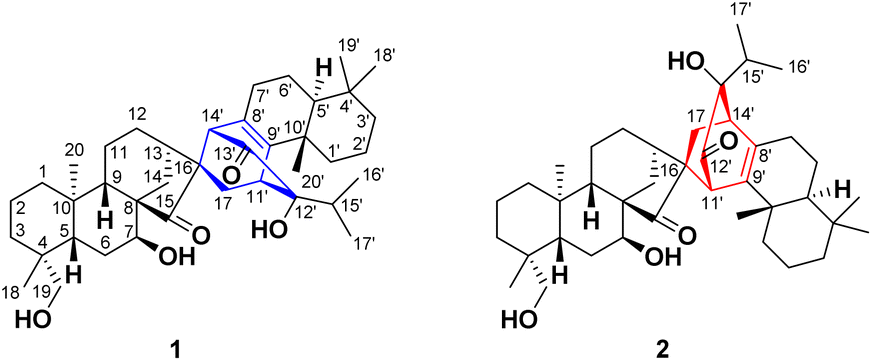 | ||
| Fig. 1 Chemical structures of compounds 1 and 2. | ||
Results and discussion
Isoxerophilin A (1) was obtained as colorless crystals. Its molecular formula C40H60O5 was determined from the HRESIMS ion peaks at m/z 643.4323 ([M + Na]+, calcd for 643.4333), which indicated 11 indices of hydrogen deficiency (HIDs). The 1H NMR spectrum of 1 (Table 1) showed signals of seven methyls, [δH 0.86 (s), 0.88 (s), 1.00 (s), 1.18 (s), 1.22 (s), 1.22 (d, J = 6.8 Hz), and 1.40 (d, J = 6.8 Hz)], one oxygenated methylene [δH 3.66 (d, J = 10.7 Hz), 3.93 (d, J = 10.7 Hz)], and one oxygenated methine [δH 4.04 (t, J = 2.7 Hz)]. The interpretation of the 13C NMR, DEPT and HSQC spectra revealed 40 carbon signals attributable to 7 methyls, 14 methylenes (one oxygenated), 8 methines (one oxygenated), and 11 non-protonated carbons (one oxygenated, two olefinic, and two ketone carbonyls). Due to signal overlaps, NMR assignment was based on a combination of 1D and 2D NMR spectra (Table 1). These functionalities accounted for three of the 11 HIDs, which indicated that 1 was a dimeric diterpenoid containing a novel octacyclic skeleton.| No. | 1 | 2 | ||
|---|---|---|---|---|
| δ C | δ H (mult., J in Hz) | δ C | δ H (mult., J in Hz) | |
| 1 | 40.4 | 1.67 (overlapped) | 40.4 | 1.70 (overlapped) |
| 0.82 (overlapped) | 0.86 (overlapped) | |||
| 2 | 19.5 | 1.61 (overlapped) | 19.6 | 1.71 (overlapped) |
| 1.54 (overlapped) | 1.45 (overlapped) | |||
| 3 | 36.8 | 2.09 (overlapped) | 37.0 | 2.10 (overlapped) |
| 1.03 (overlapped) | 1.10 (overlapped) | |||
| 4 | 39.0 | 39.1 | ||
| 5 | 46.4 | 2.14 (d, 12.7) | 46.4 | 2.17 (d, 12.8) |
| 6 | 27.3 | 2.05 (overlapped) | 27.6 | 2.08 (overlapped) |
| 1.61 (overlapped) | 1.71 (overlapped) | |||
| 7 | 73.7 | 4.04 (t, 2.7) | 73.0 | 4.11 (t, 2.8) |
| 8 | 53.9 | 55.2 | ||
| 9 | 50.2 | 1.71 (overlapped) | 49.6 | 1.77 (dd, 19.2, 10.3) |
| 10 | 40.5 | 40.6 | ||
| 11 | 30.4 | 1.32 (overlapped) | 20.2 | 1.61 (overlapped) |
| 1.24 (overlapped) | 1.05 (overlapped) | |||
| 12 | 19.3 | 1.61 (overlapped) | 27.3 | 2.08 (overlapped) |
| 1.54 (overlapped) | 1.71 (overlapped) | |||
| 13 | 39.7 | 2.48 (m) | 41.0 | 2.26 (overlapped) |
| 14 | 33.8 | 2.08 (overlapped) | 34.1 | 2.25 (overlapped) |
| 1.85 (overlapped) | 1.87 (overlapped) | |||
| 15 | 228.3 | 232.8 | ||
| 16 | 56.6 | 60.8 | ||
| 17 | 29.8 | 2.57 (dd, 12.7, 3.0) | 27.4 | 2.33 (dd, 14.0, 2.7) |
| 1.67 (overlapped) | 1.54 (dd, 14.0, 2.7) | |||
| 18 | 28.3 | 1.22 (s) | 28.5 | 1.26 (s) |
| 19 | 65.1 | 3.93 (d, 10.7) | 65.4 | 3.98 (d, 10.7) |
| 3.66 (d, 10.7) | 3.75 (d, 10.7) | |||
| 20 | 18.5 | 1.00 (s) | 18.5 | 1.10 (s) |
| 1′ | 36.0 | 1.92 (m) | 36.1 | 1.80 (m) |
| 1.52 (m) | 1.29 (m) | |||
| 2′ | 19.7 | 1.68 (overlapped) | 19.0 | 1.65 (overlapped) |
| 1.42 (overlapped) | 1.41 (overlapped) | |||
| 3′ | 42.3 | 1.42 (overlapped) | 42.5 | 1.46 (m) |
| 1.28 (overlapped) | 1.25 (overlapped) | |||
| 4′ | 33.7 | 34.0 | ||
| 5′ | 52.0 | 1.61 (overlapped) | 51.7 | 1.33 (dd, 12.4, 4.3) |
| 6′ | 18.9 | 1.60 (overlapped) | 19.5 | 1.70 (overlapped) |
| 1.46 (overlapped) | 1.46 (overlapped) | |||
| 7′ | 31.5 | 2.04 (overlapped) | 30.3 | 2.30 (dd, 11.4, 6.8) |
| 1.90 (overlapped) | 2.23 (m) | |||
| 8′ | 126.6 | 139.0 | ||
| 9′ | 149.5 | 137.7 | ||
| 10′ | 38.5 | 37.9 | ||
| 11′ | 42.6 | 3.42 (t, 2.8) | 54.0 | 3.37 (s) |
| 12′ | 76.4 | 210.7 | ||
| 13′ | 209.4 | 76.2 | ||
| 14′ | 58.1 | 3.23 (s) | 46.2 | 2.86 (t, 2.9) |
| 15′ | 34.0 | 2.03 (m) | 36.0 | 1.94 (m) |
| 16′ | 18.6 | 1.40 (d, 6.8) | 19.1 | 1.19 (d, 6.9) |
| 17′ | 18.4 | 1.22 (d, 6.8) | 18.6 | 1.43 (d, 6.9) |
| 18′ | 34.0 | 0.88 (s) | 33.6 | 0.90 (s) |
| 19′ | 22.1 | 0.86 (s) | 22.1 | 0.83 (s) |
| 20′ | 20.8 | 1.18 (s) | 21.2 | 0.91 (s) |
Its planar structure was determined based on the interpretation of 2D NMR spectra, which implied that it can be divided into units A and B (Fig. 2). The key 1H–1H COSY correlations of H2-1/H2-2/H2-3, H-5/H2-6/H-7, and H-9/H2-11/H2-12/H-13/H2-14, along with the HMBC correlations of H-7 with C-9; H-9 with C-8, C-14, and C-15; H-13 with C-16; Ha-17 with C-13; H2-19 with C-3, C-4, C-5, and C-18; and H3-20 with C-1, C-5, C-9, and C-10, established a C-20 non-oxygenated ent-kaurane scaffold (unit A). Unit B was determined to be a sempervirane scaffold from the key 1H–1H COSY correlations of H2-1′/H2-2′/H2-3′, H-5′/H2-6′/H2-7′, and H3-16′/H-15′/H3-17′, along with the key HMBC correlations of H2-6′ with C-7′ and C-8′; H-11′ with C-9′, C-12′, C-13′, and C-15′; H-14′ with C-8′, C-9′, and C-13′; H3-18′ with C-3′, C-4′, and C-5′; H3-19′ with C-4′ and C-5′; and H3-20′ with C-1′, C-5′, C-9′, and C-10′. The remaining HIDs indicated that an additional ring joined units A and B together. This deduction was further confirmed by the HMBC correlations of H-11′ with C-16, and H-14′ with C-13, C-15, C-16, and C-17. Units A and B were linked via C-16/C-14′ and C-17/C-11′ in a six-membered ring, which was presumably formed through [4 + 2] cycloaddition.
