
Shining a light on an adaptable photoinitiator: advances in photopolymerizations initiated by thioxanthones
Sajjad
Dadashi-Silab
a,
Cansu
Aydogan
a and
Yusuf
Yagci
*ab
aDepartment of Chemistry, Istanbul Technical University, 34469 Maslak, Istanbul, Turkey. E-mail: yusuf@itu.edu.tr; Fax: +90-2122856386; Tel: +90-212 285 3241
bCenter of Excellence for Advanced Materials Research (CEAMR) and Department of Chemistry, Faculty of Science, King Abdulaziz University, 21589, Jeddah, Saudi Arabia
First published on 24th July 2015
Abstract
Photochemistry plays a central role in synthetic polymer research. Aromatic ketones, examples of which include benzophenone, thioxanthone, camphorquinone, among others, are renowned for their excellent optical characteristics and have been extensively utilized for photochemical induction of polymerization processes. Of particular interest is thioxanthone due to its adaptability for bearing different functionalities and its applications in various modes of photopolymerization, in which it accomplishes photoinitiation in conjunction with other co-initiator compounds; a behavior that is referred to as bi-molecular photoinitiation. In this paper, we review the photochemistry of thioxanthone-based systems and their use in different modes of photoinitiated polymerizations. Citing examples from the literature, the development of various photoinitiating systems based on thioxanthones along with an understanding of their mechanistic behavior has been elucidated previously.
 Sajjad Dadashi-Silab | Sajjad Dadashi-Silab obtained his M.Sc. degree in Chemistry from Istanbul Technical University in 2015, studying under the supervision of Professor Yusuf Yagci. In 2012, he received his B.Sc. degree in Polymer Engineering from Amirkabir University of Technology, Tehran. His graduate research studies were mainly focused on developing new photoinitiating systems for controlled and conventional radical photopolymerizations based on heterogeneous, semiconducting photocatalysis. |
 Cansu Aydogan | Cansu Aydogan received her B.Sc. degree in Chemistry from Marmara University, in 2011. She completed her M.Sc. studies at the Department of Chemistry in Istanbul Technical University under the supervision of Professor Yusuf Yagci, in 2014. Her research in the Yagci group was essentially focused on benzoxazine chemistry during her M.Sc. She is currently continuing her PhD studies dealing with photoinitiated polymerizations. In particular, thioxanthone-based photoinitiating systems, development of novel polymeric materials, and the synthesis of hyperbranched polymers are the main interests. |
 Yusuf Yagci | Yusuf Yagci is a professor of Chemistry at Istanbul Technical University (ITU). He received his M.Sc. (1977) and PhD (1979) degrees from Liverpool University (UK). He joined ITU in 1980 where he has remained. He is the recipient of several awards including The Society of Polymer Science, Japan (SPSJ) International Award (2008), the Elginkan Foundation Technology Award (2008), OIC Ministerial COMSTEC Award (2010), and Mustafa Parlar Honorary Award (2013) He was also a member of the Turkish Academy of Sciences (1997–2011). His research interests include all aspects of synthetic polymer chemistry involving photopolymerization and controlled polymerization methods. |
Introduction
The polymer community has witnessed an increasing inclination towards light-mediated polymerization techniques in recent years. These systems essentially offer the advantage of carrying out reactions efficiently by eliminating the side effects often accompanying the thermally induced counterpart processes of special systems, just to cite one striking example.1,2 For this reason, photochemical processes are often considered as “green chemistry”.1,2 Such systems utilize light as the driving force to form suitable initiating sites by absorbing its energy and thereby promoting subsequent photochemical reactions leading to the induction of chemical reactions. Hence, choosing appropriate light absorbing species, i.e. photoinitiators or photosensitizers, becomes crucial since there has to be good matching of the optical behavior of the photoinitiators with the characteristics of the light source. Photochemical processes have been intensively used in a diverse range of chemical reactions including organic synthesis and materials science, medicinal applications, photocuring and photopolymerization systems.As the subject of this article, numerous attempts in the area of photoinitiated polymerizations have been undertaken to design new photoinitiating systems, or enhance, improve and expand the scope of existing initiators to fit the many newly emerging needs of advanced technologies.3 Various types of photoinitiating systems are being developed in academia and applied in industry. Photoinitiators, particularly those involving free radicals and related systems, have been mainly classified in two categories according to their optical behavior: (i) Type I, which are also known as α-cleavage, provide initiating radicals by bond cleavage processes upon absorption of light; (ii) Type II undergo photoexcitation followed by an electron or hydrogen transfer process and consequently form initiating species.4–6 Benzoin and its derivatives are the most widely used Type I photoinitiators for radical photopolymerization and are capable of unimolecular bond cleavage on irradiation. As Type II photoinitiators, ketone components are of great importance. Examples include benzophenone, thioxanthone, camphorquinone, etc., which exhibit bimolecular photo-behavior in the formation of initiating species. This characteristic behavior arises primarily from photoexcitation of the photoinitiator and its excited state interaction with other components called co-initiators through various transfer processes. Type II photoinitiators can be considered advantageous compared with Type I as in most cases the energy required for the photoinitiator to undergo bond cleavage is generally high and it necessitates the use of high-energy light sources, i.e. with short wavelengths. However, Type II photoinitiators exhibit absorption characteristics at higher wavelengths and can be designed and decorated to manipulate their optical behavior to extend their spectral sensitivity up to the visible range of the electromagnetic spectrum. Such a behavior carries the advantage of using low energy light sources, thus decreasing the cost of processes. Additionally, it provides the possibility of conducting photopolymerization reactions in those systems that are highly sensitive to high-energy exposure, such as those in biological and medical applications. Ultimately, in designing a photoinitiating system, the influence of the special groups used to functionalize the photoinitiators to enhance their photoactivity and lower the cost of the process should be carefully taken into consideration. More important is the influence and critical role of co-initiators which in conjunction with photoinitiators bring about initiating species. Excited state photoinitiators interact with co-initiators according to their redox potentials through reduction or oxidation processes and hence one would expect different excited state interactions in the presence of different types of co-initiator systems.
Traditionally, photoinitiated polymerizations have been employed in free radical,7,8 cationic9,10 and, in rare cases, anionic11 polymerization systems. Applications can be found across a broad spectrum of photocuring, coating, and inking and printing applications, and in medicinal and dental adhesive applications, to fabrication of multidimensional devices and so forth. Despite the tremendous breadth of implementations of these systems, they somehow all lack the ability to provide control of the polymerization process in terms of well-controlled, complex architectures with predetermined molecular weight properties. Recently, attention has been drawn to the adaptation of light-mediated processes with existing controlled polymerizations such as copper-mediated living radical polymerization techniques,12–15 reversible addition-fragmentation chain transfer,16,17 nitroxide-mediated polymerization18 and so on, or developing novel photocontrolled techniques to achieve photochemically mediated synthesis of well-controlled macromolecular architectures.19–23 For example, many attempts have been directed towards photochemical induction of the copper-mediated processes, which rely on the photochemical formation of the required copper(I) catalyst by various photochemical means.24 An important application of such photoinitiation controlled polymerization systems may be the possibility of on-demand-patterning of functionalized surfaces by providing spatiotemporal control.25
Our group has long been dealing with investigating, designing, and developing novel photochemical processes for polymer synthesis.26–28 This review intends to focus on the advent of and progress in photoinitiated polymerization techniques mediated by thioxanthone (TX) and its derivatives. TX-based photoinitiators are an efficient class of photoinitiators widely used in many free radical and cationic polymerization processes. In the first section of this article, we will describe the photochemistry of TX and analogous structures in the formation of suitable initiating sites. The following sections will give a comprehensive picture of the advent and development of various TX-based photoinitiating systems including the use of various co-initiator systems, one-component and polymeric photoinitiators. A special focus will be directed towards the mechanistic behavior of each system and their applications for different purposes in macromolecular synthesis. In addition to polymerization systems, in the last section, some miscellaneous applications of TX photosensitizers in different areas of chemistry, materials science and biology will be discussed.
Photochemistry of thioxanthone: a bimolecular photoinitiator
Photoinitiators are mainly subdivided into the two categories of Type I and Type II systems. Whilst the formation of radicals in the latter is achieved by a hydrogen abstraction or photoinduced electron transfer processes of the triple state photoinitiator in the presence of a co-initiator compound, the former undergoes unimolecular fragmentation (α- or β cleavage) upon absorption of light giving rise to the formation of two initiating radicals (Scheme 1).29 Depending on the nature of the initiating components, the formation of initiating species in Type II systems may well occur through distinct electron transfer reactions or hydrogen abstraction, or a combination of both processes. In the presence of hydrogen-donor co-initiators, like amines, it is mainly believed that an electron transfer process between the triplet state photoinitiator acting as the electron acceptor and the co-initiator as the electron donor primarily forms an ion-pair intermediate of a TX radical anion and a donor radical cation excited state complex (exciplex). Thus, a proton transfer from the α-position of the co-initiator to the photoinitiator results in the formation of a ketyl radical derived from the photoinitiator and a radical derived from the co-initiator. Direct abstraction of hydrogen is also possible as in alcohols, ethers and other compounds. Heteroatom-containing co-initiators have been proven to be efficient in the promotion of this process. Amines, thiols, alcohols, ethers, silanes and many others are among the most used co-initiators in such systems with tertiary amines being somehow the most efficient hydrogen donor. Due to the resonance stabilization and steric reasons, the highly conjugated ketyl radicals formed in the aftermath of hydrogen abstraction do not initiate polymerization, but participate in the combination or termination of the growing polymer chains. However, radicals derived from the hydrogen donor compounds are efficient in the initiation process.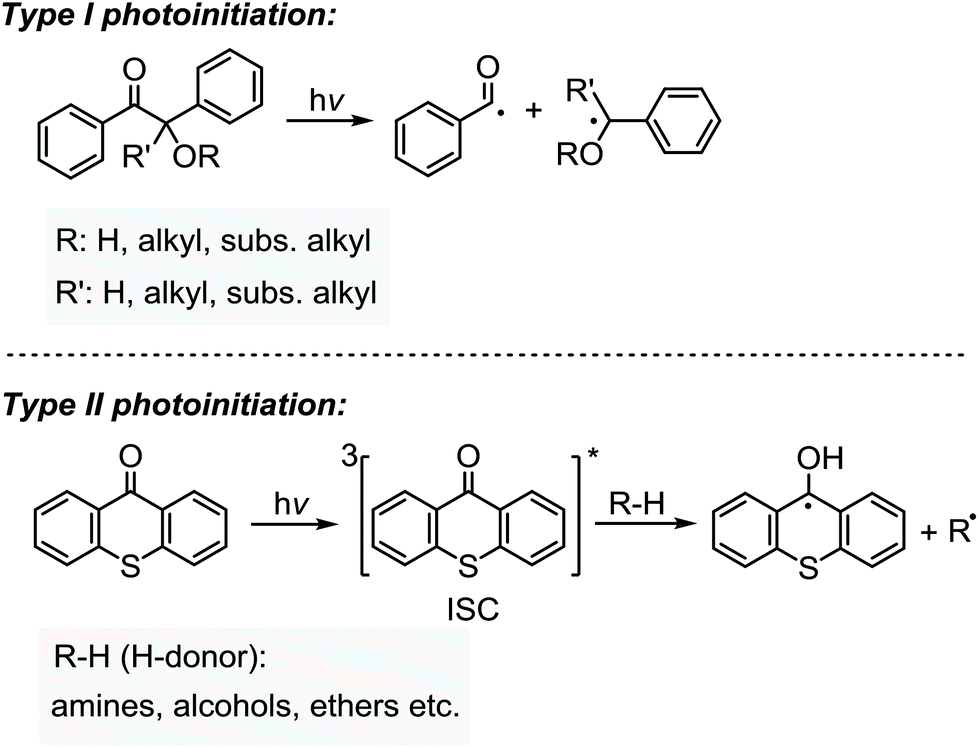 | ||
| Scheme 1 Typical representation of the photolysis of the radical photoinitiators based on Type I (top) and Type II (bottom) systems with the examples of a benzoin derivative and thioxanthone, respectively. | ||
Another possibility for the formation of initiating species is the photoinduced electron transfer reaction of a triplet state photoinitiator acting as photosensitizer, with a suitable co-initiator, in which, after a set of subsequent electron transfer and fragmentation reactions, an initiating species including both radicals and/or cations is formed. In this case, an excited state photosensitizer can undergo both oxidation or reduction reactions according to the nature of the co-initiator present and the energy of the absorbed photon. Scheme 2 illustrates the general mechanism of the photoinduced electron transfer reactions of photosensitizers and co-initiators. The excited state photosensitizer is capable of being both more oxidizing or more reducing than the ground state photosensitizer and can be quenched by the respective oxidation or reduction processes. In the oxidative quenching cycle, the excited photosensitizer acts as a reductant as an electron acceptor compound giving rise to the formation of radical anion and radical cation components. In the reductive quenching cycle, however, the excited photosensitizer acts as an oxidant and oxidizes an electron donor compound resulting in the formation of photosensitizer radical anion and co-initiator radical cation species. These radical ions can either directly initiate polymerization or, in most cases, undergo further fragmentation reactions bringing about active initiating species. There is also the possibility of reacting the radical ions with some electron donor or electron acceptor to give the ground state photosensitizer and also the corresponding radical or cation species.
