DOI:
10.1039/D1PY00193K
(Paper)
Polym. Chem., 2021,
12, 2282-2292
Conjugated microporous polymers using a copper-catalyzed [4 + 2] cyclobenzannulation reaction: promising materials for iodine and dye adsorption†
Received
12th February 2021
, Accepted 17th March 2021
First published on 17th March 2021
Abstract
A design strategy is disclosed to synthesize conjugated microporous polymers (CMPs) using a versatile copper-catalyzed [4 + 2] cyclobenzannulation reaction, which employs a diphenylethynyl terephthalaldehyde derivative 3 with a series of triptycene-based diethynyl aryl building blocks 2a–e. Investigation of the intrinsic microporosity properties of CBP1–5 using nitrogen adsorption measurements reveals Brunauer–Emmett–Teller (BET) surface areas up to 794 m2 g−1 and average pore volumes reaching 0.63 cm3 g−1. Inspection of the adsorption properties of the graphitic-like polymers CBP1–5 divulges their high iodine uptake with a maximum of 166 wt%. Moreover, the target polymers CBP1–5 prove their efficiency as selective dye adsorbents by removing up to 100% methylene blue over methyl orange from aqueous solution.
Introduction
Since the first report on the synthesis of conjugated microporous polymer (CMP) networks by Cooper et al.,1 numerous scientists have contributed to that field, leading to a steep growth in the number of publications. Conjugated microporous polymers have emerged as promising materials for a wide range of cutting-edge fields,2 mainly those related to sustainable energy applications, such as gas separation and storage,3–6 hydrogen evolution,7–9 organic solar cells (OSCs),10–12 batteries,13–16 field-effect transistors (FETs),17–21 thermoelectrics,22 sensors,23,24 optical switches,25–29 and light-emitting diodes (LEDs).30–33 The eminence of CMPs stems from the exceptional properties that they exhibit, among others, their versatile synthesis, extended π-conjugation skeleton, high stability, and large surface area.2 The chief advantage of CMP networks lies in their design that could be realized via numerous synthetic methodologies,2 which span from transition-metal catalyzed cross-coupling reactions like Suzuki–Miyaura,8,34–36 Sonogashira,37–40 Yamamoto,41–43 and direct arylation44,45 to several other synthetic approaches like oxidative coupling, condensation reactions, and cyclotrimerization, in addition to many other design strategies.46–52
Since the industrial revolution, fossil fuels have been increasingly utilized as the main feedstock in most human activities and technologies – reaching 85% of the total energy consumption in 2018 – which led to global economic developments and drastic improvements in our living standards.53 Nevertheless, the combustion of fossil fuels instigates serious impacts on the environment caused by the generation of particulate matters (PMs) and massive release of various gases into the atmosphere, among others, carbon dioxide which contributes to global warming, acid rain and smog formation.54 As the world demand for energy sources is on a sharp rise with an estimated energy consumption of 800 quadrillion British thermal units (BTUs) by 2040,55 the development of sustainable energy sources with high efficiency and low-emission deems essential.56 Amongst the alternative sources to fossil fuels, nuclear fission is considered a promising feedstock whose high efficiency in power generation is considered a major advantage, yet the formation of several radioactive gaseous byproducts, such as 85Kr, 3H, 14CO2, 123I, 125I, and 127–140I, is believed to be a major drawback. In addition to the hitherto mentioned radioactive species, fission nuclear energy generates hydrogen- and alkyl-radioactive halides which are also considered hazardous due to their interference in metabolic processes.57,58 As a result of the crucial need to develop highly efficient and cost-effective porous materials to capture radioactive iodine-containing species, several adsorbents have been developed, namely, organic cages, silica gel, zeolites, and covalent-organic and metal–organic frameworks.37,59–62
On the other hand, several industrial processes discharge water-soluble non-biodegradable biologically active dyes, which could be either cationic, like methylene blue (MB), or anionic, such as methyl orange (MO) dyes. Some of these organic dyes are highly toxic, mutagenic, and carcinogenic.63 In addition to their hazardous properties, the aforementioned pollutants cause several skin- and eye-related diseases, as well as various respiratory complications, which necessitates their removal from freshwater resources via sustainable techniques.63–65 Various methods have been reported in the literature to remove organic dyes from water, of which we note photocatalytic degradation by employing H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 66 and NaBH4,67 electrodialysis,68 membrane-based technology,69 and the use of adsorbents, where the latter technique proved to be among the most prominent owing to its high efficiency, cost-effectiveness, simplicity, and possibility of being utilized under ambient conditions.70,71
66 and NaBH4,67 electrodialysis,68 membrane-based technology,69 and the use of adsorbents, where the latter technique proved to be among the most prominent owing to its high efficiency, cost-effectiveness, simplicity, and possibility of being utilized under ambient conditions.70,71
We disclose the synthesis of five conjugated microporous polymers CBP1–5 through a copper-catalyzed [4 + 2] benzannulation72–74 from the reaction of various 1,4-diarylethynyl triptycene synthons 2a–e with 2,5-bis(phenylethynyl)terephthalaldehyde 3. The target polymers CBP1–5 were obtained in excellent yields, and their intrinsic surface areas were thoroughly investigated before carrying out studies of their iodine uptake and organic dye adsorption.
Experimental section
Materials
All the reactions were carried out under an inert atmosphere using dry argon. All chemical reagents were used without further purification as purchased from Aldrich, Merck, and HiMedia unless otherwise specified. The comonomers 2a, 2c, 2e and 3 were synthesized following the reported procedures in the literature.59,73,75–78 Anhydrous solvents, namely, hexane, DCM, THF, DCE, TEA, methanol, diethyl ether, and acetone, were further dried over molecular sieves and deoxygenated by bubbling with argon gas for 30 minutes. Thin-layer chromatography (TLC) was performed on aluminum sheets coated with silica gel 60 F254 and revealed using a UV lamp. NMR (1H: 600 MHz, 13C: 150 MHz) spectra were recorded using a Bruker BioSpin GmbH 600 MHz spectrometer using CD2Cl2 as a solvent with the chemical shifts (δ) given in ppm and referenced to tetramethylsilane (TMS). Electron impact high-resolution mass spectra (EI-HRMS) were recorded on a Thermo Scientific DFS system with a standard PFK (perfluorokerosene) as a lock mass. The analysed data are converted to accurate mass employing the Xcalibur accurate mass calculation software. UV-Vis spectra were recorded using a Shimadzu UV1800 spectrophotometer. Photoluminescence (PL) spectra were recorded on an Agilent G9800 Cary Eclipse Fluorescence spectrophotometer. An Agilent Gel Permeation Chromatograph (GPC/SEC) equipped with two columns (PL mixed-C) and calibrated against twelve monodisperse polystyrene (PS) standards, using THF as an eluent at a flow rate of 1.0 mL min−1, was employed to determine the relative weight-average (Mw) and number-average (Mn) molecular weights and polydispersity indices (Đ = Mw/Mn) of all the reported polymers. FT-IR spectra were recorded on an FT/IR-6300 type A instrument using a KBr matrix. Single-crystal data collection was done on a Rigaku R-AXIS RAPID II diffractometer using filtered Mo-Kα radiation. The structure was solved by direct methods using the CrystalStructure crystallographic software package except for refinement, which was performed using SHELXL-97. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. Brunauer–Emmett–Teller (BET) surface area and porosity measurements were evaluated using a surface area and pore size analyzer (Gemini-V, Micromeritics, USA) at the boiling point of liquid nitrogen (−196 °C). Samples were deoxygenated in a VacuPrep 061 sample degassing system at a temperature of 120 °C overnight, before the experiments. Surface areas were calculated using the Brunauer–Emmett–Teller (BET) model of isotherms, and the adsorption of N2 at small relative pressures. The total pore volume (Vt) was determined from the specific adsorption of N2 at p/p0 = 0.99. The t-plot method was used to estimate the micropore volume (Vmic) and external surface area (Sext).