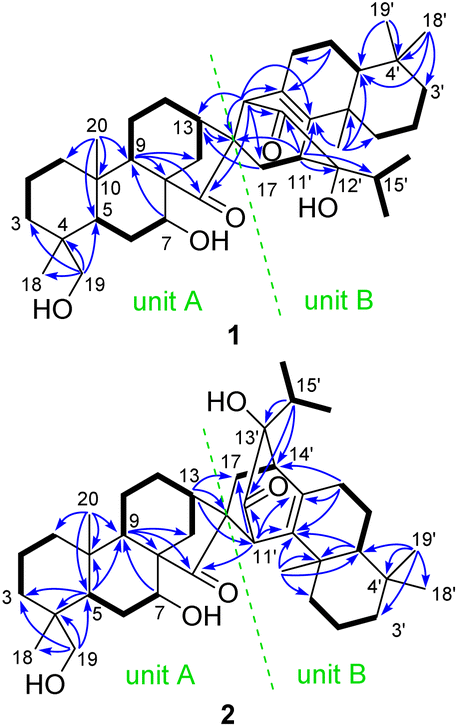 | ||
| Fig. 2 Key 1H–1H COSY (bold lines) and HMBC (blue arrows) correlations of 1 and 2. | ||
The relative configuration of 1 was partially determined by the ROESY correlations of H-7/H-14b, H-14′/H-14b, H-14′/H-13, and H3-16′/H3-20′ (Fig. S2†). Finally, its structure including the absolute configuration was established as 4R,5S,7S,8R,9S,10R,13R,16R,5′S,10′S,11′S,12′S,14′S by single crystal X-ray diffraction analysis using Cu Kα radiation with a perfect Flack parameter of 0.02(14) (CCDC 2059945,†Fig. 3). Thus, 1 was identified as a novel dimeric diterpenoid formed from ent-kaurane and sempervirane units. To the best of our knowledge, semperviranoids are rare in nature, and fewer than twenty semperviranoids have been discovered so far.14
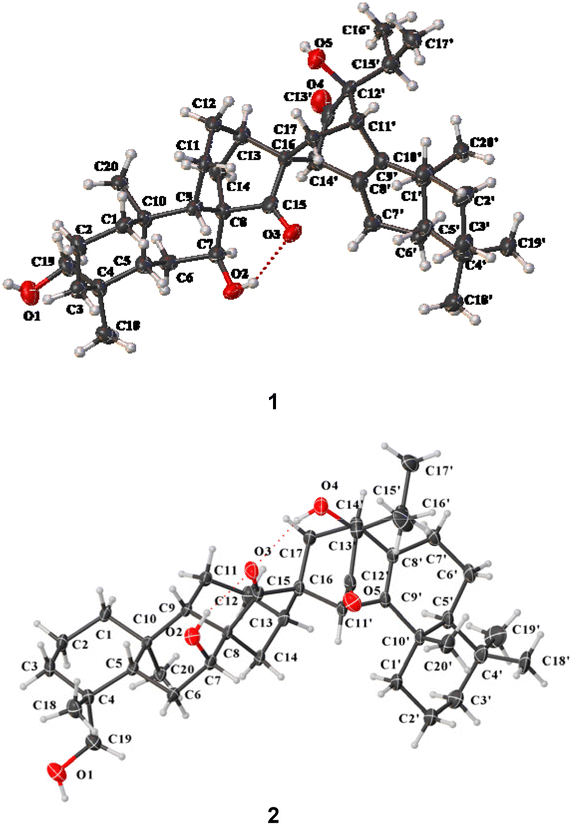 | ||
| Fig. 3 X-ray crystallographic structures of 1 and 2 (displacement ellipsoids are drawn at the 30% probability level). | ||
The molecular formula of isoxerophilin B (2) was determined to be C40H60O5 based on the HRESIMS data (m/z 643.4343 [M + Na]+, calcd 643.4338), the same as that of 1. Detailed analysis of the 2D NMR spectra established the planar structure of 2 (Fig. 2), which could be divided into units A and B. Unit A was assigned to be the same ent-kaurane scaffold as that of 1, which was supported by the HMBC correlations of H-5 with C-3, C-4, C-9 and C-10; H-7 with C-9; H-9 with C-8, C-14 and C-15; H-13 with C-16 and C-17; H2-19 with C-3, C-4, C-5, and C-18; and H3-20 with C-1, C-5, C-9, and C-10, together with the 1H–1H COSY correlations of H2-1/H2-2/H2-3, H-5/H2-6/H-7, and H-9/H2-11/H2-12/H-13/H2-14 (Fig. 2). However, unit B in 2 was assigned to be an abietane scaffold by the 1H–1H COSY correlations of H2-1′/H2-2′/H2-3′, H-5′/H2-6′/H2-7′, and H3-16′/H-15′/H3-17′, along with the HMBC correlations of H-5′ with C-9′ and C-10′; H-7′ with C-8′, C-9′, and C-14′; H-11′ with C-8′, C-9′, and C-12′; H-15′ with C-12′, C-13′, and C-14′; H3-19′ with C-3′, C-4′, C-5′, and C-18′; and H3-20′ with C-1′, C-5′, C-9′, and C-10′. An additional six-membered ring in 2 different from that in 1 was formed between units A and B presumably through biogenetic [4 + 2] cycloaddition. The linkage of C-16/C-11′ and C-17/C-14′ was supported by the HMBC correlations of H-13 with C-11′ and H-11′ with C-15 and C-17 and the 1H–1H COSY correlations of H2-17/H-14′.
The relative configuration of 2 could be partially assigned by the ROESY spectrum. The ROESY correlations of H-7/H-13α, H2-19/H3-20α, and H-11′/H-14α indicated the α-orientations of H-7, HO-19, and H-11′, respectively. To establish the whole stereochemistry, single crystals of 2 were obtained and subjected to Cu Kα X-ray diffraction analysis with a sufficient Flack parameter of 0.07(6) (CCDC 2059944†), which verified the planar structure of 2 and determined its absolute configuration to be 4R,5S,7S,8R,9S,10R,13R,16R,5′S,10′S,11′R,13′R,14′R (Fig. 3).
The intriguing structures of 1 and 2, to some extent, implied a unique biosynthetic pathway. As shown in Scheme 1, heterodimer 1 was formed by oxidatively dearomatized sempervirane B and ent-kaurane C units, while 2 was formed by oxidatively dearomatized abietane A and ent-kaurane C. To the best of our knowledge, no semperviranoids have been discovered from I. xerophilus.13 Therefore, we proposed that the abietanoid, ferruginol (4), underwent oxidative dearomatization to afford intermediate A. The Diels–Alder reaction between A and C produced heterodimer 2. On the other hand, the 1,2-shift (α-ketol rearrangement) of A gave sempervirane skeleton B, which underwent a Diels–Alder reaction with C to produce heterodimer 1.
