
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination-bond-directed synthesis of hydrogen-bonded organic frameworks from metal–organic frameworks as templates†
Jian
Su
 ,
Shuai
Yuan
,
Yi-Xun
Cheng
,
Zhi-Mei
Yang
and
Jing-Lin
Zuo
*
,
Shuai
Yuan
,
Yi-Xun
Cheng
,
Zhi-Mei
Yang
and
Jing-Lin
Zuo
*
State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210023, P. R. China. E-mail: zuojl@nju.edu.cn
First published on 11th October 2021
Abstract
Controlled synthesis of hydrogen-bonded organic frameworks (HOFs) remains challenging, because the self-assembly of ligands is not only directed by weak hydrogen bonds, but also affected by other competing van der Waals forces. Herein, we demonstrate the coordination-bond-directed synthesis of HOFs using a preformed metal–organic framework (MOF) as the template. A MOF (CuI-TTFTB) based on two-coordinated CuI centers and tetrathiafulvalene-tetrabenzoate (TTFTB) ligands was initially synthesized. CuI-TTFTB was subsequently oxidized to the intermediate (CuII-TTFTB) and hydrated to the HOF product (TTFTB-HOF). Single-crystal-to-single-crystal (SC-SC) transformation was realized throughout the MOF-to-HOF transformation so that the evolution of structures was directly observed by single-crystal X-ray diffraction. The oxidation and hydration of the CuI center are critical to breaking the Cu–carboxylate bonds, while the synergic corbelled S⋯S and π⋯π interactions in the framework ensured stability of materials during post-synthetic modification. This work not only provided a strategy to guide the design and discovery of new HOFs, but also linked the research of MOFs and HOFs.
Introduction
Hydrogen-bonded organic frameworks (HOFs) are a rising class of porous materials that are self-assembled through hydrogen-bonding interactions between organic or metal–organic linkers. With the reversible and flexible nature of hydrogen-bonding connections, HOFs show high crystallinity, easy solution processability, and facile recyclability. These merits stimulate the studies of HOFs as a multi-functional platform in the area of gas storage and separation, molecular recognition, electric/optical materials, chemical sensing, catalysis, and biomedicine.1–8 Although different methods have been explored to synthesize HOFs including slow solvent evaporation,9,10 diffusion,11–13 and solvothermal methods,14 the controllable synthesis of HOFs remains a challenge. Due to the inherently weak hydrogen bond interactions, the formation of HOFs by self-assembly is subject to the influence of other competing van der Waals force (such as π⋯π and σ⋯π interactions) between ligands and solvent molecules. A slight modification of ligands, change of solvents, or reaction temperature may significantly alter the self-assembled products.15–17 As a result, many attempted syntheses of HOFs in a one-pot reaction may end up with non-porous products formed by closely packed ligands. For comparison, metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), with coordination bonds and covalent bonds, can be rationally designed and synthesized based on reticular chemistry.18,19 Therefore, it requires a new synthetic method to control the formation of HOFs and enrich their structural diversity.Templated synthesis has been a vital strategy to construct porous materials such as MOFs and COFs. For example, templated syntheses of MOFs by post-synthetic metalation/demetallation,20,21 metal exchange,22–24 or ligand exchange25–28 have been widely used to obtain targeted functional MOFs with the same structure/topology as the MOF-template. These templated syntheses take advantage of the reversible coordination bonds which can break and reform during post-synthetic modifications. The reversible coordination bonds have also been used to template the synthesis of COFs29 and porous polymers.30–32 A representative example has been demonstrated by Yaghi and co-workers, which used the reversible formation/break of CuI-phenanthroline coordination moieties to construct COFs with woven structures.29 The copper centers are topologically independent of the weaving within the COF structure and serve as templates for bringing the threads into a woven pattern rather than the more commonly observed parallel arrangement. The weak coordinated CuI can be removed without breaking the COF structure. These works inspire us to guide the assembly of HOFs using coordination bonds.
To realize the design of coordination-bond-directed HOF synthesis, a MOF based on weak coordination bonds will be initially constructed. The weak coordination bonds will be dissociated to form hydrogen bonds while maintaining the spatial arrangement of the organic ligands and the ordered porous structure. Since the MOF-templates can be easily designed by reticular chemistry, the structure and porosity of resulting HOFs can be rationally controlled. In addition to the higher degree of control over the HOF structures, the MOF-templated synthesis may offer promise for the design and discovery of HOFs with unique topologies that are otherwise difficult to form by one-pot synthesis. However, the coordination bonds in most MOFs are usually stronger than the hydrogen bonds in HOFs, which poses a thermodynamic barrier to transform MOFs into HOFs. For example, Zhang and co-workers have reported an isostructural transformation from a hydrogen-bonded network to a MOF on Au(111)/Ag(111) surfaces by replacing the weak N–H⋯N hydrogen bonds with stronger Ag–N coordination bonds.33 But the reverse process (i.e. replacement of coordination bonds by hydrogen bonds) has never been reported to the best of our knowledge. We propose that the coordination bonds in MOFs could be broken to form hydrogen bonds assisted by the hydration of metal cations.
Herein, a MOF-template (CuI-TTFTB) has been constructed from the two-coordinated CuI ions and tetrathiafulvalene-tetrabenzoate (TTFTB) using the solvothermal method. CuI-TTFTB was stepwise oxidized to the intermediate MOF (CuII-TTFTB) and hydrated to the HOF product (TTFTB-HOF) by single-crystal-to-single-crystal (SC-SC) transformation. The synergic corbelled S⋯S and π⋯π interactions in the framework and the slow hydration reaction ensured the success of SC–SC transformation. The MOF to HOF transformation slightly increased the pore size, and significantly enhanced the electron conductivity by about 3 fold to 2.9 × 10−5 S m−1.
Results and discussion
Crystal structure of the 2-coordinated CuI-MOF
CuI-TTFTB was synthesized by the reaction between Cu(NO3)2 and H4TTFTB in which CuII was in situ reduced to CuI by N,N-dimethylformamide (DMF) solvent under solvothermal conditions. The reduction of CuII to CuI during MOF synthesis is well documented in the literature.34–36 Analysis of the X-ray diffraction data for CuI-TTFTB revealed that it crystallized in the monoclinic space group C2/c (Table S1†). The asymmetric unit (Fig. S1†) contains a half CuI ion, a half H2TTFTB2−, a half (CH3)2NH2+ counterion, and a half free N,N-dimethylformamide (DMF). The H2TTFTB2− ligand is coordinated to two CuI ions through the two carboxylate groups in which all are ligated in a monodentate mode (Fig. 1a). The CuI ion is two-coordinated with two carboxylate groups from each H2TTFTB2− (Fig. 1c). The CuI ion and two neighboring O atoms are in a linear coordination geometry with the Cu–O bond length of 1.835(2) Å, which is comparable to the reported CuI–carboxylate coordination compounds.37–40 Hydrogen bonds exist between a pair of OH from uncoordinated carboxylic acids and O from Cu-coordinated carboxylates (dO2–O3A = 2.569 Å (symmetry code A, 3/2 − x, 1/2 + y, 3/2 − z), ∠O3H3O2 = 173°, Fig. 1b). Overall, the H2TTFTB2− ligands are assembled by hydrogen bonds into a 2D network with sql topology (Fig. 1e), which are further stacked in an AB mode and linked through Cu–O coordination bonds between adjacent planes A and B (Fig. 1f). Meanwhile, the stacked 2D networks are stabilized by S⋯S and π⋯π interactions with a S⋯S distance of 3.792 Å (Fig. 1d). Microporous 1D channels were observed along the crystallographic c direction with a diameter of ∼12.2 Å (Fig. 1e, and Fig. S2 and S3†). The SQUEEZE calculations in PLATON41 give a total solvent-accessible volume of 2370.7 Å3 per unit cell, equivalent to 40.5% of the total crystal volume. The porosity of CuI-TTFTB is slightly higher than that of the transition-metal TTF-based MOF, M2(TTFTB) (37.9%).42 CuI-TTFTB could maintain its morphology in air for at least 19 days (Fig. S4†), but it dissolves in DMF possibly due to the weak hydrogen bonds.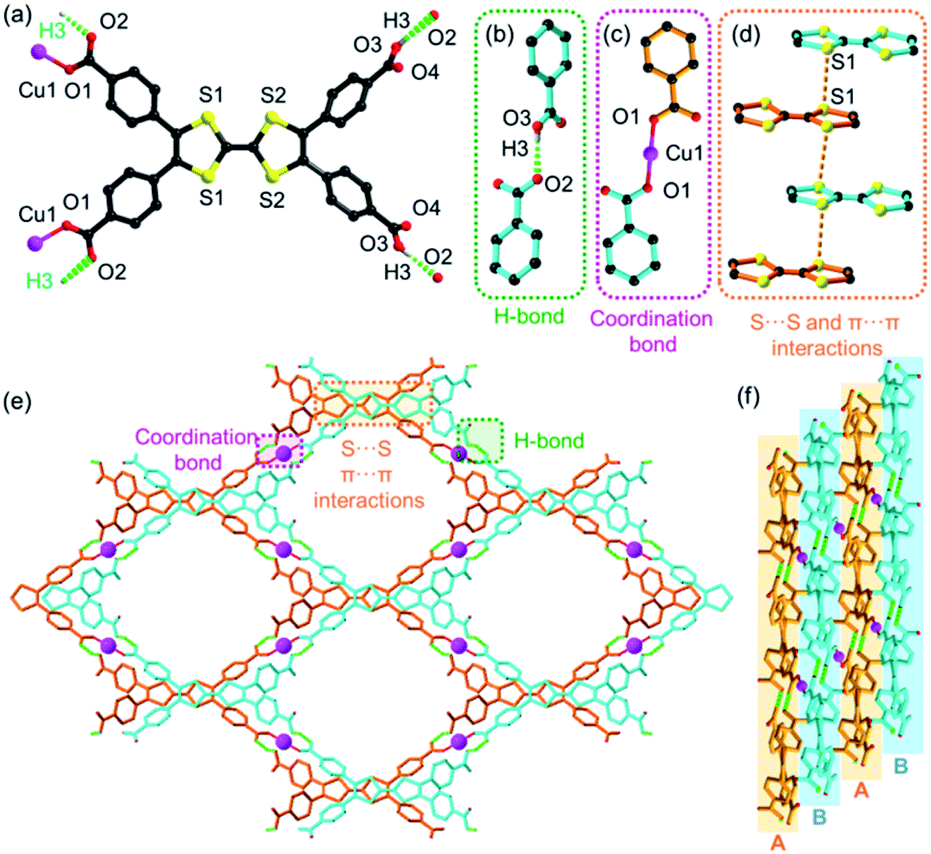 | ||
| Fig. 1 Crystal structure of CuI-TTFTB. (a) The structure and coordination environment of H2TTFTB2−. (b) The hydrogen bond between two carboxylates of H2TTFTB2−. (c) The coordination bond between CuI and H2TTFTB2−. (d) The S⋯S and π⋯π interactions between neighboring TTF moieties. (e) The structure of CuI-TTFTB viewed along the c-direction. Two sets of hydrogen-bonded networks are colored blue and yellow. The hydrogen bond within the network, coordination bond between two sets of networks, and S⋯S/π⋯π interactions are highlighted by green, violet, and orange. The molecules in the channels are omitted for clarity. (f) The structure of CuI-TTFTB viewed along the b-direction showing the AB stacking mode. Color scheme: Cu, violet; O, red; C, black; S, yellow. | ||
Crystal structure of the HOF obtained by SC-SC transformation
The structural analysis for CuI-TTFTB reveals that it can be transformed into a HOF if CuI is removed from the lattice. The transformation of CuI-TTFTB into the HOF was carried out by incubating crystals of CuI-TTFTB in water/acetone solution (1 vol%) for 14 days. The color of CuI-TTFTB crystals changed from red to dark red after the incubation (Fig. S5 and S6†) while the morphology of crystals remains visually intact. By single crystal X-ray diffraction, the resulting crystals are found to be a HOF, namely TTFTB-HOF (Fig. 2). TTFTB-HOF still crystallizes in the monoclinic space group C2/c (a = 20.495, b = 34.798, c = 8.801 Å, Table S1†) with comparable cell parameters to those of CuI-TTFTB (a = 19.165, b = 34.704, c = 8.882 Å). But the CuI has been removed from the skeleton. The (CH3)2NH2+ counterion was also absent in the crystal structure of TTFTB-HOF (Fig. S7 and S8†), and the charge was presumably balanced by the protonation of two carboxylates. Detailed structural analysis of TTFTB-HOF shows slightly rotated phenyl-carboxylate moieties and slipped TTF moieties. As a result, the hydrogen bond donor⋯acceptor lengths (O2⋯O3) reduced from 2.569 to 2.476 Å accompanied by the increase of S⋯S distance from 3.792 to 3.868 Å, indicating the enhanced hydrogen bond interaction after the removal of CuI. The pore size of TTFTB-HOF was slightly expanded by ∼1 Å compared to that of CuI-TTFTB, which is in line with the increased a-axis length from 19.165 to 20.495 Å. The ideal N2 adsorption was not obtained owing to the degraded crystallinity of TTFTB-HOF after the activation.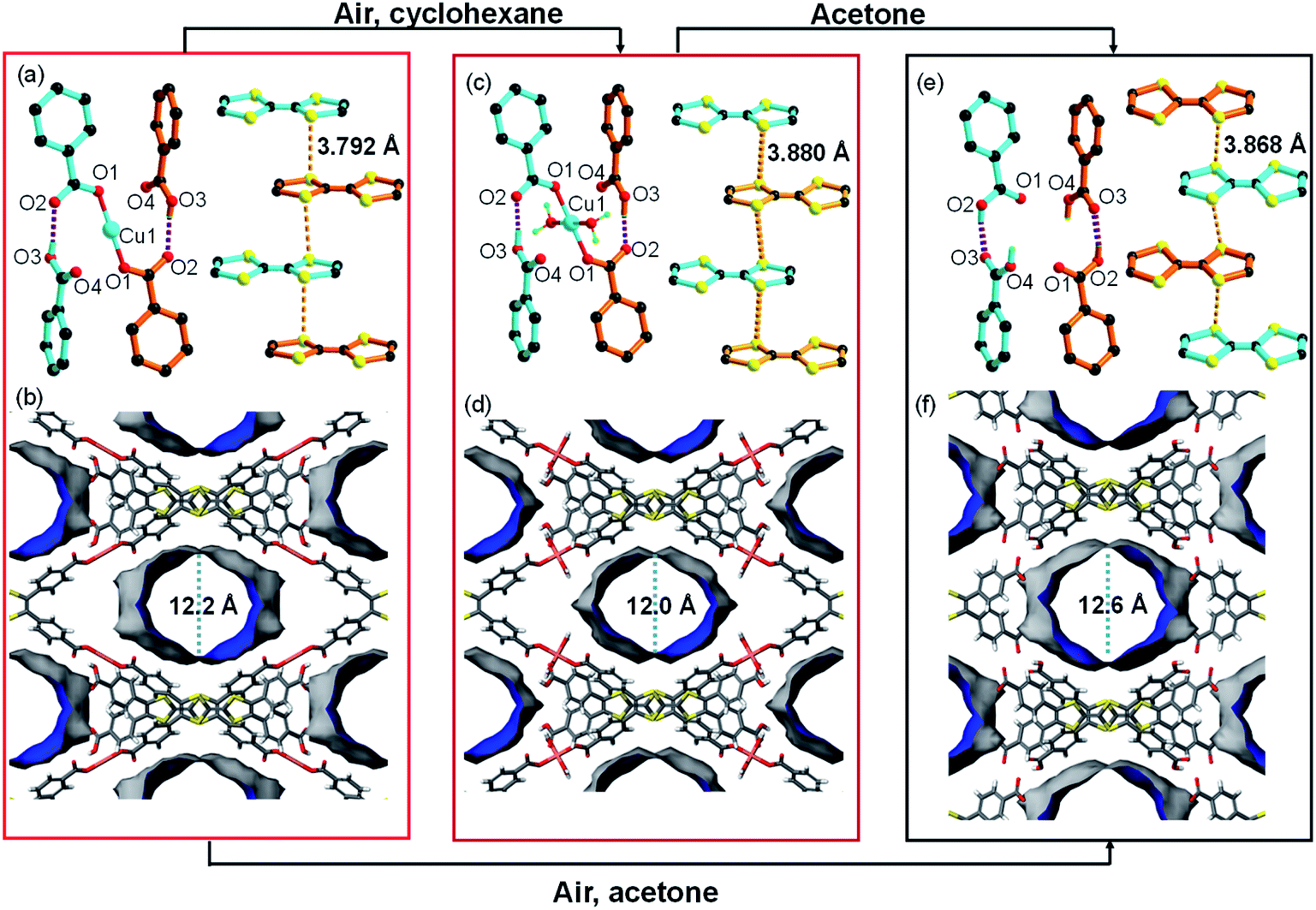 | ||
| Fig. 2 Single crystal transformation of CuI-TTFTB to CuII-TTFTB to TTFTB-HOF. The structure of the Cu center with four adjacent carboxylates and the packing of TTF moieties in CuI-TTFTB (a), CuII-TTFTB (c), and TTFTB-HOF (e). The accessible solvent surface of CuI-TTFTB (b), CuII-TTFTB (d), and TTFTB-HOF (f) showing the change of cavity sizes upon structural transformation. The molecules in the channels are omitted for clarity. The pore size and accessible solvent surface were calculated based on the N2 molecule as the probe (kinetic diameter of 3.64 Å). | ||
Comparison of reported TTFTB based-HOFs (X-TTF-1,43 PFC-77, PFC-78, and PFC-79,44 PFC = porous materials from FJIRSM, CAS) with our TTFTB-HOF in this work shows that there are several differences in the single-crystal structures. Firstly, the reported structures are all crystallized in the space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , while TTFTB-HOF is in C2/c. Secondly, the connection types between the carboxylates are different. In the reported structures, a pair of carboxylates forms a typical self-complementary dimer through two hydrogen bonds, while in our TTFTB-HOF structure, each carboxylate pair is connected by one hydrogen bond. At last, the hydrogen bonds showed different bond lengths (defined by the O⋯O distance between the carboxylates). The previously reported HOFs have hydrogen bond-lengths in the range of 2.6–2.8 Å, which are longer than that in TTFTB-HOF (2.48 Å, Table 1). Therefore, the MOF-templated synthesis of TTFTB-HOF may enrich the structural and functional diversity of HOFs.
, while TTFTB-HOF is in C2/c. Secondly, the connection types between the carboxylates are different. In the reported structures, a pair of carboxylates forms a typical self-complementary dimer through two hydrogen bonds, while in our TTFTB-HOF structure, each carboxylate pair is connected by one hydrogen bond. At last, the hydrogen bonds showed different bond lengths (defined by the O⋯O distance between the carboxylates). The previously reported HOFs have hydrogen bond-lengths in the range of 2.6–2.8 Å, which are longer than that in TTFTB-HOF (2.48 Å, Table 1). Therefore, the MOF-templated synthesis of TTFTB-HOF may enrich the structural and functional diversity of HOFs.
| Donor-H⋯Acceptor | D-H | H⋯A | D⋯A | Angle (°) |
|---|---|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: i (3/2 − x, 1/2 + y, 3/2 − z), ii (x, 1 − y, −1/2 + z). | ||||
| O2–H2⋯O3i | 0.82 | 1.70 | 2.476(6) | 156 |
| C6–H6⋯S1 | 0.93 | 2.63 | 2.998(4) | 104 |
| O17–H17⋯O2ii | 0.93 | 2.55 | 3.248(7) | 132 |
Both O2 and H2O are critical for the structural transformation. Control experiments were conducted in an Ar atmosphere or in dry acetone, which did not yield TTFTB-HOF crystals. To reveal the mechanism of CuI-TTFTB to TTFTB-HOF transformation, we replaced the water/acetone solution with nonpolar cyclohexane to slow down the hydration. An intermediate was successfully isolated, namely CuII-TTFTB. Single crystal analysis shows that CuII-TTFTB crystallizes in the monoclinic space group C2/c (a = 18.757, b = 35.559 and c = 8.899 Å, Table S1†) with slightly changed cell parameters compared with CuI-TTFTB (a = 19.165, b = 34.704 and c = 8.882 Å). Interestingly, two water molecules were added to the Cu center which formed a planar 4-coordinated Cu center (Fig. 2c). The CuI was oxidized into CuII as reflected by the 4-coordinated square planar geometry and bond lengths (Cu1–O1 = 1.868(7) Å; Cu1–O5(water) = 2.34 (3) Å). The (CH3)2NH2+ counterion was eliminated to balance the charge. The hydrogen bond lengths, defined by the O2⋯O3 distances (2.569 Å for CuI-TTFTB and 2.578 Å for CuII-TTFTB), did not noticeably change upon Cu oxidation. The packing between TTF moieties slightly slipped leading to the increase of S⋯S distance from 3.792 Å (CuI-TTFTB) to 3.880 Å (CuII-TTFTB). Compared with CuI-TTFTB, the cavity of CuII-TTFTB shrank along the a-direction and expanded along the b-direction, indicating the flexibility of the structure.
Characterization of SC–SC transformation
The Pawley refinement against powder X-ray diffraction (PXRD) patterns was carried out for the materials to determine the lattice parameters and confirm the phase purity of bulk materials (Fig. S9, S10 and Table S2†). X-ray photoelectron spectroscopy (XPS) was used to examine the oxidation state change of Cu species in CuI-TTFTB, CuII-TTFTB, and TTFTB-HOF (Fig. S11 and S12†). The Cu species in CuI-TTFTB mainly exist in +1 oxidation states with the Cu2p3/2 peak located at 931.9 eV.45 The small peak at 933.8 eV is attributed to the CuII generated by the surface oxidation of CuI-TTFTB. For CuII-TTFTB, the CuII peak is increased while the CuI peak sharply decreased, suggesting the oxidation of CuI to CuII.Interestingly, although the crystal structure of TTFTB-HOF indicated the dissociation of Cu from inorganic nodes, the Cu species were not leached out from the crystal. We propose that the dissociated CuII formed small Cu(OH)2·xH2O particles and they were trapped in the cavity of TTFTB-HOF. Although both CuII-TTFTB and TTFTB-HOF contain CuII species, TTFTB-HOF shows an up-shifted Cu2p3/2 peak (934.6 eV) compared to that of CuII-TTFTB (933.8 eV), which can be explained by the different coordination environment of the CuII center (Fig. S12†).32 Indeed, the CuII center is four-coordinated with two water and two carboxylates in CuII-TTFTB, while CuII is in a six-coordinated octahedral environment surrounded by OH in Cu(OH)2·xH2O. However, there is no characteristic diffraction for Cu(OH)2·xH2O in PXRD patterns, suggesting that Cu(OH)2·xH2O is either amorphous or forms ultra-small nanoparticles (Fig. 3a). The different coordination environments of CuII in CuII-TTFTB and TTFTB-HOF may cause shifted Cu2p3/2 peak in XPS spectra. Scanning electron microscopy combined with energy-dispersive X-ray spectroscopy (SEM/EDX) further revealed the uniform distribution of Cu and S species throughout the structural transformation (Fig. S13†). After the SC–SC transformation (CuI-TTFTB to TTFTB-HOF), the maintained crystal morphology, negligible color change, and the undetected copper element of the supernatant in the inductively coupled plasma (ICP) results, eliminating the possibility of dissolution/recrystallization.
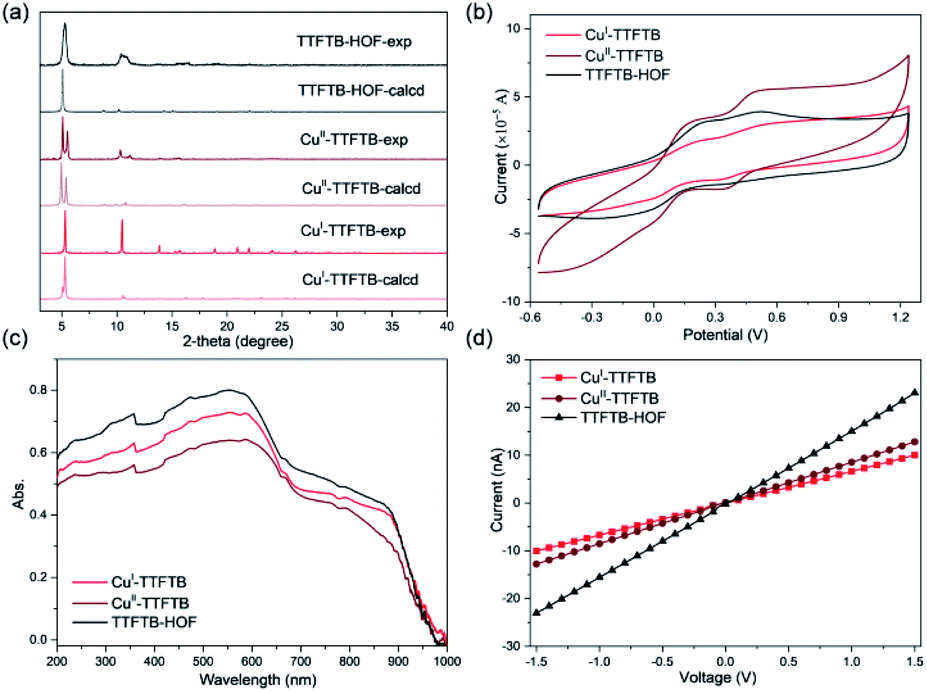 | ||
| Fig. 3 (a) Experimental and calculated PXRD patterns of CuI-TTFTB, CuII-TTFTB and TTFTB-HOF. The Mercury program allows accounting for preferred orientation of the crystallites, using the March–Dollase parameter. We input a value of 0.65 for this parameter, with orientation to (0 2 0) preferred. (b) Solid state cyclic voltammograms of CuI-TTFTB, CuII-TTFTB, and TTFTB-HOF performed at a scan rate of 400 mV s−1. The experiments were conducted in 0.1 M LiBF4 in CH3CN electrolyte. Solid-state diffuse reflectance spectra (c) and electrical conducting plots of (d) CuI-TTFTB, CuII-TTFTB and TTFTB-HOF. | ||
Mechanism of SC–SC transformation
Based on the crystal structure and XPS, we proposed a mechanism of the MOF to HOF transformation. The structural transformation was initiated by the oxidation of CuI metal nodes into CuII by O2. This triggered the coordination of two additional water molecules to the CuII center. Further hydration of the CuII center leads to the dissociation of the CuII–carboxylate coordination bond to form Cu(OH)2·xH2O and protonated carboxylic acids. The oxidation of CuI to CuII is important for the subsequent hydration reaction, as CuII has a much higher hydration enthalpy (−2161 kJ mol−1) than that of CuI (−619 kJ mol−1). This explains the observation that TTFTB-HOF cannot be formed under Ar protection. Water is also required to break the CuII–carboxylate coordination bond. CuII-TTFTB readily transformed to TTFTB-HOF in water/acetone solution (1 vol%) within 7 days, whereas this transformation did not happen in dry acetone or cyclohexane for 14 days. The slow hydration ensures the successful SC–SC transformation, while increasing the water concentration to 10 vol% leads to immediate amorphization of MOF-crystals. In addition to O2 and H2O, the intrinsic MOF structure is also important for the MOF to HOF transformation in the SC–SC manner. The closely packed TTF moieties stabilized the framework by π⋯π and S⋯S interactions, which synergic corbelled the porous structure with hydrogen bonds and maintained the structural intactness after dissociating coordination bonds. In contrast, many MOF-templates in the literature lost the crystallinity after breaking coordination bonds, resulting in amorphous polymer gels or metal oxides/hydroxides.46–49Physical properties
The electrochemical properties of CuI-TTFTB, CuII-TTFTB, and TTFTB-HOF were studied by solid state direct current (DC) cyclic voltammetry (CV) in 0.1 M LiBF4 in CH3CN (Fig. 3b). The stability of TTFTB-HOF was proved by the remaining PXRD patterns (Fig. S14 and S15†). Upon scanning anodically, two quasi-reversible one-electron processes at ∼0.2 and ∼0.5 V (vs. Fc/Fc+) were observed. These processes are attributed to the TTF/TTF˙+ and TTF˙+/TTF2+ redox couples, respectively.47 The values of the TTF/TTF˙+ and TTF˙+/TTF2+ redox couples are comparable in the three materials, suggesting that the redox properties of TTF moieties were not affected by the Cu coordination. The location of these two reversible redox processes is consistent with that of the anionic In-TTF-MOFs.49,50The solid-state absorption spectra of CuI-TTFTB, CuII-TTFTB, and TTFTB-HOF were obtained to gain insight into the influence of structural transformation on the optical and semiconducting properties. In CuI-TTFTB, there are two main absorption bands located in the region 200–650 nm (Fig. 3c). This higher energy absorption band is attributed to the π → π* or n → π* transition of the free ligand.51 In the range of 700–900 nm, the absorption band can be assigned to the partially oxidized TTF˙+. Using the UV-vis-NIR absorption data, we approximated the bandgap of CuI-TTFTB, CuII-TTFTB, and TTFTB-HOF through the Kubelka–Munk function. From the Tauc plots,52 the band gaps of CuI-TTFTB, CuII-TTFTB, and TTFTB-HOF are all approximately 1.29 eV (Fig. S16†). In general, the band gaps of these compounds are comparable to those of radical TTF-MOFs and smaller than those of their neutral counterparts.53 Correspondingly, the lower optical bandgap of these compounds is attributed to the close S⋯S interaction in the framework. The bandgap values, which are typical of semiconductors, are such that these materials could potentially be used in photo- and electro-catalysis reactions as well as in microelectronic devices. Furthermore, the electronic conductivity was measured using the I–V plots, which revealed that the electronic conductivities of CuI-TTFTB, CuII-TTFTB, and TTFTB-HOF are 1.12, 1.17, and 2.90 × 10−5 S m−1, respectively (Table S3†).
Conclusions
In conclusion, we demonstrate the coordination-bond-directed synthesis of HOFs using MOFs as templates. The MOF-to-HOF structural transformation was directly observed by single-crystal X-ray diffraction, which revealed the stepwise oxidation and hydration of the CuI center leading to the dissociation of Cu–carboxylate bonds. This structural transformation is associated with a change in pore size. The electronic conductivity enhanced by about 3 fold to 2.9 × 10−5 S m−1, as revealed by I–V measurements. Considering the diversity of MOF structures, this template strategy is expected to boost the development of HOFs. Further study is in progress in our group to explore the synthesis of functional HOF materials from a variety of MOFs with weak coordination bonds.Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
J. Z., J. S., and S. Y. conceived and designed the project. J. S., Y.·C., and Z. Y. performed the experiments. S. Y., J. S., and J. Z. drafted the original manuscript. All authors have discussed the results and given approval to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Basic Research Program of China (2018YFA0306004) and the National Natural Science Foundation of China (No. 21631006 and 21875099). Jian Su thanks the China Postdoctoral Science Foundation (No. 2019M661788).Notes and references
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- B. Wang, R. B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS PubMed.
- P. Li, M. R. Ryder and J. F. Stoddart, Acc. Mater. Res., 2020, 1, 77–87 CrossRef CAS.
- J. Yang, J. Wang, B. Hou, X. Huang, T. Wang, Y. Bao and H. Hao, Chem. Eng. J., 2020, 399, 125873 CrossRef CAS.
- J. Luo, J.-W. Wang, J.-H. Zhang, S. Lai and D.-C. Zhong, CrystEngComm, 2018, 20, 5884–5898 RSC.
- Y. Lin, X. Jiang, S. T. Kim, S. B. Alahakoon, X. Hou, Z. Zhang, C. M. Thompson, R. A. Smaldone and C. Ke, J. Am. Chem. Soc., 2017, 139, 7172–7175 CrossRef CAS PubMed.
- X. Z. Luo, X. J. Jia, J. H. Deng, J. L. Zhong, H. J. Liu, K. J. Wang and D. C. Zhong, J. Am. Chem. Soc., 2013, 135, 11684–11687 CrossRef CAS PubMed.
- Q. Huang, W. Li, Z. Mao, L. Qu, Y. Li, H. Zhang, T. Yu, Z. Yang, J. Zhao, Y. Zhang, M. P. Aldred and Z. Chi, Nat. Commun., 2019, 10, 3074 CrossRef PubMed.
- B. Wang, R. He, L. H. Xie, Z. J. Lin, X. Zhang, J. Wang, H. Huang, Z. Zhang, K. S. Schanze, J. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 12478–12485 CrossRef CAS PubMed.
- B. Wang, X. L. Lv, J. Lv, L. Ma, R. B. Lin, H. Cui, J. Zhang, Z. Zhang, S. Xiang and B. Chen, Chem. Commun., 2019, 56, 66–69 RSC.
- J. M. Taylor, P. J. Dwyer, J. W. Reid, B. S. Gelfand, D.-w. Lim, M. Donoshita, S. L. Veinberg, H. Kitagawa, V. N. Vukotic and G. K. H. Shimizu, Chem, 2018, 4, 868–878 CAS.
- Q. Yin, P. Zhao, R. J. Sa, G. C. Chen, J. Lu, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 7691–7696 CrossRef CAS PubMed.
- Y. Wang, D. Liu, J. Yin, Y. Shang, J. Du, Z. Kang, R. Wang, Y. Chen, D. Sun and J. Jiang, Chem. Commun., 2020, 56, 703–706 RSC.
- Y. Suzuki, N. Tohnai, A. Saeki and I. Hisaki, Chem. Commun., 2020, 56, 13369–13372 RSC.
- T. Takeda, M. Ozawa and T. Akutagawa, Cryst. Growth Des., 2019, 19, 4784–4792 CrossRef CAS.
- X. Jiang, X. Cui, A. J. E. Duncan, L. Li, R. P. Hughes, R. J. Staples, E. V. Alexandrov, D. M. Proserpio, Y. Wu and C. Ke, J. Am. Chem. Soc., 2019, 141, 10915–10923 CrossRef CAS PubMed.
- O. M. Yaghi, ACS Cent. Sci., 2019, 5, 1295–1300 CrossRef CAS PubMed.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Woll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60 DOI:10.1002/anie.202101644.
- M. C. Das, S. Xiang, Z. Zhang and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 10510–10520 CrossRef CAS PubMed.
- S. Yuan, Y. P. Chen, J. Qin, W. Lu, X. Wang, Q. Zhang, M. Bosch, T. F. Liu, X. Lian and H. C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 14696–14700 CrossRef CAS PubMed.
- C. K. Brozek and M. Dinca, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- L. Liu, L. Li, J. A. DeGayner, P. H. Winegar, Y. Fang and T. D. Harris, J. Am. Chem. Soc., 2018, 140, 11444–11453 CrossRef CAS PubMed.
- B. Garai, V. Bon, S. Krause, F. Schwotzer, M. Gerlach, I. Senkovska and S. Kaskel, Chem. Mater., 2020, 32, 889–896 CrossRef CAS.
- C. Liu, C. Zeng, T. Y. Luo, A. D. Merg, R. Jin and N. L. Rosi, J. Am. Chem. Soc., 2016, 138, 12045–12048 CrossRef CAS PubMed.
- J. A. Boissonnault, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2017, 139, 14841–14844 CrossRef CAS PubMed.
- L. Liu, L. Li, M. E. Ziebel and T. D. Harris, J. Am. Chem. Soc., 2020, 142, 4705–4713 CrossRef CAS PubMed.
- L. Feng, K. Y. Wang, G. S. Day and H. C. Zhou, Chem. Soc. Rev., 2019, 48, 4823–4853 RSC.
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gandara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365–369 CrossRef CAS PubMed.
- T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427–5432 CrossRef CAS PubMed.
- K. Chen and C. D. Wu, Angew. Chem., Int. Ed., 2019, 58, 8119–8123 CrossRef CAS PubMed.
- X. Wang, M. J. Dong, K. Chen, Z. K. Liu and C. D. Wu, Chem. Commun., 2021, 57, 1348–1351 RSC.
- D.-D. Zhou, J. Wang, P. Chen, Y. He, J.-X. Wu, S. Gao, Z. Zhong, Y. Du, D. Zhong and J.-P. Zhang, Chem. Sci., 2021, 12, 1272–1277 RSC.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- S. M. F. Lo, S. S. Y. Chui, L.-Y. Shek, Z. Lin, X. X. Zhang, G.-h. Wen and I. D. Williams, J. Am. Chem. Soc., 2000, 122, 6293–6294 CrossRef CAS.
- C. Hua, P. Turner and D. M. D'Alessandro, Dalton Trans., 2015, 44, 15297–15303 RSC.
- P. F. Rodesiler and E. L. Amma, J. Chem. Soc., Chem. Commun., 1974, 599 RSC.
- T. Sugiura, H. Yoshikawa and K. Awaga, Inorg. Chem., 2006, 45, 7584–7586 CrossRef CAS PubMed.
- Y. Sevryugina, O. Hietsoi and M. A. Petrukhina, Chem. Commun., 2007, 3853–3855 RSC.
- O. Hietsoi, C. Dubceac, A. S. Filatov and M. A. Petrukhina, Chem. Commun., 2011, 47, 6939–6941 RSC.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- S. S. Park, E. R. Hontz, L. Sun, C. H. Hendon, A. Walsh, T. Van Voorhis and M. Dinca, J. Am. Chem. Soc., 2015, 137, 1774–1777 CrossRef CAS PubMed.
- I. Hisaki, N. Q. E. Affendy and N. Tohnai, CrystEngComm, 2017, 19, 4892–4898 RSC.
- X. Y. Gao, Y. L. Li, T. F. Liu, X. S. Huang and R. Cao, CrystEngComm, 2021, 23, 4743–4747 RSC.
- N. T. Bui, H. Kang, S. J. Teat, G. M. Su, C. W. Pao, Y. S. Liu, E. W. Zaia, J. Guo, J. L. Chen, K. R. Meihaus, C. Dun, T. M. Mattox, J. R. Long, P. Fiske, R. Kostecki and J. J. Urban, Nat. Commun., 2020, 11, 3947 CrossRef CAS PubMed.
- T. Ishiwata, K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2017, 56, 2608–2612 CrossRef CAS PubMed.
- Z. Wang, A. Blaszczyk, O. Fuhr, S. Heissler, C. Woll and M. Mayor, Nat. Commun., 2017, 8, 14442 CrossRef CAS PubMed.
- S. Guo, Y. Zhao, H. Yuan, C. Wang, H. Jiang and G. J. Cheng, Small, 2020, 16, e2000749 CrossRef PubMed.
- J. Su, W. He, X.-M. Li, L. Sun, H.-Y. Wang, Y.-Q. Lan, M. Ding and J.-L. Zuo, Matter, 2020, 2, 711–722 CrossRef.
- J. Su, S. Yuan, H. Y. Wang, L. Huang, J. Y. Ge, E. Joseph, J. Qin, T. Cagin, J. L. Zuo and H. C. Zhou, Nat. Commun., 2017, 8, 2008 CrossRef PubMed.
- P. M. Usov, C. Fabian and D. M. D'Alessandro, Chem. Commun., 2012, 48, 3945–3947 RSC.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15, 627–637 CrossRef CAS.
- J. Su, T. H. Hu, R. Murase, H. Y. Wang, D. M. D'Alessandro, M. Kurmoo and J. L. Zuo, Inorg. Chem., 2019, 58, 3698–3706 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2082445–2082447. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03962h |
| This journal is © The Royal Society of Chemistry 2021 |