
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper coordination polymers with selective hole conductivity†
Hannes
Michaels
 ab,
Matthias J.
Golomb
c,
Byeong Jo
Kim
b,
Tomas
Edvinsson
d,
Fabio
Cucinotta
a,
Paul G.
Waddell
a,
Michael R.
Probert
a,
Steven J.
Konezny
ef,
Gerrit
Boschloo
b,
Aron
Walsh
*cg and
Marina
Freitag
*ab
ab,
Matthias J.
Golomb
c,
Byeong Jo
Kim
b,
Tomas
Edvinsson
d,
Fabio
Cucinotta
a,
Paul G.
Waddell
a,
Michael R.
Probert
a,
Steven J.
Konezny
ef,
Gerrit
Boschloo
b,
Aron
Walsh
*cg and
Marina
Freitag
*ab
aSchool of Natural and Environmental Sciences, Newcastle University, Bedson Building, NE1 7RU Newcastle upon Tyne, UK. E-mail: marina.freitag@newcastle.ac.uk
bDepartment of Chemistry – Ångström Laboratory, Uppsala University, P. O. Box 523, SE-75120 Uppsala, Sweden
cDepartment of Materials, Imperial College London, Exhibition Road, London SW7 2AZ, UK. E-mail: a.walsh@imperial.ac.uk
dDepartment of Materials Science and Engineering, Uppsala University, Solid-State Physics, P. O. Box 35, SE-751 03, Uppsala, Sweden
eEnergy Sciences Institute, Yale University, 520 West Campus Drive, West Haven, CT 06516, USA
fDepartment of Chemistry/Physics, Yale University, 225 Prospect Street, New Haven, CT 06511, USA
gDepartment of Materials Science and Engineering, Yonsei University, Seoul 03722, Korea
First published on 17th March 2022
Abstract
Emerging technologies in solar energy will be critical in enabling worldwide society in overcoming the present energy challenges and reaching carbon net zero. Inefficient and unstable charge transport materials limit the current emerging energy conversion and storage technologies. Low-dimensional coordination polymers represent an alternative, unprecedented class of charge transport materials, comprised of molecular building blocks. Here, we provide a comprehensive study of mixed-valence coordination polymers from an analysis of the charge transport mechanism to their implementation as hole-conducting layers. CuII dithiocarbamate complexes afford morphology control of 1D polymer chains linked by (CuI2X2) copper halide rhombi. Concerted theoretical and experimental efforts identified the charge transport mechanism in the transition to band-like transport with a modeled effective hole mass of 6me. The iodide-bridged coordination polymer showed an excellent conductivity of 1 mS cm−1 and a hole mobility of 5.8 10−4 cm2 (V s)−1 at room temperature. Nanosecond selective hole injection into coordination polymer thin films was captured by nanosecond photoluminescence of halide perovskite films. Coordination polymers constitute a sustainable, tunable alternative to the current standard of heavily doped organic hole conductors.
In order to solve the present energy crisis and achieve carbon net zero, emerging energy conversion and storage technologies will be vital. Currently, they are limited by the use of inefficient, unstable and expensive charge transport materials with little tunability. Large knowledge gaps still exist in charge selective and transporting materials for hybrid solar cells.1–4 As a consequence, the development of new charge transport materials is still far behind the efforts that have been made to develop light absorbing materials or other components.5–9
The requirements for charge transport materials in order to perform efficiently in hybrid solar cells are: (i) suitable energy levels adapted to the band gap of the absorbing material, (ii) high conductivity, (iii) facile processability, (iv) chemical and thermal stability, and (v) composition based on abundant elements.
1D and 2D (low dimensional, nanowires and nanosheets) coordination polymers (CPs) represent an unprecedented class of charge transporting materials, which are formed by the assembly of molecular building blocks.10–14 These building blocks consist of a metal coordination complex, preferably based on earth-abundant transition metals and organic/inorganic bridging and coordinating ligands. The tunability of these hybrid materials offers an almost infinite number of chemical, structural and physical properties. This allows the design of functional materials with a variety of useful properties on-demand.
At the transition between molecular hopping and band transport, the control of charge motion in coordination polymers remains challenging.15 The electrical conductivity of coordination polymers can be tuned from semi-conducting to a metallic character, and even superconductivity has been achieved.16–19 Conductivities of up to 2500 S cm−1 have been reported, allowing the application of CPs as e.g. transparent20 and battery electrodes,21 water splitting catalysts22,23 and gas sensors.24 The application of CPs in photovoltaic devices is an intriguing new strategy, given the superior charge transport of photon-separated electrons and holes over small organic molecules.25–28
Perovskite solar cells are an ideal benchmarking system for novel charge transport materials as a low-cost, tunable, and up to 25%-efficient photovoltaic technology.29,30 The chemical nature of halide perovskites demands a high level of compatibility and processability from charge-selective materials; the implementation of sophisticated semiconducting materials therefore remains challenging. Charge transport materials needless of cationic dopants aid the long-term stability of solar cells; furthermore, the cost of organic HTMs and vacuum-evaporated noble metal electrodes has become the main expense in the fabrication of PSCs.31 Based on carbon counter electrodes, HTM-free devices can in principle be fabricated with good efficiencies; however, the direct contact of the absorber with the carbon counter electrode allows undesired electron back-transfer (efficiencies around 16–18%).32–37
This work provides the first comprehensive study from an analysis of the charge transport mechanism in CPs for their implementation in solar cells. We introduce a series of mixed valence CuII/I coordination polymers, where the CuII units are coordinated by two dithiocarbamate groups and linked into 1D coordination polymers through rhombi of two CuI halides (Fig. 1). The tunability of the CPs through their molecular building blocks affords targeted design of amorphous semiconductor thin films, which was studied by electrochemical analysis, Raman spectroscopy and powder, single-crystal and thin-film X-ray diffraction.
