
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and photophysical properties of linear gold(I) complexes based on a CCC carbene†
Alexander S.
Romanov
 *ab,
Mikko
Linnolahti
*c and
Manfred
Bochmann
*a
*ab,
Mikko
Linnolahti
*c and
Manfred
Bochmann
*a
aSchool of Chemistry, University of East Anglia, Earlham Road, Norwich, NR4 7TJ, UK. E-mail: m.bochmann@uea.ac.uk
bSchool of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: alexander.romanov@manchester.ac.uk
cDepartment of Chemistry, University of Eastern Finland, Joensuu Campus, FI-80101 Joensuu, Finland. E-mail: mikko.linnolahti@uef.fi
First published on 8th November 2021
Abstract
The reaction between allenylpyridine (L1) and (Me2S)AuCl resulted in the quantitative formation of the (Indolizy)gold chloride complex 1 (Indolizy = indolizin-2-ylidene). The reaction of 1 with carbazole in the presence of KOtBu affords the corresponding (Indolizy)Au(Cz) complex 2. Both compounds show high air- and temperature stability. The crystal structure of 2 confirmed the linear co-planar geometry. Complex 1 shows an intense low energy absorption of mixed character in the UV-vis spectrum, ascribed to intraligand and (M + Hal)L charge transfer processes, and exhibits bright yellow phosphorescence with an excited state lifetime of 62.8 μs in the crystal and a luminescence quantum yield up to 65%. On the other hand, the carbazolate complex 2 in a polystyrene matrix shows bright red delayed fluorescence at 617 nm with a sub-microsecond excited state lifetime and a quantum yield of 21.6%.
Introduction
Coinage metal complexes with linear geometry of the type L–M–X, where L = a cyclic (alkyl)(amino)carbene (CAAC) ligand and X = halide, pseudohalide, phenolate or amide, have recently emerged as a new class of highly energy efficient light-emitting materials.1–11 Organic light-emitting diodes (OLEDs) based on these emitter types have been shown to give external quantum efficiencies of well above 20%.2,5,7 Complexes where X = aryl amide, and especially X = carbazolate (Cz), are particularly effective and have given rise to so-called “carbene–metal–amide” (CMA) materials. Luminescence of these compounds in many cases is ascribed to thermally activated delayed fluorescence (TADF), which is due to a ligand-to-ligand charge transfer (LLCT) process involving electron donation from the electron-rich amide ligand to a LUMO based mainly on the carbene p-orbital, which acts as π-electron acceptor.2,5–7,12–14 We2,3,15a and others11 have recently demonstrated that electronic and steric properties of the carbene ligand have a crucial influence on the photophysical properties of these CMA materials.15 The electronic properties of the carbene can be effectively controlled via stabilization of the vacant p-orbital of the carbene and by the number of N-atoms adjacent to the carbene-carbon atom. Gold complexes of carbenes lacking this heteroatom stabilization have been reported only recently.16 The groups of Fensterbank17 and Munoz18 reported that cycloisomerization of allenylpyridines allows the isolation of gold and platinum complexes of indolizin-2-ylidene (Indolizy) type carbenes, that is carbenes with the heteroatom in 3-position relative to the carbene-C.19 Here we report new Indolizy carbene gold complexes having chloride and carbazolide anionic ligands in order to study the effect of carbenes with remote heteroatom stabilization on the photoluminescent properties of the CMA materials.Results and discussion
Synthesis and structure
The (Indolizy)gold chloride complex 1 was prepared in nearly quantitative yield by cyclization of the pyridinylallene L1 with one equivalent of (Me2S)AuCl according to Scheme 1. Reaction of 1 with carbazole in the presence of NaOtBu as base afforded the (carbene)gold carbazolide complex 2 in high yield. The products were obtained as off-white (1) and brown (2) solids which are poorly soluble in low-polarity solvents such as hexane, Et2O or toluene but show moderate to good solubility in CH2Cl2 and 1,2-difluorobenzene (DFB). Both complexes are stable in air for at least several months. The purity was established after recrystallization by 1H and 13C{1H} NMR spectroscopy, elemental analysis and mass spectrometry. The carbene-carbon resonances were observed at δC 195.1 and 199.6 ppm for 1 and 2, respectively. The thermal stability was evaluated by thermogravimetric analysis (TGA, Fig. S1†) and differential scanning calorimetry (DSC). The complexes show a multistep decomposition process with similar decomposition temperatures (Td, 2% weight loss) of 223.4 and 212.3 °C for 1 and 2, respectively.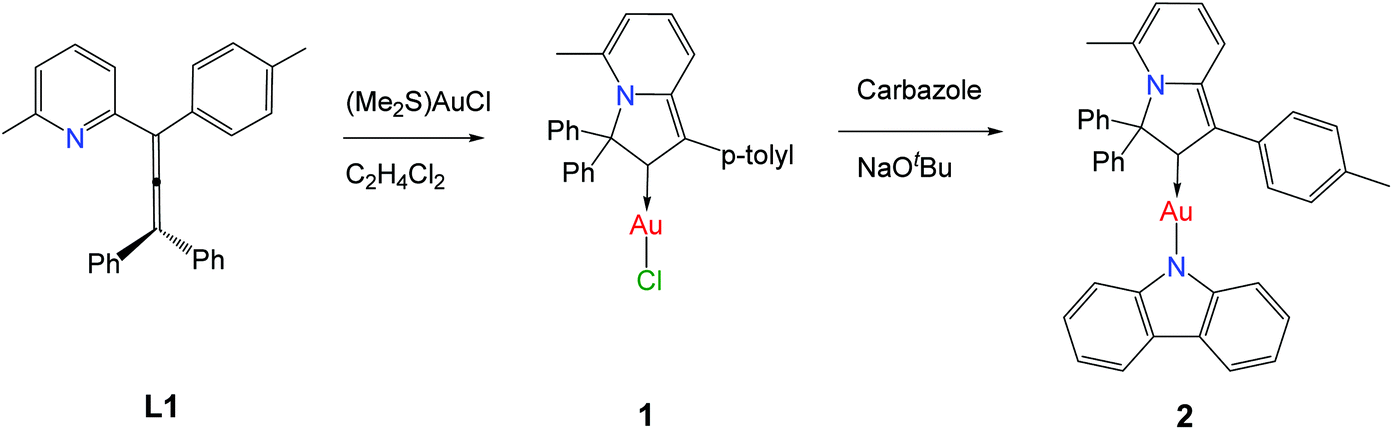 | ||
| Scheme 1 Synthesis of gold complexes 1 and 2. | ||
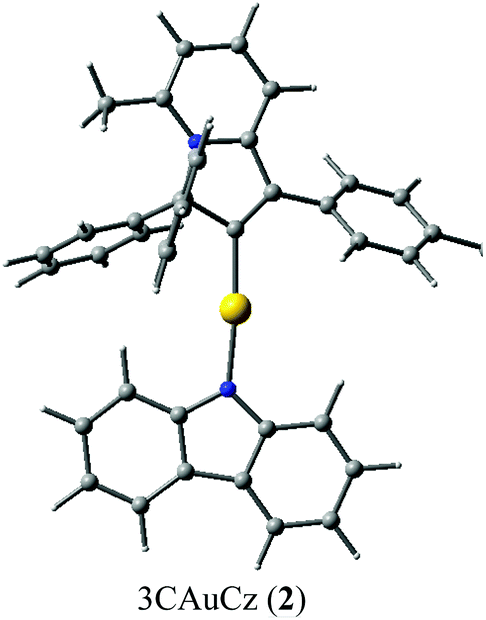
Single crystals of 2 suitable for X-ray diffraction were successfully grown from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1,2-difluorobenzene/CH2Cl2 solution layered by diethyl ether. The crystal structure (Fig. 1) shows a linear geometry, without any close Au⋯Au contacts. The Au–Ccarbene and Au–Namide bond lengths of 1.987(3) and 2.028(3) Å are similar to the distances observed in the previously reported parent CMA complex (AdCAAC)Au(Cz) (1.991(3) and 2.026(2) Å).2 These Au–C and Au–N bond lengths indicate that complex 2 can be considered as a gold carbene complex; however, the short C1–C4 distance suggests that the bonding may also be interpreted as a gold vinyl. The carbene and carbazolate ligands are nearly co-planar, which is similar to previous CMA complexes.2,3,7 The angle sum around the Cz–N atom is 356.1°, indicating a slight pyramidalisation of the N-atom; this distortion is absent in the calculated gas phase structure (vide infra). Analysis of the intermolecular contacts shows that molecules of 2 form a three-dimensional network via weak multiple C–H⋯π and short C6A–H6A⋯N2(carbazole) contacts [2.747(3) Å], where A is the symmetry operator 1/2 − x; −1/2 + y; 1/2 − z (Fig. S2†). The C6A–H6A⋯N2 contact is likely the origin for the pyramidalisation of the carbazole N2 atom in the crystalline environment and affects the photophysical properties of complex 2 in the solid state.
:1 mixture of 1,2-difluorobenzene/CH2Cl2 solution layered by diethyl ether. The crystal structure (Fig. 1) shows a linear geometry, without any close Au⋯Au contacts. The Au–Ccarbene and Au–Namide bond lengths of 1.987(3) and 2.028(3) Å are similar to the distances observed in the previously reported parent CMA complex (AdCAAC)Au(Cz) (1.991(3) and 2.026(2) Å).2 These Au–C and Au–N bond lengths indicate that complex 2 can be considered as a gold carbene complex; however, the short C1–C4 distance suggests that the bonding may also be interpreted as a gold vinyl. The carbene and carbazolate ligands are nearly co-planar, which is similar to previous CMA complexes.2,3,7 The angle sum around the Cz–N atom is 356.1°, indicating a slight pyramidalisation of the N-atom; this distortion is absent in the calculated gas phase structure (vide infra). Analysis of the intermolecular contacts shows that molecules of 2 form a three-dimensional network via weak multiple C–H⋯π and short C6A–H6A⋯N2(carbazole) contacts [2.747(3) Å], where A is the symmetry operator 1/2 − x; −1/2 + y; 1/2 − z (Fig. S2†). The C6A–H6A⋯N2 contact is likely the origin for the pyramidalisation of the carbazole N2 atom in the crystalline environment and affects the photophysical properties of complex 2 in the solid state.
 | ||
| Fig. 1 Crystal structure of 2. Ellipsoids are shown at the 50% level. Selected bond lengths [Å] and angles [°]: Au1–C1 1.987(3), Au1–N2 2.028(3), C1–C2 1.533 (5), C1–C4 1.362(5), C2–N1 1.521(4), C3–C4 1.454(4), C3–N1 1.372(4); C1–Au1–N2 178.7(1)°, Au1–N2–C41 126.2(2)°, C41–N2–C29 106.5(3)°, C29–N2–Au1 123.6(2)°; torsion angle C1–C2–N2–C41 10.9(2)°. | ||
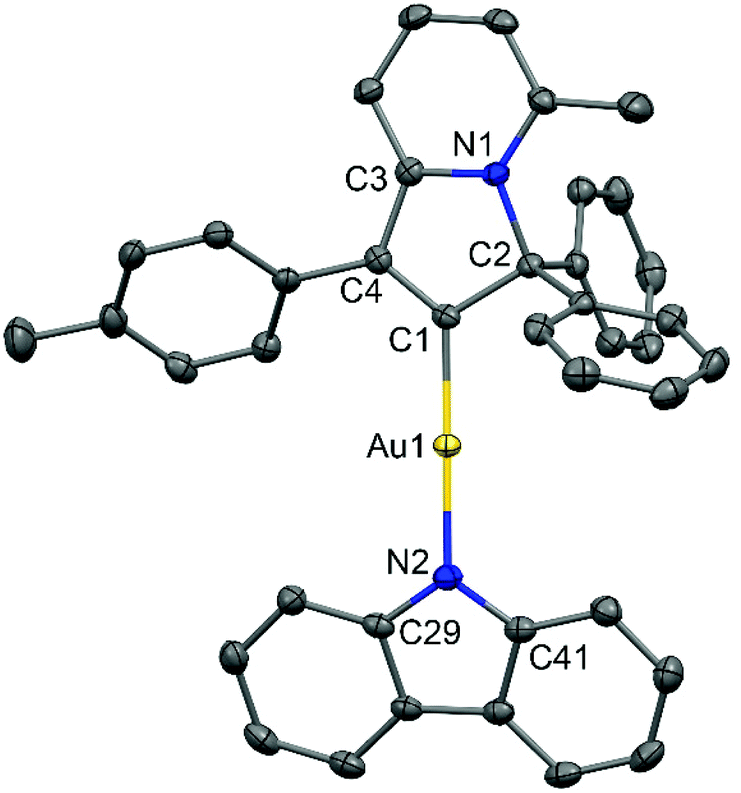
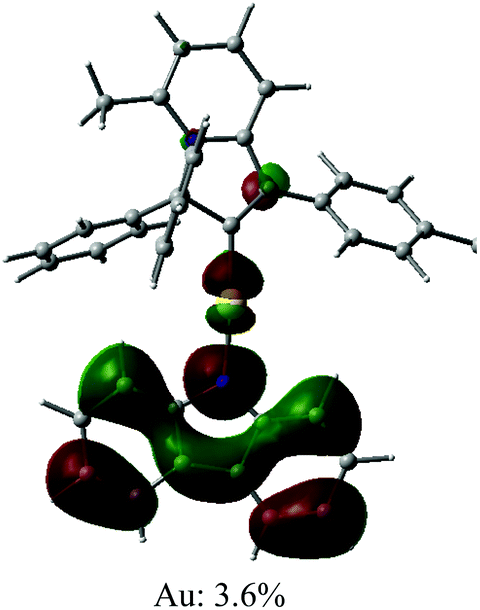
Cyclic voltammetry has been used to estimate HOMO and LUMO energy levels for 1 and 2 (DFB solution, [nBu4N]PF6 as supporting electrolyte; see Table 1 and Fig. 2). Both complexes show two irreversible reduction processes with no back peak. The reduction of 2 is slightly anodically shifted by 60 mV compared to 1. Such minor variation in the values of the reduction potentials after chemical modification of the gold complex indicates that both reductions are centered on the Indolizy carbene ligand. This is in excellent agreement with the most recent theoretical calculations17b indicating that both LUMO and LUMO+1 are localized on the carbene ligand, with an energy difference of 0.86 eV. In fact, both 1 and 2 show a 0.74 V peak separation between the first and second reduction processes (Table 1) which corroborates these theoretical results.17b Complex 1 shows one irreversible oxidation wave at +1.33 V, whereas 2 shows three well-resolved irreversible oxidations at much lower potential (Table 1). Our calculations suggest that the first oxidation process of 2 is largely localized on the Cz ligand (Table 2). The HOMO energy levels were obtained based on the onset of the first oxidation potentials (–6.38 and −5.39 eV for 1 and 2, respectively). The HOMO–LUMO gap for complex 2 is therefore 0.93 eV smaller than for 1, which has a strong impact on the luminescent properties, see below.
 | ||
| Fig. 2 Cyclic voltammograms for 1 (top) and 2 (bottom), recorded in 1,2-difluorobenzene solution (1.4 mM) with [n-Bu4N]PF6 as supporting electrolyte (0.13 M) using a glassy carbon electrode, scan rate 0.1 Vs−1. | ||
| Complex | Reduction | E LUMO eV | Oxidation | E HOMO eV | ΔE eV | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| E 2nd | E 1st | E onset red | E 1st | E onset ox | E 2nd | E 3rd | ||||
| a In 1,2-difluorobenzene solution, recorded using a glassy carbon electrode, concentration 1.4 mM, supporting electrolyte [nBu4N][PF6] (0.13 M), measured at 0.1 V s−1. EHOMO = – (Eonset ox Fc/Fc+ + 5.39) eV; ELUMO = –(Eonset red Fc/Fc+ + 5.39) eV. | ||||||||||
| 1 | –2.72 | –1.98 | –1.84 | –3.55 | +1.33 | +0.99 | — | — | –6.38 | 2.83 |
| 2 | −2.78 | −2.04 | −1.90 | –3.49 | +0.11 | +0.00 | +0.50 | +0.83 | –5.39 | 1.90 |
| Optimised geometry | HOMO | LUMO |
|---|---|---|
|
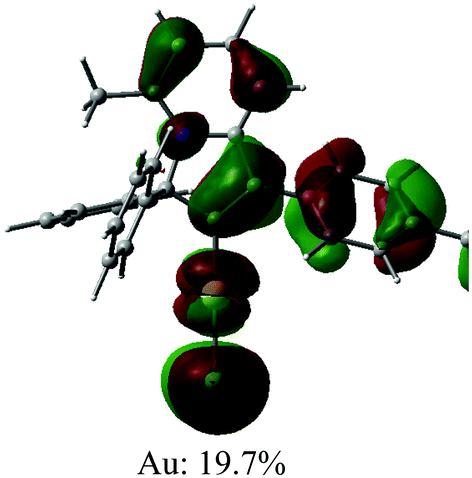
|
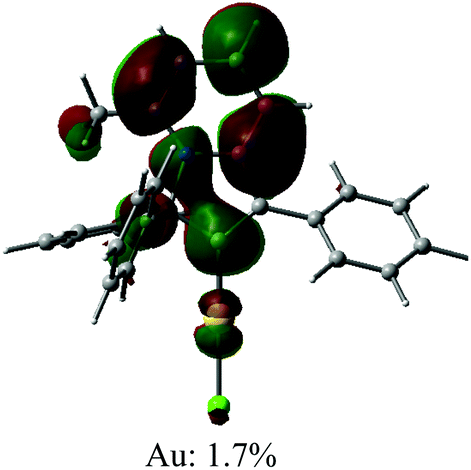
|
|
|
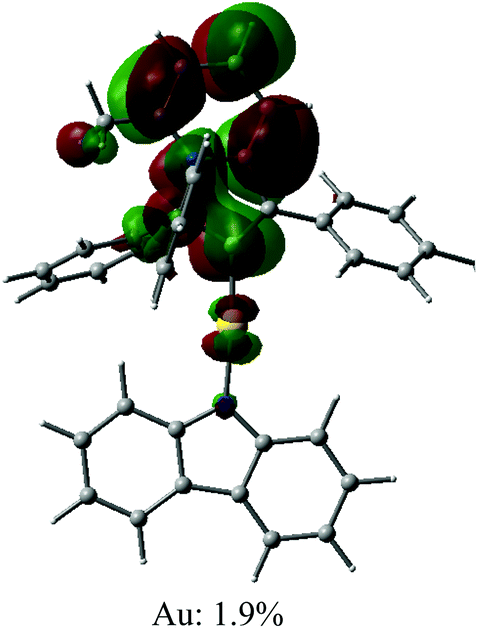
|
Photophysical properties and theoretical considerations
The UV/vis absorption spectra for gold complexes were collected in solvents of various polarity, i.e., toluene, THF and DFB (Fig. 3a and 4a), see Table S1 (ESI).† The chloride complex 1 shows an intra-ligand (IL) transition centred on the carbene ligand at ca. 300 nm which can be ascribed to an allowed π–π* transition based on strong extinction coefficients (ca. 15000 M−1 cm−1). Complex 1 shows a low-energy absorption band at ca. 400 nm, which according to theoretical calculations likely involves a combination of IL and (M + Hal)L charge transfer processes (Table 2). Complex 2 shows several high energy IL transitions associated with either the carbene (300 nm) or the carbazole (375 nm) moieties, while the lowest absorption band (425–600 nm) can be ascribed to a L(M)L charge transfer process from the HOMO (carbazole) to a LUMO (carbene) with up to 5% contribution of gold orbitals (see Table 2). This was further supported by measuring UV-vis absorption in solvents with different polarity, where 2 shows the largest blue-shift from toluene to DFB (54 nm), whereas 1 shows a much smaller blue shift of 15 nm. This negative solvatochromism is commonly observed for various CMA emitters2,10 and is associated with the opposite orientation of the dipole moments in the excited and the ground states (Table S4, ESI†). The smaller extinction coefficient for the L(M)LCT band (ca. 4200 M−1 cm−1) for 2 correlates well with the smaller HOMO–LUMO overlap integral of 0.23 for 2 vs 0.45 for 1 (Table S3, ESI†).