Synthesis
Synthesis of 2-((4-(tert-butyl)phenyl)ethynyl)benzaldehyde 1.
A Schlenk tube was charged under argon with 2-bromobenzaldehyde (0.608 mL, 5.2 mmol, 1 eq.), 4-(tert-butyl)phenylacetylene (1.4 mL, 7.8 mmol, 1.5 eq.), Pd2(dba)3 (28 mg, 0.03 mmol, 6 mol%), and CuI (20 mg, 0.1 mmol) in 3 mL of deoxygenated diisopropylamine (iPr2NH, 35 mmol) and the solution was refluxed overnight. The reaction mixture was then cooled to room temperature. After removal of the solvent under reduced pressure, the resulting mixture was dissolved in DCM and extracted with a saturated solution of NaHCO3 (50 mL × 3). The combined organic layer was washed with deionized water (100 mL × 4), and the desired product was isolated using silica gel column chromatography with DCM/hexane (15:85 v/v) as the eluent yielding a pale yellow solid (1.25 g, 92%). 1H-NMR (600 MHz, CD2Cl2, ppm): δ 10.68 (s, 1H, –CHO), 7.96 (d, 1H, J = 7.8 Hz ArH), 7.70 (d, 1H, J = 8.4, ArH) 7.63 (t, 1H, J = 7.7 Hz, ArH) 7.57 (d, 2H, J = 8.7 Hz ArH), 7.51–7.46 (m, 3H, ArH), 1.37 (s, 9H, –CH3). 13C-NMR (150 MHz, CD2Cl2, ppm): δ 192.09, 153.24, 136.45, 134.34, 133.75, 131.96, 129.06, 127.65, 127.52, 126.18, 119.85, 96.97, 84.88, 35.37, 31.44. FTIR (KBr, cm−1): 2966, 2218, 1697. EI-HRMS: m/z calculated for M˙+1 C19H19O: 263.1436, found: 263.1436.
Synthesis of CBM1 (procedure A).
A Schlenk tube was charged under argon with 2-((4-(tert-butyl)phenyl)ethynyl)benzaldehyde 1 (50 mg, 0.22 mmol, 2.2 eq.), 1,4-diethynyl triptycene 2a (30 mg, 0.1 mmol, 1 eq.), copper(II) triflate Cu(OTf)2 (7 mg, 0.02 mmol, 0.2 eq.), and trifluoroacetic acid TFA (30 μL, 0.4 mmol) in 5 mL of deoxygenated dichloroethane. The solution was heated at 100 °C overnight and the solvent was evaporated under reduced pressure. The resulting mixture was dissolved in DCM and extracted with a saturated solution of NaHCO3 (50 mL × 2). The combined organic layer was washed with deionized water (100 mL × 3), concentrated, and the desired product was isolated using silica gel column chromatography with DCM/hexane (20:80 v/v) as the eluent. White solid (48 mg, 96%). 1H-NMR (600 MHz, CD2Cl2, ppm): δ 8.13 (d, 2H, J = 8.4 Hz, ArH), 8.05 (d, 4H, J = 9.3 Hz, ArH), 8.00 (s, 2H, ArH) 7.69 (dd, 2H, J = 8.1 Hz, 1.6 Hz, ArH), 7.65 (m, 4H, ArH), 7.39 (q, 4H, J = 5.4 Hz, 2.2 Hz, ArH), 7.26 (s, 2H, ArH), 7.09 (q, 4H, J = 5.4 Hz, 2.4 Hz, ArH), 5.92 (s, 2H, triptycene-CHs). 13C-NMR (150 MHz, CD2Cl2, ppm): δ 146.06, 143.92, 138.44, 137.60, 134.12, 133.17, 128.86, 128.71, 128.65, 128.37, 128.32, 127.03, 126.98, 126.77, 126.21, 126.14, 125.95, 124.28, 51.52. EI-HRMS: m/z calculated for M˙+ C40H26: 506.2029, found: 506.2029.
Synthesis of CBM2.
CBM2 was prepared following procedure A with 2-((4-(tert-butyl)phenyl)ethynyl)benzaldehyde 1 (65 mg, 0.25 mmol, 2.2 eq.), 2c (65 mg, 0.11 mmol, 1 eq.), Cu(OTf)2 (8 mg, 0.02 mmol, 0.2 eq.), and TFA (35 μL, 0.45 mmol) in 5 mL of deoxygenated dichloroethane. Pale yellow solid (83 mg, 94%). 1H-NMR (600 MHz, CD2Cl2, ppm): δ 8.14 (s, 2H, ArH), 8.07 (t, 4H, J = 8.4 Hz, ArH), 7.95 (d, 2H, J = 7.4 Hz, ArH), 7.79 (d, 2H, J = 8.2 Hz, ArH), 7.63–7.61 (m, 6H, ArH), 7.25–7.22 (m, 8H, ArH), 7.13 (s, 4H, ArH), 5.59 (s, 2H, triptycene-CHs), 1.32 (s, 9H, –CH3,s), 1.25 (s, 9H, –CH3,s). 13C-NMR (150 MHz, CD2Cl2, ppm): δ 149.77, 146.17, 145.52, 143.96, 143.26, 139.56, 138.97, 137.59, 136.25, 135.94, 132.26, 130.75, 129.47, 127.56, 126.88, 126.22, 124.87, 124.77, 123.90, 123.38, 51.08, 34.27, 31.16. EI-HRMS: m/z calculated for M˙+ C60H50: 770.3913, found: 770.3913.
Synthesis of comonomer 2d.
A Schlenk tube was charged under argon with 1,4-diethynyl triptycene 2a (205 mg, 0.68 mmol, 1 eq.), 9-bromoanthracene (350 mg, 1.36 mmol, 2.0 eq.), bis(triphenylphosphine)palladium(II) dichloride PdCl2(PPh3)2 (23 mg, 0.034 mmol), CuI (6.4 mg, 0.034 mmol), triethylamine (284 μL, 2.0 mmol), and 15 mL of deoxygenated THF and the reaction mixture was refluxed for 16 h. The solvent was evaporated under reduced pressure and the resulting mixture was extracted with dichloromethane from a saturated aqueous solution of NaHCO3 (100 mL). The organic phase was washed with H2O (100 mL × 3). The desired product was isolated by silica gel column chromatography, with DCM/hexane (30:70 v/v) as the eluent. Yellow solid (410 mg, 93%). 1H NMR (600 MHz, CD2Cl2, ppm): δ 8.90 (d, J = 8.7 Hz, 4H, ArH), 8.62 (s, 2H, ArH) 8.17 (d, J = 8.5 Hz, 4H, ArH), 7.82–7.79 (m, 4H, ArH), 7.68–7.65 (m, 4H, ArH) 7.64 (q, J = 5.4, 3.2 Hz, 4H, ArH), 7.58 (s, 2H, ArH), 7.16 (q, J = 5.5, 3.1 Hz, 4H, ArH), 6.41 (s, 2H, triptycene–CH). 13C NMR (150 MHz, CD2Cl2, ppm): δ 147.36, 144.66, 132.75, 131.32, 128.92, 128.29, 127.061, 126.58, 125.90, 125.68, 124.07, 119.01, 116.98, 98.32, 91.14, 52.56; EI-HRMS: m/z calculated for M˙+ C52H30: 654.2342, found: 654.2342.