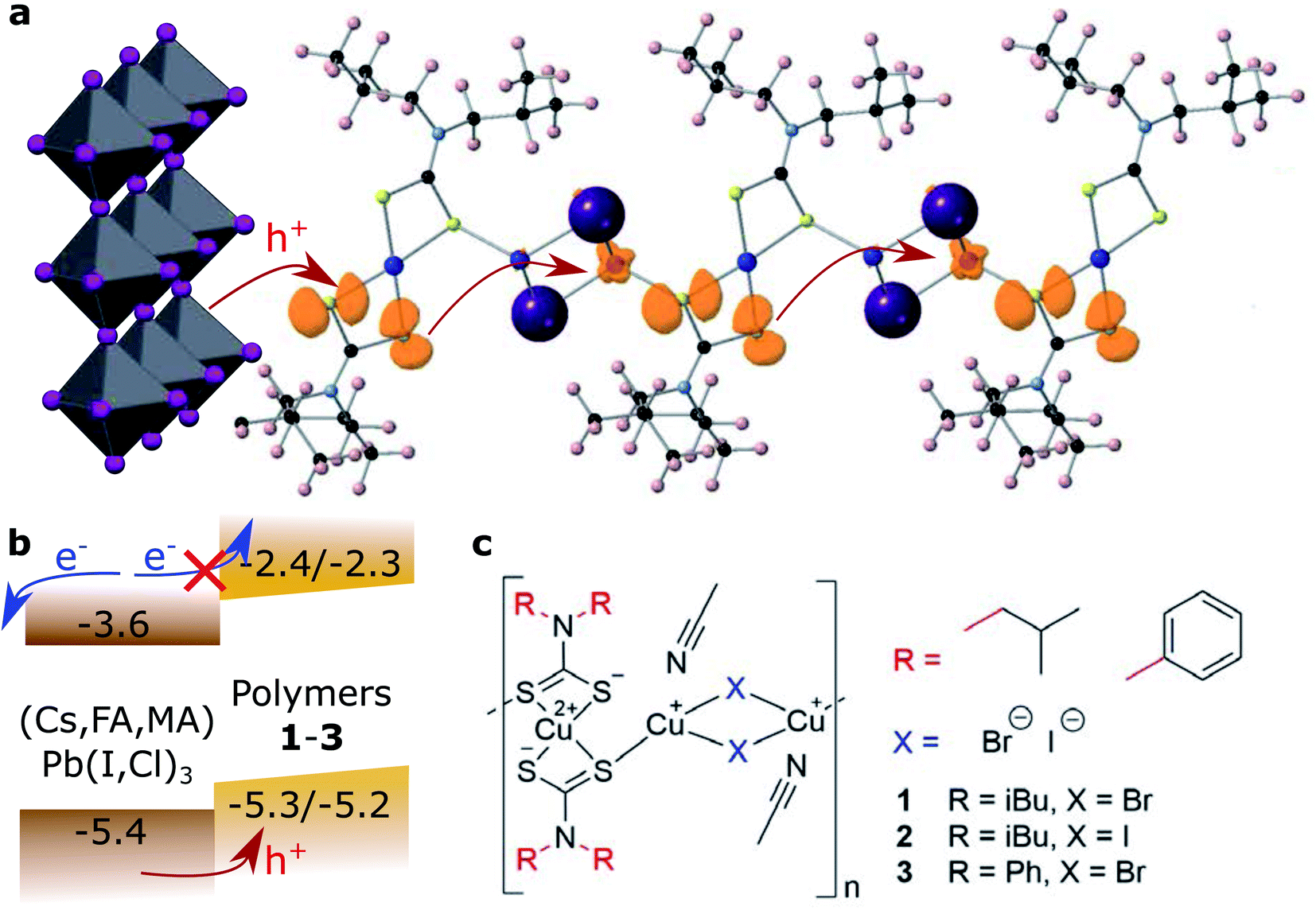 | ||
| Fig. 1 Copper coordination polymers as hole conducting materials. (a) Polarons hop along a computational model of a coordination polymer chain. (b) Energetic position of the frontier orbitals in relation to the halide perovskite valence and conduction band edges. (c) General structure of the coordination polymers presented in this work. | ||
With the synergistic effort between theory and experiment, the rapid charge transport was identified to stem from copper-halide conjugation along the polymer chains. The bridging halide ligands govern the electronic conductivity of the CPs: the iodide-bridged coordination polymer showed excellent electronic properties with a conductivity of 1 mS cm−1 and a hole mobility of 5.8 10−4 cm2 (V s)−1. The modeling of the charge transport based on the copper-halide conjugation along the polymer chains gave a modeled effective hole mass of 6me. In contrast to small molecule semiconductors relying on nearest-neighbour hopping, the CPs exhibit band-like transport at room temperature.
The CPs afford ultrafast charge extraction from halide perovskite films, shown by picosecond photoluminescence spectroscopy. The coordination polymers function as excellent hole conductors in 14%-efficient and durable carbon-based perovskite solar cells – outperforming state-of-the-art organic hole conductors (power conversion efficiency 8%).
Coordination polymers constitute sustainable, tunable alternatives to inefficient, unstable and unsustainable heavily doped organic semiconductors. This report answers a series of fundamental questions regarding heterogeneous charge-transfer in one-dimensional systems, interdependence on ligand groups and the mechanics of charge transport, and brings the anticipated wide-ranging applications of metalorganic conductive materials closer.
Results and discussion
Mixed-valence copper coordination polymers
The general preparation scheme of the presented copper coordination compounds in this work was adapted from Ngo et al.'s38 and Okubo et al.'s39 earlier reports and is documented in the ESI (Fig. S1–S8†). Briefly, the respective secondary amines, diisobutylamine or diphenylamine, were deprotonated (with LiOH·H2O or n-butyllithium) and reacted with carbon disulfide to give the respective dithiocarbamate lithium salts (Li(S2CNR2), compounds 4 and 5). These were reacted with CuCl2·2H2O to give CuII(S2CNR2)2 (compounds 6 and 7). Complexation with CuBr·S(CH3)2 or CuI then gave the respective coordination polymers 1–3. Besides the deprotonation with n-butyllithium, all steps were carried out between room temperature and 80 °C, making this class of materials feasibly accessible. We note that the molecular properties of the here presented coordination polymers, such as the average degree of polymerization, deserve more investigation than provided in this study.Cyclic voltammetry was used to analyse the redox potentials of the coordination polymers (Fig. 2a and Table 1). Two redox waveforms in each voltammogram show the different units comprised in the polymers. The CuII dithiocarbamate units are assigned to the first wave of the polymers (Fig. S10 and Table S1†), and the CuI halide units to the higher-potential wave. Due to larger frontier orbital delocalization, the aromatic diphenylamine dithiocarbamate ligand made the CuII units of polymer 3 somewhat easier to oxidize (0.60 V, all vs. SHE) than their aliphatic counterparts 1 and 2 (0.62 V). On the other hand, the iodide-bridged polymer 2 had a 60 mV more positive second redox wave (0.85 V) compared to both bromide-conjugated polymers 1 and 3 (0.79 V). The electrochemical measurements were repeated in the presence of Lewis base 4-tert-butyl pyridine (Fig. S11 and S12†). In the cyclic voltammograms of the corresponding thin films (Fig. S13†), assuming the copper(I) halide units as primary electron donors for the regeneration of light absorbing materials, we find the valence levels between −5.25 and −5.3 eV vs. the vacuum level, just above e.g. the [Cs,FA,MA]PbI3 valence band, commonly at −5.4 eV.40 The polymers thus appear to be ideal hole acceptor materials.
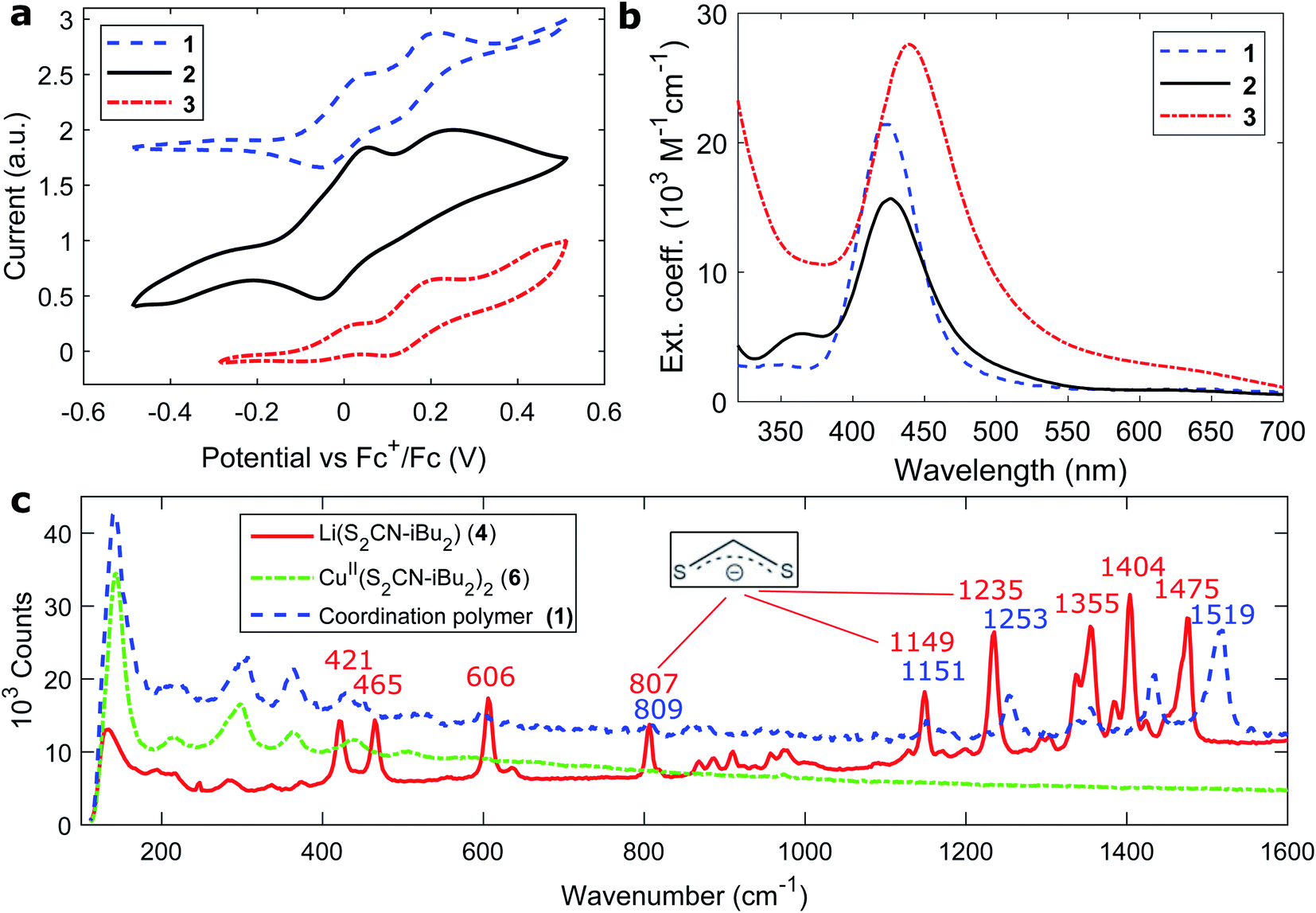 | ||
| Fig. 2 Electronic and structural characterization of the coordination polymers. (a) Cyclic voltammograms and (b) extinction coefficients of coordination polymers 1–3 (per repeating unit). (c) Raman spectra of coordination polymer 1 and its building blocks (compounds 4 and 6, Fig. S1†). | ||
The UV/Vis absorption spectra of the copper coordination polymers are shown in Fig. 2b. From all three compounds 1–3, similar spectra were recorded comprising a single visible transition around 430 nm (Table 1). A small red-shift is observed comparing the aromatic polymer to the aliphatic ones owing to less constraint of exciton localization. The absorption maximum blue-shifts about 10 nm upon polymerization (Fig. S14 and Table S1†). The optically lowest unoccupied states in the polymer lie at −2.3 to −2.4 eV vs. the vacuum level.
We are able to tailor the degree of crystallinity of the CPs through CuII dithiocarbamate ligands: the powder diffraction pattern of the aromatic polymer 3 shows a high level of crystallinity, owing to π–π stacking along the polymers (Fig. S17†). The aliphatic polymers 1 and 2 are less crystalline, indicated by broader powder diffraction peaks. All isolated crystal structures comprised residual unpolymerized trihalide salts of copper dithiocarbamate complexes (Fig. S15 and S16†); however, powder diffraction showed that the identified structures were not representing the bulk compound (Fig. S17 and S18†). Therefore, the ensemble of polymer chains in all materials takes on either polycrystalline forms or amorphous states.
Towards implementation in thin-film devices, compact carrier-selective thin films are required to avoid shunting losses.37 Amorphous semiconductor thin films of all CPs were obtained by solution processing with the aid of a Lewis base, as shown in Fig. S19.†
Fig. 2c presents the Raman spectra of polymer 1 and its building blocks. A series of vibrations could be identified from the ligand, namely C–C (421, 465 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S (807, 1149, 1235 cm−1) and CHx (1300–1500 cm−1) modes. Intriguingly, none of these vibrations were visible when examining the intermediate CuII building block, while reappearing in the final coordination polymers. A similar trend was observed from C–C ring modes in the aromatic analogues (Fig. S20†). The suppression of vibrations might be due to the crystal order in the CuII dithiocarbamate compounds as analyzed by Ngo et al.38 The Raman responses were further compared between powder and processed thin films (Fig. S21†). The Raman spectra did, however, not allow for direct observation of the insertion of copper(I) halide units. Instead, we recorded the FT-infrared spectra of all compounds from building blocks to the coordination polymers; the characteristic vibrations of each compound are listed under the respective preparation schemes for reasons of clarity (Fig. S2–S8†). From the characteristic C
S (807, 1149, 1235 cm−1) and CHx (1300–1500 cm−1) modes. Intriguingly, none of these vibrations were visible when examining the intermediate CuII building block, while reappearing in the final coordination polymers. A similar trend was observed from C–C ring modes in the aromatic analogues (Fig. S20†). The suppression of vibrations might be due to the crystal order in the CuII dithiocarbamate compounds as analyzed by Ngo et al.38 The Raman responses were further compared between powder and processed thin films (Fig. S21†). The Raman spectra did, however, not allow for direct observation of the insertion of copper(I) halide units. Instead, we recorded the FT-infrared spectra of all compounds from building blocks to the coordination polymers; the characteristic vibrations of each compound are listed under the respective preparation schemes for reasons of clarity (Fig. S2–S8†). From the characteristic C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N infrared vibration at 2224 cm−1 of coordinating acetonitrile molecules we observed the appearance of copper(I) halide blocks upon the formation of all polymers (Fig. S9†).
N infrared vibration at 2224 cm−1 of coordinating acetonitrile molecules we observed the appearance of copper(I) halide blocks upon the formation of all polymers (Fig. S9†).
Charge transport
The polymers' hole mobility was measured in the space-charge limited current domain in a FTO/PEDOT:PSS/1–3/Au sandwich. The polymer thin films were deposited with the additive of 4-tert-butylpyridine (tBP) to avoid pinhole formation,43,44 as well as in this case, inhibit crystallization (Fig. S22†). Reference measurements were taken for the well-studied Spiro-MeOTAD (Fig. S25†), both in its pristine and Li/tBP/Co-doped forms (Li: lithium bis(trifluoromethanesulfonyl)imide; Co: tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)cobalt(III) tri[hexafluorophosphate], FK209), and literature values were reproduced in good accordance (Table 2 and Fig. S23†).41,42 Evaluating the presented coordination polymers, the charge mobility in the iodide-conjugated polymer 2 was much superior to both its bromide analogues 1 and 3 (Fig. 3a and S24†). Compared to its lighter homologue, the more polarizable iodide ion forms more covalent copper-halide bonds.45 The hole mobility of iodide-conjugated 2 of 5.7 10−4 cm2 (V s)−1 was greater than that of doped Spiro-MeOTAD, despite not relying on any cationic additives (Table 2). In fact, we found that the addition of salts promotes the nucleation of polymer crystallites and counteracts the formation of uniform thin films. The bromide-conjugated 1 and 3 both showed comparably low hole mobilities. Still, the mobility of the aromatic polymer 3 was 2.5-fold higher than that of the aliphatic 1, likely from π-conjugation between polymer chains.13| RT conductivity (S cm−1) | E A (eV) | ln(A) | μ h (cm2 (V s)−1) | |
|---|---|---|---|---|
| Spiro-MeOTAD (Li/tBP/Co) | 3.7 × 10−4 (5 × 10−4) (ref. 41) | 0.371 | 0.144 | 4.0 × 10−4 (4 × 10−4) (ref. 42) |
| Spiro-MeOTAD (pristine) | 1 × 10−7 (5 × 10−7) (ref. 42) | 0.503 | −0.148 | 8 × 10−5 (1.6 × 10−4) (ref. 42) |
| 1 (tBP) | 3.9 × 10−6 | 1.29 | 2.68 | 1.1 × 10−4 |
| 2 (tBP) | 1.0 × 10−3 | 1.68 | 4.33 | 5.8 × 10−4 |
| 3 (tBP) | 4.9 × 10−6 | 2.01 | 4.34 | 2.4 × 10−4 |
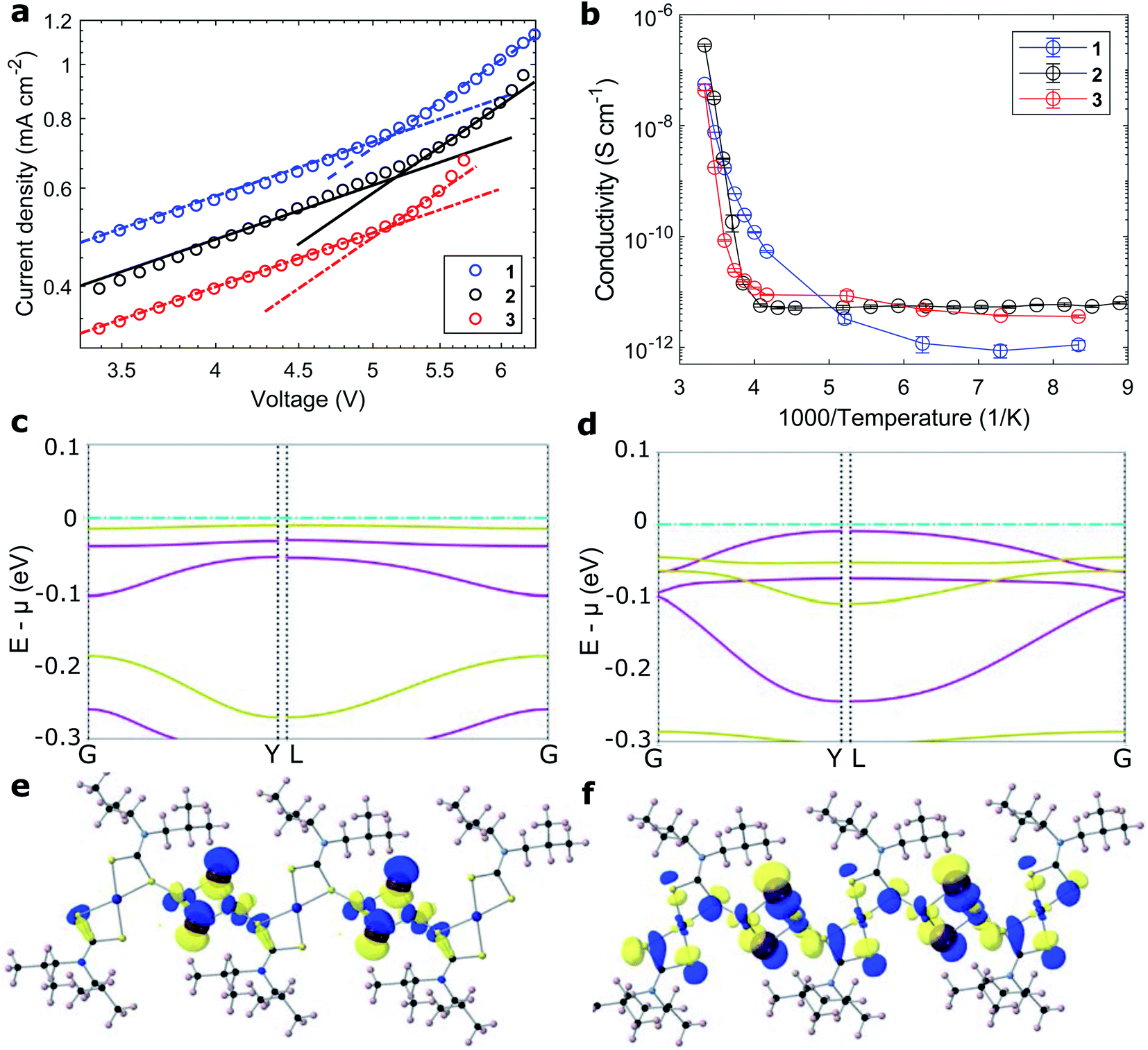 | ||
| Fig. 3 Charge transport in the coordination polymers. (a) Space-charge limited current hole mobility and (b) temperature-dependent conductivity of coordination polymers 1–3. (c and d) Band structure around the valence band of polymers 1 and 2 along high symmetry paths. (e and f) Orbital projections of the top of the valence band for 1 and 2. | ||
Conductivities were measured on fine arrays of interdigitated gold electrodes on sapphire substrates (Fig. S26†). At room temperature, the conductivity of the iodide-bridged polymer 2 of 1 10−3 S cm−1 was two orders of magnitude larger than that of both bromide analogues 1 and 3. The thermal activation of charge transport was characterized by measuring the temperature dependence of the materials' conductivity (Fig. 3b). Moving from the ambient atmosphere to vacuum (while at room temperature), the conductivities of all materials dropped by at least one order of magnitude. This is attributed to oxygen acting as an indirect vacancy dopant and promoting the charge transport, as reported for other organic/hybrid hole-conductors.46 The thermal dependence of conductivity followed an Arrhenius relationship of σ = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−EA/kBT). The extracted barriers for the activation of charge movement were between 1 and 2 eV for polymers 1–3 and as such larger than those extracted for Spiro-MeOTAD (<0.5 eV, Table 2 and Fig. S27†).47 Counterintuitively, the better-conducting polymer 2 had a larger activation energy than the less-conductive bromide analogue 1. The origin for high conductivity at room temperature stems from large pre-exponential coefficients, which were four orders of magnitude larger than that of Spiro-MeOTAD (Table 2).
exp(−EA/kBT). The extracted barriers for the activation of charge movement were between 1 and 2 eV for polymers 1–3 and as such larger than those extracted for Spiro-MeOTAD (<0.5 eV, Table 2 and Fig. S27†).47 Counterintuitively, the better-conducting polymer 2 had a larger activation energy than the less-conductive bromide analogue 1. The origin for high conductivity at room temperature stems from large pre-exponential coefficients, which were four orders of magnitude larger than that of Spiro-MeOTAD (Table 2).
To investigate the effect of conjugating halides on the movement of charges along the polymer chains, we performed calculations based on density functional theory. The structural models were created based on the crystal structure proposed in the work by Okubo et al.,39 with altered side chains attached to the polymer backbone. After a relaxation of the full unit cell, a single polymer chain was extracted as a model system for our calculations. Relaxations were performed with the PBEsol functional until forces were below the default threshold of 0.01 eV Å−1, first on a tier 1 and subsequently on a tier 2 basis set. All following electronic structure calculations then used these relaxed structures and the hybrid functional HSE06. The obtained band structures of 1 and 2 are shown in Fig. 3c and d, along with the orbital projections of their valence bands at the Γ point of the Brillouin zone (Fig. 3e, f and S28†). The superior transport properties of polymer 2 become apparent in the greater curvature of the valence band compared to polymer 1, which, in a band transport picture, is related to greater hole mobility. We used effmass48 for a quantitative analysis of the band structures. This allows for a precise calculation of the band effective mass, taking into account potential deviations from parabolic bands.49 We calculate the effective hole mass of 1 to be 94.2me, around fifteen times higher than that of 2 with 6.4me. These values qualitatively agree with the trend seen in the experimental values of the mobility; however their absolute values point to an intermediate regime between band and hopping transport. It has to be noted that these calculations assume an ordered crystalline structure and can thus only serve as an indication of charge transport behaviour in the real material. For many polymers, however, paracrystallinity does not seem to be detrimental to conduction as long as the backbone remains intact, which is often the case for high molecular weight polymers;50 we thus expect our results to be an upper, but not unrealistic limit. Nonetheless, the projected orbitals as well as the band widths of the respective valence band show higher wave function overlap of states located on copper sites in 2 than in 1.
Concerning hopping transport, an enhanced orbital overlap implies a greater coupling element between hole polarons located on copper sites and thus improved hole transport through the iodine linkers polymer. In an attempt to model polaronic transport directly, we used a number of methods to investigate the localization of polaron states at the copper sites with varying degrees of Hartree–Fock exchange in the HSE06 functional, such as substitution with Ni and initialization with the resulting wave function, or a direct manipulation of the bond lengths of the copper atom, resulting in states such as those in Fig. 1a. We additionally used constrained DFT+U to induce the localization of hole polarons on the three copper sites along the polymer chains and calculated the thermal activation barrier in a diabatic picture (Fig. S29†). The results support the experimental finding of a higher barrier for compound 2 than for compound 1 and confirm that the high mobility can either not be described by band or hopping transport alone, or that inter-chain or inter-phase charge transfer plays an important role.
Evaluating the discussed measurements and computations of electronic exchange in the presented compounds, we are able to decipher the relationship between the structure and mechanisms of charge transport in the coordination polymer compounds. Indeed, the largest fundamental difference between charge transport in the CPs compared to small organic HTMs lies in the four orders of magnitude larger preexponential factors A. The largest effect on electronic properties stems from the halide X linking the polymer chain. The pre-exponential factor in the iodide-conjugated polymer 2 was five-fold higher than that of its bromide analogue 1, and in turn showed two orders of magnitude greater electrical conductivity (1 mS cm−1). The origin is the more polarizable iodide ion, which allows greater orbital conjugation between copper ions across the halide linkers. The result is clearly visible in the computed band structures (Fig. 3c and d), where the iodide conjugated polymer 2 shows much stronger carrier dispersion. The choice of amine ligand R influenced the charge transport to a lesser extent; the aromatic polymer 3 showed a higher conductivity (4.9 10−3 mS cm−1) than aliphatic polymer 1 (3.9 10−3 mS cm−1), and an equally five-fold larger pre-exponential factor, however with a larger activation energy. As seen in Fig. 1a, the frontier orbitals surrounding the copper centers do not strongly localise onto the ligands, therefore, we can conclude that aromatic side groups facilitate the charge exchange between polymer chains rather than acting on the transport along the metal centers. In fact, the orbitals along the polymer chains appear well-conjugated (Fig. 3e and f), such that charge transfer energies of up to 2 eV seem unlikely. Rather, inter-chain hopping might be the limiting step in the charge transport. The charges need to be transferred across non-conjugated aliphatic groups, where a larger barrier appears reasonable. A picture where high-energy inter-chain hopping needs to be activated (large EA) to connect well-conjugated polymer chains (large pre-exponential factor A) seems likely. For the fabrication of thin films, the side moieties played an important role towards solubility in different solvents and wetting of various surfaces.
Due to the unusual behaviour of the investigated compounds compared to other charge transport polymers, that the conductive properties of coordination materials improve upon amorphization, as readily noted by Grätzel and coworkers,51 a study of the inter-chain properties in a static manner is unlikely to capture the important effects. At this point, we were not able to conduct dynamic studies of the materials; this is something we hope to address in the future.
Photovoltaic cells
In contrast to many charge extraction materials that have been demonstrated for photovoltaic cells, copper complexes function as extremely rapid quenchers of light absorbing materials even at driving forces as small as 100 mV.52 Here, we probed the quenching of organohalide perovskites with both steady-state and picosecond photoluminescence spectroscopy (Fig. 4a and b). Halide perovskites were selected as sophisticated light absorbing materials to demonstrate coordination polymers as a new class of charge transport materials, as these perovskites have shown to be challenging materials throughout processing stages. Processed onto a bare microscope slide, [Cs0.01FA0.91MA0.08]PbI3·0.45 MACl perovskite films show strong band-to-band emission at 790 nm (Fig. 4a), in accordance with the incident-photon-to-current-conversion efficiency (IPCE) spectrum (Fig. 4d). When films of hole-accepting coordination polymers were deposited, the relative quenched intensities (e.g. 10% for coordination polymer 2) were significantly lower than those recorded with reference HTMs Spiro-MeOTAD (18%) and poly(3-hexyl thiophene) (P3HT, (32%), Fig. S30a†).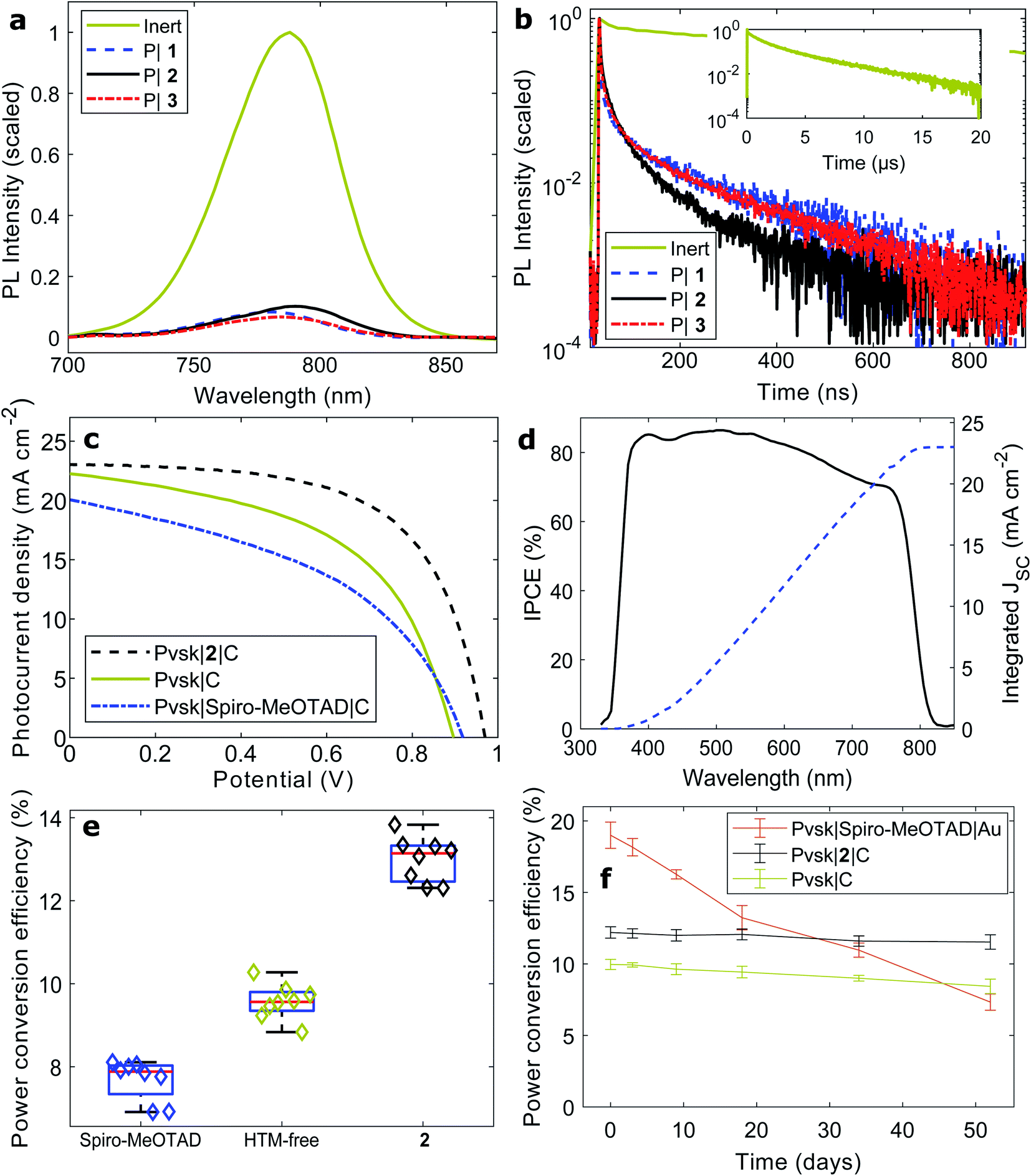 | ||
| Fig. 4 Coordination polymers as hole conductors in photovoltaic cells. (a) Steady-state and (b) time-resolved photoluminescence quenching of halide perovskite films by 1–3. (c) Current–voltage sweeps of perovskite solar cells based on a carbon counter electrode as well as hole conducting materials Spiro-MeOTAD and the best-performing coordination polymer 2. (d) Incident-photon-to-current conversion efficiency. (e) Device statistics. (f) Photovoltaic stability. | ||
Next, we measured time-resolved photoluminescence traces at the emission maximum (Fig. 4b and Table S5†). The two main photoluminescence lifetime constants of bare perovskite layers (1.3 (37%) and 4.3 μs (60%)) correspond very closely to the literature,53 and photoluminescence lifetimes above 1–2 μs are generally considered evidence of high-quality, low-recombination halide perovskite thin films.54 As in the steady-state measurements, we found clear evidence for rapid transfer of charges upon light absorption from the perovskite to the polymer layers. Carrier diffusion lengths generally exceed the film thickness in halide perovskites;55 as such, the photoluminescence lifetime constants can be translated into charge transfer rates across the interface. The quenching time constants of the copper polymers (2: 8 ns (39%) and 64 ns (61%)) were evidently faster than those of Spiro-MeOTAD and P3HT (Fig. S30b†).
Finally, the coordination polymers 1–3 were implemented in perovskite solar cells. Reference cells of the configuration FTO/SnO2/Pvsk/Spiro-MeOTAD/Au were fabricated with 21% efficiency and negligible hysteresis (Fig. S31 and Table S6†). However, when replacing the Spiro-MeOTAD layer with a copper coordination polymer, the cell performance dropped to below 1%, the cause of extremely slow charge exchange between the copper coordination polymer and the noble metal electrode, similar to platinum electrodes in dye-sensitized solar cells.
Much improved results were obtained from solar cells fabricated with carbon electrodes (Fig. 4c–e and Table 3). Compared to HTM-free devices, the current density remained largely unaltered when polymer 2 was deposited as a hole-conducting layer, while the photovoltage as well as fill factor increased to a great extent. The cells reached near 14% power conversion efficiency. In accordance with the experiments above, the iodide-conjugated 2 was found to be the best performing polymer of the three (Fig. S32–S34†). In comparison, a layer of the widely high-performance Spiro-MeOTAD did not increase the performance of carbon-based solar cells, and rather, the efficiency dropped to about 8%. The reasons potentially include both slow charge transfer kinetics at the Spiro-MeOTAD|C interface, as well as the possibility of structural damage to the Spiro-MeOTAD film when in contact with solvents during the deposition of the carbon contact. We believe that the latter mechanism is the reason for the copper polymer devices to have 1–2% conversion efficiency below those with the organic HTM P3HT (Fig. S35 and Table S8†).
| V OC (V) | J SC (mA cm−2) | FF | PCE (%) | |
|---|---|---|---|---|
| Pvsk|Spiro-MeOTAD|C | 0.913 (0.889 ± 0.03) | 19.8 (19.3 ± 68) | 0.451 (0.449 ± 0.02) | 8.11 (7.69 ± 0.49) |
| Pvsk|C | 0.894 (0.875 ± 0.01) | 22.2 (21.8 ± 0.55) | 0.525 (0.501 ± 0.02) | 10.3 (9.57 ± 0.43) |
| Pvsk|2|C | 0.970 (0.956 ± 0.01) | 23.0 (23.0 ± 0.58) | 0.620 (0.591 ± 0.02) | 13.8 (13.0 ± 0.54) |
Carbon-based devices are often subject to large hysteresis due to unbalanced, and overall lower, charge mobility compared to gold electrodes;56 however, carrier abstraction from interfaces has also been shown to play a significant role.57 The insertion of a hole conducting layer of polymer 2 leads to a significant reduction of the hysteresis (hysteresis index from 0.184 to 0.126, Fig. S36 and Table S9†). It has been shown that amine ligands in 2D-layered perovskites contribute to the passivation of exposed PbI2 lattice sites.58,59 The side chain amine moieties on the coordination polymers could therefore bind to perovskite surface states. The maximum power point-tracked conversion efficiency was 10.5% (Fig. S37†).
Ultimately, the greatest remaining challenge to overcome for all third generation photovoltaics is long-term stability. Lead halide perovskites have shown a self-healing mechanism during dark periods, and the community has recently agreed on a number of tests dedicated to quantifying the stability of such solar cells.60 Following an ISOS-D-1 dark storage test, the sensitivity to ambient humidity was tested (Fig. 4f). Over a period of 50 days, the initially most-efficient Spiro-MeOTAD|Au cells fully degraded, owing to the presence of hygroscopic cationic dopants in the Spiro-MeOTAD layer. Both the HTM-free devices (from 9.6 to 8.4%), and especially the copper polymer devices (from 12.6% to 11.9%), maintained their efficiencies as these cells function without cationic dopants.
Conclusion
A series of copper coordination polymers were presented as tunable, low-cost hole-conducting materials with a sustainability and performance edge over their organic counterparts. The charge transport was assigned to polaron hopping along the polymer chains. Therein, the conjugation of copper units by iodide anions rather than bromides provided greater orbital overlap, and the conductivity was 1 mS cm−1. The polymers proved to be rapid quenchers of halide perovskite films, and 14% power conversion efficiency was recorded based on the best-performing polymer 2 with a carbon counter electrode. The copper coordination polymers represent a unique class of sustainable, high-performance hole conducting materials.Many structural parameters of the presented compounds remain yet to be investigated, such as the degree of polymerization, the orientation of polymer chains when deposited into thin films, and the mechanism of charge exchange between polymer chains. Furthermore, it appears that, if the activation energies for polaron hopping could be lowered, either through compositional or morphological engineering, even better performing materials could be designed through a similar scheme. Sophisticated carrier transport measurements could help decipher charge motion and electrode kinetics.
Author contributions
H. M. synthesized and characterized all compounds, performed (if not otherwise listed) all structural and electronic characterization, and fabricated and characterized the solar cells; M. J. G. performed the DFT calculations; B. J. K. aided the solar cell fabrication and collected the thin-film XRD spectra; T. E. collected the Raman spectra; F. C. aided the collection of time-resolved photoluminescence spectra; P. G. W. and M. R. P. analyzed and refined the crystal structures; S. J. K. aided the measurement of temperature-dependent conductivity; G. B., S. J. K. and A. W. aided M. F. in the conceptualization of the study; M. F. conceived the idea and supervised the project; all authors contributed to the discussion of results and the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
In 2017, the group of Prof. Dr Wilfried Wurth (University of Hamburg/DESY) and Dr Giuseppe Mercurio helped characterize thin films of our coordination polymers with X-ray photoelectron spectroscopy. Prof. Wurth passed away unexpectedly in May 2019 at the age of 62 and our heartfelt condolence shall be expressed to those around him. We appreciate experimental assistance from Aneta Andruszkiewicz, Zackary Ashworth, Olle Gustafsson (Uppsala) and Dr Jamie Gould (Newcastle) and owe gratitude to Dr Pablo Docampo (Glasgow/Newcastle) for discussions and access to his laboratory. Our appreciation goes out to Prof. Andrew Houlton for helpful suggestions and discussion. M. F. acknowledges the support by the Royal Society University Research Fellowship (URF\R1\191286), Research Grant 2021 (RGS\R1\211321), Göran Gustafsson Young Researcher Prize and EPSRC New Investigator Award (EP/V035819/1). H. M. acknowledges support from the European KIT InnoEnergy PhD program. M. J. G. is funded by the Royal Society of Chemistry. A. W. is supported by a Royal Society University Research Fellowship. Via the membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work used the ARCHER UK National Supercomputing Service. We are grateful to the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1). This research was supported by the Swedish Energy Agency (42037-1 and 43294-1), the STandUP for Energy program, and the National Research Foundation of Korea (NRF-2020R1A6A3A03039130). The authors thank the Diamond Light Source for access to beamline I19 in remote-access mode (beam-time award CY22240).Notes and references
- Z. Hawash, L. K. Ono, S. R. Raga, M. V. Lee and Y. Qi, Chem. Mater., 2015, 27, 562–569 CrossRef CAS.
- L. K. Ono, S. R. Raga, M. Remeika, A. J. Winchester, A. Gabe and Y. Qi, J. Mater. Chem. A, 2015, 3, 15451–15456 RSC.
- M. L. Petrus, Y. Hu, D. Moia, P. Calado, A. M. A. Leguy, P. R. F. Barnes and P. Docampo, ChemSusChem, 2016, 9, 2699–2707 CrossRef CAS PubMed.
- J.-Y. Seo, H.-S. Kim, S. Akin, M. Stojanovic, E. Simon, M. Fleischer, A. Hagfeldt, S. M. Zakeeruddin and M. Grätzel, Energy Environ. Sci., 2018, 11, 2985–2992 RSC.
- M. K. Kashif, R. A. Milhuisen, M. Nippe, J. Hellerstedt, D. Z. Zee, N. W. Duffy, B. Halstead, F. De Angelis, S. Fantacci, M. S. Fuhrer, C. J. Chang, Y.-B. Cheng, J. R. Long, L. Spiccia and U. Bach, Adv. Energy Mater., 2016, 6, 1600874 CrossRef.
- M. K. Kashif, I. Benesperi, R. A. Milhuisen, S. Meyer, J. Hellerstedt, D. Zee, N. W. Duffy, B. Halstead, M. S. Fuhrer, J. Cashion, Y.-B. Cheng, L. Spiccia, A. N. Simonov and U. Bach, ACS Energy Lett., 2017, 2, 1855–1859 CrossRef CAS.
- T. Duong, J. Peng, D. Walter, J. Xiang, H. Shen, D. Chugh, M. Lockrey, D. Zhong, J. Li, K. Weber, T. P. White and K. R. Catchpole, ACS Energy Lett., 2018, 3, 2441–2448 CrossRef CAS.
- C.-D. Si, X.-D. Lv and S.-J. Long, Inorg. Chem. Commun., 2020, 112, 107701 CrossRef CAS.
- Z. Yu, A. Hagfeldt and L. Sun, Coord. Chem. Rev., 2020, 406, 213143 CrossRef CAS.
- N. Singh, S. Gupta and R. K. Sinha, Inorg. Chem. Commun., 2003, 6, 416–422 CrossRef CAS.
- L. Sun, C. H. Hendon, S. S. Park, Y. Tulchinsky, R. Wan, F. Wang, A. Walsh and M. Dincă, Chem. Sci., 2017, 8, 4450–4457 RSC.
- L. S. Xie, L. Sun, R. Wan, S. S. Park, J. A. DeGayner, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2018, 140, 7411–7414 CrossRef CAS PubMed.
- C. Hua, P. W. Doheny, B. Ding, B. Chan, M. Yu, C. J. Kepert and D. M. D'Alessandro, J. Am. Chem. Soc., 2018, 140, 6622–6630 CrossRef CAS PubMed.
- G. H. Morritt, H. Michaels and M. Freitag, Chem. Phys. Rev., 2022, 3, 011306 CrossRef.
- J. Calbo, M. J. Golomb and A. Walsh, J. Mater. Chem. A, 2019, 7, 16571–16597 RSC.
- G. Givaja, P. Amo-Ochoa, C. J. Gómez-García and F. Zamora, Chem. Soc. Rev., 2012, 41, 115–147 RSC.
- X. Huang, P. Sheng, Z. Tu, F. Zhang, J. Wang, H. Geng, Y. Zou, C.-a. Di, Y. Yi, Y. Sun, W. Xu and D. Zhu, Nat. Commun., 2015, 6, 7408 CrossRef CAS PubMed.
- K.-H. Low, V. A. L. Roy, S. S.-Y. Chui, S. L.-F. Chan and C.-M. Che, Chem. Commun., 2010, 46, 7328–7330 RSC.
- J. M. Williams, A. J. Schultz, U. Geiser, K. D. Carlson, A. M. Kini, H. H. Wang, W.-K. Kwok, M.-H. Whangbo and J. E. Schirber, Science, 1991, 252, 1501–1508 CrossRef CAS PubMed.
- Z. Jin, J. Yan, X. Huang, W. Xu, S. Yang, D. Zhu and J. Wang, Nano Energy, 2017, 40, 376–381 CrossRef CAS.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS PubMed.
- D. Shi, R. Zheng, M.-J. Sun, X. Cao, C.-X. Sun, C.-J. Cui, C.-S. Liu, J. Zhao and M. Du, Angew. Chem., Int. Ed., 2017, 56, 14637–14641 CrossRef CAS PubMed.
- X. Huang, H. Yao, Y. Cui, W. Hao, J. Zhu, W. Xu and D. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 40752–40759 CrossRef CAS PubMed.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- L. Qiu, X. Zheng, Y. Yang, Y. Dong, G. Dong, D. Xia, X. Liu, Q. Wu and R. Fan, ChemSusChem, 2019, 12, 2763–2772 CrossRef CAS PubMed.
- Y. Dong, J. Zhang, Y. Yang, L. Qiu, D. Xia, K. Lin, J. Wang, X. Fan and R. Fan, Angew. Chem., Int. Ed., 2019, 58, 17610–17615 CrossRef CAS PubMed.
- J. Wang, J. Zhang, Y. Yang, S. Gai, Y. Dong, L. Qiu, D. Xia, X. Fan, W. Wang, B. Hu, W. Cao and R. Fan, ACS Appl. Mater. Interfaces, 2021, 13, 5235–5244 CrossRef CAS PubMed.
- S. Wu, Z. Li, M.-Q. Li, Y. Diao, F. Lin, T. Liu, J. Zhang, P. Tieu, W. Gao, F. Qi, X. Pan, Z. Xu, Z. Zhu and A. K.-Y. Jen, Nat. Nanotechnol., 2020, 15, 934–940 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- J. M. Kadro and A. Hagfeldt, Joule, 2017, 1, 29–46 CrossRef.
- S. Liu, S. Li, J. Wu, Q. Wang, Y. Ming, D. Zhang, Y. Sheng, Y. Hu, Y. Rong, A. Mei and H. Han, J. Phys. Chem. Lett., 2019, 10, 6865–6872 CrossRef CAS PubMed.
- Y. Wang, H. Zhao, Y. Mei, H. Liu, S. Wang and X. Li, ACS Appl. Mater. Interfaces, 2019, 11, 916–923 CrossRef CAS PubMed.
- F. Meng, Y. Li, L. Gao, A. Liu, Y. Li, T. Wang, C. Zhang, M. Fan, G. Wei and T. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 34479–34486 CrossRef CAS PubMed.
- M. Que, B. Zhang, J. Chen, X. Yin and S. Yun, Mater. Adv., 2021, 2, 5560–5579 RSC.
- J. Zhou, Z. Ye, J. Hou, J. Wu, Y.-Z. Zheng and X. Tao, J. Mater. Chem. A, 2018, 6, 22626–22635 RSC.
- J. Lee, M. M. Menamparambath, J.-Y. Hwang and S. Baik, ChemSusChem, 2015, 8, 2358–2362 CrossRef CAS PubMed.
- S. C. Ngo, K. K. Banger, M. J. DelaRosa, P. J. Toscano and J. T. Welch, Polyhedron, 2003, 22, 1575–1583 CrossRef CAS.
- T. Okubo, N. Tanaka, K. H. Kim, H. Yone, M. Maekawa and T. Kuroda-Sowa, Inorg. Chem., 2010, 49, 3700–3702 CrossRef CAS PubMed.
- N.-G. Park, Mater. Today, 2015, 18, 65–72 CrossRef CAS.
- J. Burschka, A. Dualeh, F. Kessler, E. Baranoff, N.-L. Cevey-Ha, C. Yi, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2011, 133, 18042–18045 CrossRef CAS PubMed.
- H. J. Snaith and M. Grätzel, Appl. Phys. Lett., 2006, 89, 262114 CrossRef.
- E. J. Juarez-Perez, M. R. Leyden, S. Wang, L. K. Ono, Z. Hawash and Y. Qi, Chem. Mater., 2016, 28, 5702–5709 CrossRef CAS.
- S. Wang, M. Sina, P. Parikh, T. Uekert, B. Shahbazian, A. Devaraj and Y. S. Meng, Nano Lett., 2016, 16, 5594–5600 CrossRef CAS PubMed.
- A. J. Clough, J. M. Skelton, C. A. Downes, A. A. de la Rosa, J. W. Yoo, A. Walsh, B. C. Melot and S. C. Marinescu, J. Am. Chem. Soc., 2017, 139, 10863–10867 CrossRef CAS PubMed.
- U. B. Cappel, T. Daeneke and U. Bach, Nano Lett., 2012, 12, 4925–4931 CrossRef CAS PubMed.
- Y. Saygili, M. Stojanovic, H. Michaels, J. Tiepelt, J. Teuscher, A. Massaro, M. Pavone, F. Giordano, S. M. Zakeeruddin, G. Boschloo, J.-E. Moser, M. Grätzel, A. B. Muñoz-García, A. Hagfeldt and M. Freitag, ACS Appl. Energy Mater., 2018, 1, 4950–4962 CrossRef CAS.
- L. D. Whalley, effmass: An effective mass package, 2018 Search PubMed.
- L. D. Whalley, J. M. Frost, B. J. Morgan and A. Walsh, Phys. Rev. B, 2019, 99, 085207 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- Y. Cao, Y. Saygili, A. Ummadisingu, J. Teuscher, J. Luo, N. Pellet, F. Giordano, S. M. Zakeeruddin, J. E. Moser, M. Freitag, A. Hagfeldt and M. Grätzel, Nat. Commun., 2017, 8, 15390 CrossRef PubMed.
- Y. Saygili, M. Söderberg, N. Pellet, F. Giordano, Y. Cao, A. B. Muñoz-García, S. M. Zakeeruddin, N. Vlachopoulos, M. Pavone, G. Boschloo, L. Kavan, J.-E. Moser, M. Grätzel, A. Hagfeldt and M. Freitag, J. Am. Chem. Soc., 2016, 138, 15087–15096 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- J. M. Richter, M. Abdi-Jalebi, A. Sadhanala, M. Tabachnyk, J. P. Rivett, L. M. Pazos-Outón, K. C. Gödel, M. Price, F. Deschler and R. H. Friend, Nat. Commun., 2016, 7, 13941 CrossRef CAS PubMed.
- A. A. B. Baloch, F. H. Alharbi, G. Grancini, M. I. Hossain, M. K. Nazeeruddin and N. Tabet, J. Phys. Chem. C, 2018, 122, 26805–26815 CrossRef CAS.
- P. Liu, W. Wang, S. Liu, H. Yang and Z. Shao, Adv. Energy Mater., 2019, 9, 1803017 CrossRef.
- D.-H. Kang and N.-G. Park, Adv. Mater., 2019, 31, 1805214 CrossRef PubMed.
- G. Grancini, C. Roldán-Carmona, I. Zimmermann, E. Mosconi, X. Lee, D. Martineau, S. Narbey, F. Oswald, F. De Angelis, M. Graetzel and M. K. Nazeeruddin, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- C. Liu, J. Sun, W. L. Tan, J. Lu, T. R. Gengenbach, C. R. McNeill, Z. Ge, Y.-B. Cheng and U. Bach, Nano Lett., 2020, 20, 1240–1251 CrossRef CAS PubMed.
- M. V. Khenkin, E. A. Katz, A. Abate, G. Bardizza, J. J. Berry, C. Brabec, F. Brunetti, V. Bulović, Q. Burlingame, A. Di Carlo, R. Cheacharoen, Y.-B. Cheng, A. Colsmann, S. Cros, K. Domanski, M. Dusza, C. J. Fell, S. R. Forrest, Y. Galagan, D. Di Girolamo, M. Grätzel, A. Hagfeldt, E. von Hauff, H. Hoppe, J. Kettle, H. Köbler, M. S. Leite, S. F. Liu, Y.-L. Loo, J. M. Luther, C.-Q. Ma, M. Madsen, M. Manceau, M. Matheron, M. McGehee, R. Meitzner, M. K. Nazeeruddin, A. F. Nogueira, Ç. Odabaşı, A. Osherov, N.-G. Park, M. O. Reese, F. De Rossi, M. Saliba, U. S. Schubert, H. J. Snaith, S. D. Stranks, W. Tress, P. A. Troshin, V. Turkovic, S. Veenstra, I. Visoly-Fisher, A. Walsh, T. Watson, H. Xie, R. Yıldırım, S. M. Zakeeruddin, K. Zhu and M. Lira-Cantu, Nat. Energy, 2020, 5, 35–49 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2085154 and 2085155. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ta00267a |
| This journal is © The Royal Society of Chemistry 2022 |