 | ||
| Scheme 2 General representation of photoinduced electron transfer reactions of photosensitizers (PS) in the presence of electron acceptors (A) or electron donors (D). | ||
These reactions, of course, are thermodynamically feasible if the free energy change of electron transfer (ΔGet) estimated by the Rehm–Weller equation is negative:
| ΔGet = fc[Eox1/2(D/D˙+) − Ered1/2(A/A˙−)] − Es + ΔEc |
In the following sections, we will deliberate in detail the performance and efficiency of photoinitiated polymerizations by TX derivatives in conjunction with various co-initiator compounds by focusing on the mechanistic explanation of the initiation processes and photoinitiator/co-initiator.
Two-component photoinitiation
TX-based photoinitiators have become a preferable class of photoinitiators over other similar structures such as benzophenones, primarily because of their spectral characteristics. Their absorption maxima appear in the range of 380–420 nm, laying in the near UV and visible ranges, which reduces the required energy for photoexcitations and subsequent formation of initiating radicals. Although various synthesis methods for TX derivatives have been reported, the usually applied method is based on a simple condensation process of thiosalicylic acids with aromatic hydrocarbons in a concentrated sulfuric acid medium as shown in Scheme 3.30 | ||
| Scheme 3 Synthesis of thioxanthones by condensation of thiosalicylic acid and aromatic compounds in the presence of concentrated sulfuric acid. | ||
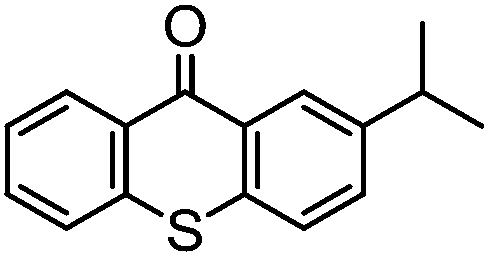
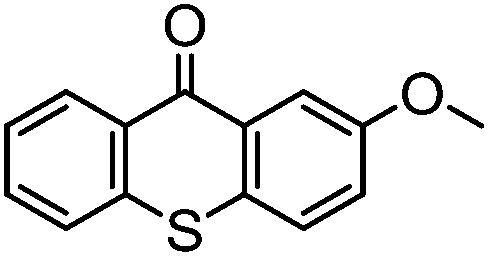
By employing functional components during the synthesis, substituted functional TX compounds can easily be obtained. Substitution of different functionalities can considerably affect the characteristics and optical behavior of TX photoinitiators. These may include further shifting of the absorption band to higher wavelengths, as well as facilitating the photoinitiators’ applicability in different media and for various purposes, or enhancement of the photoactivity of the initiator (Table 1). From the practical point of view, it is thought that some drawbacks such as yellowing of the final cured products can be considerably suppressed by using a TX-based photoinitiating system.4,6
| TX derivative | λ max (nm) | 10−6 × kq (mol−1 L s−1) H-donor |
|---|---|---|
| a BMA: bis(2-hydroxyethyl)methylamine; TEA: triethylamine; DEAE: 2-(diethylamino)ethanol; data taken from ref. 31, 33 and 42. | ||

|
∼378 | 8000 (BMA) |
| 4600 (DEAE) | ||

|
∼384 | 6000 (BMA) |
|
∼386 | 6000 (BMA) |

|
∼400 | Rapid self-quenching by hydroxyl group |
|
∼396 | 580 (DEAE) |

|
∼385 | 6000 (BMA) |
| 2500 (TEA) | ||
Through exploiting various spectroscopic techniques such as laser flash photolysis studies, the mechanism of TX-photoinitiated polymerization has been extensively investigated.31–40 The process essentially necessitates the presence of a co-initiator to accomplish the formation of initiating species; though quenching can also occur by interaction with low triplet energy monomers or solvents. Amines, for instance, due to their highly reducing properties41 are commonly used as co-initiators to carry out quenching of triplet TX through the formation of an exciplex or ion pair intermediate between the photoexcited triplet state TX and co-initiator amine, primarily by an electron transfer from the amine to TX which is followed by a proton transfer to the reduced photoinitiator TX. This brings about a free radical on the α-position of the co-initiator and a thioxanthyl ketyl radical is formed as well. It was found that the photoactivity of TX-based photoinitiators is highly dependent on the substitution pattern of the photoinitiator as well as the nature of the media and other co-compounds present in the system. Evidently, the presence and type of functionalities can considerably affect the absorption maxima, extinction coefficients and quantum yields of TX during the photoinitiation process. Most importantly, the nature of co-initiators governs the process by which initiation can occur. Table 1 lists some of the studied TX derivatives to illustrate the effect of substitution on the absorption maxima. TX derivatives seem to be more efficacious compared with unsubstituted TX in terms of higher absorption maxima and higher extinction co-efficients and applicability for various purposes.
As for the thioxanthyl ketyl radicals formed after the course of hydrogen abstraction, they are believed to rarely add across a monomer and initiate polymerization. Instead, they tend to undergo either a process of disproportionation to return the initial ground state TX and form a thioxanthole species or couple to form a pinnacol compound, though with a reduced probability due to the steric factors. Termination of the growing radical chains during the polymerization by the thioxanthyl ketyl radicals has also been reported as one of the reaction possibilities.43Scheme 4 depicts the possible reactions of thioxanthyl ketyl radicals.
 | ||
| Scheme 4 Possible reactions of thioxanthyl ketyl radicals. | ||
Co-initiators
The efficiency of the photoinitiation of polymerization assisted by TX derivatives is chiefly governed by both the properties of the TX photoinitiator as well as by the characteristics of the co-initiator used and its affinity to undergo electron or hydrogen transfer processes. Hydrogen donor compounds such as amines, thiols, alcohols etc. have been utilized as co-initiators in quenching the photoexcited triplet state TX and formation of free radicals. Of these, amines have been proven the most efficient hydrogen donor. The hydrogen atom at the α-position of a heteroatom (N, O, S) is highly likely to be abstracted by the triplet TX and go on to form initiating radicals. The following section gives a comprehensive picture of the types and efficiency of co-initiators used in the presence of TX derivatives with mechanistic explanation of the interaction and initiation processes. | ||
| Scheme 5 Photoreduction of triplet thioxanthone by amines. | ||
 | ||
| Chart 1 A list of frequently used amine co-initiators in combination with thioxanthone-based photoinitiators. | ||
 | ||
| Chart 2 Benzoxazines as co-initiators in Type II photoinitiated polymerization. | ||
| 3ITX* + R–SH → [ITX˙−⋯HS˙+–R] → ITX˙ + R–S˙ |
Lalevee and co-workers also have investigated the efficiency of thiols with different functionalities and disulfide components as co-initiators together with various photosensitizers and photoinitiators.55,56 It was suggested that an energy transfer process between the triplet state TX and the disulfide compound led to the dissociation of the S–S bond, which brought about two sulfur-centered initiating radicals derived from the disulfide:
| 3ITX* + R–S–S–R → ITX + 3R–S–S–R → ITX + 2R–S˙ |
It should also be noted that the irradiation of disulfides, though at lower wavelengths, could result in the dissociation of the S–S bond, albeit the efficiency was comparatively low as in the case of the photosensitizer/disulfide bi-component system. In the case of thiols, however, the hydrogen abstraction process, as shown above, accounted for the formation of thiyl radicals. Chart 3 shows a list of thiol and disulfide-based co-initiators used for the photoinitiation of free radical polymerizations.
 | ||
| Chart 3 Thiol and disulfide compounds used as co-initiators in photoinduced free radical polymerization in the presence of thioxanthones. | ||
Thiols have also been recognized for their part in thiol–ene reactions in the formation of polymers and networks in a radical-mediated step growth-like polymerization method. The thiol–ene polymerization mechanism is based on propagation and chain transfer processes between the thiol and double bond functional groups.57,58 Abstraction of a hydrogen atom from the participating thiol by a carbon-centered radical, results in the formation of thiyl radicals capable of adding to a double bond. There follows a radical transfer process from the carbon-centered radical to the thiol functionality. These propagation and chain transfer processes encompass the general mechanism of thiol–ene polymerization. In the photochemical process, the required initial radicals are generated by using various photoinitiator systems. Type I photoinitiators provide radicals by cleaving upon irradiation whereas in the case of Type II systems the interaction of the photoexcited triplet state photoinitiator with the thiol functionality results in hydrogen abstraction from the thiol and subsequently formation of thiyl radicals. Initial investigations in this area were carried out by Morgan and Ketley who used benzophenone as a photoinitiator to form photocured polymers of polyenes and polythiols; the photoexcited benzophenone abstracted the labile hydrogen from the thiol to generate a thiyl radical leading to crosslinking polyene and polythiol.59 Decker et al. studied photo-crosslinking of poly(styrene-co-butadiene) rubber with double bonds on the backbone (alkene) or pendant double bonds (vinyl) using the thiol–ene polymerization method in the presence of a multifunctional thiol and a photoinitiator.60 The reactivity of thiyl radicals to copolymerize with the pendant vinyl groups was found to be 10 times higher than the copolymerization with butene double bonds (Scheme 6).
 | ||
| Scheme 6 Schematic representation of photo-crosslinking of polymers containing double bonds by using multifunctional thiols. | ||
This kind of thiol–ene reaction is well known as an example of click chemistry that can be triggered via either thermal, photo, or redox processes. However, photochemical processes are preferred over the other protocols due to the advantage of spatial and temporal control over the thiol–ene addition process.28,61Scheme 7 outlines the overall mechanism of the thiol–ene click chemistry. Yagci and co-workers thoroughly investigated the influence of the type of photoinitiator on the photoinitiation of thiol–ene click chemistry for functionalization of polymers.62 Thiol- or allyl-modified polystyrenes with controlled molecular weight properties were successfully functionalized by an appropriate alkene or thiol functionality in the presence of either Type I or Type II photoinitiators with excellent yields in both systems. Type I photoinitiation appeared to be slightly more efficient than the other one as slightly higher conversion yields were obtained.