Synthesis of polymer CBP1 (procedure B).
1,4-Diethynyl triptycene 2a (145 mg, 0.48 mmol, 1 eq.), 2,5-bis(phenylethynyl)terephthalaldehyde 3 (160 mg, 0.48 mmol, 1 eq.), Cu(OTf)2 (70 mg, 0.192 mmol, 0.4 eq.), and TFA (295 μL, 3.84 mmol) were refluxed in 19 mL of deoxygenated dichloroethane in a Schlenk tube under argon. After 2 days of reaction, the precipitate was filtered and washed exhaustively with a sequence of solvents (50 mL of DCM, 50 mL of THF, 50 mL of MeOH, 50 mL of water, and 50 mL of diethyl ether) affording a deep red solid (200 mg, 98%). 1H NMR (600 MHz, CD2Cl2, ppm): δ 8.78 (br, 2H, ArH), 8.38 (br, 2H, ArH) 7.8–6.8 (m, 14H, ArH), 5.9 (br-d, 2H, triptycene–CH). 13C NMR (150 MHz, CD2Cl2, ppm): δ 145.82, 145.73, 129.09, 129.02, 127.06, 126.72, 126.28, 126.06, 125.03, 124.17, 51.68; FTIR (KBr, cm−1): 2960 (Ar–CH str.), 1680 (Ar–CH ben.), 1423 (Ar–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ben.); UV-vis: (THF, 10−8 M), λmax [nm] = 298, fluorescence: (THF, 10−8 M), λmax [nm] = 458.
C ben.); UV-vis: (THF, 10−8 M), λmax [nm] = 298, fluorescence: (THF, 10−8 M), λmax [nm] = 458.
Synthesis of CBP2.
CBP2 was prepared following procedure B with 2,5-bis(phenylethynyl)terephthalaldehyde 3 (166 mg, 0.5 mmol, 1 eq.), 2b (226 mg, 0.5 mmol, 1 eq.), Cu(OTf)2 (72 mg, 0.2 mmol, 0.4 eq.), and TFA (308 μL, 2.0 mmol) in 20 mL of deoxygenated dichloroethane. Deep red solid (275 mg, 96%). FTIR (KBr, cm−1): 3030 (Ar–CH str.), 1705 (Ar–CH ben.), 1456 (Ar–CC ben.); UV-vis: (THF, 10−8 M), λmax [nm] = 298, fluorescence: (THF, 10−8 M), λmax [nm] = 343, 370, 400.
Synthesis of CBP3.
CBP3 was prepared following procedure B with 2,5-bis(phenylethynyl)terephthalaldehyde 3 (234 mg, 0.7 mmol, 1 eq.), 2c (396 mg, 0.7 mmol, 1 eq.), Cu(OTf)2 (101 mg, 0.28 mmol, 0.4 eq.), and TFA (431 μL, 5.6 mmol) in 28 mL of deoxygenated dichloroethane. Deep red solid (450 mg, 93%). GPC (THF); Mw (g mol−1): 30187 Mn (g mol−1): 60288, Đ: 2.00; FTIR (KBr, cm−1): 3023 (Ar–CH), 2964 (aliphatic–CH str.), 1699 (Ar–CH ben.), 1460 (Ar–CC ben.); UV-vis: (THF, 10−8 M), λmax [nm] = 315, fluorescence: (THF, 10−8 M), λmax [nm] = 478.
Synthesis of CBP4.
CBP4 was prepared following procedure B with 2,5-bis(phenylethynyl)terephthalaldehyde 3 (100 mg, 0.3 mmol, 1 eq.), 2d (196 mg, 0.3 mmol, 1 eq.), Cu(OTf)2 (43 mg, 0.12 mmol, 0.4 eq.), and TFA (184 μL, 2.4 mmol) in 12 mL of deoxygenated dichloroethane. Deep red solid (225 mg, 96%). FTIR (KBr, cm−1): 3030 (Ar–CH str.), 1667 (Ar–CH ben.), 1460 (Ar–CC ben.); UV-vis: (THF, 10−8 M), λmax [nm] = 298, fluorescence: (THF, 10−8 M), λmax [nm] = 404, 430, 455.
Synthesis of CBP5.
CBP5 was prepared following procedure B with 2,5-bis(phenylethynyl)terephthalaldehyde 3 (90 mg, 0.27 mmol, 1 eq.), 2e (190 mg, 0.27 mmol, 1 eq.), Cu(OTf)2 (39 mg, 0.10 mmol, 0.4 eq.), and TFA (166 μL, 2.1 mmol) in 11 mL of deoxygenated dichloroethane. Deep red solid (218 mg, 97%). FTIR (KBr, cm−1): 3026 (Ar–CH str.), 1681 (Ar–CH ben.), 1463 (Ar–CC ben.); UV-vis: (THF, 10−8 M), λmax [nm] = 298, fluorescence: (THF, 10−8 M), λmax [nm] = 403, 429, 456.
Dye adsorption studies of CBP1–5
5 mg of CBP1–5 were immersed in a 5 mL aqueous solution of a dye (MB or MO) and the solution was stirred at room temperature. The UV-visible spectra of the solutions were then recorded after specific time intervals.
Results and discussion
Synthesis of prototypical monomers
As a proof of concept, the prototypical monomer CBM1 was prepared in 92% yield using a copper-catalyzed [4 + 2] cyclobenzannulation reaction from 1,4-diethynyltriptycene 2a76 with two equivalents of 2-((4-(tert-butyl)phenyl)ethynyl)benzaldehyde 1 in refluxing dichloroethane (Scheme 1). The structure of CBM1 was confirmed by 1H- and 13C-nuclear magnetic resonance (NMR), high-resolution mass spectroscopy (HRMS), and single crystal X-ray diffraction (XRD) (see Fig. 1 and 2 and related figures in the ESI†).
