
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIndividually separated supramolecular polymer chains toward solution-processable supramolecular polymeric materials†
Takuma
Shimada
 ab,
Yuichiro
Watanabe
c,
Takashi
Kajitani
d,
Masayuki
Takeuchi
b,
Yutaka
Wakayama
ab and
Kazunori
Sugiyasu
*c
ab,
Yuichiro
Watanabe
c,
Takashi
Kajitani
d,
Masayuki
Takeuchi
b,
Yutaka
Wakayama
ab and
Kazunori
Sugiyasu
*c
aDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, Nishi-ku, Fukuoka 819-0395, Japan
bNational Institute for Materials Science, Tsukuba, Ibaraki 305-0047, Japan
cDepartment of Polymer Chemistry, Kyoto University, Kyotodaigaku-katsura, Kyoto 615-8510, Japan. E-mail: sugiyasu.kazunori.8z@kyoto-u.ac.jp
dOpen Facility Development Office, Open Facility Center, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
First published on 4th January 2023
Abstract
Herein, we present a simple design concept for a monomer that affords individually separated supramolecular polymer chains. Random introduction of alkyl chains with different lengths onto a monomer prevented its supramolecular polymers from bundling, permitting the preparation of concentrated solutions of the supramolecular polymer without gelation, precipitation, or crystallization. With such a solution in hand, we succeeded in fabricating self-standing films and threads consisting of supramolecular polymers.
Introduction
Most supramolecular polymers can be categorized into two groups in terms of their polymerization mechanism: i.e., isodesmic polymerization or nucleation–elongation polymerization.1 In the latter mechanism, supramolecular polymerization is considered to occur in a similar manner to crystallization, except that it proceeds in one dimension exclusively. As such, seeded growth is possible (like crystallization), which has recently led to the development of living supramolecular polymerization and has opened a new door in supramolecular polymer chemistry.2–6Although supramolecular polymerization is expected to proceed one-dimensionally in the above mechanism, in reality, this is not always the case. An increase in the monomer concentration leads to an increase in the degree of supramolecular polymerization, and at higher concentrations supramolecular polymers are prone to bundle, giving rise to gelation, precipitation, or crystallization. Consequently, solution processing of supramolecular polymers has been largely unexplored.7–9 In this context, the purpose of the present study was to obtain individually separated supramolecular polymer chains at high concentrations (of the order of several mM).
Previously, Meijer and co-workers10 have shown that deviations in the temperature-dependent spectra at high concentrations from those predicted by the nucleation–elongation model are caused by clustering (bundling) of supramolecular polymers. More recently, Giuseppone and co-workers11 found that bundling of supramolecular polymers occurred either through coagulation or a secondary nucleation process, depending on the rate of cooling of the hot monomer solution. In common monomer designs for nucleated supramolecular polymerization, including the above two examples,10,11 alkyl chains are introduced at the periphery of a monomer to endow the resultant supramolecular polymers with solubility by preventing three-dimensional crystallization. Nevertheless, bundling is practically inevitable. We hypothesized that engineering the surface of a supramolecular polymer chain by modifying the peripheral alkyl chains of its monomer might be effective in modulating lateral interactions between the supramolecular polymer chains.
Results and discussion
Supramolecular polymerization and bundling
In this study, we used triphenylene-based monomers9,12 bearing six amide groups as intermonomer hydrogen-bonding sites. At the periphery of the triphenylene core, diverged through the amide linkages, eighteen octyl or octadecyl chains were introduced into monomers 18 and 118, respectively (Chart 1). Supramolecular polymerizations of monomers of this type are usually investigated in aliphatic solvents. We surmised that the solvation of the supramolecular polymers and, consequently, their bundling would be susceptible to subtle differences in the structure of the solvent molecule, so we screened linear [e.g. n-hexane (nHex)], branched [isooctane (iOc)], and cyclic [e.g. cyclohexane (cHex)] aliphatic solvents [see the ESI; Fig. S8†]. The 18 and 118 monomers were dissolved at a concentration of 50 μM in these solvents by heating, and the resultant solutions were left undisturbed at room temperature. Both monomers readily underwent one-dimensional (1D) supramolecular polymerization in nHex or iOc, and heavily bundled fibrous aggregates were observed by atomic force microscopy (AFM) (Fig. 1). In cHex, supramolecular polymerization of 18 occurred relatively slowly (see below), and 118 did not form a 1D supramolecular polymer within our experimental timescale (several months: see ESI,† Fig. S9). These results suggest that differences in the nature of the solvent molecule affect the supramolecular polymerization kinetics but do not significantly affect the lateral interaction between supramolecular polymer chains. In addition, the length of the alkyl chains (C8H17vs. C18H37) in a monomer appears not to be a critical factor from a viewpoint of the purpose of this study.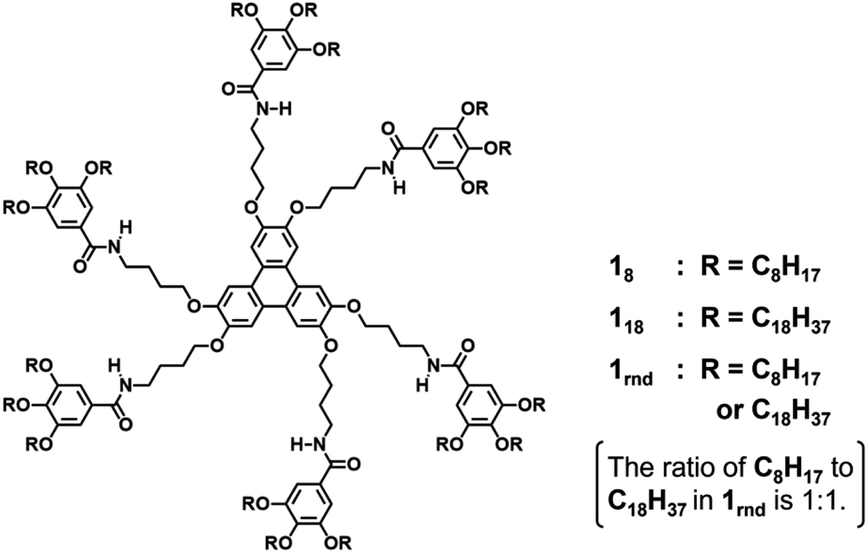 | ||
| Chart 1 Structures of the monomers used in this study. | ||
 | ||
| Fig. 1 AFM images of supramolecular polymers of (a and b) 18 and (c and d) 118 prepared in (a and c) nHex and (b and d) iOc: 50 μM; highly oriented pyrolytic graphite (HOPG) substrates; scale bar = (a and b) 200 nm and (c and d) = 1 μm. | ||
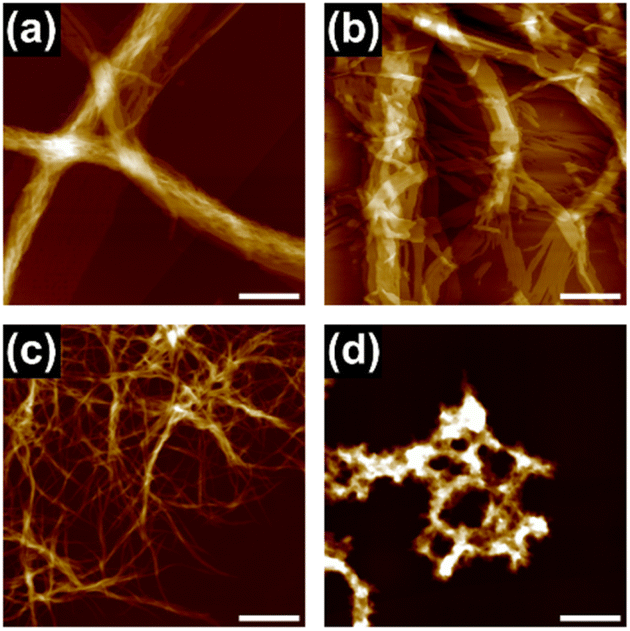
To gain an insight into the bundling process, we attempted to capture the evolution of bundles of supramolecular polymers of 18. This was possible in cHex, owing to the slow rate of supramolecular polymerization in this solvent. After cooling the hot monomer solution, aliquots of the solution were spin-coated onto a highly oriented pyrolytic graphite (HOPG) substrate at various times for AFM measurements. Even in the earliest stages (30 minutes after cooling), short supramolecular polymers had assembled laterally, indicating the occurrence of secondary nucleation (Fig. 2a).13 Over time, fibrous aggregates grew further through elongation and bundling (Fig. 2b and c). Finally, precipitates displaying birefringence appeared (Fig. S10†), thus suggesting that agglomeration observed throughout the above processes occurred in solution and not on the HOPG substrate. This result clearly indicates that the triphenylene-based supramolecular polymers have a strong propensity to undergo bundling.
 | ||
| Fig. 2 Successive AFM images of supramolecular polymers captured at (a) 30 min, (b) 60 min, and (c) 90 min after cooling a hot 50 μM solution of 18 in cHex; scale bar = 100 nm. The solution was stirred at 400 rpm at room temperature. (d) SEM image (scale bar = 5 μm) of the xerogel of 18 prepared from a 1 mM solution in cHex, (inset) photograph of the organogel. (e) XRD pattern of the xerogel of 18. | ||
At a higher concentration (1 mM), 18 formed a translucent or opaque gel, suggesting the formation of large aggregates capable of scattering visible light (ESI; Fig, S8a†), whereas 118 precipitated in most of the solvents tested (ESI; Fig. S8b†). A xerogel of 18 freeze-dried from cHex consisted of a dense network of thick fibrous aggregates with diameters of 50 nm−1.5 μm, as observed by scanning electron microscopy (SEM) (Fig. 2d). Powder X-ray diffraction (XRD) measurement of the xerogel showed clear diffraction peaks attributable to a hexagonal packing of 1D columns of 18 (Fig. 2e). Because of the crystalline nature of the aggregates, the organogels readily collapsed upon mechanical agitation, and solid materials separated out in the solvent (ESI; Fig. S11†), which made it difficult to process the supramolecular polymers to form films or threads. These results confirmed that lateral interactions between 1D supramolecular polymer chains dictates macroscopic properties of supramolecular polymers.
A new monomer design: random introduction of peripheral alkyl chains
Although octyl and octadecyl chains were individually found to be unsuitable for the purposes of this study, we surmised that a random mixture of the two types of chain might lead to a different situation. With this in mind, we designed a new monomer 1rnd in which octyl and octadecyl chains were randomly introduced (1rnd in Chart 1). We expected that the randomness of the side chains would perturb the surfaces of the supramolecular polymer chains and would prevent them from bundling (Fig. 3c). To this end, the ratio of the two types of alkyl chain in a monomer is a matter of interest, and our first choice was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, as it had been reported in a study on self-assembled monolayers (SAMs) that the greatest degree of disorder in a longer alkanethiolate was observed with equimolar amounts of a shorter alkanethiolate.14 In 1rnd, statistically there are about 8000 possible combinations (i.e., distinct monomers) in terms of the proportions and positions of the two alkyl chains. The average number of the octadecyl chains in a monomer is nine; monomers with nine such units comprise 18.5% of the mixture (Fig. 4c). Monomers with fewer than six or more than twelve octadecyl chains are in the minority (4.8% each), and the proportion substituted exclusively with octyl or octadecyl chain (i.e., 18 and 118) is negligible (0.0004%). The synthesis of 1rnd was straightforward (Fig. 4a, see ESI; Scheme S1 and S2†). First, 1:1 mixture of 1-bromooctane and 1-bromooctadecane was reacted with methyl 3,4,5-trihydroxybenzoate (methyl gallate) to afford 2rnd. Using the mixture of precursors bearing octyl or octadecyl chains (Fig. 4b), 1rnd was synthesised through common reactions. The number of aliphatic protons in 1rnd determined by 1H NMR spectroscopy agreed with the expected average value (ESI; Fig. S2†). MALDI-TOF mass spectrometry showed the presence of monomers with distinct molecular weights (ESI; Fig. S12†). These results confirmed the random introduction of octyl and octadecyl chains to the triphenylene core.
:1 molar ratio, as it had been reported in a study on self-assembled monolayers (SAMs) that the greatest degree of disorder in a longer alkanethiolate was observed with equimolar amounts of a shorter alkanethiolate.14 In 1rnd, statistically there are about 8000 possible combinations (i.e., distinct monomers) in terms of the proportions and positions of the two alkyl chains. The average number of the octadecyl chains in a monomer is nine; monomers with nine such units comprise 18.5% of the mixture (Fig. 4c). Monomers with fewer than six or more than twelve octadecyl chains are in the minority (4.8% each), and the proportion substituted exclusively with octyl or octadecyl chain (i.e., 18 and 118) is negligible (0.0004%). The synthesis of 1rnd was straightforward (Fig. 4a, see ESI; Scheme S1 and S2†). First, 1:1 mixture of 1-bromooctane and 1-bromooctadecane was reacted with methyl 3,4,5-trihydroxybenzoate (methyl gallate) to afford 2rnd. Using the mixture of precursors bearing octyl or octadecyl chains (Fig. 4b), 1rnd was synthesised through common reactions. The number of aliphatic protons in 1rnd determined by 1H NMR spectroscopy agreed with the expected average value (ESI; Fig. S2†). MALDI-TOF mass spectrometry showed the presence of monomers with distinct molecular weights (ESI; Fig. S12†). These results confirmed the random introduction of octyl and octadecyl chains to the triphenylene core.
 | ||
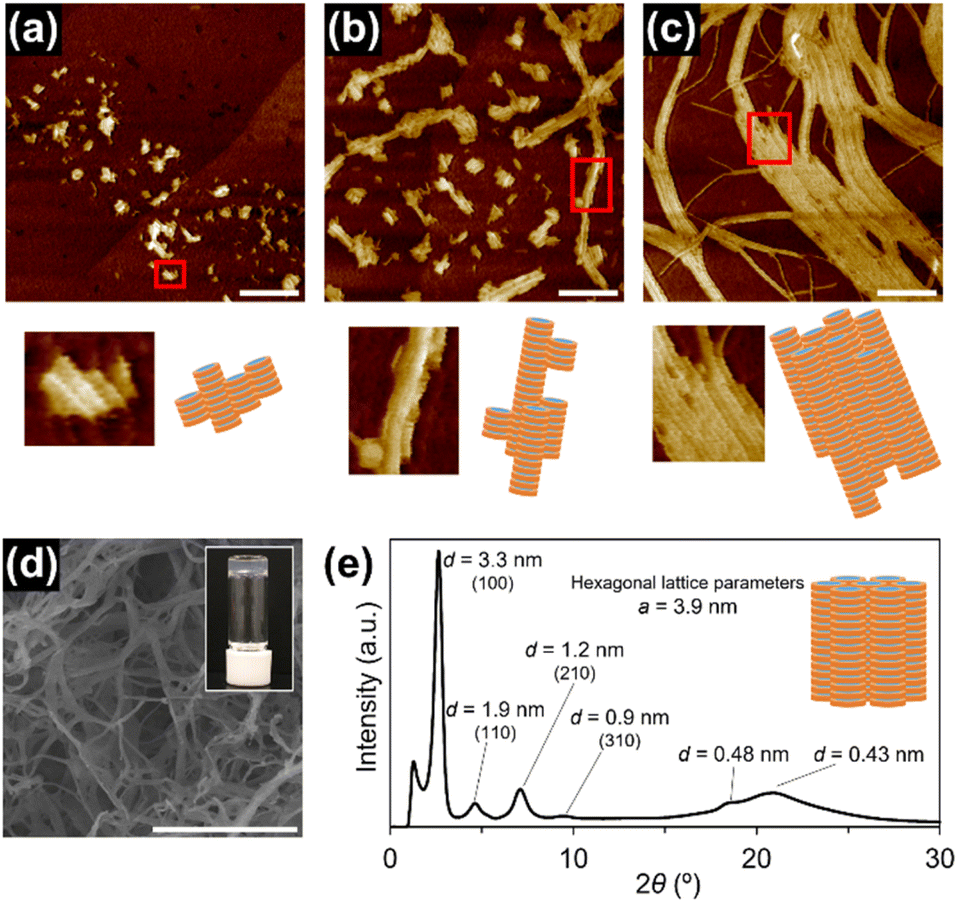
| Fig. 3 AFM images of supramolecular polymers of 1rnd prepared in (a) nHex and (b) iOc (50 μM); scale bar = 300 nm. (c) Schematic representation of supramolecular polymerization of 1rnd; bundling processes appear to be prevented owing to the surface engineering. (d) AFM image of individually separated supramolecular polymer chains of 1rnd prepared by seeded supramolecular polymerization in a cHex/iOc mixed solvent (1 mM); scale bar = 1 μm. (inset) photograph of the solution containing it. (e) Height measurements across the green line on (d). | ||
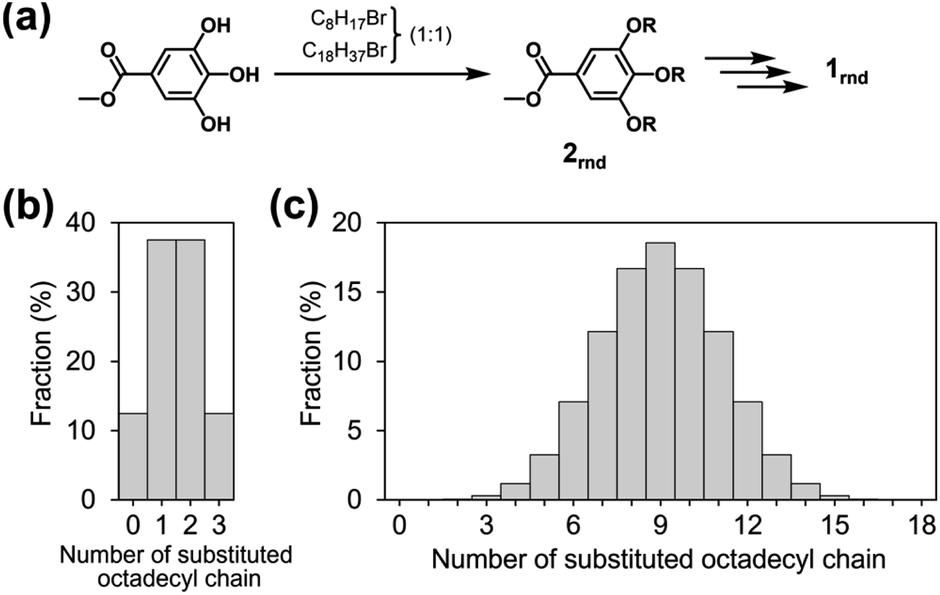 | ||
| Fig. 4 (a) Random introduction of two different alkyl chains and synthesis of 1rnd. The proportions of the octadecyl chains in (b) precursor 2rnd and (c) monomer 1rnd. | ||
Supramolecular polymerization of 1rnd was investigated in a variety of aliphatic solvents. At a relatively high concentration (1 mM), 1rnd formed transparent organogels in linear or branched aliphatic solvents. In contrast to organogels of 18 (see above), those of 1rnd were converted into homogeneous solutions upon mechanical agitation (ESI; Fig. S13†). In addition, after 2 days, the organogel reformed without any precipitation or phase separation (so-called thixotropic behaviour). AFM measurements showed that bundling was suppressed with 1rnd (50 μM in nHex and iOc; Fig. 3a and b). For comparison, we investigated the supramolecular copolymerization of 18 and 118 (1:1 molar ratio [18 + 118] = 1 mM in nHex); the resulting organogel consisted of bundled supramolecular polymer chains and did not show the abovementioned thixotropic behaviour (ESI; Fig. S14†). We infer that 18 and 118 do not copolymerize completely randomly because of the difference in their supramolecular polymerization kinetics.
As in the case of 118, 1rnd did not undergo spontaneous supramolecular polymerization in cHex. However, the addition of short supramolecular polymers of 1rnd prepared in iOc (Fig. 3b) as ‘seeds’ to the cHex solution of 1rnd initiated its 1D supramolecular polymerization (ESI; Fig. S15†). For reference, 118 was unable to form 1D supramolecular polymers with the addition of the seeds of 1rnd. Interestingly, the seeded supramolecular polymerization did not result in the formation of an organogel, but instead afforded a viscous solution, even at a higher concentration (1 mM). We infer that the supramolecular polymers propagated in a controlled fashion in terms of their primary and secondary nucleation processes through the seeded growth. Gratifyingly, very long, individually separated supramolecular polymer chains were observed in the viscous solution (Fig. 3d and e and S16; ESI†).
Solution processing of supramolecular polymer
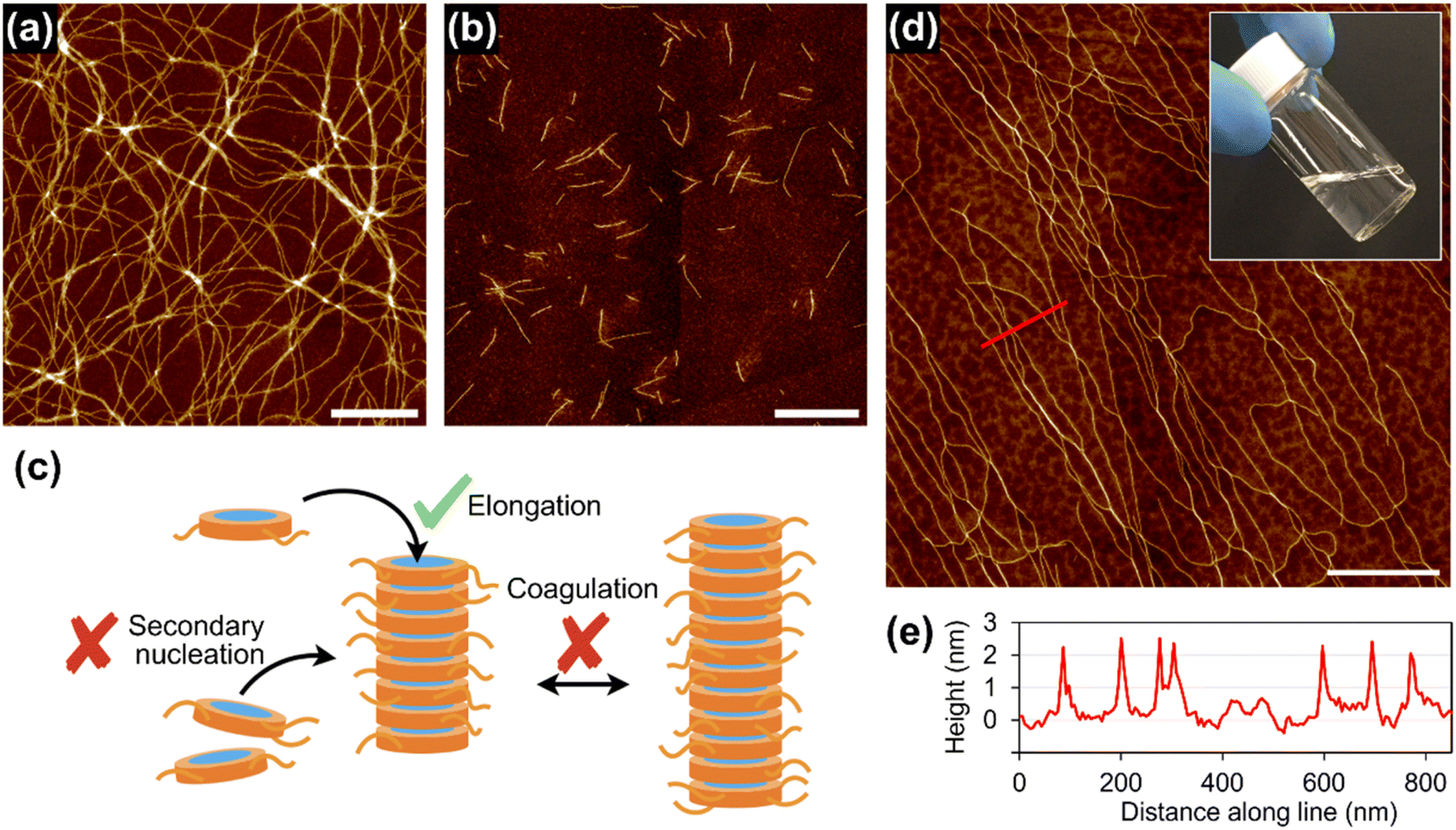
With this concentrated solution of the supramolecular polymer in hand, we were able to fabricate self-standing films (ESI; Fig. S17†) and threads. For example, a thread about 10 cm long was manually drawn from the solution by using a needle (Fig. 5a). Polarized optical microscopy (POM) under crossed polarizers revealed that supramolecular polymers of 1rnd were highly aligned along the thread (Fig. 5b and c). We carried out grazing-incident wide-angle X-ray diffraction (GI-WAXD) measurements with the X-ray beam parallel and perpendicular to the long axis of the threads (Fig. 5f). Although weak in intensity, a diffraction with a d spacing of 3.9 nm, consistent with the diameter of 1rnd, was observed by two-dimensional (2D) WAXD. The diffraction was observed as an anisotropic and an isotropic pattern with respect to the angle β (Fig. 5f) when the incident X-ray beam were perpendicular and parallel to the long axis of the thread, respectively. This result indicates that supramolecular polymers were aligned along the long axis of the thread (Fig. 5g). Remarkably, the dried thread was sufficiently flexible to permit it to be tied in a knot (Fig. 5e). It should be noted that such homogeneous solution processing was not possible with individual 18, 118, or the mixture of 18 and 118 (ESI; Fig. S14†). This result demonstrated an advantage of introducing randomness at the monomer level.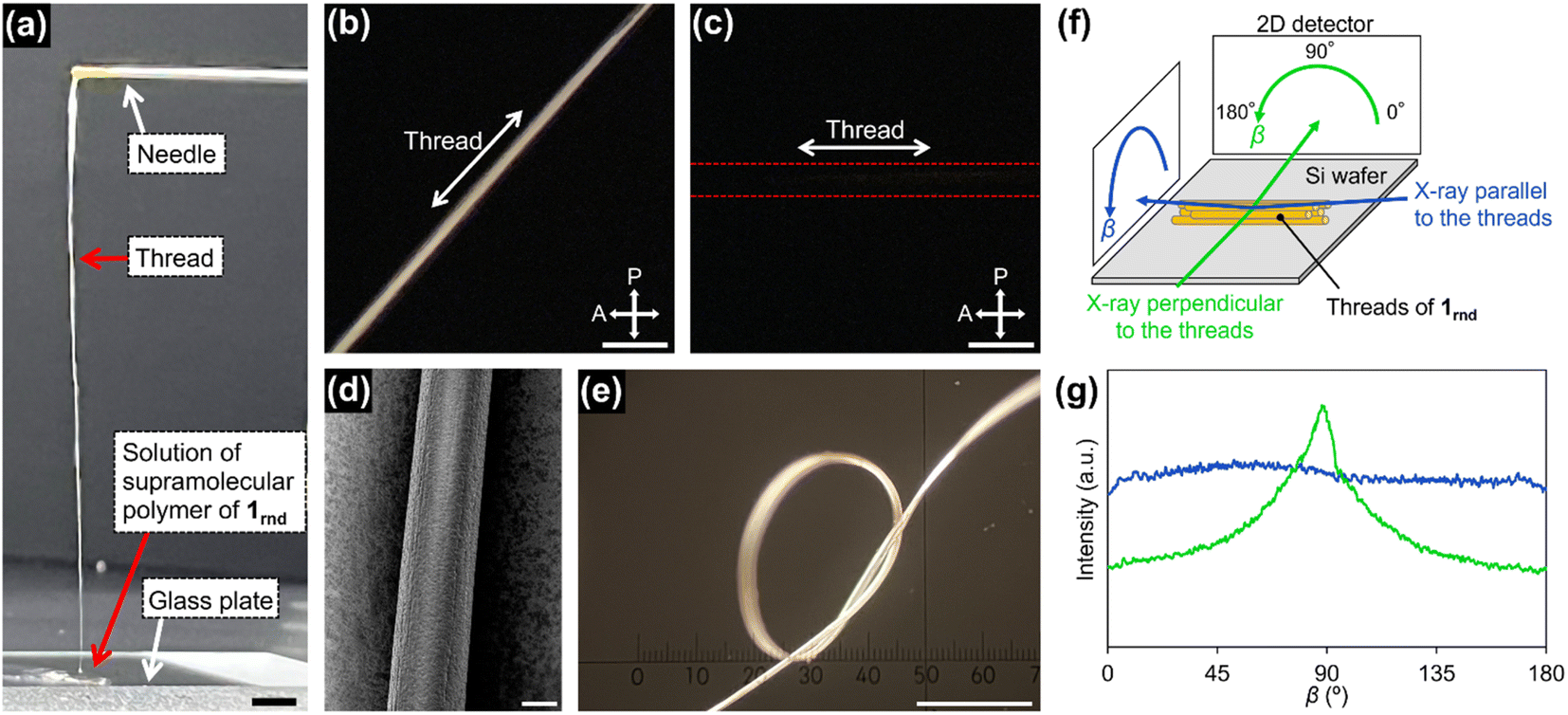 | ||
| Fig. 5 (a) Photograph of a thread drawn from a concentrated solution of 1rnd prepared by seeded polymerization (Fig. 3d): scale bar = 5 mm. POM images of the thread on a glass substrate with the thread rotated (b) 45°and (c) 0°/90°with respect to the polarizer/analyser: scale bar = 100 μm. (d) SEM image of the thread: scale bar = 20 μm. (e) Optical micrograph of a knotted thread: scale bar = 500 μm. (f) Schematic representation of the experimental setup for 2D GI-WAXD. (g) Angle-dependence of the peak intensity for 2θ = 1.7°–2.7° (ca. 3.9 nm) in the 2D GI-WAXD with respect to β, as shown in (f). | ||
Conclusions
In conclusion, we presented a simple molecular-design concept to obtain individually separated supramolecular polymer chains in organic solvents. It has previously been shown that electrostatic repulsion is effective in preventing bundling of supramolecular polymer chains; however, this type of strategy is generally limited to supramolecular polymerization in aqueous media.7 By using the randomized monomer 1rnd, we demonstrated that engineering the surface of supramolecular polymer chains is effective in disturbing lateral interactions, such as chain–chain coagulation and surface-catalyzed secondary nucleation. As a result, concentrated solutions of individually separated supramolecular polymer chains could be prepared that permitted solution processing of the supramolecular polymer. We believe that control over the lateral interaction among supramolecular polymer chains is of great importance not only for creation of higher-order mesoscopic structures5,15 but also for materialization of supramolecular polymers.16 Rheological studies on concentrated solutions, films, and threads containing the resulting supramolecular polymers are in progress.Data availability
The data supporting this study are provided in the ESI.†Author contributions
K. S. conceived the project. T. S. synthesized the monomers, characterized their supramolecular polymerization, and prepared supramolecular polymeric materials. T. K. conducted XRD measurements. All authors discussed the results. T. S. and K. S. wrote the paper with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Kazuo Tanaka (Kyoto University) for use of a polarized optical microscope. The authors thank Prof. Hiroshi Endo (Toyama Prefectural University) for a fruitful discussion. This work was supported by Grants-in-Aid Scientific Research (22H02134), a Grant-in Aid for Scientific Research on Innovative Arias “Soft Crystals” (20H04682), and a Grant-in-Aid for Transformative Research Areas (A) “Condensed Conjugation” (JP20H05868), and Data Creation and Utilization-Type Material Research and Development Project (JPMXP1122714694). Financial support from The Izumi Science and Technology Foundation, The Iketani Science and Technology Foundation, The Murata Science Foundation, Sekisui Chemical Grant Program, and The Mitsubishi Foundation are also acknowledged. T.S. thanks the Japan Society for the Promotion of Science for a research fellowship for young scientist (22J11922).Notes and references
- (a) D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471–3491 RSC; (b) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed; (c) M. Hartlieb, E. D. H. Mansfield and S. Perrier, Polym. Chem., 2020, 11, 1083–1110 RSC.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- (a) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed; (b) J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed; (c) S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed; (d) M. Endo, T. Fukui, S. H. Jung, S. Yagai, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2016, 138, 14347–14353 CrossRef CAS PubMed; (e) M. E. Robinson, A. Nazemi, D. J. Lunn, D. W. Hayward, C. E. Boott, M. Hsiao, R. L. Harniman, S. A. Davis, G. R. Whittell, R. M. Richardson, L. De Cola and I. Manners, ACS Nano, 2017, 11, 9162–9175 CrossRef CAS PubMed; (f) J. S. Valera, R. Gómez and L. Sánchez, Small, 2018, 14, 1702437 CrossRef PubMed; (g) T. Fukui, N. Sasaki, M. Takeuchi and K. Sugiyasu, Chem. Sci., 2019, 10, 6770–6776 RSC.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed.
- N. Sasaki, M. F. J. Mabesoone, J. Kikkawa, T. Fukui, N. Shioya, T. Shimoaka, T. Hasegawa, H. Takagi, R. Haruki, N. Shimizu, S. Adachi, E. W. Meijer, M. Takeuchi and K. Sugiyasu, Nat. Commun., 2020, 11, 3578 CrossRef PubMed.
- (a) S. H. Jung, D. Bochicchio, G. M. Pavan, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2018, 140, 10570–10577 CrossRef CAS PubMed; (b) W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2019, 141, 12044–12054 CrossRef CAS PubMed; (c) A. Sarkar, R. Sasmal, A. Das, S. S. Agasti and S. J. George, Chem. Commun., 2021, 57, 3937–3940 RSC.
- (a) S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. De La Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed; (b) T. Christoff-Tempesta, Y. Cho, D. Kim, M. Geri, G. Lamour, A. J. Lew, X. Zuo, W. R. Lindemann and J. H. Ortony, Nat. Nanotechnol., 2021, 16, 447–454 CrossRef CAS PubMed.
- M. Hecht, B. Soberats, J. Zhu, V. Stepanenko, S. Agarwal, A. Greiner and F. Würthner, Nanoscale Horiz., 2019, 4, 169–174 RSC.
- H. Zhang, J. Cheng, Q. Zhou, Q. Zhang and G. Zou, Soft Matter, 2020, 16, 5203–5209 RSC.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed.
- A. Osypenko, E. Moulin, O. Gavat, G. Fuks, M. Maaloum, M. A. J. Koenis, W. J. Buma and N. Giuseppone, Chem. – Eur. J., 2019, 25, 13008–13016 CrossRef CAS PubMed.
- (a) M. Ikeda, M. Takeuchi and S. Shinkai, Chem. Commun., 2003, 1354–1355 RSC; (b) Y. Tatewaki, T. Hatanaka, M. Kimura and H. Shirai, Chem. Lett., 2009, 38, 900–901 CrossRef CAS; (c) Y. Li, Y. Wang, X. Ren and L. Chen, Mater. Chem. Front., 2017, 1, 2599–2605 RSC; (d) M. L. Ślęczkowski, M. F. J. Mabesoone, P. Ślęczkowski, A. R. A. Palmans and E. W. Meijer, Nat. Chem., 2021, 13, 200–207 CrossRef PubMed; (e) M. L. Ślęczkowski, M. F. J. Mabesoone, M. D. Preuss, Y. Post, A. R. A. Palmans and E. W. Meijer, J. Polym. Sci., 2022, 60, 1871–1877 CrossRef.
- M. Törnquist, T. C. T. Michaels, K. Sanagavarapu, X. Yang, G. Meisl, S. I. A. Cohen, T. P. J. Knowles and S. Linse, Chem. Commun., 2018, 54, 8667–8684 RSC.
- P. E. Laibinis, R. G. Nuzzo and G. M. Whitesides, J. Phys. Chem., 1992, 96, 5097–5105 CrossRef CAS.
- S. Datta, Y. Kato, S. Higashiharaguchi, K. Aratsu, A. Isobe, T. Saito, D. D. Prabhu, Y. Kitamoto, M. J. Hollamby, A. J. Smith, R. Dalgliesh, N. Mahmoudi, L. Pesce, C. Perego, G. M. Pavan and S. Yagai, Nature, 2020, 583, 400–405 CrossRef CAS PubMed.
- R. Laishram, S. Sarkar, I. Seth, N. Khatun, V. K. Aswal, U. Maitra and S. J. George, J. Am. Chem. Soc., 2022, 144, 11306–11315 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06089b |
| This journal is © The Royal Society of Chemistry 2023 |