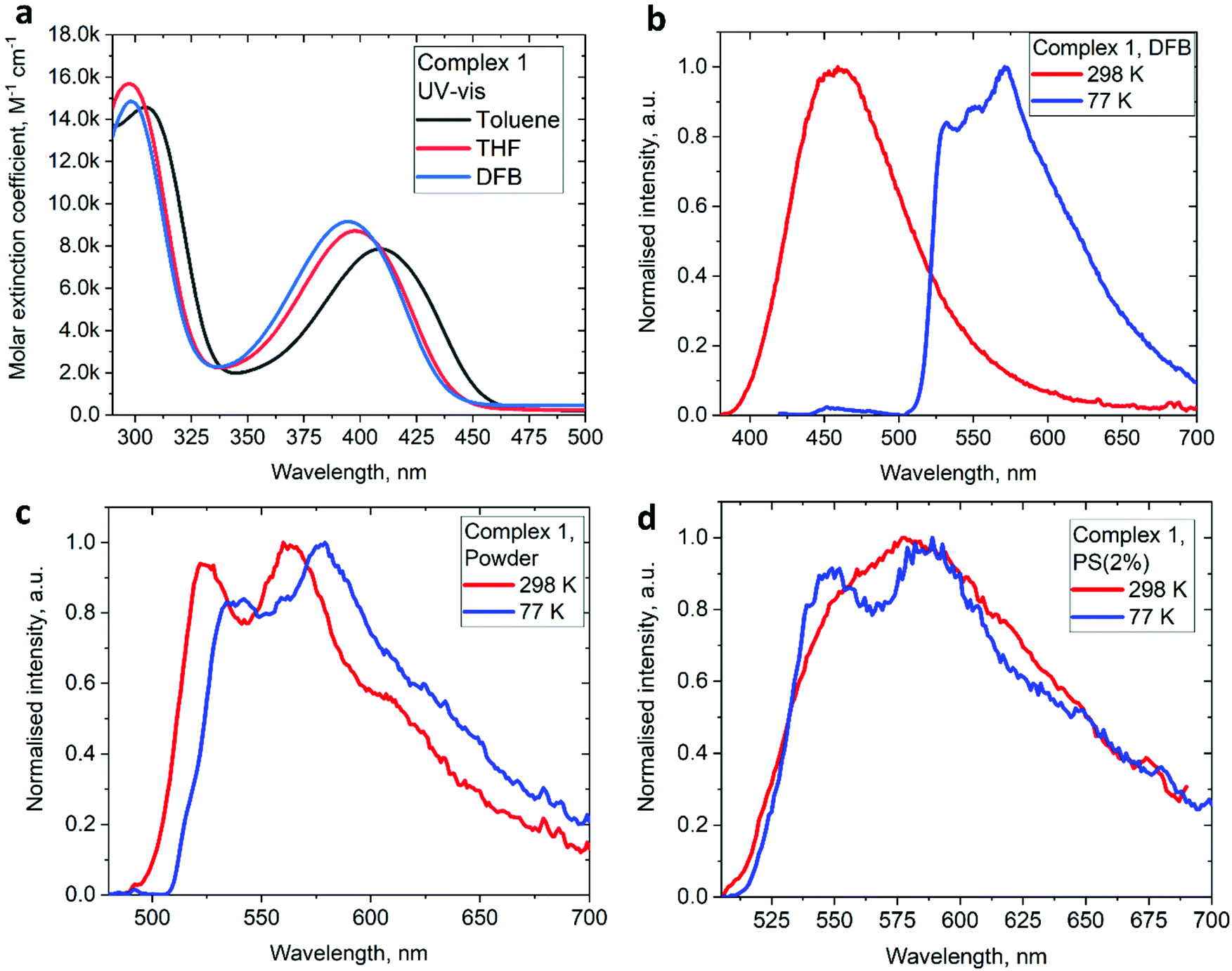 | ||
| Fig. 3 UV-Vis spectra for 1 (a) in various solvents and photoluminescence spectra (298 and 77 K) excited at 380 and 400 nm under a nitrogen atmosphere (b) DFB solution; (c) crystalline powder; (d) polystyrene film at a loading of 2 wt% of 1. | ||
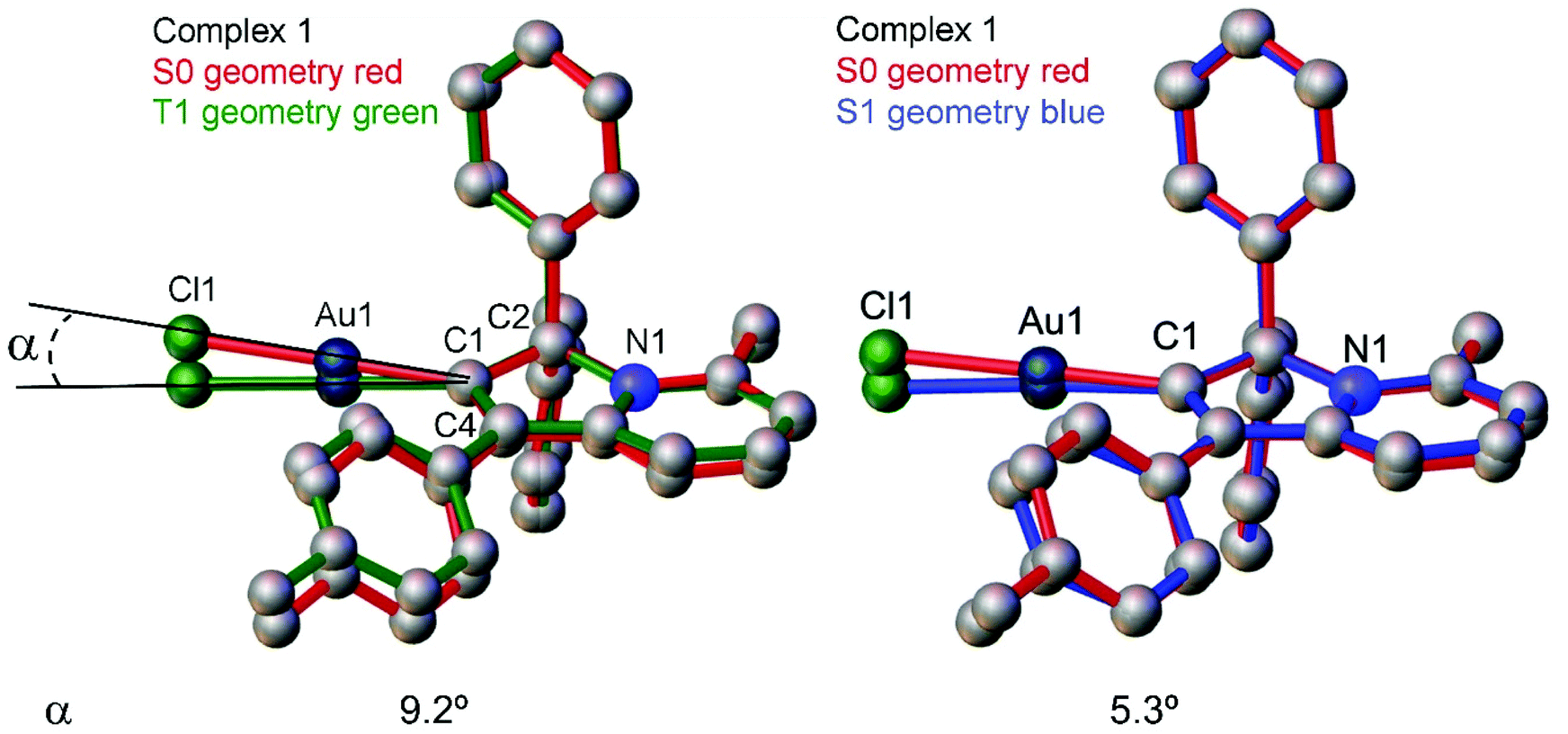 | ||
| Fig. 4 Superposition of the S0 and T1 (left), and S0 and S1 (right) geometries for complex 1 (overlay via C1, C2 and N1 atoms) determined by theoretical calculations where angle α is Au1(S0 geometry)–C1–Au1(S1 or T1 geometry). | ||
On excitation with λ = 400 nm light the chloride complex 1 displays a bright yellow photoluminescence (PL), with a high quantum yield (PLQY) of 65% in the solid state. The PL profile shows a clear vibronic progression (Fig. 3c) and a long excited state lifetime of 62.8 μs, which increases by a factor of 3 upon cooling to 77 K (Table 3). Theoretical calculations resulted in a large 0.67 eV energy difference between S1 and T1 states (Table S5†), supporting the phosphorescence nature of the PL for the chloride 1. This bright phosphorescence of 1 is in marked contrast with the majority of linear (carbene)AuCl complexes, where (M + Hal) charge transfer shows rather low extinction coefficients (equal to or well below 1000 M−1 cm−1).1,3 We suggest that the excellent photoluminescent performance of 1 in the solid state is closely related to the hybrid nature or a combination of the IL and (M + Hal)L charge transfer processes with almost one order higher extinction coefficients, thus indicating a higher allowedness of the absorption and emission properties. Our theoretical calculations support this suggestion and indicate a high oscillator strength coefficient for the vertical S0 → S1 excitation of 0.1376 (Table S5†).
| 1 | 2 | |||||
|---|---|---|---|---|---|---|
| Powder | DFB | PSa matrix | Powder | DFB | PSa matrix | |
| a Polystyrene films (2% by weight) were drop-cast from 10 mg mL−1 DFB solutions on a quartz substrate and evaporated under reduced pressure in a nitrogen-filled glove box chamber. b Triplet energy levels based on the emission max peak value in DFB glass at 77 K. c Two-component lifetime, τ average was used: τav = (B1/(B1 + B2))τ1 + (B2/(B1 + B2))τ2, where B1 and B2 are relative amplitude for τ1 and τ2, respectively. | ||||||
| λ em (nm) | 524, 564, 607 | 456 | 578 | Poorly emissive | — | 617 |
| (535, 578) | (531, 551, 572) | (550, 588) | — | (548, 612, 653) | (581, 617, 647) | |
| τ (μs) | 62.8c | 0.022c | 48.4c | — | 0.56 | |
| (157.4) | (227.2) | (153.1) | — | (112.3)c | (73.6) | |
| Φ (%, 300 K; N2) | 65.0 | 1.8 | 12.4 | — | — | 21.6 |
| k r (104 s−1) | 1.0 | 81.8 | 0.2 | — | — | 38.6 |
| k nr (104 s−1) | 0.5 | 4460 | 1.8 | — | — | 140 |
| S1/T1b (eV) | 3.11/2.41 | 2.37 (CT)/2.19 (3LE) | ||||
Next, we measured the PL behaviour in DFB solution. Complex 1 shows weak sky-blue PL at λem = 456 nm at room temperature which, given the short excited state lifetime of 22 ns, we ascribe to ligand-based fluorescence. Upon cooling to 77 K, the nature of the PL changes to yellow phosphorescence with a 227.2 μs monoexponential decay for the excited state lifetime, similar to solid samples of 1. The singlet and triplet energies for 1 were estimated by measuring the blue edge onset of the PL profile at 295 and 77 K (Table 3) to confirm the large S1/T1 energy gap of 0.7 eV. The fluorescence and phosphorescence energies are in excellent agreement with theoretical calculations (Table S6, ESI†), which predict fluorescence at 446 nm and phosphorescence at 573 nm. We optimized the geometry of complex 1 in the ground (S0) and excited states. The S1 and T1 geometries show a significant out-of-plane bending of the linear fragment C1–Au1–Cl1 from the carbene plane (C1–C2–C3–C4–N1) (Fig. 4). Such a distortion is likely responsible for the poor luminescence quantum yields in fluid media while being supressed in the crystalline state, preserving high luminescence quantum yields. These out-of-plane bending distortions for the gold chloride complex 1 are in marked difference to the previously suggested Renner–Teller type distortions of the gold centre15b which are associated with the bending of the carbene–M–ligand fragment.
Unlike 1, complex 2 shows bright red PL only in a frozen DFB glass at 77 K, with a long excited state lifetime of 112.3 μs. The PL profile exhibits a clear vibronic progression indicative of ligand-centered 3LE luminescence from the carbene moiety.
We studied the luminescence properties of 1 and 2 as dopants in amorphous polystyrene films at a loading of 2 wt% (Fig. 3d, 5b and Table 3). Complex 1 shows yellow phosphorescence at 578 nm as a broad PL profile with a PLQY value of 12%. Upon cooling to 77 K, the PL profile becomes structured, very similar to that observed in frozen DFB or in a solid powder. On cooling to 77 K the excited state lifetime of 1 increases threefold up to τ = 153.1 μs, confirming phosphorescence as dominant emission pathway. Complex 2 in PS films emits red light with a broad emission profile, peaking at 617 nm (Fig. 5b). Crystalline samples of 2 are non-emissive which is likely associated with the packing of the molecules in the unit cell, where C6A–H6A⋯N2 intermolecular contacts result in slight pyramidalisation of the carbazole N-atom. Unlike in the crystalline state, complex 2 in a PS matrix (2 wt% dopant level) shows an increased PLQY of up to 21.6% (Table 1). We explain this with the dilution effect of the dopant in a PS matrix resulting in the absence of such C6A–H6A⋯N2 intermolecular contacts which can only be observed in the rigid crystalline environment. The excited state lifetime of these PS films is 0.56 μs at 298 K, which upon cooling to 77 K increases by a factor of 130. This high temperature dependence strongly suggests emission by a TADF mechanism. The energy of the charge transfer (CT) and local excited triplet state (3LE) were estimated from the onset values of the blue emission edge of the PL spectra at 295 K and 77 K, respectively (Fig. 5b). Complex 2 shows much smaller energy difference between singlet and triplet excited states than 1, 0.18 eV, which provides further evidence in favour of a delayed luminescence mechanism for complex 2. The emission profile becomes structured upon cooling to 77 K, indicating that the luminescence at least partially originates from the localized 3LE state centred on the carbene moiety (see Fig. 5b). Analysis of the natural transition orbitals (NTOs, see Table S7†) for complex 2 indicates that the lowest-lying T1 state has mixed 3LE(carbene) and 3L(M)LCT character.
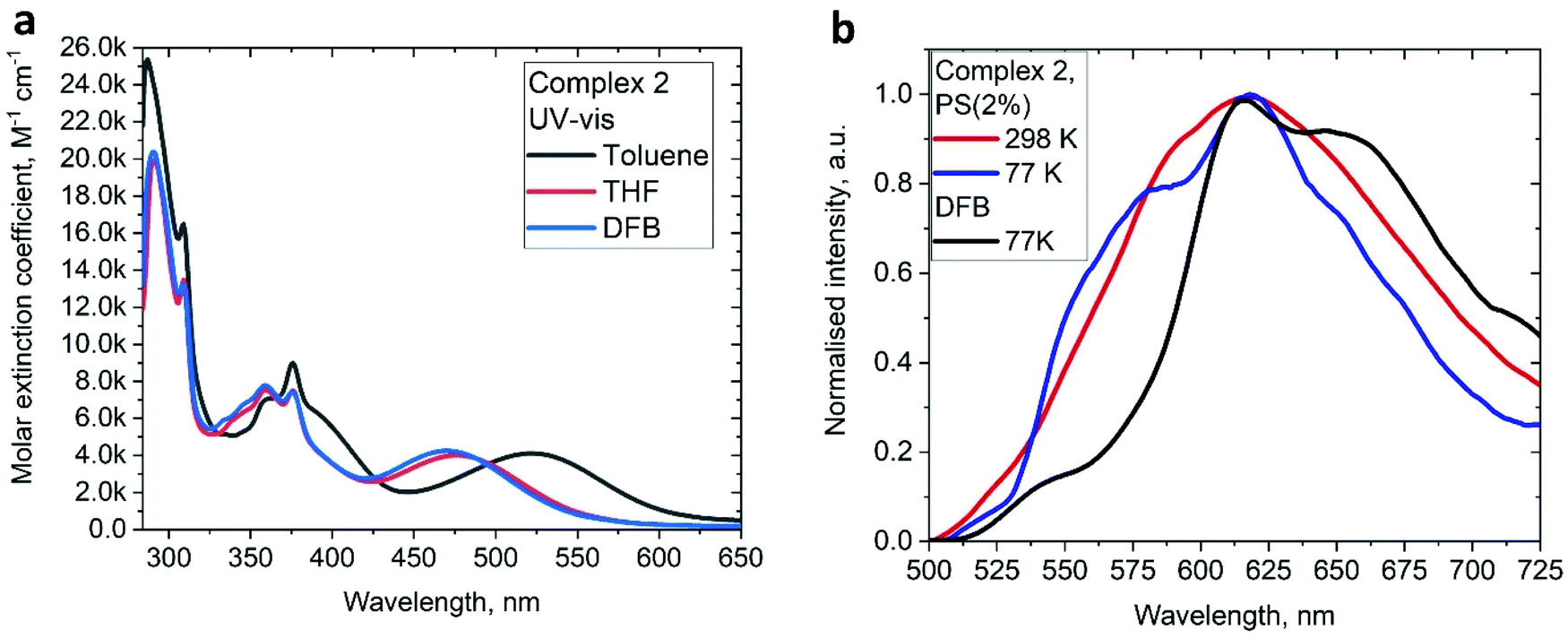 | ||
| Fig. 5 (a) UV-Vis spectra for 2 in various solvents. (b) Luminescence profiles of 2 in frozen DFB and in a polystyrene matrix (2 wt% dopant level) at 295 and 77 K. | ||
Conclusion
The first examples of linear CMA materials based on carbenes lacking heteroatom stabilisation in α-position to the carbene-C atom have become accessible by employing the indolizin-2-ylidene (Indolizy) ligand framework coordinated to gold(I). The structural features of these complexes closely resemble those of the well-known CAAC-derived carbene–metal–amide emitters systems. Complexes 1 and 2 show two irreversible reduction processes centred on the Indolizy carbene ligand. The (3C-carbene)AuCl complex shows a mixed combination of intraligand and (M + Hal)L charge transfer processes resulting in a bright yellow phosphorescence. The replacement of the chloride by a carbazolate ligand results in a significant reduction of the HOMO–LUMO energy gap by as much as 0.93 eV. As a result, the complex acts as a red emitter with a sub-microsecond excited state lifetime. Theoretical calculations explain the larger extinction coefficient for the chloride complex 1 and the smaller one for 2 due to the relatively smaller HOMO–LUMO overlap integral in the latter case. The reduced overlap integral correlates well with a smaller energy difference between singlet and triplet excited states for 2 (0.18 eV), which favours a delayed luminescence mechanism. This work demonstrates that achieving efficient luminescence by an L(M)LCT process is not restricted to N-heteroatom stabilised carbenes and provides encouragement for further structural modification of the donor and acceptor moieties to realize efficient red and near-IR emitters.Experimental
General considerations
Unless stated otherwise all reactions were carried out in air. Solvents were distilled and dried as required. The 2-ethynyl-6-methylpyridine was obtained according to a literature procedure.201H and 13C{1H} NMR spectra were recorded using a Bruker Avance DPX-500 MHz NMR spectrometer and referenced to CD2Cl2 at δ 5.32 (13C, δ 53.84) and CDCl3 at δ 7.26 (δ13C 77.16) ppm. Sodium tert-butoxide and carbazole were obtained from Sigma Aldrich. All electrochemical experiments were performed using an Autolab PGSTAT 302N computer-controlled potentiostat. Cyclic voltammetry (CV) was performed using a three-electrode configuration consisting of either a glassy carbon macrodisk working electrode (GCE) (diameter of 3 mm; BASi, Indiana, USA) combined with a Pt wire counter electrode (99.99%; GoodFellow, Cambridge, UK) and an Ag wire pseudoreference electrode (99.99%; GoodFellow, Cambridge, UK). The GCE was polished between experiments using alumina slurry (0.3 μm), rinsed in distilled water and subjected to brief sonication to remove any adhering alumina microparticles. The metal electrodes were then dried in an oven at 100 °C to remove residual traces of water, the GCE was left to air dry and residual traces of water were removed under vacuum. The Ag wire pseudoreference electrodes were calibrated to the ferrocene/ferrocenium couple in 1,2-difluorobenzene at the end of each run to allow for any drift in potential, following IUPAC recommendations.21 All electrochemical measurements were performed at ambient temperatures under an inert argon atmosphere in THF containing the complex under study (1.4 mM) and supporting electrolyte [n-Bu4N][PF6] (0.13 M). Data were recorded with Autolab NOVA software (v. 1.11). Elemental analyses were performed by London Metropolitan University. UV-visible absorption spectra were recorded using a PerkinElmer Lambda 35 UV/vis spectrometer. Photoluminescence measurements in toluene were recorded on a Fluorolog Horiba Jobin Yvon spectrofluorometer. Photoluminescence measurements of crystalline powders, PS films, time-resolved emission spectra, photoluminescence quantum yields were recorded using an integrating sphere on an Edinburgh Instruments FS5 spectrofluorometer under a nitrogen atmosphere. The emission decays for powder samples and PS films were collected on an Edinburgh Instruments FS5 spectrofluorometer using a 5 W microsecond Xe flashlamp with a repetition rate of 100 Hz. Time-resolved fluorescence data for toluene solutions were collected via a time-correlated single photon counting (TCSPC) method using a 370 nm laser as excitation source, with an instrument response function width of 60 ps. Mono- and biexponential fitting was provided by Edinburgh Instruments Fluoracle software v2.6.1. TGA-DSC analysis was performed using a Mettler-Toledo TGA-DSC, using a small amount of sample (approx. 5 mg). A background measurement, using an empty 70 μL platinum pan, was taken initially, over a 50–600 °C temperature range. Mass spectrometry data were obtained using pneumatically assisted electrospray (pNSI) or APCI(ASAP) (Atmospheric Solids Analysis Probe) on a Thermo Scientific LTQ Orbitrap XL at the National Mass Spectrometry Facility at Swansea University.Synthesis of 3-(6-methylpyridin-2-yl)-1,1-diphenylprop-2-yn-1-yl acetate
To a solution of 2-ethynyl-6-methylpyridine (2.8 g, 23.9 mmol) in 40 mL of THF was added n-BuLi (15 mL of a 1.6 M solution in hexane, 24 mmol) at −78 °C and stirred for 1 h. Benzophenone (4.56 g, 25 mmol) was added at −78 °C and slowly warmed to room temperature. After stirring at room temperature for 2 h, Ac2O (2.4 mL or 2.55 g, 25 mmol) was added and reaction mixture was stirred overnight. The reaction was hydrolized with water (30 mL), diluted with EtOAc (140 mL) and washed with water (3 × 50 mL). The organic layer was separated and dried over anhydrous MgSO4. Purification by column chromatography on silica gel using EtOAc/hexane (1
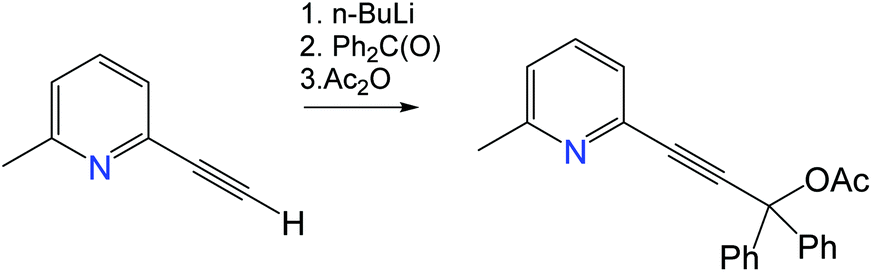 :5) as eluent afforded the product as a yellow oil. Yield: 6.2 g, 18.1 mmol (75.3%).
:5) as eluent afforded the product as a yellow oil. Yield: 6.2 g, 18.1 mmol (75.3%).
1H NMR (300 MHz, CDCl3) δ 7.56–7.61 (m, 4H, Ph), 7.54 (t, J = 9.0 Hz, 1Hb, Py), 7.37–7.24 (m, 6H Ph + 1Hc Py overlaps with residual solvent CHCl3), 7.10 (d, J = 9.0 Hz, 1Ha, Py), 2.55 (s, 3H, pyridine CH3), 2.18 (s, 3H, CH3). HRMS C23H19NO2 theoretical [M + H]+ = 342.1489, HRMS (pNSI in CH2Cl2/MeOH + NH4OAc): = 342.1489.
Synthesis of 2-(3,3-diphenyl-1-(p-tolyl)propa-1,2-dien-1-yl)-6-methylpyridine (L1)
A solution of (p-tolyl)MgBr (73 mL of 1 M in THF, 73 mmol, 5 equiv.) was added to a solution of ZnCl2 (73 mL of 1 M in THF, 73 mmol, 5 equiv.) at 0 °C under nitrogen and stirred for 1 h. The solution of pyridyl acetate from the previous step (5 g, 14.6 mmol, 1 equiv.) and Pd(PPh3)4 (0.170 g, 0.146 mmol, 0.01 equiv.) in THF (40 mL) were added dropwise to the mixture at −40 °C and warmed to room temperature to give a dark red solution. After stirring overnight, the mixture was concentrated to ca. 100 mL and quenched with a saturated aqueous NH4Cl solution (50 mL). The product was extracted with EtOAc (250 mL). The organic layer was washed with brine (2 × 100 mL) and dried over anhydrous MgSO4. Purification by silica gel chromatography with EtOAc/hexane (1
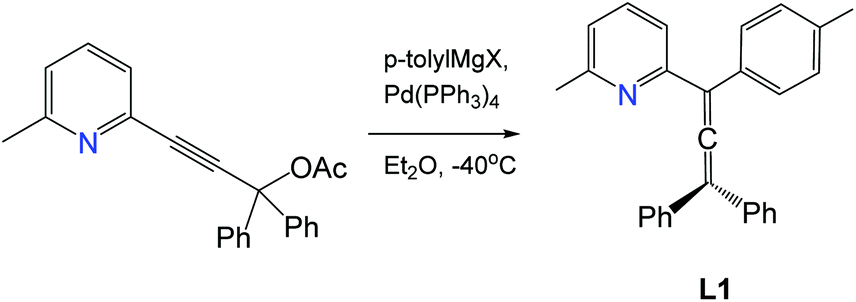 :6) as eluent provided the product as yellow oil. Yield: 2.45 g, 6.5 mmol (44.5%).
:6) as eluent provided the product as yellow oil. Yield: 2.45 g, 6.5 mmol (44.5%).
1H NMR (300 MHz, CDCl3) δ 7.53 (t, J = 9.0 Hz, 1H), 7.50–7.47 (m, 4H), 7.41–7.29 (m, 9H), 7.12 (d, J = 6.0 Hz, 2H), 7.05 (d, J = 9.0 Hz, 1H), 2.57 (s, 3H, pyridine CH3), 2.35 (s, 3H, p-tolyl CH3). HRMS C28H23N theoretical [M + H]+ = 374.1903, HRMS (pNSI in CH2Cl2/MeOH + NH4OAc): = 374.1896.
Synthesis of (5-methyl-3,3-diphenyl-1-(p-tolyl)-3H-indolizin-2-ylidene)gold(I) chloride (1)
A solution of allene L1 (1.6 g, 4.3 mmol) and (Me2S)AuCl (1.2 g, 4.1 mmol) in C2H4Cl2 (25 mL) was stirred overnight, giving a solution with a colourless residue. This was concentrated to 10 mL and additional product was precipitated with 100 mL of pentane. The product was centrifuged, washed with pentane and dried in vacuum to afford an off-white crystalline solid. Yield: 2.44 g (4 mmol; 97.5%).1H NMR (300 MHz, CD2Cl2) δ 8.07 (t, J = 6.0 Hz, 1Hb, Py), 7.65 (d, J = 6 Hz, 1Hc, Py) overlapping with 7.63 (d, J = 9.0 Hz, 2Hd, tolyl), 7.48–7.40 (m, 10H, 2C6H5), 7.29 (d, J = 9.0 Hz, 2He, tolyl), 7.15 (d, J = 6.0 Hz, 1Ha, Py), 2.40 (s, 3H, p-tolyl CH3), 2.21 (s, 3H, pyridine CH3). 13C NMR (75 MHz, CD2Cl2) δ 195.1 (Carbene C), 160.1 (NCipso(Py)), 154.6 (NCipso(CH3Py)), 145.0 (C–Hb), 138.6 (p-tolyl Cipso), 134.6 (Cipso), 133.0 (Cipso, Ph), 132.2 (Cipso), 129.7 (C–He), 129.4 (C–H, Ph), 129.2 (C–H, Ph), 129.0 (C–H, Ph), 128.9 (C–Hd), 122.2 (C–Ha), 117.2 (C–Hc), 97.0 (NCipso(Ph)2), 22.1 (Py-CH3), 21.4 (tolyl-CH3).
Anal. Calcd for C28H23AuClN (605.12): C, 55.50; H, 3.83; N, 2.31. Found: C, 55.67; H, 3.90; N, 2.23. HRMS C28H24N theoretical [M–AuCl]+ = 374.1909, HRMS (APCI(ASAP): = 374.1903.
Synthesis of (5-methyl-3,3-diphenyl-1-(p-tolyl)-3H-indolizin-2-ylidene)gold(I)(carbazolate) (2)
A mixture of 1 (160 mg, 0.29 mmol), NaOtBu (29 mg, 0.29 mmol) and carbazole (50 mg, 0.29 mmol) in dry THF (30 mL) was stirred overnight. The volatiles were removed and the residue extracted with CH2Cl2. The organic layer was washed with water and dried over MgSO4. The solution was filtered off. All volatiles were evaporated. The residue was washed with hexane and dried in vacuum to give a brown-red analytically pure powder. Yield: 0.18 g (0.27 mmol, 93%).1H NMR (300 MHz, CD2Cl2): δ 8.04 (t, J = 6.0 Hz, 1Hb, Py), 7.95 (d, J = 7.8 Hz, 2Hi, carbazole), 7.86 (d, J = 7.8 Hz, 2Hd, tolyl), 7.73 (d, J = 6 Hz, 1Hc, Py), 7.64–7.62 (m, 4H, Ph) 7.47–7.44 (m, 6H, Ph), 7.37 (d, J = 7.8 Hz, 2He, tolyl), 7.31 (d, J = 8.0 Hz, 2Hf, carbazole), 7.17 (d, J = 8.0 Hz, 2Hg, carbazole), 7.08 (d, J = 7.2 Hz, 1Ha, Py), 6.91 (t, J = 7.8 Hz, 2Hh, carbazole), 2.35 (s, 3H, p-tolyl CH3), 2.24 (s, 3H, pyridine CH3) ppm. 13C NMR (75 MHz, CD2Cl2) δ 199.6 (Carbene C), 160.1 (NCipso(Py)), 154.6 (NCipso(CH3Py)), 150.2 (NCipso(carbazole), 144.9 (C–Hb), 138.5 (p-tolyl Cipso), 134.9, 133.5, 132.6, 129.6, 129.5, 129.2, 129.16, 129.15 (C–Hd), 124.1 (Cipso (carbazole)), 123.5 (C–Hg (carbazole)), 121.8 (C–Hb), 119.5 (C–Hi (carbazole)), 117.5 (C–Hc), 115.7 (C–Hh (carbazole)), 114.2 (C–Hf (carbazole)), 97.6 (NCipso(Ph)2), 22.1 (Py-CH3), 21.5 (tolyl-CH3) ppm. Anal. Calcd for C40H31N2Au (736.22): C, 65.22; H, 4.24; N, 3.80. Found: C, 65.31; H, 4.31; N, 3.74. HRMS C40H31AuN2 theoretical [M–Au]+ = 539.2487, HRMS (APCI(ASAP): = 539.2495.
Computational details
The ground states of the complexes were studied by density functional theory (DFT) and the excited states by time-dependent DFT (TD-DFT) using the Tamm-Dancoff approximation.22,23 Calculations were carried by the global hybrid MN15 functional of the Minnesota series by Truhlar and coworkers, which has especially good performance for noncovalent interactions and excitation energies.24 The def2-TZVP basis set25,26 was employed with relativistic effective core potential of 60 electrons for description of the core electrons of Au.27 We have previously employed the selected methodology with success for closely related molecules.28,29 Overlap integrals and HOMO–LUMO centroid distances have been calculated using Multiwfn program.30 All calculations were carried out by Gaussian 16.31Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Research Council and the Royal Society. M. B. is an ERC Advanced Investigator Award holder (grant no. 338944-GOCAT). A. S. R. acknowledges support from the Royal Society (grant no. URF\R1\180288 and RGF\EA\181008). M.L. acknowledges the Academy of Finland Flagship Programme, Photonics Research and Innovation (PREIN), decision 320166. (TD) DFT computations were made possible by use of the Finnish Grid and Cloud Infrastructure resources (urn:nbn:fi:research-infras-2016072533). Mass spectrometry data were obtained at the National Mass Spectrometry Facility at Swansea University.References
- A. S. Romanov, D. Di, Le Yang, J. Fernandez-Cestau, C. R. Becker, C. E. James, B. Zhu, M. Linnolahti, D. Credgington and M. Bochmann, Chem. Commun., 2016, 52, 6379–6382 RSC and Chem. Commun., 2018, 54, 3672 Search PubMed.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi-Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- A. S. Romanov, C. R. Becker, C. E. James, D. Di, D. Credgington, M. Linnolahti and M. Bochmann, Chem. – Eur. J., 2017, 23, 4625–4637 CrossRef CAS PubMed.
- A. S. Romanov and M. Bochmann, J. Organomet. Chem., 2017, 847, 114–120 CrossRef CAS.
- P. J. Conaghan, S. M. Menke, A. S. Romanov, A. J. Pearson, E. W. Evans, M. Bochmann, N. C. Greenham and D. Credgington, Adv. Mater., 2018, 30, 1802285 CrossRef PubMed.
- C. R. Hall, A. S. Romanov, M. Bochmann and S. R. Meech, J. Phys. Chem. Lett., 2018, 9, 5873–5876 CrossRef CAS PubMed.
- A. S. Romanov, S. T. E. Jones, L. Yang, P. J. Conaghan, D. Di, M. Linnolahti, D. Credgington and M. Bochmann, Adv. Opt. Mater., 2018, 6, 1801347 CrossRef.
- M. Gernert, U. Meller, M. Haehnel, J. Pflaum and A. Steffen, Chem. – Eur. J., 2017, 23, 2206–2216 CrossRef CAS.
- R. Hamze, R. Jazzar, M. Soleilhavoup, P. I. Djurovich, G. Bertrand and M. E. Thompson, Chem. Commun., 2017, 53, 9008–9011 RSC.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. C. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed.
- S. Shi, M. C. Jung, C. Coburn, A. Tadle, D. Sylvinson M. R., P. I. Djurovich, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2019, 141, 3576–3588 CrossRef CAS PubMed.
- J. Föller and C. M. Marian, J. Phys. Chem. Lett., 2017, 8, 5643–5647 CrossRef.
- E. J. Taffet, Y. Olivier, F. Lam, D. Beljonne and G. D. Scholes, J. Phys. Chem. Lett., 2018, 9, 1620–1626 CrossRef CAS PubMed.
- S. Thompson, J. Eng and T. J. Penfold, J. Chem. Phys., 2018, 149, 014304 CrossRef CAS PubMed.
- (a) F. Chotard, V. Sivchik, M. Linnolahti, M. Bochmann and A. S. Romanov, Chem. Mater., 2020, 32, 6114–6122 CrossRef CAS; (b) T.-Y. Li, D. Sylvinson, M. Ravinson, R. Haiges, P. I. Djurovich and M. E. Thompson, J. Am. Chem. Soc., 2020, 142, 6158–6172 CrossRef; (c) M. Gernert, L. Balles-Wolf, F. Kerner, U. Müller, A. Schmiedel, M. Holzapfel, C. M. Marian, J. Pflaum, C. Lambert and A. Steffen, J. Am. Chem. Soc., 2020, 142, 8897–8909 CrossRef CAS PubMed.
- (a) G. Seidel and A. Fürstner, Angew. Chem., Int. Ed., 2014, 53, 4807–4811 CrossRef CAS PubMed; (b) M. W. Hussong, F. Rominger, P. Kramer and B. F. Straub, Angew. Chem., Int. Ed., 2014, 53, 9372–9375 CrossRef CAS; (c) R. J. Harris and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2014, 53, 9369–9371 CrossRef CAS PubMed; (d) M. Joost, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2014, 53, 14512–14516 CrossRef CAS PubMed.
- (a) A. Vanitcha, G. Gontard, N. Vanthuyne, E. Derat, V. Mouries-Mansuy and L. Fensterbank, Adv. Synth. Catal., 2015, 357, 2213–2218 CrossRef CAS; (b) T. Martinez, A. Vanitcha, C. Troufflard, N. Vanthuyne, J. Forte, G. Gontard, G. Lemiere, V. Mouries-Mansuy and L. Fensterbank, Angew. Chem., Int. Ed., 2021, 60, 19879–19888 CrossRef CAS PubMed.
- H. K. Maliszewska, D. L. Hughes and M. P. Munoz, Dalton Trans., 2020, 49, 4034–4038 RSC.
- Indolizy gold complexes can be described by two mesomeric structures, as Au-carbene, or as zwitterionic gold vinyls.
- D. Alagille, R. M. Baldwin, B. L. Roth, J. T. Wroblewski, E. Grajkowska and G. D. Tamagnan, Bioorg. Med. Chem., 2005, 13, 197–209 CrossRef CAS PubMed.
- G. Gritzner and J. Kůta, Electrochim. Acta, 1984, 29, 869–873 CrossRef.
- F. Furche and D. Rappoport, Density functional methods for excited states: equilibrium structure and electronic spectra, in Computational Photochemistry, ed. M. Olivuccim, Elsevier, Amsterdam, 2005, pp. 93–128 Search PubMed.
- G. M. J. Peach and D. J. Tozer, J. Phys. Chem. A, 2012, 116, 9783–9789 CrossRef PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- F. Chotard, A. S. Romanov, D. L. Hughes, M. Linnolahti and M. Bochmann, Eur. J. Inorg. Chem., 2019, 4234–4240 CrossRef CAS.
- A. S. Romanov, S. T. E. Jones, Q. Gu, P. J. Conaghan, B. H. Drummond, J. Feng, F. Chotard, L. Buizza, M. Foley, M. Linnolahti, D. Credgington and M. Bochmann, Chem. Sci., 2020, 11, 435–446 RSC.
- T. Lu and F. J. Chen, Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV, X-ray crystallographic and theoretical calculations data. CCDC 2110631 for 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03393j |
| This journal is © The Royal Society of Chemistry 2021 |