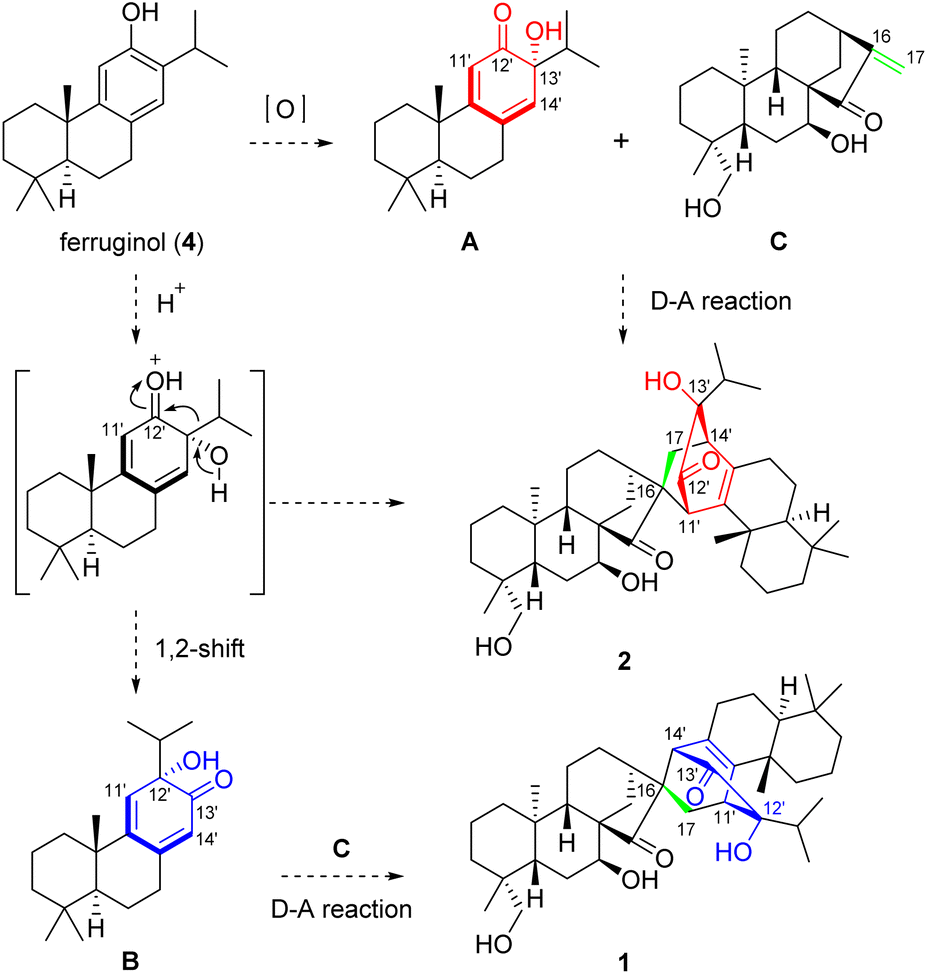 | ||
| Scheme 1 Plausible biogenetic pathways of 1 and 2. | ||
Inspired by the proposed biogenetic pathways of 1 and 2, we initiated a concise semisynthesis of isoxerophilin-like dimers for a further biological study. As shown in Scheme 2, isoxerophilin analogues 11–20 were prepared through sequential oxidative dearomatization of 4, followed by intermolecular [4 + 2] cycloaddition with ent-kauranoids 6–10.2c These samples are from the compound library of our laboratory, respectively. 8,11,13-Abietatriene (5) was prepared as previously described in the literature starting from natural dehydroabietic acid (3) through reduction (LiAlH4), tosylation (tosyl chloride in pyridine), and reduction (Zn powder-NaI in DMF).15 Treatment of 5 with phthaloyl peroxide16 afforded 4 in 65% yield in a single step, instead of Friedel–Crafts acetylation, Baeyer–Villiger oxidation and methanolysis protocols.15
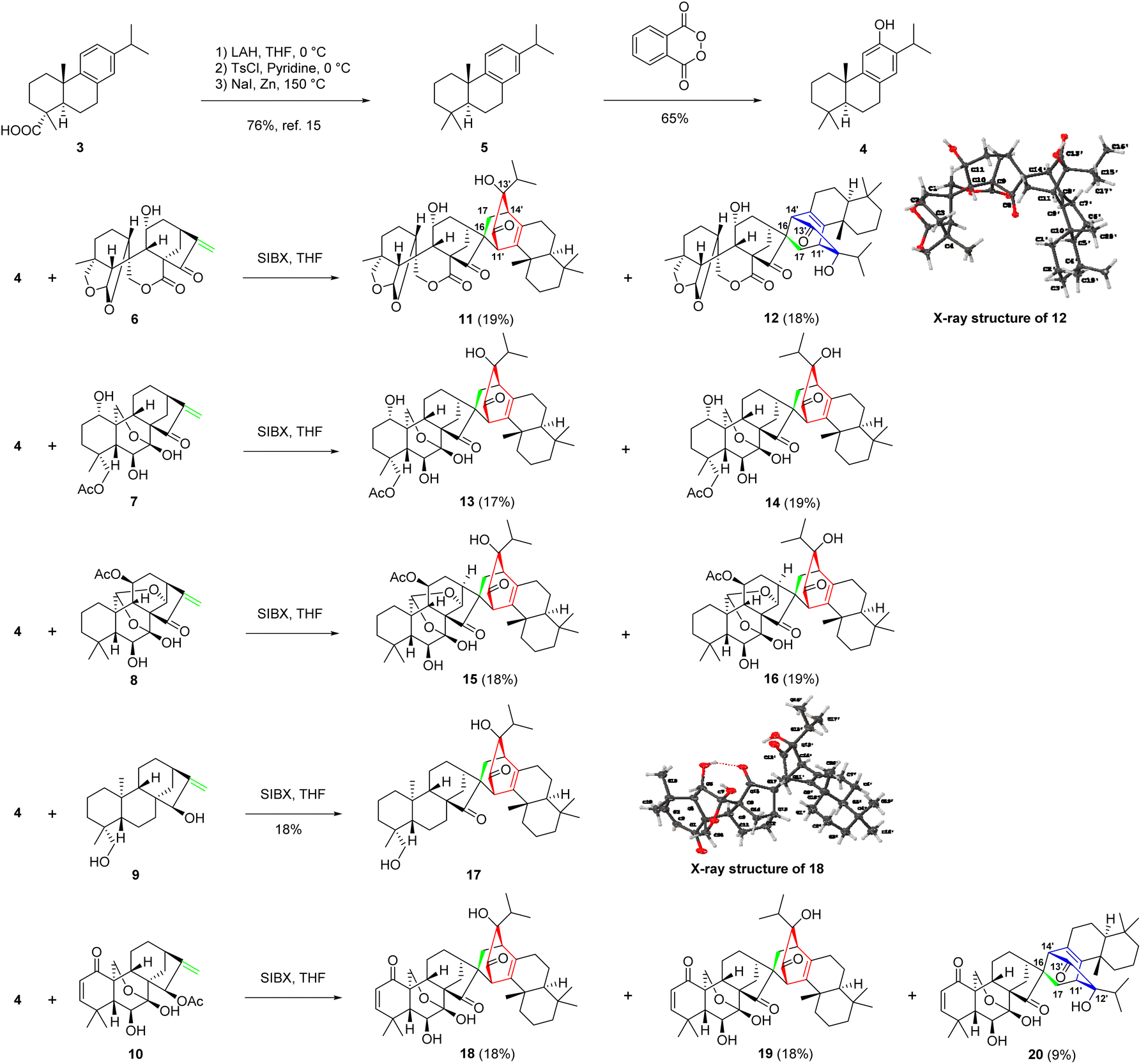 | ||
| Scheme 2 Bioinspired synthesis of natural-like diterpenoid heterodimers 11–20. | ||
With ferruginol (4) in hand, we then explored the key tandem oxidative dearomatization/intermolecular Diels–Alder reaction for the synthesis of isoxerophilin-like heterodimers. When 4 and ent-kauranoids 6–8 were treated with Pb(OAc)4 in acetic acid, respectively, only the oxidatively dearomatized acetate ketals were detected. We then changed the oxidant to SIBX (stabilized 2-iodoxybenzoic acid).17 To our delight, the tandem reaction occurred smoothly to give the desired dimers 11–16 (Scheme 2). Furthermore, oxidation of HO-15 in ent-kauranoid 9 to enone and sequential oxidative dearomatization of 4 occurred in one pot in the presence of SIBX, followed by a tandem intermolecular Diels–Alder reaction to give isoxerophilin B analogue 17. In the presence of SIBX, allylic acetate 10 (maoecrystal B) was hydrolyzed and then oxidized to enone as a dienophile for oxidative dearomatization and an intermolecular Diels–Alder tandem reaction, producing heterodimers 18–20.
The structures of 12 and 18 were confirmed by single crystal X-ray diffraction analysis (CCDC 2129366† for 12 and CCDC 2129362† for 18). The bioinspired semisynthesis provided evidence for the proposed biogenetic pathways of 1 and 2, featuring oxidative dearomatization and intermolecular [4 + 2] cycloaddition, coupled with isopropyl migration of ferruginol for 1.
NKG2D (natural killer group 2 member D) is a typical activating receptor expressed on NK cells and plays an important role in initial surveillance and elimination of cancer through interacting with NKG2D ligands represented by ULBPs and MICA/B on cancer cells.18,19 The reduction and shedding of NKG2D ligands resulting in NK-mediated immune escape have been frequently observed during tumor progression.20 Therefore, induction of NKG2D ligands on tumor cells renders cancer cells susceptible to immune destruction and is considered to be a promising immunotherapy strategy.
With ample samples of the synthesized analogues 11–20, flow cytometric analysis was undertaken to evaluate the surficial level of the NKG2D ligand MICA/B on hepatocellular carcinoma SMMC-7721 cells. As shown in Fig. 4, 12 markedly increased the cell surficial engagement of MICA/B with a mean fluorescence intensity (MFI) up to 164% at a concentration of 40 μM without dominant cytotoxicity,21 while the NKG2D ligand-inducing activity was weak in the following serial dilution. But it is of great interest to optimize the structure of 12 to improve the NKG2D ligand-inducing activity. It is worthwhile to conduct further pharmacological studies on its antitumor immune responses and mechanisms of action.
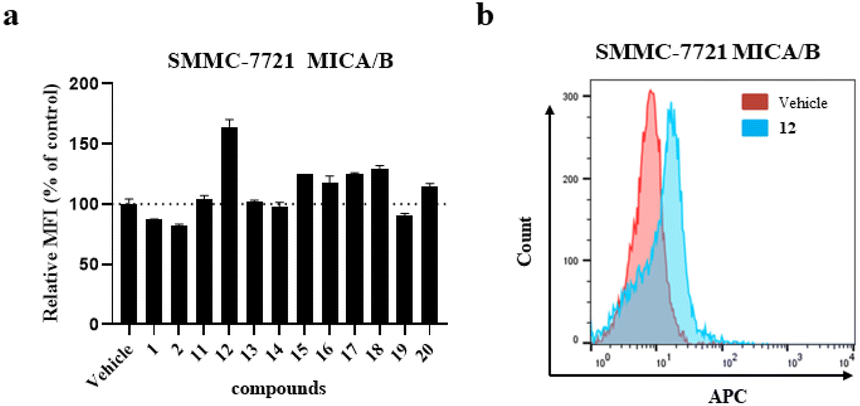 | ||
| Fig. 4 (a) The mean fluorescence intensity of the NKG2D ligand MICA/B on SMMC-7721 cells treated with the indicated compounds is presented as a histogram. The results are presented as the means of duplicate experiments ± SD. (b) SMMC-7721 cells were treated with the indicated compound 12 or the vehicle for 24 h, and then the cell surface expression was detected by flow cytometry. | ||
Conclusions
In summary, isoxerophilins A (1) and B (2), the first examples of dimeric diterpenoids formed from ent-kaurane and sempervirane/abietane units, were obtained from the roots of I. xerophilus cultivated in Kunming Botanical Garden. We developed a concise approach for the synthesis of isoxerophilin analogues 11–20via a tandem oxidation dearomatization/intermolecular Diels–Alder reaction. The transformation of abietane ferruginol into a sempervirane scaffold was achieved in the developed SIBX-mediated tandem reaction. The bioinspired semisynthesis provided evidence for the putative biogenetic pathways of natural isoxerophilins A and B. Moreover, it facilitated biological identification of a novel structural template with promising tumor immunotherapy activity.Experimental section
General experimental procedures
See the ESI.†Plant material
The rhizomes of Isodon xerophilus were collected from the cultivated plants at Kunming Botanical Garden in Yunnan Province of China in October 2017, and were identified by Prof. Xi-Wen Li from Kunming Institute of Botany. A voucher specimen (KIB 2014101601-2) has been deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.Extraction and isolation
The air-dried and powdered rhizomes of I. xerophilus (8.0 kg) were extracted with 70% aqueous acetone (4 × 40 L, 3 days each) at room temperature and filtered. The filtrate was evaporated under reduced pressure and partitioned by liquid–liquid extraction between EtOAc and H2O. The EtOAc extract (120.0 g) was chromatographed over silica gel (2.4 kg, 80–100 mesh) and eluted with CHCl3–Me2CO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–0:1, v/v; gradient system). Four major fractions were obtained from the silica gel column fractions, A–D. Fr. B (CHCl3–Me2CO, 8:2, v/v; 36.0 g), a gum, was decolorized using MCI gel and eluted with MeOH–H2O (90:10, v/v) to provide two fractions B1 (26.0 g) and B2 (8.0 g). Fr. B1 was subjected to RP-18 silica gel CC (MeOH–H2O, 30:70–100:0, v/v; gradient) to provide four fractions, B1-1–B1-4. Fr. B1-3 (3.0 g) was purified by repeated silica gel CC (petroleum ether–Me2CO, 10:1–1:1, v/v) to give three subfractions (B1-3-1–B1-3-3) based on TLC analysis. Fr. B1-3-2 (300.0 mg) was purified using Sephadex LH-20 with CHCl3–MeOH (1:1, v/v) to afford fractions B1-3-2-1–B1-3-2-3; Fr. B1-3-2-2 was subjected to semi-preparative HPLC (3 mL min−1, detector UV λmax 210 nm, CH3CN–H2O, 20:80, v/v) to afford compound 1 (1.0 mg) and compound 2 (6.0 mg).
ε) 203 (3.43), 248 (2.85), 333.5 (2.21) nm; ECD (MeOH) λmax (Δε) 196 (0.34), 238 (−0.26), 255 (−0.22), 308 (0.16), 336 (−0.03) nm; IR (KBr) νmax 3473, 3424, 2959, 2922, 2852, 1702, 1628, 1466, 1384, 1261, 1097, 1024, 842, 802, cm−1; 1H and 13C NMR data, see Table S1;† positive HRESIMS [M + Na]+m/z 643.4323 (calcd for C40H60O5Na, 643.4333).
ε) 203 (3.40), 239 (2.93), 318 (2.24) nm; ECD (MeOH) λmax (Δε) 209 (−0.14), 245 (0.37), 327 (−0.43) nm; IR (KBr) νmax 3440, 2926, 2854, 1717, 1630, 1384, 1169, 1053, 911, 878, 721, 668 cm−1; 1H and 13C NMR data, see Tables 1, S1 and S3;† positive HRESIMS [M + Na]+m/z 643.4343 (calcd for C40H60O5Na, 643.4338).
:0–0:1, v/v; gradient system). Four major fractions were obtained from the silica gel column fractions, A–D. Fr. B (CHCl3–Me2CO, 8:2, v/v; 36.0 g), a gum, was decolorized using MCI gel and eluted with MeOH–H2O (90:10, v/v) to provide two fractions B1 (26.0 g) and B2 (8.0 g). Fr. B1 was subjected to RP-18 silica gel CC (MeOH–H2O, 30:70–100:0, v/v; gradient) to provide four fractions, B1-1–B1-4. Fr. B1-3 (3.0 g) was purified by repeated silica gel CC (petroleum ether–Me2CO, 10:1–1:1, v/v) to give three subfractions (B1-3-1–B1-3-3) based on TLC analysis. Fr. B1-3-2 (300.0 mg) was purified using Sephadex LH-20 with CHCl3–MeOH (1:1, v/v) to afford fractions B1-3-2-1–B1-3-2-3; Fr. B1-3-2-2 was subjected to semi-preparative HPLC (3 mL min−1, detector UV λmax 210 nm, CH3CN–H2O, 20:80, v/v) to afford compound 1 (1.0 mg) and compound 2 (6.0 mg).
ε) 203 (3.43), 248 (2.85), 333.5 (2.21) nm; ECD (MeOH) λmax (Δε) 196 (0.34), 238 (−0.26), 255 (−0.22), 308 (0.16), 336 (−0.03) nm; IR (KBr) νmax 3473, 3424, 2959, 2922, 2852, 1702, 1628, 1466, 1384, 1261, 1097, 1024, 842, 802, cm−1; 1H and 13C NMR data, see Table S1;† positive HRESIMS [M + Na]+m/z 643.4323 (calcd for C40H60O5Na, 643.4333).