 | ||
| Scheme 7 Light-induced thiol–ene click reaction. | ||
 | ||
| Scheme 8 Proposed mechanism of quenching triplet thioxanthone by phenolic co-initiators. | ||
 | ||
| Scheme 9 Phosphorus-containing compounds as co-initiators in Type II photoinitiated polymerization. | ||
 | ||
| Chart 4 Germane and stannane co-initiators. | ||
These radicals were efficient in the photoinitiation of both free radical polymerization and free radical promoted cationic polymerization. In the latter case, the resulting radicals were oxidized to form active cationic species with the help of an iodonium salt to initiate the cationic polymerization.81–84
Taking diphenyliodonium, triphenylsulfonium, and N-ethoxy-2-methylpyridinium as examples of commonly used onium salts, the Gibbs free energy for electron transfer with TX calculated by the equation above is given in Table 2. Iodonium and pyridinium salts appear to be more efficient than sulfonium salts as the energy of electron transfer in sulfonium salts is rather positive.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
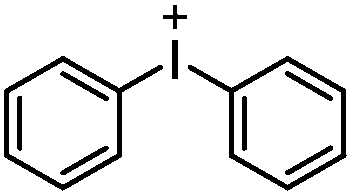


A wide range of efficient onium salts and photosensitizers were developed by Crivello, Fouassier, and Yagci. Mechanistic investigations of quenching triplet TX-based photosensitizers by onium salts further proved the involvement of the electron transfer process and formation of TX radical cation and onium radical species. Additionally, in the presence of a hydrogen-donor, the radical cation can abstract a hydrogen atom yielding a Brønsted acid and ground state photosensitizer (Scheme 10). These radical cation or Brønsted acid species are believed to initiate the cationic polymerization of the related monomers including cyclohexene oxide, isobutyl vinyl ether, tetrahydrofuran, N-vinylcarbazole etc. In addition to TX derivatives, other triplet photosensitizers such as benzophenone, anthracene, perylene, phenothiazine, dyes and analogous structures have been shown to undergo a similar quenching with onium salts.86–96 Some prominent onium salts which have been employed in photoinitiated polymerizations are depicted in Chart 5.
 | ||
| Scheme 10 Photosensitized cationic polymerization using onium salts. | ||
 | ||
| Chart 5 Some onium salts utilized in photopolymerization initiated by Type II photoinitiators. | ||
Onium salts have also appeared in three-component photoinitiating systems consisting of a photosensitizer, an electron donor (mostly an amine or a silane), and an electron acceptor (onium salt) component.97–104 The three-component photoinitiation systems are aimed to enhance the rate and efficiency of the photopolymerization process in two simultaneously occurring reaction pathways. Direct interaction of the onium salt with the triplet photosensitizer, as mentioned above, generates a neutral onium-centered radical and a photoinitiator radical cation by an electron transfer process, which can then correspondingly initiate free radical or cationic polymerizations. This also requires the simultaneous interaction of the amine co-initiator with the triplet photoinitiator, which forms an initiating radical through sequential electron and proton transfer reactions. The second pathway, on the other hand, involves participation of all three initiating components wherein two of the three components react with one another after which the third component then reacts with the resultant of the prior reaction. Plainly, the radical cation formed in the course of the electron transfer between onium salt and photoinitiator can abstract a hydrogen atom from the amine co-initiator to form an aminoalkyl radical while regenerating the ground state photosensitizer.
The reverse also holds true. The irreversible oxidation of the generated radicals in amine–photoinitiator interactions, especially non-propagating ketyl radicals, by onium salt leads to a neutral onium radical for the initiation of radical polymerization as well as a protonic acid capable of initiating cationic polymerization while regenerating the initial ground state photosensitizer. Both free radical101,105,106 and cationic84,107,108 polymerizations are applicable in three-component photoinitiation systems since they form appropriate active radical and cation centers to initiate target radical or cationic polymerizations. Regeneration of the initial photosensitizer, the possibility of the formation of both radical and cationic species by various interactions for the initiation of different modes of polymerization, and converting terminating agents to active initiating sites (oxidation of ketyl radicals to form initiating radical or cationic species) all make three-component photoinitiation systems highly efficacious and promising for many target applications. Scheme 11 illustrates the reaction mechanisms involved in the three-component photoinitiation systems of free radical and cationic polymerizations.
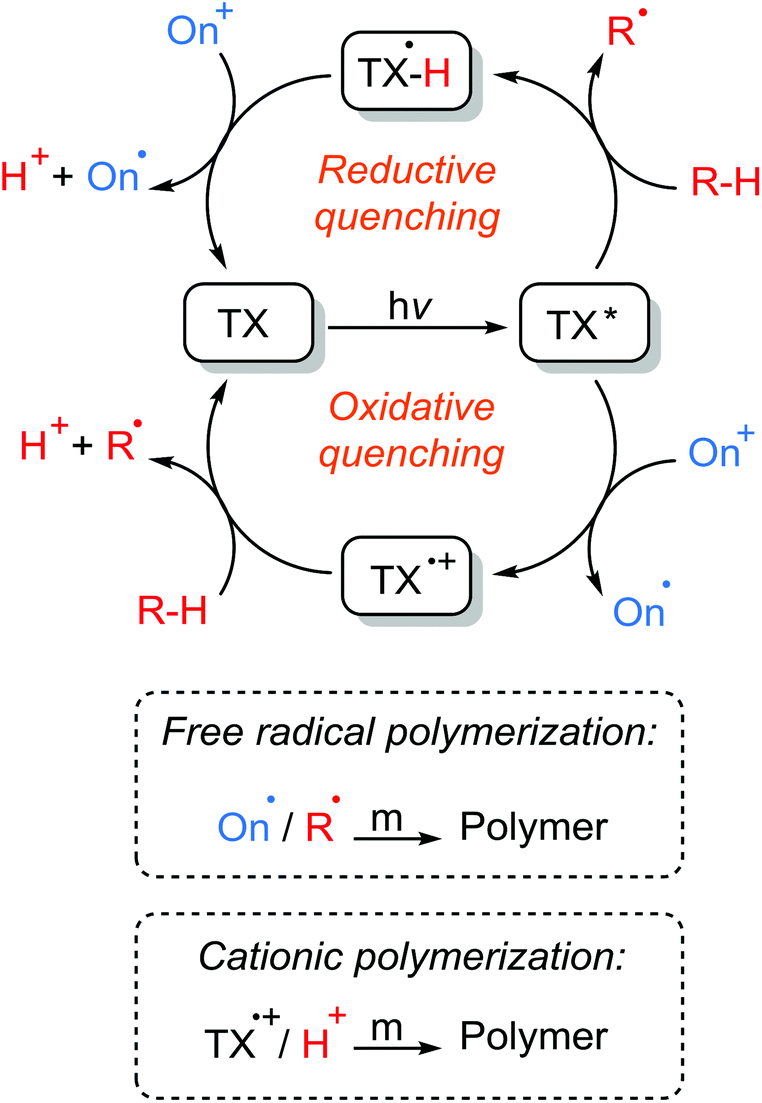 | ||
| Scheme 11 Three-component photoinitiation of free radical and/or cationic polymerizations in the presence of thioxanthone photosensitizer, amine hydrogen donor (R–H), and onium salt (On+) components: triplet thioxanthone is quenched through either oxidization by the onium salt or reduction by the amine hydrogen donor. | ||
One-component photoinitiation
The research focus in Type II photoinitiation has been to design new classes of photoinitiators capable of acting as both photoinitiator and co-initiator at the same time. Chemical incorporation of various co-initiators into the structure of photoinitiators makes one-component photoinitiators exhibit double functionality. Without the need for additional co-initiators, one-component Type II photoinitiators form initiating species through intramolecular and/or intermolecular interactions between the triplet state chromophore core and co-initiator part of the photoinitiator. Some disadvantages have been encountered using individual co-initiators, which may cause strong odors or yellowing and migration of the photolysis byproducts in cured films. These disadvantages associated with additional co-initiators can be suppressed to some extent by linking the co-initiator functionality to the photoinitiator structure. In this regard, amine-, thiol-, and ether-like hydrogen donors have been incorporated into photoinitiator structures to make one-component photoinitiating systems.As part of preliminary work in one-component TX photoinitiators, 2-mercaptothioxanthone (TX-SH), a thiol substituted derivative of TX, has been extensively utilized as an efficient one-component Type II photoinitiator.109–111 It was synthesized by reacting thiosalicylic acid with thiophenol as the starting materials in a concentrated sulfuric acid media. As revealed by laser flash photolysis studies, an intermolecular interaction between triplet 3TX-SH* and ground state TX-SH molecules results in the formation of thiyl radicals through consecutive electron transfer and hydrogen atom abstraction processes. Intramolecular interaction is unlikely to happen due to the rigidity of the spacer group between the carbonyl and thiol functionalities and therefore the dominant reaction is through an intermolecular hydrogen abstraction process (Scheme 12). Thiyl radicals are insensitive towards oxygen inhibition and can initiate polymerization in both the absence and presence of air. While initiating a polymerization process, TX functionalities are incorporated into the polymer chain and thus can further contribute in the photoinitiation processes.112 Another striking advantage of the TX-SH photoinitiator involves its applicability to styrene-based monomer formulations. Indeed, aromatic ketone/amine combinations appear to be effective photoinitiator systems for the polymerization of acrylates or methacrylates, whereas they have low reactivity toward styrenic monomers by virtue of the high quenching rate of the monomer and the reduced reactivity of the resulting α-amino radicals with styrene. It has been proven that the efficiency of the TX-SH photoinitiator for the polymerization of styrene is far higher than that with TX and amine co-initiator combinations.
 | ||
| Scheme 12 Photoinitiation mechanism of the formation of initiating radicals by 2-mercaptothioxanthone. | ||
TX-SH was also reported to behave as an efficient photosensitizer for Type I photoinitiators. As demonstrated by Jockusch, Arsu, and co-workers, the energy transfer between excited triplet TX-SH and acylphosphine oxide derivatives as α-cleavable Type I photoinitiators, leads to excitation of acylphosphine oxide photoinitiators and subsequently bond cleavage to yield initiating radicals. In addition to facilitating the energy transfer and excitation of acylphosphine oxide photoinitiators by improving the efficiency of light absorption, TX-SH serves as an oxygen insensitive species thus enabling photopolymerizations in the presence of air without any significant inhibitory effect of oxygen.113
The versatility of the synthesis route for TX derivatives has made it possible to incorporate desired functionalities into the structure of TX using functional aromatic compounds as starting materials. Carboxylic acid-functionalized TX photoinitiators have been prepared and utilized efficiently for free radical photopolymerization systems.114 Acting as a one-component Type II photoinitiator, the mechanism of the formation of initiating free radicals in carboxylic acid-functionalized TX occurs through the abstraction of the acidic hydrogen by the triplet excited state TX core, which further results in a decarboxylation process to evolve carbon dioxide yielding initiating radicals. Intermolecular hydrogen abstraction has been reported as the dominant reaction pathway (Scheme 13). This decarboxylation was proved visually by a model reaction employing a solution of sodium carbonate in the presence of phenolphthalein, which was connected to a tube containing carboxylic acid TX solution. The experiment was designed so that the evolving carbon dioxide gas resulting in decarboxylation on irradiation could be directed to the other tube containing sodium carbonate solution. The pink colored sodium carbonate solution turned into a colorless solution upon contact with the evolved carbon dioxide.
 | ||
| Scheme 13 Photoinitiation mechanism of the formation of initiating radicals by carboxylic acid-functionalized thioxanthone. | ||
Several other carboxylic acid-functionalized TX compounds able to act as one-component Type II photoinitiators have been reported with different spacer lengths capable of affecting the efficiency of photoinitiation process.115,116 For example, thioacetic acid TX was shown to undergo intramolecular electron transfer yielding an ion exciplex intermediate that resulted in hydrogen abstraction and subsequently decarboxylation processes.117 The flexibility of carboxylic acid moieties due to longer spacer length favors intramolecular electron and hydrogen transfer processes (Scheme 14). This is supported by the short triplet lifetime of thioacetic acid TX (65 ns) due to the involvement of intramolecular quenching processes, as compared to unsubstituted TX. It has been also found that the photoinitiation efficiency can be increased further by introducing a second acetic acid substituent to the photoinitiator.118
 | ||
| Scheme 14 Photoinitiation mechanism of thioacetic acid thioxanthone. | ||
Arsu and co-workers investigated the influence of substitution pattern and position on the photoinitiation activity of carboxylic acid-functionalized TX derivatives.119 Photoinitiators with carboxylic acid functionalities at the 1-position of TX appeared to be inefficient for photoinitiation of polymerization in certain environments. With these compounds, intramolecular hydrogen bonding results in rapid excited state quenching and deactivation. Additionally, it was observed that the nature of solvents could significantly affect the initiation mechanism. Apparently, using acetonitrile as the solvent, intramolecular hydrogen bonding occurs which prevents the possibility of any electron transfer and formation of initiating radicals. No polymerization occurred in this solvent. However, when the solvent was replaced with hydrogen-bond-disrupting solvents such as dimethylformamide or dimethyl sulfoxide, which form intermolecular hydrogen bonds, the polymerization efficiently proceeded through consecutive electron and proton transfer and decarboxylation processes. For reactions in acetonitrile, the addition of a small amount of hydrogen-bond-disrupting solvent to suppress intramolecular hydrogen bonding seemed necessary for successful polymerization.
On the other hand, photoinitiators with substitutions at the 2-position were reported to efficiently form initiating radicals and initiate polymerization process due to a sterically unfavorable intramolecular hydrogen-bonding interaction.
An anthracene incorporated TX (TX-A) exhibits a photoinitiation mechanism different from that of the other hydrogen-abstraction Type II photoinitiators.120,121 The anthracene moiety is a well-known photosensitizer by itself and when combined with TX, shifts the absorption maxima to higher wavelengths up to visible light. The proposed mechanism of the photoinitiation relies on the photoexcitation of the TX-A photoinitiator and quenching of the triplet excited state primarily by molecular oxygen to form singlet oxygen species. Singlet oxygen then reacts with the anthracene moiety of TX-A to generate an endoperoxide intermediate. The endoperoxide thus formed undergoes photochemical or thermal decomposition resulting in the formation of initiating radicals. Free radical polymerization of methacrylate and styrene based monomers were efficiently initiated using TX-A as one-component photoinitiator in the presence of oxygen. Scheme 15 depicts the photoinitiation mechanism by TX-A. The necessity of oxygen molecules for the TX-A photoinitiator to form free radicals makes this photoinitiator highly advantageous in terms of the possibility of overcoming oxygen inhibition problems in free radical systems. Substituted TX-A photoinitiators have also been synthesized and efficiently used in photopolymerization reactions.122,123
 | ||
| Scheme 15 Proposed photoinitiation mechanism by thioxanthone–anthracene photoinitiator in the presence of oxygen. | ||
A panchromatic behavior was observed when using thiosalicylic acids with N-phenylglycine to form a glycine-functionalized TX photoinitiator. Allonas, Arsu and co-workers demonstrated that the absorbance of the glycine-functionalized TX (Chart 6) was shifted to longer wavelengths up to 600 nm covering a wide range of the UV and visible light parts of the electromagnetic spectrum. This shift to longer wavelengths was attributed to the formation of hydrogen bonding facilitated by glycine functionalities. Photopolymerizations were successfully achieved using different light sources at 392, 473, 532, and 635 nm wavelengths with increasing photoinitiation efficiency with decreasing irradiation wavelength, consistent with increasing the absorption coefficient at shorter wavelengths.124
 | ||
| Chart 6 Glycine-functionalized thioxanthone showing panchromatic characteristics. | ||
Several other nitrogen-containing one-component photoinitiators bearing abstractable hydrogen sites have also been synthesized and utilized for the photoinitiation of polymerization (Chart 7).125–127
 | ||
| Chart 7 Some nitrogen-containing thioxanthone-based compounds as one-component Type II photoinitiators. | ||
Carbazole functionalities were also taken advantage of for forming one-component TX photoinitiators with extended conjugation. In this regard, for example, carbazole and ethylcarbazol functionalized TX photoinitiators (Chart 8) have been reported.84,128–130 Due to the extended conjugation contributed by the incorporation of carbazole moieties, these photoinitiators exhibit a strong absorption band in the visible region (>400 nm) where both TX and carbazole chromophores have no significant absorption characteristics. Bearing abstractable hydrogen sites, free radical polymerization was achieved using carbazole TX photoinitiators without the use of hydrogen donors; though supplying additional co-initiators contributed to enhancing the efficiency of the process. Ethylcarbazole-attached TX was reported to be highly soluble in a diverse range of both polar and nonpolar solvents due to the presence of the ethyl group of the carbazole functionality.
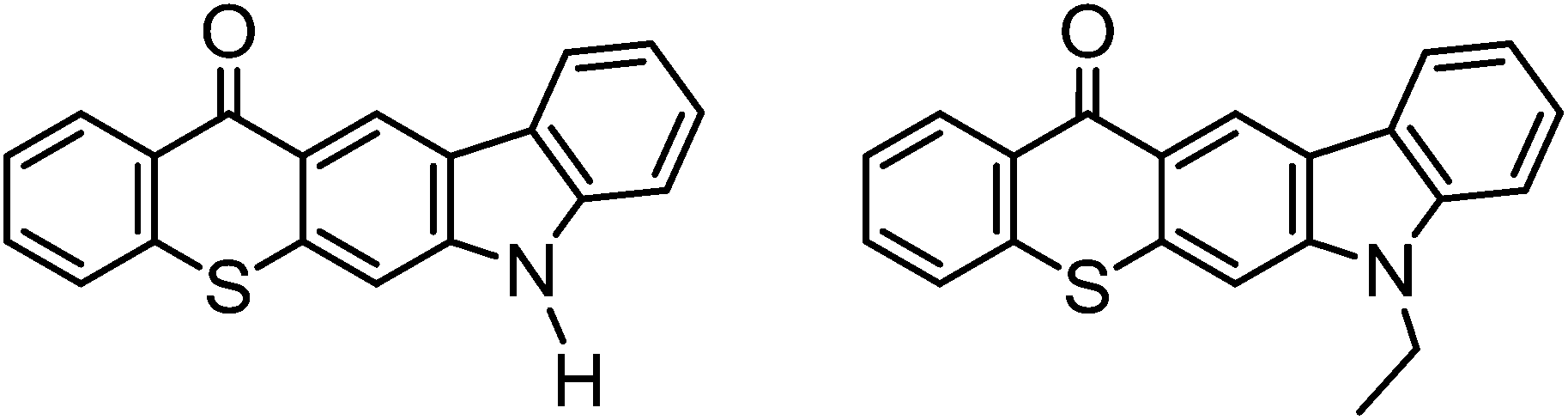 | ||
| Chart 8 Carbazole–thioxanthones as one-component photoinitiators. | ||
Additionally, free radical promoted cationic photopolymerization was achieved using ethylcarbazole TX in conjunction with onium salt-based oxidants. The free radicals formed in the course of the photolysis of carbazole TX interacted with the onium salt compounds while being oxidized to form the corresponding cationic species. Applicability of the initiation process to suitable cyclic and vinylic monomers was demonstrated.84,128
Water-soluble TX photoinitiators
In order for photoinitiators to be water compatible, water-solubilizing groups have to be incorporated in the skeleton of oil-soluble photoinitiators. Various methodologies have been proposed to introduce hydrophilic substituents to convert non-water-soluble photoinitiators to water-soluble. In this regard, ammonium derivatives, sulfonic and other acidic groups, or several water-soluble macromolecules, among others, have been successfully taken advantage of for making water-soluble photoinitiators (Chart 9).131 The photochemistry of these water-soluble photoinitiators is principally similar to their oil-soluble counterparts and they follow the same trend in interacting with co-initiators for the formation of initiating radicals.32 There might be some expected changes in the optical properties of water-solubilized photoinitiators causing extension of light absorption maxima to higher wavelengths.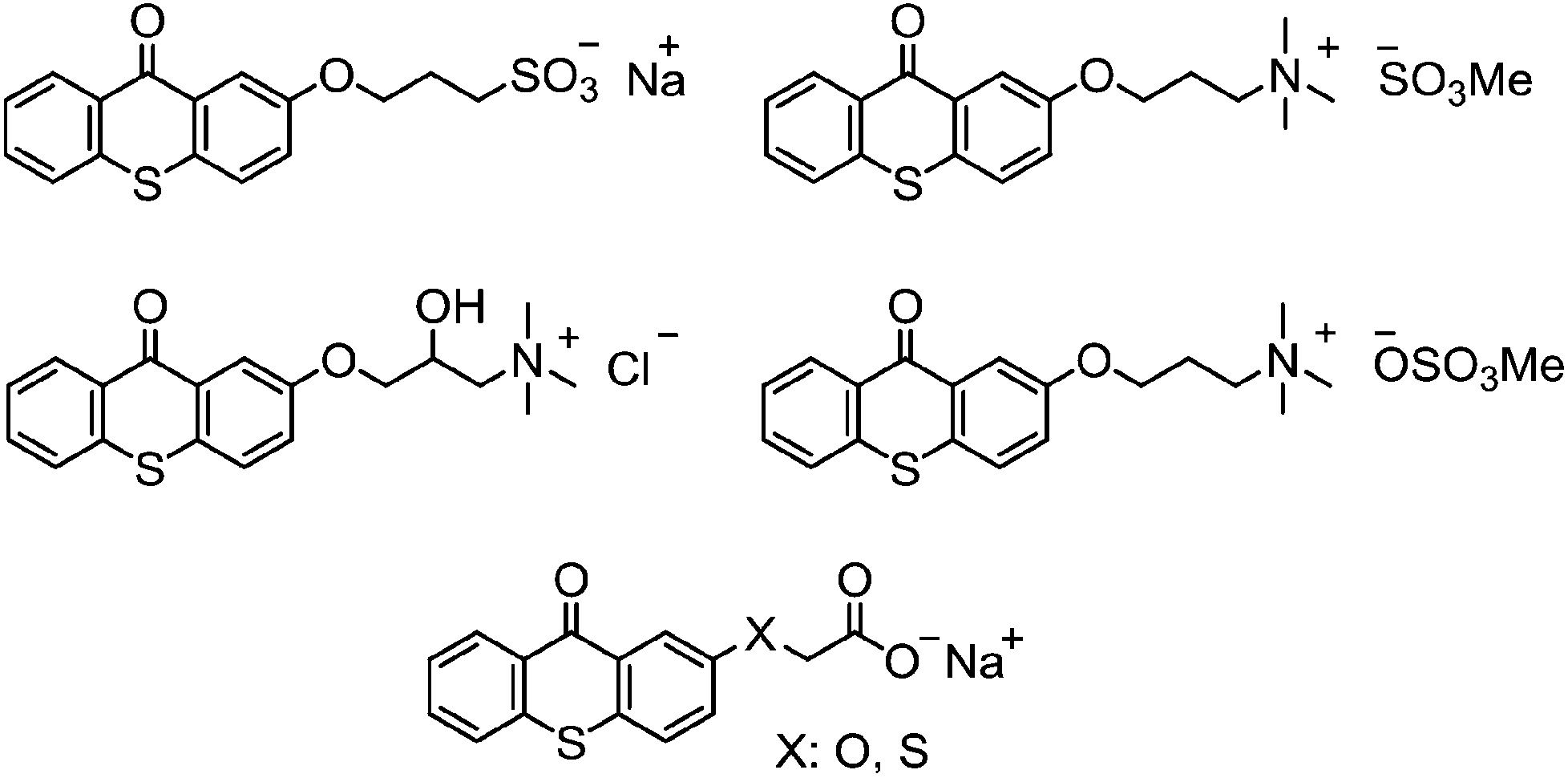 | ||
| Chart 9 A selection of some-water soluble thioxanthone-based photoinitiators. | ||
Complexation of photoinitiators with water-soluble agents has also been used to make water-soluble photoinitiators.132 Cyclodextrins consisting of a hydrophobic cavity and a hydrophilic outer space have been used to form host–guest complexes with oil-soluble photoinitiators.
Two-photon initiated photopolymerization
TX derivatives have also found their place in two-photon initiated polymerization systems. Two-photon initiated photopolymerizations are distinguished from commercial one-photon induced photopolymerizations by their distinctive ability to absorb the energy of two photons of the irradiating light as opposed to one-photon absorbing commercial photoinitiators. Two-photon photopolymerizations are especially unique and powerful techniques for the micro-fabrication of three-dimensional (3D) subtle, fine objects.Crucial for the highly efficient two-photon absorbing behavior of photoinitiators is the presence of electron-donor and/or electron-acceptor groups with extended conjugation properties. Malval et al. reported the enhancement of the two-photon absorption characteristics of TX by introducing an anthracene group to TX in a chevron-shaped architecture which in combination with an amine co-initiator was capable of exhibiting excellent two-photon absorption properties for the fabrication of 3D structures.133 Gryko and co-workers recently studied two-photon absorption characteristics of the π-expanded donor–acceptor photoinitiators of TX moieties.134 Dialkylamino groups as electron-donor components were substituted to TX with a significant enhancement of the optical properties. Those possessing arylethylene and arylethynyl linkages (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or C
C or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, respectively) showed favorable two-photon absorption properties with remarkable two-photon absorption cross section compared to the bare TX or non-conjugated photoinitiators.
C bonds, respectively) showed favorable two-photon absorption properties with remarkable two-photon absorption cross section compared to the bare TX or non-conjugated photoinitiators.
Macrophotoinitiators
To empower the (photo)activity of photoinitiators and incorporate different functionalities into their structure, an efficient way is to design and construct polymeric photoinitiators, also known as macrophotoinitiators.135 Polymeric photoinitiators bearing photoinitiator sites are prepared through various polymerization methods of suitable monomers, including step-growth136–141 and addition142 (co)polymerization, functionalization by click chemistry techniques, dendrimerization and other functionalization methodologies. In addition to their enhanced photoactivity by virtue of incorporating functional groups, the main idea of the formation of polymeric photoinitiators would probably deal with overcoming some of the disadvantages encountered with small molecule photoinitiators, which cause migration or volatility, yellowing and other drawbacks in the final cured products. Yin and co-workers reported a string of TX-containing macrophotoinitiators by step-growth polymerization methods. Containing in-chain TX and amine groups, these macrophotoinitiators were reported to act as one-component photoinitiators.138–140,143,144Using glucamine as the co-monomer, which contains tertiary amine groups as suitable hydrogen-donor sites, made the resulting TX macrophotoinitiators water-soluble.140 Examples of some TX macrophotoinitiators prepared by step-growth polymerization are collected in Chart 10.
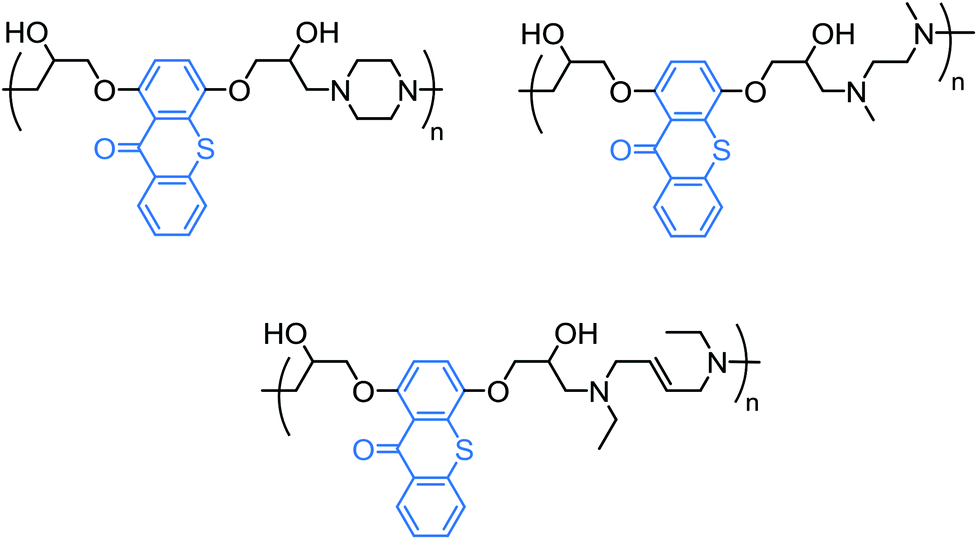 | ||
| Chart 10 A selection of thioxanthone-based, one-component polymeric photoinitiators prepared by step-growth polymerization techniques. | ||
Alternatively, functionalization of supramolecular or hyper-branched polymeric structures containing amine or etheric functional groups as hydrogen donating groups145 with photoinitiator moieties have been used to form one-component macrophotoinitiators.146,147 As an example, dendritic or hyperbranched supramolecular amines were reacted with an epoxy functional TX molecule resulting in the formation of dendritic structures with their TX end functional groups exhibiting photoinitiation activity much higher than small molecular weight photoinitiating systems.148–150 In this context, hyper-branched poly(ethylene imine) or dendritic poly(propylene imine) have been utilized for the formation of TX-bearing dendritic macrostructures (Scheme 16).
 | ||
| Scheme 16 Dendritic poly(propylene imine) end-functionalized with thioxanthone moieties as a one-component Type II macrophotoinitiator. | ||
Post-modification of polystyrene as a polymeric backbone by treating it with thiosalicylic acid, which leads to the thioxanthonation of polystyrene, has been reported to prepare macrophotoinitiators with pendant TX groups.151 Additionally, this process can bring about water-soluble polystyrene-based polymeric photoinitiators by performing a sulfonation during the thioxanthonation process in a one-pot manner.152 An amine co-initiator was necessary for polymerization to occur.
Click chemistry techniques, examples of which include copper-catalyzed azide–alkyne cycloaddition (CuAAC) or Diels–Alder click reactions, offer unique possibilities for the preparation of functional polymeric photoinitiators, which efficiently enable the incorporation of suitable photoinitiator sites into polymeric supports. For example, using both CuAAC and Diels–Alder techniques together, an alkyne-functionalized maleimide group was linked to a polystyrene backbone with side-chain azide groups using CuAAC technique (Scheme 17).153 The maleimide group possessing alkyne as well as protected norborene functionalities was used as a double-functional click linker to facilitate incorporation of TX groups to the azidated polystyrene. TX-A group was then clicked to the polystyrene through the maleimide linker using a Diels–Alder click reaction occurring between the antracene and norborene functionalities. The resulting TX-incorporated polystyrene with double click chemistry techniques showed similar optical characteristics to that of bare TX with excellent photoinitiation efficiency. However, different absorption characteristics compared to the precursor TX-A were observed probably due to the loss of the aromaticity of the anthracene group in the course of the Diels–Alder click reaction.
 | ||
| Scheme 17 Synthesis of polystyrene-based macrophotoinitiator with thioxanthone groups using double click reactions. | ||
Owing to its good solubility in aqueous media and its possession of etheric hydrogen abstraction sites in its structure, poly(ethylene glycol) (PEG) has been widely used as a building block to prepare macrophotoinitiators. Using Diels–Alder click chemistry, for instance, antracene-TX was linked to a maleimide-functionalized PEG support yielding a water-soluble, one-component polymeric photoinitiator (Scheme 18).154 A variety of water-soluble monomers were polymerized using this PEG-TX photoinitiator through a grafting method due to the formation of initiating free radicals onto the PEG backbone as a result of the etheric hydrogen abstraction process by the triplet state TX.
 | ||
| Scheme 18 Synthesis of thioxanthone end-functionalized poly(ethylene glycol) by Diels–Alder click reaction. | ||
Counterion incorporation of TX to poly(ethylene oxide) (PEO) support through a straightforward acid–base salt formation has also been reported. Reacting an α-amino functional PEO with a carboxylic acid functionalized TX group led to the formation of a polymeric salt with ionically attached TX groups (Scheme 19).155 Polymerization of water-soluble monomers was initiated upon counterion sensitization to triplet state TX abstracting a hydrogen atom from the PEO segment.
 | ||
| Scheme 19 Synthesis of thioxanthone end-functionalized poly(ethylene oxide) by counterion exchange. | ||
More recently, poly(vinyl alcohol) (PVA) was used as a polymeric support for the incorporation of a TX chromophore (Scheme 20).156 An aldehyde functional TX was initially synthesized and then linked to PVA through a simple acetylation process yielding PVA with pendant TX groups. Bearing several abstractable hydrogen sites, this macrophotoinitiator was reported to act as a one-component photoinitiator with efficient photoactivity and applicability in both organic and aqueous media for the photopolymerization of vinyl monomers including acrylamide and methyl methacrylate as model water soluble and oil-soluble monomers, respectively.
 | ||
| Scheme 20 Synthesis of poly(vinyl alcohol) thioxanthone macrophotoinitiator. | ||
Heterogeneous TX networks
Porous materials having high accessible active surface area have been widely used in a broad range of photocatalysis applications. With respect to polymerization processes, semiconducting nanoparticles or organic porous networks have been reported as heterogeneous photoinitiators for various modes of polymerization techniques.157–161 Given the heterogeneous nature of this type of macrophotoinitiator, which makes them easily separable from the reaction media eliminating any contamination problems associated with by-products, heterogeneous photochemical processes become of great importance. They often offer the advantage of being reused in promoting reactions multiple times while preserving their reactivity, which is a desirable and significant behavior from the point of view of economic and environmental issues.In this regard, the groups of Thomas and Yagci recently developed a new strategy to generate TX-based heterogeneous networks as potential macrophotoinitiators for conducting photopolymerization.162 Using various cross-coupling processes such as Sonogashira–Hagihara or Friedel–Crafts alkylation techniques (the latter being also referred to as “knitting” process), conjugated microporous networks of TX with specific surface areas of up to 750 m2 g−1 were obtained employing TX along with other suitable co-monomers. Dibromothioxanthone and triethynylbenzene were subjected to the Sonogashira–Hagihara coupling reaction to form a microporous network of TX with a pore size of 1.4 nm and a microporosity of 500 m2 g−1. Using the Friedel–Crafts alkylation method, TX and benzene (or triphenylmethane) were “knitted” together. It was found that these macrophotoinitiators being two or three-dimensional networks had strong absorption characteristics in the visible region which were reasoned to be due to the strong π interactions of the highly conjugated nature of the networks. Free radical and cationic photopolymerizations were achieved in the presence of different co-initiators under visible or sun light irradiation. In free radical polymerization of vinyl monomers, a hydrogen abstraction from the amine co-initiator produced an initiating free radical species whereas in the presence of an onium salt co-initiator (diphenyl iodonium hexafluoruphosphate, Ph2I+ PF6−) electron transfer reactions brought about the initiating species. In the latter case, as explained earlier, the photoexcited TX moieties reduce the onium salt through electron transfer reactions forming a diphenyl iodonium radical which further decomposes to give a phenyl radical capable of initiating polymerization of vinyl monomers. Ring-opening cationic polymerization of cyclic ethers was achieved by a free radical promoted cationic process in the presence of the iodonium salt and an amine (dimethylaniline) or an ether (tetrahydrofuran) hydrogen donor. The so-formed radical species via hydrogen abstraction processes were responsible for the reduction of the onium salt to generate suitable cationic species as well as the other above-mentioned mechanisms, which form active cationic components to initiate ring-opening cationic polymerization. Reusability was found for all three types of microporous TX networks in both free radical and cationic polymerizations.
Miscellaneous applications
In addition to their widespread use in photopolymerization systems, TX photoinitiators have found substantial applicability in other areas of materials science and (photo)chemistry. In this section, we briefly evaluate the fields in which the photochemical behavior of TX derivatives has been taken advantage of for carrying out target reactions.One interesting area concerns the photochemical formation of metal nanoparticles such as silver (Ag) or gold (Au) based on photochemical reduction of metal ions by electron-donor radicals from different photoinitiators. Malval et al. studied the photo-generation of silver nanoparticles by carboxylate derivatives of TX (TX–O–CH2–COO− Na+ and TX–S–CH2–COO− Na+) in the presence of amine hydrogen donors.163 While silver nanoparticles form by reduction of silver cations by photogenerated radicals as a result of interaction of triplet state TX with amine co-initiator, these nanoparticles can be capped by the carboxylate functionality of TXs acting as a ligand to stabilize nanoparticles. It was found that the substitution pattern had a significant impact on the formation, stability and morphology of the final nanoparticles. For example, when using a bare TX, a rapid aggregation of silver metal was observed. However, carboxylate-derivatives of TX suppressed this aggregation significantly owing to their ligand effect, uniformly capping the generated nanoparticles through carboxylate functional groups, which in turn resulted in the formation of uniform individual nanoparticles. This capping behavior was considerable in the case of TX–S–CH2–COO− as compared with TX–O–CH2–COO−, by which homogeneous silver nanoparticles with narrow size distributions were obtained. Similar redox processes can also be achieved by cleavage type photoinitiators providing that they yield electron donor radicals.164–166
Our group recently reported a new TX-based copper catalyst for the photochemical conduction of the CuAAC click reaction.167 CuAAC is known to be promoted by photochemical processes in which the required copper(I) catalyst for CuAAC is achieved by photoreduction of copper(II) species.28 Using a TX carboxylate which was converted to its sodium salt by treatment in sodium hydroxide media, copper(II) ions were bonded to the TX (Cu(TX)2) by an ion exchange process between TX sodium and copper(II) triflate (Scheme 21). The reduction of copper(II) species in the obtained Cu(TX)2 catalyst was observed by irradiation of the catalyst in a dimethylformamide solution without the use of any additional ligand or hydrogen donor compounds. The solubility of the catalyst was due to the solubility of the TX part, which in turn eliminated the need for additional ligands. This was attributed to the intramolecular photoinduced electron transfer reactions between the triplet TX and copper(II) ions which resulted in the reduction of copper(II). CuAAC was successfully catalyzed in a ligand-free manner under soft irradiation conditions using the Cu(TX)2 photocatalyst providing the advantage of temporarily controlling the process.
 | ||
| Scheme 21 Synthesis of copper(II) thioxanthone carboxylate photocatalyst (a) and its use as a ligand-free photocatalyst for copper(I)-catalyzed azide–alkyne cycloaddition (b). | ||
Alonso and Bach developed a photosensitization approach for the induction of enantioselective [2 + 2] photocyloaddition reactions using a chiral TX as the organocatalyst compound.168 They reported that the intramolecular [2 + 2] photocycloaddition of quinolones, which generally exhibit absorbance in the UV region around 300 nm, could be efficiently realized under visible light irradiation of TX by a triplet energy transfer process to the quinolones to sensitize their intermolecular cycloaddition (Scheme 22). The reactions were conducted in a non-hydrogen donating media (i.e. trifluorotoluene) so as to eliminate the probability of hydrogen abstraction and further decomposition of TX. After the reaction, though not completely, TX photoinitiator was successfully recovered signifying the preservation of TX functionality after the triplet charge transfer process.
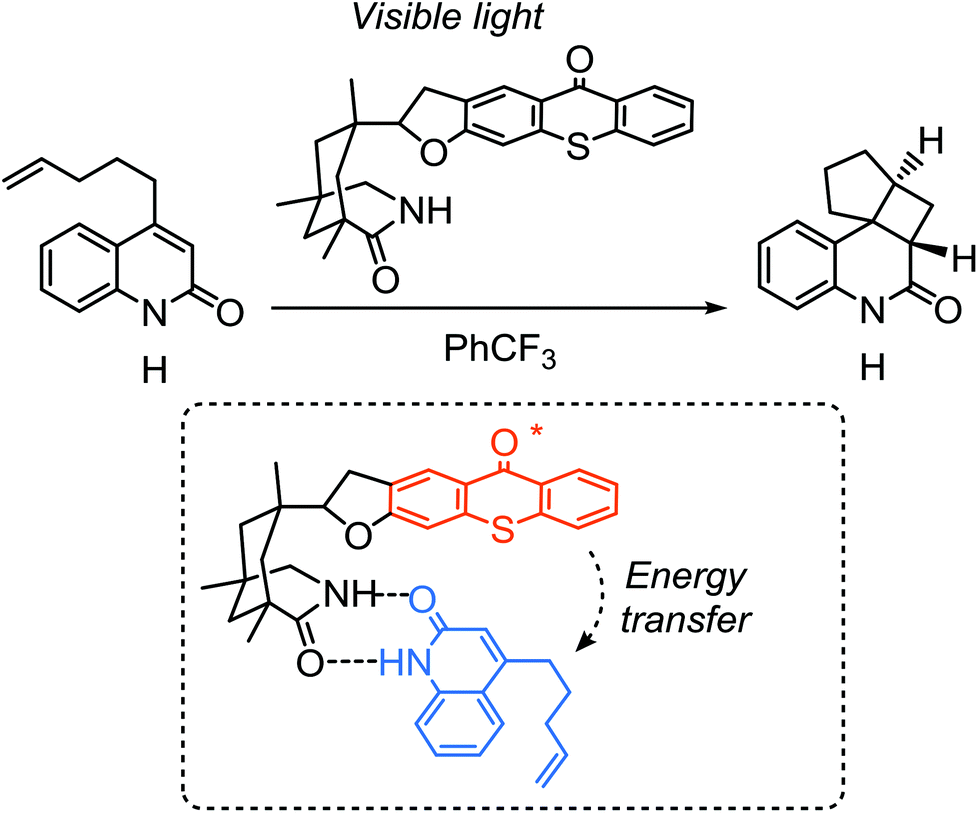 | ||
| Scheme 22 Photosensitization of [2 + 2] photocyloaddition by a chiral thioxanthone photosensitizer through energy transfer process. | ||
TX derivatives are also known for their excellent biological and pharmaceutical activities.169 Some biologically relevant applications concerning drug development, anti-cancer and tumor treatment170–172 or protein damage173 induced and mediated by TX photosensitizers have been reported in this context.
Conclusion
In this paper, we have reviewed recent advances of photopolymerizations initiated by TX derivatives. We have discussed in detail the conceptual aspects of the photochemistry of TX molecules, explaining how they would behave under light and interact with the additional reaction components (i.e. in the presence of co-initiators) to form suitable initiating sites of interest. In this regard, the applicability of various co-initiators in combination with TXs has been explained by providing a comprehensive picture of their mechanistic behavior. Developments of specially designed TX-based photoinitiators involving one- and two-component, water-soluble, and polymeric photoinitiators obtained through various processes have been exemplified for macromolecular synthesis. In addition, some miscellaneous applications of TX photoinitiators in promoting and photosensitizing other chemical reactions and biological events have been reviewed.The field of photopolymerization is gaining more and more research interest across a broad range of polymers and a wide realm of the chemistry and science communities. For more than three decades, our laboratory has been engaged in designing, developing, and taking advantages of photochemical strategies for the synthesis of polymers having specific characteristics. A great deal of effort has gone into “softening” polymerization conditions or enhancing the efficacy of photoinitiation reactions. This would be through, for instance, the use of photoinitiating systems insensitive to oxygen inhibition problems, especially in large-scale industrial applications or photoinitiating systems efficiently working in the visible range of the electromagnetic spectrum with newly emerging light sources like Light Emitting Diodes (LED) or natural sunlight, which would significantly contribute to lowering the cost of such processes as well as suppressing some of the disadvantages that high energy sources may cause to some special systems.
References
- A. Albini and M. Fagnoni, Green Chem., 2004, 6, 1–6 RSC.
- S. Protti and M. Fagnoni, Photochem. Photobiol. Sci., 2009, 8, 1499–1516 CAS.
- J. V. Crivello and E. Reichmanis, Chem. Mater., 2014, 26, 533–548 CrossRef CAS.
- R. S. Davidson, Exploring the Science, Technology and Applications of U.V. and E.B. Curing, SITA Technology Ltd., London, 1998 Search PubMed.
- J.-P. Fouassier and J. Lalevee, Photoinitiators for Polymer Synthesis: Scope, Reactivity and Efficiency, Wiley-VCH, Weinheim, 2012 Search PubMed.
- J. V. Crivello and K. Dietliker, Photoinitiators for free radical Cationic and anionic photopolymerization, John Wiley & Sons, Chichester, 1998 Search PubMed.
- P. Xiao, J. Zhang, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevee, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS PubMed.
- J. Z. Shao, Y. Huang and Q. U. Fan, Polym. Chem., 2014, 5, 4195–4210 RSC.
- Y. Yagci and I. Reetz, Prog. Polym. Sci., 1998, 23, 1485–1538 CrossRef CAS.
- Y. Yagci and W. Schnabel, Macromol. Symp., 1988, 13–4, 161–174 CrossRef PubMed.
- C. Kutal, P. A. Grutsch and D. B. Yang, Macromolecules, 1991, 24, 6872–6873 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 2271–2275 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- V. Nikolaou, A. Anastasaki, F. Alsubaie, A. Simula, D. J. Fox and D. M. Haddleton, Polym. Chem., 2015, 6, 3581–3585 RSC.
- D. Konkolewicz, K. Schroeder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Chem. Sci., 2015, 6, 1341–1349 RSC.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D. L. Versace, J. Lalevee and J. P. Fouassier, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- M. A. Tasdelen, M. Ciftci, M. Uygun and Y. Yagci, in Progress in Controlled Radical Polymerization: Mechanisms and Techniques, ed. K. Matyjaszewski, B. S. Sumerlin and N. V. Tsarevsky, 2012, vol. 1100, pp. 59–72 Search PubMed.
- S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981–994 CrossRef CAS PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- A. J. Perkowski, W. You and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 7580–7583 CrossRef CAS PubMed.
- S. Dadashi-Silab, M. A. Tasdelen and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2878–2888 CrossRef CAS PubMed.
- J. E. Poelma, B. P. Fors, G. F. Meyers, J. W. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 6844–6848 CrossRef CAS PubMed.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- M. A. Tasdelen and Y. Yagci, Aust. J. Chem., 2011, 64, 982–991 CrossRef CAS.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- H. F. Gruber, Prog. Polym. Sci., 1992, 17, 953–1044 CrossRef CAS.
- S. Archer and C. M. Suter, J. Am. Chem. Soc., 1952, 74, 4296–4309 CrossRef CAS.
- G. Amirzadeh and W. Schnabel, Macromol. Chem. Phys., 1981, 182, 2821–2835 CrossRef CAS PubMed.
- N. S. Allen, F. Catalina, P. N. Green and W. A. Green, Eur. Polym. J., 1986, 22, 347–350 CrossRef CAS.
- N. S. Allen, F. Catalina, P. N. Green and W. A. Green, Eur. Polym. J., 1986, 22, 793–799 CrossRef CAS.
- N. S. Allen, F. Catalina, P. N. Green and W. A. Green, Eur. Polym. J., 1986, 22, 871–875 CrossRef CAS.
- N. S. Allen, F. Catalina, P. N. Green and W. A. Green, J. Photochem., 1987, 36, 99–112 CrossRef CAS.
- N. S. Allen, F. Catalina, J. Lucgardette, P. N. Green, W. A. Green and O. Fatinikun, J. Oil Colour Chem. Assoc., 1987, 70, 332–336 CAS.
- N. S. Allen, F. Catalina, B. Moghaddam, P. N. Green and W. A. Green, Eur. Polym. J., 1986, 22, 691–697 CrossRef CAS.
- N. S. Allen, F. Catalina, P. N. Green and W. A. Green, Eur. Polym. J., 1985, 21, 841–848 CrossRef CAS.
- N. S. Allen, N. G. Salleh, M. Edge, M. Shah, C. Ley, F. Morlet-Savary, J. P. Fouassier, F. Catalina, A. Green, S. Navaratnam and B. J. Parsons, Polymer, 1999, 40, 4181–4193 CrossRef CAS.
- T. Corrales, C. Peinado, F. Catalina, M. G. Neumann, N. S. Allen, A. M. Rufs and M. V. Encinas, Polymer, 2000, 41, 9103–9109 CrossRef CAS.
- S. G. Cohen, A. Parola and G. H. Parsons, Chem. Rev., 1973, 73, 141–161 CrossRef CAS.
- M. V. Encinas, A. M. Rufs, T. Corrales, F. Catalina, C. Peinado, K. Schmith, M. G. Neumann and N. S. Allen, Polymer, 2002, 43, 3909–3913 CrossRef CAS.
- D. G. Anderson, R. S. Davidson and J. J. Elvery, Polymer, 1996, 37, 2477–2484 CrossRef CAS.
- S. F. Yates and G. B. Schuster, J. Org. Chem., 1984, 49, 3349–3356 CrossRef CAS.
- M. A. Tasdelen, B. Kiskan and Y. Yagci, Macromol. Rapid Commun., 2006, 27, 1539–1544 CrossRef CAS PubMed.
- N. N. Ghosh, B. Kiskan and Y. Yagci, Prog. Polym. Sci., 2007, 32, 1344–1391 CrossRef CAS PubMed.
- C. R. Morgan, F. Magnotta and A. D. Ketley, J. Polym. Sci., Part A: Polym. Chem., 1977, 15, 627–645 CrossRef CAS PubMed.
- S. Inbar, H. Linschitz and S. G. Cohen, J. Am. Chem. Soc., 1982, 104, 1679–1682 CrossRef CAS.
- S. C. Ligon, B. Husar, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 557–589 CrossRef CAS PubMed.
- P. G. Stone and S. G. Cohen, J. Am. Chem. Soc., 1982, 104, 3435–3440 CrossRef CAS.
- E. Andrzejewska, D. Zych-Tomkowiak, M. Andrzejewski, G. L. Hug and B. Marciniak, Macromolecules, 2006, 39, 3777–3785 CrossRef CAS.
- E. Andrzejewska, D. Zych-Tomkowiak, M. B. Bogacki and M. Andrzejewski, Macromolecules, 2004, 37, 6346–6354 CrossRef CAS.
- S. Suzuki, P. Emilie, T. Urano, S. Takahara and T. Yamaoka, Polymer, 2005, 46, 2238–2243 CrossRef CAS PubMed.
- V. V. Krongauz and C. P. Chawla, Polymer, 2003, 44, 3871–3876 CrossRef CAS.
- J. Lalevee, L. Zadoina, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2494–2502 CrossRef CAS PubMed.
- J. Lalevee, F. Morlet-Savary, M. El Roz, X. Allonas and J. P. Fouassier, Macromol. Chem. Phys., 2009, 210, 311–319 CrossRef CAS PubMed.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Macromolecules, 2003, 36, 7964–7969 CrossRef CAS.
- N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3311–3319 CrossRef CAS PubMed.
- C. R. Morgan, F. Magnotta and A. D. Ketley, J. Polym. Sci., Part A: Polym. Chem., 1977, 15, 627–645 CrossRef CAS PubMed.
- C. Decker and T. N. T. Viet, Macromol. Chem. Phys., 1999, 200, 1965–1974 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS PubMed.
- E. C. Lathioor and W. J. Leigh, Photochem. Photobiol., 2006, 82, 291–300 CrossRef CAS PubMed.
- W. J. Leigh, E. C. Lathioor and M. J. StPierre, J. Am. Chem. Soc., 1996, 118, 12339–12348 CrossRef CAS.
- M. Yamaji, J. Oshima and M. Hidaka, Chem. Phys. Lett., 2009, 475, 235–239 CrossRef CAS PubMed.
- D. Das and D. N. Nath, J. Phys. Chem. A, 2008, 112, 11619–11626 CrossRef CAS PubMed.
- M. Dossot, M. Sylla, X. Allonas, A. Merlin, P. Jacques and J. P. Fouassier, J. Appl. Polym. Sci., 2000, 78, 2061–2074 CrossRef CAS.
- A. Valdebenito and M. V. Encinas, J. Photochem. Photobiol., A, 2008, 194, 206–211 CrossRef CAS PubMed.
- J. Lalevee, F. Morlet-Savary, M. A. Tehfe, B. Graff and J. P. Fouassier, Macromolecules, 2012, 45, 5032–5039 CrossRef CAS.
- M. El-Roz, J. Lalevee, X. Allonas and J. P. Fouassier, Macromol. Rapid Commun., 2008, 29, 804–808 CrossRef CAS PubMed.
- J. Lalevee, X. Allonas and J. P. Fouassier, J. Org. Chem., 2007, 72, 6434–6439 CrossRef CAS PubMed.
- J. Lalevee and J. P. Fouassier, Polym. Chem., 2011, 2, 1107–1113 RSC.
- M. El-Roz, J. Lalevee, F. Morlet-Savary, X. Allonas and J. P. Fouassier, Macromolecules, 2009, 42, 4464–4469 CrossRef CAS.
- D. L. Versace, M. A. Tehfe, J. Lalevee, V. Casarotto, N. Blanchard, F. Morlet-Savary and J.-P. Fouassier, J. Phys. Org. Chem., 2011, 24, 342–350 CrossRef CAS PubMed.
- J. Lalevee, N. Blanchard, B. Graff, X. Allonas and J. P. Fouassier, J. Organomet. Chem., 2008, 693, 3643–3649 CrossRef CAS PubMed.
- M. El-Roz, M.-A. Tehfe, J. Lalevee, B. Graff, X. Allonas and J. P. Fouassier, Macromolecules, 2010, 43, 2219–2227 CrossRef CAS.
- J. Lalevee, M. El-Roz, F. Morlet-Savary, B. Graff, X. Allonas and J. P. Fouassier, Macromolecules, 2007, 40, 8527–8530 CrossRef CAS.
- M. A. Tehfe, M. El-Roz, J. Lalevee, F. Morlet-Savary, B. Graff and J. P. Fouassier, Eur. Polym. J., 2012, 48, 956–962 CrossRef PubMed.
- J. Lalevee, A. Dirani, M. El-Roz, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3042–3047 CrossRef CAS PubMed.
- M. El-Roz, J. Lalevee, X. Allonas and J. P. Fouassier, Macromolecules, 2009, 42, 8725–8732 CrossRef CAS.
- B. Aydogan, Y. Y. Durmaz, M. U. Kahveci, M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Symp., 2011, 308, 25–34 CrossRef CAS PubMed.
- Y. Y. Durmaz, N. Moszner and Y. Yagci, Macromolecules, 2008, 41, 6714–6718 CrossRef CAS.
- Y. Yagci, Y. Y. Durmaz and B. Aydogan, Chem. Rec., 2007, 7, 78–90 CrossRef CAS PubMed.
- G. Yilmaz, S. Beyazit and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1591–1596 CrossRef CAS PubMed.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- J. V. Crivello and J. L. Lee, Macromolecules, 1981, 14, 1141–1147 CrossRef CAS.
- G. Manivannan, J. P. Fouassier and J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1999–2001 CrossRef CAS PubMed.
- Y. Yagci, I. Lukac and W. Schnabel, Polymer, 1993, 34, 1130–1133 CrossRef CAS.
- J. P. Fouassier, D. Burr and J. V. Crivello, J. Macromol. Sci., Pure Appl. Chem., 1994, A31, 677–701 CrossRef CAS PubMed.
- S. Denizligil, R. Resul, Y. Yagci, C. McArdle and J. P. Fouassier, Macromol. Chem. Phys., 1996, 197, 1233–1240 CrossRef CAS PubMed.
- A. Kunze, U. Muller, K. Tittes, J. P. Fouassier and F. MorletSavary, J. Photochem. Photobiol., A, 1997, 110, 115–122 CrossRef CAS.
- J. V. Crivello and M. Jang, J. Photochem. Photobiol., A, 2003, 159, 173–188 CrossRef CAS.
- M. R. Rodrigues and M. G. Neumann, Macromol. Chem. Phys., 2001, 202, 2776–2782 CrossRef CAS.
- M. R. Rodrigues and M. G. Neumann, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 46–55 CrossRef CAS.
- M. U. Kahveci, M. A. Tasdelen and Y. Yagci, Polymer, 2007, 48, 2199–2202 CrossRef CAS PubMed.
- D. Dossow, Q. Q. Zhu, G. Hizal, Y. Yagci and W. Schnabel, Polymer, 1996, 37, 2821–2826 CrossRef CAS.
- J. P. Fouassier and J. Lalevee, RSC Adv., 2012, 2, 2621–2629 RSC.
- J. P. Fouassier, D. Ruhlmann, Y. Takimoto, M. Harada and M. Kawabata, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2245–2248 CrossRef CAS PubMed.
- D. Kim and J. W. Stansbury, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 887–898 CrossRef CAS PubMed.
- A. Erddalane, J. P. Fouassier, F. MorletSavary and Y. Takimoto, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 633–642 CrossRef CAS.
- J. P. Fouassier, X. Allonas, J. Lalevee and M. Visconti, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4531–4541 CrossRef CAS.
- Y. Yagci and Y. Hepuzer, Macromolecules, 1999, 32, 6367–6370 CrossRef CAS.
- K. S. Padon and A. B. Scranton, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2057–2066 CrossRef CAS.
- K. Kawamura, C. Ley, J. Schmitt, M. Barnet and X. Allonas, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4325–4330 CrossRef CAS PubMed.
- W. D. Cook and F. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5030–5041 CrossRef CAS PubMed.
- W. D. Cook and F. Chen, Polym. Chem., 2015, 6, 1325–1338 RSC.
- W. D. Cook, S. H. Chen, F. Chen, M. U. Kahveci and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5474–5487 CrossRef CAS PubMed.
- J. Lalevee, M. El-Roz, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2008–2014 CrossRef CAS PubMed.
- L. Cokbaglan, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2003, 36, 2649–2653 CrossRef CAS.
- F. Scigalski and K. Jankowski, Polym. Bull., 2015, 72, 255–263 CrossRef CAS.
- S. Niu, R. Schneider, L. Vidal, S. Hajjar-Garreau and L. Balan, J. Nanopart. Res., 2014, 16 Search PubMed.
- F. Karasu, N. Arsu and Y. Yagci, J. Appl. Polym. Sci., 2007, 103, 3766–3770 CrossRef CAS PubMed.
- S. Keskin, S. Jockusch, N. J. Turro and N. Arsu, Macromolecules, 2008, 41, 4631–4634 CrossRef CAS.
- D. S. Esen, G. Temel, D. K. Balta, X. Allonas and N. Arsu, Photochem. Photobiol., 2014, 90, 463–469 CrossRef CAS PubMed.
- M. Aydin, N. Arsu and Y. Yagci, Macromol. Rapid Commun., 2003, 24, 718–723 CrossRef CAS PubMed.
- G. Yilmaz, B. Aydogan, G. Temel, N. Arsu, N. Moszner and Y. Yagci, Macromolecules, 2010, 43, 4520–4526 CrossRef CAS.
- M. Aydin, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2005, 38, 4133–4138 CrossRef CAS.
- F. Karasu, N. Arsu, S. Jockusch and N. J. Turro, Macromolecules, 2009, 42, 7318–7323 CrossRef CAS.
- F. Karasu, N. Arsu, S. Jockusch and N. J. Turro, J. Org. Chem., 2013, 78, 9161–9165 CrossRef CAS PubMed.
- D. K. Balta, N. Arsu, Y. Yagci, A. K. Sundaresan, S. Jockusch and N. J. Turro, Macromolecules, 2011, 44, 2531–2535 CrossRef CAS.
- D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- D. K. Balta and N. Arsu, J. Photochem. Photobiol., A, 2013, 257, 54–59 CrossRef CAS PubMed.
- D. K. Balta, G. Temel, G. Goksu, N. Ocal and N. Arsu, Macromolecules, 2012, 45, 119–125 CrossRef CAS.
- H. Tar, D. S. Esen, M. Aydin, C. Ley, N. Arsu and X. Allonas, Macromolecules, 2013, 46, 3266–3272 CrossRef CAS.
- Q. Wu, Y. Xiong, Q. Liang and H. Tang, RSC Adv., 2014, 4, 52324–52331 RSC.
- S. K. Dogruyol, Z. Dogruyol and N. Arsu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4037–4043 CrossRef CAS PubMed.
- S. K. Dogruyol, Z. Dogruyol and N. Arsu, J. Lumin., 2013, 138, 98–104 CrossRef CAS PubMed.
- D. Tunc and Y. Yagci, Polym. Chem., 2011, 2, 2557–2563 RSC.
- G. Yilmaz, A. Tuzun and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5120–5125 CrossRef CAS PubMed.
- N. Karaca, D. K. Balta, N. Ocal and N. Arsu, J. Lumin., 2014, 146, 424–429 CrossRef CAS PubMed.
- D. J. Lougnot, C. Turck and J. P. Fouassier, Macromolecules, 1989, 22, 108–116 CrossRef CAS.
- D. K. Balta, E. Bagdatli, N. Arsu, N. Ocal and Y. Yagci, J. Photochem. Photobiol., A, 2008, 196, 33–37 CrossRef CAS PubMed.
- J.-P. Malval, M. Jin, F. Morlet-Savary, H. Chaumeil, A. Defoin, O. Soppera, T. Scheul, M. Bouriau and P. L. Baldeck, Chem. Mater., 2011, 23, 3411–3420 CrossRef CAS.
- R. Nazir, E. Balčiūnas, D. Buczyńska, F. Bourquard, D. Kowalska, D. Gray, S. Maćkowski, M. Farsari and D. T. Gryko, Macromolecules, 2015, 48, 2466–2472 CrossRef CAS.
- R. S. Davidson, J. Photochem. Photobiol., A, 1993, 69, 263–275 CrossRef.
- J. Wei, H. Wang, X. Jiang and J. Yin, Macromol. Chem. Phys., 2006, 207, 1752–1763 CrossRef CAS PubMed.
- J. Wei, H. Wang and J. Yin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 576–587 CrossRef CAS PubMed.
- X. S. Jiang and J. Yin, Macromol. Rapid Commun., 2004, 25, 748–752 CrossRef CAS PubMed.
- X. S. Jiang and H. Yin, Polymer, 2004, 45, 5057–5063 CrossRef CAS PubMed.
- X. Jiang and J. Yin, Macromol. Chem. Phys., 2008, 209, 1593–1600 CrossRef CAS PubMed.
- X. S. Jiang, X. W. Luo and J. Yin, J. Photochem. Photobiol., A, 2005, 174, 165–170 CrossRef CAS PubMed.
- H. Wang, J. Wei, X. Jiang and J. Yin, J. Photochem. Photobiol., A, 2007, 186, 106–114 CrossRef CAS PubMed.
- X. S. Jiang, H. J. Xu and H. Yin, Polymer, 2004, 45, 133–140 CrossRef CAS PubMed.
- X. S. Jiang, J. Luo and J. Yin, Polymer, 2009, 50, 37–41 CrossRef CAS PubMed.
- M. A. Tasdelen, A. L. Demirel and Y. Yagci, Eur. Polym. J., 2007, 43, 4423–4430 CrossRef CAS PubMed.
- Y. Chen, J. Loccufier, L. Vanmaele and H. Frey, J. Mater. Chem., 2007, 17, 3389–3392 RSC.
- Y. Chen, J. Loccufier, L. Vanmaele, E. Barriau and H. Frey, Macromol. Chem. Phys., 2007, 208, 1694–1706 CrossRef CAS PubMed.
- X. S. Jiang and J. Yin, Macromolecules, 2004, 37, 7850–7853 CrossRef CAS.
- Y. Wen, X. Jiang, R. Liu and J. Yin, Polymer, 2009, 50, 3917–3923 CrossRef CAS PubMed.
- X. Jiang, W. Wang, H. Xu and J. Yin, J. Photochem. Photobiol., A, 2006, 181, 233–237 CrossRef CAS PubMed.
- G. Temel, N. Arsu and Y. Yagci, Polym. Bull., 2006, 57, 51–56 CrossRef CAS.
- G. Temel and N. Arsu, J. Photochem. Photobiol., A, 2009, 202, 63–66 CrossRef CAS PubMed.
- B. Gacal, H. Akat, D. K. Balta, N. Arsu and Y. Yagci, Macromolecules, 2008, 41, 2401–2405 CrossRef CAS.
- H. Akat, B. Gacal, D. K. Balta, N. Arsu and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2109–2114 CrossRef CAS PubMed.
- G. Yilmaz, G. Acik and Y. Yagci, Macromolecules, 2012, 45, 2219–2224 CrossRef CAS.
- S. Kork, G. Yilmaz and Y. Yagci, Macromol. Rapid Commun., 2015, 36, 923–928 CrossRef CAS PubMed.
- B. Kiskan, J. S. Zhang, X. C. Wang, M. Antonietti and Y. Yagci, ACS Macro Lett., 2012, 1, 546–549 CrossRef CAS.
- S. Dadashi-Silab, M. A. Tasdelen, B. Kiskan, X. Wang, M. Antonietti and Y. Yagci, Macromol. Chem. Phys., 2014, 215, 675–681 CrossRef CAS PubMed.
- S. Dadashi-Silab, M. A. Tasdelen, A. M. Asiri, S. B. Khan and Y. Yagci, Macromol. Rapid Commun., 2014, 35, 454–459 CrossRef CAS PubMed.
- S. Dadashi-Silab, A. M. Asiri, S. B. Khan, K. A. Alamry and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1500–1507 CrossRef CAS PubMed.
- S. Dadashi-Silab, Y. Yar, H. Yagci Acar and Y. Yagci, Polym. Chem., 2015, 6, 1918–1922 RSC.
- S. Dadashi-Silab, H. Bildirir, R. Dawson, A. Thomas and Y. Yagci, Macromolecules, 2014, 47, 4607–4614 CrossRef CAS.
- J.-P. Malval, M. Jin, L. Balan, R. Schneider, D.-L. Versace, H. Chaumeil, A. Defoin and O. Soppera, J. Phys. Chem. C, 2010, 114, 10396–10402 CAS.
- Y. Yagci, M. Sangermano and G. Rizza, Chem. Commun., 2008, 2771–2773 RSC.
- M. Sangermano, Y. Yagci and G. Rizza, Macromolecules, 2007, 40, 8827–8829 CrossRef CAS.
- J. Amici, M. Sangermano, E. Celasco and Y. Yagci, Eur. Polym. J., 2011, 47, 1250–1255 CrossRef CAS PubMed.
- S. Dadashi-Silab and Y. Yagci, Submitted.
- R. Alonso and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 4368–4371 CrossRef CAS PubMed.
- A. M. Paiva, M. M. Pinto and E. Sousa, Curr. Med. Chem., 2013, 20, 2438–2457 CrossRef CAS.
- T. H. Corbett, C. Panchapor, L. Polin, N. Lowichik, S. Pugh, K. White, J. Kushner, J. Meyer, J. Czarnecki, S. Chinnukroh, M. Edelstein, P. LoRusso, L. Heilbrun, J. P. Horwitz, C. Grieshaber, R. Perni, M. Wentland, S. Coughlin, S. Elenbaas, R. Philion and J. Rake, Invest. New Drugs, 1999, 17, 17–27 CrossRef CAS.
- J. P. Stevenson, D. DeMaria, D. Reilly, J. D. Purvis, M. A. Graham, G. Lockwood, M. Drozd and P. J. O'Dwyer, Cancer Chemother. Pharmacol., 1999, 44, 228–234 CrossRef CAS PubMed.
- E. Izbicka, R. Lawrence, K. Davidson, J. B. Rake and D. D. Von Hoff, Invest. New Drugs, 1998, 16, 221–225 CrossRef CAS.
- H. P. Zhu, W. F. Wang and S. D. Yao, Invest. New Drugs, 2006, 24, 465–470 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |