
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong-range PEG stapling: macrocyclization for increased protein conformational stability and resistance to proteolysis†
Qiang
Xiao
,
Dallin S.
Ashton
,
Zachary B.
Jones
,
Katherine P.
Thompson
and
Joshua L.
Price
 *
*
Department of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84602, USA. E-mail: jlprice@chem.byu.edu
First published on 13th August 2020
Abstract
We previously showed that long-range stapling of two Asn-linked O-allyl PEG oligomers via olefin metathesis substantially increases the conformational stability of the WW domain through an entropic effect. The impact of stapling was more favorable when the staple connected positions that were far apart in primary sequence but close in the folded tertiary structure. Here we validate these criteria by identifying new stabilizing PEG-stapling sites within the WW domain and the SH3 domain, both β-sheet proteins. We find that stapling via olefin metathesis vs. the copper(I)-catalyzed azide/alkyne cycloaddition (CuAAC) results in similar energetic benefits, suggesting that olefin and triazole staples can be used interchangeably. Proteolysis assays of selected WW variants reveal that the observed staple-based increases in conformational stability lead to enhanced proteolytic resistance. Finally, we find that an intermolecular staple dramatically increases the quaternary structural stability of an α-helical GCN4 coiled-coil heterodimer.
Introduction
Macrocyclization is one of the most useful strategies for increasing the stability of peptides, proteins, and binding complexes in supramolecular chemistry and chemical biology.1–4 Covalent constraints can preorganize a peptide or protein into a shape that resembles its folded or bound conformation, thereby “pre-paying” part of the cost associated with folding or binding, through a combination of entropic and enthalpic effects.1–4 Disulfide bonds can play this role in synthetic peptides or proteins;5–9 however, correct disulfide connectivity sometimes requires creative protecting group strategies and the disulfides themselves are not stable in reducing environments, making disulfide-stapled peptides and proteins unsuitable as therapeutics with intracellular targets. Efforts to address these limitations have led to a growing number of chemoselective ligation reactions10 (i.e., stapling reactions) that are tolerant of water and are selective for a particular reactive partner in the presence of diverse biological nucleophiles and electrophiles. Thiol alkyl-11–15 or arylation16 takes advantage of the nucleophilicity of Cys but results in thioether staples that are stable to reducing conditions. For example, azobenzene-linked bis-electrophiles can provide photoisomerization-based conformational control.17,18 Tris-electrophiles can connect three different Cys residues,19,20 thereby stabilizing existing tertiary structures21 or providing access to new macrocyclic topologies not possible with disulfides alone.22 These Cys-centric approaches are generally limited to side-chain/side-chain crosslinks; in contrast, other approaches facilitate stapling in both side-chain and backbone contexts.23 For example, lactam staples can be prepared via conventional peptide coupling chemistry24–29 or by diverse chemoselective strategies, including the Ugi reaction;30,31 direct thioester aminolysis;32 native chemical ligation;33–35 KAHA ligation;36 traceless Staudinger ligation;37 and a variety of enzymatic methods.38–40 Other creative stapling strategies include C–H activation;41–43 the Petasis reaction;44 the Glaser reaction;45 oxime46,47 or hydrazone48 formation; the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC);49–55 and olefin metathesis.56–61We are interested in understanding the origin and determinants of protein stabilization via macrocyclization/stapling in diverse structural contexts. The WW domain is a triple-stranded antiparallel β-sheet protein;62 positions 16 and 19 within WW are close in sequence and in tertiary structure: both are located within a reverse turn that connects first and second β-strands. Each is also a location where Asn-PEGylation is substantially stabilizing.63 In WW variant 16/19-o23, residues 16 and 19 are occupied by Asn residues that have been modified with two- and three-unit O-allyl PEGs, respectively (Fig. 1).64 Bis-PEGylated 16/19-o23 is −0.75 ± 0.02 kcal mol−1 more stable than its non-PEGylated counterpart 16/19-00; crosslinking of the O-allyl PEGs via olefin metathesis results in stapled variant s16/19-o23, which is −0.29 ± 0.02 kcal mol−1 more stable than 16/19-o23. This stabilizing change in folding free energy (ΔΔG) comes from a favorable change in entropy (i.e., −TΔΔS), partially offset by an unfavorable change in enthalpy (ΔΔH), an observation that is consistent with the anticipated impact of macrocyclization on protein folding as described above. However, the ΔΔG associated with stapling (−0.29 ± 0.02 kcal mol−1; compare s16/19-o23vs.16/19-o23) is much smaller than the ΔΔG associated with bis-PEGylation (−0.75 ± 0.02 kcal mol−1; compare s16/19-o23vs.16/19-00). Incorporating PEGs of longer and shorter lengths within this staple failed to improve the observed staple-based stabilization.64
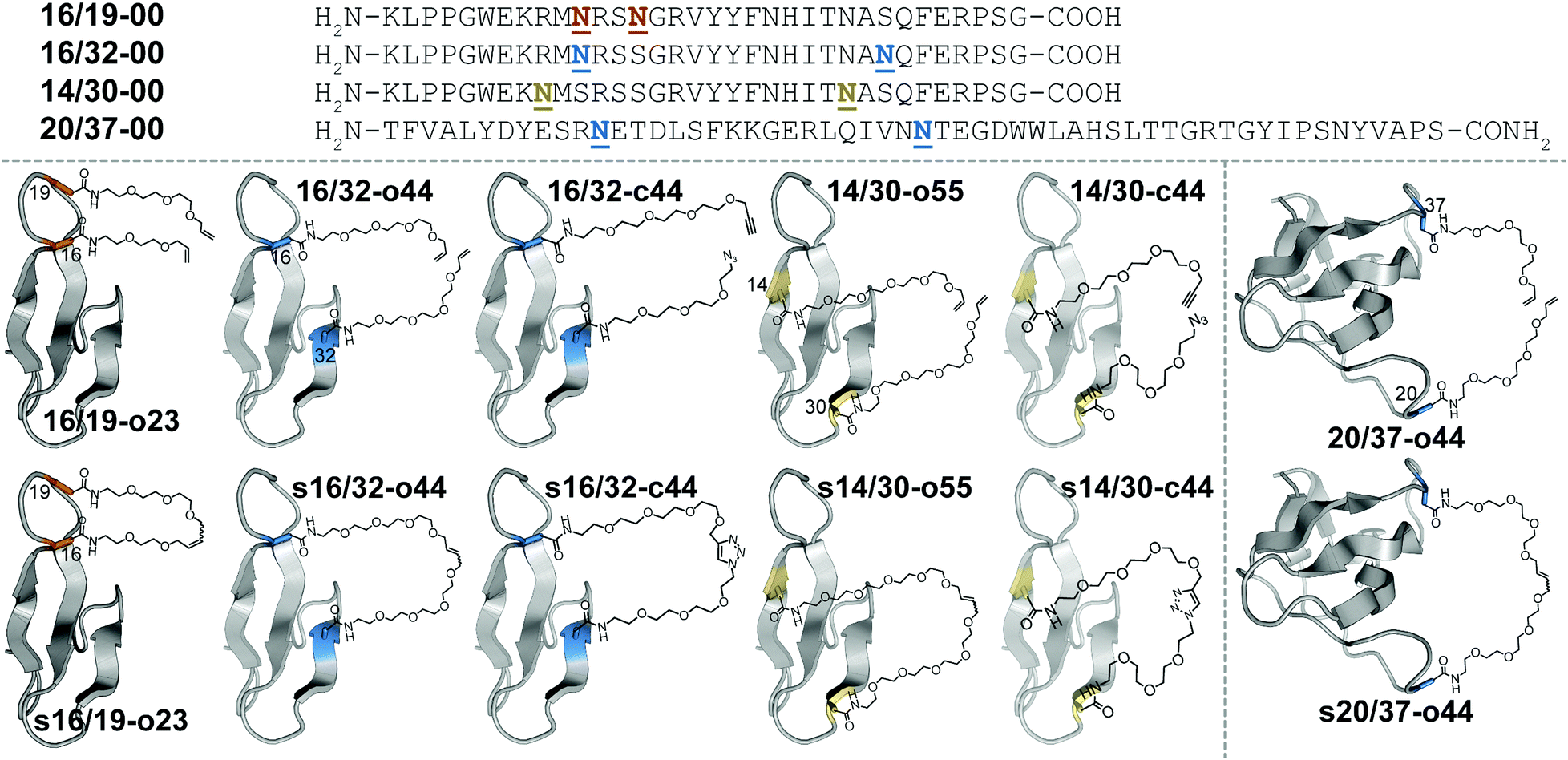 | ||
Fig. 1 Sequences and structures of olefin-stapled WW variants s16/19-o23, s16/32-o44 and s14/30-o55; triazole-stapled WW variants s16/32-c44 and s14/30-c44; and olefin-stapled SH3 variant s20/37-o44, and their non-stapled and non-PEGylated counterparts. ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) represents a PEG-modified Asn residue; the PEG oligomer(s) within each variant have the number of ethylene oxide units and the olefin, azide, alkyne, or triazole functional groups as indicated in the structural drawings. represents a PEG-modified Asn residue; the PEG oligomer(s) within each variant have the number of ethylene oxide units and the olefin, azide, alkyne, or triazole functional groups as indicated in the structural drawings. | ||
We wondered whether the limited energetic benefits of stapling positions 16 and 19 reflected their proximity in primary sequence (3 residues apart) as well as in tertiary structure (4.0 Å between Cβ's of these positions in the crystal structure of the parent WW domain65): positions 16 and 19 may be similarly close in both folded and unfolded conformations of non-stapled WW, such that covalently linking them together has only marginal benefits. We hypothesized that stapling between positions that are farther apart in primary sequence but still relatively close in the folded tertiary structure would have a more favorable impact. Position 32 at the C-terminal end of the third β-strand in WW is a stabilizing Asn-PEGylation site;63 it is on the same face of WW as is position 16 (9.4 Å between Cβ's of these positions), but is much farther from position 16 in primary sequence (i.e. 16 residues) than is position 19. Bis-PEGylated WW variant 16/32-o44 (with Asn-linked four-unit O-allyl PEGs at positions 16 and 32) is −0.40 ± 0.05 kcal mol−1 more stable than non-PEGylated 16/32-00. Olefin-stapled s16/32-o44 is −1.11 ± 0.04 kcal mol−1 more stable than non-stapled 16/32-o44 due to a favorable entropic effect offset by a smaller unfavorable change in enthalpy.64 The ΔΔG and −TΔΔS values associated with stapling of positions 16 and 32 are much more favorable than we observed for positions 16 and 19, presumably because the staple increases the proximity of positions 16 and 32 in the unfolded ensemble, thereby reducing the entropic cost of their proximity in the folded conformation.64
These published observations suggest that substantial separation in primary sequence but proximity in tertiary structure are important criteria for identifying stabilizing PEG stapling sites within proteins. Here we validate these criteria by identifying new PEG stapling sites within WW and the Src SH3 domain. We also explore the stabilizing impact of stapling via olefin metathesis vs. CuAAC at selected locations within WW and demonstrate that staple-based stabilization is associated with enhanced resistance to proteolytic degradation. Finally, we show that intermolecular PEG stapling increases the quaternary structural stability of an α-helical GCN4 coiled-coil heterodimer.