ε) 203 (3.40), 239 (2.93), 318 (2.24) nm; ECD (MeOH) λmax (Δε) 209 (−0.14), 245 (0.37), 327 (−0.43) nm; IR (KBr) νmax 3440, 2926, 2854, 1717, 1630, 1384, 1169, 1053, 911, 878, 721, 668 cm−1; 1H and 13C NMR data, see Tables 1, S1 and S3;† positive HRESIMS [M + Na]+m/z 643.4343 (calcd for C40H60O5Na, 643.4338).
X-ray crystal structure analysis
Single crystals of 1, 2, 12, and 18 can be obtained by slow solvent diffusion. Their intensity data were collected on a Bruker APEX DUO diffractometer using graphite-monochromated Cu Kα radiation. The structures of these compounds were determined using direct methods (SHELXS97), expanded using difference Fourier techniques, and refined by the program and full-matrix least squares calculations. The non-hydrogen atoms were refined anisotropically, and hydrogen atoms were fixed at calculated positions. Crystallographic data for the structures of 1 (deposition number CCDC 2059945†), 2 (deposition number CCDC 2059944†), 12 (deposition number CCDC 2129366†), and 18 (deposition number CCDC 2129362†) have been deposited in the Cambridge Crystallographic Data Centre database.:4, v/v), C40H60O5·CH4O, M = 652.92, a = 7.9127(2) Å, b = 18.4253(6) Å, c = 24.4103(7) Å, α = 90°, β = 90°, γ = 90°, V = 3558.87(18) Å3, T = 100(2) K, space group P212121, Z = 4, μ(Cu Kα) = 0.624 mm−1, 17388 reflections measured, 6014 independent reflections (Rint = 0.1114). The final R1 values were 0.0703 (I > 2σ(I)). The final wR(F2) values were 0.1482 (I > 2σ(I)). The final R1 values were 0.1559 (all data). The final wR(F2) values were 0.1885 (all data). The goodness of fit on F2 was 1.102. Flack parameter = 0.02(14).
:2, v/v), C40H60O5, M = 620.88, a = 10.6576(2) Å, b = 7.68660(10) Å, c = 21.0221(4) Å, α = 90°, β = 98.7830(10)°, γ = 90°, V = 1701.95(5) Å3, T = 293(2) K, space group P21, Z = 2, μ(Cu Kα) = 0.606 mm−1, 16771 reflections measured, 5769 independent reflections (Rint = 0.0412). The final R1 values were 0.0492 (I > 2σ(I)). The final wR(F2) values were 0.1406 (I > 2σ(I)). The final R1 values were 0.0495 (all data). The final wR(F2) values were 0.1412 (all data). The goodness of fit on F2 was 1.039. Flack parameter = 0.07(6).
:4, v/v), C40H54O8·CH4O·H2O, M = 712.89, a = 25.253(2) Å, b = 7.4272(6) Å, c = 20.3947(19) Å, α = 90°, β = 104.462(4)°, γ = 90°, V = 3704.0(6) Å3, T = 100.(2) K, space group C2, Z = 4, μ(Cu Kα) = 0.731 mm−1, 33150 reflections measured, 7223 independent reflections (Rint = 0.0765). The final R1 values were 0.0583 (I > 2σ(I)). The final wR(F2) values were 0.1591 (I > 2σ(I)). The final R1 values were 0.0639 (all data). The final wR(F2) values were 0.1665 (all data). The goodness of fit on F2 was 1.062. Flack parameter = 0.21(8).
:2, v/v), C40H54O7, M = 646.83, a = 11.8280(11) Å, b = 7.7673(8) Å, c = 18.926(2) Å, α = 90°, β = 93.109(6)°, γ = 90°, V = 1736.2(3) Å3, T = 100.(2) K, space group P21, Z = 2, μ(Cu Kα) = 0.663 mm−1, 23902 reflections measured, 6476 independent reflections (Rint = 0.2120). The final R1 values were 0.1050 (I > 2σ(I)). The final wR(F2) values were 0.2852 (I > 2σ(I)). The final R1 values were 0.1867 (all data). The final wR(F2) values were 0.3370 (all data). The goodness of fit on F2 was 1.096. Flack parameter = 0.2(3).
Synthesis of compound 4
Phthaloyl peroxide was prepared according to the literature.16a To a stirred solution of 5 (200.0 mg, 0.74 mmol, 1.00 equiv.) in HFIP (6.00 mL) was added phthaloyl peroxide (157.90 mg, 0.96 mmol, 1.30 equiv.) at 40 °C. After 24 hours, the reaction was cooled and the solvent was evaporated. Under positive N2 pressure, degassed MeOH (3.0 mL) and saturated aqueous NaHCO3 solution (0.2 mL) were added to the crude reaction mixture. Following complete consumption of the phthaloyl ester the reaction mixture was diluted with aqueous NaHCO3 solution and extracted with EtOAc. The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography on silica gel (pentane/acetone = 10:1) to afford 4 (137.6 mg, 0.48 mmol, 65%) as a pale yellow oil. 4: Rf = 0.70 (pentane); 1H NMR (600 MHz, CDCl3) δH: 6.83 (s, 1H), 6.63 (s, 1H), 4.53 (s, 1H), 3.11 (p, J = 6.9 Hz, 1H), 2.81 (dddd, J = 28.2, 17.0, 9.1, 6.2 Hz, 2H), 2.17 (dt, J = 13.2, 3.5 Hz, 1H), 1.85 (ddt, J = 12.1, 7.5, 2.3 Hz, 1H), 1.80–1.54 (m, 3H), 1.46 (dq, J = 13.4, 2.9 Hz, 1H), 1.42–1.29 (m, 2H), 1.23 (t, J = 6.8 Hz, 6H), 1.17 (s, 3H), 0.94 (s, 3H), 0.91 (s, 3H); 13C NMR (150 MHz, CDCl3) δC: 150.8, 148.8, 131.5, 127.4, 126.8, 111.1, 50.5, 41.8, 39.0, 37.7, 33.6, 33.5, 29.9, 26.9, 25.0, 22.9, 22.7, 21.8, 19.5, 19.4. All analytical data agree with literature values.15
General synthetic procedures for the preparation of compounds 11–20
To a solution of 4 (31.77 μmol, 1.00 equiv.) and ent-kauranoid (2.00 equiv.) in THF (320 μL, 0.10 M) at 0 °C was added SIBX (80 wt%, 1.30–2.00 equiv.) in one portion. The resulting suspension was stirred at 40 °C for 14 h and then was poured into ice-cold water (2.00 mL). This mixture was treated with aqueous 1.00 M NaOH, which was added dropwise until pH = 9.0. The resultant mixture was extracted with EtOAc three times. The combined organic phases were washed with water, dried over Na2SO4, and evaporated under vacuum. The residue was purified by silica column chromatography and then further purified by semi-preparative HPLC (3.00 mL min−1, detector UV λmax 230 nm, MeCN/H2O = 60:40) to afford the dimeric products.
Compound 11 (2.00 mg, 19%): white amorphous powder, Rf = 0.49 (silica, EtOAc–petroleum ether, 1:5); [α]26D = −6.3 (c = 0.100 in MeOH); 1H and 13C NMR data, see Tables S1 and S3;† HRESIMS (m/z): [M + NH4]+ calcd for C40H58O8N, 680.4157, found 680.4162.
Compound 12 (1.80 mg, 18%): colorless crystals, m.p. 276.0–281.0 °C; Rf = 0.49 (silica, EtOAc–petroleum ether, 1:5); [α]26D = −12.1 (c = 0.060 in MeOH); 1H and 13C NMR data, see Tables S1 and S3;† HRESIMS (m/z): [M + Na]+ calcd for C40H54O8Na, 685.3711, found 685.3716.
The 1H–1H COSY spectrum of 11 indicated the presence of two spin systems: H2-17/H-14′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-11′ and C-17/C-14′ bonds could be determined by the HMBC correlations from H-11′ to C-13, C-15, C-16, C-17, C-8′, C-9′, and C-12′ and H-14′ to C-16, C-7′, C-8′, C-9′, C-12′, C-13′, and C-15′ (Fig. S1†). Additionally, the ROESY correlations of H-11′/H-13, H-11′/H-14b, and H3-16′/H3-20′ indicated that the configurations of C-16 and C-13′ were R and R, respectively (Fig. S2†). Finally, the structure of 11 was established as shown.
The 1H–1H COSY spectrum of 12 indicated the presence of two spin systems: H2-17/H-11′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-14′ and C-17/C-11′ bonds could be determined by the HMBC correlations from H-11′ to C-16, C-8′, C-9′, C-12′, and C-13′; from H-14′ to C-13, C-15, C-16, C-17, C-8′, C-9′, and C-13′; and from H2-17 to C-12′ (Fig. S1†). Additionally, the ROESY correlations of H-14′/H2-14 and H3-20′/H3-16′(H3-17′) indicated that the configurations of C-16 and C-12′ were R and S, respectively (Fig. S2†). Finally, the structure of 12 was established as shown.
Compound 13 (2.00 mg, 17%): white amorphous powder, Rf = 0.51 (silica, EtOAc–petroleum ether, 1:5); [α]26D = −83.1 (c = 0.100 in MeOH); 1H and 13C NMR data, see Tables S1 and S3;† HRESIMS (m/z): [M + Na]+ calcd for C42H60O9Na, 731.4130, found 731.4127.
Compound 14 (1.80 mg, 19%): white amorphous powder, Rf = 0.51 (silica, EtOAc–petroleum ether, 1:5); [α]26D = +10.7 (c = 0.040 in MeOH); 1H and 13C NMR data, see Tables S1 and S3;† HRESIMS (m/z): [M + H]+ calcd for C42H61O9, 709.4310, found 709.4318.
The 1H–1H COSY spectrum of 13 indicated the presence of two spin systems: H2-17/H-14′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-11′ and C-17/C-14′ bonds could be determined by the HMBC correlations from H-11′ to C-13, C-16, C-8′, C-9′, C-12′, and C-13′ and from H-14′ to C-16, C-7′, C-8′, C-9′, C-12′, C-13′ and C-15′ (Fig. S1†). Additionally, the ROESY correlations of H-11′/H-14a and H3-16′/H-7′b indicated that the configurations of C-16 and C-12′ were R and R, respectively (Fig. S2†). Finally, the structure of 13 was established as shown.
The 1H–1H COSY spectrum of 14 indicated the presence of two spin systems: H2-17/H-14′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-11′ and C-17/C-14′ bonds could be determined by the HMBC correlations from H-11′ to C-15, C-16, C-17, C-8′, C-9′, C-12′, and C-13′ and from H-14′ to C-16, C-7′, C-8′, C-9′, C-12′, C-13′, and C-15′ (Fig. S1†). Additionally, the ROESY correlations of H-14a/H-11′ and H-15′/H-17a indicated that C-16 and C-13′ had R and S configurations, respectively (Fig. S2†). Finally, the structure of 14 was established as shown.
Compound 15 (2.00 mg, 18%): white amorphous powder, Rf = 0.51 (silica, EtOAc–petroleum ether, 1:5); [α]26D = −14.0 (c = 0.060 in MeOH); 1H and 13C NMR data, see Tables S2 and S3;† HRESIMS (m/z): [M + Na]+ calcd for C42H58O9Na, 729.3973, found 729.3982.
Compound 16 (2.30 mg, 20%): white amorphous powder, Rf = 0.51 (silica, EtOAc–petroleum ether, 1:5); [α]26D = −6.80 (c = 0.130 in MeOH); 1H and 13C NMR data, see Tables S2 and S3;† HRESIMS (m/z): [M + Na]+ calcd for C42H58O9Na, 729.3973, found 729.3972.
The 1H–1H COSY spectrum of 15 indicated the presence of two spin systems: H2-17/H-14′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-11′ and C-17/C-14′ bonds could be determined by the HMBC correlations from H-11′ to C-13, C-15, C-16, C-17, C-8′, C-9′, C-10′, C-12′, and C-13′ and from H-14′ to C-16, C-7′, C-8′, C-9′, C-12′, C-13′, and C-15′ (Fig. S1†). Additionally, the ROESY correlations of H-11′/H-14a and H3-16′(H3-17′)/H-7′b indicated that C-16 and C-13′ had R and R configurations, respectively (Fig. S2†). Finally, the structure of 15 was established as shown.
The 1H–1H COSY spectrum of 16 indicated the presence of two spin systems: H2-17/H-14′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-11′ and C-17/C-14′ bonds could be determined by the HMBC correlations from H-11′ to C-13, C-15, C-16, C-17, C-8′, C-9′, C-10′, C-12′, and C-13′ and from H-14′ to C-16, C-7′, C-8′, C-9′, C-12′, C-13′, and C-15′ (Fig. S1†). Additionally, the ROESY correlations of H-11′/H-14a and H3-16′(H3-17′)/H-17a(H-17b) indicated that C-16 and C-13′ had R and S configurations, respectively (Fig. S2†). Finally, the structure of 16 was established as shown.
Compound 17 (1.50 mg, 18%): white amorphous powder, Rf = 0.52 (silica, EtOAc–petroleum ether, 1:5); [α]24D = −2.17 (c = 0.060 in MeOH); 1H and 13C NMR data, see Tables S2 and S3;† HRESIMS (m/z): [M + Na]+ calcd for C40H60O4Na, 627.4384, found 627.4376.
The 1H–1H COSY spectrum of 17 indicated the presence of two spin systems: H2-17/H-14′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-11′ and C-17/C-14′ bonds could be determined by the HMBC correlations from H-11′ to C-13, C-16, C-17, C-8′, C-9′, C-10′, C-12′, and C-13′ and from H-14′ to C-16, C-7′, C-8′, C-9′, C-12′, and C-13′ (Fig. S1†). Additionally, the ROESY correlations of H-11′/H-14b, H3-16′/H-7′b, and H3-17′/H-7′a indicated that C-16 and C-13′ had R and R configurations, respectively (Fig. S2†). Finally, the structure of 17 was established as shown.
:40) to afford compounds 18 (4.00 mg, 18%), 19 (4.00 mg, 18%), and 20 (2.00 mg, 9%).
Compound 18: colorless crystals, m.p. 238.0–240.0 °C; Rf = 0.51 (silica, EtOAc–petroleum ether, 1:5); [α]26D = −58.2 (c = 0.075 in MeOH); 1H and 13C NMR data, see Tables S2 and S3;† HRESIMS (m/z): [M + H]+ calcd for C40H55O7, 647.3942, found 647.3944.
Compound 19: white amorphous powder, Rf = 0.51 (silica, EtOAc–petroleum ether, 1:5); [α]26D = −15.9 (c = 0.074 in CHCl3); 1H and 13C NMR data, see Tables S2 and S3;† HRESIMS (m/z): [M + H]+ calcd for C40H55O7, 647.3942, found 647.3948.
Compound 20: white amorphous powder, Rf = 0.51 (silica, EtOAc–petroleum ether, 1:5); [α]26D = −19.1 (c = 0.070 in MeOH); Tables S2 and S3;† HRESIMS (m/z): [M + Na]+ calcd for C40H54O7Na, 669.3762, found 669.3762.
The 1H–1H COSY spectrum of 18 indicated the presence of two spin systems: H2-17/H-14′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-11′ and C-17/C-14′ bonds could be determined by the HMBC correlations from H-11′ to C-13, C-16, C-17, C-8′, C-9′, C-10′, C-12′, and C-13′ and from H-14′ to C-16, C-7′, C-8′, C-9′, C-12′, and C-15′ (Fig. S1†). Additionally, the ROESY correlations of H-11′/H-14a and H3-16′/H-20′ indicated that C-16 and C-13′ had R and R configurations, respectively (Fig. S2†). Finally, the structure of 18 was established as shown.
The 1H–1H COSY spectrum of 19 indicated the presence of two spin systems: H2-17/H-14′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-11′ and C-17/C-14′ bonds could be determined by the HMBC correlations from H-11′ to C-13, C-15, C-16, C-17, C-9′, C-10′, and C-12′ and from H-14′ to C-16, C-7′, C-9′, C-12′, and C-15′ (Fig. S1†). Additionally, the ROESY correlations of H-11′/H-14a and H3-16′/H-17a indicated that C-16 and C-13′ had R and S configurations, respectively (Fig. S2†). Finally, the structure of 19 was established as shown.
The 1H–1H COSY spectrum of 20 indicated the presence of two spin systems: H2-17/H-11′ and H3-16′/H-15′/H3-17′ (Fig. S1†). The linkage of 4 and 6 through C-16/C-14′ and C-17/C-11′ bonds could be determined by the HMBC correlations from H-11′ to C-8′, C-9′, C-12′, and C-13′; from H-14′ to C-13, C-16, C-17, C-8′, C-9′, C-10′, and C-12′; and from H2-17 to C-12′ (Fig. S1†). Additionally, the ROESY correlations of H-14′/H-14a and H3-20′/H3-16′ indicated that C-16 and C-13′ had R and S configurations, respectively (Fig. S2†). Finally, the structure of 20 was established as shown.
Bioassay
According to the previously reported procedures,21 SMMC-7721 cells were treated with compounds or the vehicle for 24 h, and then the cell surface expression of MICA/B was detected by flow cytometry. The mean fluorescence intensity of MICA/B was normalized to the vehicle group, and the results are presented as the means of duplicate experiments ± SD. The results are shown in Table S4.†Author contributions
P.-T. P. designed the project; J.-M. D. performed the experiment of isolation; J.-M. D. and B.-C. Y. performed the experiment of synthesis; L.-M. K. completed the biological evaluation; X.-N. L. analyzed the single-crystal X-ray diffraction data; J.-M. D., B.-C. Y., L.-M. K., K. H. and X.-R. L. analyzed the experimental data; and J.-M. D. and B.-C. Y. wrote the initial draft. All the authors discussed the results and contributed to the preparation of the final paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Gang Xu for providing samples of ferruginol for biomimetic synthesis and the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany for bioassays. This project was financially supported by the National Science Fund for Distinguished Young Scholars (82325047), the Second Tibetan Plateau Scientific Expedition and Research Program (STEP) (2019QZKK0502), the NSFC-Joint Foundation of Yunnan Province (U2002221), the Major Projects for Fundamental Research of Yunnan Province (202201BC070002), the CAS “Light of West China” Program, the CAS Interdisciplinary Innovation Team (Pema-Tenzin Puno), the National Natural Science Foundation of China (22007089), the Natural Science Foundation of Yunnan Province (202001AT070074) and the CAS Youth Innovation Promotion Association (B.-C. Yan).References
- (a) D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed; (b) C. Schlesinger, L. Tapmeyer, S. D. Gumbert, D. Prill, M. Bolte, M. U. Schmidt and C. Saal, Absolute configuration of pharmaceutical research compounds determined by X-ray powder diffraction, Angew. Chem., Int. Ed., 2018, 57, 9150–9153 CrossRef CAS; (c) J. X. Zhao, Y. Y. Yu, S. S. Wang, S. L. Huang, Y. Shen, X. H. Gao, L. Sheng, J. Y. Li, Y. Leng, J. Li and J. M. Yue, Structural elucidation and bioinspired total syntheses of ascorbylated diterpenoid hongkonoids A–D, J. Am. Chem. Soc., 2018, 140, 2485–2492 CrossRef CAS PubMed.
- (a) Y. Chang, C. Sun, C. Wang, X. Huo, W. Zhao and X. Ma, Biogenetic and biomimetic synthesis of natural bisditerpenoids: hypothesis and practices, Nat. Prod. Rep., 2022, 39, 2030–2056 RSC; (b) L. G. Lin, C. O. L. Ung, Z. L. Feng, L. Huang and H. Hu, Naturally occurring diterpenoid dimers: source, biosynthesis, chemistry and bioactivities, Planta Med., 2016, 82, 1309–1328 CrossRef CAS PubMed; (c) M. Liu, W. G. Wang, H. D. Sun and J. X. Pu, Diterpenoids from Isodon species: an update, Nat. Prod. Rep., 2017, 34, 1090–1140 RSC.
- B. Huang, C. J. Xiao, Z. Y. Huang, X. Y. Tian, X. Cheng, X. Dong and B. Jiang, Hispidanins A–D: four new asymmetric dimeric diterpenoids from the rhizomes of Isodon hispida, Org. Lett., 2014, 16, 3552–3555 CrossRef CAS.
- (a) W. H. Lin, J. M. Fang and Y. S. Cheng, Diterpenes and related cycloadducts from Taiwania cryptomerioides, Phytochemistry, 1996, 42, 1657–1663 CrossRef CAS; (b) W. H. Lin, J. M. Fang and C. Y. S. Cheng, Cycloadducts of terpene quinones from Taiwania cryptomerioides, Phytochemistry, 1997, 46, 169–173 CrossRef CAS.
- W. L. Wang, D. R. Zhu, J. Luo, F. Y. Liu, C. Chen, T. Y. Zhu, L. N. Li, C. Han, J. G. Luo and L. Y. Kong, Taiwanoids A–D, four dimeric diterpenoids featuring tetracyclic [7.75, 9. 4.05, 10. 08, 9] octodecane from Taiwania cryptomerioidesim, Org. Biomol. Chem., 2018, 16, 9059–9063 RSC.
- (a) M. S. Sarwar, Y. X. Xia, Z. M. Liang, S. W. Tsang and H. J. Zhang, Mechanistic pathways and molecular targets of plant-derived anticancer ent-kaurane diterpenes, Biomolecules, 2020, 10, 144 CrossRef CAS PubMed; (b) Q. L. Xiao, F. Xia, X. W. Yang, Y. Liao, L. X. Yang, Y. K. Wei, X. Li and G. Xu, New dimeric and seco-abietane diterpenoids from Salvia wardii, Nat. Prod. Bioprospect., 2015, 5, 77–82 CrossRef CAS.
- (a) H. J. Zhang, Y. M. Zhang, J. G. Luo, J. Luo and L. Y. Kong, Anti-inflammatory diterpene dimers from the root barks of Aphanamixis grandifolia, Org. Biomol. Chem., 2015, 13, 7452–7458 RSC; (b) S. Aquila, Z. Y. Weng, Y. Q. Zeng, H. D. Sun and J. L. Rios, Inhibition of NF-κB activation and iNOS induction by ent-kaurane diterpenoids in LPS-stimulated RAW264.7 murine macrophages, J. Nat. Prod., 2009, 72, 1269–1272 CrossRef CAS PubMed.
- G. H. Tang, Y. Zhang, Y. C. Gu, S. F. Li, Y. T. Di, Y. H. Wang, C. X. Yang, G. Y. Zuo, S. L. Li, H. P. He and X. J. Hao, Trigoflavidols A–C, degraded diterpenoids with antimicrobial activity, from Trigonostemon flavidus, J. Nat. Prod., 2012, 75, 996–1000 CrossRef CAS.
- J. Liu, X. F. He, G. H. Wang, E. F. Merino, S. P. Yang, R. X. Zhu, L. S. Gan, H. Zhang, M. B. Cassera, H. Y. Wang, D. G. I. Kingston and J. M. Yue, Aphadilactones A–D, four diterpenoid dimers with DGAT inhibitory and antimalarial activities from a Meliaceae plant, J. Org. Chem., 2014, 79, 599–607 CrossRef CAS PubMed.
- S. X. Huang, W. L. Xiao, L. M. Li, S. H. Li, Y. Zhou, L. S. Ding, L. G. Lou and H. D. Sun, Bisrubescensins A–C: three new dimeric ent-kauranoids isolated from Isodon rubescens, Org. Lett., 2006, 8, 1157–1160 CrossRef CAS.
- J. H. Yang, W. G. Wang, X. Du, F. He, H. B. Zhang, X. N. Li, Y. Li, J. X. Pu and H. D. Sun, Heterodimeric ent-kauranoids from Isodon tenuifolius, J. Nat. Prod., 2014, 77, 2444–2453 CrossRef CAS.
- L. H. Huang, Q. Fu, J. M. Dai, B. C. Yan, D. W. Wang, P. T. Puno and J. B. Yue, High-content screening of diterpenoids from Isodon, species as autophagy modulators and the functional study of their antiviral activities, Cell Biol. Toxicol., 2021, 37, 695–713 CrossRef CAS PubMed.
- (a) A. J. Hou, M. L. Li, B. Jiang, Z. W. Lin, S. Y. Ji, Y. P. Zhou and H. D. Sun, New 7,20: 14,20-diepoxy ent-kauranoids from Isodon xerophilus, J. Nat. Prod., 2000, 63, 599–601 CrossRef CAS; (b) A. J. Hou, H. Yang, B. Jiang, Q. S. Zhao, Y. Z. Liu, Z. W. Lin and H. D. Sun, Two new ent-kaurane diterpenoids from Isodon xerophilus, Chin. Chem. Lett., 2000, 11, 795–798 CAS; (c) A. J. Hou, H. Yang, Y. Z. Liu, Q. S. Zhao, Z. W. Lin and H. D. Sun, Novel ent-kaurane diterpenoids from Isodon xerophilus, Chin. J. Chem., 2001, 19, 365–370 CrossRef CAS; (d) A. J. Hou, Q. S. Zhao, M. L. Li, B. Jiang, Z. W. Lin, H. D. Sun, Y. P. Zhou, Y. Lu and Q. T. Zheng, Cytotoxic 7,20-epoxy ent-kauranoids from Isodon xerophilus, Phytochemistry, 2001, 58, 179–183 CrossRef CAS PubMed; (e) S. H. Li, X. M. Niu, L. Y. Peng, H. J. Zhang, P. Yao and H. D. Sun, ent-Kaurane diterpenoids from the Leaves of Isodon xerophilus, Planta Med., 2002, 68, 946–948 CrossRef CAS; (f) X. M. Niu, S. H. Li, Z. Na, Z. W. Lin and H. D. Sun, Two novel ent-abietane diterpenoids from Isodon xerophilus, Helv. Chim. Acta, 2004, 87, 1951–1957 CrossRef CAS; (g) L. M. Li, Z. Y. Weng, S. X. Huang, J. X. Pu, S. H. Li, H. Huang, B. B. Yang, Y. Han, W. L. Xiao, M. L. Li, Q. B. Han and H. D. Sun, Cytotoxic ent-kauranoids from the medicinal plant Isodon xerophilus, J. Nat. Prod., 2007, 70, 1295–1301 CrossRef CAS PubMed; (h) L. M. Li, J. X. Pu, W. L. Xiao and H. D. Sun, ent-Abietane diterpenoids from Isodon xerophilus, Arch. Pharmacal Res., 2011, 34, 875–879 CrossRef CAS; (i) D. D. Luo, Y. J. Yi, K. Peng, T. R. Liu, J. Y. Yang, S. Liu, W. Z. Zhao, X. J. Qu, W. G. Yu, Y. C. Gu and S. B. Wan, Oridonin derivatives as potential anticancer drug candidates triggering apoptosis through mitochondrial pathway in the liver cancer cells, Eur. J. Med. Chem., 2019, 178, 365–379 CrossRef CAS PubMed; (j) J. M. Dai, K. Hu, B. C. Yan, X. R. Li, X. N. Li, H. D. Sun and P. T. Puno, ent-Kaurane-based diterpenoids, dimers, and meroditerpenoids from Isodon xerophilus, J. Nat. Prod., 2020, 83, 3717–3725 CrossRef CAS PubMed.
- (a) B. Zhou, Y. H. Ren, Y. S. Han, T. Mesplède and J. M. Yue, Diverse types of diterpenoids with an aromatized C ring from the twigs of Podocarpus imbricatus, J. Nat. Prod., 2020, 83, 2416–2424 CrossRef CAS; (b) Y. Fu, X. Ding, X. Zhang, X. Shao, J. Zhao, Y. Xu, X. Luo and W. Zhao, Diterpenoids from the root bark of Pinus massoniana and evaluation of their phosphodiesterase type 4D inhibitory activity, J. Nat. Prod., 2020, 83, 1229–1237 CrossRef CAS; (c) L. Mangoni and R. Caputo, Sempervirol, a novel type of diterpene phenol, Tetrahedron Lett., 1967, 8, 673–675 CrossRef.
- (a) M. Tada and K. Ishimaru, Efficient ortho-oxidation of phenol and synthesis of anti-MRSA and anti-VRE compound abietaquinone methide from dehydroabietic acid, Chem. Pharm. Bull., 2006, 54, 1412–1417 CrossRef CAS PubMed; (b) P. A. Peixoto, M. El Assal, I. Chataigner, F. Castet, A. Cornu, R. Coffinier, C. Bosset, D. Deffieux, L. Pouységu and S. Quideau, Bispericyclic Diels–Alder dimerization of ortho-quinols in natural product (bio)synthesis: bioinspired chemical 6-step synthesis of (+)-maytenone, Angew. Chem., Int. Ed., 2021, 60, 14967–14974 CrossRef CAS; (c) C. Thommen, C. K. Jana, M. Neuburger and K. Gademann, Syntheses of taiwaniaquinone F and taiwaniaquinol A via an unusual remote C–H functionalization, Org. Lett., 2013, 15, 1390–1393 CrossRef CAS PubMed.
- (a) C. Yuan, Y. Liang, T. Hernandez, A. Berriochoa, K. N. Houk and D. Siegel, Metal-free oxidation of aromatic carbon–hydrogen bonds through a reverse-rebound mechanism, Nature, 2013, 499, 192–196 CrossRef CAS PubMed; (b) Z. Qiu and C. J. Li, Transformations of less-activated phenols and phenol derivatives via C–O cleavage, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, New reactions of IBX: oxidation of nitrogen- and sulfur-containing substrates to afford useful synthetic intermediates, Angew. Chem., Int. Ed., 2003, 42, 4077–4082 CrossRef CAS; (b) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, o-Iodoxybenzoic acid (IBX) as a viable reagent in the manipulation of nitrogen- and sulfur-containing substrates: scope, generality, and mechanism of IBX-mediated amine oxidations and dithiane deprotections, J. Am. Chem. Soc., 2004, 126, 5192–5201 CrossRef CAS; (c) C. Zhao, H. Zheng, P. Jing, B. Fang, X. Xie and X. She, Tandem oxidative dearomatization/intramolecular Diels–Alder reaction for construction of the tricyclic core of palhinine A, Org. Lett., 2012, 14, 2293–2295 CrossRef CAS.
- C. Li, B. Ge, M. Nicotra, J. N. H. Stern, H. D. Kopcow, X. Chen and J. L. Strominger, JNK MAP kinase activation is required for MTOC and granule polarization in NKG2D-mediated NK cell cytotoxicity, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3017–3022 CrossRef CAS PubMed.
- D. S. Bae, Y. K. Hwang and J. K. Lee, Importance of NKG2D-NKG2D ligands interaction for cytolytic activity of natural killer cell, Cell. Immunol., 2012, 276, 122–127 CrossRef CAS.
- N. Nausch and A. Cerwenka, NKG2D ligands in tumor immunity, Oncogene, 2008, 27, 5944–5958 CrossRef CAS.
- (a) H. Zhu, B. Wang, L. Kong, T. An, G. Li, H. Zhou, L. Gong, Z. Zhao, Y. Gong, H. Sun, P. Puno and Y. Li, Parvifoline AA promotes susceptibility of hepatocarcinoma to natural killer cell-mediated cytolysis by targeting peroxiredoxin, Cell Chem. Biol., 2019, 26, 1122–1132 CrossRef CAS PubMed; (b) Y. Zhu, Z. Zhao, M. Xue, D. Wang, G. Su, X. Ju, Q. Yang, S. Zhang, D. Fan, H. Zhu, M. Yu, Y. Li, L. Kong and H. Zhou, Ciclopirox olamine sensitizes leukemia cells to natural killer cell-mediated cytolysis by upregulating NKG2DLs via the Akt signaling pathway, Biochem. Biophys. Res. Commun., 2003, 659, 10–19 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2059945, 2059944, 2129366 and 2129362. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01679j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |