DOI:
10.1039/D2PY01161A
(Paper)
Polym. Chem., 2022,
13, 6312-6321
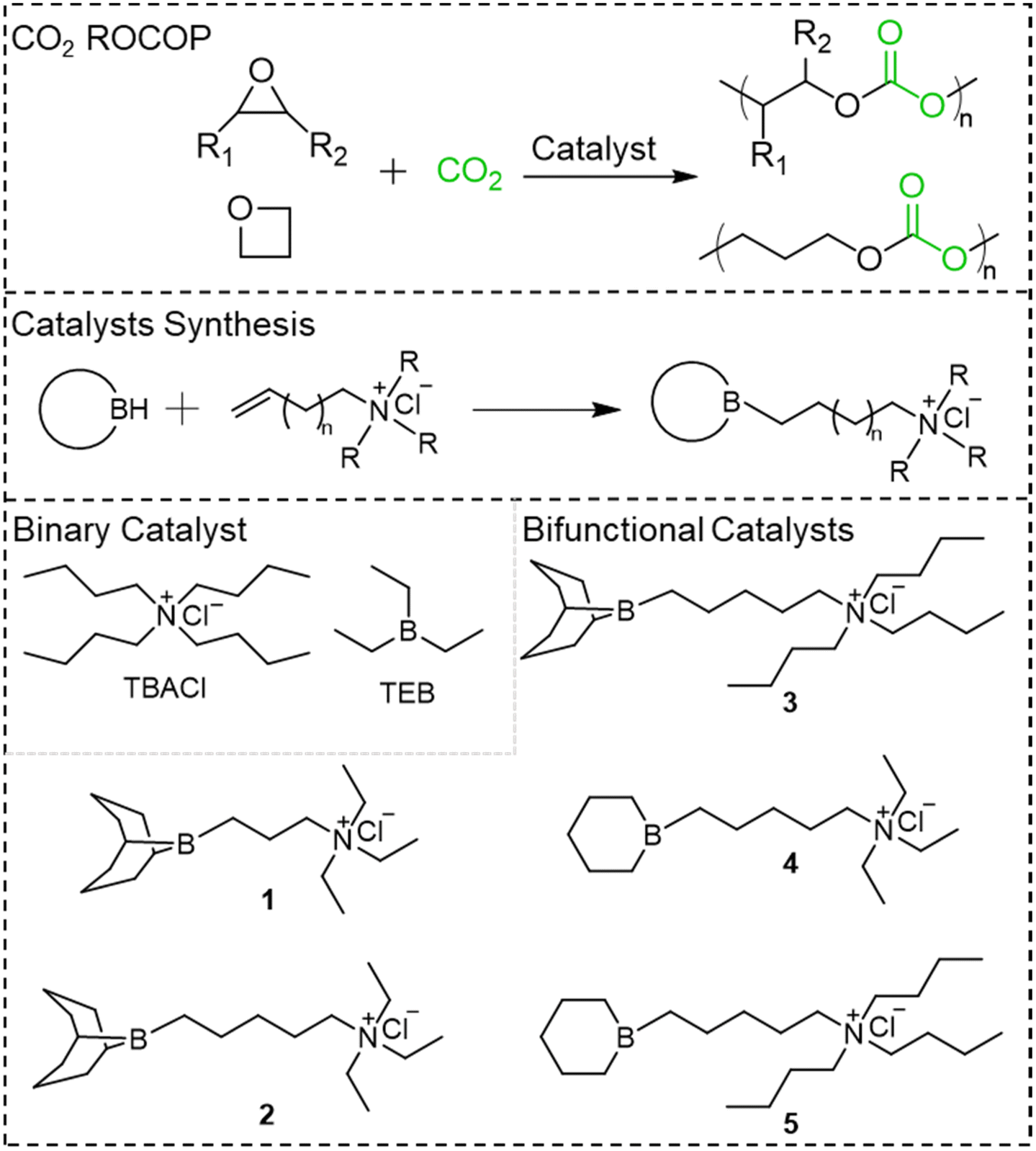
Borinane-based organoboron catalysts for alternating copolymerization of CO2 with cyclic ethers: improved productivity and facile recovery†
Received
8th September 2022
, Accepted 25th October 2022
First published on 1st November 2022
Abstract
The design and synthesis of organoboron catalysts for effective alternating ring-opening copolymerization (ROCOP) of CO2 with cyclic ethers are described. Such organoboron catalysts include an ammonium salt which is connected to a boron center, separated from the ammonium cation by a few carbon–carbon bonds. The type of boron center whether it was carried by borinane or by 9-BBN, the distance separating the boron center from the ammonium cation, and the size of the substituents of the cation were the various parameters considered when assessing the performance of these organoboron catalysts. Of all the boron-based catalysts, the borinane-based catalyst 5 enabled the copolymerization of CO2 and epoxides with the highest productivity: 271.5 g of poly(propylenecarbonate) per g of catalyst and 5.7 kg of poly(cyclohexene carbonate) per g of catalyst, respectively. In addition, 5 could be easily recycled via a simple precipitation process and also used as a difunctional initiator when combined with a diacid. Density Functional Theory computation gives a deep insight into the mechanism underlying the bifunctional catalyst-mediated CO2 copolymerization and reveals the importance of the type of boron centers in the overall performance.
Introduction
With the concept of carbon neutrality gaining momentum, reducing CO2 emission and developing technologies for its capture become significant research topics, aiming at achieving a net-zero carbon emission world. Apart from the catalytic transformation of CO2 to a C1 source for small molecules, the direct copolymerization of CO2 with cyclic ethers provides an effective pathway for the synthesis of polycarbonates and the transformation of CO2 into valuable materials.1–9 For instance, the alternating ROCOP of CO2 and propylene oxide (PO) affords polymeric materials incorporating no less than 43 wt% of CO2 that can serve as substitutes for polyethers which are normally generated from fossil resources. Interest in CO2-based polycarbonates is also spurred by their promising applications contemplated for materials such as films, coatings, adhesives, and elastomers.3,10,11 For instance, low molar mass polycarbonate diols are seen as promising and eco-friendly precursors for the preparation of polyurethane (PU) which represents a 22 megaton market.12 Generally, specific catalysts are necessary for the coupling of CO2 and epoxides towards the selective synthesis of polycarbonates. The past 50 years have witnessed the rapid development of metal catalysts for effective ROCOP of epoxides with CO2 and its terpolymerization with other monomers. Transition metals including Zn,13–16 Fe,17,18 Co,19–28 and Cr,26,29 main group metals including Mg30,31 and Al,32–34 and lanthanides including La and Lu,35,36 associated with sophisticated organic ligands, have been designed and shown to successfully trigger the CO2 copolymerization with epoxides. Some state-of-the-art metallic catalysts have demonstrated exceptional activities by dramatically reducing the activation energy of CO2/epoxide coupling. Nevertheless, the multi-step synthesis of specific ligands and the tedious removal of metal residues (especially for toxic cobalt and chromium catalysts) have been a challenge for a large scale implementation of ROCOP of CO2 and epoxides using these metallic catalysts.
For a long time, these metallic systems were the only available means to efficiently copolymerize CO2 with epoxides towards polycarbonates. In 2016, our group reported the first metal-free catalyst based on triethylborane (TEB) associated with onium salts as an efficient system to copolymerize CO2 with epoxides.37 TEB in such systems plays a double role in this copolymerization: (1) it forms an “ate complex” with the growing oxyanion which helps to decrease the nucleophilicity of the latter reactive species and thus prevent side reactions38 and (2) it also activates the monomer. Since then, TEB was widely used not only in the copolymerization of CO2 or COS with epoxides, but also in the copolymerization of epoxides with anhydrides, isocyanates, etc.39–45 One advantage associated with the use of the TEB/onium salt system is the absence of any metallic residues in the polycarbonates synthesized, thus making this organic system particularly attractive for the synthesis of polycarbonates for biomedical applications and others. Moreover, CO2-based polycarbonates with various architectures were further designed and synthesized.46–48 When applied to synthesize small molar mass polycarbonates a high amount of TEB—equivalent to that of the initiator—is needed, which is a drawback of this TEB/onium salt system. We thus developed a method to recycle both TEB and onium salt to make the process of synthesizing polycarbonate polyols more economic.43 But such a recovery process required two additional chemicals and high labor costs. Efforts have thus been made by other groups to limit the use of TEB when copolymerizing CO2 and PO. Using stoichiometric amounts of trialkylborane and tertiary amine as a Lewis pair, Zhang et al.39 reported the synthesis of alternating polycarbonate through zwitterionic chain growth. In this case, a tertiary amine was incorporated into the poly(propylene carbonate) chain, and thus, only the borane could be eventually recycled for further polymerization. Recently, Wu et al.49–52 and Lin et al.53 reported a series of 9-borabicyclo[3.3.1]nonane (9-BBN) based bifunctional catalysts for the effective copolymerization of CO2 with CHO and epichlorohydrin. In the case of a monomer of industrial relevance such as PO, those catalysts could regulate the content of carbonate and afford a carbonate linkage content of 95.2%.54 Based on our experience in boron-mediated ROCOP of CO2 and PO, we envision that a perfect alternating carbonate linkage and increased reactivity could be achieved by fine tuning the steric hindrance and Lewis acidity of boron centers in such bifunctional systems. Overall, it is still a challenge to bring about the organocatalytic copolymerization of CO2 and PO to access perfectly alternating polycarbonates and effective catalyst recovery.
Even though TEB and the organic cation that contributes to the initiating system can be recycled in high yield, we confirm that its activity in the ROCOP of CO2 with PO is lower than that of the state-of-the-art metal catalysts. To further improve the activity of these boron-based catalytic systems and simplify the recycling process, we synthesized a series of borinane-based bifunctional catalysts specifically aimed at the ROCOP of CO2 with cyclic ethers (Scheme 1). It is worthy to note that these catalysts are oxygen sensitive. Since the steric hindrance around boron centers and the size of the substituents of the onium cation are crucial to the copolymerization of CO2 and PO, we investigated the relationship between the structures of these catalysts and their catalytic performance. For instance, the productivity achieved by 5 increased up to 271.5 g of PPC per g of catalyst. In addition, this borinane-based bifunctional catalyst showed applicability for a broad range of monomers including various epoxides and oxetane, resulting in the synthesis of C2 and C3-based polycarbonates. Furthermore, the same bifunctional catalyst 5 could be easily recycled by the addition of a diacid followed by a simple filtration procedure. The catalyst formed was then used to generate polycarbonate diols through bidirectional ROCOP of CO2 with PO. Density Functional Theory (DFT) computation revealed the catalytic pathway and polymerization mechanism. The outstanding catalytic performance and reusability of such borinane-based bifunctional catalysts make them viable candidates for the industrial production of PPC polyols.
 |
| | Scheme 1 Catalyst mediated CO2 copolymerization with epoxides and oxetane; binary and bifunctional catalysts synthesized and screened in this work. | |
Synthesis and characterization of bifunctional catalysts
Whether they are part of a binary (TEB/onium salts)37 or of a bifunctional system (this study), the type of organoboranes plays a crucial role in the ROCOP of CO2 with epoxides. Steric hindrance around boron centers and the Lewis acidity are two criteria influencing the overall performance of boron-based catalysts. For instance, bulky triphenylborane has a higher Lewis acidity than TEB but is unfit for the ROCOP of PO with CO2. Another criterion that was shown to influence the activity of these catalysts is the spatial proximity between boron centers and the growing active species: we thus endeavored to construct bifunctional organoboron catalysts that feature the same steric characteristics as those exhibited by TEB and thus synthesized the six-membered cyclic borinane. As shown in Scheme S1,† pure borinane was synthesized via Brown's method55 with a yield of up to 95%. Once a small quantity of borinane was obtained it was used as seeds for amplification: 2 moles of borinane was used for hydroboration of 1 mole of pentadiene followed by a treatment with 1 mole of borane to afford 3 moles of borinane without any purification procedure. The corresponding ammonium salts were synthesized via the Menshutkin reaction by quaternization of a tertiary amine with alkenyl bromide (Scheme S2†). Upon hydroboration of the alkenyl group carried by the above ammonium salts with borinane, bifunctional catalysts 4 and 5 could be easily prepared in quantitative yield, and the structures of the obtained catalysts were determined by 1H NMR, 13C NMR, and 11B NMR spectroscopy (Fig. S16–21†). As for bifunctional catalysts 1–3 fitted with a boron center carried by 9-BBN, they were prepared using a method derived from the work of Wu.49
The relationship between the catalyst structure and catalytic performance
In the case of the binary system made of TEB and ammonium salts, theoretically two moles of TEB are necessary for an effective ROCOP of CO2 and epoxides resulting in a polymer with a high content of carbonates. As shown in Table 1 entries 1 and 2, with just one mole ratio of TEB for one mole of ammonium salt, the linear vs. cyclic carbonate selectivity is poor. As for bifunctional systems that benefit from the synergistic effect present in a unimolecular compound of a boron center connected to the ammonium cation of the initiating site, we systematically assessed the influence of their structure and catalytic performance. A comprehensive study of the effect of the type of boron, of the spatial proximity between the boron center and the ammonium cation, and of the size of cation substituents on the ROCOP of CO2 with PO is thus carried out (Table 1). Catalyst 1, with its bulky 9-BBN and a short trimethylene linker between the ammonium cation and the boron center, actually promoted the formation of cyclic carbonate (entries 3 and 4, Table 1). Upon extending the linker length between the boron center and the ammonium cation in 2 the linear vs. cyclic carbonate could dramatically be improved (entries 5 and 6, Table 1), indicating that the distance between the boron center and N+ is a key parameter for the high selectivity of linear polycarbonates. Compared to catalyst 2, catalyst 3 with its bulkier cation N+ due to its larger substituents gives an even better selectivity (entries 7 and 8, Table 1) which reveals that the effect of coulombic interaction between the ammonium cation and the growing anion is also a key parameter favoring the formation of linear polycarbonates. In an attempt to improve the performance of these bifunctional catalysts, we decreased the steric hindrance around the boron center and replaced the bulky 9-BBN with the newly designed borinane. As expected catalyst 4 with its less crowded boron center showed higher TOF values (entries 9 and 10, Table 1) than its analogue catalyst 2 but its linear vs. cyclic carbonate selectivity had to be improved. Observing that the tetrabutyl substituents of ammonium cation disfavor the back biting reaction, we then synthesized 5 with borinane as the boron center. As expected with 5 used as the catalyst both the polymer selectivity and the overall reactivity were significantly improved. The major difference between 3 and 5 can actually be attributed to the steric hindrance of the boron center. The distance between Cl− and B is equal to 2.07 Å (Fig. 1A) in 5 against 2.12 Å in 3 (Fig. 1B). With the synergistic effect due to borinane, the length of the linker between B and N+, and the size of the cation substituents, 5 surpasses all other bifunctional catalysts in productivity when used for the ROCOP of CO2 with PO. As shown in Fig. 1C, catalyst 5 affords the highest productivity (271.5 g PPC per g 5) for the preparation of PPC over other organocatalysts when compared under the same N+ to B ratio (Table S1†); the high productivity exhibited by 5 in the alternating ROCOP of CO2 and PO translates a higher atom utilization of 5.
 |
| | Fig. 1 DFT computation optimized structures of (A) catalyst 5 and (B) catalyst 3. (C) The productivity of PPC obtained using different catalysts. | |
Table 1 ROCOP of CO2 and PO using different catalystsa
| Entry |
Cat |
M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cat :Cat |
Conv.b (%) |
Sel.b |
CO2b (%) |
TONc |
TOFd (h−1) |
M
n,thero.e (kg mol−1) |
M
n,GPCf (kg mol−1) |
Đ
|
|
Polymerizations were run under neat conditions, 10 bar CO2 for 12 h unless specified.
Conversion, polymer selectivity and the ratio of carbonate linkage were determined by 1H NMR.
Turnover number (TON) = moles of epoxides consumed/moles of catalysts.
Turnover frequency (TOF) = TON/time (h).
Determined by 1H NMR.
Determined by GPC in THF using standard PS as calibration.
TBACl:TEB = 1:1.
Polymerization was run for 24 h.
Polymerization was run for 48 h.
|
| 1g |
TEB/TBACl |
100:1 |
99 |
79 |
100 |
100 |
8.3 |
8.1 |
11.1 |
1.03 |
| 2g |
TEB/TBACl |
500:1 |
51 |
43 |
100 |
255 |
21.3 |
11.2 |
10.4 |
1.06 |
| 3 |
1
|
100:1 |
99 |
0 |
0 |
100 |
8.3 |
— |
— |
— |
| 4 |
1
|
500:1 |
25 |
0 |
0 |
125 |
10.4 |
— |
— |
— |
| 5 |
2
|
100:1 |
83 |
79 |
100 |
83 |
6.9 |
6.7 |
6.5 |
1.04 |
| 6 |
2
|
500:1 |
31 |
62 |
100 |
155 |
12.9 |
9.8 |
7.2 |
1.16 |
| 7 |
3
|
100:1 |
77 |
97 |
100 |
77 |
6.4 |
7.6 |
7.1 |
1.05 |
| 8 |
3
|
500:1 |
32 |
95 |
100 |
160 |
13.3 |
15.5 |
11.9 |
1.04 |
| 9 |
4
|
100:1 |
99 |
50 |
100 |
100 |
8.3 |
5.1 |
6.5 |
1.04 |
| 10 |
4
|
500:1 |
45 |
73 |
100 |
225 |
17.9 |
16.8 |
15.2 |
1.03 |
| 11 |
5
|
100:1 |
99 |
99 |
85 |
100 |
8.3 |
9.5 |
9.6 |
1.12 |
| 12 |
5
|
500:1 |
65 |
99 |
100 |
325 |
27.1 |
33.2 |
27.0 |
1.05 |
| 13h |
5
|
1000:1 |
44 |
91 |
100 |
440 |
18.3 |
40.8 |
36.9 |
1.13 |
| 14i |
5
|
2000:1 |
55 |
90 |
100 |
1100 |
22.9 |
101 |
50.1 |
1.34 |
Kinetic study
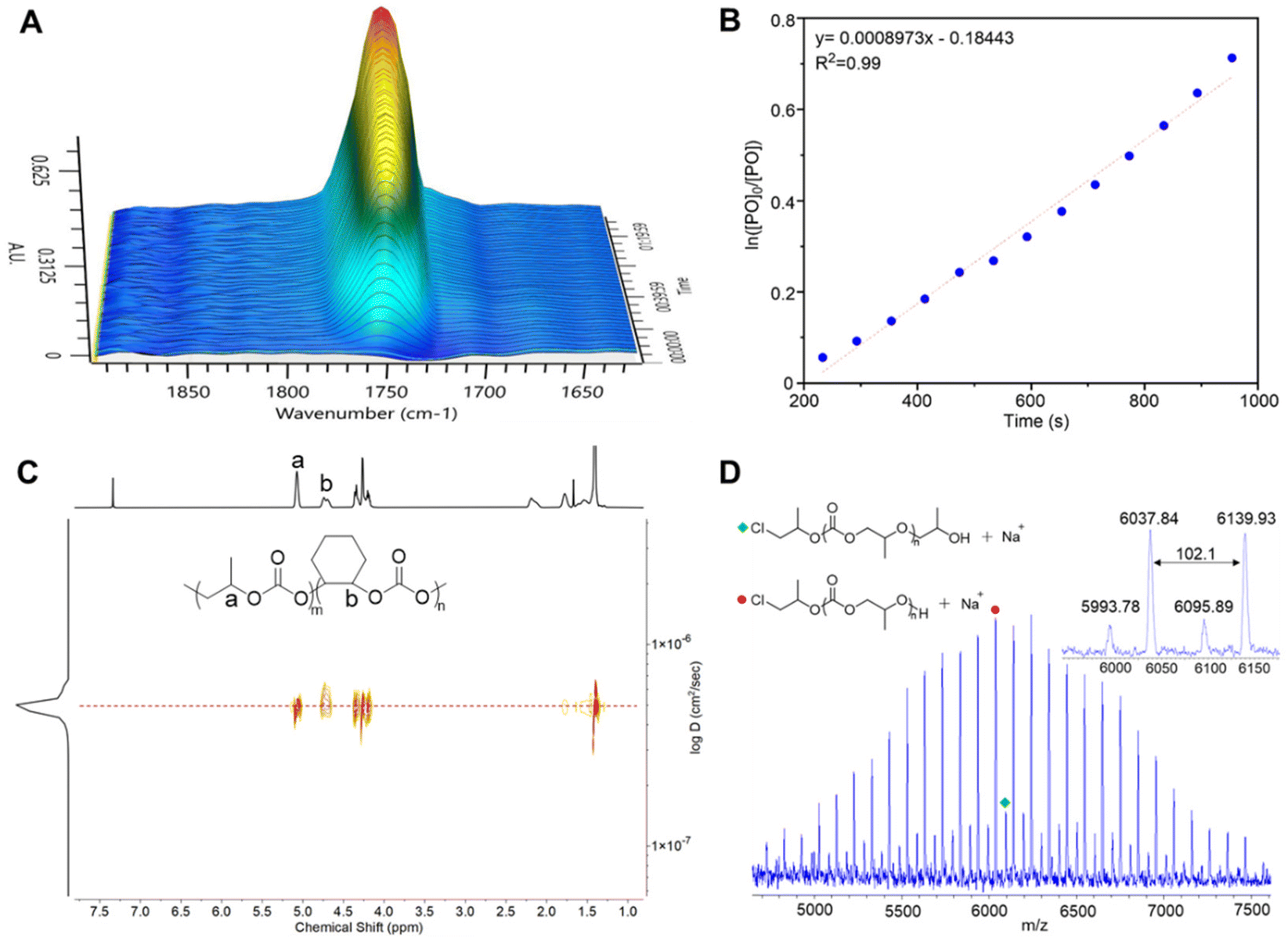
To gain a deeper insight into the ROCOP of CO2 and PO, the in situ FT-IR technique was used to monitor the course of copolymerization mediated by 5. As shown in Fig. 2A, the region of 1750 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration shows the formation of polycarbonate as a function of time. The absence of infrared absorption around 1800 cm−1 attributed to cyclic carbonate confirms the high selectivity of the ROCOP of CO2 with PO using 5 as a catalyst. By plotting ln([PO]0/[PO]) data obtained by IR vs. time (Fig. 2B) a rate constant, reflecting the consumption of both PO and CO2, could be deduced as kobs: 9.0 × 10−4 s−1 at 60 °C. To further understand the copolymerization process, an experiment monitoring the increase of molar mass with time was further performed using 5 as a catalyst with PO:5 = 500:1. The Mn of PPC increased linearly with PO conversion (Fig. S22†) suggesting the living character of PO/CO2 copolymerization mediated by 5. In all cases, the obtained PPC showed unimodal SEC peaks and narrow distribution with Đ < 1.1 (Table S2 and Fig. S23†). In addition, a well-defined block copolymer PPC-b-PCHC (Fig. 2C and Fig. S24, S25†) could be obtained by chain extension of PPC with CHO that further confirms the livingness of the ROCOP in the presence of catalyst 5. Notably, 1H NMR of the resulting PPC showed the integrals of CH, CH2, and CH3 at 5.0, 4.2, and 1.3 ppm with ratios of 1:2:3, demonstrating the alternating structure of the obtained PPC (Fig. S26†). As shown in Fig. 2D, the matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry further confirms the perfect alternating structure of PPC. Moreover, unlike the case of bromide initiated copolymerization of epichlorohydrin and CO2 which exhibits α-alkylene, ω-OH chains,50 the chloride bearing catalyst 5 afforded solely α-chloride, ω-OH ended PPC copolymer chains confirming the absence of any side reactions. The other advantage of catalyst 5 over metal catalysts is that it gives achromatous polycarbonates (Fig. S27†) without metallic residues.
O stretching vibration shows the formation of polycarbonate as a function of time. The absence of infrared absorption around 1800 cm−1 attributed to cyclic carbonate confirms the high selectivity of the ROCOP of CO2 with PO using 5 as a catalyst. By plotting ln([PO]0/[PO]) data obtained by IR vs. time (Fig. 2B) a rate constant, reflecting the consumption of both PO and CO2, could be deduced as kobs: 9.0 × 10−4 s−1 at 60 °C. To further understand the copolymerization process, an experiment monitoring the increase of molar mass with time was further performed using 5 as a catalyst with PO:5 = 500:1. The Mn of PPC increased linearly with PO conversion (Fig. S22†) suggesting the living character of PO/CO2 copolymerization mediated by 5. In all cases, the obtained PPC showed unimodal SEC peaks and narrow distribution with Đ < 1.1 (Table S2 and Fig. S23†). In addition, a well-defined block copolymer PPC-b-PCHC (Fig. 2C and Fig. S24, S25†) could be obtained by chain extension of PPC with CHO that further confirms the livingness of the ROCOP in the presence of catalyst 5. Notably, 1H NMR of the resulting PPC showed the integrals of CH, CH2, and CH3 at 5.0, 4.2, and 1.3 ppm with ratios of 1:2:3, demonstrating the alternating structure of the obtained PPC (Fig. S26†). As shown in Fig. 2D, the matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry further confirms the perfect alternating structure of PPC. Moreover, unlike the case of bromide initiated copolymerization of epichlorohydrin and CO2 which exhibits α-alkylene, ω-OH chains,50 the chloride bearing catalyst 5 afforded solely α-chloride, ω-OH ended PPC copolymer chains confirming the absence of any side reactions. The other advantage of catalyst 5 over metal catalysts is that it gives achromatous polycarbonates (Fig. S27†) without metallic residues.
 |
| | Fig. 2 (A) In situ FTIR surface image monitoring the formation of polycarbonate (1750 cm−1). (B) The plot of ln([PO]0/[PO]) versus time under the polymerization conditions: PO:5 = 100:1, CO2 10 Bar, and 60 °C. (C) DOSY NMR of the PPC-b-PCHC block copolymer. (D) MALDI-TOF MS of the polycarbonate obtained in the presence of catalyst 5. | |
Monomer scope
The success of 5-catalyzed alternating ROCOP of CO2 and PO prompted us to explore its catalytic performance with respect to other epoxides and even a four-membered cyclic ether (Table 2). In the case of CHO, its copolymerization with CO2 in the presence of 5 afforded well-defined PCHC with a molar mass of up to 160 kg mol−1. Moreover, upon further reducing the loading of 5 a TON value of 15000 (entry 3, Table 2) could be obtained which means that one gram of 5 produced at least 5.7 kg of PCHC. To the best of our knowledge, a productivity of 5.7 kg of PCHC per g of catalyst establishes a new record value compared to both organocatalysts and metallic catalysts reported so far. In the case of long-chain substituted epoxides, namely butyl oxide (BO) and octene oxide (OO), they exhibited comparable TOF values to the ones measured for PO, indicating a very similar rate of copolymerization in the presence of 5. In contrast, the ROCOP of unsubstituted epoxide, EO, with CO2 gives a TOF value 2 times higher than that of PO (entry 4, Table 2). Not surprisingly, the polymerization rate of allyl glycidyl ether (AGE) was slower compared to that of PO which might be caused by the interaction of the double bond carried by AGE with the boron centers of 5. In addition to the C2 polycarbonate, the direct copolymerization of oxetane with CO2 was also successfully carried out, resulting in the formation of the C3 polycarbonate. The state-of-the-art metallic catalysts were shown to produce poly(oxetane carbonate) with a molar mass lower than 15 kg mol−1 (ref. 56) and in the case of TEB57 associated with a specific ammonium salt the molar mass was lower than 10 kg mol−1. Due to the low ring strain, the ROCOP of CO2 and oxetane is a challenge. Interestingly, 5 enabled an efficient ROCOP of CO2 and oxetane giving rise to poly(oxetane carbonate) with a molar mass of up to 16.1 kg mol−1 and narrow molar mass distribution (entry 10, Table 2). In all cases, the obtained polycarbonates showed alternating structures (Fig. S28–32†). Overall, 5 exhibits its ability as a universal and high performance catalyst for the ROCOP of CO2 and cyclic ethers.
Table 2 ROCOP of CO2 and various cyclic ethers in the presence of catalyst 5a
| Entry |
M |
M:Cat |
T (°C) |
Time (h) |
Conv.b (%) |
Sel.b |
CO2b (%) |
TONc |
TOFd (h−1) |
M
n,GPCe (kg mol−1) |
Đ
|
|
Polymerizations were run under neat conditions, 10 bar CO2 for 12 h unless specified.
Conversion, polymer selectivity and the ratio of carbonate linkage were determined by 1H NMR.
Turnover number (TON) = moles of epoxides consumed/moles of catalysts. Turnover frequency (TOF) = TON/time (h).
Determined by 1H NMR.
Determined by GPC in THF using standard PS as calibration.
|
| 1 |
CHO |
5000:1 |
80 |
12 |
98 |
99 |
100 |
4900 |
408 |
159.7 |
1.40 |
| 2 |
CHO |
10000:1 |
80 |
12 |
36 |
99 |
100 |
3600 |
300 |
59.9 |
1.18 |
| 3 |
CHO |
20000:1 |
120 |
24 |
75 |
99 |
100 |
15000 |
625 |
80.8 |
1.25 |
| 4 |
EO |
1000:1 |
40 |
12 |
95 |
99 |
100 |
950 |
79 |
43.9 |
1.10 |
| 5 |
BO |
500:1 |
60 |
18 |
82 |
98 |
100 |
410 |
23 |
29.2 |
1.08 |
| 6 |
OO |
500:1 |
60 |
18 |
76 |
95 |
100 |
380 |
21 |
41.5 |
1.06 |
| 7 |
AGE |
500:1 |
60 |
12 |
42 |
90 |
100 |
210 |
18 |
7.2 |
1.16 |
| 8 |
Oxetane |
50:1 |
90 |
12 |
98 |
85 |
85 |
49 |
4.1 |
4.5 |
1.25 |
| 9 |
Oxetane |
150:1 |
90 |
12 |
95 |
70 |
93 |
143 |
11.9 |
9.3 |
1.27 |
| 10 |
Oxetane |
250:1 |
60 |
24 |
87 |
90 |
97 |
218 |
9.1 |
16.1 |
1.23 |
Catalyst recycling and synthesis of polycarbonate diols
The formation of the “ate complex” is crucial in the TEB mediated ROCOP of CO2 and epoxides. On the one hand, the “ate complex” decreases the nucleophilicity of growing oxyanions and prevents the occurrence of intramolecular back biting reactions. On the other hand, an excess of TEB is necessary to activate epoxides and favor propagation. Based on the considerations two or more mole ratios of TEB (versus initiator) were normally used for an effective copolymerization of CO2 with epoxides. In the latter case, utilizing protonic acids to protonate and thus terminate the living chains, in order to release TEB from the “ate complex” and recycle it, was not possible because acids can not only terminate the polymer chains but also react with free TEB. To effectively reuse TEB our group developed a recovery procedure with no less than 7 steps (Fig. S34†) to recycle >95% of TEB and the initiator (ammonium salt).43 In sharp contrast, the bifunctional catalyst 5 carries exactly one boron for one growing chain initiated by Cl− which means there is no free boron apart from the one involved in the growing “ate complex”. Hence, we hypothesized that upon deactivating the growing chains with a protonic acid a carboxylate salt could be formed and isolated by precipitation in a suitable solvent such as toluene, which is a good solvent for the copolymer and a non-solvent for the formed carboxylate salt. In the case of the bifunctional catalyst 5, the recovery procedure was extremely simple and could be realized in two steps (Fig. 3A). Furthermore, using a dicarboxylic acid as the deactivating agent a difunctional catalyst 6 could be isolated and further used to generate polycarbonate telechelics fitted with terminal hydroxyl groups. 1H (Fig. 2B and Fig. S35†), 13C (Fig. S36†) and 11B (Fig. S37†) NMR characterization studies confirm the structure of the recycled difunctional catalyst 6. Thanks to a very simple recovery process, 5 could be reused for at least 5 times with merely <5% loss for each recovery (Fig. 3C). Accordingly, the molar mass of the resulting PPC polymers slightly increased (Fig. S38†) due to minor loss of 5 during recycling. Nevertheless, the MALDI-TOF results demonstrate the high chain end fidelity of both hetero-functionalized α-Cl,ω-OH PPC (Fig. S39†) and α-, ω-OH PPC diol (Fig. 3D) catalyzed by 6.
 |
| | Fig. 3 (A) Simplified recovery procedure of 5. (B) 1H NMR of the pristine catalyst and the corresponding recycled catalyst. (C) Catalyst recovery efficiency at different times. (D) MALDI-TOF mass spectrometry of the recycled polycarbonate. | |
DFT computation assisted mechanism study
Based on our experience of TEB mediated ROCOP of CO2 and PO, the proposed mechanism occurring in the presence of 5 is depicted in Fig. 4. At the initial state, the chloride anion (Cl−) is stabilized by coordination with its boron center and through coulombic interaction with N+. In step I, upon addition of PO the interaction between boron and Cl− is disrupted and the oxygen atom of PO interacts directly with the borinane entity. This activation subsequently induced the ring-opening of PO via a nucleophilic attack of Cl− to methine carbon of PO. Because of the strong nucleophilicity of the newly formed alkoxyl anion, CO2 can easily be inserted into the boron–oxyanion coordination bond (step III). Step IV corresponds to the activation of another PO monomer at the expense of the boron–oxyanion “ate complex” followed by its ring-opening by the carbonate anion. Whether the boron center is carried by 9-BBN or the borinane the overall mechanism underlying the ROCOP of CO2 is essentially the same. To gain a deeper insight into the mechanism of such a ROCOP of CO2 with PO mediated by the bifunctional catalyst 5 and understand the influence of the boron center on the copolymerization process, quantum calculations with DFT were performed (Fig. 5, S40, and S41†). When comparing catalysts 5 and 3, a minute difference in energy barrier is observed for the transition state TS1 which indicates that the ring-opening of PO is more favorable with 5 (15.5 kcal mol−1) than that with 3 (16 kcal mol−1). However, 5 formed a more stable intermediate (INT2) with the growing alkoxy anion because of a stronger coordination of the latter to the boron center, due to the less steric hindrance of borinane. The insertion of CO2 for both types of catalysts exhibits a small energy barrier. Once CO2 is inserted into the boron-alkoxy anion the carbonate anion that is formed coordinates with the boron center (TS2). The bulky 9-BBN-based catalyst 3 faces a considerably higher energy barrier of 20 kcal mol−1 compared to that of 8.7 kcal mol−1 observed for catalyst 5. Next, the newly formed carbonate anion is then keen to ring-open PO once the latter gets activated by the boron center. Our previous study revealed that the nucleophilic attack of bare PO by the TEB coordinated carbonate anion has a much higher energy barrier (39.9 kcal mol−1).38 As a consequence, two or more mole ratios of TEB to initiator were generally necessary for the TEB mediated ROCOP of CO2 and epoxides. The synergistic effect resulting from the presence of both the boron center and the cation of the growing chain in the same compound lowers the energy barrier of 5 (30.8 kcal mol−1, INT4–TS3) and 3 (32.2 kcal mol−1) for monomer addition in comparison with that observed for one mole TEB participating in the copolymerization. Typically, the process (INT4–TS3) which is similar to the initiation step includes the disassociation of the carbonate anion with the boron center (INT 5), PO activation (INT 6), and the ring-opening of PO by the carbonate anion (TS2). The DFT computational study clearly demonstrates the advantage of using less steric hindrance borinane-based bifunctional systems which are more prone to overcome the energy barrier of the addition of PO by the carbonate anion; this study also demonstrates the superiority of borinane-based bifunctional systems over the bulky 9-BBN based catalyst.45
 |
| | Fig. 4 Proposed mechanism of the catalyst 5 mediated ROCOP of CO2 and PO. | |
 |
| | Fig. 5 Gibbs free energy profile comprising intermediates (INT1–INT7) and transition states (TS1–TS3) for the ROCOP of CO2 and PO catalyzed by 5 and 3 respectively. | |
Conclusions
In summary, we described in this contribution the rational design and synthesis of a series of bifunctional catalysts that include either borinane or 9-BBN on the one hand and ammonium salts on the other in a single molecule. The relationship between their structure and their catalytic performance was disclosed through systematic CO2 copolymerization with epoxides. Thanks to the large size of the cation, less steric hindrance of borinane compared to bulky 9-BBN, the appropriate length of the linker connecting boron to cation N+, and also the synergistic effect due to the presence in a single compound of a boron center and ammonium salt, 5 was proven to be an excellent catalyst for the alternating ROCOP of a broad range of monomers including several epoxides and oxetane with CO2. The kinetic study demonstrates that CO2-based copolymerization is living with no side reaction being detected, establishing 5 as an outstanding catalyst for the synthesis of well-defined polycarbonates. In the presence of 5, high polycarbonate productivities of 271.5 g g−1 and 5.7 kg g−1 could be achieved for PO and CHO respectively in their copolymerization with CO2. Significantly, borinane-based bifunctional catalysts could be recycled several times with a recovery efficiency of ≥95%. Finally, the DFT computational study provides a deep understanding of the mechanism and copolymerization process of CO2 with epoxides mediated by such bifunctional catalysts. Overall, the outstanding performance and dramatically simplified recovery procedure herald 5 as a very viable candidate for the industrial preparation of polycarbonate polyols.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This research work is supported by KAUST under baseline funding (BAS/1/1374-01-01).
References
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef PubMed.
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2022, 61, e202112835 Search PubMed.
- Y. Xu, L. Lin, M. Xiao, S. Wang, A. T. Smith, L. Sun and Y. Meng, Prog. Polym. Sci., 2018, 80, 163–182 CrossRef.
- Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- Y. Li, Y.-Y. Zhang, L.-F. Hu, X.-H. Zhang, B.-Y. Du and J.-T. Xu, Prog. Polym. Sci., 2018, 82, 120–157 CrossRef CAS.
- X. B. Lu, W. M. Ren and G. P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- H. Cao and X. Wang, SusMat, 2021, 1, 88–104 CrossRef.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Pena Carrodeguas, G. Trott, A. Santmarti, K. Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed.
- G.-W. Yang, Y.-Y. Zhang, Y. Wang, G.-P. Wu, Z.-K. Xu and D. J. Darensbourg, Macromolecules, 2018, 51, 1308–1313 CrossRef CAS.
- H. Sardon, D. Mecerreyes, A. Basterretxea, L. Avérous and C. Jehanno, ACS Sustainable Chem. Eng., 2021, 9, 10664–10677 CrossRef CAS.
- D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487–12496 CrossRef CAS.
- K. Nakano, K. Nozaki and T. Hiyama, J. Am. Chem. Soc., 2003, 125, 5501–5510 CrossRef CAS PubMed.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- S. D. A. Christopher, M. Byrne, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef PubMed.
- K. Nakano, K. Kobayashi, T. Ohkawara, H. Imoto and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 8456–8459 CrossRef CAS PubMed.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC.
- S. Sujith, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306–7309 CrossRef PubMed.
- A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef PubMed.
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef PubMed.
- X.-B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574–3577 CrossRef PubMed.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed.
- K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS PubMed.
- E. K. Noh, S. J. Na, S. Sujith, S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS PubMed.
- S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
- A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS PubMed.
- A. C. Deacy, A. Phanopoulos, W. Lindeboom, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 17929–17938 CrossRef CAS.
- K. Ni and C. M. Kozak, Inorg. Chem., 2018, 57, 3097–3106 CrossRef CAS PubMed.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- Y. Xiao, Z. Wang and K. Ding, Macromolecules, 2006, 39, 128–137 CrossRef CAS.
- N. Kindermann, À. Cristòfol and A. W. Kleij, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS.
- W. Wu, X. Sheng, Y. Qin, L. Qiao, Y. Miao, X. Wang and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2346–2355 CrossRef CAS.
- H. Cao, Y. Qin, C. Zhuo, X. Wang and F. Wang, ACS Catal., 2019, 9, 8669–8676 CrossRef CAS.
- D. Cui, M. Nishiura and Z. Hou, Macromolecules, 2005, 38, 4089–4095 CrossRef CAS.
- H. Nagae, R. Aoki, S. N. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492–2496 CrossRef CAS PubMed.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- D.-D. Zhang, X. Feng, Y. Gnanou and K.-W. Huang, Macromolecules, 2018, 51, 5600–5607 CrossRef CAS.
- Y. Wang, J.-Y. Zhang, J.-L. Yang, H.-K. Zhang, J. Kiriratnikom, C.-J. Zhang, K.-L. Chen, X.-H. Cao, L.-F. Hu, X.-H. Zhang and B. Z. Tang, Macromolecules, 2021, 54, 2178–2186 CrossRef CAS.
- J. L. Yang, H. L. Wu, Y. Li, X. H. Zhang and D. J. Darensbourg, Angew. Chem., Int. Ed., 2017, 56, 5774–5779 CrossRef PubMed.
- J. Zhang, L. Wang, S. Liu and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202111197 Search PubMed.
- M. Jia, D. Zhang, G. W. de Kort, C. Wilsens, S. Rastogi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2020, 53, 5297–5307 CrossRef PubMed.
- N. Patil, S. Bhoopathi, V. Chidara, N. Hadjichristidis, Y. Gnanou and X. Feng, ChemSusChem, 2020, 13, 5080–5087 CrossRef PubMed.
- S. Ye, W. Wang, J. Liang, S. Wang, M. Xiao and Y. Meng, ACS Sustainable Chem. Eng., 2020, 8, 17860–17867 CrossRef CAS.
- M. Jia, N. Hadjichristidis, Y. Gnanou and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 1593–1598 CrossRef CAS PubMed.
- N. G. Patil, S. K. Boopathi, P. Alagi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2019, 52, 2431–2438 CrossRef CAS.
- P. Alagi, G. Zapsas, N. Hadjichristidis, S. C. Hong, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 6144–6152 CrossRef CAS.
- D. Augustine, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2020, 53, 895–904 CrossRef CAS.
- G. W. Yang, Y. Y. Zhang, R. Xie and G. P. Wu, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed.
- G. W. Yang, C. K. Xu, R. Xie, Y. Y. Zhang, X. F. Zhu and G. P. Wu, J. Am. Chem. Soc., 2021, 143, 3455–3465 CrossRef CAS PubMed.
- Y.-Y. Zhang, C. Lu, G.-W. Yang, R. Xie, Y.-B. Fang, Y. Wang and G.-P. Wu, Macromolecules, 2022, 55, 6443–6452 CrossRef CAS.
- G. W. Yang, Y. Y. Zhang and G. P. Wu, Acc. Chem. Res., 2021, 54, 4434–4448 CrossRef CAS PubMed.
- J. Schaefer, H. Zhou, E. Lee, N. S. Lambic, G. Culcu, M. W. Holtcamp, F. C. Rix and T.-P. Lin, ACS Catal., 2022, 12, 11870–11885 CrossRef CAS.
- G.-W. Yang, C.-K. Xu, R. Xie, Y.-Y. Zhang, C. Lu, H. Qi, L. Yang, Y. Wang and G.-P. Wu, Nat. Synth., 2022 DOI:10.1038/s44160-022-00137-x.
- H. C. Brown and G. G. Pai, J. Organomet. Chem., 1983, 250, 13–22 CrossRef CAS.
- D. J. Darensbourg, A. I. Moncada, W. Choi and J. H. Reibenspies, J. Am. Chem. Soc., 2008, 130, 6523–6533 CrossRef CAS PubMed.
- C.-J. Zhang, S.-Q. Wu, S. Boopathi, X.-H. Zhang, X. Hong, Y. Gnanou and X.-S. Feng, ACS Sustainable Chem. Eng., 2020, 8, 13056–13063 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details for the synthesis of catalysts, polymerization conditions, and DFT computation. 1H, 13C, and 11B NMR spectra and GPC characterization data shown in Schemes S1 and S2, Tables S1 and S2, Fig. S1–S41. See DOI: https://doi.org/10.1039/d2py01161a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Xiaoshuang
Feng
* and
Xiaoshuang
Feng