 |
| | Scheme 1 Synthesis of prototypical monomers CBM1–2. | |
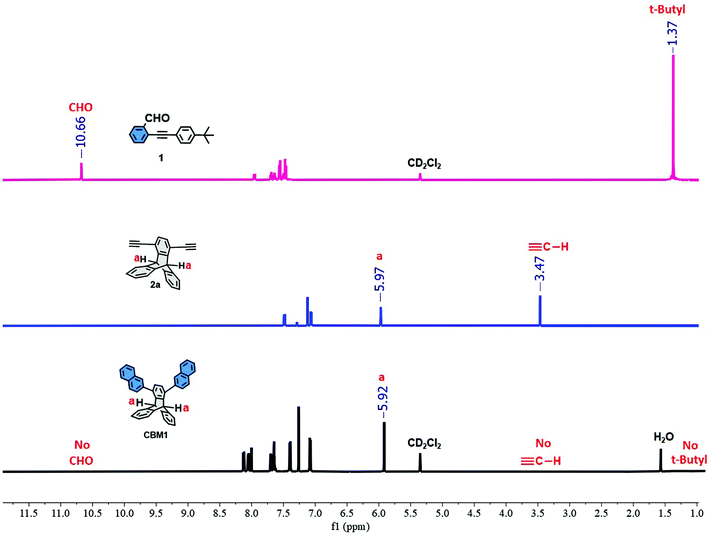 |
| | Fig. 1 Comparative 1H-NMR of 1, 2a, and CBM1. | |
 |
| | Fig. 2 Single crystal structures of CBM1 (left) and CBM2 (right). | |
Fig. 1 portrays the comparative 1H-NMR spectra of synthons 1, 2a and CBM1 where the spectrum of the latter prototypical monomer clearly confirms the presence of the desired peaks and the disappearance of all peaks related to the starting materials 1 and 2a, namely, the fingerprint chemical shifts of the carbaldehyde group at 10.66 ppm and the t-butyl group at 1.37 ppm for synthon 1 in addition to the proton peak of the ethynyl group of 2a at 3.47 ppm. The chemical shifts ranging from 7.0 ppm to 8.1 ppm are attributed to the aromatic protons of CBM1 while the peak detected at 5.9 ppm portrays the characteristic tertiary triptycene hydrogens (cf. peaks labeled e in Fig. 1). The distinctive peaks of the starting materials completely disappear in the 1H-NMR spectrum of CBM1, namely that of the terminal hydrogen of the ethynyl groups detected at 3.6 ppm in 2a79 in addition to the proton peaks of the aldehyde and t-butyl groups in synthon 1 detected at 10.6 ppm and 1.3 ppm, respectively (see Fig. S1 and S2 in the ESI†). On the other hand, the 13C-NMR spectrum of CBM1 displays all the expected aromatic peaks in the range of 146.0–124.2 ppm in addition to the chemical shift of the sp3 carbon of the triptycene unit at 51.52 ppm (see Fig. S7 in the ESI†). Furthermore, the high purity of CBM1 was confirmed by electron-impact high-resolution mass spectrometry (EI-HRMS, see Fig. S12 in the ESI†). Similarly, the replacement of the terminal sp-hybridized hydrogen atoms in 2a by the more sterically demanding p-tert-butylphenyl group (i.e.2c) resulted in the formation of the prototypical monomer CBM2 in 94% yield (Scheme 1). The structure of the latter compound was confirmed by 1H- and 13C-NMR as well as EI-HRMS (see Fig. 2 and related figures in the ESI†).
Single crystals of CBM1–2 were grown by depositing a layer of hexane on top of a solution of the monomers in dichloromethane. The molecular structures of CBM1–2 were investigated by single crystal X-ray diffraction, thus confirming the deviation of the pending naphthyl rings from planarity and leading, consequently, to highly branched structures in space (Fig. 2).
Synthesis of polymers CBP1–5
Synthesis of the target conjugated polymers CBP1–5 (Scheme 2) was carried out using reaction conditions similar to those employed to make the prototypical monomers CBM1–2 described in Scheme 1. The copper-catalyzed [4 + 2] cyclobenzannulation reaction of various 1,4-diethynyl triptycene derivatives 2a–e (with R = H, aryl) and 2,5-bis(phenylethynyl)terephthalaldehyde 3 afforded the target conjugated polymers CBP1–5 in excellent yields (Scheme 2). These latter polymers were decorated with various side groups like simple hydrogen atoms (CBP1), phenyl (CBP2) and p-tert-butylphenyl (CBP3) groups, as well as with the more sterically hindered anthracene (CBP4) and pyrene (CBP5) moieties.
 |
| | Scheme 2 Synthesis of polymers CBP1–5. | |
It is worthwhile to note that the degree of polymerization (DP) of the target conjugated polymers CBP1–5 was improved by optimizing both the reactants’ concentration and reaction time (Table 1). Therefore, the polymerization reaction of a 0.01 M solution of 2a with an equimolar amount of 3 in the presence of copper(II) triflate Cu(OTf)2 and trifluoroacetic acid (TFA) in refluxing dichloroethane (DCE) for 24 hours afforded CBP1 in 58% yield. Gel permeation chromatography (GPC) analysis of the latter target polymer revealed a number average molecular weight Mn of ≈5.5 kDa and polydispersity index Đ = Mw/Mn of ∼4.0 (Table 1, entry 1). When the reaction time was prolonged to 48 h, the Mn value slightly increased to ∼7.4 kDa whereas the polydispersity index Đ decreased to ∼3.8 (Table 1, entry 2). A further increase of the reaction time to 96 h did not improve the number average molecular weight Mn whose value stagnated at ∼7.0 kDa but recorded a very high polydispersity index Đ of 5.7 (Table 1, entry 3). It is noteworthy that the formation of CBP1 was further confirmed by 1H- and 13C-NMR spectroscopy (see Fig. S5 and S10 in the ESI†). However, the increasing polydispersity index that is accompanied by a relatively low number average molecular weight Mn, which does not improve even when prolonging the reaction time, could be explained by the low concentration of the reactants, thus leading to a non-homogeneous distribution of the polymers chains in solution during the reaction. Therefore, increasing the molar concentrations of monomers 2a and 3 to 0.025 M and maintaining the reaction for 48 h afforded polymer CBP1 as an insoluble brown powder in quantitative yield (Table 1, entry 4). The improvement of the degree of polymerization of the cyclobenzannulation reactions was further confirmed by preparing the target polymer CBP3 which contains tert-butyl side groups allowing for its GPC analysis that revealed a number average molecular weight Mn of ≈30 kDa with a polydispersity index Đ of ∼2.0. After optimization of the reaction conditions, the conjugated polymers CBP1–5 were all obtained in excellent yields but were found to be insoluble in common organic solvents, which limited their characterization to FTIR, UV-vis absorption and emission spectroscopies (see Fig. 3 and 4 and related figures in the ESI†).
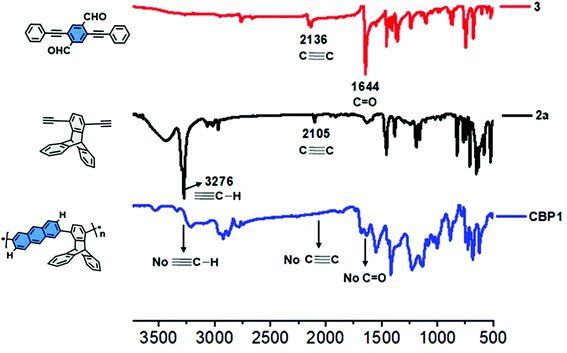 |
| | Fig. 3 Comparative FT-IR spectra of 3 (red), 2a (black), and CBP1 (blue). | |
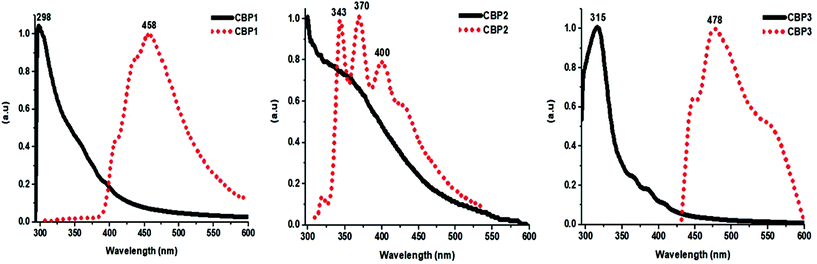 |
| | Fig. 4 Normalized UV-VIS absorption (solid lines) and emission (dotted lines) spectra of CBP1–3 (CM = 10−8 M in THF). | |
Table 1 Optimization conditions of the cyclobenzannulation polymerization reaction
| Entry |
Polymera |
Time (h) |
Yield (%) |
C
Mb [M] |
M
n (KDa) |
M
w (KDa) |
Đ
|
|
Cu(OTf)2(0.4 eq.), TFA (8.0 eq.), and DCE.