Results and discussion
Positions 14 and 30 in WW are 16 residues apart in primary sequence and occupy the same face of WW (11.7 Å between Cβ's of these positions65), similar to the relationship between positions 16 and 32. We wondered whether PEG stapling of positions 14 and 30 would be similarly stabilizing. A potential complicating issue is our previous observation that individual Asn-PEGylation has a minimal impact on WW conformational stability at position 14 and at position 30, whereas individual Asn-PEGylation is stabilizing at position 16 and position 32.63 We wondered whether PEG stapling of positions 14 and 30 would continue to be stabilizing in the absence of strong PEG-based stabilization at these positions. Interestingly, bis-PEGylated WW variant 14/30-o55 (with Asn-linked five-unit O-allyl PEGs at positions 14 and 30) is −0.34 ± 0.02 kcal mol−1 more stable than its non-PEGylated counterpart 14/30-00. Olefin-stapled s14/30-o55 is −0.49 ± 0.05 kcal mol−1 more stable than 14/30-o55, a more favorable value than we observed previously for stapling of positions 16 and 19 (ΔΔG = −0.29 ± 0.02 kcal mol−1), but less favorable than for stapling of positions 16 and 32 (ΔΔG = −1.11 ± 0.04 kcal mol−1). The small magnitude and high uncertainty in the values of ΔΔH and −TΔΔS for s14/30-o55vs.14/30-o55 (see Table 1) make it difficult to assess the entropic vs. enthalpic origin of the staple-based stabilization at positions 14 and 30. However, these results suggest that close proximity in tertiary structure and substantial separation in primary sequence are the most important criteria for identifying locations where PEG stapling will be stabilizing, though optimal staple-based stabilization may depend moderately on the intrinsic impact of PEGylation at the prospective staple sites. It is also noteworthy that the relatively flexible linkers (containing 5–10 ethylene oxide units) within s16/19-o23, s16/32-o44, and s14/30-o55 can provide such a substantial level (−0.3 to −1.2 kcal mol−1) of staple-based stabilization.| Protein | T m (°C) | ΔG (kcal mol−1) | Impact of stapling | ||
|---|---|---|---|---|---|
| ΔΔG (kcal mol−1) | ΔΔH (kcal mol−1) | −TΔΔS (kcal mol−1) | |||
| a Folding free energies for each variant are given ± std. error in kcal mol−1 at the melting temperature of its non-stapled non-PEGylated counterpart. WW variants 16/32-00, 14/30-00 and SH3 variant 20/37-00 and their derivatives were analyzed at 50 μM protein concentration in 20 mM sodium phosphate buffer (pH 7). GCN4 disulfide-bound heterodimer d27/29′-c40 and its triazole-stapled counterpart sd27/29′-c40 were analyzed at 15 μM protein concentration in 20 mM sodium phosphate buffer (pH 7) + 4.0 M GdnHCl. GCN4 noncovalent heterodimer 27/29′-c40 and its triazole-stapled counterpart s27/29′-c40 were analyzed at 15 μM protein concentration in 20 mM sodium phosphate buffer (pH 7) + 0.5 M GdnHCl. | |||||
| 16/32-00 | 49.2 ± 0.6 | 0.00 ± 0.04 | |||
| 16/32-o44 | 54.0 ± 0.2 | −0.40 ± 0.02 | |||
| s16/32-o44 | 71.7 ± 0.3 | −1.51 ± 0.04 | −1.11 ± 0.04 | 2.1 ± 0.9 | −3.2 ± 0.9 |
| 16/32-c44 | 54.2 ± 0.2 | −0.44 ± 0.02 | |||
| s16/32-c44 | 71.4 ± 0.1 | −1.68 ± 0.03 | −1.24 ± 0.03 | 8.3 ± 0.8 | −9.6 ± 0.8 |
| 14/30-00 | 28.6 ± 0.2 | 0.00 ± 0.01 | |||
| 14/30-o55 | 33.2 ± 0.1 | −0.34 ± 0.01 | |||
| s14/30-o55 | 39.5 ± 0.6 | −0.83 ± 0.05 | −0.49 ± 0.05 | −1.2 ± 0.9 | 0.7 ± 0.9 |
| 14/30-c44 | 30.5 ± 0.2 | −0.13 ± 0.01 | |||
| s14/30-c44 | 41.3 ± 0.1 | −0.73 ± 0.01 | −0.61 ± 0.02 | 1.9 ± 0.4 | −2.5 ± 0.4 |
| 20/37-00 | 61.1 ± 0.3 | 0.00 ± 0.02 | |||
| s20/37-o44 | 76.0 ± 0.8 | −0.94 ± 0.06 | −0.93 ± 0.07 | 9.9 ± 1.3 | −10.8 ± 1.3 |
| d27/29′-c40 | 41.1 ± 0.2 | 0.00 ± 0.02 | |||
| sd27/29′-c40 | 48.2 ± 0.1 | −0.65 ± 0.01 | −0.65 ± 0.02 | 1.3 ± 0.6 | −1.9 ± 0.6 |
| 27/29′-c40 | 34.8 | — | |||
| s27/29′-c40 | 82.0 ± 0.2 | — | — | — | — |
We next sought to apply these criteria to a larger and more structurally complex protein. We previously found that Asn-PEGylation at position 20 within the Src SH3 domain (hereafter called SH3) substantially increases the conformational stability of the PEGylated SH3 variant relative to its non-PEGylated counterpart.63 Positions 20 and 37 occur within two different unstructured loops in SH3 and are far apart in primary sequence (i.e. 17 residues); however, they occupy the same face of folded SH3 tertiary structure (17.0 Å between Cβ's of these positions in the crystal structure of the parent SH366). We hypothesized that metathesis-based PEG stapling of these two positions would increase the conformational stability of SH3. Accordingly, we prepared bis-PEGylated SH3 variant 20/37-o44, in which positions 20 and 37 are each occupied by four-unit Asn-linked O-allyl PEGs. The thermal unfolding behavior of variant 20/37-o44 was not consistent with two-state folding, which precluded detailed analysis of its conformational stability. In contrast, olefin-stapled variant s20/37-o44 (Tm = 76.0 ± 0.8 °C) is −0.93 ± 0.07 kcal mol−1 more stable than non-PEGylated 20/37-00 (Tm = 61.1 ± 0.3 °C). The unusual thermal unfolding behavior of 20/37-o44 prevents us from determining how much of the favorable ΔΔG value for s20/37-o44vs.20/37-00 comes from bis-PEGylation vs. olefin stapling. However, these observations hint at the intriguing potential for olefin-based PEG stapling to rescue two-state folding and restore conformational stability to proteins with unusual thermal unfolding behavior.
We next wondered whether stapling of Asn-linked PEGs via CuAAC (i.e., click stapling) would provide levels of stabilization similar to what we observed previously for olefin stapling. To explore this possibility, we prepared WW variant 16/32-c44, in which an O-propargyl four-unit Asn-PEG occupies position 16, with an azide-terminated four-unit Asn-PEG at position 32 (Fig. 1). Click stapling results in variant s16/32-c44, which is −1.24 ± 0.03 kcal mol−1 more stable than its non-stapled counterpart. Similarly, click-stapled WW variant s14/30-c44 is −0.61 ± 0.02 kcal mol−1 more stable than non-stapled 14/30-c44 (see Table 1). These results demonstrate that the impact of stapling is tolerant of variations in the nature of the staple, with click stapling slightly more stabilizing than olefin stapling at the positions we investigated.
We previously showed that PEG-based increases in WW conformational stability are associated with increased levels of protection from proteolysis. We wondered whether this would be true for PEG-stapled WW variants. We explored this possibility by exposing 50 μM solutions of WW variants 16/19-00, 16/19-o23, s16/19-o23, 16/32-00, 16/32-o44, and s16/32-o44 to proteinase K (17 μg mL−1) and monitoring the amount of full-length protein remaining in solution at regular intervals by analytical HPLC. We fit the resulting data to a monoexponential decay function to obtain apparent proteolysis rate constants. The results of this analysis are shown in Fig. 2A and B. PEGylated olefin-stapled variant s16/19-o23 is more resistant to proteolysis than its PEGylated but non-stapled counterpart 16/19-o23, which is, in turn, more resistant to proteolysis than non-PEGylated non-stapled 16/19-00. Similarly, PEGylated olefin-stapled variant s16/32-o44 is more resistant to proteolysis than PEGylated but non-stapled 16/32-o44, which is more resistant to proteolysis than non-stapled non-PEGylated 16/32-00. For each variant, we calculated a proteolytic resistance factor r, which is the ratio between the apparent rate constant for a PEGylated olefin-stapled variant or its PEGylated but non-stapled counterpart relative to the parent non-PEGylated non-stapled variant. Variants with smaller values of r are more resistant to proteolysis than the corresponding parent variant. We then plotted the natural logarithm of r against the corresponding difference in free energy for the compound relative to its non-PEGylated non-stapled parent variant (Fig. 2C). ln r varies linearly with ΔΔG as indicated by least-squares regression (R2 = 0.996), indicating that more stabilized WW variants experience greater levels of proteolytic resistance, independent of whether the increased stability comes primarily from PEGylation, olefin-stapling, or a combination of the two.
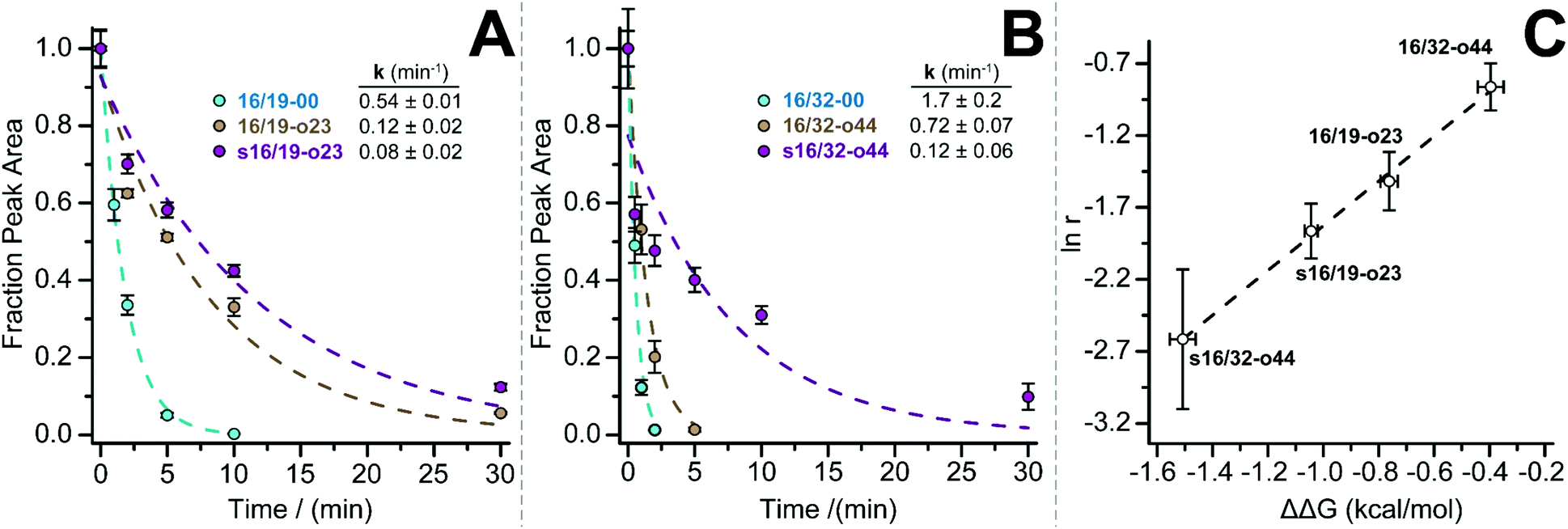 | ||
| Fig. 2 Proteolysis of (A) 16/19-00 (blue circles), 16/19-o23 (brown circles), and s16/19-o23 (magenta circles) and of (B) 16/32-00 (blue circles), 16/32-o44 (brown circles), and s16/32-o44 by proteinase K (17 μg mL−1) at 50 μM protein concentration in 20 mM sodium phosphate buffer (pH 7) as monitored by HPLC. Data points represent the average of three replicate experiments. Solid lines represent fits of the data to a mono-exponential decay function, which was used to determine apparent proteolysis rate constants. (C) Plot of the impact of PEGylation or PEG stapling on proteolytic resistance (as assessed by the natural logarithm of r, the ratio of apparent proteolysis rate constant for PEGylated or PEG-stapled WW variants relative to their non-stapled non-PEGylated counterparts) vs. the impact of PEGylation or PEG stapling on WW conformational stability (ΔΔG). Dotted line represents fit of the ln r vs. ΔΔG data to a linear equation. Slope = 1.55 ± 0.07; intercept = −0.28 ± 0.07; R2 = 0.996. | ||
In the examples described above, we installed olefin or click staples between two positions in the same monomeric protein (WW or SH3). We wondered whether the extent of stabilization observed in these monomeric systems might extend to intermolecular staples between subunits of quaternary structure. We explored this possibility within an α-helical coiled coil, one of the best understood tertiary/quaternary structural motifs in proteins.67–69 Coiled-coil primary sequence is comprised of a seven-residue repeating unit called a heptad; the first and fourth residues within this unit (i.e. positions a and d of an abcdefg heptad) are typically occupied by nonpolar residues, with charged residues at e and g positions and polar or charged residues at b, c, and f positions.70,71 Burial of non-polar residues at a and d positions provides the major driving force for folding; the shape of these a and d residues can specify coiled-coil oligomerization state (dimer, trimer, tetramer, etc.).72–77 Complementary electrostatic interactions between an e residue on one helix and an g residue on the other provide specificity for homo- vs. hetero-oligomerization78–81 and for parallel vs. antiparallel orientation.82–84
Others have already begun to apply intermolecular stapling to α-helical coiled coils, but with limited focus on the thermodynamic consequences of stapling. Arora and coworkers previously used CuAAC to install a bis-triazole staple in place of native interhelical e/g and e/e salt bridges within antiparallel85 and parallel86 coiled-coil heterodimers comprised of two nine-residue peptides. The staple enabled a surprisingly large extent of helicity at such a short oligomer length, though its precise energetic contribution to coiled-coil conformational stability was not assessed. Karlström and coworkers87 installed a single interhelical staple between a Cys residue and a chloroacetamide-modified Lys within a monomeric three-helix bundle HER2 affibody. Of the three locations they tested, only one Cys–Lys staple led to a substantial increase in conformational stability relative to a non-stapled reference compound (i.e. a 5 °C increase in melting temperature); the origin of this disparity was not explored in detail. Jiang, Liu, and coworkers88 formed an isopeptide bond between each of three identical e-position Glu residues within a helix-bundle trimer and a g-position Lys from the previous heptad on an adjacent helix. The resulting triply-stapled helix bundle had no cooperative thermal unfolding transition below 90 °C and was resistant to aggregation and proteolysis, though no comparison with its non-stapled counterpart was reported.
We explored the quantitative impact of interhelical stapling on α-helical coiled-coil conformational stability in the context of a previously characterized coiled-coil tertiary structure based on the GCN4 homodimer, in which acidic peptide A and basic peptide B are covalently connected via a disulfide bond to form parallel monomeric coiled coil A/B.89 In disulfide-bound A/B, e-position Glu residues in peptide A engage in interhelical salt bridges with g-position Lys residues in peptide B (similarly, e-position Lys residues in B interact with g-position Glu residues in A). Each e/g pair is oriented such that a g residue on one helix is close to the e residue from the previous heptad on the other helix. For example, Oε2 of e-position Glu27 in A is only 4.3 Å from Nζ of g-position Lys22′ in B but is 9.7 Å of from Nζ of g-position Lys29′. Whereas Arora85,86 and Liu88 used stapling to replace e/g salt bridges within parallel coiled coils, we wondered how a longer-range staple might influence coiled-coil conformational stability.
We addressed this question by preparing peptide 27-c4 (a variant of acidic peptide A in which e-position Glu27 has been replaced with an azide-terminated Asn-PEG comprised of four ethylene oxide units) and peptide 29′-c0 (a variant of basic peptide B in which g-position Lys29 has been replaced with propargylglycine), which are shown in Fig. 3. We chose these positions because Glu27 in A and Lys29 in B are not involved in a salt bridge with each other in the parent disulfide-bound coiled-coil monomer A/B. We mixed 27-c4 and 29′-c0 in an equimolar ratio in the presence of air to form monomeric disulfide-bound d27/29′-c40; we then prepared its click-stapled counterpart sd27/29′-c40via CuAAC. CD data for d27/29′-c40 and sd27/29′-c40 are consistent with the formation of an α-helical coiled-coil tertiary structure. The disulfide bond makes both variants monomeric even though one is stapled and the other is not; this facilitates direct comparison of their folding free energies. In the presence of 4 M GdnHCl, triazole-stapled sd27/29′-c40 (Tm = 48.2 ± 0.1 °C) is −0.65 ± 0.01 kcal mol−1 more stable than non-stapled d27/29′-c40 (Tm = 41.1 ± 0.2 °C, see Table 1); we used 4 M GdnHCl because these variants were too stable to characterize via variable temperature CD in the absence of denaturant (i.e., their thermal unfolding transitions were not complete even at 94 °C). These results indicate that a long-range interhelical staple between non-interacting e and g positions can increase the conformational stability of a coiled coil to a similar extent as we observed above for click and olefin staples within the β-sheet-rich WW and SH3 domains.
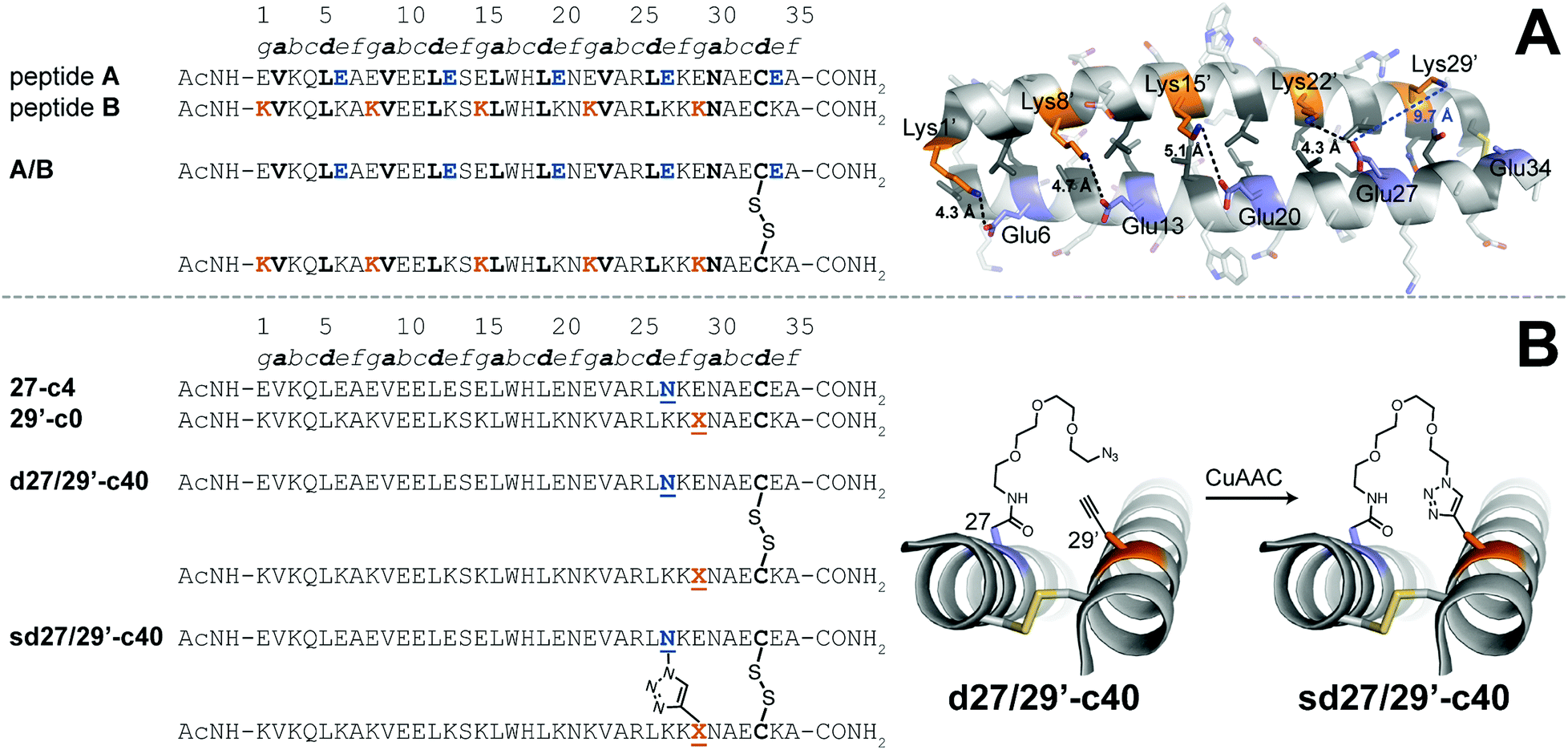 | ||
Fig. 3 (A) Sequences of acidic peptide A and basic peptide B, along with disulfide-bonded parallel coiled-coil monomer A/B. Ribbon diagram of the published crystal structure of A/B (PDB ID: 1KD9), with side chains shown as sticks. e-Position Glu residues on peptide A are colored blue; g-position Lys residues on peptide B are colored orange; non-polar a- and d-position residues on peptides A and B are colored dark grey. Black dotted lines indicate distances between the Oε2 of Glu and Nζ of Lys within each of four e/g′ interhelical salt bridges (i.e., Glu6/Lys1′, Glu13/Lys8′, Glu20/Lys15′, and Glu27/Lys22′). The blue dotted line indicates the distance between Oε2 of Glu27 and Nζ of Lys29′, which are not involved in an interhelical salt bridge with each other (B) Sequences of acidic variant 27-c4, basic variant 29′-c0, disulfide-bound coiled-coil monomer d27/29′-c40, and its triazole-stapled counterpart sd27/29′-c40. ![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) represents propargyl glycine and represents an azide-terminated Asn-PEG, with the structures as shown. represents propargyl glycine and represents an azide-terminated Asn-PEG, with the structures as shown. | ||
We wondered how much this interhelical staple would stabilize a heterodimeric coiled-coil quaternary structure in which the individual helices were not disulfide-bound. Accordingly, we prepared peptides 27A-c4 and 29A′-c0, variants of 27-c4 and 29′-c0, respectively, in which Ala occupies position 33 instead of Cys. Peptides 27A-c4 and 29A′-c0 combine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to form noncovalent heterodimeric coiled coil 27/29′-c40; the CD spectrum of 27/29′-c40 is consistent with coiled-coil quaternary structure; variable temperature CD data in the presence of 0.5 M GdnHCl indicate that 27/29′-c40 undergoes a cooperative thermal unfolding transition with Tm = 34.8 °C. Click stapling converts noncovalent heterodimer 27/29′-c40 into stapled monomeric s27/29′-c40, which has a similar CD spectrum, and undergoes a cooperative thermal unfolding transition with Tm = 82.0 ± 0.2 °C. The folding free energies of 27/29′-c40 and s27/29′-c40 are not directly comparable because of their distinct association states: the ΔG of noncovalent heterodimer 27/29′-c40 is concentration dependent, whereas the ΔG of stapled monomeric s27/29′-c40 is not. However, stapling increases the melting temperature of s27/29′-c40 by 49.2 °C relative to noncovalent heterodimer 27/29′-c40 in 0.5 M GdnHCl, suggesting that the stabilizing impact of intermolecular interhelical stapling is substantial.
:1 ratio to form noncovalent heterodimeric coiled coil 27/29′-c40; the CD spectrum of 27/29′-c40 is consistent with coiled-coil quaternary structure; variable temperature CD data in the presence of 0.5 M GdnHCl indicate that 27/29′-c40 undergoes a cooperative thermal unfolding transition with Tm = 34.8 °C. Click stapling converts noncovalent heterodimer 27/29′-c40 into stapled monomeric s27/29′-c40, which has a similar CD spectrum, and undergoes a cooperative thermal unfolding transition with Tm = 82.0 ± 0.2 °C. The folding free energies of 27/29′-c40 and s27/29′-c40 are not directly comparable because of their distinct association states: the ΔG of noncovalent heterodimer 27/29′-c40 is concentration dependent, whereas the ΔG of stapled monomeric s27/29′-c40 is not. However, stapling increases the melting temperature of s27/29′-c40 by 49.2 °C relative to noncovalent heterodimer 27/29′-c40 in 0.5 M GdnHCl, suggesting that the stabilizing impact of intermolecular interhelical stapling is substantial.
Conclusion
Here we have shown that PEG stapling enhances WW conformational stability best when the staple sites are distant in primary sequence, close in tertiary structure, and are each individually stabilized by Asn-PEGylation. We applied these criteria to the SH3 domain, where PEG stapling increased the stability of the stapled variant by −0.9 kcal mol−1 relative to its non-PEGylated non-stapled counterpart. We found that staple-based stabilization is associated with increased proteolytic resistance and is tolerant of variation in linker chemistry, with triazole and olefin linkers providing similar energetic benefits. We also found that an intermolecular PEG staple between non-interacting e- and g-positions in a GCN4-derived α-helical coiled-coil heterodimer dramatically increases the stability of the stapled coiled-coil relative to its non-stapled counterpart.We previously found that staples comprised of PEGs shorter than a certain threshold can actually decrease protein conformational stability, presumably because the PEGs are too short to accommodate the distance between the staple sites in the folded tertiary structure.64 We originally expected longer PEG staples to have a less stabilizing impact; we reasoned that a longer PEG would not be as effective at restricting the conformational freedom of the staple sites. Our results were not consistent with this hypothesis: we found that incremental increases to PEG length beyond the minimum threshold do not dramatically change the stabilizing impact of stapling or its entropic origin.64 In agreement with these previous results, we herein observed substantial levels of entropy-derived stabilization despite the length of the PEG staples: eight ethylene oxide units in s16/32-o44, s16/32-c44, and 20/37-o44; ten in 14/30-o55; and four in s27/29′-c40. It is possible that the length and flexibility of the PEG staple is responsible for its versatility in the secondary, tertiary, and quaternary structural contexts investigated here (β-sheet tertiary structures, α-helical coiled-coil quaternary structure). This versatility should be useful in applying PEG stapling to the stabilization of therapeutic proteins. In any case, it will be interesting to see whether the stabilizing impact of longer staples is a unique feature of PEG stapling or whether it also extends to stapling with other linkers (e.g., hydrocarbons).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by NIH grant number 2 R15 GM116055-02.References
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1986, 25, 1039–1057 CrossRef.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- V. J. Hruby, Life Sci., 1982, 31, 189–199 CrossRef CAS.
- D. Y. Jackson, D. S. King, J. Chmielewski, S. Singh and P. G. Schultz, J. Am. Chem. Soc., 1991, 113, 9391–9392 CrossRef CAS.
- M. Gongora-Benitez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef CAS PubMed.
- S. F. Betz, Protein Sci., 1993, 2, 1551–1558 CrossRef CAS.
- T. Zhang, E. Bertelsen and T. Alber, Nat. Struct. Biol., 1994, 1, 434–438 CrossRef CAS PubMed.
- A. A. Dombkowski, K. Z. Sultana and D. B. Craig, FEBS Lett., 2014, 588, 206–212 CrossRef CAS.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Chem. Rev., 2019, 119, 9971–10001 CrossRef CAS PubMed.
- F. M. Brunel and P. E. Dawson, Chem. Commun., 2005, 2552–2554, 10.1039/B419015G.
- F. Zhang, O. Sadovski, S. J. Xin and G. A. Woolley, J. Am. Chem. Soc., 2007, 129, 14154–14155 CrossRef CAS PubMed.
- N. Bionda, A. L. Cryan and R. Fasan, ACS Chem. Biol., 2014, 9, 2008–2013 CrossRef CAS.
- L. Peraro, T. R. Siegert and J. A. Kritzer, Methods Enzymol., 2016, 580, 303–332 CAS.
- E. J. Moore, D. Zorine, W. A. Hansen, S. D. Khare and R. Fasan, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12472–12477 CrossRef CAS.
- A. J. Rojas, C. Zhang, E. V. Vinogradova, N. H. Buchwald, J. Reilly, B. L. Pentelute and S. L. Buchwald, Chem. Sci., 2017, 8, 4257–4263 RSC.
- D. G. Flint, J. R. Kumita, O. S. Smart and G. A. Woolley, Chem. Biol., 2002, 9, 391–397 CrossRef CAS.
- G. A. Woolley, Acc. Chem. Res., 2005, 38, 486–493 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS.
- C. Heinis and G. Winter, Curr. Opin. Chem. Biol., 2015, 26, 89–98 CrossRef CAS.
- M. Pelay-Gimeno, T. Bange, S. Hennig and T. N. Grossmann, Angew. Chem., Int. Ed., 2018, 57, 11164–11170 CrossRef CAS.
- B. Dang, H. Wu, V. K. Mulligan, M. Mravic, Y. Wu, T. Lemmin, A. Ford, D. A. Silva, D. Baker and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10852–10857 CrossRef CAS PubMed.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS.
- M. Chorev, E. Roubini, R. L. Mckee, S. W. Gibbons, M. E. Goldman, M. P. Caulfield and M. Rosenblatt, Biochemistry, 1991, 30, 5968–5974 CrossRef CAS.
- A. M. Leduc, J. O. Trent, J. L. Wittliff, K. S. Bramlett, S. L. Briggs, N. Y. Chirgadze, Y. Wang, T. P. Burris and A. F. Spatola, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11273–11278 CrossRef CAS.
- K. Fujimoto, N. Oimoto, K. Katsuno and M. Inouye, Chem. Commun., 2004, 1280–1281, 10.1039/B403615H.
- N. E. Shepherd, G. Abbenante and D. P. Fairlie, Angew. Chem., Int. Ed., 2004, 43, 2687–2690 CrossRef CAS PubMed.
- E. Vaz, W. C. Pomerantz, M. Geyer, S. H. Gellman and L. Brunsveld, ChemBioChem, 2008, 9, 2254–2259 CrossRef CAS PubMed.
- M. Kajino, K. Fujimoto and M. Inouye, J. Am. Chem. Soc., 2011, 133, 656–659 CrossRef CAS PubMed.
- A. V. Vasco, C. S. Perez, F. E. Morales, H. E. Garay, D. Vasilev, J. A. Gavin, L. A. Wessjohann and D. G. Rivera, J. Org. Chem., 2015, 80, 6697–6707 CrossRef CAS.
- A. V. Vasco, Y. Méndez, A. Porzel, J. Balbach, L. A. Wessjohann and D. G. Rivera, Bioconjugate Chem., 2019, 30, 253–259 CrossRef CAS.
- C. Wang, X. Li, F. Yu, L. Lu, X. Jiang, X. Xu, H. Wang, W. Lai, T. Zhang, Z. Zhang, L. Ye, S. Jiang and K. Liu, Sci. Rep., 2016, 6, 32161 CrossRef CAS PubMed.
- L. Zhang and J. P. Tam, J. Am. Chem. Soc., 1997, 119, 2363–2370 CrossRef CAS.
- J. A. Camarero and T. W. Muir, Chem. Commun., 1997, 1369–1370, 10.1039/A702083J.
- J. A. Camarero and T. W. Muir, J. Am. Chem. Soc., 1999, 121, 5597–5598 CrossRef CAS.
- F. Rohrbacher, A. Zwicky and J. W. Bode, Chem. Sci., 2017, 8, 4051–4055 RSC.
- R. Kleineweischede and C. P. Hackenberger, Angew. Chem., Int. Ed., 2008, 47, 5984–5988 CrossRef CAS.
- J. M. Antos, M. W. Popp, R. Ernst, G. L. Chew, E. Spooner and H. L. Ploegh, J. Biol. Chem., 2009, 284, 16028–16036 CrossRef CAS PubMed.
- G. K. T. Nguyen, A. Kam, S. Loo, A. E. Jansson, L. X. Pan and J. P. Tam, J. Am. Chem. Soc., 2015, 137, 15398–15401 CrossRef CAS PubMed.
- H. Luo, S. Y. Hong, R. M. Sgambelluri, E. Angelos, X. Li and J. D. Walton, Chem. Biol., 2014, 21, 1610–1617 CrossRef CAS.
- L. Mendive-Tapia, S. Preciado, J. Garcia, R. Ramon, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- A. F. M. Noisier, J. Garcia, I. A. Ionut and F. Albericio, Angew. Chem., Int. Ed., 2017, 56, 314–318 CrossRef CAS.
- M. M. Lorion, N. Kaplaneris, J. Son, R. Kuniyil and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 1684–1688 CrossRef CAS PubMed.
- M. G. Ricardo, D. Llanes, L. A. Wessjohann and D. G. Rivera, Angew. Chem., Int. Ed., 2019, 58, 2700–2704 CrossRef CAS PubMed.
- P. A. Cistrone, A. P. Silvestri, J. C. J. Hintzen and P. E. Dawson, ChemBioChem, 2018, 19, 1031–1035 CrossRef CAS PubMed.
- C. M. Haney, M. T. Loch and W. S. Horne, Chem. Commun., 2011, 47, 10915–10917 RSC.
- J. M. Smith, J. R. Frost and R. Fasan, Chem. Commun., 2014, 50, 5027–5030 RSC.
- E. Cabezas and A. C. Satterthwait, J. Am. Chem. Soc., 1999, 121, 3862–3875 CrossRef CAS.
- M. Roice, I. Johannsen and M. Meldal, QSAR Comb. Sci., 2004, 23, 662–673 CrossRef CAS.
- S. Cantel, A. Le Chevalier Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D’Ursi, A. M. Papini and M. Chorev, J. Org. Chem., 2008, 73, 5663–5674 CrossRef CAS PubMed.
- Y. H. Lau, Y. T. Wu, P. de Andrade, W. R. J. D. Galloway and D. R. Spring, Nat. Protoc., 2015, 10, 585–594 CrossRef CAS PubMed.
- C. M. Haney, H. M. Werner, J. J. McKay and W. S. Horne, Org. Biomol. Chem., 2016, 14, 5768–5773 RSC.
- P. T. Tran, C. O. Larsen, T. Rondbjerg, M. De Foresta, M. B. A. Kunze, A. Marek, J. H. Loper, L. E. Boyhus, A. Knuhtsen, K. Lindorff-Larsen and D. S. Pedersen, Chem. – Eur. J., 2017, 23, 3490–3495 CrossRef CAS PubMed.
- M. Scrima, A. Le Chevalier-Isaad, P. Rovero, A. M. Papini, M. Chorev and A. M. D'Ursi, Eur. J. Org. Chem., 2010, 446–457 CrossRef CAS.
- S. A. Kawamoto, A. Coleska, X. Ran, H. Yi, C.-Y. Yang and S. Wang, J. Med. Chem., 2012, 55, 1137–1146 CrossRef CAS PubMed.
- H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS PubMed.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- R. N. Chapman, G. Dimartino and P. S. Arora, J. Am. Chem. Soc., 2004, 126, 12252–12253 CrossRef CAS PubMed.
- A. Patgiri, A. L. Jochim and P. S. Arora, Acc. Chem. Res., 2008, 41, 1289–1300 CrossRef CAS PubMed.
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3–33 CAS.
- M. Jäger, H. Nguyen, J. C. Crane, J. W. Kelly and M. Gruebele, J. Mol. Biol., 2001, 311, 373–393 CrossRef PubMed.
- P. B. Lawrence, Y. Gavrilov, S. S. Matthews, M. I. Langlois, D. Shental-Bechor, H. M. Greenblatt, B. K. Pandey, M. S. Smith, R. Paxman, C. D. Torgerson, J. P. Merrell, C. C. Ritz, M. B. Prigozhin, Y. Levy and J. L. Price, J. Am. Chem. Soc., 2014, 136, 17547–17560 CrossRef CAS PubMed.
- Q. Xiao, N. A. Becar, N. P. Brown, M. S. Smith, K. L. Stern, S. R. E. Draper, K. P. Thompson and J. L. Price, Org. Biomol. Chem., 2018, 16, 8933–8939 RSC.
- R. Ranganathan, K. P. Lu, T. Hunter and J. P. Noel, Cell, 1997, 89, 875–886 CrossRef CAS PubMed.
- H. Yu, M. K. Rosen and S. L. Schreiber, FEBS Lett., 1993, 324, 87–92 CrossRef CAS.
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS PubMed.
- A. N. Lupas and M. Gruber, Adv. Protein Chem., 2005, 70, 37–78 CrossRef CAS.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112 CrossRef CAS PubMed.
- A. D. McLachlan and M. Stewart, J. Mol. Biol., 1975, 98, 293–304 CrossRef CAS.
- E. K. O'Shea, J. D. Klemm, P. S. Kim and T. Alber, Science, 1991, 254, 539–544 CrossRef PubMed.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401–1407 CrossRef CAS PubMed.
- F. K. Junius, J. P. Mackay, W. A. Bubb, S. A. Jensen, A. S. Weiss and G. F. King, Biochemistry, 1995, 34, 6164–6174 CrossRef CAS PubMed.
- K. J. Lumb and P. S. Kim, Biochemistry, 1995, 34, 8642–8648 CrossRef CAS PubMed.
- T. L. Vincent, P. J. Green and D. N. Woolfson, Bioinformatics, 2013, 29, 69–76 CrossRef CAS PubMed.
- A. R. Thomson, C. W. Wood, A. J. Burton, G. J. Bartlett, R. B. Sessions, R. L. Brady and D. N. Woolfson, Science, 2014, 346, 485–488 CrossRef CAS PubMed.
- J. M. Fletcher, G. J. Bartlett, A. L. Boyle, J. J. Danon, L. E. Rush, A. N. Lupas and D. N. Woolfson, ACS Chem. Biol., 2017, 12, 528–538 CrossRef CAS.
- E. K. O’Shea, K. J. Lumb and P. S. Kim, Curr. Biol., 1993, 3, 658–667 CrossRef.
- N. A. Schnarr and A. J. Kennan, J. Am. Chem. Soc., 2002, 124, 9779–9783 CrossRef CAS PubMed.
- A. Kashiwada, H. Hiroaki, D. Kohda, M. Nango and T. Tanaka, J. Am. Chem. Soc., 2000, 122, 212–215 CrossRef CAS.
- T. Kiyokawa, K. Kanaori, K. Tajima, M. Kawaguchi, T. Mizuno, J. I. Oku and T. Tanaka, Chem. – Eur. J., 2004, 10, 3548–3554 CrossRef CAS PubMed.
- D. A. D. Parry, R. D. B. Fraser and J. M. Squire, J. Struct. Biol., 2008, 163, 258–269 CrossRef CAS PubMed.
- G. Grigoryan and A. E. Keating, Curr. Opin. Struct. Biol., 2008, 18, 477–483 CrossRef CAS PubMed.
- W. M. Dawson, G. G. Rhys and D. N. Woolfson, Curr. Opin. Chem. Biol., 2019, 52, 102–111 CrossRef CAS PubMed.
- M. G. Wuo, A. B. Mahon and P. S. Arora, J. Am. Chem. Soc., 2015, 137, 11618–11621 CrossRef CAS PubMed.
- M. G. Wuo, S. Hong, A. Singh and P. S. Arora, J. Am. Chem. Soc., 2018, 140, 16284–16290 CrossRef CAS PubMed.
- J. Lindgren and A. Karlström, ChemBioChem, 2014, 15, 2132–2138 CrossRef CAS PubMed.
- C. Wang, X. Li, F. Yu, L. Lu, X. Jiang, X. Xu, H. Wang, W. Lai, T. Zhang, Z. Zhang, L. Ye, S. Jiang and K. Liu, Sci. Rep., 2016, 6, 32161 CrossRef CAS PubMed.
- A. E. Keating, V. N. Malashkevich, B. Tidor and P. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14825–14830 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods; compound characterization data, including mass spectra, HPLC chromatograms, and NMR spectra where applicable; CD spectra; global fits of variable temperature CD data; proteolysis assay data. See DOI: 10.1039/d0cb00075b |
| This journal is © The Royal Society of Chemistry 2020 |