
β-Fluorinated porpholactones and metal complexes: synthesis, characterization and some spectroscopic studies†
Ji-Yun
Hu
 a,
Zhuo-Yan
Wu
a,
Ke
Chai
b,
Zi-Shu
Yang
a,
Yin-Shan
Meng
a,
Yingying
Ning
a,
Jing
Zhang
*b and
Jun-Long
Zhang
*a
a,
Zhuo-Yan
Wu
a,
Ke
Chai
b,
Zi-Shu
Yang
a,
Yin-Shan
Meng
a,
Yingying
Ning
a,
Jing
Zhang
*b and
Jun-Long
Zhang
*a
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, P.R. China. E-mail: Zhangjunlong@pku.edu.cn
bCollege of Materials Science and Optoelectronics Technology, University of Chinese Academy of Sciences, Beijing, 100049, P. R. China. E-mail: zhangj271@ucas.ac.cn
First published on 24th July 2017
Abstract
We describe the synthesis of β-fluorinated porpholactones by oxidation of the fluorinated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the pyrrolic subunit in porphyrin using the “RuCl3 + Oxone®” protocol. The electron-withdrawing effects of β-fluorine on the absorption, fluorescence, phosphorescence and redox properties of the β-fluoroporpholactones were studied. A potential application of the β-fluoroporpholactones was demonstrated in sensitizing near-IR (NIR) emissive ytterbium, giving a high quantum yield (58%) and long luminescence time (525 μs) in CD2Cl2.
C bond of the pyrrolic subunit in porphyrin using the “RuCl3 + Oxone®” protocol. The electron-withdrawing effects of β-fluorine on the absorption, fluorescence, phosphorescence and redox properties of the β-fluoroporpholactones were studied. A potential application of the β-fluoroporpholactones was demonstrated in sensitizing near-IR (NIR) emissive ytterbium, giving a high quantum yield (58%) and long luminescence time (525 μs) in CD2Cl2.
Introduction
Introducing electron-withdrawing groups into porphyrinate compounds is a preferred way to modulate the photophysical and redox potential properties as well as the reactivities of the molecules.1–6 Among all the electron-withdrawing substituents, fluorine is a quite special one because of its strongest electronegativity (χ = 4), small atom radius (1.47 Å) and unique biological properties imparted when introduced into organic molecules.7,8 In fact, introducing fluorine atoms, unlike its heavier counterparts chlorine and bromine,9–12 into the β positions of tetrapyrrole molecules was once a difficult task and has been pursued intensively by many chemists.13–19 For example, β-octafluorine-tetraaryl porphyrins were reported by the DiMagno group in 1997,15,16 and then β-octafluorocorrole17,18 and octafluoroporphyrin19 were reported by the Chang group in 2003 and 2016 respectively, using the condensation of 3,4-difluoropyrrole20,21 and an aldehyde in the presence of an acid catalyst. A significant electronic effect of β fluorination has been found on the reactivity of metal complexes. For instance, enhanced water splitting catalytic performance was observed on cobalt(III) hangman β-octafluorocorrole by Nocera and co-workers.22 Increased low temperature preferential oxidation of carbon monoxide was achieved with rhodium(III) β-octafluoroporphyrin by the DiMagno group.23 These excellent studies stimulated us to expand such an approach to more tetrapyrrole derivatives.Porpholactone, in which a porphyrin pyrrole unit is formally replaced by an oxazolone moiety, is an important type of porphyrinoid. Since the first report on porpholactone by Crossley et al.,24 several strategies to convert a meso-arylporphyrin to porpholactone have been reported.25–29 Their potential applications have been demonstrated in optical materials, biology and catalysis in recent years.30–39 However, much less attention has been paid on the electronic structure of the macrocycle, and no β-fluorinated porpholactone has been reported yet, probably due to the limited synthetic methodology. A practical method to β-fluoroporpholactone would be direct modification of the β-fluoroporphyrin and the bottleneck of this approach lies in the high stability of the C–F bond (bond dissociation energy ∼105 kcal mol−1).40,41 Inspired by recent work on the electrophilic activation of the C–F bond using high-valent metal oxo species,42–46 we report here the transformation of β-fluoroporphyrin to β-fluoroporpholactone on β C–F bond cleavage catalysed by ruthenium under oxidative conditions. This protocol enables us to obtain the first perfluorinated tetraarylporpholactone and to investigate the electronic effect of β-fluorine on porpholactones as well as the potential application in sensitizing near-IR emissive ytterbium.
Results and discussion
Synthesis of β-fluoroporpholactone
Our previous work showed that the “RuCl3 + Oxone®” protocol was efficient in catalytically oxidizing porphyrin to porpholactone in one step.28 The in situ generated high-valent [RuO] was believed to be the active intermediate. Enlightened by high-valent [FeO] and [CuO] in the hydroxylation of the C–F bond, we wonder whether the [RuO] species could show similar reactivity towards the β C–F bond of porphyrin. We started the investigation from the oxidation of 2,3,7,8,12,13,17,18-octafluoro-5,10,15,20-tetrakis(pentafluorophenyl)porphyrin (1a). To our delight, after the optimization of the reaction conditions, porpholactone (2a, 43%) and porphodilactones (3a, a mixture of cis and trans isomers, 10%) were obtained with the “RuCl3 + Oxone®” protocol (Scheme 1).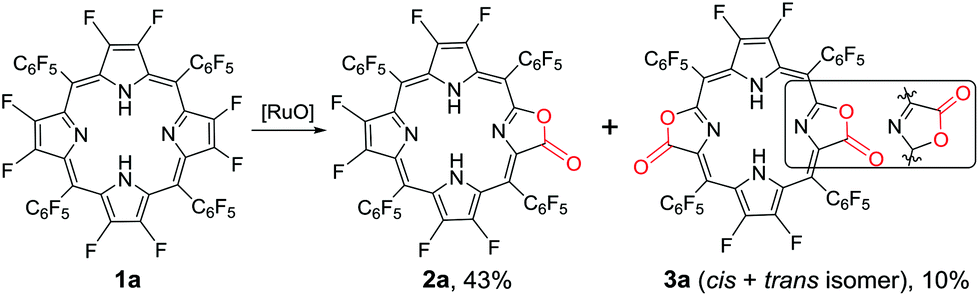 | ||
| Scheme 1 Oxidation of 1a by the “RuCl3 + Oxone®” protocol. Conditions: 1a 0.05 mmol, RuCl3·nH2O 0.01 mmol, 2,2′-bipyridine 0.01 mmol, Oxone® 1 mmol, NaOH 1 mmol, 1,2-dichloroethane/H2O = 1/1 (v/v) 60 mL, 80 °C, 1 h. See the ESI† for details. | ||
2a was fully characterized by NMR (1H/19F/13C), HR-ESI-MS, IR, and UV-Vis spectrometry (see details in the ESI†). The characteristic carbon signal of the carbonyl moiety appears at 164.2 ppm in the 13C NMR spectrum and at 1796 cm−1 in the FT-IR spectrum. The UV-vis absorption spectra of the products are shown in Fig. 1. A slight red shift of ca. 3 nm from 393 nm to 396 nm and a narrowed Soret band are observed upon introducing the lactone moiety on 1a to give 2a (Fig. 1). Four Q bands appear for 2a as a result of the lowered symmetry compared to 1a. These results are in line with our previously reported porpholactones.28
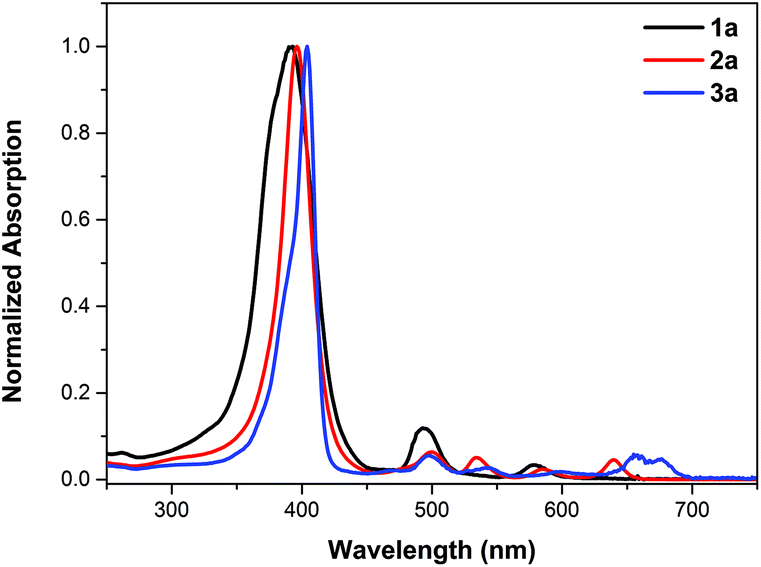 | ||
| Fig. 1 UV-vis absorption spectra of porphyrin 1a (black line), porpholactone 2a (red line) and the mixture of porphodilactones 3a (blue line) in CH2Cl2. | ||
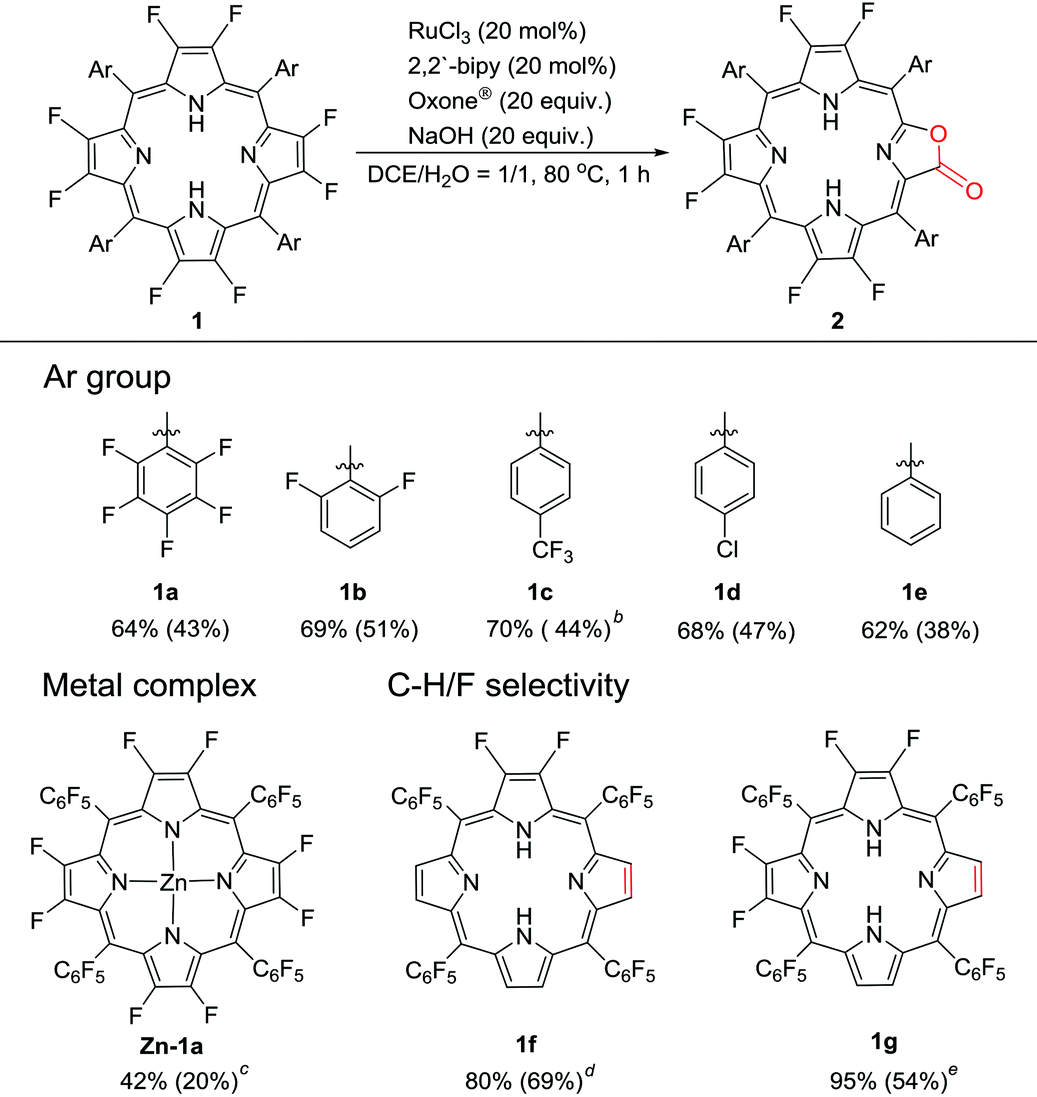
To examine the generality of this methodology, we used several β-octafluoroporphyrins with different meso-phenyl groups as substrates, including 2,6-difluorophenyl (1b), p-trifluoromethylphenyl (1c), p-chlorophenyl (1d) and phenyl (1e). Typical isolated yields are around 40%–50% with the optimal conversions of around 60%–70% (Table 1). The catalytic protocol shows good regioselectivity, leaving the meso-aryl C–F bond and trifluromenthyl group intact. Moreover, the metal complex Zn-1a could also be oxidized to the corresponding metalloporpholactone Zn-2a, albeit with low yield (20%) due to the significant disruption of the porphyrin macrocycle. The chemo-selectivity between β-C–F and C–H bonds was investigated using 2,3-difluoro-5,10,15,20-tetrapentafluorophenylporphyrin (1f) and 2,3,7,8-tetrafluoro-5,10,15,20-tetrapentafluorophenylporphyrin (1g) as substrates. The isolated products showed that the non-fluorinated pyrrole unit adjacent to the difluoropyrrole unit was selectively oxidized for both compounds, indicating that the C–H bond is more susceptible to oxidization than the C–F bond. Interestingly, for 1g, the lactonized products are two isomers in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, regarding the relative orientation of the lactone moiety with respect to the pyrrolic unit.
:1 ratio, regarding the relative orientation of the lactone moiety with respect to the pyrrolic unit.
A preliminary reaction mechanism study was carried out according to the “breaking and mending strategy” developed by Brückner and co-workers.30 A cis-Diol-substituted chlorin (Ch-H), formed from the reaction of porphyrin with the mild oxidant OsO4, could be converted to porpholactone in the presence of a strong oxidant such as MnO4− (Scheme 2A).26,47–49 In this work, we attempted the dihydroxylation of 1b with OsO4, and however we could not obtain the desired dihydroxyl product (Ch-F) but 2b in 17% yield after the reaction for four days at room temperature (Scheme 2B).47 It is well known that α-fluoroalcohols are highly unstable and undergo facile HF elimination to give carbonyl compounds.50,51 Herrmann et al. reported that the catalytic oxidation of fluorinated olefins with OsO4 gave dione products.52 However, Crossley's seminal first paper on porpholactones showed that the oxidation of dione (β,β′-dioxoporphyrin) gave only a trace amount of porpholactone.24 We therefore hypothesize that Ch-F is prone to oxidization to porpholactone even by the weak oxidant OsO4 without the intermediation of dione, in contrast to its hydrogenated congener Ch-H with high stability.
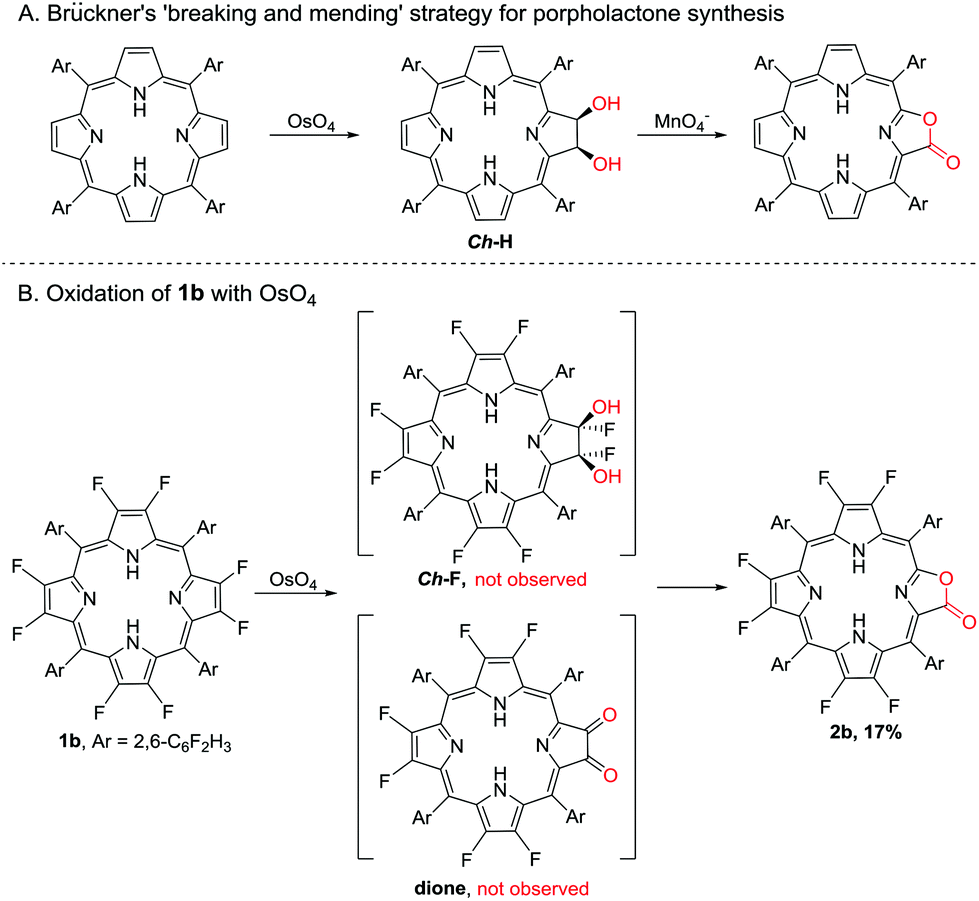 | ||
| Scheme 2 The reactivity of porphyrin towards OsO4. | ||
Metalation of perfluoroporpholactone 2a
To demonstrate the coordination ability of β-fluorinated porpholactones towards transition metals, in this work, we chose 2a as an example and investigated the metalation with zinc(II) and palladium(II) ions. The new porpholactone metal complexes Zn-2a and Pd-2a were synthesized according to literature methods in 95% and 53% yield respectively (Scheme 3).33 Metalation with zinc commenced immediately and was complete within minutes upon mixing 2a and zinc acetate in 1:1 CH2CH2:CH3OH solution. Palladium insertion was accomplished by refluxing 2a and palladium acetylacetonate in benzonitrile. Complexes Zn-4a, Zn-5a, Pd-4a and Pd-5a were prepared as well for the discussion of electrochemical and photophysical properties in the context (Scheme 3). These complexes were characterized by multinuclear NMR, HR-ESI-MS and IR spectrometry (see the ESI†).
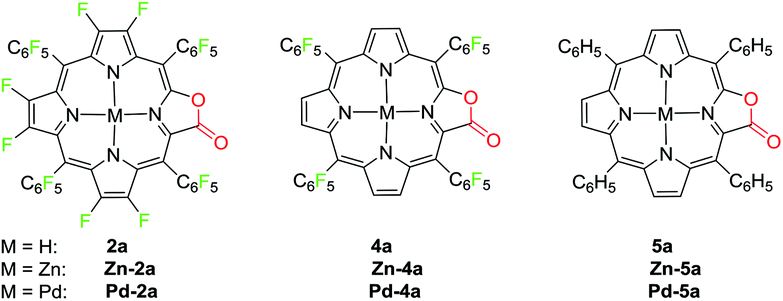 | ||
| Scheme 3 Porpholactone compounds studied in this work. | ||
Importantly, a single crystal of Zn-2a suitable for X-ray analysis was obtained in CHCl3/hexane (CCDC 1501201†). As shown in Fig. 2, the macrocyclic ring is essentially flat and the hexa-coordinate Zn (with two axial coordinate pyridine molecules) lies in the mean plane of four nitrogen atoms. The CO bond length of Zn-2a is 1.27(1) Å, similar to that of its hydrogenated counterpart Zn-5a (1.25(4) Å).26 The structure confirms the oxazolone moiety of porphyrin in Zn-2a, consistent with the above structure determination of β-fluorinated porpholactones.
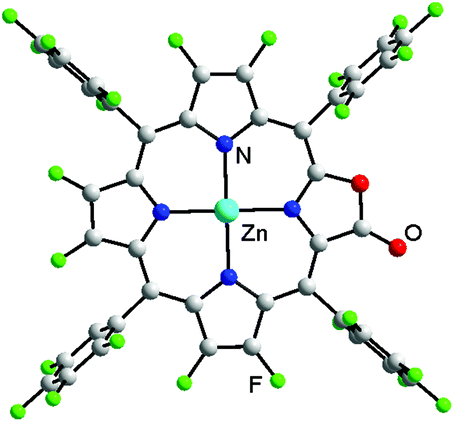 | ||
| Fig. 2 Perspective drawing of the crystal structure of Zn-2a (the two axial pyridines and solvent molecules are omitted for clarity; the lactone moiety is crystallographically disordered, see the ESI†). | ||
Photophysical and electrochemical features
To show how fluorine groups at β positions regulate the electronic structures of porpholactones, the spectroscopic properties of the representative porpholactone free bases as well as the spectroscopic and electrochemical properties of metal complexes (Pd and Zn) were studied (Scheme 3). The absorption and emission data are given in Table 2 and the spectra are depicted in Fig. 3.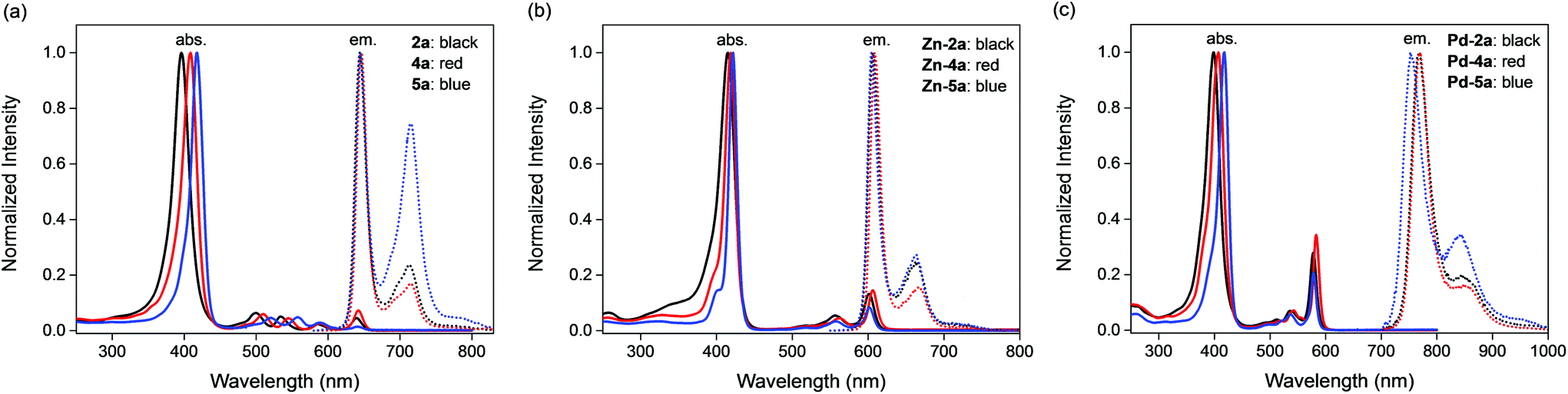 | ||
| Fig. 3 UV-vis absorption (solid line) and emission spectra (dashed line) of (a) porpholactone free bases, (b) zinc complexes and (b) palladium complexes in CH2Cl2 solution at 298 K. The emission spectra of palladium complexes were recorded in degassed solution. | ||
| Compound | Abs. [nm] | Em. [nm] (τ [ns or μs])a | Stokes shift [cm−1] | QYb | k obs [s−1] | k f [s−1] |
|---|---|---|---|---|---|---|
| a The emission lifetimes are in the nanosecond scale for free base and zinc complexes and are in the microsecond scale for palladium complexes. b Referenced to TPP in toluene (Φ = 0.11) for free bases and to ZnTPP in toluene (Φ = 0.033) for metal complexes. Errors for quantum yield values (±10%) are estimated. c Ref. 28. d Ref. 54. e Ref. 33. | ||||||
| 2a | 396, 500, 534, 585, 640 | 644 (5.8), 714 | 97 | 0.075 | 1.7 × 108 | 1.3 × 107 |
|
4ac |
409, 510, 545, 589, 642 | 643 (6.9), 710 | 24 | 0.13 | 1.4 × 108 | 1.9 × 107 |
|
5ac |
418, 520, 558, 589, 641 | 644 (8.1), 709 | 73 | 0.065 | 1.2 × 108 | 0.8 × 107 |
| Zn-2a | 415, 556, 601 | 606 (0.84), 665 | 137 | 0.025 | 11.9 × 108 | 3.0 × 107 |
|
Zn-4ac |
417, 542, 606 | 609 (0.86), 667 | 27 | 0.040 | 11.6 × 108 | 4.7 × 107 |
| Zn-5a | 422, 557, 602 | 605 (0.93), 664 | 82 | 0.035 | 10.8 × 108 | 3.8 × 107 |
| Pd-2a | 399, 536, 578 | 768 (367), 853 | 4280 | 0.046 | 2.7 × 103 | 12.5 |
|
Pd-4ad |
407, 542, 583 | 770 (472), 847 | 4166 | 0.060 | 2.3 × 103 | 12.7 |
|
Pd-5ae |
418, 537, 578 | 752 (300), 844 | 4003 | 0.061 | 3.3 × 103 | 20.3 |
Fluorine substituents at both meso and β positions are effective in tuning the UV-vis absorption of porpholactones. A progressive blue shift occurs from 5a to 2a. Compound 2a exhibits a Soret band at 396 nm in CH2Cl2, while 4a at 409 nm and 5a at 418 nm. The four Q bands of the three compounds show similar blue shifts upon fluorination except that the Qx(0,0) bands nearly appear at the same wavelength of around 641 nm. The corresponding Zn and Pd complexes show similar blue-shifted Soret bands upon fluorination, ranging from 415–422 nm and 399–418 nm respectively. However, compared to Zn-4a (542, 606 nm) and Pd-4a (542, 583 nm), the two Q bands of Zn-2a (556, 601 nm) and Pd-2a (536, 578 nm) shift to lower energy, occurring at nearly identical wavelengths to those of Zn-5a (557, 602 nm) and Pd-5a (537, 578 nm) respectively.
The fluorescence spectra of the three porpholactone molecules and their zinc complexes are very similar, peaking at 644 and 605 nm respectively with well-resolved vibronic progressions. They all display good mirror image symmetry with their respective Q-band absorptions in intensity and spacing with a small Stokes shift (<150 cm−1), indicating that the compounds do not undergo significant reorganization of nuclear coordinates in the excited states. The fluorescence lifetimes of the free bases decrease slightly from 8.1 to 5.8 μs in going from 5a to 2a. The corresponding zinc complexes also show a reduced fluorescence lifetime, yet to much less degree, from 0.93 to 0.84 ns upon perfluorination. The fluorescence quantum yields show an increase upon the first fluorination of meso-phenyl groups but a decrease upon further fluorination of β positions for both free bases and their zinc complexes. The deactivation rates of the excited state (kobs) are calculated from lifetimes and are listed in Table 2. Increasing magnitudes of kobs are observed for both free bases and zinc compounds upon fluorination. The radiative rates calculated from the S1 lifetimes and the quantum yields of fluorescence (kf = Φf/τf) are also listed in Table 2. The radiative rate order follows the same order observed for the fluorescence quantum yield.
The three palladium complexes show intense phosphorescence at room temperature in degassed CH2Cl2 solutions (Fig. 3c). The emission profile of Pd-2a resembles that of Pd-4a with the maximum peak at 768 nm, showing a 16 nm blue-shift to that of Pd-5a at 752 nm. A similar trend was found in the corresponding platinum porphyrin complex series in an early report by Chang et al.53 The lifetime of Pd-2a is 367 μs, longer than that of Pd-5a (300 μs), but shorter than that of Pd-4a (472 μs). In our hands the lifetime of Pd-5a and Pd-4a are longer than literature reports (157 and 296 μs respectively),33,54 presumably resulted from the lower air concentration. The quantum yields of phosphorescence change little after meso-phenyl fluorination for Pd-4a (0.060), but diminish on further β fluorination for Pd-2a (0.046).
The electrochemical properties of the porpholactone free bases and the zinc complexes were investigated by cyclic voltammetry at 298 K using 0.1 M nBu4NPF6 as the supporting electrolyte in CH2Cl2 and the data are tabulated in Table 3 (see the ESI† for figures). It is clearly shown that the fluorine substituents at both β positions and meso-phenyl groups are effective in tuning the redox potentials of the porpholactone compounds. For free bases, there is no oxidation peak observed for 2a up to a potential of 1.8 V, while there is one irreversible peak for 4a at 1.61 V and two quasi-reversible peaks for 5a at 1.54 and 1.30 V respectively. The six fluorine substituents on the porphyrin ring bring about a 0.3 V shift to the anode of the two reversible reduction waves (−0.23 and −0.71 V for 2avs. −0.54 and −0.99 V for 4a), comparable to the 0.35 V shift brought by the twenty fluorines at meso-phenyl groups (vs. −0.89 and −1.34 V for 5a). Besides, a third reduction peak appears at −1.66 V for 2a, indicating the substantially decreased electron density of 2a.
| Compound | E ox2 | E ox1 | E red1 | E red2 | E red3 |
|---|---|---|---|---|---|
| a Cyclic voltammetry conditions: compound concentration, 1 mM; sweep rate, 0.1 V s−1; 0.1 M nBu4PF6; Ag/AgCl reference. b Ref. 15. c Ref. 36. | |||||
| 2a | — | — | −0.23 | −0.71 | −1.66 |
| 4a | — | +1.61 | −0.54 | −0.99 | — |
| 5a | +1.54 | +1.30 | −0.89 | −1.34 | — |
|
Zn-1ab |
— | +1.70 | −0.63 | −1.04 | — |
| Zn-2a | — | +1.59 | −0.51 | −0.94 | — |
|
Zn-4ac |
+1.61 | +1.33 | −0.72 | −1.16 | — |
| Zn-5a | +1.10 | +0.87 | −1.14 | −1.54 | — |
Similar trends were observed in the corresponding zinc complexes. Compared to Zn-4a and Zn-5a, Zn-2a shows a positive shift in potentials for both the porphyrinato oxidation and reduction, implying a decreased electron density of the macrocyclic ring by fluorination at meso-phenyl and β-pyrrolic positions. For Zn-2a, there is only one oxidation peak at 1.59 V up to a potential of 1.8 V, whereas there are two reversible oxidative peaks for Zn-4a (1.61 and 1.33 V) and Zn-5a (1.10 and 0.87 V) respectively. The two reduction waves of Zn-2a (−0.51 and −0.94 V) are anodically shifted from those of Zn-4a (−0.72 and −1.16 V) and Zn-5a (−1.14 and −1.54 V). Therefore, Zn-2a is easily reduced and resistant to oxidation relative to Zn-4a and Zn-5a. Compared to Zn-1a, introducing the lactone moiety results in catholically shifted oxidation waves and anodically shifted reduction waves.
NIR emissive ytterbium complex with enhanced quantum yield
To demonstrate the potential application of β-fluorinated porpholactones, we used them as an antenna ligand for NIR emissive lanthanides. NIR emitters such as Er, Nd and Yb complexes have many attractive potential applications in telecommunications, light-emitting devices and biological imaging but suffer from low quantum yields.55–58 Our previous studies demonstrate that both porphyrin and porpholactone are good antenna ligands for Yb(III).59,60 Replacing β hydrogens with fluorine substituents significantly increases the NIR emission efficiency of Yb as a result of the minimized non-radiative process caused by C–H bond vibrational quenching.60 In this work, ytterbium complexes Yb-2a and Yb-4a, sandwiched by porpholactone and the partly deuterated Kläui ligand were prepared according to our previous procedure (Scheme 4).60–63 These complexes were characterized by NMR, HR-ESI-MS, IR, and UV-Vis spectrometry and single crystal X-ray analysis (CCDC 1540175 (Yb-2a) and 1540176 (Yb-4a), see details in the ESI†).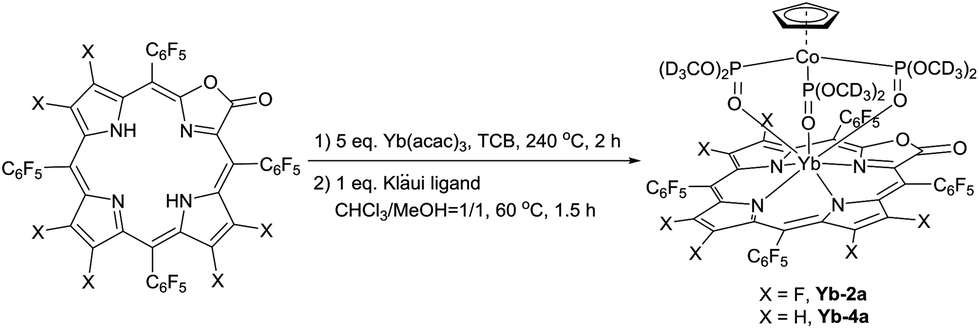 | ||
| Scheme 4 Synthesis of ytterbium porpholactone complexes. | ||
As shown in Fig. 4a, the luminescence lifetime (τ) of Yb-2a (525 μs) is much longer than that of Yb-4a (75 μs) in CD2Cl2. The emission quantum yield (Φ) of Yb-2a (58%) is also much higher than that of Yb-4a (6.8%) in CD2Cl2 (Fig. 4b, referenced to Yb(TPP)(LOEt), Φ = 2.4% in CH2Cl2, estimated uncertainty in Φ is 15%).64Yb-2a even shows comparable luminescence properties to our previously best results with Yb-1a (τ = 714 μs, Φ = 63% in CD2Cl2),60 indicating that β-lactonization little affects the sensitization ability of porphyrin towards Yb. It is worth noting that, for the easy modification of the β-oxazolone moiety demonstrated by Brückner and us,38,65–68Yb-2a is anticipated to introduce flexibility to functionalize the Yb complexes. On the other hand, the remarkably higher value of the luminescence lifetime and quantum yield of Yb-2a than that of Yb-4a demonstrates the key role of the C–F bond in improving the NIR emission quantum yield of Yb by suppressing the vibrational quenching.69
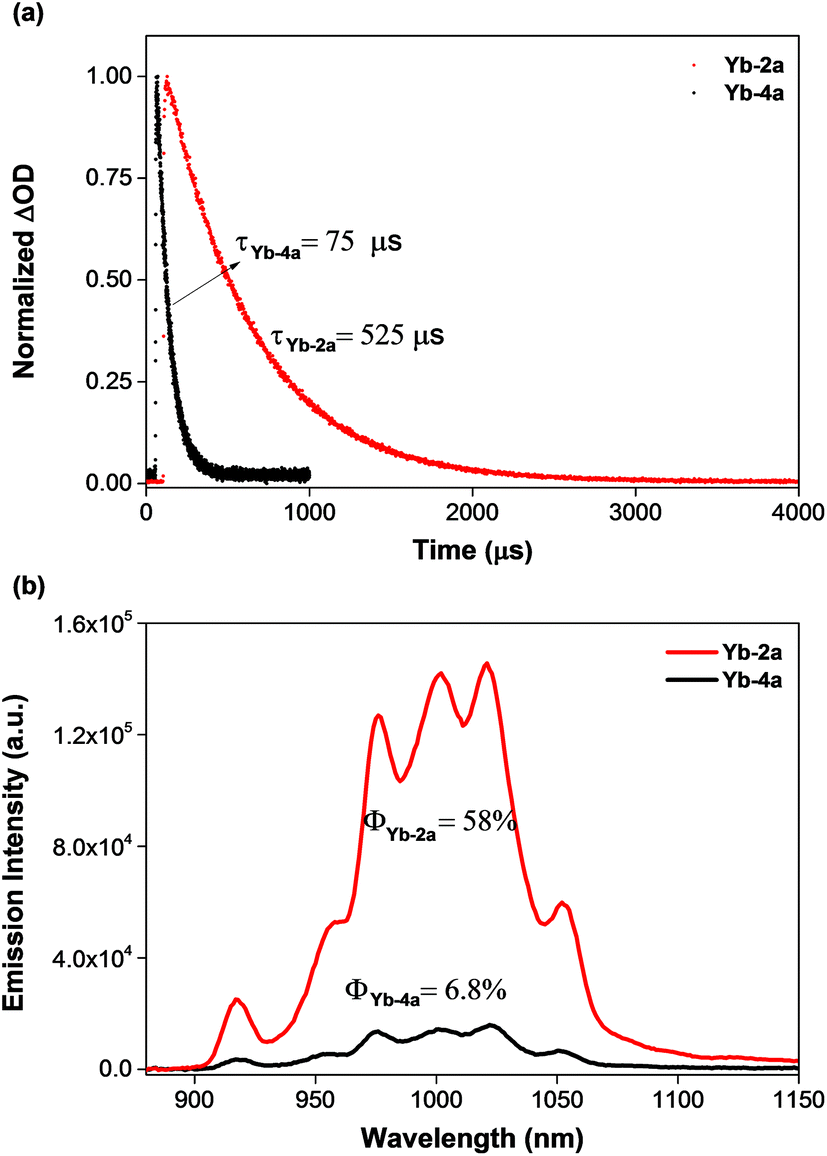 | ||
| Fig. 4 (a) Decay of the NIR luminescence in the ytterbium complexes in CD2Cl2; (b) emission spectra of the ytterbium complexes under the same conditions (λex = 425 nm, absorbance = 0.10) in CD2Cl2. The estimated uncertainty in quantum yields is 15%. Experimental relative errors: τ, ±2%; Φ, ±10%. | ||
Conclusion
In summary, we report here the synthesis of a new type of porpholactone with β-fluorine substituents, via the lactonization of β-fluorinated porphyrin. A high-valent ruthenium oxo intermediate generated from the protocol “RuCl3 + Oxone®” was proposed as the active intermediate in the cleavage of the pyrrolic C–F bond. The perfluorinated porpholactone 2a exhibits a similar coordination ability towards transition metals to porphyrin analogues. Comparative studies of photophysical and electrochemical properties with non-fluorinated counterparts show that β-fluorination leads to blue-shifted absorption bands and anodically shifted the redox potential of the porpholactone. As an example of the potential application of these molecules, we demonstrated that 2a is an excellent antenna ligand for sensitizing NIR-emissive ytterbium, achieving a high luminescence quantum yield and a long lifetime.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Basic Research Support Foundation of China (Grants 2015CB856301) and the National Scientific Foundation of China (Grants 21571007, 21271013 and 21321001).Notes and references
- B. Meunier, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS.
- D. Dolphin, T. G. Traylor and L. Y. Xie, Acc. Chem. Res., 1997, 30, 251–259 CrossRef CAS.
- C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC.
- A. Giraudeau, H. Callot, J. Jordan, I. Ezhar and M. Gross, J. Am. Chem. Soc., 1979, 101, 3857–3862 CrossRef CAS.
- H. Fujii, J. Am. Chem. Soc., 1993, 115, 4641–4648 CrossRef CAS.
- T. G. Traylor, C. Kim, J. L. Richards, F. Xu and C. L. Perrin, J. Am. Chem. Soc., 1995, 117, 3468–3474 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- N. V. S. D. K. Bhupathiraju, W. Rizvi, J. D. Batteas and C. M. Drain, Org. Biomol. Chem., 2016, 14, 389–408 CAS.
- P. Bhyrappa and V. Krishnan, Inorg. Chem., 1991, 30, 239–245 CrossRef CAS.
- T. G. Traylor and S. Tsuchiya, Inorg. Chem., 1987, 26, 1338–1339 CrossRef CAS.
- T. Wijesekera, A. Matsumoto, D. Dolphin and D. Lexa, Angew. Chem., Int. Ed. Engl., 1990, 29, 1028–1030 CrossRef.
- P. Ochsenbein, K. Ayougou, D. Mandon, J. Fischer, R. Weiss, R. N. Austin, K. Jayaraj, A. Gold, J. Terner and J. Fajer, Angew. Chem., Int. Ed. Engl., 1994, 33, 348–350 CrossRef.
- J. Leroy and A. Bondon, Eur. J. Org. Chem., 2008, 417–433 CrossRef CAS.
- S. G. DiMagno, J. C. Biffinger and H. Sun, in Fluorine in Heterocyclic Chemistry Volume 1: 5-Membered Heterocycles and Macrocycles, ed. V. Nenajdenko, Springer International Publishing, Cham, 2014, pp. 589–620 Search PubMed.
- E. K. Woller and S. G. DiMagno, J. Org. Chem., 1997, 62, 1588–1593 CrossRef CAS.
- J. Leroy, A. Bondon, L. Toupet and C. Rolando, Chem. – Eur. J., 1997, 3, 1890–1893 CrossRef CAS.
- H.-Y. Liu, T.-S. Lai, L.-L. Yeung and C. K. Chang, Org. Lett., 2003, 5, 617–620 CrossRef CAS PubMed.
- E. Steene, A. Dey and A. Ghosh, J. Am. Chem. Soc., 2003, 125, 16300–16309 CrossRef CAS PubMed.
- C. Kashi, C.-C. Wu, C.-L. Mai, C.-Y. Yeh and C. K. Chang, Angew. Chem., Int. Ed., 2016, 55, 5035–5039 CrossRef CAS PubMed.
- J. Leroy and C. Wakselman, Tetrahedron Lett., 1994, 35, 8605–8608 CrossRef CAS.
- E. K. Woller, V. V. Smirnov and S. G. DiMagno, J. Org. Chem., 1998, 63, 5706–5707 CrossRef CAS.
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- J. C. Biffinger, S. Uppaluri, H. Sun and S. G. DiMagno, ACS Catal., 2011, 1, 764–771 CrossRef CAS PubMed.
- M. J. Crossley and L. G. King, J. Chem. Soc., Chem. Commun., 1984, 920–922 RSC.
- M. Gouterman, R. J. Hall, G. E. Khalil, P. C. Martin, E. G. Shankland and R. L. Cerny, J. Am. Chem. Soc., 1989, 111, 3702–3707 CrossRef CAS.
- C. Brückner, J. Ogikubo, J. R. McCarthy, J. Akhigbe, M. A. Hyland, P. Daddario, J. L. Worlinsky, M. Zeller, J. T. Engle, C. J. Ziegler, M. J. Ranaghan, M. N. Sandberg and R. R. Birge, J. Org. Chem., 2012, 77, 6480–6494 CrossRef PubMed.
- H. Lv, B. Yang, J. Jing, Y. Yu, J. Zhang and J.-L. Zhang, Dalton Trans., 2012, 41, 3116–3118 RSC.
- Y. Yu, H. Lv, X. Ke, B. Yang and J.-L. Zhang, Adv. Synth. Catal., 2012, 354, 3509–3516 CrossRef CAS.
- M. Sharma, E. Meehan, B. Q. Mercado and C. Brückner, Chem. – Eur. J., 2016, 22, 11706–11718 CrossRef CAS PubMed.
- C. Brückner, Acc. Chem. Res., 2016, 49, 1080–1092 CrossRef PubMed.
- L. Costa, J. Costa and A. Tomé, Molecules, 2016, 21, 320 CrossRef PubMed.
- L. Liang, H. Lv, Y. Yu, P. Wang and J.-L. Zhang, Dalton Trans., 2012, 41, 1457–1460 RSC.
- W.-P. To, Y. Liu, T.-C. Lau and C.-M. Che, Chem. – Eur. J., 2013, 19, 5654–5664 CrossRef CAS PubMed.
- Y. Ishigami, W. Waskitoaji, M. Yoneda, K. Takada, T. Hyakutake, T. Suga, M. Uchida, Y. Nagumo, J. Inukai, H. Nishide and M. Watanabe, J. Power Sources, 2014, 269, 556–564 CrossRef CAS.
- J. Tang, J.-J. Chen, J. Jing, J.-Z. Chen, H. Lv, Y. Yu, P. Xu and J.-L. Zhang, Chem. Sci., 2014, 5, 558–566 RSC.
- X.-S. Ke, Y. Chang, J.-Z. Chen, J. Tian, J. Mack, X. Cheng, Z. Shen and J.-L. Zhang, J. Am. Chem. Soc., 2014, 136, 9598–9607 CrossRef CAS PubMed.
- J. L. Worlinsky, S. Halepas, M. Ghandehari, G. Khalil and C. Brückner, Analyst, 2015, 140, 190–196 RSC.
- X.-S. Ke, J. Tang, J.-J. Chen, Z.-Y. Zhou and J.-L. Zhang, ChemPlusChem, 2015, 80, 237–252 CrossRef CAS.
- Z.-Y. Wu, T. Wang, Y.-S. Meng, Y. Rao, S. Gao, J. Zheng, B.-W. Wang and J.-L. Zhang, Chem. Sci., 2017 10.1039/C7SC02073B.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef PubMed.
- J.-Y. Hu and J.-L. Zhang, Top. Organomet. Chem., 2015, 52, 143–196 CrossRef CAS.
- S. Sahu, M. G. Quesne, C. G. Davies, M. Dürr, I. Ivanović-Burmazović, M. A. Siegler, G. N. L. Jameson, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2014, 136, 13542–13545 CrossRef CAS PubMed.
- J. Serrano-Plana, I. Garcia-Bosch, R. Miyake, M. Costas and A. Company, Angew. Chem., Int. Ed., 2014, 53, 9608–9612 CrossRef CAS PubMed.
- C. Colomban, E. V. Kudrik, P. Afanasiev and A. B. Sorokin, J. Am. Chem. Soc., 2014, 136, 11321–11330 CrossRef CAS PubMed.
- S. Sahu, B. Zhang, C. J. Pollock, M. Dürr, C. G. Davies, A. M. Confer, I. Ivanović-Burmazović, M. A. Siegler, G. N. L. Jameson, C. Krebs and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 12791–12802 CrossRef CAS PubMed.
- G. de Ruiter, N. B. Thompson, M. K. Takase and T. Agapie, J. Am. Chem. Soc., 2016, 138, 1486–1489 CrossRef CAS PubMed.
- C. Brückner, S. J. Rettig and D. Dolphin, J. Org. Chem., 1998, 63, 2094–2098 CrossRef.
- J. R. McCarthy, H. A. Jenkins and C. Brückner, Org. Lett., 2003, 5, 19–22 CrossRef CAS PubMed.
- K. K. Lara, C. R. Rinaldo and C. Brückner, Tetrahedron, 2005, 61, 2529–2539 CrossRef CAS.
- S. Andreades and D. C. England, J. Am. Chem. Soc., 1961, 83, 4670–4671 CrossRef CAS.
- C. J. Willis, Coord. Chem. Rev., 1988, 88, 133–202 CrossRef CAS.
- W. A. Herrmann, S. J. Eder and W. Scherer, Angew. Chem., Int. Ed. Engl., 1992, 31, 1345–1347 CrossRef.
- S.-W. Lai, Y.-J. Hou, C.-M. Che, H.-L. Pang, K.-Y. Wong, C. K. Chang and N. Zhu, Inorg. Chem., 2004, 43, 3724–3732 CrossRef CAS PubMed.
- X.-S. Ke, H. Zhao, X. Zou, Y. Ning, X. Cheng, H. Su and J.-L. Zhang, J. Am. Chem. Soc., 2015, 137, 10745–10752 CrossRef CAS PubMed.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS.
- X.-S. Ke, B.-Y. Yang, X. Cheng, S. L.-F. Chan and J.-L. Zhang, Chem. – Eur. J., 2014, 20, 4324–4333 CrossRef CAS PubMed.
- J.-Y. Hu, Y. Ning, Y.-S. Meng, J. Zhang, Z.-Y. Wu, S. Gao and J.-L. Zhang, Chem. Sci., 2017, 8, 2702–2709 RSC.
- C.-P. Wong, R. F. Venteicher and W. D. Horrocks Jr., J. Am. Chem. Soc., 1974, 96, 7149–7150 CrossRef CAS PubMed.
- W.-K. Wong, A. Hou, J. Guo, H. He, L. Zhang, W.-Y. Wong, K.-F. Li, K.-W. Cheah, F. Xue and T. C. Mak, J. Chem. Soc., Dalton Trans., 2001, 3092–3098 RSC.
- H. He, J. Guo, Z. Zhao, W. K. Wong, W. Y. Wong, W. K. Lo, K. F. Li, L. Luo and K. W. Cheah, Eur. J. Inorg. Chem., 2004, 2004, 837–845 CrossRef.
- T. J. Foley, B. S. Harrison, A. S. Knefely, K. A. Abboud, J. R. Reynolds, K. S. Schanze and J. M. Boncella, Inorg. Chem., 2003, 42, 5023–5032 CrossRef CAS PubMed.
- J. Akhigbe, J. Haskoor, M. Zeller and C. Bruckner, Chem. Commun., 2011, 47, 8599–8601 RSC.
- J. Ogikubo, E. Meehan, J. T. Engle, C. J. Ziegler and C. Brückner, J. Org. Chem., 2012, 77, 6199–6207 CrossRef CAS PubMed.
- Y. Yu, B. Czepukojc, C. Jacob, Y. Jiang, M. Zeller, C. Brückner and J.-L. Zhang, Org. Biomol. Chem., 2013, 11, 4613–4621 CAS.
- X.-S. Ke, J. Tang, Z.-S. Yang and J.-L. Zhang, J. Porphyrins Phthalocyanines, 2014, 18, 950–959 CrossRef CAS.
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293, 19–47 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, complete characterization data, and NMR, IR, ESI-HRMS spectra. CCDC 1501201, 1540175 and 1540176. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qi00375g |
| This journal is © the Partner Organisations 2017 |