Molar concentration ×10−2 of 3.
|
| 1 |
CBP1
|
24 |
58 |
1.0 |
5.5 |
22 |
4.0 |
| 2 |
CBP1
|
48 |
79 |
1.0 |
7.4 |
28 |
3.8 |
| 3 |
CBP1
|
96 |
75 |
1.0 |
7.0 |
41 |
5.8 |
| 4 |
CBP1
|
48 |
100 |
2.5 |
Insoluble |
— |
— |
| 5 |
CBP2
|
48 |
87 |
2.5 |
Insoluble |
— |
— |
| 6 |
CBP3
|
48 |
100 |
2.5 |
30 |
60 |
2.0 |
| 7 |
CBP4
|
48 |
85 |
2.5 |
Insoluble |
— |
— |
| 8 |
CBP5
|
48 |
100 |
2.5 |
Insoluble |
— |
— |
Fig. 3 shows the comparative FT-IR absorption spectra of comonomers 3 and 2a besides their corresponding target polymer CBP1. The characteristic stretching vibrations of the alkynyl (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) and carbonyl (CO) groups in 3 are observed at 2136 cm−1 and 1644 cm−1, respectively. Similarly, the characteristic stretching vibrations of the terminal alkyne (C–H) and alkynyl (CC) in 2a can be detected at 3276 cm−1 and 2105 cm−1. All the stretching vibrations for the distinctive functional groups of synthons 2a and 3 (i.e C–H, CC and CO) completely disappeared in the FT-IR absorption spectrum of the target conjugated polymer CBP1, thereby proving the complete cyclobenzannulation reaction. It is noteworthy that the FT-IR spectrum of CBP1 discloses more pronounced aromatic CC and C–H bending and stretching vibrations in the regions of 1600–1400 cm−1 and 3050–2960 cm−1, respectively, when compared to the corresponding stretching vibrations recorded for the starting materials 2a and 3. This further confirms the full conversion of the aldehyde and ethynyl moieties into the corresponding aryl groups.
C) and carbonyl (CO) groups in 3 are observed at 2136 cm−1 and 1644 cm−1, respectively. Similarly, the characteristic stretching vibrations of the terminal alkyne (C–H) and alkynyl (CC) in 2a can be detected at 3276 cm−1 and 2105 cm−1. All the stretching vibrations for the distinctive functional groups of synthons 2a and 3 (i.e C–H, CC and CO) completely disappeared in the FT-IR absorption spectrum of the target conjugated polymer CBP1, thereby proving the complete cyclobenzannulation reaction. It is noteworthy that the FT-IR spectrum of CBP1 discloses more pronounced aromatic CC and C–H bending and stretching vibrations in the regions of 1600–1400 cm−1 and 3050–2960 cm−1, respectively, when compared to the corresponding stretching vibrations recorded for the starting materials 2a and 3. This further confirms the full conversion of the aldehyde and ethynyl moieties into the corresponding aryl groups.
The photophysical properties of the target polymers were measured by means of UV-Vis absorption and fluorescence spectroscopies (see Fig. 4 and Fig. S18 in the ESI†). CBP1, 2, 4 and 5 display similar features with a strong UV absorption band at 298 nm whereas CBP3, i.e. the polymer that contains tertiary butyl groups, discloses a strong absorption band at 315 nm, thus revealing a red shift by 17 nm (Fig. 4). The emission spectrum of CBP1 portrays a broad peak whose maximum is detected at ∼458 nm, where the latter could be attributed to the fingerprint region of the newly formed anthracene units (Fig. 4). On the other hand, while the emission spectrum of CBP2 displays three distinctive peaks at 343 nm, 370 nm, and 400 nm, the fluorescence of CBP3 discloses a broader red-shifted peak with an emission maximum at 478 nm. Interestingly, both target polymers CBP4 and CBP5 portray similar emission spectra with characteristic peaks at 404 nm, 430 nm, and 455 nm (see Fig. S18 in the ESI†).
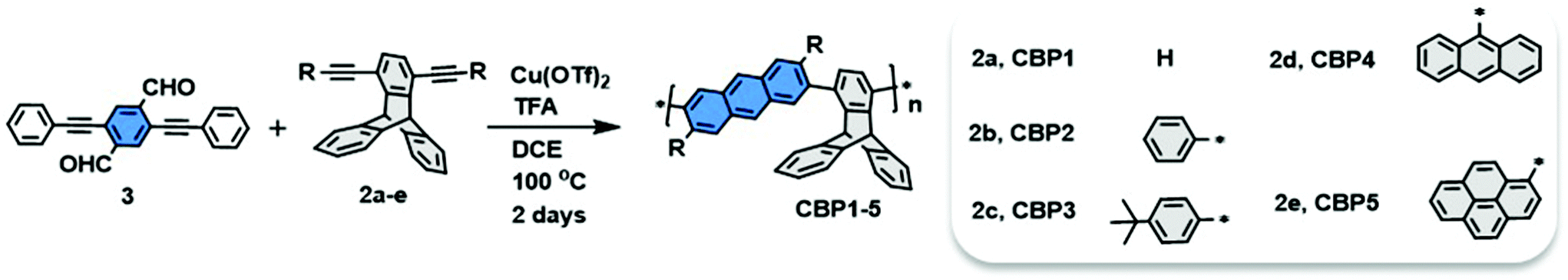
BET surface area studies
Microporosity properties of CBP1–5 were investigated by carrying out N2 adsorption experiments at 77 K and a low relative pressure (Fig. 5). Table 2 shows the Brunauer–Emmett–Teller (BET) surface areas and pore volumes derived from these isotherms. The conjugated polymer CBP1 shows a scarce BET surface area of ∼3.0 m2 g−1 (Table 2, entry 1). Conjugated polymer CBP2, i.e. bearing phenyl side groups, shows a surface area of 143 m2 g−1 (Table 2, entry 2). The polymers bearing anthracene (CBP4) and pyrene (CBP5) side groups reveal surface areas of 98 m2 g−1 and 203 m2 g−1, respectively (Table 2, entries 4 and 5). Interestingly, the conjugated polymer bearing the bulky tert-butylphenyl side groups, CBP3, displays the largest surface area of 794 m2 g−1 (Table 2, entry 3). It is worthwhile to note the hysteresis of N2 adsorption/desorption isotherms for CBP3 with significant N2 uptake at a low relative pressure (p/p0), which is considered as the primary indicator of microporosity (Fig. 5).
