 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of alkali-metal cation on oxygen adsorption at Pt single-crystal electrodes in non-aqueous electrolytes
Julia
Fernández-Vidal†
 a,
Laurence J.
Hardwick
a,
Gema
Cabello‡
a and
Gary A.
Attard
*b
a,
Laurence J.
Hardwick
a,
Gema
Cabello‡
a and
Gary A.
Attard
*b
aStephenson Institute for Renewable Energy, Department of Chemistry, University of Liverpool, Peach Street, L69 7ZF Liverpool, UK
bDepartment of Physics, University of Liverpool, Crown Street, L69 7ZD Liverpool, UK. E-mail: attard@liverpool.ac.uk
First published on 11th July 2023
Abstract
The effect of Group 1 alkali-metal cations (Na+, K+, and Cs+) on the oxygen reduction and evolution reactions (ORR and OER) using dimethyl sulfoxide (DMSO)-based electrolytes was investigated. Cyclic voltammetry (CV) utilising different Pt-electrode surfaces (polycrystalline Pt, Pt(111) and Pt(100)) was undertaken to investigate the influence of surface structure upon the ORR and OER. For K+ and Cs+, negligible variation in the CV response (in contrast to Na+) was observed using Pt(111), Pt(100) and Pt(poly) electrodes, consistent with a weak surface-metal/superoxide complex interaction. Indeed, changes in the half-wave potentials (E1/2) and relative intensities of the redox peaks corresponding to superoxy (O2−) and peroxy (O22−) ion formation were consistent with a solution-mediated mechanism for larger cations, such as Cs+. Support for this finding was obtained via in situ shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS). During the ORR and in the presence of Cs+, O2− and weakly adsorbed caesium superoxide (CsO2) species were detected. Because DMSO was found to strongly interact with the surface at potentials associated with the ORR, CsO2 was readily displaced at more negative potentials via increased solvent adsorption at the surface. This finding highlights the important impact of the solvent during ORR/OER reactions.
Introduction
Lithium–oxygen (Li–O2) batteries have become of particular interest due to their high theoretical specific energy (3505 W h kg−1 for the non-aqueous system) compared to the commonly used Li-ion batteries.1 Studies of Li–O2 systems in non-aqueous electrolytes have shown that on Au, Pt, and C surfaces, lithium peroxide (Li2O2) is the main product of the oxygen reduction reaction (ORR).2–5 The precipitation of solid Li2O2 during the ORR passivates the electrode surface and blocks the electrode active sites, reducing any catalytic effect of the surface, which leads to the occurrence of parasitic reactions associated with the generation of singlet oxygen.2,6–8In 2013, Hartmann et al.9 and Ren et al.10 demonstrated that the irreversibility presented by the two-electron reduction of O2 to Li2O2 could be improved by the formation of an oxygen/superoxide redox couple that enables a single-electron reversible oxygen reduction reaction/oxygen evolution reaction (ORR/OER) process with high specific energy (wth = 1605 W h kg−1 for Na2O2)9 and avoids the formation of solid peroxide species that passivate the electrode surface.
In non-aqueous Li+-containing electrolytes, control of the product stability and reduced oxygen species11,12 through electrolyte solvation can be achieved by using solvents with increased Gutmann donor numbers.13,14 High Gutmann-donor-number solvents allow the formation of Li+/O2− ion pairs that react within the double layer, offering a preferred solution-based mechanism that avoids the passivation of the electrode and enhances the sluggish kinetics of the ORR.15 The reversible formation of superoxide (O2−) and peroxide (O22−) species has been detected on charge/discharge as the main products, together with some minor degradation observed over time for Li systems in dimethyl sulfoxide (DMSO).16,17 DMSO presents a relatively high Gutmann donor number of 29.8 kcal mol−1,18,19 based on the nucleophilic properties of the solvent and high stability.20
The size of the cations has been demonstrated to affect the ORR activity.27–32 In aqueous systems, the cation hydration energy and non-covalent interactions between hydrated alkali-metal cations and OHads have been highlighted in this regard.21–23 In Li-containing aprotic electrolytes, the ORR occurs almost exclusively with Li2O2 as the reaction product.3,4 Conversely, as one moves down the Group 1 metals, the reaction mechanism favours the formation of significant amounts of both O22− and O2− species.24,25 Indeed, within aprotic electrolytes, the ORR pathway has also been proved to depend on the cation size.9,10,26 Studies of the Group 1 alkali metals have mostly focused on the smaller cations with investigations being predominantly confined to Li+ and Na+.24,27–31 K–O2 batteries32–34 have attracted recent interest due to the reduced overpotential reported in the absence of catalysts and redox mediators compared to Li–O2 batteries.10 Nevertheless, despite the ongoing investigations into the ORR, observation of mechanistic details at the interface for different cations remains elusive.35–37
Pt has been demonstrated to be one of the most efficient catalysts for the ORR in both aqueous and aprotic electrolytes on model systems.38–40 In the past decade, in situ shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) has proven to be a powerful method to detect surface intermediates.41,42 Specifically, in situ SHINERS studies on Pt single-crystal electrodes have been successfully used to investigate the adsorption of ORR intermediates in aqueous electrolytes43–45 and also the Na–O2 non-aqueous system.4,24 However, despite the success of this approach, there is still a paucity of combined single-crystal/in situ SHINERS investigations involving non-aqueous electrolytes to afford a fundamental molecular level insight into the ORR processes associated with metal–oxygen battery systems, probably due to the experimental challenges that such studies present. Hence, in this work, we perform a qualitative study on the effect that the cation size has on the ORR and OER using such an approach. From SHINERS measurements, the key role that DMSO solvent molecules play in the displacement of adsorbed Cs-cation–superoxide moieties, formed in the initial oxygen reduction step, is demonstrated. Together with cyclic voltammetry (CV) data, showing negligible surface structural sensitivity towards reduction processes involving caesium and oxygen, a strong contrast with the analogous sodium-cation/oxygen redox system is revealed.
Experimental
Chemicals
Dimethyl sulfoxide (DMSO) (>99.9% Hi-Dry®, ROMIL), dried and purified by distillation, was stored inside a glovebox over molecular sieves to ensure an H2O content <4 ppm. Alkali-metal salts NaClO4 (≥98.0%), KClO4 (≥99.99%), and CsClO4 (≥99.995%), from Sigma-Aldrich, were dried under vacuum at 80 °C overnight and stored inside a glovebox prior to use. Up to N5-purity Ar (≥99.998%, BOC) and O2 (≥99.999%, BOC) gases, dried by passing through a desiccant (P2O5)-filled drying tube, were used for purging and bubbling the electrolytes, respectively, whilst not contaminating the glovebox atmosphere.Pt single-crystal preparation
Clavilier46 half-bead Pt(111) and (100) single crystals were annealed with a butane flame and then cooled in a bubbler containing Milli-Q® water and a steady flow of 5% H2 in Ar to produce a well-ordered surface, and subsequently quenched with a drop of Milli-Q® water (18.2 MΩ cm resistivity). The single crystal was then dipped in 1 mM NaBr (≥99.0%, Sigma-Aldrich) solution, to protect the surface via the formation of an irreversibly adsorbed bromine adlayer (Fig. 1).47 The electrode was then dried under vacuum, transferred to a glovebox, and cycled 5–10 times to achieve a clean electrode surface, free of Br adatoms on the surface.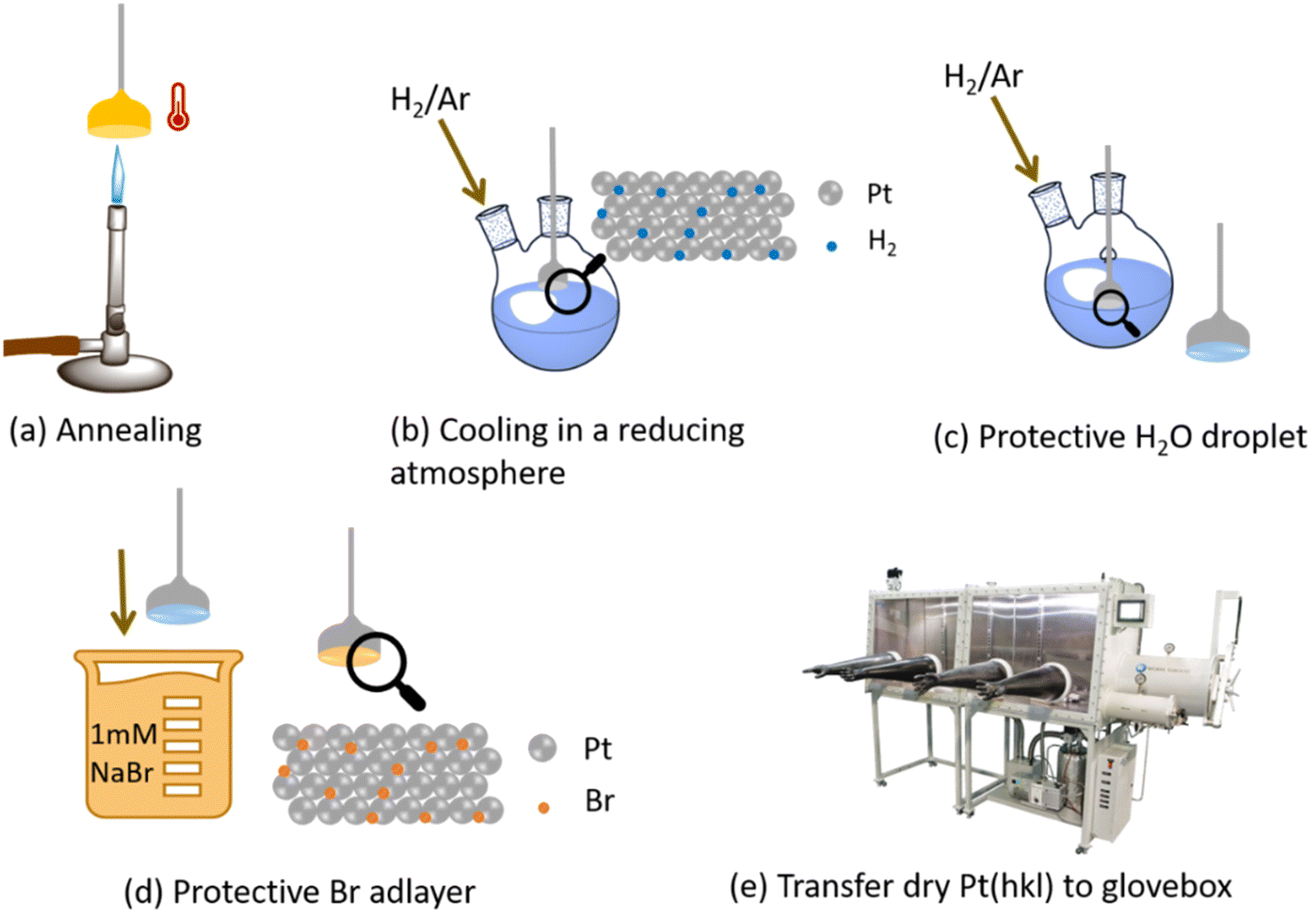 | ||
| Fig. 1 Schematic description of single-crystal preparation for non-aqueous electrochemical studies. Reproduced from Galloway et al.24 with permission from the Royal Society of Chemistry. | ||
Polycrystalline Pt preparation
Pt polycrystalline disk electrodes embedded in polyether ether ketone (PEEK) were polished using three different suspensions of decreasing-grain-size alumina (0.1 μm, 0.3 μm and 0.05 μm), then rinsed with ethanol and Milli-Q® water, and finally sonicated in water. The Pt(poly) electrodes were dried at 80 °C overnight under vacuum and then introduced into a glovebox (argon atmosphere; O2, H2O <0.1 ppm).Meniscus configuration
Pt half-bead single crystals were used under a meniscus configuration to ensure that the voltammetric profile corresponded exclusively to the well-oriented face of the electrode. The meniscus configuration avoids voltammetric features originating from the poly-oriented sides of the electrode, as depicted in Fig. 2a. To address the role of the meniscus48 in O2 diffusion during the ORR/OER, Pt(poly) electrodes have also been used in the traditional configuration, inserted flat in the electrolyte, as represented in Fig. 2b.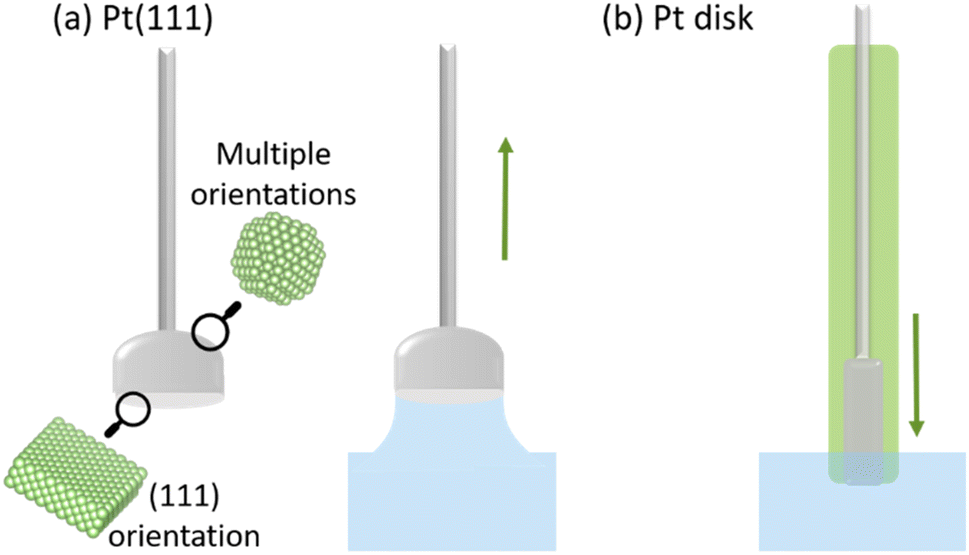 | ||
| Fig. 2 (a) Schematic of a Pt single-crystal electrode with a (111) facet on the flat side under meniscus configuration. Hard-sphere model made with VESTA software.49 (b) Schematic of a Pt polycrystalline disk electrode in contact with the electrolyte. | ||
Electrochemistry
Electrochemical experiments were conducted using a BioLogic (VSP-100) potentiostat in an Ar glovebox with moisture levels <1 ppm. Cyclic voltammetry experiments in the present study were carried out in a standard 3-electrode electrochemical cell. A silver wire was used as a pseudo-reference electrode. The Ag/Ag+ couple was calibrated with the ferrocene/ferrocenium redox couple (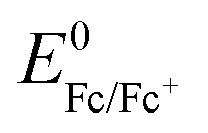 = +680 mV vs. SHE in DMSO).50 All potentials are displayed vs. the calibrated Ag/Ag+ redox couple to allow accurate comparison between experiments.
= +680 mV vs. SHE in DMSO).50 All potentials are displayed vs. the calibrated Ag/Ag+ redox couple to allow accurate comparison between experiments.
Synthesis of shell-isolated nanoparticles for enhanced Raman spectroscopy
The synthesis of the Au core was carried out using the Turkevich–Frens citrate-reduction method.51–53 200 mL of a solution containing 0.01% HAuCl4·nH2O (99.995%, Sigma-Aldrich) were heated under vigorous stirring until boiling. 1.5 mL of a freshly prepared 1% Na3C6H5O7·2H2O (≥99.5%, Sigma-Aldrich) solution were immediately added to achieve a nanoparticle size of approximately ∼55 nm. The protocol developed by Liz-Marzán and co-workers54,55 and used by Li et al.56 for SHINERS was followed for SiO2 shell synthesis. Suitability of the silica shells for SHINERS was checked via the detection or absence of a Raman-active molecule (pyridine) on Au and Si substrates, respectively.42In situ Raman measurements
Raman measurements were carried out with a Raman spectrometer (Renishaw inVia) with an inverted microscope via a 50× objective (Leica), using a 633 nm laser with an exposure time of 10 s and 5 acquisitions. Pt wire was used in the spectro-electrochemical cell as a counter electrode and Ag wire as a reference. Long sealing screw caps were used to both isolate the cell and avoid any contaminants from dissolving into the electrolyte.Results and discussion
Pt(111) cyclic voltammetry in 0.1 M NaClO4 in DMSO electrolyte is displayed in Fig. 3. All the voltammetric features observed correspond exclusively to the adsorption of oxygen species onto the Pt electrode surface, as can be gleaned by the lack of faradaic current in the Ar-purged electrolyte.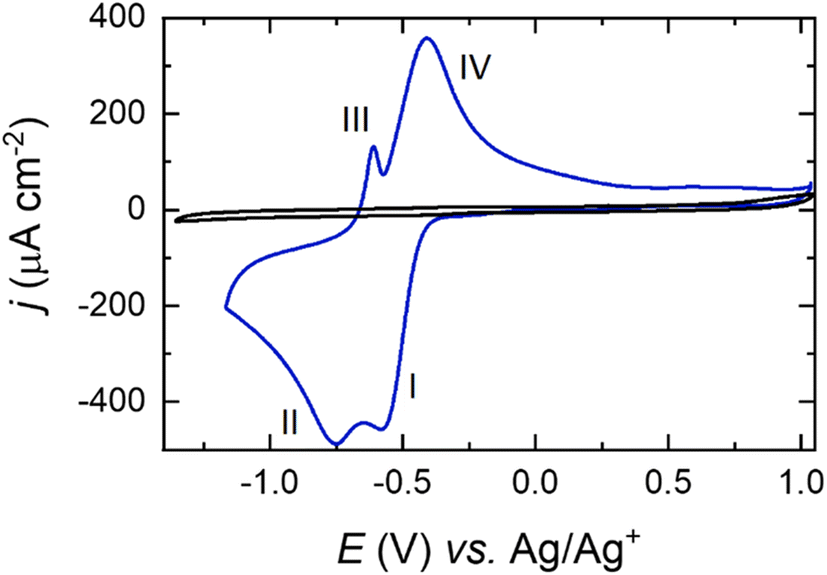 | ||
| Fig. 3 Cyclic voltammograms of a Pt(111) electrode in a solution containing 0.1 M NaClO4 in DMSO, for Ar- (black) and O2- (blue) saturated solutions. Scan rate: 50 mV s−1 at 23 °C. | ||
The Na–O2 system on Pt basal planes has been previously studied in detail and the reaction mechanisms have been identified through in situ Raman microscopy.24 The electrochemical behaviour of different single-crystal facets demonstrated that both O2− and O22− species are formed during the ORR in the presence of Na+. In agreement with the literature,11 for Pt(111) the peak at −0.55 V (I) observed in the reduction sweep in Fig. 3 corresponds to the reduction of O2via formation of metal–O2−, according to:
| O2 + Na+ + e− → NaO2 | (1) |
The second reduction peak at around −0.75 V (II) corresponds to a second electron reduction that forms the metal–O22−:57
| NaO2 + Na+ + e− → Na2O2 | (2) |
In the oxidation sweep, two peaks at −0.60 V (III) and −0.40 V (IV) are observed, which constitute the OER:
| Na2O2 → NaO2 + Na+ + e− | (3) |
| NaO2 → O2 + Na+ + e− | (4) |
As with Na+, for K+ and Cs+, the ORR proceeds via two quasi-reversible electrochemical processes with two main reduction steps involving the formation of O2− and O22− species (Fig. 4).57–61
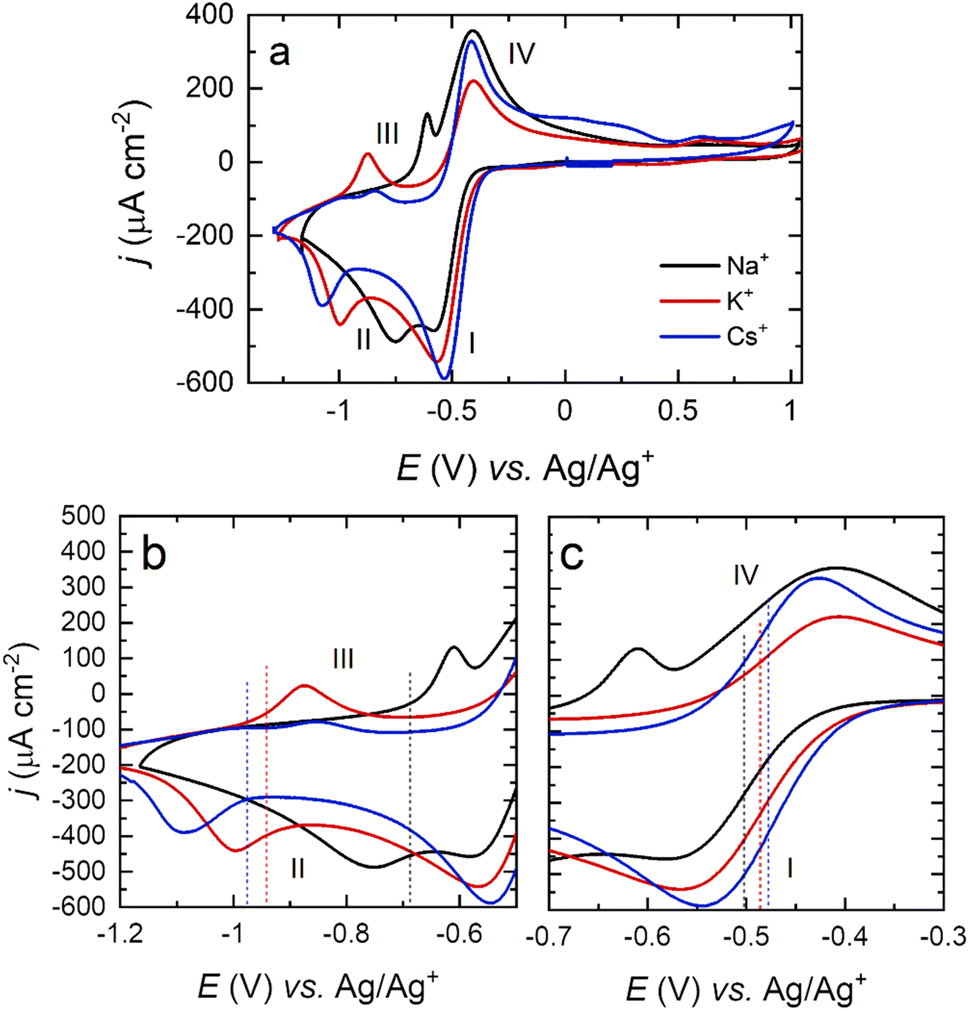 | ||
| Fig. 4 (a) Cyclic voltammograms of a Pt(111) electrode in an O2-saturated DMSO solution containing 0.1 M NaClO4 (black), KClO4 (red) and CsClO4 (blue). Scan rate: 50 mV s−1. Expanded view of the peaks relating to the formation of O22− (b) and O2− (c). Dotted lines represent the calculated E1/2 of the ORR half reactions for Na+ (black), K+ (red) and Cs+ (blue). | ||
The half-wave potential (E1/2) corresponding to the ion-pair formation between the alkali metal and O2− shifts towards more negative potentials for smaller cations (Na+ > K+ > Cs+ – Table 1), while the E1/2 corresponding to the formation of O22− follows the opposite trend (Table 2).
| Group 1 metal ion | Ionic radii (Å) |
|---|---|
| Na+ | 1.02 |
| K+ | 1.38 |
| Cs+ | 1.67 |
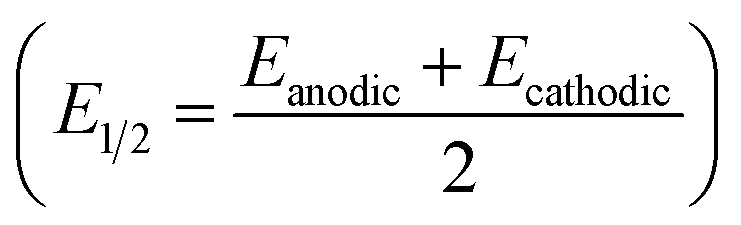 presented in Fig. 4 for Pt(111) and Fig. 5 for Pt(poly)
presented in Fig. 4 for Pt(111) and Fig. 5 for Pt(poly)
| NaO2 | KO2 | CsO2 | Na2O2 | K2O2 | Cs2O2 | |
|---|---|---|---|---|---|---|
| Pt(111) | −0.50 V | −0.48 V | −0.47 V | −0.69 V | −0.93 V | −0.97 V |
| Pt(poly) | −0.53 V | −0.52 V | −0.50 V | −0.71 V | −0.90 V | −0.93 V |
With a Na+-containing electrolyte, Galloway et al.24 studied the adsorption of species using different Pt(hkl) surfaces and demonstrated the surface specificity of Na+-ORR on Pt electrodes. In contrast, in the presence of cations larger than Cs+, such as tetraalkylammonium (TAA+) (ionic radius of 3.22 Å for tetramethylammonium (TMA+)),63,64 the ORR is a highly reversible (less negative E1/2) one-electron process that involves the O2/O2− couple,65–67 according to:
| O2 + TAA+ + e− → TAA+⋯O2− | (5) |
Under steady-state conditions, Welford et al.68 reported that the formation of O2− is limited by the diffusion of O2 through the three-phase boundary O2|electrode|electrolyte through the hanging meniscus in TAA+-containing electrolytes with Au(111). These results suggest that the O2− species formation occurs exclusively via a solution-preferred mechanism limited by the diffusion of O2, i.e., with low surface specificity compared to Na+.
In this paper, we suggest that the differences in the voltammetric response among alkali cations (Fig. 4) are due to a surface-mediated ORR for smaller cations, while larger cations prefer a solution-based mechanism dominated by the formation of O2−. When O2 is reduced to O2−, larger cations stabilise O2− through ion-pair formation (ionic bonding), which allows a less negative E1/2 for CsO2 formation than for NaO2.11,69 The polarisability of the ionic bond decreases (the bond becomes more covalent in nature) for smaller alkali metals,26 and therefore, such entities present increased peak separation in the voltammetric response and a more negative E1/2. The fact that surface-adsorbed NaO2 formed at the surface may also block metal active sites, thus inhibiting further ORR, may also play a part in the increase in ORR overpotential observed for Na+ relative to Cs+ in Fig. 4.
Differences in the peak current intensities among alkali metals are also observed in Fig. 4. The ratio of current between the O2− (I) and the O22− (II) peaks is close to 1 for Na+ and decreases for larger cations in the order Na+ > K+ > Cs+. This asymmetry could be related to the lifetime of the formed O2− complex. O2− in the presence of large cations could readily be solvated back into solution, which would reduce the number of species on the surface to undergo the second reduction to O22− (II), which in turn results in lower currents and a more negative potential for the onset of the second reduction to O22−. In addition, the separation of peaks I and II may also be affected. Upon the formation of strongly adsorbed NaO2 at the surface of Pt(111), quantitative and rapid activation towards the formation of Na2O2via Pt surface catalysis might be taking place. This would result in two effects in agreement with the data presented: (i) the intensity ratio of peak I to peak II being close to unity and (ii) the difference in potential between peaks I and II reflecting the extent of surface interaction of the adsorbed MO2 complex (smallest peak separation for Na+). Additionally, the intensity of the peak related to the oxidation of O22− (III) also decays for larger cations (Na+ > K+ > Cs+). In this sense, larger cations complexing with O22− seem to have a more labile/dissociating nature compared to smaller cations, which would explain the lower intensity for the Cs2O2 electrooxidation (III) (upon formation, the complex decays or diffuses away prior to the positive-going sweep, leading to a lower intensity).
In order to further study the surface-dependence of the ORR among different alkali cations without the possibility of O2 diffusion influencing the results, cyclic voltammetry in O2-saturated DMSO containing Na+, K+, and Cs+ was recorded, using a Pt polycrystalline disk electrode immersed in the electrolyte (no meniscus, Fig. 5).
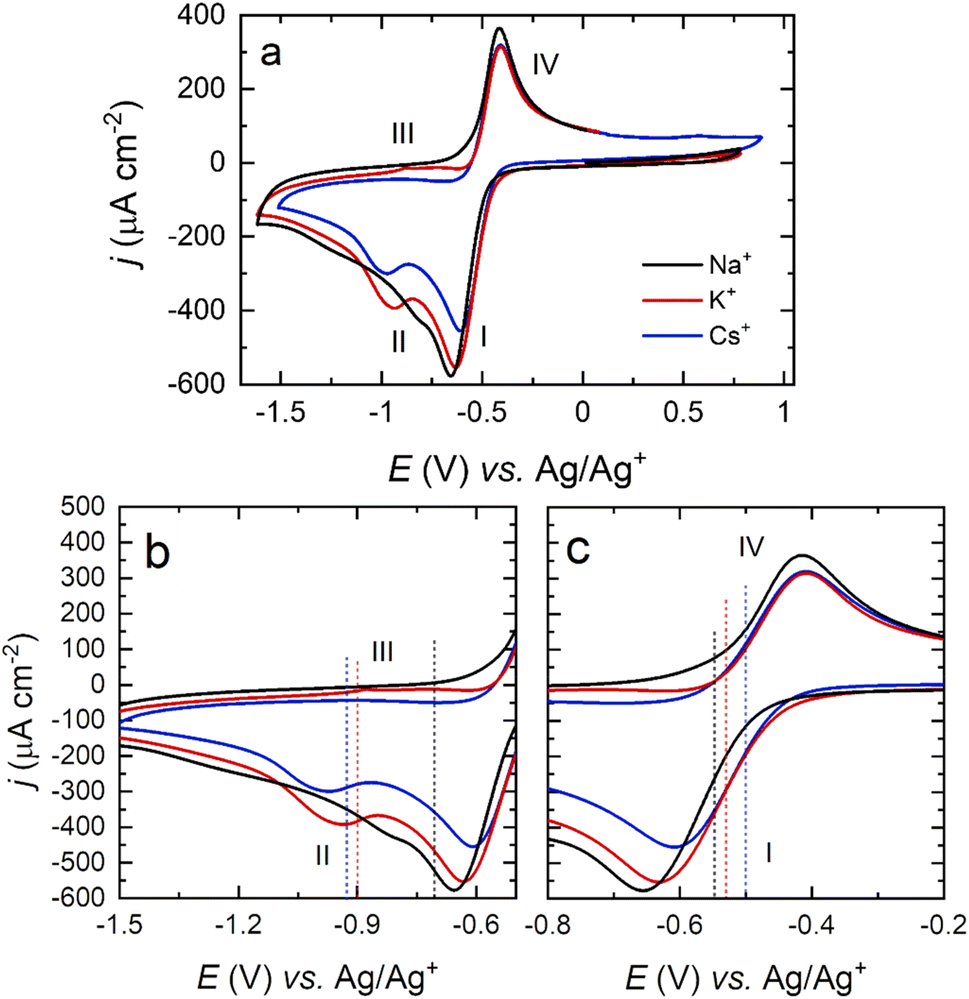 | ||
| Fig. 5 (a) Cyclic voltammograms of a Pt(poly) electrode in an O2-saturated DMSO solution containing 0.1 M NaClO4 (black), KClO4 (red) and CsClO4 (blue), at 50 mV s−1 scan rate. Expanded view of the potential range corresponding to the formation of O22− (b) and O2− (c). Dotted lines represent the E1/2 of the ORR half reactions for Na+ (black), K+ (red) and Cs+ (blue). | ||
The trends in the position of E1/2 for the first and second reduction of O2 as a function of metal cation are comparable both for Pt(111) and Pt(poly) electrodes (Table 2), with a small variation of ca. 30–40 mV associated with differences in surface structure.
While the current density ratio between the O2− (I) and O22− (II) peaks is equivalent between Pt(111) and Pt(poly)using larger cations, i.e., K+ and Cs+, the ratio in Na+-containing DMSO differs, with a ratio close to 1 for Pt(111) and a ratio >1 for Pt(poly). Other previous studies have highlighted that both the surface and electrolyte have an impact on the Na+-ORR mechanism.30 In DMSO, Pt(110) and (111) facets support the formation of Na2O2 as the main reduction product on the surface, while on Pt(100) and polycrystalline surfaces, the formation of NaO2 is less favoured and desorption and solvation is reported,4,13 attributed to a more limited surface interaction of oxygen with electrode surfaces that do not contain {111} and {110} terrace sites.24 This finding is consistent with the lower intensity of the redox peak associated with the formation of Na2O2 on Pt(poly) surfaces compared with Pt(111) and Pt(110). The NaO2 surface complex in this case is less strongly interacting with the surface, so the activation of the second reduction step by the Pt(100) surface is much less marked, resulting in a lower intensity for peak II.
The difference in the relative current densities of the M+–O22− complex on the forward and reverse sweeps shows the same trend as for Pt(111) (Cs+ > K+ > Na+), consistent with the dissociation/lability of the O22− complex for larger cations, which suggests that Cs+-ORR chemistry would be mostly dominated by solution-phase processes rather than being surface controlled.
While careful control of the crystal facet influences the reaction pathway, this is highly dependent on the cation of choice, as can be observed in Fig. 6. Shifts in the peak potential between the (111) and (100) facets for Na+–O2 OER/ORR are significant, whilst being negligible for Cs+, differing by as much as ±0.2 V for the first reduction and ±0.3 V for the second reduction when Na+ cations are present. These results again suggest that the surface structure has less effect on the ORR for larger alkali metals. The relative strength of adsorption for the peroxy- and superoxy-ion pairs would govern this behaviour, with those moieties formed by Cs+ cations giving rise to the weakest surface interactions. This assertion is borne out during spectroscopic measurements, to be outlined below.
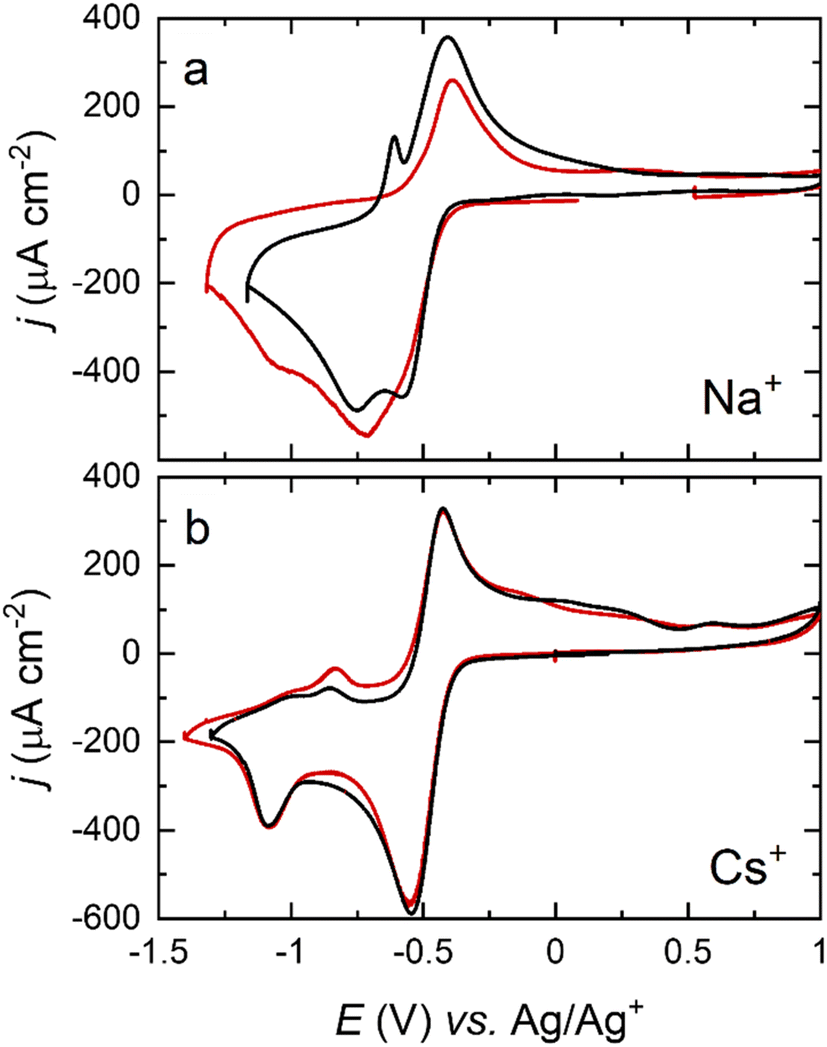 | ||
| Fig. 6 Cyclic voltammograms of a Pt(111) (black) and a Pt(100) (red) electrode in an O2-saturated DMSO solution containing (a) 0.1 M NaClO4 and (b) 0.1 M CsClO4. Scan rate: 50 mV s−1. | ||
The change in nature for the ionic bond26 between alkali cations and the O2− directly depends on the cation size, and increased polarisability of the ionic bond for larger alkali metals may affect the stability of superoxide complexes. To investigate the stability of O2− as a function of potential in the presence of a larger cation, in situ SHINERS spectra were collected with a Pt(111) electrode in 0.1 M CsClO4/DMSO (Fig. 7a). The intensity of the band centred at 1160 cm−1, corresponding to ν(O–O) in O2−, can only be observed at −0.55 V, which correlates with the onset potential for the first reduction of O2 to O2−. The band fades at more negative potentials, associated with the desorption of O2− from the surface of the electrode. These results differ from those previously reported for Na+ in DMSO, where the band associated with ν(O–O) can be observed even up to −1.0 V (at 1154 cm−1).24
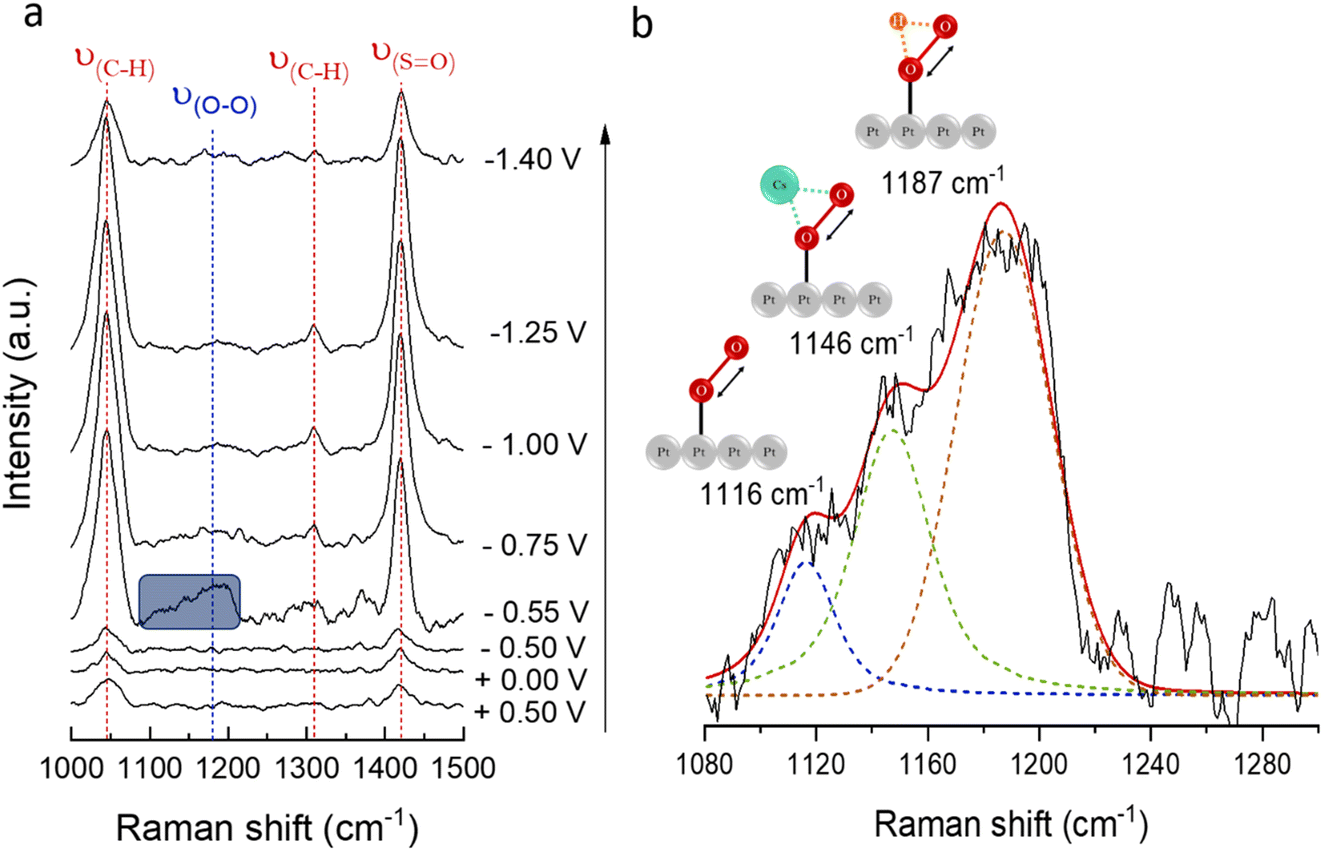 | ||
| Fig. 7 (a) Potential-dependent in situ SHINERS spectra obtained from a Pt(111) electrode in a solution containing 0.1 M CsClO4/DMSO, purged with Ar and, subsequently, saturated with O2. (b) Voigt model fitting of ν(O–O) at −0.55 V. | ||
The deconvolution of the band centred at 1160 cm−1 is shown in Fig. 7b. The broad ν(O–O) band has contributions from free O2− at 1116 cm−1, CsO2 at 1146 cm−1, and HO2 (most likely from minor traces of H2O in the electrolyte) at 1187 cm−1.26,70
The ν(O–O) frequency shifts depend on the strength of the bond between the alkali cation and the O2−. Hence, the higher frequency observed for ν(O–O) (NaO2) at 1161 cm−1,71,72 compared to ν(O–O) (CsO2) at 1146 cm−1,13,24,26,73 may be ascribed to the ionic radii of the cations, i.e., smaller cations display a substantial electrostatic force that strongly attracts the valence electrons from O2− and, therefore, present a stronger bonding interaction with the O2−. Additionally, while no free O2− has been detected in the presence of Na+ (at equal alkali-metal concentrations),24 spectroscopic evidence of the presence of free O2− (1116 cm−1) in Cs+-containing electrolytes (Fig. 7b) demonstrates the dissociation of O2− back into solution in the presence of larger cations.
Bands at 1040 and 1310 cm−1 correspond to the methyl-group rocking modes in DMSO and that at 1420 cm−1 to ν(S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).74–76 The bands associated with DMSO vibrations increase at −0.55 V, coinciding with the formation of O2− species at the electrode|electrolyte interface. Enhanced band intensity for DMSO at this potential may be due to the solvation of O2−, i.e., Cs+(DMSO)4(O2−). The further uptake of DMSO onto the Pt(111) surface following desorption of Cs+(DMSO)4(O2−) species is also indicated by the significant increase in DMSO bands after this point. Increased DMSO band intensity in the potential range where ORR occurs has not been observed for Na+,24 probably due to DMSO less successfully competing for Pt sites with more strongly adsorbed NaO2, i.e., smaller cation–O2− complexes are more strongly adsorbed on the Pt(111) surface, while for larger cation–O2− complexes, adsorption is weaker, and they are readily desorbed back into solution. The lower intensity of the DMSO bands at −1.4 V could result from a potential-dependent reorientation of DMSO on the surface, similar to that reported for the reorientation of acetonitrile on roughened Au surfaces.77
O).74–76 The bands associated with DMSO vibrations increase at −0.55 V, coinciding with the formation of O2− species at the electrode|electrolyte interface. Enhanced band intensity for DMSO at this potential may be due to the solvation of O2−, i.e., Cs+(DMSO)4(O2−). The further uptake of DMSO onto the Pt(111) surface following desorption of Cs+(DMSO)4(O2−) species is also indicated by the significant increase in DMSO bands after this point. Increased DMSO band intensity in the potential range where ORR occurs has not been observed for Na+,24 probably due to DMSO less successfully competing for Pt sites with more strongly adsorbed NaO2, i.e., smaller cation–O2− complexes are more strongly adsorbed on the Pt(111) surface, while for larger cation–O2− complexes, adsorption is weaker, and they are readily desorbed back into solution. The lower intensity of the DMSO bands at −1.4 V could result from a potential-dependent reorientation of DMSO on the surface, similar to that reported for the reorientation of acetonitrile on roughened Au surfaces.77
Conclusions
The ORR and OER have been studied on Pt(111) and Pt(100) single crystals and on a polycrystalline Pt disk, using a non-aqueous solvent, dimethyl sulfoxide (DMSO), in the presence of alkali-metal perchlorate salts. Voltammetric results show that the cation size strongly affects the mechanism of the ORR in O2-saturated DMSO-based electrolyte.In contrast to Na+, surface structure has been proved not to significantly influence the reaction pathway involving Cs+ cations. This behaviour is ascribed to the relative surface adsorption strengths of superoxy and peroxy ion-pair complexes formed when utilising different cations. We conclude that because no changes were detected in the voltammetric profile of the ORR in the presence of Cs+ cations using polycrystalline Pt, Pt(111) and Pt(100) electrodes, this supports a preferred solution-based mechanism with a lower impact of the surface structure compared to smaller cations, such as Na+. Shifts in the E1/2 and changes in the intensities of redox peaks I to IV as a function of cation size are also consistent with this finding. Moreover, the separation in potential between peaks I and II is suggested to be a marker for the extent of interaction between the electrode surface and MO2. Because of this, it is speculated that activation of this surface complex to facilitate the second electron transfer to form peroxy species will be promoted as cation size decrease, i.e., the ORR pathway preference changes as the cation size increases, from O22− formation in the case of Na+ (and indeed Li+) to O2− formation for larger cations, i.e., K+ and Cs+.
To assess this proposal, the stability of the O2− complex adsorbed at the surface was studied with in situ Raman spectroscopy in Cs+-containing electrolytes and results demonstrated the formation of CsO2 in a limited potential range, commensurate with the first reduction peak, but this species readily desorbs into solution. The labile nature of the surface O2− bond is consistent with the lack of structural sensitivity in CV, suggesting a solution-based ORR/OER mechanism for larger cations. This may disfavour subsequent reduction to O22− when using Cs+ compared to Na+ and this is made manifest by the relative magnitudes of peroxy- and superoxy-redox peaks highlighted above. Furthermore, in situ Raman spectroscopy indicates that the displacement of CsO2 species may be ascribed to uptake of DMSO at the interface, interacting strongly with the surface and causing displacement of ion pairs at potentials where the ORR occurs. This study highlights the possibility of using large alkali-metal cations, such as Cs+, in the electrolyte of Li and Na–O2 cells to tailor the initial ORR pathway towards a one-electron transfer and thus, contributing to preventing the formation of surface-blocking peroxide intermediates.
Author contributions
All authors have contributed to writing, reviewing, and editing the final version of the manuscript. JFV contributed with investigation, methodology, data curation, conceptualisation, formal analysis, visualisation, and writing of the original draft. LJH contributed with funding acquisition, conceptualisation, formal analysis, supervision, resources, and project administration. GC contributed to investigation and methodology. GAA contributed to visualisation, supervision, formal analysis, and conceptualisation.Conflicts of interest
All authors declare no conflict of interest.Acknowledgements
JFV acknowledges PhD funding from the UKRI Engineering and Physical Sciences Research Council (EPSRC). LJH and GC acknowledge funding support from the UK Faraday Institution (EPSRC EP/S003053/1) through the Degradation Project (grant numbers FIRG001 and FIRG024). We acknowledge discussions with Dr Tom Galloway.References
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Bardé and P. G. Bruce, The lithium–oxygen battery with ether-based electrolytes, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef CAS PubMed.
- D. Sharon, D. Hirshberg, M. Afri, A. A. Frimer, M. Noked and D. Aurbach, Aprotic Metal–Oxygen Batteries: Recent Findings and Insights, J. Solid State Electrochem., 2017, 21, 1861–1878 CrossRef CAS.
- T. A. Galloway, G. Attard and L. J. Hardwick, An Electrochemical Investigation of Oxygen Adsorption on Pt Single Crystal Electrodes in a Non-Aqueous Li+ Electrolyte, Electrochem. Commun., 2020, 119, 106814 CrossRef CAS.
- T. A. Galloway, L. Cabo-Fernandez, I. M. Aldous, F. Braga and L. J. Hardwick, Shell Isolated Nanoparticles for Enhanced Raman Spectroscopy Studies in Lithium–Oxygen Cells, Faraday Discuss., 2017, 205, 469–490 RSC.
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Charging a Li–O2 Battery Using a Redox Mediator, Nat. Chem., 2013, 5, 489–494 CrossRef CAS.
- P. Reinsberg, A. Weiß, P. P. Bawol and H. Baltruschat, Electrochemical Reaction Order of the Oxygen Reduction Reaction in Li+-Containing DMSO, J. Phys. Chem. C, 2017, 121, 7677–7688 CrossRef CAS.
- A. I. Belova, D. G. Kwabi, L. V. Yashina, Y. Shao-Horn and D. M. Itkis, Mechanism of Oxygen Reduction in Aprotic Li-air Batteries: The Role of Carbon Electrode Surface Structure, J. Phys. Chem. C, 2017, 121, 1569–1577 CrossRef CAS.
- W. Wang and Y. C. Lu, The Potassium–Air Battery: Far from a Practical Reality?, Acc. Mater. Res., 2021, 2, 515–525 CrossRef CAS.
- P. Hartmann, C. L. Bender, M. Vračar, A. K. Dürr, A. Garsuch, J. Janek and P. Adelhelm, A Rechargeable Room-Temperature Sodium Superoxide (NaO2) Battery, Nat. Mater., 2013, 12, 228–232 CrossRef CAS.
- X. Ren and Y. Wu, A Low-Overpotential Potassium–Oxygen Battery Based on Potassium Superoxide, J. Am. Chem. Soc., 2013, 135, 2923–2926 CrossRef CAS.
- C. J. Bondue, P. P. Bawol, A. A. Abd-El-Latif, P. Reinsberg and H. Baltruschat, Gaining Control over the Mechanism of Oxygen Reduction in Organic Electrolytes: The Effect of Solvent Properties, J. Phys. Chem. C, 2017, 121, 8864–8872 CrossRef CAS.
- C. J. Bondue, P. Reinsberg, A. A. Abd-El-Latif and H. Baltruschat, Oxygen Reduction and Oxygen Evolution in DMSO Based Electrolytes: The Role of the Electrocatalyst, Phys. Chem. Chem. Phys., 2015, 17, 25593–25606 RSC.
- I. M. Aldous and L. J. Hardwick, Solvent-Mediated Control of the Electrochemical Discharge Products of Non-Aqueous Sodium–Oxygen Electrochemistry, Angew. Chem., Int. Ed., 2016, 55, 8254–8257 CrossRef CAS PubMed.
- P. M. Radjenovic and L. J. Hardwick, Time-resolved SERS study of the oxygen reduction reaction in ionic liquid electrolytes for non-aqueous lithium-oxygen cells, Faraday Discuss., 2018, 206, 379–392 RSC.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. Freunberger, P. Ashok, B. Praveen, K. Dholakia, J. Tarascon and P. Bruce, The Role of LiO2 Solubility in O2 Reduction in Aprotic Solvents and its Consequences for Li–O2 Batteries, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- N. Mozhzhukhina, L. P. Méndez De Leo and E. J. Calvo, Infrared Spectroscopy Studies on Stability of Dimethyl Sulfoxide for Application in a Li–Air Battery, J. Phys. Chem. C, 2013, 117, 18375–18380 CrossRef CAS.
- M. A. Schroeder, N. Kumar, A. J. Pearse, C. Liu, S. B. Lee, G. W. Rubloff, K. Leung and M. Noked, DMSO-Li2O2 Interface in the Rechargeable Li–O2 Battery Cathode: Theoretical and Experimental Perspectives on Stability, ACS Appl. Mater. Interfaces, 2015, 7, 11402–11411 CrossRef CAS.
- V. Gutmann, The Donor–Acceptor Approach to Molecular Interactions, Springer, US, 1978 Search PubMed.
- V. Gutmann, Solvent Effects on the Reactivities of Organometallic Compounds, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS.
- A. D. Goolsby and D. T. Sawyer, The Electrochemical Reduction of Superoxide Ion and Oxidation of Hydroxide Ion in Dimethyl Sulfoxided, Anal. Chem., 1968, 40, 83–86 CrossRef CAS.
- T. Kumeda, H. Tajiri, O. Sakata, N. Hoshi and M. Nakamura, Effect of hydrophobic cations on the oxygen reduction reaction on single-crystal platinum electrodes, Nat. Commun., 2018, 9, 4378 CrossRef.
- D. Strmcnik, K. Kodama, D. Van Der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, The Role of Non-Covalent Interactions in Electrocatalytic Fuel-Cell Reactions on Platinum, Nat. Chem., 2009, 1, 466–472 CrossRef CAS.
- P. H. Reinsberg, A. Koellisch and H. Baltruschat, On the Importance of Ion Pair Formation and the Effect of Water in Potassium–Oxygen Batteries, Electrochim. Acta, 2019, 313, 223–234 CrossRef CAS.
- T. A. Galloway, J.-C. Dong, J.-F. Li, G. Attard and L. J. Hardwick, Oxygen Reactions on Pt{hkl} in a Non-Aqueous Na+ Electrolyte: Site Selective Stabilisation of a Sodium Peroxy Species, Chem. Sci., 2019, 10, 2956–2964 RSC.
- S. Ma, W. C. McKee, J. Wang, L. Guo, M. Jansen, Y. Xu and Z. Peng, Mechanistic Origin of Low Polarization in Aprotic Na–O2 Batteries, Phys. Chem. Chem. Phys., 2017, 19, 12375–12383 RSC.
- P. M. Radjenovic and L. J. Hardwick, Evaluating Chemical Bonding in Dioxides for the Development of Metal–Oxygen Batteries: Vibrational Spectroscopic Trends of Dioxygenyls, Dioxygen, Superoxides and Peroxides, Phys. Chem. Chem. Phys., 2019, 21, 1552–1563 RSC.
- P. G. Bruce, L. J. Hardwick and K. M. Abraham, Lithium–Air and Lithium–Sulfur Batteries, MRS Bull., 2011, 36, 506–512 CrossRef CAS.
- T. Zhang, Z. Xu, Y. Guo, C. Liang, J. Wang and J. Yang, Building High Performance Silicon–Oxygen and Silicon–Sulfur Battery by In-Situ Lithiation of Fibrous Si/C Anode, J. Alloys Compd., 2019, 806, 335–342 CrossRef CAS.
- S. K. Das, S. Lau and L. A. Archer, Sodium–Oxygen Batteries: A New Class of Metal–Air Batteries, J. Mater. Chem. A, 2014, 2, 12623–12629 RSC.
- C. L. Bender, D. Schröder, R. Pinedo, P. Adelhelm and J. Janek, One- or Two-Electron Transfer? The Ambiguous Nature of the Discharge Products in Sodium-Oxygen Batteries, Angew. Chem., Int. Ed., 2016, 55, 4640–4649 CrossRef CAS PubMed.
- H. Yadegari, Q. Sun and X. Sun, Sodium–Oxygen Batteries: A Comparative Review from Chemical and Electrochemical Fundamentals to Future Perspective, Adv. Mater., 2016, 28, 7065–7093 CrossRef CAS PubMed.
- P. H. Reinsberg, A. Koellisch, P. P. Bawol and H. Baltruschat, K–O2 Electrochemistry: Achieving Highly Reversible Peroxide Formation, Phys. Chem. Chem. Phys., 2019, 21, 4286–4294 RSC.
- L. Xue, Y. Li, H. Gao, W. Zhou, X. Lü, W. Kaveevivitchai, A. Manthiram and J. B. Goodenough, Low-Cost High-Energy Potassium Cathode, J. Am. Chem. Soc., 2017, 139, 2164–2167 CrossRef CAS PubMed.
- N. Xiao, X. Ren, W. D. McCulloch, G. Gourdin and Y. Wu, Potassium Superoxide: A Unique Alternative for Metal–Air Batteries, Acc. Chem. Res., 2018, 51, 2335–2343 CrossRef CAS PubMed.
- P. K. Shen and Z. Zhao, Mechanism of Oxygen Reduction Reaction, in Electrochemical Oxygen Reduction: Fundamental and Applications, ed. P. K. Shen, Springer, 2021, pp. 11–22 Search PubMed.
- N. M. Marković, T. J. Schmidt, V. Stamenković and P. N. Ross, Oxygen Reduction Reaction on Pt and Pt Bimetallic Surfaces: A Selective Review, Fuel Cells, 2001, 1, 105–116 CrossRef.
- A. M. Gómez-Marín and J. M. Feliu, New insights into the oxygen reduction reaction mechanism on Pt (111): a detailed electrochemical study, ChemSusChem, 2013, 6, 1091–1100 CrossRef.
- J. Zhang, Y. Mo, M. B. Vukmirovic, R. Klie, K. Sasaki and R. R. Adzic, Platinum Monolayer Electrocatalysts for O2 Reduction: Pt Monolayer on Pd(111) and on Carbon-Supported Pd Nanoparticles, J. Phys. Chem. B, 2004, 108, 10955–10964 CrossRef CAS.
- X. Zhang, H. Li, J. Yang, Y. Lei, C. Wang, J. Wang, Y. Tang and Z. Mao, Recent Advances in Pt-based Electrocatalysts for PEMFCs, RSC Adv., 2021, 11, 13316–13328 RSC.
- C. Song and J. Zhang, Electrocatalytic Oxygen Reduction Reaction, in PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications, 2008 Search PubMed.
- A. Haryanto and C. W. Lee, Shell Isolated Nanoparticle Enhanced Raman Spectroscopy for Mechanistic Investigation of Electrochemical Reactions, Nano Converg., 2022, 9, 9 CrossRef CAS PubMed.
- Y.-J. Zhang, H. Ze, P.-P. Fang, Y.-F. Huang, A. Kudelski, J. Fernández-Vidal, L. J. Hardwick, J. Lipkowski, Z.-Q. Tian and J.-F. Li, Shell-isolated nanoparticleenhanced Raman spectroscopy, Nat. Rev. Methods Primers, 2023, 1–22 Search PubMed.
- J.-C. C. Dong, X.-G. G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. L. Yang, D.-Y. Y. Wu, J. M. Feliu, C. T. Williams, Z. Q. Tian and J. F. Li, In situ Raman spectroscopic evidence for oxygen reduction reaction intermediates at platinum single-crystal surfaces, Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- Y.-F. Huang, P. J. Kooyman and M. T. M. Koper, Intermediate Stages of Electrochemical Oxidation of Single-Crystalline Platinum Revealed by In Situ Raman Spectroscopy, Nat. Commun., 2016, 7, 12440 CrossRef CAS PubMed.
- R. Rizo, J. Fernández-Vidal, L. J. Hardwick, G. A. Attard, F. J. Vidal-Iglesias, V. Climent, E. Herrero and J. M. Feliu, Investigating the Presence of Adsorbed Species on Pt Steps at Low Potentials, Nat. Commun., 2022, 13, 2550 CrossRef CAS PubMed.
- J. Clavilier, R. Faure, G. Guinet and R. Durand, Preparation of monocrystalline Pt microelectrodes and electrochemical study of the plane surfaces cut in the direction of the {111} and {110} planes, J. Electroanal. Chem., 1980, 107, 205–209 CrossRef CAS.
- A. M. Bittner, J. Wintterlin, B. Beran and G. Ertl, Bromine adsorption on Pt(111), (100), and (110)-an STM study in air and in electrolyte, Surf. Sci., 1995, 335, 291–299 CrossRef CAS.
- H. Oguz, N. Yokoi and S. Kinoshita, The Height and Radius of the Tear Meniscus and Methods for Examining These Parameters, Cornea, 2000, 19, 497–500 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for Three-Dimensional Visualization of Crystal, Voltammetric and Morphology Data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- C. G. Zoski, Handbook of Electrochemistry, Elsevier, 2007 Search PubMed.
- J. Turkevich, Colloidal gold. Part I, Gold Bull., 1985, 18, 86–91 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hiller, Synthesis of Gold Nanoparticles Turkevich method, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- G. Frens, Controlled Nucleation for the Regulation of the Particle Size in Monodisperse Gold Suspensions, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Synthesis of Nanosized Gold–Silica Core–Shell Particles, Langmuir, 1996, 12, 4329–4335 CrossRef.
- C. Hanske, M. N. Sanz-Ortiz and L. M. Liz-Marzán, Silica-Coated Plasmonic Metal Nanoparticles in Action, Adv. Mater., 2018, 30, 1707003 CrossRef PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Shell-Isolated Nanoparticle-Enhanced Raman Spectroscopy, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, Elucidating the Mechanism of Oxygen Reduction for Lithium–Air Battery Applications, J. Phys. Chem. C, 2009, 113, 20127–20134 CrossRef CAS.
- K. M. Abraham and Z. A. Jiang, Polymer Electrolyte-Based Rechargeable Lithium/Oxygen Battery, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef CAS.
- K. M. Abraham, Z. Jiang and B. Carroll, Highly Conductive PEO-like Polymer Electrolytes, Chem. Mater., 1997, 9, 1978–1988 CrossRef CAS.
- H. S. Choe, B. G. Carroll, D. M. Pasquariello and K. M. Abraham, Characterization of Some Polyacrylonitrile-Based Electrolytes, Chem. Mater., 1997, 9, 369–379 CrossRef CAS.
- S. S. Zhang, D. Foster and J. Read, The Effect of Quaternary Ammonium on Discharge Characteristic of a Non-Aqueous Electrolyte Li/O2 Battery, Electrochim. Acta, 2011, 56, 1283–1287 CrossRef CAS.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides, Acta Crystallogr. A, 1976, 32, 751–767 CrossRef.
- J. Palomo and P. N. Pintauro, Competitive Absorption of Quaternary Ammonium and Alkali Metal Cations into a Nafion Cation-Exchange Membrane, J. Membr. Sci., 2003, 215, 103–114 CrossRef CAS.
- D. H. Aue, H. M. Webb and M. T. Bowers, A Thermodynamic Analysis of Solvation Effects on the Basicities of Alkylamines. An Electrostatic Analysis of Substituent Effects, J. Am. Chem. Soc., 1976, 98, 318–329 CrossRef CAS.
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, Influence of Nonaqueous Solvents on the Electrochemistry of Oxygen in the Rechargeable Lithium–Air Battery, J. Phys. Chem. C, 2010, 114, 9178–9186 CrossRef CAS.
- Y.-T. Lu, A. R. Neale, C.-C. Hu and L. J. Hardwick, Trapped Interfacial Redox Introduces Reversibility in the Oxygen Reduction Reaction in a Non-Aqueous Ca2+ Electrolyte, Chem. Sci., 2021, 12, 8909–8919 RSC.
- C. J. Bondue, A. A. Abd-El-Latif, P. Hegemann and H. Baltruschat, Quantitative Study for Oxygen Reduction and Evolution in Aprotic Organic Electrolytes at Gas Diffusion Electrodes by DEMS, J. Electrochem. Soc., 2015, 162, A479–A487 CrossRef CAS.
- P. J. Welford, B. A. Brookes, V. Climent and R. G. Compton, The hanging meniscus contact: geometry induced diffusional overpotential. The reduction of oxygen in dimethylsulphoxide at Au(111), J. Electroanal. Chem., 2001, 513, 8–15 CrossRef CAS.
- I. I. Vol’nov, Peroxides, Superoxides, and Ozonides of Alkali and Alkaline Earth Metals, Springer, US, 1966 Search PubMed.
- X. Li and A. A. Gewirth, Oxygen Electroreduction through a Superoxide Intermediate on Bi-Modified Au Surfaces, J. Am. Chem. Soc., 2005, 127, 5252–5260 CrossRef CAS PubMed.
- J. B. Bates and M. H. Brooker, Determination of Longitudinal Optical Mode Frequencies by Polarized Specular Reflectance Infrared Spectroscopy, J. Phys. Chem. Solids, 1971, 32, 2403–2407 CrossRef CAS.
- J. B. Bates, M. H. Brooker, A. S. Quist and G. E. Boyd, Raman Spectra of Molten Alkali Metal Carbonates, J. Phys. Chem., 1972, 76, 1565–1571 CrossRef CAS.
- J. B. Bates, M. H. Brooker and G. E. Boyd, Raman Spectra of O-2 and O-3 Ions in Alkali-Metal Superoxides and Ozonides, Chem. Phys. Lett., 1972, 16, 391–395 CrossRef CAS.
- V. M. Wallace, N. R. Dhumal, F. M. Zehentbauer, H. J. Kim and J. Kiefer, Revisiting the Aqueous Solutions of Dimethyl Sulfoxide by Spectroscopy in the Mid- and Near-Infrared: Experiments and Car-Parrinello Simulations, J. Phys. Chem. B, 2015, 119, 14780 CrossRef CAS.
- Q. Yu and S. Ye, In Situ Study of Oxygen Reduction in Dimethyl Sulfoxide (DMSO) Solution: A Fundamental Study for Development of the Lithium–Oxygen Battery, J. Phys. Chem. C, 2015, 119, 12236–12250 CrossRef CAS.
- C. Xu, A. Ge, K. Kannari, B. Peng, M. Xue, B. Ding, K. Inoue, X. Zhang and S. Ye, The Decisive Role of Li2 O2 Desorption for Oxygen Reduction Reaction in Li–O2 Batteries, ACS Energy Lett., 2023, 8, 1289–1299 CrossRef CAS.
- I. M. Aldous and L. J. Hardwick, Influence of tetraalkylammonium cation chain length on gold and glassy carbon electrode interfaces for alkali metal–oxygen batteries, J. Phys. Chem. Lett., 2014, 5, 3924–3930 CrossRef CAS PubMed.
Footnotes |
| † Present address: Leiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. |
| ‡ Present address: SLB Cambridge Research, High Cross, Madingley Road, CB3 0EL Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2024 |