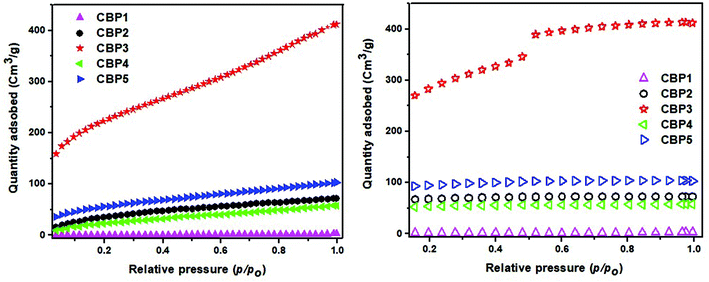 |
| | Fig. 5 Nitrogen adsorption (left) and desorption isotherms (right) of CBP1–5 measured at 77 K. | |
Table 2 Summary of BET results of polymers CBP1–5
| Entry |
Polymer |
BET surface area (m2 g−1) |
Pore volume (cm3 g−1) |
| 1 |
CBP1
|
3.0 |
0.005 |
| 2 |
CBP2
|
143 |
0.11 |
| 3 |
CBP3
|
794 |
0.63 |
| 4 |
CBP4
|
98 |
0.09 |
| 5 |
CBP5
|
203 |
0.16 |
Iodine adsorption studies
The graphitic-like structures of CBP1–5 polymers and their porous properties with BET surface areas reaching up to 794 m2 g−1 render them potential candidates as adsorbent materials, which have prompted us to explore their iodine uptake properties. Therefore, gravimetric analysis of iodine vapor adsorption59,80 was carried out by taking a 20 mg sample from each of the polymers CBP1–5 and exposing it to iodine vapors in an open glass vial, which was in turn placed inside a sealed glass vessel that contains excess iodine crystals heated at 80 °C under atmospheric pressure. The target polymers CBP1–4 revealed iodine uptake values ranging between 101 wt% and 145 wt% after 24 hours of exposure to iodine vapors (Table 3). Interestingly, ∼102 wt% was adsorbed by CBP5 after the first hour of exposure to iodine vapors, and which reached ∼166 wt% after 24 hours (Fig. 6). Furthermore, it has been noticed that the iodine adsorption values do not increase even when the target polymers CBP1–5 were kept under excess vapors of iodine for 48 hours, which suggests the saturation of CBP1–5 after 24 hours of exposure to iodine. The record value of 166 wt% of iodine uptake by CBP5 is considered to be promising when compared to the iodine adsorbent materials reported in the literature, namely, conjugated microporous polymers (200 wt%),59 hierarchically porous adamantane-based macromolecules (202 wt%),61 conjugated microporous polymers with thiophene units (222 wt%),81 nitrogen-rich triptycene-based materials (180 wt%),82 calix[4]arene-based 2D macromolecules (114 wt%),83 porphyrin and pyrene-based conjugated microporous polymers (130 wt%),84 JLUE covalent organic polymers (∼90 wt%),57 triazine-based covalent frameworks (177 wt%),47 and fluorine-enriched polymers (141 wt%).37
 |
| | Fig. 6 Wt% iodine adsorption (left) and desorption (right) graphs of CBP5. | |
Table 3 Summary of iodine adsorption and desorption of polymers CPP1–5
| Entry |
Polymer |
Wt% I2 adsorption |
Wt% I2 desorption |
| In 24 h |
In 180 min |
| 1 |
CBP1
|
145 |
137 |
| 2 |
CBP2
|
101 |
101 |
| 3 |
CBP3
|
135 |
135 |
| 4 |
CBP4
|
140 |
132 |
| 5 |
CBP5
|
166 |
166 |
The complete desorption efficiency of the iodine-loaded polymers (I2@CBP1–5) was recorded at different time intervals where the iodine vapors adsorbed by CBP1–5 were released by simple heating of the latter polymers in the air at 120 °C (Fig. 6 and Table 3). The reusability of the polymers was also investigated using CBP5 as a model candidate where a sample of CBP5 fully loaded with iodine vapors (I2@CBP5) was heated at 120 °C for 24 hours, in order to ensure the complete release of the adsorbate from the polymer backbone. The reactivated CBP5 was then exposed to iodine vapors and its uptake was recorded gravimetrically using the procedure described above, revealing an uptake pattern similar to that of a freshly prepared polymer. The iodine adsorption–desorption cycles were repeated several times showing no change in iodine uptake, and thus, confirming the possibility of regenerating the polymers.
Dye adsorption studies
Dye adsorption tests were investigated by soaking a sample of the conjugated target polymers CBP1–5 in various dyes’ aqueous solutions, in particular, methyl orange (MO), an anionic pigment, and methylene blue (MB), a cationic dye (see Fig. 7 and Fig. S19–22 in the ESI†). The efficiency of MB and MO dye removal from water by polymers CBP1–5 was investigated by recording the UV-visible absorbance spectra of the dyes’ aqueous solutions at different time intervals. The dye adsorption experiments were carried out by stirring a 5 mg sample of polymers CBP1–5 in a 5 mL aqueous solution of MB (5 mg L−1) at ambient temperature. A conspicuous decrease in the intensity of the MB absorbance maximum peak at 663 nm was observed, which confirms the adsorption of MB by polymers CBP1–5. Interestingly, more than 90% of MB dye was removed after stirring either CBP2 or CBP5 for 4 hours in the dye solution, which reached 100% MB adsorption when kept in solution overnight at room temperature. On the other hand, target polymers CBP1, 3 and 4 were found to adsorb up to ∼60%, ∼40%, and ∼90%, respectively, of MB when they were kept in the aqueous solution overnight at room temperature (see Fig. S19–S22 in the ESI†). MB adsorption could be noticed physically where the color of the solution faded from blue to colorless (Fig. 7, inset), and which proves the efficient removal of this dye from water by polymers CBP1–5. In contrast to its high uptake of MB, target polymer CBP5 was found to adsorb an insignificant amount of MO even after stirring it overnight at room temperature in the dye aqueous solution (Fig. 7).
 |
| | Fig. 7 MB (left) and MO (right) adsorption by CBP5 at various time intervals (inset: photographs showing the color change upon dye adsorption). | |
Selective adsorption and separation of dyes is one of the ways to assess the performance of a material as an adsorbent of a particular dye and/or a group of dyes. Therefore, the adsorption selectivity of polymer CBP5 over cationic/anionic dyes was explored by stirring 5 mg of polymer CBP5 at ambient temperature in a 5 ml aqueous solution of an equal mixture of the cationic MB (5 mg L−1) and anionic MO (5 mg L−1) dyes. The UV-visible spectra were recorded at different time intervals to investigate the adsorption of the two dyes present in solution (Fig. 8). Interestingly, the green solution mixture of MB and MO turned orange only after 5 minutes from the addition of CBP5 (Fig. 8 inset) where the UV-Visible absorption spectroscopy disclosed that more than 80% of the cationic MB dye was removed as evidenced by a sharp decrease in the characteristic maximum absorption peak of MB at ∼663 nm (Fig. 8). It is noteworthy that even when the solution is stirred overnight, only ∼10% of MO was adsorbed by CBP5, which strongly suggests that CBP5 can be employed as a selective sorbent material for cationic dyes such as MB over anionic dyes like MO.
 |
| | Fig. 8 Selective dye removal capability of CBP5 from a mixed solution of MB and MO (inset: photographs showing the color change after MB adsorption). | |
Conclusion
A novel synthetic methodology which consists of employing a copper-catalyzed [4 + 2] cyclobenzannulation polymerization reaction is disclosed leading to the formation of five conjugated graphitic-like polymers CBP1–5 in excellent yields. The conjugated polymers reveal high BET surface areas reaching up to 794 m2 g−1 and average pore volumes of 0.63 cm3 g−1. Iodine uptake tests of CBP1–5 demonstrate adsorption rates in the range of ∼101–166 wt%. Dye adsorption from aqueous solution using CBP1–5 show excellent and selective dye adsorbent properties reaching up to 100% removal of the toxic methylene blue over methyl orange at ambient temperature. The modular synthetic strategy, in addition to the promising microporous properties, and the use of these polymers as iodine and cationic dye adsorbents qualify these polymers to be considered as prominent materials for environmental and energy applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project was partially supported by the Kuwait Foundation for the Advancement of Sciences (KFAS) under project codes PN18-14SC-03 and P314-34SC-01. We would like to thank the general facilities projects GS01/01, GS01/03, GS01/05, GS03/01, and GS03/08 at Kuwait University.
References
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- H. Hong, Z. Guo, D. Yan and H. Zhan, Microporous Mesoporous Mater., 2020, 294, 109870 CrossRef CAS.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- S.-J. Yang, X. Ding and B.-H. Han, Macromolecules, 2018, 51, 947–953 CrossRef CAS.
- D. Reinhard, W.-S. Zhang, Y. Vaynzof, F. Rominger, R. R. Schröder and M. Mastalerz, Chem. Mater., 2018, 30, 2781–2790 CrossRef CAS.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- Y. Zhao, W. Ma, Y. Xu, C. Zhang, Q. Wang, T. Yang, X. Gao, F. Wang, C. Yan and J.-X. Jiang, Macromolecules, 2018, 51, 9502–9508 CrossRef CAS.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- J. Yang, P. Cong, L. Chen, X. Wang, J. Li, A. Tang, B. Zhang, Y. Geng and E. Zhou, ACS Macro Lett., 2019, 8, 743–748 CrossRef.
- K. Aoshima, M. Nomura and A. Saeki, ACS Omega, 2019, 4, 15645–15652 CrossRef CAS PubMed.
- H. Huang, H. Bin, Z. Peng, B. Qiu, C. Sun, A. Liebman-Pelaez, Z.-G. Zhang, C. Zhu, H. Ade, Z. Zhang and Y. Li, Macromolecules, 2018, 51, 6028–6036 CrossRef CAS.
- S. Gu, S. Wu, L. Cao, M. Li, N. Qin, J. Zhu, Z. Wang, Y. Li, Z. Li, J. Chen and Z. Lu, J. Am. Chem. Soc., 2019, 141(24), 9623–9628 CrossRef CAS PubMed.
- S. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2018, 58, 849–853 CrossRef PubMed.
- C.-F. Yao, K.-L. Wang, H.-K. Huang, Y.-J. Lin, Y.-Y. Lee, C.-W. Yu, C.-J. Tsai and M. Horie, Macromolecules, 2017, 50, 6924–6934 CrossRef CAS.
- H. Wang, Z. Cheng, Y. Liao, J. Li, J. Weber, A. Thomas and C. F. J. Faul, Chem. Mater., 2017, 29, 4885–4893 CrossRef CAS.
- V. Lakshmi, C.-H. Liu, M. Rajeswara Rao, Y. Chen, Y. Fang, A. Dadvand, E. Hamzehpoor, Y. Sakai-Otsuka, R. S. Stein and D. F. Perepichka, J. Am. Chem. Soc., 2020, 142, 2155–2160 CrossRef CAS PubMed.
- S.-H. Kang, A. Jeong, H. R. Lee, J. H. Oh and C. Yang, Chem. Mater., 2019, 31, 3831–3839 CrossRef CAS.
- Y. Du, H. Yao, L. Galuska, F. Ge, X. Wang, H. Lu, G. Zhang, X. Gu and L. Qiu, Macromolecules, 2019, 52, 4765–4775 CrossRef CAS.
- F. Chen, Y. Jiang, Y. Sui, J. Zhang, H. Tian, Y. Han, Y. Deng, W. Hu and Y. Geng, Macromolecules, 2018, 51, 8652–8661 CrossRef CAS.
- T. M. Swager, Macromolecules, 2017, 50, 4867–4886 CrossRef CAS.
- X. Yan, M. Xiong, J.-T. Li, S. Zhang, Z. Ahmad, Y. Lu, Z.-Y. Wang, Z.-F. Yao, J.-Y. Wang, X. Gu and T. Lei, J. Am. Chem. Soc., 2019, 141, 20215–20221 CrossRef CAS PubMed.
- R. Sun, S. Feng, B. Zhou, Z. Chen, D. Wang and H. Liu, ACS Macro Lett., 2020, 9, 43–48 CrossRef CAS.
- A. Hirose, K. Tanaka, R. Yoshii and Y. Chujo, Polym. Chem., 2015, 6, 5590–5595 RSC.
- T. Ikai, T. Yoshida, K.-I. Shinohara, T. Taniguchi, Y. Wada and T. M. Swager, J. Am. Chem. Soc., 2019, 141, 4696–4703 CrossRef CAS PubMed.
- B. Alameddine, N. Baig, S. Shetty and S. Al-Mousawi, Polymer, 2019, 178, 121589 CrossRef CAS.
- N. Baig, S. Shetty, S. Al-Mousawi, F. Al-Sagheer and B. Alameddine, Mater. Today Chem., 2018, 10, 213–220 CrossRef CAS.
- K. Aravindu, E. Cloutet, C. Brochon, G. Hadziioannou, J. Vignolle, F. Robert, D. Taton and Y. Landais, Macromolecules, 2018, 51, 5852–5862 CrossRef CAS.
- Z. Wang, C. Wang, Y. Fang, H. Yuan, Y. Quan and Y. Cheng, Polym. Chem., 2018, 9, 3205–3214 RSC.
- B. Alameddine, S. Shetty, N. Baig and S. Al-Mousawi, Mater. Today Chem., 2019, 14, 100190 CrossRef CAS.
- A. Leventis, J. Royakkers, A. G. Rapidis, N. Goodeal, M. K. Corpinot, J. M. Frost, D.-K. Bučar, M. O. Blunt, F. Cacialli and H. Bronstein, J. Am. Chem. Soc., 2018, 140, 1622–1626 CrossRef CAS PubMed.
- A. Ichige, H. Saito, J. Kuwabara, T. Yasuda, J.-C. Choi and T. Kanbara, Macromolecules, 2018, 51, 6782–6788 CrossRef CAS.
- M. J. Sung, S. Yoon, S.-K. Kwon, Y.-H. Kim and D. S. Chung, ACS Appl. Mater. Interfaces, 2016, 8, 31172–31178 CrossRef CAS PubMed.
- S. Shetty, N. Baig, A. Hassan, S. Al-Mousawi, N. Das and B. Alameddine, Microporous Mesoporous Mater., 2020, 303, 110256 CrossRef CAS.
- J. M. Tobin, J. Liu, H. Hayes, M. Demleitner, D. Ellis, V. Arrighi, Z. Xu and F. Vilela, Polym. Chem., 2016, 7, 6662–6670 RSC.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- G. Li, C. Yao, J. Wang and Y. Xu, Sci. Rep., 2017, 7, 13972 CrossRef PubMed.
- J. Choi, J. H. Ko, C. W. Kang, S. M. Lee, H. J. Kim, Y.-J. Ko, M. Yang and S. U. Son, J. Mater. Chem. A, 2018, 6, 6233–6237 RSC.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. I. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738–8741 CrossRef CAS PubMed.
- J.-X. Jiang, A. Trewin, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1777–1781 RSC.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- H. Bohra, P. Li, C. Yang, Y. Zhao and M. Wang, Polym. Chem., 2018, 9, 1972–1982 RSC.
- S. Hayashi, Y. Togawa, S.-I. Yamamoto, T. Koizumi, K. Nishi and A. Asano, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3862–3867 CrossRef CAS.
- A. Alam, S. Mishra, A. Hassan, R. Bera, S. Dutta, K. Das Saha and N. Das, ACS Omega, 2020, 5, 4250–4260 CrossRef CAS PubMed.
- T. Geng, W. Zhang, Z. Zhu, G. Chen, L. Ma, S. Ye and Q. Niu, Polym. Chem., 2018, 9, 777–784 RSC.
- Z. Guo, P. Sun, X. Zhang, J. Lin, T. Shi, S. Liu, A. Sun and Z. Li, Chem. – Asian J., 2018, 13, 2046–2053 CrossRef CAS PubMed.
- S. Bai, Q. Hu, Q. Zeng, M. Wang and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 11319–11327 CrossRef CAS PubMed.
- X. Ding and B.-H. Han, Angew. Chem., Int. Ed., 2015, 54, 6536–6539 CrossRef CAS PubMed.
- Z. J. Wang, R. Li, K. Landfester and K. A. I. Zhang, Polymer, 2017, 126, 291–295 CrossRef CAS.
- Y. Wei, W. Chen, X. Zhao, S. Ding, S. Han and L. Chen, Polym. Chem., 2016, 7, 3983–3988 RSC.
- BP Statistical Review of World Energy, Report https://www.bp.com/en/global/corporate/energy-economics/statistical-review-of-world-energy.html, British Petroleum, 2019.
-
M. Stephenson, Energy and Climate Change: An Introduction to Geological Controls, Interventions and Mitigations, 2018, ISBN: 978-0-12-812021-7 Search PubMed.
-
B. Zohuri, in Nuclear Reactor Technology Development and Utilization, ed. S. U.-D. Khan and A. Nakhabov, Woodhead Publishing, 2020, pp. 61–120, DOI:10.1016/B978-0-12-818483-7.00002-0.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- H. Guan, D. Zou, H. Yu, M. Liu, Z. Liu, W. Sun, F. Xu and Y. Li, Front. Mater., 2019, 6, 12 CrossRef.
- H. Ma, J.-J. Chen, L. Tan, J.-H. Bu, Y. Zhu, B. Tan and A. C. Zhang, ACS Macro Lett., 2016, 5, 1039–1043 CrossRef CAS.
- N. Baig, S. Shetty, S. Al-Mousawi and B. Alameddine, Polym. Chem., 2020, 11, 3066–3074 RSC.
- M. Janeta, W. Bury and S. Szafert, ACS Appl. Mater. Interfaces, 2018, 10, 19964–19973 CrossRef CAS PubMed.
- D. Chen, Y. Fu, W. Yu, G. Yu and C. Pan, Chem. Eng. J., 2018, 334, 900–906 CrossRef CAS.
- C. Pei, T. Ben, S. Xu and S. Qiu, J. Mater. Chem. A, 2014, 2, 7179–7187 RSC.
- A. Narula and C. P. Rao, ACS Omega, 2019, 4, 5731–5740 CrossRef CAS PubMed.
- M. Yan, Y. Wu, Y. Yan, X. Yan, F. Zhu, Y. Hua and W. Shi, ACS Sustainable Chem. Eng., 2016, 4, 757–766 CrossRef CAS.
- A. Kumar and S. Priyanka, New J. Chem., 2019, 43, 14997–15013 RSC.
- L. Cheng, M. Wei, L. Huang, F. Pan, D. Xia, X. Li and A. Xu, Ind. Eng. Chem. Res., 2014, 53, 3478–3485 CrossRef CAS.
- S. Jain, S. Mishra and T. K. Sarma, ACS Sustainable Chem. Eng., 2018, 6, 9771–9783 CrossRef CAS.
- J. Lin, F. Lin, X. Chen, W. Ye, X. Li, H. Zeng and B. Van der Bruggen, Ind. Eng. Chem. Res., 2019, 58, 11003–11012 CrossRef CAS.
- G. Han, T.-S. Chung, M. Weber and C. Maletzko, Environ. Sci. Technol., 2018, 52, 3676–3684 CrossRef CAS PubMed.
- H. Liu and H. Liu, J. Mater. Chem. A, 2017, 5, 9156–9162 RSC.
- H. Liu, R. Sun, S. Feng, D. Wang and H. Liu, Chem. Eng. J., 2019, 359, 436–445 CrossRef CAS.
- D. Lehnherr, J. M. Alzola, E. B. Lobkovsky and W. R. Dichtel, Chem. – Eur. J., 2015, 21, 18122–18127 CrossRef CAS PubMed.
- X. Yang, L. Yuan, Z. Chen, Z. Liu and Q. Miao, Org. Lett., 2018, 20, 6952–6956 CrossRef CAS PubMed.
- S. J. Hein, D. Lehnherr, H. Arslan, F. J. Uribe-Romo and W. R. Dichtel, Acc. Chem. Res., 2017, 50, 2776–2788 CrossRef CAS PubMed.
- Z. Zhu and T. M. Swager, Org. Lett., 2001, 3, 3471–3474 CrossRef CAS PubMed.
- B. Alameddine, N. Baig, S. Shetty, F. Al-Sagheer and S. Al-Mousawi, Asian J. Org. Chem., 2018, 7, 378–382 CrossRef CAS.
- N. Baig, S. Shetty, S. Al-Mousawi, F. Al-Sagheer and B. Alameddine, React. Funct. Polym., 2019, 139, 153–161 CrossRef CAS.
- Z. Xie, B. Yang, L. Liu, M. Li, D. Lin, Y. Ma, G. Cheng and S. Liu, J. Phys. Org. Chem., 2005, 18, 962–973 CrossRef CAS.
- B. Alameddine, N. Baig, S. Shetty, S. Al-Mousawi and F. Al-Sagheer, Polymer, 2018, 154, 233–240 CrossRef CAS.
- Y. H. Abdelmoaty, T.-D. Tessema, F. A. Choudhury, O. M. El-Kadri and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2018, 10, 16049–16058 CrossRef CAS PubMed.
- X. Qian, Z.-Q. Zhu, H.-X. Sun, F. Ren, P. Mu, W. Liang, L. Chen and A. Li, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS.
- R. M. Weiss, A. L. Short and T. Y. Meyer, ACS Macro Lett., 2015, 4, 1039–1043 CrossRef CAS.
- D. Shetty, T. Skorjanc, J. Raya, S. K. Sharma, I. Jahovic, K. Polychronopoulou, Z. Asfari, D. S. Han, S. Dewage, J.-C. Olsen, R. Jagannathan, S. Kirmizialtin and A. Trabolsi, ACS Appl. Mater. Interfaces, 2018, 10, 17359–17365 CrossRef CAS PubMed.
- K. C. Park, J. Cho and C. Y. Lee, RSC Adv., 2016, 6, 75478–75481 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2059925 and 2059926. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1py00193k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab